

Abstract
Anti‐myelin‐associated glycoprotein (MAG) neuropathy is a disabling autoimmune peripheral neuropathy that is caused by circulating monoclonal IgM autoantibodies directed against the human natural killer‐1 (HNK‐1) epitope. This carbohydrate epitope is highly expressed on adhesion molecules such as MAG, a glycoprotein present in myelinated nerves. We previously showed the therapeutic potential of the glycopolymer poly(phenyl disodium 3‐O‐sulfo‐β‐d‐glucopyranuronate)‐(1→3)‐β‐d‐galactopyranoside (PPSGG) in selectively neutralizing anti‐MAG IgM antibodies in an immunological mouse model and ex vivo with sera from anti‐MAG neuropathy patients. PPSGG is composed of a biodegradable backbone that multivalently presents a mimetic of the HNK‐1 epitope. In this study, we further explored the pharmacodynamic properties of the glycopolymer and its ability to inhibit the binding of anti‐MAG IgM to peripheral nerves. The polymer selectively bound anti‐MAG IgM autoantibodies and prevented the binding of patients’ anti‐MAG IgM antibodies to myelin of non‐human primate sciatic nerves. Upon PPSGG treatment, neither activation nor inhibition of human and murine peripheral blood mononuclear cells nor alteration of systemic inflammatory markers was observed in mice or ex vivo in human peripheral blood mononuclear cells. Intravenous injections of PPSGG to mice immunized against the HNK‐1 epitope removed anti‐MAG IgM antibodies within less than 1 hr, indicating a fast and efficient mechanism of action as compared to a B‐cell depletion with anti‐CD20. In conclusion, these observations corroborate the therapeutic potential of PPSGG for an antigen‐specific treatment of anti‐MAG neuropathy.

Read the Editorial Highlight for this article on page 465.
Keywords: antigen‐specific treatment, anti‐MAG IgM autoantibodies, demyelinating peripheral neuropathy, monoclonal gammopathy of neurological significance, myelin‐associated glycoprotein
Anti‐MAG (myelin‐associated glycoprotein) neuropathy is a disabling autoimmune peripheral neuropathy that is caused by circulating monoclonal IgM autoantibodies directed against the HNK‐1 (human natural killer‐1) epitope. This glycoepitope is highly expressed on adhesion molecules such as MAG and is localized mainly in paranodal loops and Schmidt–Lantermann incisures of the peripheral nervous system. The glycopolymer PPSGG selectively bound anti‐MAG IgM autoantibodies and prevented the binding of patients’ autoantibodies to myelin of primate sciatic nerves in the IFA (indirect fluorescents assay) anti‐MAG IgM assay (right panel). These findings corroborate the therapeutic potential of PPSGG for an antigen‐specific treatment of anti‐MAG neuropathy.

Read the Editorial Highlight for this article on page 465.
Abbreviations
- BSA
bovine serum albumin
- BTU
Bühlmann titer units
- CANOMAD
chronic ataxic neuropathy ophthalmoplegia M‐protein agglutination disialosyl antibodies syndrome
- CIDP
chronic inflammatory demyelinating polyneuropathy
- DC
dendritic cells
- EBV
Epstein–Barr virus
- ELISA
enzyme‐linked immunosorbent assay
- FBS
fetal bovine serum
- HNK‐1
human natural killer‐1
- LPS
lipopolysaccharide
- MAG
myelin‐associated glycoprotein
- MFI
mean fluorescence intensity
- MGUS
monoclonal gammopathy of unknown significance
- PBMC
peripheral blood mononuclear cells
- PBS
phosphate‐buffered saline
- PPSGG
poly(phenyl disodium 3‐O‐sulfo‐β‐d‐glucopyranuronate)‐(1→3)‐β‐d‐galactopyranoside
- RPMI
Roswell Park Memorial Institute medium
- SGPG/SGLPG
sulfoglucuronyl paragloboside/sulfoglucuronyl‐lactosaminyl‐paragloboside
1. INTRODUCTION
Anti‐myelin‐associated glycoprotein (MAG) neuropathy is a rare form of acquired demyelinating polyneuropathy associated with IgM monoclonal gammopathy of neurological significance commonly referred as monoclonal gammopathy of undetermined significance (Talamo, Mir, Mir, Pandey, Sivik, & Raheja, 2015). Typically, the disease onset occurs after the age of 50 years and is 2.7 times more frequent in men than in women (Vallat et al. 2016) with a prevalence of about 1 in 100,000 (Heesters, van der Poel, Poel, Das, & Carroll, 2016; Mahdi‐Rogers & Hughes, 2014; Mygland & Monstad, 2001; Talamo et al., 2015). This gammopathy of neurological significance leads to the production of monoclonal anti‐MAG IgM antibodies that recognize the human natural killer‐1 (HNK‐1) epitope, which is highly expressed on adhesion molecules such as MAG in the peripheral nervous system (Quarles, 2007; Steck, Stalder, Stalder, & Renaud, 2006). MAG, a member of the sialic acid‐binding Ig‐like lectins (siglecs), is located in the periaxonal membranes of oligodendroglial cells of the central nervous system and in Schwann cells of the peripheral nervous system (PNS), where it is localized in the paranodal loops and Schmidt‐Lanterman incisures (Dalakas, 2010; Erb et al., 2006; Kelm et al., 1994). MAG is a mediator for the formation and maintenance of the myelin sheaths (Tang et al., 1997). Anti‐MAG IgM autoantibodies recognize the HNK‐1 epitope, the sulfated trisaccharide 3‐O‐sulfo‐β‐d‐GlcA‐(1→3)‐β‐d‐Gal‐(1→4)‐d‐GlcNAc, which is expressed mainly on the glycoprotein MAG but also on PMP22 and glycolipids such as sulfoglucuronyl‐paragloboside, and sulfoglucuronyl‐lactosaminyl‐paragloboside (Quarles, 2007). Nerve biopsies from affected patients show demyelination and widening of myelin lamellae (Ritz et al., 1999; Steck, Murray, Murray, Meier, Page, & Perruisseau, 1983). There is strong evidence that the deposition of anti‐MAG IgM on myelin sheath and myelin lamellae is responsible for the demyelination, which clinically manifests itself as a peripheral neuropathy affecting primarily sensory nerves (Gabriel et al., 1998; Latov et al., 1981; Willison et al., 1988). Patients suffer from sensory ataxia with impaired gait, paresthesia, distal muscle weakness, and tremor (Braun, Frail, Frail, & Latov, 1982; Dalakas, 2010; Steck et al., 1983). Progression of the chronic neuropathy is linked to the presence of anti‐MAG IgM antibodies and there is evidence that the reduction in anti‐MAG IgM titers leads to clinical improvements (Benedetti et al., 2007; Gabriel et al., 1996; Nobile‐Orazio et al., 1988; Pestronk et al., 2003; Renaud et al., 2006; Steck et al., 2006). However, the causes and the exact mechanisms behind the expansion of anti‐MAG IgM producing B‐cell clones are not fully understood.
Most off‐label treatments aim to reduce pathogenic autoantibody titers by depleting autoantibody‐producing B‐cell clones, interfere with antibody‐effector mechanisms, or physically remove autoantibodies from the circulation. Most frequently, the anti‐CD20 monoclonal antibody rituximab is used to treat anti‐MAG neuropathy patients (Dalakas, 2010; Lunn & Nobile‐Orazio, 2016; Steck et al., 2006). However, all of these treatment options lack selectivity, efficiency, or can induce severe adverse effects in some patients. For example, acute worsening of the condition related to an IgM flair and concurrent rise in anti‐MAG IgM plasma titers has been described after rituximab treatment (Broglio & Lauria, 2005; Sala, Robert‐Varvat, Paul, Camdessanché, & Antoine, 2014).
In a previous study, we described the glycopolymer poly(phenyl disodium 3‐O‐sulfo‐β‐d‐glucopyranuronate)‐(1→3)‐β‐d‐galactopyranoside (PPSGG) designed to specifically target the pathogenic anti‐MAG IgM autoantibodies as a potential drug candidate for the treatment of anti‐MAG neuropathy (Herrendorff et al., 2017). The glycopolymer is composed of a biodegradable poly‐l‐lysine backbone (≈ 80 kDa) presenting multiple copies of a mimetic of the sulfated HNK‐1 trisaccharide to facilitate a multivalent interaction with anti‐MAG IgM autoantibodies. We previously showed in a competitive ELISA that PPSGG prevented the binding of patients’ anti‐MAG IgM autoantibodies to MAG at low nanomolar concentrations. Furthermore, we demonstrated a highly efficient reduction in anti‐MAG IgM antibody titers in an immunological mouse model for anti‐MAG neuropathy. In this study, the pharmacodynamic properties and potential immunomodulatory effects of PPSGG were further investigated. PPSGG was found to selectively and efficiently inhibit the binding of patients’ anti‐MAG IgM to sciatic nerve myelin of non‐human primates. Furthermore, the pharmacodynamic profile of the glycopolymer was characterized, including a dose titration study in mice to predict the estimated dose required in anti‐MAG neuropathy patients. Finally, it was shown that PPSGG removes the anti‐MAG IgM autoantibodies in a highly selective fashion without the activation of murine or human immune cells.
2. METHODS
2.1. Patients and control blood samples
All experiments using human samples were performed in accordance with the Declaration of Helsinki (1964, Association H. E. C. o. E. o. t. W. M. 1964). Patients and control subjects signed the written informed consent for their participation in this study. The study was not pre‐registered and the protocol was approved by the Ethical Committees EKNZ, and CER‐VD (Commission cantonale d'éthique de la recherche sur l'être humain), Switzerland (Study Nr. 01551). Heparinized whole blood of patients with clinically confirmed anti‐MAG neuropathy and healthy control subjects were obtained from the Department of Neurology of the University Hospital Basel (Basel, Switzerland), the Lausanne University Hospital (Lausanne, Switzerland), and Centre Hospitalier de la Timone, AP‐HM (Assistance Public‐Hôpitaux de Marseille), (Marseille, France). The blood samples of the anti‐MAG patients were taken during regular visit at the center and the control samples were taken in the morning.
2.2. Experimental animals
The wild type BALB/cJRj (Ref. C‐BJ‐56‐M/F; Janvier Labs) mice (5–10 weeks old male and female (n = 61) used in this study were bred at the University Hospital Basel and kept under specific pathogen‐free conditions in a controlled environment (12‐hr light–dark cycle at 20°C) with food and water ad libitum. Unless stated, experiments were performed in the morning without blinding of the experimenter. Mice were allocated arbitrarily into the different groups and the dosing was increased from low to high. No anesthetic or medication was required to reduce treatment related effects and no animal was excluded or died during the studies. Mice were sacrificed by CO2 suffocation and decapitation after the last blood sampling (Figure 1). The power calculation was based on the previous study (Herrendorff et al., 2017) where the null hypothesis was disproved and showed a concentration‐dependent relationship of the PPSGG and the anti‐MAG IgM binding (>80% relative reduction in the anti‐MAG IgM titers in the dose range of 2–10 mg/kg in mice). Therefore, we assume an alternative hypothesis (µA − µB ≠ 0) for the primary endpoint that states that the anti‐MAG IgM antibodies can be neutralized and removed by PPSGG in vivo compared to vehicle (phosphate‐buffered saline, PBS) treatment (one‐tailed) to have a power of >80% and a type I error of 5%, we would need six subjects per group to prove the effect is significant (2 independent means, one‐sided, µA = 100, µB = 20, κ = nA/nB = 1, σ = 20). The study was not pre‐registered and all animal experiments were performed in accordance with authorization No. 2778 of the Animal Research Authorities of the Canton of Basel‐Stadt, Switzerland.
Figure 1.
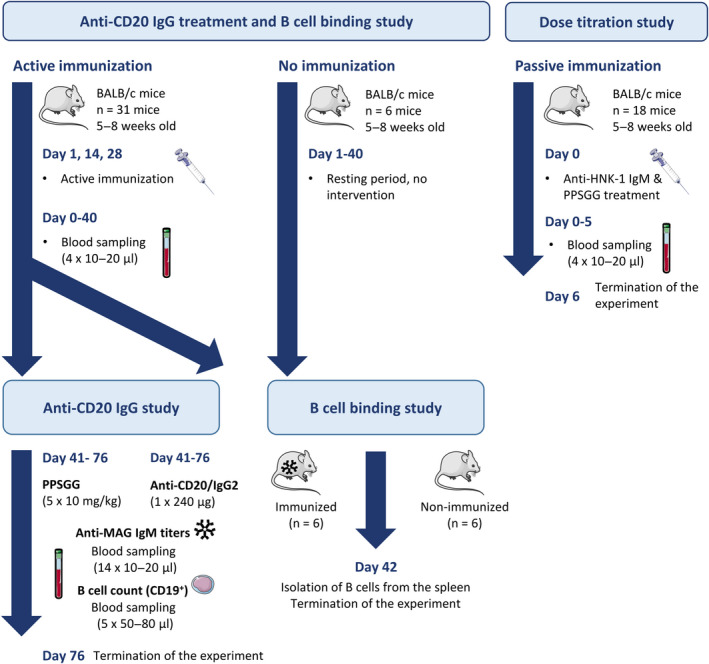
Flow chart of the animal experiments. Overview of the timelines of the animal experiments (n = 61). Images adapted from SMART Servier Medical ART
2.3. Isolation and culture of human PBMC, B cells, and DC
Peripheral blood mononuclear cells (PBMC) were isolated from heparinized blood samples by gradient centrifugation according to the protocol provided by GE‐Healthcare (Ficoll‐Paque PLUS). B cells (CD19+) and monocytes (CD14+, CD16‐) were isolated from PBMC by negative selection using the EasyEights™ EasySep™ Magnet (18103), the EasySep™ Human B Cell Isolation Kit (17954), and the EasySep™ Human Monocyte Isolation Kit (19359) all provided by StemCell™ Technologies.
B cells were maintained for up to 2 days in Roswell Park Memorial Institute medium (RPMI) 1640 medium (R8758, Roswell Park Memorial Institute medium; Sigma‐Aldrich), supplemented with 10% fetal bovine serum (FBS, 10270–106), 1% antibiotic‐antimycotic solution (AA, 15240062), and 1% l‐glutamine solution (l‐Gln, 11500626) all provided by Gibco, Thermo Fisher Scientific.
Monocytes were seeded and differentiated to dendritic cells (DC) in complete RPMI 1640 medium containing 10% FBS, 1% AA, 1% l‐Gln and supplemented with 800 IU/ml recombinant GM‐CSF (Granulocyte macrophage colony‐stimulating factor) and 500 IU/ml recombinant IL‐4 (R&D Systems) for 6 days at 37°C and 5% CO2.
2.4. Ex vivo Epstein–Barr transformation of B cells
To prolong the viability of primary human B cells (n = 4 anti‐MAG neuropathy patients) ex vivo, the cells were transformed with Epstein‐Barr virus (EBV) (Hollyoake, Stühler, Stühler, Farrell, Gordon, & Sinclair, 1995). Marmoset B95.8 Epstein–Barr virus‐producing cells (RRID:CVCL_1953; Vircell) were maintained in complete RPMI 1640 medium containing 10% FBS, 1% AA, and 1% l‐Gln. The supernatant containing the EBV was collected after 20 days by centrifuging cells (1400g, 15 min) and filtering the supernatant with a 0.45 μm and a 0.2 μm Filtropur S filter (83.1826; Sarstedt). Freshly isolated human PBMC (1x106 cells) were resuspended in 3 ml EBV supernatant and incubated for 24 hr before adding 7 ml complete RPMI 1640 medium supplemented with 20 ng/ml cyclosporin A (30024; Sigma‐Aldrich). The cell growth was monitored and the medium changed every 2–3 days to achieve an efficient immortalization. After 14 days of culture, cells were maintained in complete RPMI 1640 medium without cyclosporin A, at a density of approximately 5 × 105 to 1 × 106 cells/mL.
2.5. Indirect immunofluorescence assay
Prior to the indirect immunofluorescence assay, the anti‐MAG IgM titers were determined by ELISA (see below) in Bühlmann titer units (BTU). Anti‐MAG neuropathy patients’ sera (n = 5) and control neuropathy patient sera (n = 1 chronic ataxic neuropathy ophthalmoplegia M‐protein agglutination disialosyl antibodies syndrome (CANOMAD) patient and n = 1 chronic inflammatory demyelinating polyneuropathy (CIDP) patient with IgM monoclonal gammopathy of unknown significance but no MAG or ganglioside activity) were used. Both control sera were previously tested for their positive reaction on sciatic nerves and negative for anti‐MAG and anti‐ganglioside antibodies. The CANOMAD patient sera was tested positive for anti‐diasialosyl antibodies, however, the exact target of the antibodies in case of the CIDP patient is unknown.
Undiluted sera were incubated for 30 min with different concentrations of PPSGG (62.5, 125 and 250 μg/ml in PBS. The samples were then diluted 1:20 in PBS and incubated for 30 min with samples of sciatic nerve cynomolgus monkeys (Macaca fascicularis). The non‐human primate sciatic nerve samples were part of the ImmuGlo™ anti‐MAG IFA kit (REF 1172, LOT 1905438B) and obtained from IMMCO Diagnostics. Data were analyzed and quantified by ImageJ software from ten fields of observation for each experimental condition.
2.6. Anti‐MAG autoantibodies ELISA
Sera from control subjects, commercially available (n = 1, H4522; Sigma‐Aldrich) and non‐commercial human serum (n = 6), and anti‐MAG neuropathy patients (n = 8) were used to determine the reactivity of PPSGG to other immunoglobulins by ELISA. Maxisorp MP 96 wells plates (442404; Nunc, Sigma‐Aldrich) were coated overnight at 4°C with 50 µl PPSGG (0.1 µg/ml in PBS), then washed four times with 300 µl ELISA buffer (0.1% Tween 20 in PBS) and blocked with 100 µl of 5% bovine serum albumin (BSA, A9647; Sigma‐Aldrich) in ELISA buffer for 2 hr at 20‐25°C. The sera samples were diluted 1:1,000 (2.5% BSA in ELISA buffer) and added to the plates after another washing step. After incubating for 2 hr at 20‐25°C, the plates were washed four times with 300 μl ELISA buffer and 100 µl of HRP‐labeled anti‐human IgM (1:2’000 in 1% BSA in ELISA buffer, RRID:AB_258318; Sigma‐Aldrich), 100 µl HRP (horseradish peroxidase)‐labeled anti‐human IgG (1:10’000 in 1% BSA in ELISA buffer, RRID:AB_258388; Sigma‐Aldrich), or the anti‐human IgM antibody conjugated to HRP (B‐MAG‐ELM; Bühlmann Laboratories) were added and incubated for 2 hr at 20‐25°C, respectively. To develop the assay, 50 µl of undiluted 3,3’,5,5’‐tetramethylbenzidine (B‐TMB; Bühlmann Laboratories) was added and incubated at 20‐25°C on a plate rotator (600 RPM) for 30 min. The stop solution (50 µl of 0.25 M H2SO4) was added and the optical density at 450 nm (OD450) was determined within <5 min with the Spectra Max 190 (Molecular Devices).
2.7. PPSGG dose titration study in mice
To estimate the dose of PPSGG required to bind and exhaustively eliminate anti‐MAG IgM autoantibodies in anti‐MAG neuropathy patients, a dose titration study was performed in mice. Therefore, naïve BALB/c mice were intravenously injected with 60 µg (n = 10 mice) or 120 µg (n = 8 mice) of purified, sodium azide‐ and BSA‐free anti‐HNK‐1 IgM (ab187274, Abcam), followed by 1–20 µg intravenous doses of PPSGG, 10–20 min after IgM administration. Blood samples were taken 5–10 min after PPSGG administration and the serum was collected and analyzed for unbound anti‐HNK‐1 IgM. Additional serum samples were taken and analyzed at a later time point (24 hr after administration) to confirm the removal of the injected anti‐HNK‐1 IgM antibodies by PPSGG. The detection of the free anti‐HNK‐1 IgM in mouse sera was performed as described elsewhere (Herrendorff et al., 2017).
2.8. CD20+ cell depletion in mice
BALB/c mice were immunized with purified bovine sulfoglucuronyl paragloboside/sulfoglucuronyl‐lactosaminyl‐paragloboside mixed with keyhole limpet hemocyanin (KLH, H8283; Sigma‐Aldrich) and TiterMax Gold (T2684; Sigma‐Aldrich) (Herrendorff et al., 2017). After mouse anti‐MAG IgM titers plateaued, single intravenous injections (n = 6 mice) of 240 µg of anti‐mouse CD20 IgG (RRID:AB_2629619; Biolegend) or 240 µg of Ultra‐LEAF Purified Rat IgG2b (RRID:AB_11149687; Biolegend) were performed (n = 3 mice). Blood samples were taken prior and at various time points after the treatment and collected in ethylenediaminetetraacetic acid (EDTA)‐coated tubes from the vena saphena. To lyse the erythrocytes, 1 ml of ammonium‐chloride‐potassium lysing buffer (A10492‐01; Gibco, Thermo Fisher Scientific) was added at 20‐25°C, and after 5 min cells were washed with PBS and centrifuged (170 g, 5 min) before repeating the lysis step once more. Whole blood (40 µl) was stained with 1 µl of anti‐mouse CD19‐PECy7 (RRID:AB_657664; eBioscience) for 45 min at 4°C (protected from light). The samples were then diluted with PBS to a final volume of 200 µl and stained with 1 µl of DAPI (4’,6‐diamidino‐2‐phenylindol, D3571; Invitrogen) prior to the analysis of the cells by flow cytometry with the LSR Fortessa (Becton Dickinson). At least 10’000 events were recorded and data were further gated and analyzed with FlowJo v 10.2 (FlowJo LLC).
2.9. Binding of PPSGG to murine and human B cells
The binding of PPSGG to B cells and their potential activation was studied by flow cytometry and fluorescence microscopy. Fluorescently labeled PPSGG (PPSGG, 5% loading with sulforhodamine 101) was used to assess binding to the cells and Fluo‐4 AM was used to determine the intracellular calcium (Ca2+) levels as a marker for B cell activation through the B cell receptor (Baba & Kurosaki, 2016). Isolated human PBMC, human B cells, murine splenocytes, and murine B cells were stained with anti‐CD19 PE‐Cy7 (mouse anti‐human IgG1, RRID:AB_2535491; rat anti‐mouse IgG2a, RRID:AB_657664) and anti‐F4/80 eFluor 450 (rat anti‐mouse IgG2a, RRID:AB_1548747), Fluo‐4 AM (F14201; Thermo Fisher Scientific), propidium iodide (PI, P4170; Sigma Aldrich), and Hoechst 33258 solution (94403; Sigma Aldrich). The cells were loaded with 3 μM Fluo‐4 AM (cell‐permeable, fluorescent Ca2+ indicator) for 30 min in the dark at 37°C and 5% CO2 before the antibodies and PPSGG‐S were added and incubated for another 30 min. The cells were washed once with extracellular solution (135 mM NaCl, 5 mM KCl, 5 mM HEPES, 10 mM D‐glucose, 2 mM CaCl2, pH 7.35) and kept in the dark for the immediate recording.
Isolated murine spleen cells and B cells (n = 6 naïve mice and n = 6 immunized mice) or PBMC and human B cells (n = 4 anti‐MAG neuropathy patients and n = 3 healthy control donors) were incubated at approximately 1 × 106 cells/mL with 10 μM or 1 μM PPSGG‐S. The fluorescently labeled antibodies were added at a concentration of 1 μL/mL. For assessing B‐cell activation, a control measurement was recorded to set the gate for the baseline signal and after addition of 100 μM unlabeled PPSGG the sample was measured every 2 min on a LSR Fortessa (BD Biosciences).Murine cells treated with 3 μM Ionomycin acting as a Ca2+ ionophore and EBV transformed human B cells showing increased intracellular Ca2+ levels were used as positive controls. At least 10’000 events were recorded and data were analyzed with FlowJo.
The same setup was used to measure binding and activation with a fluorescence microscopy (Applied Precision Widefield DV Core microscope; GE Life Sciences). B cells were plated in Ibidi 8 well µ‐Slides (80826; Ibidi GmbH) and loaded with 3 µM Fluo‐4 AM and incubated with PPSGG‐S at 10 µM or 1 µM. The cells were washed once with extracellular solution and the plate was placed in a humidified atmosphere (37°C and 5% CO2) on the microscope using 475 nm and 575 nm solid‐state illumination and a 60×/1.42 oil‐immersion objective (Plan Apo N). Five randomly taken spots per sample were identified and recorded. According to the flow cytometry experiments, Fluo‐4 AM loaded samples were recorded before and after addition of 100 µM PPSGG every 5 min for 20 min. The images were processed with softWoRx (GE Life Sciences) and analyzed with FIJI software. The PPSGG‐S binding and the relative changes in intracellular Ca2+ concentration was assessed by measuring fluorescence intensity of each individual cell after background subtraction.
2.10. Dendritic cell activation and intracellular cytokine analysis by flow cytometry
Non‐adherent monocyte‐derived DC (n = 5 healthy control donors) were reseeded at day 6 to a 96‐well plate (1 × 105 cells per well) in complete RPMI 1640 media and incubated for 1 hr or 18 hr with lipopolysaccharide (LPS) (100 ng/ml), PPSGG (30 μg/ml), or both LPS and PPSGG. Afterward, the DC were stained for 20 min at 20‐25°C with CD14 FITC (RRID:AB_2571928), HLA‐DR PerCP (RRID:AB_893574), CD40 BV605 (RRID:AB_2564243), CD83 APC (RRID:AB_314519), CD11b Alexa Fluor 700 (RRID:AB_493705), CD1c APC‐Cy7 (RRID:AB_10643413), together with Fc receptor block (anti‐CD16/32 Ab, RRID:AB_312801) in PBS supplemented with 1% BSA. The mean fluorescence intensity (MFI) 16’481 ± 398 cells per sample was assessed by flow cytometry (LSR Fortessa; BD Biosciences).
2.11. Human interleukin‐1β ELISA
Supernatant from PBMC (n = 5 healthy control donors) stimulated for 6 hr with lipopolysaccharide (LPS, 100 ng/ml) or PPSGG (30 μg/ml) in complete RPMI 1640 media (10% FBS, 1% AA, and 1% L‐glut) was collected and stored at −20°C until use. Total production of IL‐1β was assessed using the eBioscience™ Human IL‐1 beta ELISA Ready‐SET‐Go!™ (Invitrogen™ 88726122) according to the protocol provided by the manufacturer (Thermo Fisher).
2.12. Intracellular cytokine analysis by flow cytometry
For the intracellular cytokine analysis, freshly isolated PBMC (n = 5 healthy control donors) were reseeded in complete RPMI 1640 media and stimulated for 2 hr with LPS (100 ng/ml) or PPSGG (30 μg/ml) at 37°C and subsequently incubated with a mix of brefeldin A and monensin (both diluted 2’000×) for an additional 4 hr or 18 hr at 37°C. Cells were stained with HLA‐DR PerCP (RRID:AB_893574), CD8 BV510 (RRID:AB_2561378), CD14 Alexa Fluor 700 (RRID:AB_2566715), CD4‐PE (RRID:AB_1937246), CD11c PE‐Alexa Fluor 594 (RRID:AB_2564082), and CD19 PE‐Cy7 (RRID:AB_2564202) together with Fc receptor block (anti‐CD16/32 Ab, RRID:AB_312801). PBMC were then fixed with 4% paraformaldehyde in PBS and permeabilized with PBS solution containing 0.5% BSA, 0.5% Tween 20, followed by intracellular staining with IL‐6 FITC (RRID:AB_315151), IL‐12/IL‐23 p40 Pacific Blue (RRID:AB_2124517), TNFα BV605 (RRID:AB_11203719), and IL‐10 APC (RRID:AB_315456). Data were collected on a Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo.
2.13. Statistical analysis
Unless stated, results are given as mean ± SD of three experiments and analyzed by Prism GraphPad Software (Version 7.04). No test for outliers was conducted and no data point was excluded in the study. The normal distribution was assessed by D'Agostino and Pearson normality test. G*Power software (Version 3.1.9.4) was used to determine sample size within the animal approval (No. 2778). No additional sample calculation was performed for the experiments in this publication. Comparisons between two conditions were performed using either Student's t test or one‐way ANOVA with Dunnett's multiple comparison posttest with a 0.05 confidence level accepted for statistical significance (*p ≤ .05, **p ≤ .01, ***p ≤ .001).
3. RESULTS
3.1. Inhibition of patients’ anti‐MAG IgM binding to myelin of non‐human primates and highly selective binding of anti‐MAG IgM by PPSGG
To assess the potential of PPSGG to inhibit patients’ anti‐MAG IgM binding to myelin of peripheral nerves, sciatic nerve preparation of cynomolgus monkeys were incubated with patients’ sera and the myelin reactivity was determined by indirect immunofluorescence measurements (Figure 2). The five analyzed sera of anti‐MAG neuropathy patients exhibited a wide range of anti‐MAG IgM titers (6’975, 19’333, 36’383, 61’684, 249’600 BTU). As controls, the sera samples of a chronic ataxic neuropathy (CANOMAD) patient and a CIDP patient, with IgM monoclonal gammopathy and positive reaction on sciatic nerve but no MAG or ganglioside activity were used. They exhibited anti‐MAG IgM titers below the threshold of 1’000 BTU in the ELISA. The normalized fluorescence intensity of all the anti‐MAG neuropathy patients’ samples decreased by 80% (±5% SD), when the sera were incubated with 250 μg/ml PPSGG. The incubation of the anti‐MAG neuropathy patients’ samples with 62.5 μg/ml PPSGG already showed a decrease of 78% (±7% SD) in the normalized fluorescence intensity. The serum of patient 4 exhibiting low anti‐MAG titer (19’333 BTU) showed residual myelin binding compared to patient 2 (61’638 BTU) or 3 (36’383 BTU) with higher anti‐MAG IgM titers at a PPSGG concentration of 62.5 μg/ml. The anti‐MAG neuropathy patient 1 with the highest titer showed almost a complete decrease in the normalized fluorescence intensity when the serum was incubated with 125 μg/ml PPSGG. The control neuropathy patients showed no significant reduction of the normalized fluorescence intensity when the undiluted sera were incubated with 250 μg/ml PPSGG.
Figure 2.
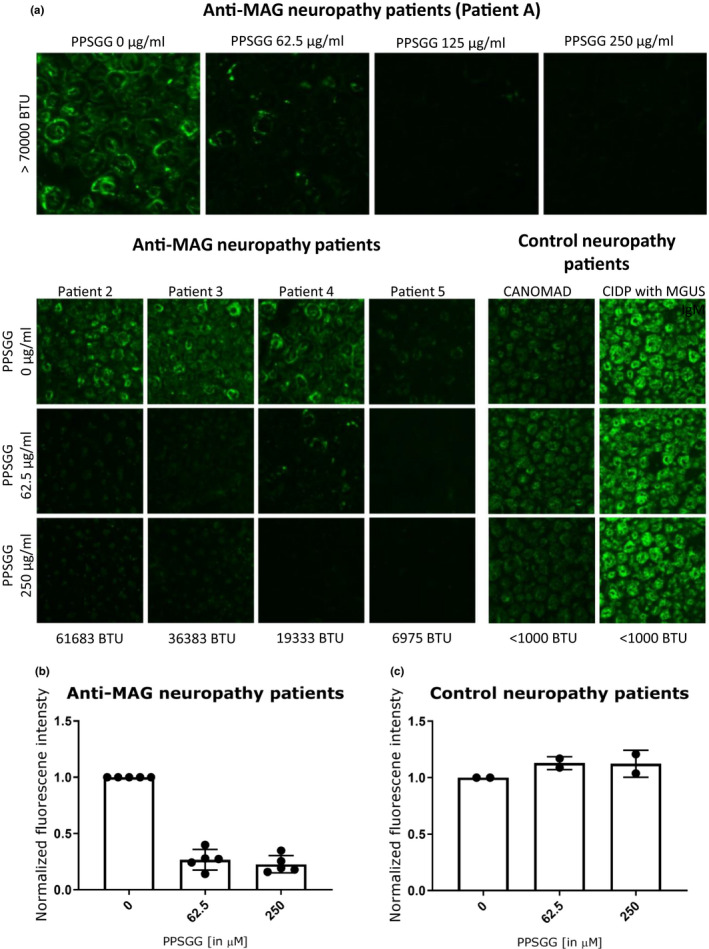
Selective blocking of the binding of anti‐myelin‐associated glycoprotein (MAG) IgM from patients sera to sciatic nerve myelin of non‐human primates. (a) After incubation of undiluted serum (with or without poly(phenyl disodium 3‐O‐sulfo‐β‐d‐glucopyranuronate)‐(1→3)‐β‐d‐galactopyranoside, PPSGG), the samples were diluted 1 in 20 in phosphate‐buffered saline and the myelin‐reactivity tested by indirect immunofluorescence. (b) Five anti‐MAG neuropathy patients’ and two control neuropathy patients’ (chronic ataxic neuropathy ophthalmoplegia M‐protein agglutination disialosyl antibodies syndrome and chronic inflammatory demyelinating polyneuropathy with IgM monoclonal gammopathy of unknown significance but no MAG or ganglioside activity) sera were tested in the indirect immunofluorescence assay. A concentration of 125 µg/ml PPSGG completely blocked the binding of anti‐MAG IgM to primate sciatic nerve myelin. However, (c) PPSGG had no inhibitory activity on the myelin reactivity of the control neuropathy patients
To confirm the selective blocking of anti‐MAG IgM autoantibodies by PPSGG, the binding of control subject sera (commercial and non‐commercial), and anti‐MAG neuropathy patients’ sera to PPSGG was tested. PPSGG showed only reactivity to IgM present in the serum samples of anti‐MAG neuropathy patients. No reactivity of PPSGG to IgM of healthy control samples was detected. Moreover in neither control subject sera nor anti‐MAG neuropathy patients’ sera significant binding of IgG was detected (Figure 3). No significant difference in PPSGG reactivity between the commercial and non‐commercial human serum was observed.
Figure 3.
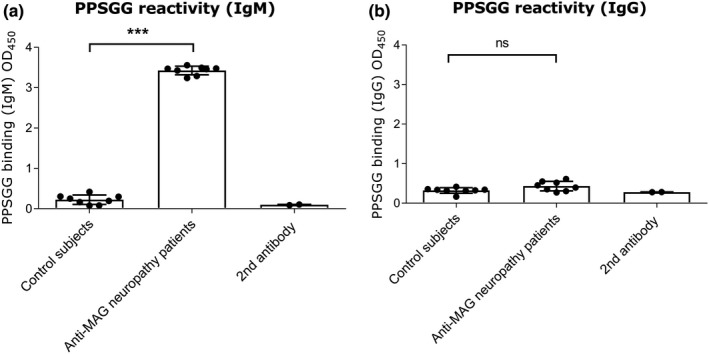
Reactivity of immunoglobulins (IgG and IgM) from healthy subjects and anti‐myelin‐associated glycoprotein (MAG) neuropathy patients to poly(phenyl disodium 3‐O‐sulfo‐β‐d‐glucopyranuronate)‐(1→3)‐β‐d‐galactopyranoside (PPSGG). A PPSGG‐binding ELISA was performed with healthy controls (healthy control serum samples n = 7), commercially available normal human serum (n = 1), and anti‐MAG neuropathy patient samples (n = 8). Only IgM (a) and no IgG (b) binding in sera samples of anti‐MAG neuropathy patient exhibited PPSGG activity in the ELISA. Data are indicated by mean and standard deviation (one‐way ANOVA, *p ≤ .05, **p ≤ .01, ***p ≤ .001)
3.2. Fast and efficient removal of circulating anti‐HNK‐1 IgM by PPSGG in BALB/c mice, while CD20+ cell depletion showed no significant effect on the anti‐MAG IgM titers in the immunological mouse model for anti‐MAG neuropathy
To estimate the PPSGG dose required to reduce anti‐MAG IgM autoantibody titers in anti‐MAG neuropathy patients, we performed a dose‐finding study in mice (Figure 4). Therefore, naïve BALB/c mice were injected with different concentrations of murine anti‐HNK‐1 IgM (60, 120 µg/kg) followed by intravenous PPSGG injection (1–20 µg). Anti‐MAG IgM titer analysis in mouse plasma samples taken 5–10 min after PPSGG administration showed that the administration of a 10 µg PPSGG dose was sufficient to bind 93.28% (±0.1% SD) of the 60 µg anti‐HNK1 IgM in vivo, as indicated by the measured baseline anti‐MAG IgM titers. A dose range of 3–5 µg was still sufficient to reduce the anti‐HNK1 IgM level by ≥ 88.56% (±1.64% SD). The lowest dose of 1 µg PPSGG still reduced the 60 µg anti‐HNK1 IgM level by 23.08% (±0.64% SD) compared to the pre‐treatment level. At the dose of 120 µg anti‐MAG IgM, 10 µg PPSGG reduced the anti‐MAG IgM titers by 97.85% (±0.50% SD), 4 µg PPSGG reduced the titers by 88.74% (±8.68% SD), and 2 µg PPSGG reduced the titers by 42.71% (±3.03% SD) compared to the pre‐treatment titers. To check whether anti‐MAG IgM antibodies were permanently removed, mice were sampled at a later time points (24 and 96 hr after PPSGG administration) and showed persistent reduction of the titers.
Figure 4.
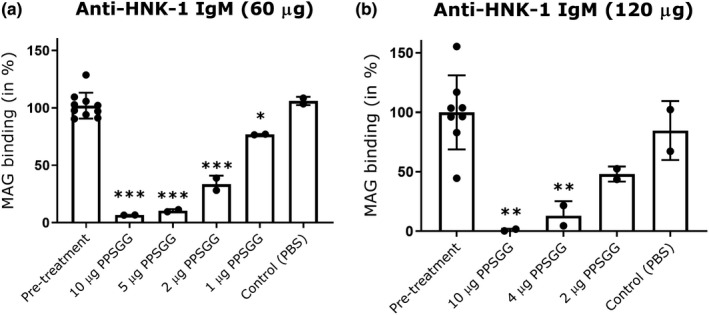
Poly(phenyl disodium 3‐O‐sulfo‐β‐d‐glucopyranuronate)‐(1→3)‐β‐d‐galactopyranoside (PPSGG) dose titration study in mice. The intravenous injection of 60 µg (n = 10 mice) (a) or 120 µg anti‐human natural killer‐1 (HNK‐1) IgM (n = 8 mice) (b) was followed by the injection of different doses of PPSGG (<10 min). Blood samples were taken 5 to 15 min after PPSGG administration and the amount of PPSGG required to bind most of the administered mouse anti‐HNK‐1 IgM in circulation was determined by ELISA. The intravenous injection of 10 µg was sufficient to deplete ≥ 92% of the total IgM. Data are indicated by mean and standard deviation (one‐way ANOVA with Dunnett's multiple comparisons test, *p ≤ .05, **p ≤ .01, ***p ≤ .001)
To further verify the mechanism of anti‐MAG IgM removal in vivo, we compared PPSGG treatment to treatment with a CD20+ B cell depleting monoclonal antibody (Figure 5). Therefore, wild type BALB/c mice were immunized against the HNK‐1 bearing glycolipids sulfoglucuronyl paragloboside/sulfoglucuronyl‐lactosaminyl‐paragloboside. Once the mice exhibited a constant production of anti‐MAG IgM antibodies, they were treated either with a single intravenous injection of mouse anti‐CD20 IgG, rat IgG2b κ isotype control antibody, or with weekly injections of PPSGG (10 mg/kg). The number of B cells and anti‐MAG IgM titers were followed up by flow cytometry and ELISA in the blood and serum, respectively. Flow cytometry of PBMC revealed the significant depletion of circulating B cells in the anti–CD20 antibody treated group, while no significant changes in the number of circulating B cell were observed in the IgG2 κ isotype control antibody group or PPSGG treated group. However, neither B‐cell depletion nor IgG2 κ isotype control antibody changed the anti‐MAG IgM antibody titers significantly during the follow‐up period of 5 weeks. Only the mice treated once per week with 10 mg/kg PPSGG exhibited a significant reduction of the anti‐MAG IgM titers.
Figure 5.
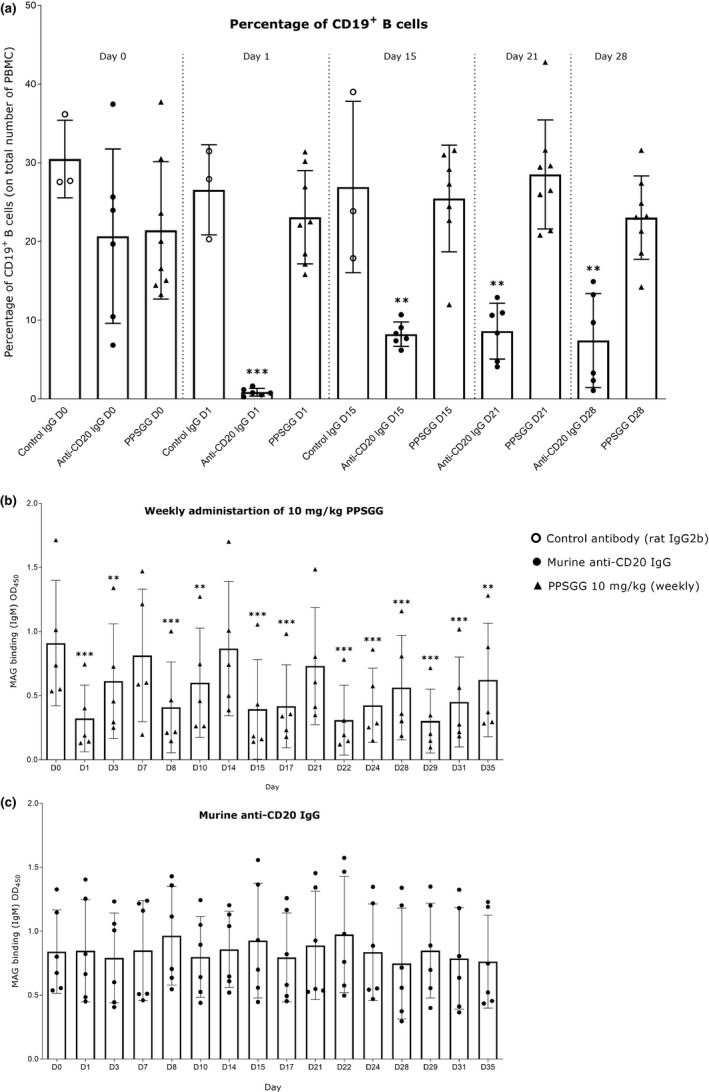
Effects of anti‐CD20 antibody treatment, control antibody treatment, and poly(phenyl disodium 3‐O‐sulfo‐β‐d‐glucopyranuronate)‐(1→3)‐β‐d‐galactopyranoside (PPSGG) treatment on anti‐myelin‐associated glycoprotein (MAG) IgM antibodies in the immunological BALB/c mouse model for anti‐MAG neuropathy. (a) Single injection of 240 µg of anti‐CD20 antibody (n = 6) depleted almost all circulating CD19+ B cells, whereas weekly PPSGG treatment (10 mg/kg, n = 8) or single treatment with a control rat IgG2b antibody (n = 3) did not affect the levels of circulating CD19+ B cells. Weekly PPSGG treatment (10 mg/kg, D1, D8, D15, D22, D28) lowered the anti‐MAG IgM antibody titers significantly, compared to pre‐treatment levels at D0 and depicted by the decreased binding of the anti‐human natural killer‐1 IgM to MAG (n = 5) (b). A single injection of 240 µg of anti‐CD20 antibody (c) and of 240 µg (n = 6) of a control antibody (Figures S1–S5 in supplementary information) did not affect the anti‐MAG IgM antibody titers in mice. Data are indicated by mean and standard deviation (one‐way ANOVA, *p ≤ .05, **p ≤ .01, ***p ≤ .001).
3.3. No murine or human leukocyte activation after incubation with PPSGG
As PPSGG presents multiple copies of the HNK‐1 mimetic, it can be seen as multivalent soluble antigen which could be recognized by B cells and potentially trigger a humoral response (Heesters et al., 2016). Therefore, we assessed if there is specific binding of PPSGG to B cells of immunized and non‐immunized mice, or to B cells of anti‐MAG neuropathy patients and of healthy controls (Figure 6). Independent of the technique, neither the flow cytometry data nor the fluorescent microscopy data showed a significant difference between control (non‐immunized) and anti‐MAG IgM and IgG producing (immunized) B cells isolated from the murine spleen. Nonetheless, the percentage of B cells with surface PPSGG staining was highly dependent on the concentration of PPSGG. It was increased when murine B cells were incubated with 10 μM PPSGG compared to 1 μM PPSGG. A sulforhodamine 101 labeled thioglycerol capped poly‐l‐lysine backbone was used as control and showed no binding to isolated B cells at 1 or 10 μM. The effect of PPSGG on intracellular Ca2+ signaling as a marker for B cell activation was assessed by flow cytometry and fluorescence microscopy (Donjerković & Scott, 2000). B cells of immunized and non‐immunized mice did not exhibit an increase in the relative intracellular Ca2+ levels monitored over the time course of 8 min by flow cytometry and 20 min by fluorescence microscopy, even after incubation with 100 μM PPSGG. B cells of anti‐MAG neuropathy patients did not show enhanced binding of PPSGG compared to B cells of control subjects, when they were incubated for 30 min with 1 or 10 μM PPSGG.
Figure 6.
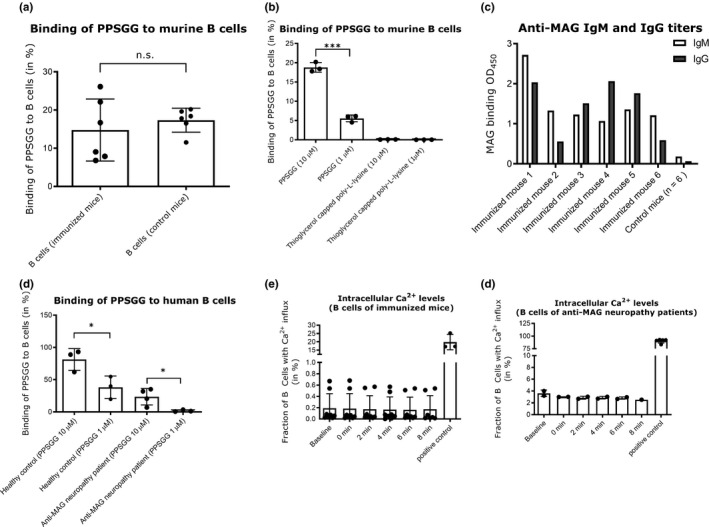
Concentration‐dependent binding of poly(phenyl disodium 3‐O‐sulfo‐β‐d‐glucopyranuronate)‐(1→3)‐β‐d‐galactopyranoside (PPSGG) to murine and human B cells without increasing intracellular Ca2+levels. Binding of fluorescently labeled PPSGG to B cells of immunized mice (n = 6), control mice (n = 6), anti‐myelin‐associated glycoprotein (MAG) neuropathy patients (n = 4), and healthy human controls (n = 3) were assessed by flow cytometry and fluorescent microscopy. B cells were either incubated with 10 μM and 1 μM PPSGG‐S or as control with 10 μM and 1 μM fully thioglycerol capped poly‐L‐lysine backbone labeled with sulforhodamie 101 (a, b). B cells were binding PPSGG in a dose‐dependent manner and showed no binding to thioglycerol capped poly‐L‐lysine backbone. There was no difference in binding of PPSGG in immunized versus control mice. To confirm that mice were immunized successfully, anti‐MAG IgM and IgG titers were analysed in sera samples by ELISA (c). Anti‐MAG neuropathy patient B cells and B cells from healthy controls were incubated with PPSGG for 30 min. Anti‐MAG neuropathy patient B cells showed lower binding of PPSGG compared to the healthy controls (d). To determine changes in intracellular Ca2+ concentration induced by PPSGG binding, murine and human B cells were treated with 100 μM PPSGG after loading with 3 μM Fluo‐4 AM. The changes in intracellular Ca2+ levels were monitored for the duration of 8 min, in 2 min intervals after a baseline measurement and the subsequent addition of 100 μM PPSGG. Neither B cells from immunized mice (e) nor from anti‐MAG neuropathy patients (f) exhibited increased intracellular Ca2+ concentration after PPSGG incubation. Data are shown as mean values ± SD (unpaired, two‐tailed t test, *p ≤ .05, **p ≤ .01, ***p ≤ .001)
To confirm the presence of anti‐MAG IgM producing B cells in the obtained peripheral blood samples from anti‐MAG neuropathy patients, PBMC of four patients (Nr. 23, 27, 49, and 67) were transformed with EBV to increase viability of B cells ex vivo. Before the transformation, the anti‐MAG IgM levels of patients 23, 27, and 49 were higher compared to patient 67. Fourteen days after transformation, the cell supernatants were analyzed by ELISA for their anti‐MAG IgM titers. The cells of patient 49 had the highest anti‐MAG IgM titer and maintained the ability to secrete anti‐MAG IgM in vitro (Figure 7). Besides B cells activation, we assessed whether PPSGG can induce DC activation by measuring the expression of co‐stimulatory molecules (Figure 8). Human monocyte‐derived DC (CD1c+, CD14‐, and CD11b+) were incubated for 18 hr with 30 μg/ml PPSGG, 100 ng/ml LPS, or with both PPSGG (30 μg/ml) and LPS (100 ng/ml) and HLA‐DR, CD40, and CD83 expression was determined for each condition. LPS was used as a positive control for DC activation and the co‐incubation of PPSGG as well as to assess potential synergistic effects. We observed that MFI for CD40 and CD83 increased upon LPS exposure, but were unaffected by PPSGG treatment. A trend toward an increased MFI for HLA‐DR was observed in the LPS‐treated groups, but PPSGG had no significant influence on the expression of HLA‐DR. To assess whether potential pro‐ or anti‐inflammatory reactions were induced by PPSGG, freshly isolated PBMC from healthy human donors were treated with 30 μg/ml PPSGG, 100 ng/ml LPS, or a combination of both for 6 or 20 hr, respectively (Figure 9). Subsequently, TNF‐α, IL‐6, IL‐10 and IL‐12 production by CD14+ monocytes and HLA‐DR+ CD11c+ DC was assessed by flow cytometry or ELISA. No significant induction of TNF‐α, IL‐6, IL‐12, and IL‐1β was observed, when the CD14+ monocytes or CD11c+ DC were incubated with PPSGG. A significant increase in the production of TNF‐α, IL‐6, IL‐12, and IL‐1β was only observed upon treatment with LPS, whereas no impact was observed when co‐incubating PPSGG with LPS. In addition, to assess potential inflammatory or allergic‐like reactions induced by the administration of PPSGG in BALB/c mice, a cytokine and chemokine array including IFNγ, IL‐2, IL‐6, and TNFα was performed. Neither a single nor multiple administrations (daily for five consecutive days) of 10 mg/kg PPSGG, led to a significant alteration of the studied cytokines and chemokines compared to the pre‐treatment titers (Figures S1–S5 in supplementary information).
Figure 7.
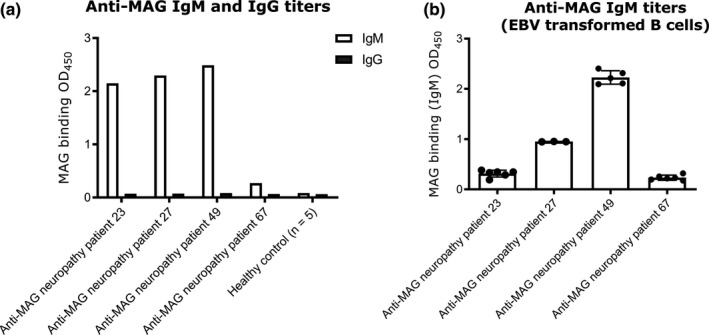
Anti‐myelin‐associated glycoprotein (MAG) IgM titers before and after transformation of B cells with Epstein‐Barr virus (EBV). Antibody titers in the sera of anti‐MAG patients and in the supernatants of EBV transformed B cells were determined by ELISA. Patients 23, 27, and 49 show high titers of anti‐MAG IgM antibodies. Patient 49 exhibited the highest titers in the supernatant after the EBV transformation of the B cells, indicating that isolated B cells maintainted their ability to secret anti‐MAG IgM after the transformation with EBV. Results are shown as mean ± SD
Figure 8.
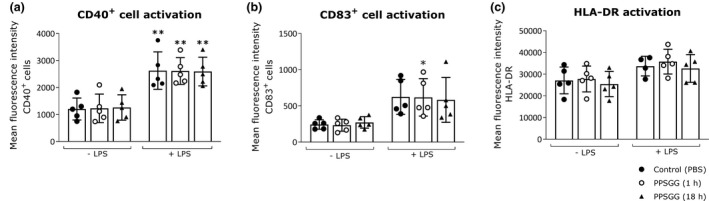
No indication of dendritic cells (DC) activation upon treatment with poly(phenyl disodium 3‐O‐sulfo‐β‐d‐glucopyranuronate)‐(1→3)‐β‐d‐galactopyranoside (PPSGG) ex vivo. Human monocyte‐derived DC (n = 5 donors) were incubated overnight (18 hr) with lipopolysaccharide (LPS) (100 ng/ml), PPSGG (30 µg/ml), or the combination of both for 1 hr or 18 hr. DC (CD1c+, CD14‐, CD11b+) were gated and the mean fluorescence intensity (MFI) for CD40 (a), CD83 (b), and HLA‐DR (c) was assessed by flow cytometry. An increase in MFI was observed for CD40 and CD83 in LPS‐treated groups, but no increase of activation markers was related to PPSGG treatment. Results are shown as mean ± SD (one‐way ANOVA with Sidak posttest, *p ≤ .05, **p ≤ .01, ***p ≤ .001)
Figure 9.
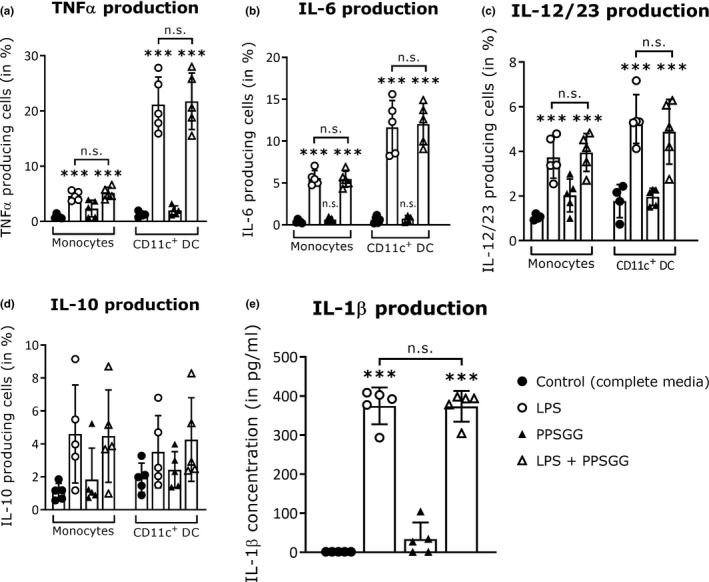
No significant cytokine production in peripheral blood mononuclear cells (PBMC) upon to poly(phenyl disodium 3‐O‐sulfo‐β‐d‐glucopyranuronate)‐(1→3)‐β‐d‐galactopyranoside (PPSGG) treatment ex vivo. Freshly isolated human PBMC (n = 5 donors) were treated with lipopolysaccharide (LPS) (100 ng/ml), PPSGG (30 µg/ml), or the combination of both for 6 hr or in case of IL‐10 production for 20 hr. The production of TNFα (a), IL‐6 (b), IL‐10 (c), and IL‐12 (d) by CD14+ monocytes and HLA‐DR+ CD11c+ Dendritic cells (DC) was subsequently assessed by flow cytometry. Release of IL‐1β in the supernatant of incubated cells was assessed by ELISA (e). No significant increase in the production of any measured cytokine was observed related to PPSGG treatment. Results are shown as mean ± SD (one‐way ANOVA with Sidak posttest, *p ≤ .05, **p ≤ .01, ***p ≤ .001)
4. DISCUSSION
Anti‐MAG neuropathy is a disabling demyelinating peripheral neuropathy with an autoimmune etiology caused by monoclonal IgM autoantibodies recognizing the HNK‐1 epitope present on MAG as well as on other glycoproteins of the peripheral nervous system. Previously we have shown the efficient removal of anti‐MAG autoantibodies by PPSGG in a mouse model for anti‐MAG neuropathy (Herrendorff et al., 2017), reflecting the immunological aspects of gammopathy with continuous production of polyclonal IgM and IgG anti‐MAG antibodies. The study suggested a therapeutic potential for PPSGG as a fast‐acting antigen‐specific agent which selectively targets and eliminates the disease‐causing anti‐MAG IgM autoantibodies. Such a therapeutic could be highly beneficial compared to unselective immunomodulatory or immunosuppressive agents currently used to treat anti‐MAG neuropathy (Dalakas, 2018; Nobile‐Orazio, Bianco, Bianco, & Nozza, 2017). In addition to the results of the immunological mouse model, we assessed the therapeutic potential of PPSGG in a monkey neurological target model. Furthermore, the selectivity of anti‐MAG IgM binding and potential modulatory effects of PPSGG on the immune system were studied. Finally, a dose titration study in mice was conducted in preparation for first‐in‐patient clinical trials.
In ex vivo binding studies of patients’ anti‐MAG IgM autoantibodies to myelin in cynomolgus monkey sciatic nerve preparations, we could demonstrate that myelin reactivity was completely blocked when undiluted sera of anti‐MAG neuropathy patients were treated with PPSGG at low concentrations (250 μg/ml PPSGG), independent of anti‐MAG IgM autoantibody titers (>70’000—6’975 BTU). The applied concentration corresponds to a dose of approximately 10 mg/kg based on the total serum volume of humans. Although, the serum sample of patient 4 had only a low anti‐MAG IgM autoantibody titer of 19’333 BTU, the sample still exhibited residual myelin binding at the lowest PPSGG concentration (62.5 μg/ml PPSGG) in contrast to the samples of patient A, B, and C with higher titers (>70’000—36’383 BTU). Importantly, this dose of 62.5 μg/ml, corresponding to a dose of approximately 2 mg/kg humans, reduced myelin binding to almost the same degree as the higher dose (corresponding to 10 mg/kg).
These results are in line with the finding that anti‐MAG IgM autoantibodies of different patients can have different affinities to the HNK‐1 epitope and potentially explain why high anti‐MAG IgM titers do not necessarily correlate with disease severity (Delmont et al., 2019; Ogino, Tatum, Tatum, & Latov, 1994). Selective inhibition of IgM binding to myelin was confirmed by the use of sera from two control neuropathy patients in the indirect fluorescent assay. Both, the CANOMAD and CIDP patients’ sera exhibited IgM binding to myelin but no HNK‐1 reactivity (<1’000 BTU). Neither of the control neuropathy patients’ sera was inhibited by incubation with PPSGG. To further confirm selective binding of anti‐MAG IgM, we showed that PPSGG does not bind to other immunoglobulins of the IgM or IgG subclass present in normal human sera.
To corroborate that the dose range of 2–10 mg/kg would be sufficient to deplete circulating anti‐MAG autoantibodies in anti‐MAG neuropathy patients, a dose‐finding study in mice, using passive immunization with a mouse anti‐HNK‐1 IgM was performed. The intravenous injection of 5 µg PPSGG turned out to be sufficient to bind 89.43% (±1.33 SD) of the 60 µg anti‐MAG IgM and 10 µg PPSGG was sufficient to bind 93.28% (±0.50 SD) of the 120 µg anti‐HNK‐1 IgM. Based on these findings, a dose of approximately 80 mg PPSGG would be sufficient to bind and remove 1 g of anti‐MAG IgM, whereas 40 mg of PPSGG would be sufficient to bind and remove most of 1 g anti‐MAG IgM autoantibodies in humans. Since the anti‐HNK‐1/MAG IgM antibody levels were reduced for up to 2 weeks post‐administration of 60 µg anti‐HNK‐1 IgM and 10 µg PPSGG (Figures S1–S5 in supplementary information) and remained undetectable, and given the short half‐life of PPSGG, we conclude that it is a removal rather than a neutralization of the antibodies. The pentameric IgM has a molecular weight of 900–1000 kDa and is therefore approximately five times heavier than PPSGG with a calculated average weight of 194 kDa. Based on the results of our in vivo experiment, the binding stoichiometry of PPSGG:anti‐HNK‐1 IgM is around 1:1 to 1:2. In general, the paraprotein (monoclonal anti‐MAG IgM) levels in anti‐MAG patients are in the range of 1–10 g/L, whereas in healthy subjects the total IgM levels are between 0.5 and 3 g/L (Cook & Macdonald, 2007; Lunn & Nobile‐Orazio, 2016). Based on a range of 1–10 g/L of monoclonal anti‐MAG IgM (3–30 g total paraprotein), 240 mg to 2,400 mg of PPSGG are expected to be sufficient to completely remove the circulating anti‐MAG IgM antibodies and at doses of 120–1200 mg most of the paraprotein load should be removed. Finally, based on an estimated population average of 4 g/L paraprotein, a dose of 1,000 mg PPSGG is expected to lead to a complete response for the majority of anti‐MAG neuropathy patients.
To further evaluate the therapeutic potential of the glycopolymer PPSGG, its efficacy was compared with the B‐cell depleting treatment by an anti‐CD20 IgG antibody in actively immunized mice. Anti‐CD20 B‐cell depletion did not affect the anti‐MAG IgM titers in mice, whereas PPSGG effectively reduced the antibody titers. The former was surprising given the half‐life of a murine IgM (2–4 days), as it was expected that the significant reduction of B cells would reduce IgM titers during the following 5 weeks (Vieira & Rajewsky, 1988). Weekly treatment of immunized mice with PPSGG did not affect the number of circulating B cells (CD19+). This is consistent with the observation that PPSGG did not induce or lead to the release of the examined murine and human cytokines and chemokines in vivo and ex vivo, and did not activate B cells in immunized mice or anti‐MAG neuropathy patients ex vivo. Given these findings, we conclude that the cells responsible for the production of anti‐MAG IgM in the immunized mouse model are not circulating B cells but rather mature plasma cells or late stage B cells (CD20−) residing in the bone marrow or a small population of anti‐CD20 treatment resistant B cells in the spleen (Ahuja, Anderson, Anderson, Khalil, & Shlomchik, 2008; DiLillo et al., 2008; Häusler et al., 2018; Hofmann, Clauder, Clauder, & Manz, 2018).
In contrast to the immunological murine model, B cells clones that produce anti‐MAG IgM were successfully isolated from peripheral blood samples of anti‐MAG neuropathy patients and transformed with EBV. They maintained the ability to secrete anti‐MAG IgM antibodies ex vivo, showing that at least for some patients, anti‐MAG IgM‐producing B cells (CD20+) can be found in the periphery. Based on the observed limited efficacy of anti‐CD20 IgG antibody treatment, it is likely that the disease has heterogeneous characteristics in terms of the anti‐MAG IgM producing cells (Hammarlund et al., 2017; Leger et al., 2013; Nutt, Hodgkin, Hodgkin, Tarlinton, & Corcoran, 2015).
Since the immunized mice produce anti‐MAG antibodies of both the IgM and IgG subtype, we assumed that the mice would have memory B cells (CD20‐) residing in peripheral lymphoid organs and in the circulation (Herrendorff et al., 2017). Nevertheless, we were unable to observe a specific binding of the fluorescently labeled glycopolymer to B cells isolated from the spleen of immunized mice, where the binding of PPSGG could potentiality trigger an immune modulation. In B cells challenged with an antigen specific for the recognition by its B‐cell receptors, a local clustering of these receptors has been observed and the activation of these B cells can be monitored by the changes in intracellular Ca2+ levels (Scharenberg, Humphries, Humphries, & Rawlings, 2007; Vig & Kinet, 2009).
Since neither in murine nor in human B cells significant changes of the intracellular Ca2+ were observed, the sustained antibody reduction seen in our previous studies is unlikely driven by B cells directly. Furthermore, as there was no induction of peripheral inflammatory cytokines and chemokines neither in vivo after single and multiple PPSGG administrations in naive mice, nor ex vivo in various subpopulation of isolated human and murine leukocytes, we assume that the treatment of anti‐MAG neuropathy patients with PPSGG should not give rise to side effects by inducing acute inflammatory markers. However, some caution should be kept as some of these experiments were performed in the absence of its target (anti‐MAG autoantibodies) and because of the limitations of the in vivo and ex vivo models.
5. CONCLUSION
In contrast to treatments depleting anti‐CD20+ cells like rituximab or obinutuzumab, PPSGG binds and removes anti‐MAG IgM autoantibodies highly selectively within minutes. Our findings support a mechanism of action of PPSGG leading to a fast and selective in vivo depletion of pathogenic anti‐MAG IgM autoantibodies, independent of the response of anti‐MAG neuropathy patient to CD20+ B cells depleting treatments. This supports the use of PPSGG as a standalone therapy or in combination with immunosuppressive therapeutic agents, possibly at reduced doses, so that patients would benefit from both fast removal and long‐term suppression of anti‐MAG IgM autoantibody production.
CONFLICT OF INTEREST
R.H., P.H., A.J.S., and B.E. are co‐founders of a University of Basel spin‐off, Polyneuron Pharmaceuticals AG, whose activity is related to the subject matter of this article. A.J.S. and B.E. are members of the advisory board, and R.H. is a member of the board of directors. R.H., A.J.S., and B.E. are named as co‐inventors on relevant patent applications.
AUTHOR CONTRIBUTIONS
B.A., D.D., E.S., A.B., and T.O. performed experiments. E.D., T.D. M.T., P.T., and T.K. provided sera and information from patients. B.A., D.D., E.S., A.B., J.B., and P.H. analysed and interpreted data. B.A., D.D., E.S., J.B., and P.H. drafted the manuscript. B.A., A.J.S., B.E., R.H., and P.H. edited and revised the manuscript. All authors approved final version of the manuscript. R.H. and P.H. contributed to conception and design of research.
Supporting information
Figure S1–S5
ACKNOWLEDGMENTS
This work was funded by the Swiss Commission for Technology and Innovation, the Neuromuscular Research Association Basel, and the Gebert Rüf Stiftung.
All experiments were conducted in compliance with the ARRIVE guidelines.
Aliu B, Demeestere D, Seydoux E, et al. Selective inhibition of anti‐MAG IgM autoantibody binding to myelin by an antigen‐specific glycopolymer. J Neurochem. 2020;154:486–501. 10.1111/jnc.15021
Butrint Aliu and Delphine Demeestere contributed equally to this work.
Read the Editorial Highlight for this article on page 465.
Contributor Information
Butrint Aliu, Email: b.aliu@unibas.ch.
Pascal Hänggi, Email: pascalrhaenggi@gmail.com.
REFERENCES
- Ahuja, A. , Anderson, S. M. , Khalil, A. , & Shlomchik, M. J. (2008). Maintenance of the plasma cell pool is independent of memory B cells. Proceedings of the National Academy of Sciences of the United States of America, 105(12), 4802–4807. 10.1073/pnas.0800555105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Association H. E. C. o. E. o. t. W. M. (1964). Human experimentation: Code of Ethics of the World Medical Association (Declaration of Helsinki). Canadian Medical Association Journal, 91(11), 619. [PMC free article] [PubMed] [Google Scholar]
- Baba, Y. , & Kurosaki, T. (2016). Role of calcium signaling in B cell activation and biology. Current Topics in Microbiology and Immunology, 393, 143–174. [DOI] [PubMed] [Google Scholar]
- Benedetti, L. , Briani, C. , Grandis, M. , Vigo, T. , Gobbi, M. , Ghiglione, E. , … Schenone, A. (2007). Predictors of response to rituximab in patients with neuropathy and anti‐myelin associated glycoprotein immunoglobulin M. J Peripher Nerv Syst, 12(2), 102–107. 10.1111/j.1529-8027.2007.00129.x [DOI] [PubMed] [Google Scholar]
- Braun, P. E. , Frail, D. E. , & Latov, N. (1982). Myelin‐associated glycoprotein is the antigen for a monoclonal IgM in polyneuropathy. Journal of Neurochemistry, 39(5), 1261–1265. 10.1111/j.1471-4159.1982.tb12563.x [DOI] [PubMed] [Google Scholar]
- Broglio, L. , & Lauria, G. (2005). Worsening after rituximab treatment in anti‐mag neuropathy. Muscle and Nerve, 32(3), 378–379. 10.1002/mus.20386 [DOI] [PubMed] [Google Scholar]
- Cook, L. , & Macdonald, D. H. (2007). Management of paraproteinaemia. Postgraduate Medical Journal, 83(978), 217–223. 10.1136/pgmj.2006.054627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalakas, M. C. (2010). Pathogenesis and treatment of anti‐MAG neuropathy. Current Treatment Options in Neurology, 12(2), 71–83. 10.1007/s11940-010-0065-x [DOI] [PubMed] [Google Scholar]
- Dalakas, M. C. (2018). Advances in the diagnosis, immunopathogenesis and therapies of IgM‐anti‐MAG antibody‐mediated neuropathies. Therapeutic Advances in Neurological Disorders, 11, 1756285617746640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmont, E. , Attarian, S. , Antoine, J. C. , Paul, S. , Camdessanché, J. P. , Grapperon, A. M. , … Boucraut, J. (2019). Relevance of anti‐HNK1 antibodies in the management of anti‐MAG neuropathies. Journal of Neurology, 266(8), 1973–1979. [DOI] [PubMed] [Google Scholar]
- DiLillo, D. J. , Hamaguchi, Y. , Ueda, Y. , Yang, K. , Uchida, J. , Haas, K. M. , … Tedder, T. F. (2008). Maintenance of long‐lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. The Journal of Immunology, 180(1), 361–371. [DOI] [PubMed] [Google Scholar]
- Donjerković, D. , & Scott, D. W. (2000). Activation‐induced cell death in B lymphocytes. Cell Research, 10(3), 179–192. [DOI] [PubMed] [Google Scholar]
- Erb, M. , Flueck, B. , Kern, F. , Erne, B. , Steck, A. J. , & Schaeren‐Wiemers, N. (2006). Unraveling the differential expression of the two isoforms of myelin‐associated glycoprotein in a mouse expressing GFP‐tagged S‐MAG specifically regulated and targeted into the different myelin compartments. Molecular and Cellular Neurosciences, 31(4), 613–627. [DOI] [PubMed] [Google Scholar]
- Gabriel, J. M. , Erne, B. , Bernasconi, L. , Tosi, C. , Probst, A. , Landmann, L. , & Steck, A. J. (1998). Confocal microscopic localization of anti‐myelin‐associated glycoprotein autoantibodies in a patient with peripheral neuropathy initially lacking a detectable IgM gammopathy. Acta Neuropathologica, 95(5), 540–546. [DOI] [PubMed] [Google Scholar]
- Gabriel, J. M. , Erne, B. , Miescher, G. C. , Miller, S. L. , Vital, A. , Vital, C. , & Steck, A. J. (1996). Selective loss of myelin‐associated glycoprotein from myelin correlates with anti‐MAG antibody titre in demyelinating paraproteinaemic polyneuropathy. Brain, 119, 775–787. [DOI] [PubMed] [Google Scholar]
- Hammarlund, E. , Thomas, A. , Amanna, I. J. , Holden, L. A. , Slayden, O. D. , Park, B. , … Slifka, M. K. (2017). Plasma cell survival in the absence of B cell memory. Nature Communications, 8(1), 1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusler, D. , Häusser‐Kinzel, S. , Feldmann, L. , Torke, S. , Lepennetier, G. , Bernard, C. C. A. , … Weber, M. S. (2018). Functional characterization of reappearing B cells after anti‐CD20 treatment of CNS autoimmune disease. Proceedings of the National Academy of Sciences of the United States of America, 115(39), 9773–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesters, B. A. , van der Poel, C. E. , Das, A. , & Carroll, M. C. (2016). Antigen presentation to B cells. Trends in Immunology, 37(12), 844–854. [DOI] [PubMed] [Google Scholar]
- Herrendorff, R. , Hänggi, P. , Pfister, H. , Yang, F. , Demeestere, D. , Hunziker, F. , … Ernst, B. (2017). Selective in vivo removal of pathogenic anti‐MAG autoantibodies, an antigen‐specific treatment option for anti‐MAG neuropathy. Proceedings of the National Academy of Sciences of the United States of America, 114(18), E3689–E3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, K. , Clauder, A. K. , & Manz, R. A. (2018). Targeting B cells and plasma cells in autoimmune diseases. Frontiers in Immunology, 9, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollyoake, M. , Stühler, A. , Farrell, P. , Gordon, J. , & Sinclair, A. (1995). The normal cell cycle activation program is exploited during the infection of quiescent B lymphocytes by Epstein‐Barr virus. Cancer Research, 55(21), 4784–4787. [PubMed] [Google Scholar]
- Kelm, S. , Pelz, A. , Schauer, R. , Filbin, M. T. , Tang, S. , de Bellard, M. E. , … Bradfield, P. (1994). Sialoadhesin, myelin‐associated glycoprotein and CD22 define a new family of sialic acid‐dependent adhesion molecules of the immunoglobulin superfamily. Current Biology, 4(11), 965–972. [DOI] [PubMed] [Google Scholar]
- Latov, N. , Braun, P. E. , Gross, R. B. , Sherman, W. H. , Penn, A. S. , & Chess, L. (1981). Plasma cell dyscrasia and peripheral neuropathy: Identification of the myelin antigens that react with human paraproteins. Proceedings of the National Academy of Sciences of the United States of America, 78(11), 7139–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger, J.‐M. , Viala, K. , Nicolas, G. , Creange, A. , Vallat, J.‐M. , Pouget, J. , … Grp, R. S. (2013). Placebo‐controlled trial of rituximab in IgM anti‐myelin‐associated glycoprotein neuropathy. Neurology, 80(24), 2217–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn, M. P. , & Nobile‐Orazio, E. (2016). Immunotherapy for IgM anti‐myelin‐associated glycoprotein paraprotein‐associated peripheral neuropathies. Cochrane Database Systematic Review, (10), CD002827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdi‐Rogers, M. , & Hughes, R. A. (2014). Epidemiology of chronic inflammatory neuropathies in southeast England. European Journal of Neurology, 21(1), 28–33. [DOI] [PubMed] [Google Scholar]
- Mygland, A. , & Monstad, P. (2001). Chronic polyneuropathies in Vest‐Agder, Norway. European Journal of Neurology, 8(2), 157–165. [DOI] [PubMed] [Google Scholar]
- Nobile‐Orazio, E. , Baldini, L. , Barbieri, S. , Marmiroli, P. , Spagnol, G. , Francomano, E. , & Scarlato, G. (1988). Treatment of patients with neuropathy and anti‐MAG IgM M‐proteins. Annals of Neurology, 24(1), 93–97. [DOI] [PubMed] [Google Scholar]
- Nobile‐Orazio, E. , Bianco, M. , & Nozza, A. (2017). Advances in the treatment of paraproteinemic neuropathy. Current Treatment Options in Neurology, 19(12), 43. [DOI] [PubMed] [Google Scholar]
- Nutt, S. L. , Hodgkin, P. D. , Tarlinton, D. M. , & Corcoran, L. M. (2015). The generation of antibody‐secreting plasma cells. Nature Reviews Immunology, 15(3), 160–171. 10.1038/nri3795 [DOI] [PubMed] [Google Scholar]
- Ogino, M. , Tatum, A. H. , & Latov, N. (1994). Affinity studies of human anti‐MAG antibodies in neuropathy. Journal of Neuroimmunology, 52(1), 41–46. 10.1016/0165-5728(94)90160-0 [DOI] [PubMed] [Google Scholar]
- Pestronk, A. , Florence, J. , Miller, T. , Choksi, R. , Al‐Lozi, M. T. , & Levine, T. D. (2003). Treatment of IgM antibody associated polyneuropathies using rituximab. Journal of Neurology, Neurosurgery and Psychiatry, 74(4), 485–489. 10.1136/jnnp.74.4.485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarles, R. H. (2007). Myelin‐associated glycoprotein (MAG): Past, present and beyond. Journal of Neurochemistry, 100(6), 1431–1448. 10.1111/j.1471-4159.2006.04319.x [DOI] [PubMed] [Google Scholar]
- Renaud, S. , Fuhr, P. , Gregor, M. , Schweikert, K. , Lorenz, D. , Daniels, C. , … Steck, A. J. (2006). High‐dose rituximab and anti‐MAG‐associated polyneuropathy. Neurology, 66(5), 742–744. 10.1212/01.wnl.0000201193.00382.b3 [DOI] [PubMed] [Google Scholar]
- Ritz, M. F. , Erne, B. , Ferracin, F. , Vital, A. , Vital, C. , & Steck, A. J. (1999). Anti‐MAG IgM penetration into myelinated fibers correlates with the extent of myelin widening. Muscle and Nerve, 22(8), 1030–1037. [DOI] [PubMed] [Google Scholar]
- Sala, E. , Robert‐Varvat, F. , Paul, S. , Camdessanché, J. P. , & Antoine, J. C. (2014). Acute neurological worsening after Rituximab treatment in patients with anti‐MAG neuropathy. Journal of the Neurological Sciences, 345(1–2), 224–227. [DOI] [PubMed] [Google Scholar]
- Scharenberg, A. M. , Humphries, L. A. , & Rawlings, D. J. (2007). Calcium signalling and cell‐fate choice in B cells. Nature Reviews Immunology, 7(10), 778–789. 10.1038/nri2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck, A. J. , Murray, N. , Meier, C. , Page, N. , & Perruisseau, G. (1983). Demyelinating neuropathy and monoclonal IgM antibody to myelin‐associated glycoprotein. Neurology, 33(1), 19–23. [DOI] [PubMed] [Google Scholar]
- Steck, A. J. , Stalder, A. K. , & Renaud, S. (2006). Anti‐myelin‐associated glycoprotein neuropathy. Current Opinion in Neurology, 19(5), 458–463. 10.1097/01.wco.0000245368.36576.0d [DOI] [PubMed] [Google Scholar]
- Talamo, G. , Mir, M. A. , Pandey, M. K. , Sivik, J. K. , & Raheja, D. (2015). IgM MGUS associated with anti‐MAG neuropathy: A single institution experience. Annals of Hematology, 94(6), 1011–1016. 10.1007/s00277-014-2294-7 [DOI] [PubMed] [Google Scholar]
- Tang, S. , Shen, Y. J. , DeBellard, M. E. , Mukhopadhyay, G. , Salzer, J. L. , Crocker, P. R. , & Filbin, M. T. (1997). Myelin‐associated glycoprotein interacts with neurons via a sialic acid binding site at ARG118 and a distinct neurite inhibition site. Journal of Cell Biology, 138(6), 1355–1366. 10.1083/jcb.138.6.1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallat, J.‐M. , Magy, L. , Ciron, J. , Corcia, P. , Le Masson, G. , & Mathis, S. (2016). Therapeutic options and management of polyneuropathy associated with anti‐MAG antibodies. Expert Review of Neurotherapeutics., 16(9), 1111–1119. [DOI] [PubMed] [Google Scholar]
- Vieira, P. , & Rajewsky, K. (1988). The half‐lives of serum immunoglobulins in adult mice. European Journal of Immunology, 18(2), 313–316. 10.1002/eji.1830180221 [DOI] [PubMed] [Google Scholar]
- Vig, M. , & Kinet, J. P. (2009). Calcium signaling in immune cells. Nature Immunology, 10(1), 21–27. 10.1038/ni.f.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willison, H. J. , Trapp, B. D. , Bacher, J. D. , Dalakas, M. C. , Griffin, J. W. , & Quarles, R. H. (1988). Demyelination induced by intraneural injection of human antimyelin‐associated glycoprotein antibodies. Muscle and Nerve, 11(11), 1169–1176. 10.1002/mus.880111111 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1–S5