

Abstract
Chiral β‐aminoalkylzinc halides were prepared starting from optically pure commercial β‐amino‐alcohols. These amino‐alcohols were converted to the corresponding N‐pyrrolyl‐protected alkyl iodides which undergo a zinc insertion in the presence of LiCl (THF, 25 °C, 10–90 min). Subsequent Negishi cross‐coupling or acylation reactions with acid chlorides produced amino‐derivatives with retention of chirality. Diastereoselective CBS‐reductions of some prepared N‐pyrrolyl‐ketones provided 1,3‐subsituted N‐pyrrolyl‐alcohols with high diastereoselectivity. Additionally, a deprotection procedure involving an ozonolysis allowed the conversion of the pyrrole‐ring into a formamide without loss of optical purity.
Keywords: amines, cross-coupling, heterocycles, organozinc reagents, palladium
Chiral β‐aminoalkylzinc halides for enantioselective amine synthesis: Chiral β‐N‐pyrrolyl‐alkyl iodides prepared from commercial β‐amino‐alcohols undergo zinc insertion in the presence of LiCl (THF, 25 °C, 10–90 min). Negishi cross‐coupling or acylation quenching reactions produced a range of chiral β‐N‐pyrrolyl derivatives that may be converted into 1,3‐amino‐alcohols and other chiral amino‐derivatives. Subsequent ozonolysis allowed the conversion of the pyrrole moiety into formamides without loss of optical purity.

Introduction
The preparation of chiral amino‐derivatives is of great synthetic importance for the development of new pharmaceuticals and agrochemicals.1 In this context, the preparation of organometallic intermediates bearing an amino group in β‐position may provide a powerful tool for the synthesis of functionalized amino‐derivatives. Organozinc compounds are known to tolerate a broad range of functional groups2 and are therefore well suited for synthesis of chiral amino‐derivatives. Jackson has demonstrated the utility of serine derived chiral organozinc reagents for the preparation of novel amino acids using Negishi cross‐coupling reactions.3 Furthermore, chiral β‐aminoalkylzinc reagents derived from the corresponding amino‐alcohols have been already used for the synthesis of functionalized amines.4, 5
In this work, we have envisioned to prepare stable zinc organometallics bearing an amino group in β‐position as key organometallic intermediates. Thus, 2‐amino‐alcohols of type 1, readily available by reduction of the corresponding natural amino‐acid, will afford after protection of the amino group the corresponding amino‐alcohol of type 2 and after functional group interconversion the corresponding alkyl iodides of type 3.
Treatment of these functionalized alkyl iodides (3) with zinc dust in the presence of LiCl should provide 2‐aminoalkylzinc halides (4) which should readily react with various electrophiles in the presence of an appropriate transition‐metal catalyst (Scheme 1 a).
Scheme 1.

Preparation of chiral β‐aminoalkylzinc halides of type 4 from optically pure amino‐alcohols of type 1. [a] Zinc insertion conditions: LiCl (1.0 equiv), Zn dust (1.5 equiv, activated with DBE/TMSCl) in THF at 25 °C. [b] Complexed with LiCl. [c] Determined by titration against iodine.7
Results and Discussion
Preliminary results showed that the choice of the protecting group was essential for the success of the proposed reaction sequence (Scheme 1 a). Whereas most electron‐withdrawing amino‐protecting groups6 will enhance elimination side reactions for the resulting organozinc reagent of type 4, most donating amino‐protecting groups on the other hand will favor aziridination of the starting iodide 3. After some experimentation, we have found that the use of a pyrrole ring as protecting group is an excellent compromise which allows to accomplish the proposed sequence with high efficiency (Scheme 1 b).
Therefore, the treatment of commercially available 1,2‐amino‐alcohols (1 a–e) derived from optically enriched amino‐acids8 with 2,5‐dimethoxytetrahydrofuran in the presence of NaOAc/AcOH in 1,2‐dichloroethane (DCE) and water (90 °C, 1–16 h) produced the pyrrol‐1‐yl‐alcohol derivatives 2 a–e in 71–92 % yield.9 An Appel reaction10 (PPh3, I2, imidazole in dichloromethane (DCM)) allowed the conversion of the alcohols 2 a–e to the corresponding primary iodides 3 a–e in 71–91 % yield. Direct insertion of zinc powder (1.5 equiv, >325 mesh) previously activated with 1,2‐dibromoethane (DBE) and Me3SiCl (TMSCl) to the alkyl iodides 3 a–e in presence of LiCl11 proceeded smoothly in THF at 25 °C (10–90 min) furnishing the desired alkylzinc halides (4 a–e) in 71–93 % yield. These alkylzinc reagents showed no tendency to undergo elimination reactions and were stable at 25 °C for several days without decomposition.
In preliminary experiments, 2‐(1H‐pyrrol‐1‐yl)ethylzinc iodide (4 a) was used to determine the optimum cross‐coupling and acylation conditions. Negishi cross‐coupling reactions12 of 4 a were best performed using 2 mol % Pd(OAc)2 and 4 mol % SPhos13 as catalyst with both electron‐rich or electron‐poor aryl‐ and heteroaryl bromides or iodides in THF (25 °C, 16 h) affording the pyrrole derivatives 5 a–h in 83–98 % yield. Negishi acylation reactions14 of 4 a were also readily performed using aryl‐, heteroaryl‐ and cyclopropyl‐substituted acyl chlorides in the presence of 4 mol % Pd(PPh3)4 in THF (25–50 °C, 16 h) providing the β‐pyrrolyl‐ketones (5 i–l) in 73–87 % yield (Scheme 2). No significant amount of β‐hydride elimination was observed in these cross‐coupling reactions.
Scheme 2.

Palladium‐catalyzed Negishi cross‐coupling and acylation reactions of 4 a with various aryl halides and acid chlorides leading to functionalized pyrrole derivatives 5 a–l. [a] Yield of analytically pure isolated products.
With these optimized conditions in hand, we have extended these reactions to the chiral organozinc reagents 4 b–e. Various chiral arylated pyrroles (6 a–d, 7 a–e, 8 a–d, 9 a–e) were obtained after Negishi cross‐couplings with aryl iodides and bromides in 75–99 % isolated yield with full retention of configuration (ee=99 %, Scheme 3).
Scheme 3.

Negishi cross‐coupling reactions of chiral β‐N‐pyrrolyl alkylzinc reagents 4 b–e with various functionalized aryl halides. [a] Yield of analytically pure isolated products.
Similarly, the reagents 4 b–e were used in palladium‐catalyzed acylation reactions with several aryl‐, heteroaryl‐ and alkyl‐substituted acyl chlorides providing the desired chiral ketones (7 f, 7 i,j and 8 e–h) in 66–86 % yield. Furthermore, as a cheap alternative to palladium‐catalysis and to prevent cross‐coupling side reactions, copper(I) iodide was used as a catalyst for acylations.15 Chiral halogenated ketones (6 e–g, 7 g–h, 9 f) and a 2‐thienyl‐ketone (6 h) were obtained in 59–80 % yield (Scheme 4). All ketones, whether prepared by using palladium‐ or copper‐derivatives as catalyst, were obtained with full retention of configuration (ee=99 %).
Scheme 4.

Palladium‐ and copper‐catalyzed acylation reactions using chiral organozinc reagents 4 b–e with various acyl chlorides. [a] Palladium‐catalyzed acylation. [b] Copper‐catalyzed acylation. [c] Yield of analytically pure isolated products.
To further extend these reactions, we have prepared an indolyl‐substituted zinc reagent.16 Starting from tryptophanol (R)‐1 f, successive pyrrole‐protection leading to (R)‐2 f and iodination using our standard procedures17 provided the optically pure alkyl iodide (R)‐3 f which was converted to the corresponding organozinc reagent (R)‐4 f. Interestingly, the presence of a secondary amine of the indole side‐chain did not interfere with the zinc insertion and the resulting organozinc reagent (R)‐4 f was prepared in 91 % yield.18 Palladium‐catalyzed Negishi cross‐coupling reactions with various aryl and heteroaryl iodides or bromides were performed and functionalized indole derivatives 10 a–f were obtained in 68–98 % yield without racemization (Scheme 5).
Scheme 5.

Preparation of a chiral pyrrole‐protected alkylzinc reagent ((R)‐4 f) derived from tryptophanol (R)‐1 f bearing an unprotected indolyl‐moiety and its use in Negishi cross‐coupling reactions.
Additionally, a tyrosine‐based alkylzinc compound was prepared.16 For this purpose, the acidic phenolic hydroxyl group of l‐tyrosine was protected by methylation with MeI. Starting from l‐tyrosine, a straightforward synthesis pathway including pyrrole formation, phenol protection, methyl esterification and subsequent reduction of the methyl ester was performed in 40 % yield over three steps providing the pyrrol‐1‐yl‐alcohol (S)‐2 g (Scheme 6). A subsequent conversion into the alkyl iodide (S)‐3 g using Appel‐reaction conditions was proceeding in 95 % yield and 92 % ee. After oxidative addition of zinc dust into this alkyl iodide, the corresponding chiral N‐pyrrolyl‐alkylzinc halide (S)‐4 g was obtained after 15 min at 25 °C in 99 % yield. Negishi cross‐coupling reactions using electron‐rich and electron‐poor aryl iodides afforded the functionalized tyrosine derivatives (S)‐11 a–c in 93–96 % yield. Additional copper‐ and palladium‐catalyzed acylation reactions were performed providing ketones (S)‐11 d–f bearing halogens in ortho‐, meta‐ or para‐position in 75–84 % yield (Scheme 6). All products were obtained with retention of configuration with 92 % ee as determined previously for the starting alkyl iodide (S)‐3 g.
Scheme 6.

Preparation of a chiral N‐pyrrolyl‐alkylzinc iodide ((S)‐4 g) derived from l‐tyrosine and its use in Negishi cross‐coupling and acylation reactions.
In addition to the organozinc reagents derived from amino‐acids, we have also prepared a chiral zinc reagent derived from trans‐(1S,2S)‐2‐aminocyclohexanol ((S,S )‐1 h). After protection as a pyrrole ring, the corresponding N‐pyrrolyl‐alkyl alcohol (S,S )‐2 h was obtained in 83 % yield and further Appel‐iodination led to the desired cycloalkyl iodide (S,R )‐3 h at 50 °C in 76 % yield after 30 h. Following zinc insertion of (S,R)‐3 h provided the alkylzinc reagent (S)‐4 h in 79 % yield. The stereochemistry of the carbon bearing zinc is lost due to the radical insertion reaction.19 However, cross‐coupling reactions of the organozinc reagent (S)‐4 h using Pd(OAc)2 and SPhos as catalyst with different substituted electron‐rich and electron‐deficient electrophiles provided the cross‐coupling products 12 a–d in 84–97 % yield (Scheme 7). Remarkably, using this palladium system, the trans‐cross‐coupling products were formed selectively (dr=99:1) and all products exhibit a high optical purity (99 % ee).19
Scheme 7.

Preparation of a chiral N‐pyrrolyl‐alkylzinc reagent starting from the 1,2‐cis substituted iodocyclohexyl derivative (S,R )‐3 h providing exclusively trans‐configured products using palladium‐catalysis.
Reaction of (S)‐4 h with 4‐fluorobenzoyl chloride using Pd(PPh3)4 as acylation catalyst led solely to the trans‐ketone (S,S )‐12 e in 93 % yield (dr=99:1, 99 % ee). The same reaction using copper(I) iodide as catalyst resulted in a diastereomeric mixture (dr=83:17).
Furthermore, this method was extended to a N‐pyrrolyl‐alkylzinc reagent derived from trans‐(1R,2R)‐2‐aminocyclopentanol ((R,R )‐1 i). After protection of the amino group as a pyrrole ring, the corresponding N‐pyrrolyl‐alcohol (R,R )‐2 i was obtained in 77 % yield. Its iodination was rather difficult due to the increased formation of an elimination product and the cis‐iodide20 (R,S )‐3 i was obtained at 50 °C in only 43 % yield after 16 h reaction time. Subsequent insertion of zinc dust in the presence of LiCl led to the secondary organozinc reagent (R)‐4 i at 25 °C after 10 min reaction time in 96 % yield (Scheme 8).
Scheme 8.

Preparation of the 2‐N‐pyrrolyl‐cyclopentlyzinc reagent (R)‐4 i. Exclusively trans‐configured products of type 13 were obtained after palladium‐catalyzed Negishi cross‐coupling.
A stereo‐converging reaction occurred when using the diastereomeric mixture of organozinc reagent (R)‐4 i in palladium‐catalyzed Negishi cross‐coupling reactions with aryl iodides bearing various functional groups in ortho‐, meta‐ or para‐position providing selectively the trans‐products 13 a‐f in 75–93 % yield with high enantiopurity (Scheme 8). Determination of the crystal structure of (S,R)‐13 a by X‐ray analysis confirmed the trans‐configuration.20
A post‐functionalization of some chiral pyrrole‐containing products was performed providing chiral amino‐alcohol derivatives. Thus, the acylation products (R) ‐8 f and (S)‐8 f were selectively reduced to optically pure 1,3‐substituted N‐pyrrolyl‐alcohols using the Corey–Bakshi–Shibata asymmetric reduction.21 Therefore, the CBS catalyst ((R)‐ or (S)‐2‐methyl‐CBS‐oxazaborolidine) was mixed with BH3⋅SMe2 at 0 °C and the ketone 8 f was slowly added. The desired alcohols 14 a–d were obtained in 92–98 % yield with a diastereomeric ratio of 99:1 and an enantiomeric excess of 99 % (Scheme 9).
Scheme 9.

Enantioselective CBS‐reduction of the ketones (R)‐8 f and (S)‐8 f provides a pathway towards the 1,3‐substituted N‐pyrrolyl‐alcohols 14 a–d.
To confirm the absolute configuration, the alcohol (R,S )‐14 d was esterified with the (R)‐ as well as the (S)‐Mosher acid (MTPA) and the resulting two diastereomeric esters were analyzed by NMR spectroscopy. The chemical shifts were compared and evaluated as reported in the literature (see Supporting Information).22 The analysis confirmed the configuration previously predicted by the CBS‐model. Thus, the (R)‐CBS catalyst provided the (S)‐alcohols (14 b, 14 c) and vice versa the (S)‐CBS catalyst led to the (R)‐alcohols (14 a, 14 d).
A deprotection procedure for the pyrrole group was developed based on literature procedures.23 Ozonolysis converted pyrroles into solid formamides, which were easier to handle than the corresponding alkylamines. Thus, treatment of a pyrrole derivative with ozone at −78 °C in a DCM/MeOH mixture for 5 min, followed by a reductive work‐up with methyl sulfide led to a mixture of the corresponding formamide and diformylamine. This mixture was then stirred in a diluted potassium hydroxide solution in ethanol (0.1 m) for 1 h leading selectively to formamides of type 15 (Table 1).24
Table 1.
Deprotection of the pyrroles via ozonolysis and subsequent reductive and basic workup leading to the formamides 15 a–g with retention of configuration.
|
| ||
|---|---|---|
|
Entry |
Functionalized pyrrole |
Formamide 15 a–15 g |
|
1 |
|
|
|
|
5 g |
15 a, 71 % yield |
|
2 |
|
|
|
|
(S)‐8 c, 99 % ee |
(S)‐15 b, 74 % yield, 99 % ee |
|
3 |
|
|
|
|
(S)‐8 d, 99 % ee |
(S)‐15 c, 65 % yield, 99 % ee |
|
4 |
|
|
|
|
(S)‐11 a, 92 % ee |
(S)‐15 d, 62 % yield, 92 % ee |
|
5 |
|
|
|
|
(S,R )‐12 c, dr=99:1, 99 % ee |
(S,R )‐15 e, 74 % yield, dr=99:1, 99 % ee |
|
6 |
|
|
|
|
(R,S )‐13 f, dr=99:1, 99 % ee |
(R,S )‐15 f, 71 % yield, dr=99:1, 99 % ee |
|
7 |
|
|
|
|
(S,R )‐14 a, dr=99:1, 99 % ee |
(R,S )‐15 g, 76 % yield, dr=99:1, 99 % ee |








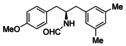






This deprotection protocol was applied to selected cross‐coupling products (5 g, 8 c, 8 d, 11 a, 12 c, 13 f) as well as the CBS‐reduced alcohol (S,R )‐14 a and the corresponding formamide derivatives 15 a–g were obtained 62–76 % yield (Table 1). The stereocenters were not affected by this treatment and full retention of configuration was achieved for all compounds.
Additionally, the crystal structures of (S,R )‐15 e and (R,S )‐15 g were obtained by X‐ray diffraction of single‐crystals. The structure of (S,R )‐15 e (Figure 1) confirmed the trans‐configuration of the formamide group to the aryl moiety which was determined for pyrrole derivative (S,R )‐12 c via NMR‐spectroscopy.
Figure 1.

Molecular structure of the deprotected formamide (S,R )‐15 e. Thermal ellipsoids are drawn at 50 % probability level. The fluorine atoms of both CF3 groups are disordered.
Furthermore, the crystal structure of (R,S )‐15 g (Figure 2) showed an anti‐configuration. This was in accordance with the results of the Mosher's ester analysis using NMR‐spectroscopy described above.
Figure 2.

Molecular structure of the deprotected formamide (R,S )‐15 g. Thermal ellipsoids are drawn at 50 % probability level.
Conclusions
In conclusion, we have developed a straightforward preparation of pyrrole‐protected β‐aminoalkylzinc halides starting from optically pure β‐amino‐alcohols. The pyrrole moiety was a suitable protecting group for the preparation of such organozinc reagents. These β‐aminoalkylzinc reagents underwent palladium‐catalyzed Negishi cross‐coupling reactions and acylation reactions with a broad range of functionalized electrophiles providing a range of chiral amino‐derivatives. Similarly, secondary cycloalkylzinc halides were prepared from the corresponding iodides. Subsequent cross‐coupling and acylation reactions produced various chiral 1,2‐cyclohexyl‐ and cyclopentyl‐amines. An amino‐ketone prepared via acylation reaction was reduced using an asymmetric CBS‐reduction providing optically pure 1,3‐substituted N‐pyrrolyl‐alcohols. A deprotection procedure was also developed involving an ozonolysis with subsequent reductive work‐up. This allowed to convert these pyrrole derivatives into diastereomerically and enantiomerically enriched formamides.
Experimental Section
Typical procedure for the preparation of N‐pyrrolyl‐alcohols of type 2 from amino‐alcohols of type 1: Preparation of (S)‐3‐phenyl‐2‐(1H‐pyrrol‐1‐yl)propan‐1‐ol ((S)‐2d)
(S)‐2‐Amino‐3‐phenyl‐1‐propanol (S)‐1 d (3.8 g, 25 mmol, 1.0 equiv) and NaOAc⋅3H2O (3.4 g, 25 mmol,1.0 equiv) were dissolved in a DCE/water mixture (1:1, 75 mL) and glacial acetic acid (12.5 mL). 2,5‐Dimethoxytetrahydrofuran (cis/trans mixture, 3.3 g, 25 mmol, 1.0 equiv) was added and the two‐phase mixture was refluxed at 90 °C under vigorous stirring for 16 h. The organic phase was separated, the aqueous phase was extracted three times with EtOAc and the combined organic phases were neutralized with sat. aq. NaHCO3 solution, washed with brine and dried over MgSO4. After evaporation of solvents, the residue was purified by flash column chromatography (i‐hexanes/ethyl acetate 8:2+1 vol% Et3N) and the corresponding pyrrol‐1‐yl‐alcohol (S)‐2 d (4.3 g, 21 mmol, 85 % yield) was obtained.
Typical procedure for the preparation of alkyl iodides of type 3: Preparation of (S)‐1‐(1‐iodo‐3‐phenylpropan‐2‐yl)‐1H‐pyrrole ((S)‐3d)
In a dry Schlenk flask, PPh3 (6.1 g, 23 mmol, 1.1 equiv) and imidazole (1.6 g, 23 mmol, 1.1 equiv) were dissolved in dry DCM (44 mL) and cooled to 0 °C. Iodine (5.9 g, 23 mmol, 1.1 equiv) was added in three portions over a period of 10 min. The corresponding pyrrol‐1‐yl‐alcohol (S)‐2 d (4.3 g, 21 mmol, 1.0 equiv) dissolved in dry DCM (40 mL) was added dropwise to the reaction mixture at 0 °C over a period of 30 min. The mixture was stirred for 30 min at this temperature and then warmed to 25 °C and stirred additionally for 1 h. After quenching with sat. aq. Na2S2O3 solution, the organic phase was separated and the aqueous phase extracted three times with DCM. The combined organic phases were dried over CaCl2 and solvents were evaporated. Purification by flash column chromatography (i‐hexanes/ethyl acetate 99:1) afforded the corresponding alkyl iodide (S)‐3 d (5.5 g, 18 mmol, 84 % yield, 99 % ee).
Typical procedure for the zinc insertion into pyrrole‐containing alkyl iodides: Preparation of (S)‐(3‐phenyl‐2‐(1H‐pyrrol‐1‐yl)propyl)zinc(II) iodide ((S)‐4d)
In a Schlenk flask, LiCl (340 mg, 8.0 mmol 1.0 equiv) and zinc dust (785 mg, 12 mmol, 1.5 equiv) were dried in vacuo. Under argon atmosphere, freshly distilled dry THF (12 mL) was added and the zinc dust was activated using 1,2‐dibromoethane (35 μl, 5 mol %) and Me3SiCl (50 μL, 5 mol %). The suspension was slightly heated with a heat‐gun until a gas formation started. Afterwards, the alkyl iodide (S)‐3 d (2.5 g, 8.0 mmol, 1.0 equiv) was added slowly and the mixture stirred at 25 °C. The reaction progress was monitored by gas chromatographic analysis of small aliquots quenched with sat. aq. NH4Cl solution. After 10 min, the solution was passed through a syringe filter and the concentration of the organozinc reagent (S)‐4 d was determined by titration against iodine (13 mL, 0.47 m, 6.5 mmol, 82 % yield).7
Typical procedure for palladium‐catalyzed cross‐coupling reactions: Preparation of ethyl (S)‐4‐(3‐phenyl‐2‐(1H‐pyrrol‐1‐yl)propyl)benzoate ((S)‐8c)
In a dry Schlenk flask, Pd(OAc)2 (2.3 mg, 10 μmol, 2 mol %) and SPhos (8.2 mg, 20 μmol, 4 mol %) were dissolved in freshly distilled dry THF (1 mL) and stirred for 10 min at 25 °C. Ethyl 4‐iodobenzoate (140 mg, 0.5 mmol, 1.0 equiv) was added and to this mixture, the alkylzinc reagent (S)‐4 d in THF (1.3 mL, 0.47 m, 0.6 mmol, 1.2 equiv) was added dropwise. The reaction mixture was stirred for 16 h at 25 °C and then quenched with sat. aq. NH4Cl solution (1 mL). The mixture was extracted three times with EtOAc, the combined organic phases were dried over MgSO4 and solvents were evaporated. Purification via flash column chromatography (i‐hexanes/ethyl acetate 95:5) provided the cross‐coupling product (S)‐8 c (162 mg, 0.49 mmol, 96 % yield, 99 % ee).
Typical procedure for palladium‐catalyzed acylation reactions: Preparation of (S)‐1‐(3‐chlorophenyl)‐4‐phenyl‐3‐(1H‐pyrrol‐1‐yl)butan‐1‐one ((S)‐8f)
In a dry Schlenk flask, Pd(PPh3)4 (23 mg, 20 μmol, 4 mol %) was dissolved in freshly distilled dry THF (1 mL) and 3‐chlorobenzoyl chloride (88 mg, 0.5 mmol, 1.0 equiv) was added. To this mixture, the alkylzinc reagent (S)‐4 d in THF (1.3 mL, 0.47 m, 0.6 mmol, 1.2 equiv) was added dropwise. The reaction mixture was stirred for 16 h at 50 °C and then quenched with sat. aq. NH4Cl solution (1 mL). The mixture was extracted three times with EtOAc, the combined organic phases were dried over MgSO4 and solvents were evaporated. Purification via flash column chromatography (i‐hexanes/ethyl acetate 95:5) provided the acylation product (S)‐8 f (138 mg, 0.43 mmol, 85 % yield, 99 % ee).
Typical procedure for deprotection of the pyrrole‐group: Preparation of ethyl (S)‐4‐(2‐formamido‐3‐phenylpropyl)benzoate ((S)‐15b)
In a Schlenk flask, (S)‐8 c (334 mg, 1.0 mmol, 1.0 equiv) was dissolved in DCM (8 mL) and methanol (2 mL) and then cooled to −78 °C. Ozone was passed through the solution for 5 min and afterwards flushed with nitrogen for 10 min. Dimethyl sulfide (621 mg, 10 mmol, 10 equiv) was added and the reaction mixture stirred for 4 h at 25 °C. After evaporation of the solvents, the residue was stirred for 1 h in 10 mL KOH in ethanol (0.1 m) and afterwards ethanol was removed under reduced pressure. The crude product was purified by via flash column chromatographic purification (i‐hexanes/ethyl acetate 1:1) and formamide derivative (S)‐15 b (230 mg, 0.74 mmol, 74 % yield, 99 % ee) was obtained.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the LMU Munich and the DFG for financial support. Furthermore, we like to thank Albemarle (Frankfurt a. M.) and the BASF (Ludwigshafen) for the generous gift of chemicals.
M. Leroux, W.-Y. Huang, Y. Lemke, T. J. Koller, K. Karaghiosoff, P. Knochel, Chem. Eur. J. 2020, 26, 8951.
References
- 1.
- 1a. Nugent T. C., El-Shazly M., Adv. Synth. Catal. 2010, 352, 753; [Google Scholar]
- 1b. Nugent T. C., Chiral Amine Synthesis Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim: 2010; [Google Scholar]
- 1c. Breuer M., Ditrich K., Habicher T., Hauer B., Kesseler M., Stürmer R., Zelinski T., Angew. Chem. Int. Ed. 2004, 43, 788; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 806. [Google Scholar]
- 2.
- 2a. Dagousset G., François C., León T., Blanc R., Sansiaume-Dagousset E., Knochel P., Synthesis 2014, 46, 3133; [Google Scholar]
- 2b. Bégouin J.-M., Gosmini C., J. Org. Chem. 2009, 74, 3221; [DOI] [PubMed] [Google Scholar]
- 2c. Krasovskiy A., Malakhov V., Gavryushin A., Knochel P., Angew. Chem. Int. Ed. 2006, 45, 6040; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6186; [Google Scholar]
- 2d. Lacasse M.-C., Poulard C., Charette A. B., J. Am. Chem. Soc. 2005, 127, 12440; [DOI] [PubMed] [Google Scholar]
- 2e. Gosmini C., Rollin Y., Nedelec J. Y., Perichon J., J. Org. Chem. 2000, 65, 6024; [DOI] [PubMed] [Google Scholar]
- 2f. Charette A. B., Beauchemin A., Marcoux J.-F., J. Am. Chem. Soc. 1998, 120, 5114; [Google Scholar]
- 2g. Knochel P., Singer R. D., Chem. Rev. 1993, 93, 2117. [Google Scholar]
- 3.
- 3a. Ross A. J., Dreiocker F., Schäfer M., Oomens J., Meijer A. J. H. M., Pickup B. T., Jackson R. F. W., J. Org. Chem. 2011, 76, 1727; [DOI] [PubMed] [Google Scholar]
- 3b. Ross A. J., Lang H. L., Jackson R. F. W., J. Org. Chem. 2010, 75, 245; [DOI] [PubMed] [Google Scholar]
- 3c. Carrillo-Marquez T., Caggiano L., Jackson R. F. W., Grabowska U., Rae A., Tozer M. J., Org. Biomol. Chem. 2005, 3, 4117; [DOI] [PubMed] [Google Scholar]
- 3d. Deboves H. J. C., Grabowska U., Rizzo A., Jackson R. F. W., J. Chem. Soc. Perkin Trans. 1 2000, 4284; [Google Scholar]
- 3e. Jackson R. F. W., James K., Wythes M. J., Wood A., J. Chem. Soc. Chem. Commun. 1989, 644. [Google Scholar]
- 4. Duddu R., Eckhardt M., Furlong M., Knoess H. P., Berger S., Knochel P., Tetrahedron 1994, 50, 2415. [Google Scholar]
- 5.
- 5a. Goddard C. M. L., Massah A. R., Jackson R. F. W., Tetrahedron 2010, 66, 9175; [Google Scholar]
- 5b. Tang S., Li S., Zhou D., Zeng H., Wang N., Sci. China Chem. 2013, 56, 1293. [Google Scholar]
- 6.An exception to this is the electron-withdrawing protecting group TFA which has been reported to stabilize the resulting organozinc compound (see Reference [5a]).
- 7. Krasovskiy A., Knochel P., Synthesis 2006, 5, 0890. [Google Scholar]
- 8. Dickman D. A., Meyers A. I., Smith G. A., Gawley R. E., Org. Synth. 1985, 63, 136. [Google Scholar]
- 9.
- 9a. Gourlay B. S., Molesworth P. P., Ryan J. H., Smith J. A., Tetrahedron Lett. 2006, 47, 799; [Google Scholar]
- 9b. Jefford C. W., de Villedon de Naide F., Sienkiewicz K., Tetrahedron: Asymmetry 1996, 7, 1069. [Google Scholar]
- 10.
- 10a. Morozova V., Skotnitzki J., Moriya K., Karaghiosoff K., Knochel P., Angew. Chem. Int. Ed. 2018, 57, 5516; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5614; [Google Scholar]
- 10b. Lange G. L., Gottardo C., Synth. Commun. 1990, 20, 1473; [Google Scholar]
- 10c. Garegg P. J., Samuelsson B., J. Chem. Soc. Perkin Trans. 1 1980, 2866; [Google Scholar]
- 10d. Appel R., Angew. Chem. Int. Ed. Engl. 1975, 14, 801; [Google Scholar]; Angew. Chem. 1975, 87, 863. [Google Scholar]
- 11.
- 11a. Blümke T. D., Piller F. M., Knochel P., Chem. Commun. 2010, 46, 4082; [DOI] [PubMed] [Google Scholar]
- 11b. Feng C., Easter Q. T., Blum S. A., Organometallics 2017, 36, 2389; [Google Scholar]
- 11c. Feng C., Cunningham D. W., Easter Q. T., Blum S. A., J. Am. Chem. Soc. 2016, 138, 11156; [DOI] [PubMed] [Google Scholar]
- 11d. Jess K., Kitagawa K., Tagawa T. K. S., Blum S. A., J. Am. Chem. Soc. 2019, 141, 9879. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Negishi E., Valente L. F., Kobayashi M., J. Am. Chem. Soc. 1980, 102, 3298; [Google Scholar]
- 12b. Negishi E., Acc. Chem. Res. 1982, 15, 340; [Google Scholar]
- 12c. Hadei N., Kantchev E. A. B., O'Brie C. J., Organ M. G., Org. Lett. 2005, 7, 3805; [DOI] [PubMed] [Google Scholar]
- 12d. Valente C., Belowich M. E., Hadei N., Organ M. G., Eur. J. Org. Chem. 2010, 4343; [Google Scholar]
- 12e. Haas D., Hammann J. M., Greiner R., Knochel P., ACS Catal. 2016, 6, 1540. [Google Scholar]
- 13.
- 13a. Barder T. E., Walker S. D., Martinelli J. R., Buchwald S. L., J. Am. Chem. Soc. 2005, 127, 4685; [DOI] [PubMed] [Google Scholar]
- 13b. Yang Y., Oldenhuis N. J., Buchwald S. L., Angew. Chem. Int. Ed. 2013, 52, 615; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 643. [Google Scholar]
- 14.
- 14a. Rieke R. D., Kim S.-H., Tetrahedron Lett. 2011, 52, 3094; [Google Scholar]
- 14b. Negishi E., Bagheri V., Chatterjee S., Luo F.-T., Miller J. A., Stoll A. T., Tetrahedron Lett. 1983, 24, 5181. [Google Scholar]
- 15.
- 15a. Jung H.-S., Kim S.-H., Tetrahedron Lett. 2015, 56, 1004; [Google Scholar]
- 15b. Nakamura E., Kuwajima I., J. Am. Chem. Soc. 1984, 106, 3368. [Google Scholar]
- 16. Massah A. R., Dreiocker F., Jackson R. F. W., Pickup B. T., Oomens J., Meijer A. J. H. M., Schäfer M., Phys. Chem. Chem. Phys. 2011, 13, 13255. [DOI] [PubMed] [Google Scholar]
- 17.For the experimental procedure, see Supporting Information.
- 18. Knoess H. P., Furlong M. T., Rozema M. J., Knochel P., J. Org. Chem. 1991, 56, 5974. [Google Scholar]
- 19. Thaler T., Haag B., Gavryushin A., Schober K., Hartmann E., Gschwind R. M., Zipse H., Mayer P., Knochel P., Nat. Chem. 2010, 2, 125. [DOI] [PubMed] [Google Scholar]
- 20.Structure and configuration was determined by single-crystal x-ray analysis; see Supporting Information.
- 21.
- 21a. Corey E. J., Helal C. J., Angew. Chem. Int. Ed. 1998, 37, 1986; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 2092; [Google Scholar]
- 21b. Corey E. J., Shibata S., Bakshi R. K., J. Org. Chem. 1988, 53, 2861. [Google Scholar]
- 22.
- 22a. Hoye T. R., Jeffrey C. S., Shao F., Nat. Protoc. 2007, 2, 2451; [DOI] [PubMed] [Google Scholar]
- 22b. Seco J. M., Quiñoá E., Riguera R., Chem. Rev. 2004, 104, 17. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Tokumaru E., Tengeiji A., Nakahara T., Shiina I., Chem. Lett. 2015, 44, 1768; [Google Scholar]
- 23b. Kashima C., Maruyama T., Fujioka Y., Harada K., J. Chem. Soc. Perkin Trans. 1 1989, 1041. [Google Scholar]
- 24.
- 24a. Yinglin H., Hongwen H., Synthesis 1990, 615; [Google Scholar]
- 24b. Wüst F., Skaddan M. B., Leibnitz P., Spies H., Katzenellenbogen J. A., Johannsen B., Bioorg. Med. Chem. 1999, 7, 1827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary