

Abstract
Earth‐abundant metal complexes have been attracting increasing attention in the field of photo(redox)catalysis. In this work, the synthesis and full characterisation of four new heteroleptic CuI complexes are reported, which can work as photosensitizers. The complexes bear a bulky diphosphine (DPEPhos=bis[(2‐diphenylphosphino)phenyl] ether) and a diimine chelating ligand based on 1‐benzyl‐4‐(quinol‐2′yl)‐1,2,3‐triazole. Their absorption has a relative maximum in the visible‐light region, up to 450 nm. Thus, their use in photocatalytic systems for the reduction of CO2 with blue light in combination with the known catalyst [NiII(cyclam)]Cl2 was tested. This system produced CO as the main product through visible light (λ=420 nm) with a TON up to 8 after 4 hours. This value is in line with other photocatalytic systems using the same catalyst. Nevertheless, this system is entirely noble‐metal free.
Keywords: [Ni(cyclam)]2+, CuI complexes, earth-abundant metal, photocatalytic CO2 reduction, photosensitizers
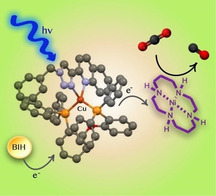
New photosensitizers based on earth‐abundant CuI absorb blue light and undergo reductive quenching with an electron donor (BIH). A second electron transfer to a Nickel catalyst triggers the catalytic reduction of CO2 to CO.
Introduction
Solar light is essential for natural life. The exploitation of this perennial primary resource for energetic human needs is a must for sustainable development of processes.1 The most common use of solar energy is its conversion into thermal or electrical energy.2 Nevertheless, the best way to store energy is in the form of fuels, namely in chemical bonds.3 This is what nature does with photosynthesis, and many research efforts have been carried on for decades in order to achieve an artificial photosynthetic system. In nature, water and carbon dioxide are the raw materials to start with, in order to produce biomass and the solar photons give the energy that activates these elaborated processes. Ideally, the exploitation of the solar energy will lead to a circular economy,4 where the fuels are produced from the waste generated by their use, without the assistance of other types of resources if not wholly renewable. Carbon dioxide is one of the major waste products of combustion, and it is also a greenhouse gas. Striving for the closure of the carbon cycle means then achieving the formation of fuels from CO2.5 Although nature reduces carbon dioxide in the so‐called dark reactions, for example, Calvin cycle, to form carbohydrates and biomass, in artificial photosynthesis, researchers aim at a direct photoreduction of CO2, employing photocatalytic systems.6 Usually, the major components of an artificial photosynthetic system consist of a photosensitizer (PS) and a catalyst (CAT), which sometimes can be the same unit.7 A sacrificial electron donor (e‐D) is needed to close the catalytic cycle and regenerate the ground‐state of the photosensitizer. To overcome the kinetic and thermodynamic stability of CO2, heterogeneous as well as homogeneous systems have been developed, from semiconductor materials8 (e.g., TiO2, SiC) to molecular photocatalysts,9 mainly based on expensive and rare metal complexes, such as Ru,10 Re11 or Ir.12
Many milestones have been reached so far. However, endeavours to achieve up‐scalable systems are still needed. In fact, from a practical point of view, the development of PS or CAT based on cost‐effective and earth‐abundant material will trigger viable industrial applications. More recently, molecular systems entirely based on earth‐abundant metals have been designed,13 and their results are inspiring in order to search accessible solutions intensively. Nevertheless, to meet global demand, recycling is always required, and substantial investments in this direction are of utmost importance. Notably, CuI‐based coordination complexes have been considered potential alternatives to RuII‐polypyridine complexes, and only recently, their potential use in photochemistry is rising their consideration as photosensitizers or photoredox catalysts.14, 15 Copper is a 3d transition metal, present in the earth's crust at a concentration of about 67 ppm.16 CuI complexes have a d10 electronic configuration; therefore metal central (MC) transitions are forbidden, while they exhibit metal‐to‐ligand charge‐transfer (MLCT) transitions, often absorbing at relatively low energy, in the visible region. This is an attractive property for their exploitation as photosensitizers. However, they may suffer from a Jahn–Teller distortion in their excited state, decreasing the charm of their photophysical properties. This effect is usually reduced by the use of bulky substituents and the use of sterically hindered ancillary ligands, such as chelating diphosphines. On the other hand, easy ligand replacements make these compounds not very stable in the presence of a coordinating solvent. Although this might be advantageous in photoredox catalysis, so that the complex can coordinate reactant and/or substrates,17 stability of the photosensitizer is highly desirable in photosensitised reactions. In this work, we present four new photosensitizers based on CuI heteroleptic complexes, a mononuclear and three dinuclear, of general formula [Cu(NN)(PP)]BF4, where NN is the diimine ligand and PP is the chelating phosphine. The bulky ancillary diphosphine is for all compounds the bis[(2‐diphenylphosphino)phenyl] ether (DPEPhos) and the chromophoric chelating diimines are 5‐(quinol‐2′‐yl)‐1H‐1,2,3‐triazole derivatives. These complexes absorb the blue light and are quite stable even in a coordinating solvent such as acetonitrile. Their photo‐ and redox properties will be hereafter illustrated, and their performance as photosensitizers will be evaluated by employing these new complexes in combination with a known catalyst,18 [NiII(cyclam)]Cl2 (cyclam: 1,4,8,11‐tetra‐azacyclotetradecane) for the photoreduction of CO2.
Results and Discussion
The diimine ligands, responsible for the colour of the final CuI complexes, are synthesised through a Cu alkyne‐azide cycloaddition (CuAAC)19 from 2‐ethynylquinoline and benzyl azide, in case of the monochelating diimine (ligand 3 in Scheme 1), or α,α′‐diazido‐ (ortho, meta and para)‐xylenes, (6 a, 6 b and 6 c, respectively) in case of the bis‐chelating ligands (4 a: o‐substituted; 4 b: m‐substituted; 4 c: p‐substituted). In particular, the diazido‐xylenes were formed in situ from their bromo‐derivatives and NaN3 at room temperature. The starting alkyne was synthesised through a Sonogashira cross‐coupling reaction from 2‐bromoquinoline and trimethylsilylacetylene in good yield (for details, please see experimental part). For the majority of the ligands, no further purification was needed, and the diimines were used in the next step for the coordination of CuI. Following an established procedure,20 mono‐ and dinuclear heteroleptic complexes were synthesised under inert atmosphere (Ar) in dry dichloromethane (DCM), (Scheme 1). The complexes were obtained in very high yield (up to 95 %). Recrystallisation from dichloromethane/cyclohexane solution gave crystals 1, 2 a and 2 c of good quality for X‐ray analysis. The crystals of 2 b were obtained by slow diffusion of diethyl ether in a concentrated solution of dioxane. The structure of the mononuclear CuI complex 1 is reported in Figure 1.
Scheme 1.
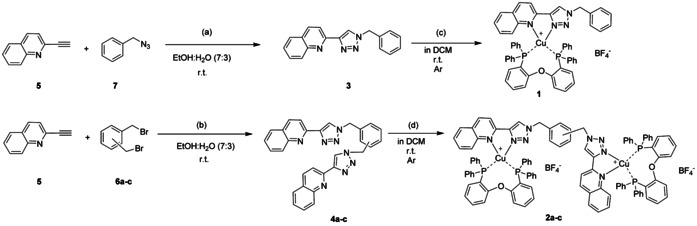
Synthetic procedures for the synthesis of the monochelating, and bis‐chelating ligands and CuI complexes thereof (1: mononuclear complex, 2: dinuclear complexes with xylenes as bridging unit: 2 a (o‐xylene); 2 b (m‐xylene); 2 c (p‐xylene)). (a) CuSO4; ascorbic acid; Na2CO3; (b) NaN3; CuSO4, ascorbic acid; Na2CO3; (c) DPEPhos; Cu(CH3CN)4BF4; (d) double amount of equivalents as for (c).
Figure 1.
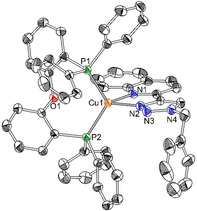
ORTEP drawing of crystals 1 shown at the 50 % probability level. Hydrogen atoms, counterion and solvent molecules were omitted for clarity.
The structure of the dinuclear complexes 2 a and 2 b are shown in Figure 2, while the structure of complex 2 c is reported in the Supporting Information (Figure S35). The crystal system for compound 1 is monoclinic with P21/n space group, while the dinuclear complexes 2 a–c have a triclinic system and a P space group. The distances between Cu and triazoles (Cu−N2 in 1; Cu1−N2 and Cu2−N7 for 2 a–c) are very similar in each complex (1: 2.069 Å; 2 a: 2.092 Å; 2 b: 2.083 Å; 2 c: 2.046 Å). This is true also for the distances between Cu and N atom of the quinoline moiety, besides for 2 b that has a slightly longer distance (1: 2.093 Å; 2 a: 2.095 Å; 2 b: 2.110 Å; 2 c: 2.098 Å). The distances between the atom of copper and the phosphorus atoms are also similar for the four compounds (1: 2.249 Å; 2 a: 2.240 Å; 2 b: 2.226 Å; 2 c: 2.236 Å, for Cu1−P1 and Cu1−P2, and 2 a: 2.225 Å; 2 b: 2.223 Å; 2 c: 2.267 Å, for Cu2−P3 and Cu2−P4). The angles between the copper atom and the chelating nitrogen atoms of the diimine ligand are similar (1: 79.8°; 2 a: 79.9°; 2 b: 79.6°; 2 c: 79.7°). Interestingly, in the dinuclear complexes, the angles between the two planes formed by Cu and the two N‐atoms of the diimine ligand (N2‐Cu1‐N1 and N8‐Cu2‐N7) vary depending on the compound from being parallel or almost in 2 c and 2 a, respectively, to a 60° angle for 2 b (2 a: 7.14°; 2 b: 63.3°; 2 c: 180°).
Figure 2.
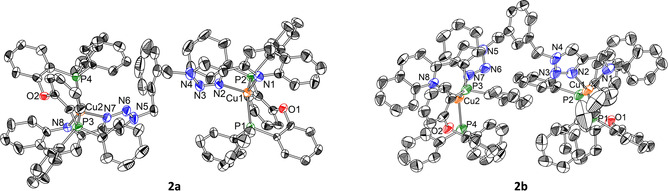
ORTEP drawing of crystals 2 a (left) and 2 b (right) shown at the 50 % probability level. Hydrogen atoms, counterions and solvent molecules were omitted for clarity.
Photophysical characterisation of the CuI complexes
The new photosensitizers were fully characterised in spectroscopic dichloromethane and acetonitrile solutions. Absorption spectra are very similar among the four complexes, as it is expected since they all bear the same core ligands (Figure 3).
Figure 3.
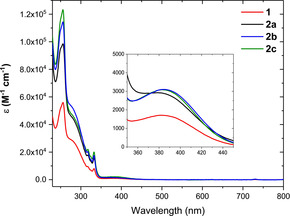
Absorption spectra in acetonitrile. Inset: zoom‐in in the range of the 1MLCT.
The main difference is on the absorptivity coefficients ϵ, where the mononuclear species 1 presents almost the half values in comparison to the ϵ for the species 2 a–c since it contains only one chromophoric unit. In particular, they display intense ligand centre (LC) absorption bands in the high‐energy UV region, below 320 nm, which are assigned to a π–π* transitions centred on the chelating diphosphine ligand. LC absorption bands centred on the diimine ligands are also occurring up to 360 nm, where two sharp signals can be recognised at 320 and 340 nm ca. The characteristic 1MLCT band of CuI complexes can be recognised above 370 nm with a relative maximum at 398 nm for 1, 388 nm for complexes 2 a and 2 b, 387 nm for 2 c.
These transitions are assigned to charge transfer from the metal core to the diimine ligand. Compared to the absorption bands of previously reported CuI complexes based on pyrid‐2‐yl‐triazoles,21 the MLCT band is bathochromic shifted, allowing the complexes to absorb blue light (up to 450 nm). This is rationalised by the increased π‐conjugation of the quinoline compared with the pyridine ring.22 In acetonitrile solutions, no remarkable change could be observed in the absorption spectra of all complexes.
The photophysical properties are reported in Table 1. The emissions of the complexes in solution are broad and structureless, typical for MLCT emissions. Moreover, when the measurements are done in air, the presence of the oxygen quenches the emission obviously, indicating that a triplet excited state is involved. Therefore, all the emission and excitation spectra were then recorded in an inert atmosphere (Ar). The excitation spectra correspond nicely to the absorption signals.
Table 1.
Photophysical properties of the new CuI photosensitizers.[a,b]
|
Sample |
λ abs [nm] |
λ em [nm] |
PLQY[c] |
τ [d] [μs] |
|---|---|---|---|---|
|
1 |
395[a] |
633 |
0.014 |
2.25 |
|
|
398[b] |
640[b] |
<0.001[b] |
0.107[b] |
|
2 a |
386[a] |
635 |
0.013 |
1.26 |
|
|
388[b] |
646[b] |
<0.001[b] |
0.109[b] |
|
2 b |
388[a] |
630 |
0.017 |
2.65 |
|
|
388[b] |
650[b] |
<0.001[b] |
0.125[b] |
|
2 c |
388[a] |
636 |
0.023 |
2.31 |
|
|
387[b] |
643[b] |
<0.001[b] |
0.128[b] |
[a] In Ar‐saturated DCM solution. [b] In Ar‐saturated ACN. [c] Photoluminescence quantum yields were measured with the relative method using Ru(bpy)3Cl2 in aerated water solution as standard (PLQY=0.040).23 [d] Lifetimes were measured with TCSPC using Nanoled for excitation (λ exc=366 nm)
The energy of the emission for the dinuclear complexes are the same as the mononuclear complex 1 (maximum emission λ≈635 nm in DCM). In fact, they bear the same chromophoric unit Cu(NN)(PP) and any particular cooperation effect in dinuclear compounds 2 a, 2 b and 2 c cannot be observed, since the coordinated metal parts are bridged with benzyl groups, namely with no electronic communication.
In acetonitrile solutions (ACN), the MLCT emission bands undergo a bathochromic shift (λ≈648 nm), caused by a stronger stabilisation of the excited state in this polar solvent. In DCM solutions, the complexes have small quantum yields (1 % for 2 a, 1 % for 1 and 2 b, 2 % for 2 c) and relatively long excited‐state lifetimes (τ higher than 2 μs for compounds 1, 2 b and 2 c; τ≈1 μs for 2 a). Nevertheless, in acetonitrile, all complexes turned out to present lifetimes of just a hundred nanoseconds, as for the photoluminescence quantum yields (PLQY) that are very low (less than 0.001). This can be rationalised by the possible quenching of the charge transfer excited state induced by the polar solvent (ACN).24 The emission and excitation spectra in ACN for the four compounds are displayed in Figure 4. Emission and excitation spectra in DCM can be found in the Supporting Information (Figure S19). Despite their low quantum yield and lifetime in acetonitrile, their use as photosensitizers was tested. In fact, they present unexpected stability in this coordinating solvent. While other heteroleptic complexes are known to release ligands in solution, with simultaneous formation of the corresponding homoleptic complexes, this phenomenon was not observed for our CuI complexes. Their 1H NMR spectra in deuterated ACN presented the same pattern even after a long time and also under irradiation, there are no changes in their UV/Vis spectra (see stability tests in the Supporting Information, Figures S10–S13 and S20–S23).
Figure 4.
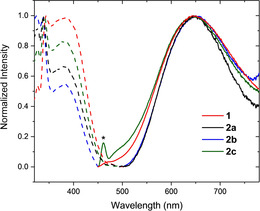
Excitation (dashed plots) and emission (solid plots) spectra in Ar‐saturated acetonitrile solution. (*: solvent Raman peak). Emissions were recorded exciting at 400 nm.
Electrochemical characterisation
Cyclic voltammetry was performed in acetonitrile solution with 0.1 m tetrabutylammonium hexafluorophosphate (TBAPF6) and a disk of Pt as the working electrode. The redox properties of the four new heteroleptic CuI complexes are reported versus Fc/Fc+ couple25 in Table 2. All the species present two irreversible oxidation processes. In particular, the first oxidation (around 1 V) is usually attributed to the CuI/CuII process. The second one is due to the oxidation of the chelating phosphine (1.9 V and 1.7 V for 2 c and 2 b, respectively and 1.2 V and 1.3 for 1 and 2 a). In the reduction part, one irreversible process is visible in the electrochemical window for complexes 1 and 2 c, at −2.1 V and −2.6 V respectively, while for complexes 2 a and 2 b the irreversible processes can be recognised at −1.8 V and −1.95 V respectively. The cyclic voltammetry plots recorded at a scan rate of 100 mV s−1 are displayed in the supporting information (Figures S24–S27). The redox properties of the complexes in their excited state in acetonitrile were calculated using the spectroscopic and electrochemical data.26 In their excited state, all the complexes are better oxidants and better reductants at the same time. However, if a reductive or an oxidative quenching might occur, it is essential to see if the thermodynamics is feasible, that is the driving force of the electron transfer (ΔG) must be negative. We have investigated one of the most used sacrificial electron donors: 1,3‐dimethyl‐2‐phenylbenzimidazoline (BIH).27 We prepared it according to reported literature procedure and determined its oxidation potential versus ferrocene/ferrocenium couple (−0.204 V; Figure S28). The driving force for the reductive quenching for the new heteroleptic CuI complexes is exergonic, indicating that the photoinduced electron transfer from BIH to the complexes is energetically feasible (Table S1).
Table 2.
Redox properties of the new CuI photosensitizer in ACN (0.1 m TBAPF6).[a]
|
Sample |
E ox [V] |
E red [V] |
E ox* [V][b] |
E red* [V][b] |
|---|---|---|---|---|
|
1 |
0.95; 1.2 |
−2.09 |
−1.75 |
0.61 |
|
2 a |
0.90; 1.3 |
−1.8; −2.5 |
−1.81 |
0.85 |
|
2 b |
1.00; 1.7 |
−1.95 |
−1.31 |
0.75 |
|
2 c |
0.99; 1.9 |
−2.6 |
−1.71 |
0.06 |
[a] Estimated by cyclic voltammetry, at a scan rate of 100 mV s−1, and reported versus ferrocene/ferrocenium couple. [b] Redox potentials of the excited states, calculated from the formulas E ox*=E ox−E 00; E red*=E red+E 00, where E 00 (≈2.7 eV) is defined as the energy of the transition from the lowest excited state in thermal equilibrium to the zero vibrational level of the ground state. It was quantitatively estimated, according to equations reported in ref. 26a and 26b.
Photoactivated CO2 reduction
The application of these new CuI complexes as photosensitizers in the photochemical reduction of carbon dioxide was evaluated in combination with [Ni(cyclam)]2+, a NiII coordination complex that can reduce CO2 both photochemically18d, 18f, 18g, 18h, 28 and electrochemically.18a, 18e, 18i, 29 The photocatalytic experiments were carried out dissolving all the components in a 4 mL mixed solution of ACN and triethanolamine (TEOA) in 5:1 volume ratio. The concentration of the photosensitizers was fixed at 1 mm for the mononuclear complex 1, and at 0.5 mm for the dinuclear complexes 2 a–c. At these concentrations, the optical density of the photocatalytic solutions at the excitation wavelength (420 nm) is 1.2 when the PS was the mononuclear complex 1 and 0.9 when the dinuclear complexes 2 a–c were used (Figure S33). The sacrificial electron donor BIH was used in 20 mm concentration unless otherwise specified. In order to evaluate the photocatalytic system with the new PS, the observed product is carbon monoxide, and the results are reported in Table 3. All the new CuI complexes showed the ability to photosensitise the reduction of CO2. Although [Ni(cyclam)]2+ is known to produce also molecular hydrogen, this could not be detected in our experiments. Upon absorption of light, the excited‐state of the photosensitizers is populated and undergoes reductive quenching by BIH. Stern–Volmer (SV) bimolecular quenching experiments confirmed the favoured thermodynamics already proven with the redox potentials of PS*, resulting in apparent quenching rate constants close to the diffusion limit (k(1)=2.0×109 s−1 m −1; k(2 a)=2.8×109 s−1 m −1; k(2 b)=4.3×109 s−1 m −1; k(2 c)=1.91×109 s−1 m −1). The linear responses of the SV experiments are displayed in the Supporting Information, Figures S29–S32. The absorption spectra were also recorded. We observed, by adding BIH, an enhancement of the absorption band at 350 nm, most probably due to the increased BIH concentration, while the MLCT absorption band remains constant. Notably, at high concentration of BIH (>5 mm), the emission of the complexes 1 and 2 a–c centred at ≈640 nm is quenched and a new emission appears at 550 nm. The presence of oxygen does not influence the intensity of this emission. Thus, we assume it must be a singlet state. Further investigations to assign the nature of this new emission band are currently ongoing and are out of the main aim of the present work.
Table 3.
Photocatalytic CO2 reactions and control experiments in ACN/TEOA (5:1) after 4 h of irradiation at 420 nm.[a]
|
Entry |
PS |
[CAT] |
[BIH] |
CO [μmol] |
TONCAT |
|---|---|---|---|---|---|
|
1 |
1 |
0.1 mm |
20 mm |
1.73 |
4.3 |
|
2 |
2 a |
0.1 mm |
20 mm |
1.94 |
4.9 |
|
3 |
2 b |
0.1 mm |
20 mm |
3.24 |
8.1 |
|
4 |
2 c |
0.1 mm |
20 mm |
1.83 |
4.6 |
|
5 |
1 |
0.1 mm |
10 mm |
1.02 |
2.6 |
|
6 |
2 a |
0.1 mm |
10 mm |
1.40 |
3.5 |
|
7 |
2 b |
0.1 mm |
10 mm |
2.00 |
5.0 |
|
8 |
2 c |
0.1 mm |
10 mm |
0.70 |
1.8 |
|
9[b] |
1 |
0.1 mm |
10 mm |
1.13[b] |
2.8[b] |
|
10 |
2 b |
0 |
10 mm |
n.d. |
n.d. |
|
11[c] |
2 b |
0.1 mm |
10 mm |
n.d.[c] |
n.d. [c] |
|
12 |
2 b |
0.1 mm |
0 |
n.d. |
n.d. |
|
13[d] |
2 b |
0.1 mm |
10 mm |
n.d.[d] |
n.d.[d] |
|
14 |
none |
0.1 mm |
10 mm |
n.d. |
n.d. |
|
15[e] |
1 |
0.1 mm |
10 mm |
n.d. |
n.d. |
|
16 |
Cu(ACN)4 |
0.1 mm |
10 mm |
n.d. |
n.d. |
|
17 |
1 |
0.1 mm |
20 mm |
2.9[f] |
7.3[f] |
|
18 |
2 a |
0.1 mm |
20 mm |
2.6[f] |
6.5[f] |
[a] From headspace analysis, reactions were repeated twice. [b] ACN/TEOA (4:1,v/v); [c] With trimethylamine instead of TEOA. [d] Without CO2 in Ar atmosphere; [e] in dark (n.d.=not detected). [f] After 10 h irradiation.
Photoinduced electron transfer from the BIH to PS* occurs, and the reduced form of PS is oxidised back by the catalyst. After 4 h of irradiation at 420 nm, analyses of the headspace were performed by gas‐chromatography, equipped with a thermal conductivity detector (TCD). When the mononuclear CuI complex 1 was used, 1.73 μmol of CO were produced. Very similar results were obtained when PS was the binuclear complex 2 c. These values correspond to a turn‐over number in respect to the CAT (TONCAT) of 4.3 and 4.6 respectively. Interestingly, when 2 b photosensitised the photocatalytic reduction of CO2, the amount of produced CO is almost double (3.24 μmol), corresponding to a TONCAT of 8.1. This result is excellent compared to homogeneous systems using [Ni(cyclam)]2+ as the CAT (see Table S4).
Furthermore, to the best of our knowledge, our system is the first one that exploits earth‐abundant CuI photosensitizers in combination with this catalyst. When a lower concentration of BIH (10 mm) was used, the efficiency of the systems was lower (entries 5–9 in Table 3). A weak improvement could be achieved increasing the amount of TEOA (in ACN, 4:1 instead of 5:1), where 1.13 μmol instead of 1.02 μmol were obtained (entry 9 compared to entry 5 in Table 3). Control experiments under Ar atmosphere (entry 13, Table 3) did not give any production of CO. Moreover, each component of our photocatalytic system herein presented was demonstrated to be necessary for the photoinduced reduction of carbon dioxide, since control experiments carried out without BIH (entry 12), without PS (entry 14), and without CAT (entry 10) produced no CO. The same result was also obtained when the reaction was carried out in the dark (entry 15). The new PSs are very stable in ACN solution under irradiation, as it can be seen in Figures S20–S23, where the absorption spectra present no modification even after 5 h irradiation. However, a decrease of the MLCT absorption of the PSs could be observed, and the degradation of PS started already after 30 min irradiation of the mixed photocatalytic reactions. It is not to exclude that TEOA may replace the chromophoric ligand; in fact, a mass corresponding to DPEPhosCu(TEOA) (750 Da) was observed when HR‐ESI mass analyses were performed on the homogenous photocatalytic solutions. Interestingly, the degradation of the PS happens only in the presence of BIH and TEOA, when the atmosphere is carbon dioxide. We assume that TEOA forms with CO2 a zwitterionic carbonate species30 that induces complexation with the reduced form of PS, developed after reductive quenching by BIH. A detailed mechanism with the identification of the intermediate species is in process. Nevertheless, when the photocatalytic reaction was performed with Cu(ACN)4BF4 instead of any of the new photosensitizers, CO could not be observed, even after addition of one equivalent of DPEPhos (entry 16, Table 3), so the new complexes are necessary for the successful photocatalytic reduction of CO2. Longer reaction times increase the amount of produced CO, so after 10 h of irradiation, the produced CO was 2.9 μmol (TONCAT 7.3) when 1 was used and 2.6 μmol (TONCAT 6.5) when 2 a was used (entries 17 and 18 in Table 3). For those experiments, the results were plotted as function of irradiation time in Figure S34 of the Supporting Information. Increasing the concentration of the photosensitizer 1 and the catalysts to 3 mm and 0.3 mm, respectively, the amount of CO was also higher (6.0 μmol). However, the TONCAT was almost constant (5.1) after 4 h. The quantum efficiency of the photocatalytic system was calculated, measuring the irradiated light intensity by chemical actinometry (see the experimental part for details).The calculated quantum yields for the typical photocatalytic systems used (entries 1 to 4, Table 3) are 1.0 % (1), 1.2 % (2 a); 2.1 % (2 b), 1.1 % (2 c). The results herein reported have been promising and stimulated further research in our working group, in order to develop new photosensitizers and photocatalysts based on earth‐abundant metal complexes.
Conclusions
This work reports the synthesis, full characterisation and employment in CO2‐reduction of four new coordination metal complexes based on earth‐abundant CuI. Their MLCT absorption falls in the visible region (up to 450 nm), and they are very stable even in coordinating solvent such as acetonitrile. Thus, we have studied their photophysical and electrochemical properties in order to prove their use as photosensitizers. In particular, we presented the homogeneous photocatalytic reduction of CO2 with [NiII(cyclam)]Cl2 as the catalyst. After 4 hours of irradiation with LED lamps, the production of CO could be observed in the presence of the reported complexes. To the best of our knowledge, this is the first example reported of CuI‐based photosensitizers with [NiII(cyclam)]Cl2. Moreover, the TONs obtained (up to 8) are in line with those results produced in previous works with noble‐metal based photosensitizers. These results motivate further development of new earth‐abundant metal‐based photosensitizers and catalysts in our research team.
Experimental Section
Experimental details and general remarks: All starting materials were purchased from commercial suppliers and used as received unless otherwise noted.
Synthesis of 2‐ethynylquinoline (5): In a Schlenk‐tube, under inert atmosphere (Ar), 2‐bromoquinoline (210 mg, 1.0 mmol, 1.0 equiv) was dissolved in 20 mL diisopropylamine (DIPA), dried over CaH2. Trimethylsilylacetylene (700 μl, 4,98 mmol, 5.0 equiv) was added, followed by Pd(PPh3)2Cl2 (7.4 mg, 1 mol %) and CuI (3.6 mg, 2 mol %). The muddy brown reaction mixture was stirred at room temperature for 4 hours. The 2‐(trimethylsilylethynyl)quinoline is a viscous brown oil that was purified by column chromatography in silica, eluent mixture CH2Cl2/C6H12 (1:1), R f=0.25. This intermediate was then dissolved in methanol and K2CO3 (2 equiv) was added for the deprotection of the triple bond. After completion of the reaction (2 h), 5 mL of water was added, and the organic products were extracted with dichloromethane. The organic phase was washed with brine, dried over Na2SO4, filtrated and the solvent was removed under reduced pressure. The desired product was purified by a silica gel chromatographic column using cyclohexene/EtOAc (1:1) as eluent, R f=0.75. (92 mg, 0.60 mmol) Yield: 60 %. 1H NMR (300 MHz, CDCl3): δ= 8.08 (d, J=8.4 Hz, 2 H), 7.76 (d, J=9 Hz, 1 H), 7.72 (ddd, J1=8.6 Hz, J2=6.9 Hz, J3=1.5 Hz, 1 H), 7.53 (dd, J 1=9 Hz, J2=, 6 Hz, 2 H), 3.24 ppm (s, 1 H).
Synthesis of the ligands: The mono‐chelating ligand 3 was synthesised following a modified procedure from literature,19b by dissolving 1 equivalent of 5 (120 mg, 0.80 mmol) with benzylazide (107 mg, 0.80 mmol, 1.0 equiv) followed by sodium ascorbate (63.4 mg, 0.32 mmol, 0.4 equiv), CuSO4(⋅5 H2O) (25.5 mg, 0.16 mmol, 0.2 equiv) and sodium carbonate (57.2 mg, 0.54 mmol, 0.675 equiv) into a solution of ethanol and water (7:3), giving 200 mg (0.696 mmol, yield 87 %) of desired product. 1H NMR (500 MHz, CDCl3): δ=8.26 (d, J=8.6 Hz, 1 H), 8.19 (s, 1 H), 8.15 (d, J=8.6 Hz, 1 H), 7.93 (d, J=8.5 Hz, 1 H), 7.73 (dd, J1=8.1 Hz, J2= 1.4 Hz, 1 H), 7.60 (ddd, J1=8.4 Hz, J2=6.9 Hz, J3=1.5 Hz, 1 H), 7.42 (ddd, J1=8.0 Hz, J2= 6.8 Hz, J3=1.0 Hz, 1 H), 7.38–7.25 (m, 5 H), 5.54 ppm (s, 2 H).; 13C NMR (125 MHz, CDCl3): δ=150.44, 149.08, 148.01, 136.89, 134.38, 129.74, 129.21, 128.97, 128.89, 128.32, 127.81, 127.78, 126.37, 122.71, 118.70,( 77.31, 77.26, 76.80, CDCl3) 54.48 ppm.
The bis‐chelating ligands 4 a–c were prepared following the general procedure showed hereafter. In a 100 mL flask, 1 equivalent (120 mg, 0.80 mmol) of 2‐ethynylquinoline 5 were dissolved in 10 mL of a solution of ethanol and water (7:3). The desired isomer of dibromomethylbenzene (103 mg, 0.39 mmol, 0.5 equiv) was added, followed by NaN3 (101 mg, 1.55 mmol, 2 equiv), sodium ascorbate (61 mg, 0.4 equiv), CuSO4 (⋅5 H2O) (0.04 g, 0.2 equiv) and sodium carbonate (50 mg, 0.675 equiv). The reaction mixture becomes yellow‐orange and stirred overnight at room temperature. The reaction was stopped by addition of a solution of NH4OH (10 %). The precipitate was filtered off, dissolved in dichloromethane and washed with NH4OH (10 % solution) three times. The organic phases were washed with brine, dried over MgSO4. The solvent was removed under vacuum in a rotatory evaporator. The product was obtained without further purification needed.
4 a: The desired product was obtained as a light yellow powder (149 mg, 0.30 mmol) Yield: 38 %. 1H NMR (300 MHz, CDCl3): δ=8.78 (s, 2 H), 8.42 (d, J=7.2, 2 H), 8.18 (d, J=8.1 Hz, 2 H). 7.98 (d, J=7.8 Hz, 2 H), 7.93 (d, J=8.7 Hz, 2 H), 7.72 (dd, J1=8.1 Hz, J2=7.2 Hz, 2 H), 7.56 (dd, J1=7.8 Hz, J2=7.2 Hz, 2 H), 7.41 (m, CH 2 H), 7.38 (m, 2 H), 6.00 ppm (s, 4 H). 13C NMR (126 MHz, CDCl3): δ=150.17, 149.21, 148.05, 147.97, 136.83, 136.50, 133.22, 130.91, 130.14, 129.68, 129.25, 128.99, 128.22, 127.79, 127.73, 127.31, 126.70, 126.37, 126.15, 122.77, 118.61, (77.29, 77.03, 76.78, CDCl3), 53.44 ppm.
4 b: The desired product was obtained as a light yellow powder (253 mg, 0.51 mmol) Yield: 64 %.1H NMR (300 MHz, CDCl3): δ=8.46 (s, 2 H), 8.28(d, J=8.7 Hz, 2 H), 8.22(d, J=8.7 Hz, 2 H). 8.00 (d, J=8.1 Hz, 2 H), 7.8 (d, J=6 Hz, 2 H), 7.67 (dd, J1=7.2 Hz, J2=7.8 Hz, 2 H), 7.50 (dd, J1=6.3 Hz, J2=8.1 Hz, 2 H), 7.37 (m, 4 H) 5.64 ppm (s, 4 H). 13C NMR (126 MHz, CDCl3): δ=150.29, 149.25, 147.99, 136.93, 135.67, 130.16, 129.77, 128.99, 128.66, 127.87, 127.84, 127.79, 126.42, 122.86, 118.68, (77.29, 77.03, 76.78, CDCl3), 54.04, 53.46 ppm.
4 c: The desired product was obtained as a light yellow powder (174 mg, 0.35 mmol) Yield: 44 %.1H NMR (300 MHz, CDCl3): δ=8.36 (s, 2 H), 8.33 (d, J not visible, under singlet, 2 H), 8.27 (d, J=8.7 Hz, 2 H). 8.05 (d, J=8.4 Hz, 2 H), 7.83 (d, J=8.1 Hz, 2 H), 7.69 (dd, J1=9 Hz, J2=9 Hz, 2 H), 7.52 (dd, J1=6.9 Hz, J2=8.1, 2 H), 7.40 (s, 4 H), 5.54 ppm (s, 4 H). 13C NMR (126 MHz, CDCl3): δ=150.31, 149.24, 148.01, 136.89, 136.50, 135.30, 134.55, 129.74, 129.04, 128.99, 128.93, 128.74, 127.79, 126.40, 122.77, 118.64, (77.32, 77.07, 76.82, CDCl3), 53.93 ppm.
Synthesis of the CuI complexes: The general procedure was followed as previously reported,21a by dissolving in 20 mL of dry dichloromethane 3 (183 mg, 0.64 mmol, 1.0 equiv) with DPEPhos (344 mg, 0.64 mmol, 1.0 equiv) and Cu(CH3CN)4BF4 (200 mg, 0.64 mmol, 1.0 equiv) under Ar atmosphere. The desired product 1 (449 mg, 0.46 mmol) was obtained with a yield of 72 %. 1H NMR (500 MHz, CD3CN): δ=7.92 (s, 1 H), 7.86 (d, J=8.4 Hz, 1 H), 7.71–7.19 (3d, 3 H), 6.76–6.42 (m, 25 H), 6.34‐ 6.25 (m, 6 H), 6.03–5.95 (m, 4 H), 4.99 ppm (s, 2 H). 13C‐NMR (125 MHz, CDCl3): δ=158.55, 147.76, 145.97, 146.60, 139.00, 134.51, 134.56, 131.89, 131.57, 130.93, 130.66, 130.26, 129.62, 129.47, 128.88, 128.68, 128.53, 128.29, 127.72, 127.11, 125.80, 124.88, 120.08, 119.86, 54.97 ppm. 31P NMR (162 MHz CDCl3) δ=−12.73 ppm. HRMS (ESI) m/z (C54H42CuN4OP2): 887.21 (calc.), 887.21 (found). Elemental analysis (C54H42CuN4OP2BF4): C=65.51, H=4.43, N=5.74 (calc.); C=65.53, H=4.357, N=5.54 (found).
2 a: Under Ar atmosphere, 4 a (138 mg, 0.28 mmol, 1.0 equiv) was dissolved in dry dichloromethane, followed by DPEPhos (301 mg, 0.56 mmol, 2.0 equiv) and Cu(CH3CN)4BF4 (176 mg, 0.56 mmol, 2.0 equiv) to give the desired product 2 a (477 mg, 0.25 mmol). Yield: 91 %. 1H NMR (500 MHz, CDCl3): δ= 9.07 (s, 2 H), 8.39 (d, J=5.1 Hz, 2 H), 8.11 (d, J=5.1 Hz, 2 H), 7.94 (d, J=5.1 Hz, 2 H), 7.81 (d, J=5.1 Hz, 2 H), 7.48 (m, 8 H), 7.35 (dd, J1=4.5 Hz, J2=4.5 Hz, 2 H) 7.28–7.24 (m, 12 H), 7.22–7.19 (m, 8 H), 7.10 (m, 4 H), 7.04 (m, 4 H), 6.93 (m, 6 H) 6.84 (m, 8 H), 6.72–6.69 (m, 4 H), 6.60–6.57(m, 8 H), 5.85 ppm (s, 4 H). 13C NMR (125 MHz, CDCl3): δ=158.62, 147.47, 146.16, 145.67, 139.04, 134.53, 134.09, 132.97, 131.97, 131.73, 131.03, 130.64, 130.35, 129.89, 129.49, 128.34, 128.32, 127.73, 127.35, 125.69, 124.69, 124.23, 120.13, 119.72, 51.52 ppm. 31P NMR (162 MHz, CDCl3): δ=−12.60 ppm. HRMS (ESI) m/z (C102H78Cu2P4N8O2 BF4): (z=1) 1785.38 (calc), 1785.40 (found) Elemental analysis (C102H78Cu2N8O2P4B2F8): C=65.43, H=4.20, N=5.98 (calc.); C=65.10, H=4.321, N=5.91 (found).
2 b: Under Ar atmosphere, 4 b (227 mg, 0.46 mmol, 1.0 equiv) was dissolved in dry dichloromethane with DPEPhos (495 mg, 0.92 mmol, 2.0 equiv) and Cu(CH3CN)4BF4 (288 mg, 0.92 mmol, 2.0 equiv) to give the desired product 2 b (534 mg, 0.28 mmol). Yield: 62 %. 1H NMR (500 MHz, CD3CN): δ=8.50 (s, 2 H), 8.46 (d, 2 H), 7.94 (m, 4 H), 7.80 (m, 2 H), 7.46–6.60 (m, 64 H), 5.58 ppm (s, 4 H). 13C NMR (125 MHz, CD3CN): δ=158.78, 149.87, 149.06, 147.54, 146.125, 135.89, 134.50, 132.52, 131.72, 130.43, 130.26, 129.00, 128.92, 128.76, 128.39, 127.86, 125.29, 124.75, 124.05, 123.93, 123.81, 120.77, 119.15, 55.76 ppm. 31P NMR (162 MHz, CD3CN): δ=−13.19 ppm. HRMS m/z (C102H78Cu2P4N8O2BF4): 1785.38 (calc.), 1785.39 (found). Elemental analysis (C102H78Cu2N8O2P4B2F8): C=65.43, H=4.20, N=5.98 (calc.); C=65.29, H=4.242, N=5.93 (found).
2 c: Under Ar atmosphere, 4 c (163 mg, 0.33 mmol, 1.0 equiv) was dissolved in dry dichloromethane with DPEPhos (355 mg, 0.66 mmol, 2.0 equiv) and Cu(CH3CN)4BF4 (207 mg, 0.66 mmol, 2.0 equiv) to give the desired product 2 c (580 mg, 0.31 mmol). Yield: 94 % 1H NMR (500 MHz, CD3CN): δ=8.48 (m, 4 H), 7.92 (m, 4 H), 7.78 (s, 2 H), 7.43–6.58 (m, 64 H), 5.66 ppm (s, 4 H). 13C NMR (125 MHz, CD3CN): δ=158.73, 147.49, 146.53, 146.3, 139.59, 135.63, 134.42, 132.47, 131.28, 130.41, 129.10, 128.83, 128.36, 127.88, 125.20, 124.80, 123,98, 123.87, 123.75, 120.69, 119.08, 100.26, 55.27 ppm. 31P NMR (162 MHz, CD3CN): δ=−13.30 ppm. HRMS m/z (C102H78BCu2F4N8O2P4): 1785.38 (calc), 1785.39 (found). Elemental analysis (C102H78Cu2N8O2P4B2F8): C=65.43, H=4.20, N=5.98 (calc.); C=65.28, H=3.956, N=6.19 (found).
Photocatalytic reactions: In a 12 mL vial, closed with an aluminium cap equipped with a rubber septum, 4 mL of solution, containing photosensitizers (1 mm for 1 and 0.5 mm for 2 a, 2 b and 2 c), catalyst (0.1 mm), and electron donor (20 mm), were introduced and kept under dark. CO2 gas (99.995 %, from Air Linde) was bubbled inside for 20 minutes. The vials were irradiated in the photo‐reactor LZC‐IC2 from Luzchem with 4 fluorescent lamps (8 W each) at 420 nm. The total photon flux of the system was measured by K3Fe(C2O4)3 actinometry31 and the amount is ∼2.5×10−8 E s−1. All solutions were stirred continuously during irradiation at a temperature of 295 K. The produced CO was analysed by manual injection of 200 μL of the headspace with a gas‐tight syringe into our Gas‐chromatograph with a thermal conductivity detector (TCD) from PerkinElmer Arnel (Clarus 590, HayeSep N and Molecular sieves columns) using He as carrier gas. Calibration curves were performed with 5 known standard injections of CO. Each reaction was tested after 4 hours unless otherwise noted. The quantum yields for the photocatalytic CO2 reduction reactions were determined by using Equation (1):
| (1) |
in which the fap is the fraction of the absorbed photons of the photocatalytic system at the excitation wavelength (1–10‐A). The number of CO molecules was determined from the moles of CO measured from the headspace of the reaction, using a calibrated GC‐TCD. The factor 2 is because CO is a 2 electron reduced product of CO2. The number of incident photons was estimated using K3Fe(C2O4)3 actinometry.
Photophysics: UV/Vis absorption spectra were recorded for all the compounds in a solution of CH2Cl2 and CH3CN (concentrations ≈10−5 m). The instrument used was a Lambda 750 double‐beam UV/Vis‐NIR spectrometer. Emission and excitation spectra were recorded with a Fluoromax 4 from Horiba Jobin. All the solutions were air‐free by letting Ar bubbling inside for 10 minutes at least. Lifetime experiments were performed by time‐correlated single‐photon counting method (TCSPC) with a DeltaTime kit for DeltaDiode source on FluoroMax systems, including DeltaHub and DeltaDiode controller. NanoLED 370 was used as the excitation source (λ=366 nm).
Electrochemistry: Cyclic voltammetry experiments were performed with a Gamry Interface 1010B in a three electrodes electrochemical cell. The electrochemical cell was equipped with a Pt‐disc working electrode, Ag/AgNO3 reference electrode and a Pt wire as the auxiliary electrode. All the experiments were performed in acetonitrile (0.1 m TBAPF6) solution, under Ar atmosphere, unless otherwise specified. Ferrocene (Fc) was added after each experiment as an internal standard, according to IUPAC recommendation. The redox properties are reported versus Fc/Fc+ couple.
Crystallography: Deposition numbers 1988207 (1), 1988208 (2 a), 1988209 (2 b), and 1988210 (2 c) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service. Single‐crystal X‐ray diffraction data were collected on a STADI VARI diffractometer with monochromated Ga Kα (λ=1.34143 Å) or Mo Kα (λ=0.71073) radiation at low temperature. Using Olex2,32 the structures were solved with the ShelXT33 structure solution program using Intrinsic Phasing and refined with the ShelXL34 refinement package using Least Squares minimisation. Refinement was performed with anisotropic temperature factors for all non‐hydrogen atoms; hydrogen atoms were calculated on idealised positions.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Claudia Bizzarri received her B.Sc. and M.Sc. in Chemistry from the University of Rome “Tor Vergata” (Italy). In 2011 she obtained her Ph.D. working on luminescent iridium(III) and copper(I) complexes for optoelectronic applications under the supervision of Prof. Luisa De Cola at the University of Münster (Germany). In 2007, she was a visiting student at the University of Minnesota Duluth (USA) and in 2010 at the University College London (UK). After a couple of years as a Process Engineer at AIXTRON SE (Aachen, Germany), she was a post‐doc at the CAT Catalytic Center of the University RWTH Aachen. Since the end of 2016, Claudia Bizzarri is an independent Junior Group Leader at the Karlsruhe Institute of Technology. Her research interests focus on photoactive compounds based on earth‐abundant metals. Her coordination metal complexes find applications in the field of artificial photosynthesis and bioimaging.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to thank generous funding from the Deutsche Forschungsgemeinschaft (DFG), Collaborative Centre SFB/TRR 88 “3MET” (Bizzarri, Bruschi and Gracia: Project B9; Weis: Project C6). L.L. thanks the Erasmus plus program from the University of Bologna. Prof. Stefan Bräse (KIT) is deeply acknowledged for his scientific support. Prof. H.A. Wagenknecht is greatly acknowledged for allowing us to measure with his photophysical equipment.
L.-L. Gracia, L. Luci, C. Bruschi, L. Sambri, P. Weis, O. Fuhr, C. Bizzarri, Chem. Eur. J. 2020, 26, 9929.
Dedicated to Prof. Luisa De Cola on the occasion of her 60th birthday
References
- 1.
- 1a. Armaroli N., Balzani V., Angew. Chem. Int. Ed. 2007, 46, 52–66; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 52–67; [Google Scholar]
- 1b. Armaroli N., Balzani V., Chem. Eur. J. 2016, 22, 32–57; [DOI] [PubMed] [Google Scholar]
- 1c. Lewis N. S., Nocera D. G., Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Crabtree G. W., Lewis N. S., Physics Today 2007, 60, 37–42; [Google Scholar]
- 2b. Grätzel M., Inorg. Chem. 2005, 44, 6841–6851. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Gray H. B., Nature Chem. 2009, 1, 7;21378780 [Google Scholar]
- 3b. Balzani V., Pacchioni G., Prato M., Zecchina A., Rendiconti Lincei. Sci. Fis.Nat. 2019, 30, 443–452. [Google Scholar]
- 4. Keijer T., Bakker V., Slootweg J. C., Nature Chem. 2019, 11, 190–195. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Iaquaniello G., Centi G., Salladini A., Palo E., Perathoner S., Chem. Eur. J. 2018, 24, 11831–11839; [DOI] [PubMed] [Google Scholar]
- 5b. Centi G., Quadrelli E. A., Perathoner S., En. & Environm.l Sci. 2013, 6, 1711; Environm.l Sci. 2013, 6, 1711. [Google Scholar]
- 6. Berardi S., Drouet S., Francas L., Gimbert-Surinach C., Guttentag M., Richmond C., Stoll T., Llobet A., Chem. Soc. Rev. 2014, 43, 7501–7519. [DOI] [PubMed] [Google Scholar]
- 7. Yui T., Tamaki Y., Sekizawa K., Ishitani O., Top. Curr. Chem. 2011, 303, 151–184. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Liu X., Inagaki S., Gong J., Angew. Chem. Int. Ed. 2016, 55, 14924–14950; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15146–15174; [Google Scholar]
- 8b. Sorcar S., Thompson J., Hwang Y., Park Y. H., Majima T., Grimes C. A., Durrant J. R., In S.-I., En. & Environm. Sci. 2018, 11, 3183–3193; Environm. Sci. 2018, 11, 3183–3193. [Google Scholar]
- 9.
- 9a. Yamazaki Y., Takeda H., Ishitani O., J. Photochem. Photobiol. C 2015, 25, 106–137; [Google Scholar]
- 9b. Elgrishi N., Chambers M. B., Wang X., Fontecave M., Chem. Soc. Rev. 2017, 46, 761–796. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Kuramochi Y., Itabashi J., Toyama M., Ishida H., ChemPhotoChem 2018, 2, 314–322; [Google Scholar]
- 10b. Kuramochi Y., Itabashi J., Fukaya K., Enomoto A., Yoshida M., Ishida H., Chem. Sci. 2015, 6, 3063–3074; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Rau S., Walther D., Vos J. G., Dalton Trans. 2007, 915–919; [DOI] [PubMed] [Google Scholar]
- 10d. Matlachowski C., Braun B., Tschierlei S., Schwalbe M., Inorg. Chem. 2015, 54, 10351–10360. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Yamazaki Y., Ohkubo K., Saito D., Yatsu T., Tamaki Y., Tanaka S., Koike K., Onda K., Ishitani O., Inorg. Chem. 2019, 58, 11480–11492; [DOI] [PubMed] [Google Scholar]
- 11b. Tamaki Y., Koike K., Ishitani O., Chem. Sci. 2015, 6, 7213–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Genoni A., Chirdon D. N., Boniolo M., Sartorel A., Bernhard S., Bonchio M., ACS Catal. 2017, 7, 154–160; [Google Scholar]
- 12b. Thoi V. S., Kornienko N., Margarit C. G., Yang P., Chang C. J., J. Am. Chem. Soc. 2013, 135, 14413–14424. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Takeda H., Cometto C., Ishitani O., Robert M., ACS Catal. 2017, 7, 70–88; [Google Scholar]
- 13b. Shipp J. D., Carson H., Spall S. J. P., Parker S. C., Chekulaev D., Jones N., Mel′nikov M. Y., Robertson C. C., Meijer A. J. H. M., Weinstein J. A., Dalton Trans. 2020, 49, 4230–4243. ; [DOI] [PubMed] [Google Scholar]
- 13c. Rosas-Hernández A., Steinlechner C., Junge H., Beller M., Green Chem. 2017, 19, 2356–2360; [DOI] [PubMed] [Google Scholar]
- 13d. Takeda H., Ohashi K., Sekine A., Ishitani O., J. Am. Chem. Soc. 2016, 138, 4354–4357; [DOI] [PubMed] [Google Scholar]
- 13e. Chen L., Qin Y., Chen G., Li M., Cai L., Qiu Y., Fan H., Robert M., Lau T. C., Dalton Trans. 2019, 48, 9596–9602; [DOI] [PubMed] [Google Scholar]
- 13f. Guo Z., Cheng S., Cometto C., Anxolabéhère-Mallart E., Ng S.-M., Ko C.-C., Liu G., Chen L., Robert M., Lau T.-C., J. Am. Chem. Soc. 2016, 138, 9413–9416; [DOI] [PubMed] [Google Scholar]
- 13g. Rao H., Bonin J., Robert M., ChemSusChem 2017, 10, 4447–4450; [DOI] [PubMed] [Google Scholar]
- 13h. Rao H., Bonin J., Robert M., Chem. Commun. 2017, 53, 2830–2833; [DOI] [PubMed] [Google Scholar]
- 13i. Takeda H., Kamiyama H., Okamoto K., Irimajiri M., Mizutani T., Koike K., Sekine A., Ishitani O., J. Am. Chem. Soc. 2018, 140, 17241–17254; [DOI] [PubMed] [Google Scholar]
- 13j. Steinlechner C., Roesel A. F., Oberem E., Päpcke A., Rockstroh N., Gloaguen F., Lochbrunner S., Ludwig R., Spannenberg A., Junge H., Francke R., Beller M., ACS Catal. 2019, 9, 2091–2100; [Google Scholar]
- 13k. Sakaguchi Y., Call A., Cibian M., Yamauchi K., Sakai K., Chemical Commun. 2019, 55, 8552–8555; [DOI] [PubMed] [Google Scholar]
- 13l. Zhang X., Cibian M., Call A., Yamauchi K., Sakai K., ACS Catal. 2019, 9, 11263–11273. [Google Scholar]
- 14.
- 14a. Reiser O., Acc. Chem. Res. 2016, 49, 1990–1996; [DOI] [PubMed] [Google Scholar]
- 14b. Hernandez-Perez A. C., Collins S. K., Acc. Chem. Res. 2016, 49, 1557–1565; [DOI] [PubMed] [Google Scholar]
- 14c. Minozzi C., Caron A., Grenier-Petel J.-C., Santandrea J., Collins S. K., Angew. Chem. Int. Ed. 2018, 57, 5477–5481; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5575–5579. [Google Scholar]
- 15.
- 15a. Paria S., Reiser O., ChemCatChem 2014, 6, 2477–2483; [Google Scholar]
- 15b. Hossain A., Bhattacharyya A., Reiser O., Science 2019, 364, eaav9713. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Armaroli N., Accorsi G., Cardinali F., Listorti A., Photochemistry and Photophysics of Coordination Compounds I (Eds.: V. Balzani, S. Campagna), Springer, Berlin, 2007, pp. 69–115; [Google Scholar]
- 16b. https://periodictable.com/Elements/029/data.html.
- 17.
- 17a. Hossain A., Engl S., Lutsker E., Reiser O., ACS Catal. 2019, 9, 1103–1109; [Google Scholar]
- 17b. Bagal D. B., Kachkovskyi G., Knorn M., Rawner T., Bhanage B. M., Reiser O., Angew. Chem. Int. Ed. 2015, 54, 6999–7002; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7105–7108. [Google Scholar]
- 18.
- 18a. Jiang C., Nichols A. W., Walzer J. F., Machan C. W., Inorg. Chem. 2020, 59, 1883–1892; [DOI] [PubMed] [Google Scholar]
- 18b. Schneider C. R., Lewis L. C., Shafaat H. S., Dalton Trans. 2019, 48, 15810–15821; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18c. Behnke S. L., Manesis A. C., Shafaat H. S., Dalton Trans. 2018, 47, 15206–15216; [DOI] [PubMed] [Google Scholar]
- 18d. Kuehnel M. F., Sahm C. D., Neri G., Lee J. R., Orchard K. L., Cowan A. J., Reisner E., Chem. Sci. 2018, 9, 2501–2509; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18e. Froehlich J. D., Kubiak C. P., J. Am. Chem. Soc. 2015, 137, 3565–3573; [DOI] [PubMed] [Google Scholar]
- 18f. Méndez M. A., Voyame P., Girault H. H., Angew. Chem. Int. Ed. 2011, 50, 7391–7394; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7529–7532; [Google Scholar]
- 18g. Kimura E., Wada S., Shionoya M., Okazaki Y., Inorg. Chem. 1994, 33, 770–778; [Google Scholar]
- 18h. Kimura E., Bu X., Shionoya M., Wada S., Maruyama S., Inorg. Chem. 1992, 31, 4542–4546; [Google Scholar]
- 18i. Beley M., Colli J.-P., Ruppert R., Sauvage J.-P., J. Chem. Soc. Chem. Commun. 1984, 1315–1316. [Google Scholar]
- 19.
- 19a. Himo F., Lovell T., Hilgraf R., Rostovtsev V. V., Noodleman L., Sharpless K. B., Fokin V. V., J. Am. Chem. Soc. 2005, 127, 210–216; [DOI] [PubMed] [Google Scholar]
- 19b. Feldman A. K., Colasson B., Fokin V. V., Organic Lett. 2004, 6, 3897–3899. [DOI] [PubMed] [Google Scholar]
- 20. Bizzarri C., Strabler C., Prock J., Trettenbrein B., Ruggenthaler M., Yang C.-H., Polo F., Iordache A., Brüggeller P., Cola L. D., Inorg. Chem. 2014, 53, 10944–10951. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Bizzarri C., Arndt A. P., Kohaut S., Fink K., Nieger M., J. Organomet. Chem. 2018, 871, 140–149; [Google Scholar]
- 21b. Bizzarri C., Fléchon C., Fenwick O., Cacialli F., Polo F., Gàlvez-Lòpez M. D., Yang C.-H., Scintilla S., Sun Y., Fröhlich R., De Cola L., ECS J. Solid State Sci. Technol. 2016, 5, R83–R90. [Google Scholar]
- 22. Keller S., Alkan-Zambada M., Prescimone A., Constable E. C., Housecroft C. E., Crystals 2020, 10, 255. [Google Scholar]
- 23.
- 23a. Brouwer A. M., Pure Appl. Chem. 2011, 83, 2213–2228; [Google Scholar]
- 23b. Ishida H., Bünzli J.-C., Beeby A., Pure Appl. Chem. 2016, 88, 701–711. [Google Scholar]
- 24. Cuttell D. G., Kuang S.-M., Fanwick P. E., McMillin D. R., Walton R. A., J. Am. Chem. Soc. 2002, 124, 6–7. [DOI] [PubMed] [Google Scholar]
- 25. Gritzner G., Kuta J., Pure Appl. Chem. 1984, 56, 461. [Google Scholar]
- 26.
- 26a. Opperman K. A., Mecklenburg S. L., Meyer T. J., Inorg. Chem. 1994, 33, 5295–5301; [Google Scholar]
- 26b. Jasimuddin S., Yamada T., Fukuju K., Otsuki J., Sakai K., Chem. Commun. 2010, 46, 8466–8468. [DOI] [PubMed] [Google Scholar]
- 27. Pellegrin Y., Odobel F., Comptes Rendus Chimie 2017, 20, 283–295. [Google Scholar]
- 28. Neri G., Forster M., Walsh J. J., Robertson C. M., Whittles T. J., Farràs P., Cowan A. J., Chem. Commun. 2016, 52, 14200–14203. [DOI] [PubMed] [Google Scholar]
- 29. Fisher B. J., Eisenberg R., J. Am. Chem. Soc. 1980, 102, 7361–7363. [Google Scholar]
- 30. Sampaio R. N., Grills D. C., Polyansky D. E., Szalda D. J., Fujita E., J. Am. Chem. Soc. 2020, 142, 2413–2428. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Kuhn H. J., Braslavsky S. E., Schmidt R., Pure Appl. Chem. 2004, 76, 2105; [Google Scholar]
- 31b. Wegner E. E., Adamson A. W., J. Am. Chem. Soc. 1966, 88, 394–404. [Google Scholar]
- 32. Dolomanov O. V., Bourhis L. J., Gildea R. J., Howard J. A. K., Puschmann H., J. Appl. Crystallogr. 2009, 42, 339–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sheldrick G., Acta Crystallogr. Sect. A 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sheldrick G., Acta Crystallogr. Sect. C 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary