

Abstract
Mitochondria have a multitude of functions, including energy generation and cell signaling. Recent evidence suggests that mitochondrial dynamics (i.e. the balance between mitochondrial fission and fusion) also regulate immune functions. Here, we reveal that lipopolysaccharide (LPS) stimulation increases mitochondrial numbers in mouse bone marrow‐derived macrophages (BMMs) and human monocyte‐derived macrophages. In BMMs, this response requires Toll‐like receptor 4 (Tlr4) and the TLR adaptor protein myeloid differentiation primary response 88 (MyD88) but is independent of mitochondrial biogenesis. Consistent with this phenomenon being a consequence of mitochondrial fission, the dynamin‐related protein 1 (Drp1) GTPase that promotes mitochondrial fission is enriched on mitochondria in LPS‐activated macrophages and is required for the LPS‐mediated increase in mitochondrial numbers in both BMMs and mouse embryonic fibroblasts. Pharmacological agents that skew toward mitochondrial fusion also abrogated this response. LPS triggered acute Drp1 phosphorylation at serine 635 (S635), followed by sustained Drp1 dephosphorylation at serine 656 (S656), in BMMs. LPS‐induced S656 dephosphorylation was abrogated in MyD88‐deficient BMMs, suggesting that this post‐translational modification is particularly important for Tlr4‐inducible fission. Pharmacological or genetic targeting of Tlr4‐inducible fission had selective effects on inflammatory mediator production, with LPS‐inducible mitochondrial fission promoting the expression and/or secretion of a subset of inflammatory mediators in BMMs and mouse embryonic fibroblasts. Thus, triggering of Tlr4 results in MyD88‐dependent activation of Drp1, leading to inducible mitochondrial fission and subsequent inflammatory responses in macrophages.
Keywords: Dynamin‐related protein 1, lipopolysaccharide, macrophage, mitochondrial dynamics, mitochondrial fission, Toll‐like receptor
Here, we demonstrate that lipopolysaccharide (LPS) acts via a Tlr4–MyD88–Drp1 axis to initiate mitochondrial fission and selective inflammatory responses in macrophages. We also show that LPS promotes sequential dynamin‐related protein 1 (Drp1) serine phosphorylation (S635) and dephosphorylation (S656), with the latter modification requiring the adaptor molecule myeloid differentiation primary response 88 (MyD88). We did not observe LPS‐inducible mitochondrial DNA replication in the same cells, suggesting that LPS‐inducible mitochondrial fission can occur independently of mitochondrial DNA replication.

Introduction
Innate immune cells such as macrophages act as danger‐sensing sentinels, responding to environmental perturbations to ensure maintenance of homeostasis. These cells use pattern recognition receptors such as Toll‐like receptors (TLRs) to recognize and respond to an array of endogenous molecules released during cellular stress, as well as microbial products such as lipopolysaccharide (LPS). 1 Functional consequences of TLR activation encompass production of mediators that orchestrate inflammation (e.g. cytokines and chemokines) and expression of molecules that sculpt adaptive immunity (e.g. costimulatory molecules). TLRs signal through the activation of canonical Toll/interleukin‐1 (IL‐1) receptor‐containing adaptor proteins [e.g. myeloid differentiation primary response 88 (MyD88), MyD88 adaptor‐like (MAL), Toll/IL‐1R domain‐containing adaptor‐inducing interferon‐β (TRIF) and TRIF‐related adaptor molecule (TRAM)] 1 as well as noncanonical non‐Toll/IL‐1 receptor adaptors (e.g. SCIMP). 2 With respect to Toll/IL‐1 receptor‐containing adaptors, LPS signaling via TLR4 engages both the MAL/MyD88 and the TRAM/TRIF signaling arms. In the MyD88‐dependent pathway, MyD88 interacts with other death domain‐containing proteins, such as IL‐1 receptor‐associated kinase 4 and 1, forming an oligomeric complex referred to as the Myddosome. 3 In addition to triggering transcription factor activation for inducible gene expression, downstream TLR signaling components can also translocate to mitochondria to reprogram their functions. For example, TLR activation results in increased mitochondrial reactive oxygen species generation in macrophages. 4
Further links between TLRs and mitochondria have emerged through discoveries showing that TLR signaling triggers major changes in metabolic pathways in murine macrophages. 5 Hallmarks of this remodeling are increased glycolysis and flux through the tricarboxylic acid cycle to generate acetyl coenzyme A for inducible gene expression. 6 At later time points, the tricarboxylic acid cycle is disrupted, leading to the accumulation of tricarboxylic acid cycle intermediates such as succinate that have been implicated in specific inflammatory responses. 7 Mitochondria are incredibly dynamic organelles; under different conditions, they can exist as a complex network (driven by fusion) or as fragmented organelles (driven by fragmentation/fission). 8 For example, severe cellular stress and impaired oxidative phosphorylation are associated with mitochondrial fission, whereas nutrient starvation and increased oxidative phosphorylation are linked to mitochondrial fusion. 9 Mitochondrial dynamics are controlled by several dynamin‐related GTPases, including dynamin‐related protein 1 (DRP1), mitofusins 1 and 2, and optic atrophy 1. 8 While the mitofusins and specific optic atrophy 1 isoforms promote mitochondrial tubulation, 10 DRP1 is essential for the fission response. 11 Cytoplasmic DRP1 is recruited by mitochondrial fission factor 12 and other mitochondrial proteins 13 to the mitochondrial outer membrane in combination with the endoplasmic reticulum, where it promotes fission by constricting the mitochondria. 14 DRP1 activity is regulated by several post‐translational modifications, with phosphorylation at S616 (mouse S635) 15 and dephosphorylation at S637 (mouse S656) 16 , 17 , 18 being particularly implicated in its regulation. The exact roles of these and other DRP1 modifications in its activation or inhibition are likely to be cell type and signal dependent.
Previous studies have provided some biochemical 19 , 20 and genetic 21 , 22 evidence to support a role for LPS in regulating mitochondrial dynamics. However, another study reported that LPS stimulates mitochondrial biogenesis in macrophages, 23 and the relationship between the effects of LPS on mitochondrial numbers, mitochondrial fission and mitochondrial biogenesis remains unclear. Moreover, little is known about the specific receptors and downstream signaling pathways associated with LPS‐inducible fission. Here, we address these questions, providing compelling evidence for a Tlr4–MyD88–Drp1 axis as a mediator of LPS‐inducible fission in macrophages and of sequential Drp1 activation during this process. Our study also adds further weight to the emerging concept that mitochondrial fission is linked to specific inflammatory responses. 24 , 25
Results
LPS triggers Tlr4‐ and MyD88‐dependent mitochondrial fission in macrophages
We began by examining mitochondrial morphology in bone marrow‐derived macrophages (BMMs) following LPS stimulation. LPS appeared to promote mitochondrial fragmentation (Figure 1a, b), which was quantified by assessing mitochondrial numbers with two independent methods in an unbiased fashion, the maxima method (assessing the maximum intensity point; Figure 1c, Supplementary figure 1a) and the skeletal method (assessing the connectivity of the mitochondrial network; Figure 1d, Supplementary figure 1b). Because of the consistency in the maxima method across different experiments, this method of quantification was used for all subsequent analyses. The specificity of MitoTracker staining was demonstrated by costaining for the mitochondrial proteins heat‐shock protein 60 and Tom20 (Supplementary figure 1c, d). Some LPS responses, including with respect to signaling, 26 gene expression 27 and cell metabolism, 28 are divergent between primary mouse and human macrophages. Thus, we next assessed LPS‐inducible mitochondrial fragmentation in human monocyte‐derived macrophages. Here we found that, as observed in BMMs, LPS increased mitochondrial numbers in these cells (Figure 1e, Supplementary figure 1e, f). Given this conservation in response between human and mouse macrophages, we next used BMMs to explore molecular mechanisms involved. We first investigated whether the effect of LPS is mediated by its transmembranal receptor Tlr4 29 or the cytosolic LPS receptor caspase‐11. 30 , 31 Here we found that the LPS‐mediated increase in mitochondrial numbers was defective in Tlr4 −/− BMMs, but not in Casp1/11 −/− BMMs (Figure 1f). Moreover, the response was absolutely dependent on the TLR adaptor protein MyD88 (Figure 1g).
Figure 1.
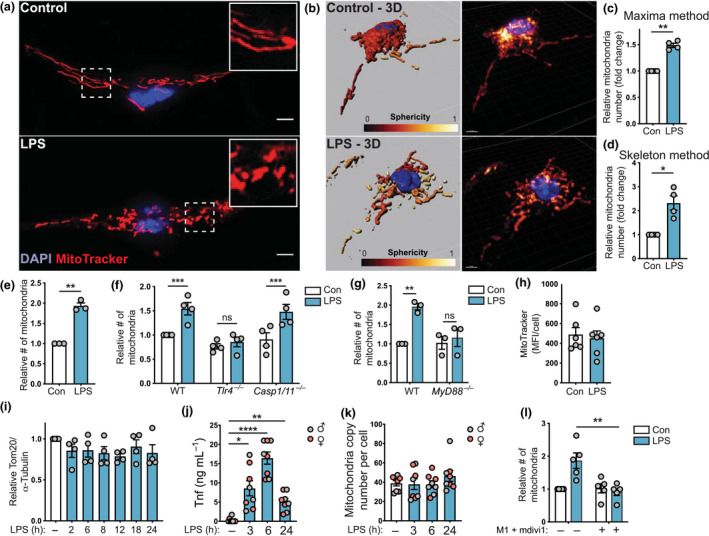
LPS triggers Tlr4‐ and MyD88‐dependent mitochondrial fission in macrophages. (a–l) Unless otherwise indicated, BMMs were stimulated with LPS (100 ng mL−1) or left untreated for 6 h, prior to performing indicated analyses. Mitochondrial numbers were determined relative to the unstimulated control in each experiment. (a) BMMs were stained with MitoTracker (mitochondria, red) and DAPI (nuclear DNA, blue). (b) The mitochondrial network was reconstituted in 3D using Imaris software. (c, d) Relative mitochondrial number observed in BMMs using (c) a maxima analysis method and (d) a skeletal analysis method (same data sets used for each analysis). Data are combined from four independent experiments (mean ± s.e.m., one sample t‐test). (e) Relative mitochondrial numbers in human monocyte‐derived macrophages (mean ± s.e.m., three independent experiments, one sample t‐test). (f) Relative mitochondrial numbers in WT, Tlr4 −/− and Caspase 1/11 −/− BMMs [mean ± s.e.m., four independent experiments, two‐way ANOVA with Šidák’s multiple comparisons test]. (g) Relative mitochondrial numbers in WT and MyD88 −/− BMMs (mean ± s.e.m., three independent experiments, two‐way ANOVA with Šidák’s multiple comparisons test). (h) Median fluorescence intensity (MitoTracker) per cell (mean ± s.e.m., six independent experiments, t‐test). (i) Mitochondrial protein Tom20 levels (relative to tubulin) in BMMs stimulated with LPS for the indicated times. Cells were lysed, after which Tom20 and tubulin levels were assessed by western blot (mean ± s.e.m., four independent experiments, one‐way ANOVA with Dunnett’s multiple comparisons test). (j, k) BMMs from male (gray circles) and female (red circles) mice were treated with LPS for 3, 6 or 24 h. (j) Tnf production was assessed by ELISA and (k) mitochondrial DNA copy number relative to nuclear DNA was determined (mean ± s.e.m., BMMs harvested from eight different mice, one‐way ANOVA with Dunnett’s multiple comparisons test). (l) BMMs were pretreated for 1 h with DMSO or M1 + mdivi1, after which cells were stimulated with LPS and relative mitochondrial numbers were calculated (mean ± s.e.m., five independent experiments, two‐way ANOVA with Šidák’s multiple comparisons test). In all panels, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Scale bars, 5 µm. BMMs, bone marrow‐derived macrophages; Con, control; DAPI, 4′,6‐diamidino‐2‐phenylindole; DMSO, dimethyl sulfoxide; LPS, lipopolysaccharide; MFI, mean fluorescent intensity; ns, not significant; Tnf, tumor necrosis factor; WT, wild type.
We next considered if the increase in mitochondrial numbers was linked to increased mitochondrial biogenesis and/or reduced mitophagy. The median fluorescence intensity of MitoTracker‐stained cells was similar in control and LPS‐treated cells (Figure 1h) and LPS treatment did not alter Tom20 protein levels (Figure 1i, see also Figure 3). This suggests that the mitochondrial mass is comparable in control and LPS‐treated BMMs. Next, we assessed the effect of LPS on mitochondrial DNA copy number in BMMs. Whereas LPS induced tumor necrosis factor (TNF) production in BMMs (Figure 1j), the ratio of mitochondrial genes (non‐NUMT, a specific region of mitochondrial DNA) 23 to nuclear genes (nuclear DNA B2m) remained unchanged over an LPS time course (Figure 1k). Collectively, these data (Figure 1h–k) suggest that, although LPS induces fragmentation of the mitochondrial network, it does not increase mitochondrial mass and/or mitochondrial DNA replication. Finally, we further validated our methodology by confirming that the mitochondrial fusion‐promoting compound M1 32 in combination with the Drp1 inhibitor mdivi1 33 blocked the LPS‐inducible increase in mitochondrial numbers in BMMs (Figure 1l). We conclude that LPS acts via Tlr4 and MyD88 to skew toward mitochondrial fission in macrophages.
Figure 3.
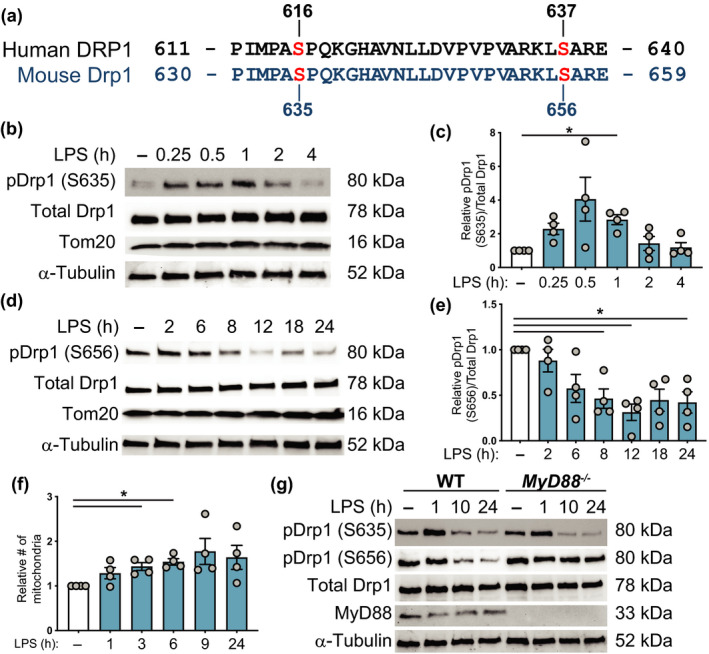
LPS induces acute Drp1 phosphorylation at S635 and sustained dephosphorylation at S656 in macrophages. (a) Amino acid sequence comparison for the indicated residues of RefSeq human DRP1 variant 1 (isoform 1) and RefSeq mouse Drp1 variant 6 (isoform e), with indicated serine (S) phosphorylation sites on each sequence. (b) BMMs were stimulated with LPS for the indicated time points, after which whole cell extracts were collected and analyzed by western blot for phospho‐Drp1 (S635), total Drp1, Tom20 and α‐tubulin. Immunoblots are representative of four independent experiments. (c) Quantitative analysis of phospho‐Drp1 (S635) relative to total Drp1 levels, with all data plotted relative to the unstimulated control cells (−). Data are combined from four independent experiments [mean ± s.e.m., one‐way ANOVA with Dunnett’s multiple comparisons test]. (d) Cells were stimulated with LPS for the indicated time points, after which whole cell extracts were collected and analyzed by western blot for phospho‐Drp1 (S656), total Drp1 and α‐tubulin. Immunoblots are representative of four independent experiments. (e) Quantitative analysis of phospho‐Drp1 (S656) relative to total Drp1 levels, with all data plotted relative to the unstimulated control cells (−). Data are combined from four independent experiments (mean ± s.e.m., one‐way ANOVA with Dunnett’s multiple comparisons test). (f) Mitochondrial numbers (relative to unstimulated control cells) at different time points following LPS stimulation (mean ± s.e.m., four independent experiments, one‐way ANOVA with Dunnett’s multiple comparisons test). (g) Wild‐type and MyD88 −/− BMMs were stimulated with LPS as indicated. Whole cell extracts were prepared, and then analyzed by western blot for phospho‐Drp1 (S635), phospho‐Drp1 (S656), total Drp1, MyD88 and α‐tubulin. The immunoblot is representative of four experiments. In all panels, *P < 0.05. BMMs, bone marrow‐derived macrophages; DRP1, dynamin‐related protein 1; LPS, lipopolysaccharide; WT, wild type.
LPS‐inducible mitochondrial fragmentation is dependent on Drp1
To further confirm that LPS induces mitochondrial fission, we next assessed the involvement of the fission‐promoting GTPase Drp1 using genetic approaches. Both small interfering RNA‐mediated Drp1 silencing in BMMs (Supplementary figure 2a, b) and Drp1 deletion in mouse embryonic fibroblast (MEF) cells 34 (Supplementary figure 2c) revealed that LPS‐induced mitochondrial fragmentation required Drp1 (Figure 2a, b). This was further confirmed by quantifying LPS‐inducible mitochondrial numbers in these cells (Figure 2c, d). Drp1 is recruited from the cytoplasm to mitochondria to initiate mitochondrial fission, 35 and we also observed that LPS increased the association of Drp1 with mitochondria (Figure 2e, f, Supplementary figure 2d). Thus, LPS promotes Drp1 recruitment to mitochondria to initiate fission.
Figure 2.
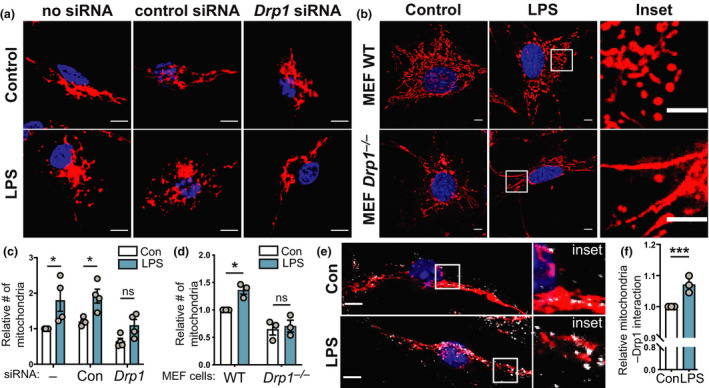
LPS‐inducible mitochondrial fragmentation requires Drp1. (a–f) The indicated cell populations were stimulated with LPS (100 ng mL−1) or left untreated for 6 h, prior to performing indicated analyses. Mitochondrial numbers were determined relative to the unstimulated control in each experiment. (a) BMMs were electroporated with no siRNA or siRNAs targeting either a control gene (Hdac1) or Drp1, incubated for 18 h and then treated with LPS. Cells were stained with MitoTracker (mitochondria, red) and DAPI (nuclear DNA, blue). (b) WT and Drp1 −/− MEF cells were treated with LPS, before being stained with MitoTracker (mitochondria, red) and DAPI (nuclear DNA, blue). Images in a and b are representative of a minimum of three independent experiments. (c, d) Quantification of the relative number of mitochondria in a and b. (c) Mean ± s.e.m. of four independent experiments (calculated using two‐way ANOVA with Šidák’s multiple comparisons test) and (d) mean ± s.e.m. of three independent experiments (calculated using two‐way ANOVA with Šidák’s multiple comparisons test). (e) BMMs were LPS treated before being stained with MitoTracker (mitochondria, red), DAPI (nuclear DNA, blue) and anti‐Drp1 (white). Images are representative of three independent experiments. (f) Relative colocalization of Drp1 with MitoTracker, with data displayed relative to unstimulated control cells (mean ± s.e.m., three independent experiments, one sample t‐test). In all panels, *P < 0.05, ***P < 0.001. Scale bars, 5 µm. BMMs, bone marrow‐derived macrophages; Con, control; DAPI, 4′,6‐diamidino‐2‐phenylindole; LPS, lipopolysaccharide; MEF, mouse embryonic fibroblast; ns, not significant; siRNA, small interfering RNA; WT, wild type.
LPS regulates Drp1 phosphorylation in BMMs
We next examined LPS‐regulated post‐translational modifications on Drp1. Specific post‐translational modifications on Drp1, particularly phosphorylation of S616 15 (S585 in rat, S635 in mouse) and dephosphorylation of S637 (S656 in mouse) 16 , 17 , 18 (Figure 3a), have been associated with its activation as well as its translocation to the mitochondria in different cell types. In BMMs, LPS promoted acute phosphorylation (approximately 15–60 min after stimulation) of Drp1 at S635, whereas the levels of total Drp1 remained unchanged (Figure 3b, c). LPS also triggered dephosphorylation of Drp1 at S656, with this effect being apparent at approximately 6 h after stimulation and being sustained for 24 h (Figure 3d, e). Because Drp1 phosphorylation at S635 was a rapid response, we next examined mitochondrial fission across an LPS time course. Indeed, a trend for increased mitochondrial numbers was apparent as early as 1 h after LPS stimulation, with the response plateauing at approximately 6–9 h after LPS treatment (Figure 3f). To gain further insight into post‐translational modifications on Drp1 that are likely important for LPS‐inducible fission, we monitored the Drp1 phosphorylation status in MyD88 −/− BMMs, because the LPS‐inducible mitochondrial fission response was defective in these cells (Figure 1g). Whereas the transient phosphorylation of Drp1 on S635 still occurred in MyD88 −/− BMMs, S656 dephosphorylation was abrogated (Figure 3g, Supplementary figure 2e, f). Thus, LPS triggers rapid Drp1 phosphorylation at S635 independently of MyD88, whereas dephosphorylation of Drp1 at S656 requires this TLR adaptor.
Mitochondrial fission regulates a subset of Tlr4‐inducible inflammatory responses
Some studies have linked mitochondrial dynamics to inflammatory responses, particularly in microglial cells. 20 , 22 Thus, we next investigated whether LPS‐inducible mitochondrial fission regulates inflammatory responses in BMMs. Pretreating BMMs with M1 and mdivi1 that skew mitochondrial dynamics toward fusion 32 , 33 resulted in reduced LPS‐inducible production of the proinflammatory cytokines Il‐12p40 and Il‐6 (Figure 4a, b). These effects were selective, as the combination of M1 and mdivi1 did not affect inducible production of nitric oxide in BMMs (Figure 4c). The inhibitory effect on Il‐12p40 and Il‐6 in BMMs was also apparent at the messenger RNA (mRNA) level (Figure 4d, e), whereas the marginal reduction of LPS‐inducible Nos2 mRNA (required for nitric oxide production) was not statistically significant (Figure 4f). LPS‐inducible Ccl2 was also unaffected (Supplementary figure 3a), confirming that M1/mdivi1 did not globally affect LPS responses in BMMs. Given that mdivi1 can have off‐target effects, 36 we next examined Drp1 dependence for LPS responses using Drp1‐deficient MEF cells. In this system, we examined LPS‐regulated gene expression, because levels of secreted Il‐12p40 and Il‐6 were too low for accurate quantification. Here we found that Drp1 deletion abrogated LPS‐inducible Il12b mRNA expression (Figure 4g), with a trend toward reduced Il6 (Figure 4h). By contrast, Nos2 and Ccl2 were not significantly affected (Figure 4i, Supplementary figure 3b). The proinflammatory mediators Il‐1β and Tnf are regulated not only by transcriptional and translational mechanisms, but also by post‐translational cleavage events. Tnf is produced as a transmembranal precursor that is cleaved by the metalloproteinase TNF‐α‐converting enzyme, 37 whereas release of Il‐1β from cells requires inflammasome‐dependent cleavage by proinflammatory caspases. 38 Experimentally, the microbial product nigericin is widely used to initiate inflammasome activation and processing of pro‐Il‐1β for secretion of bioactive Il‐1β. Here we found that antagonizing fission significantly reduced nigericin‐triggered Il‐1β release from LPS‐primed BMMs (Figure 4j), whereas LPS‐inducible Il1b mRNA expression was only modestly affected in the same cells (Supplementary figure 3c). Similarly, there was a clear trend for M1/mdivi1 to reduce LPS‐induced Tnf secretion in BMMs (Figure 4k), with only a very minor effect on Tnf mRNA levels being apparent (Supplementary figure 3d). We conclude that LPS triggers Drp1 activation for inducible fission and inflammatory responses in macrophages, possibly through multiple mechanisms.
Figure 4.
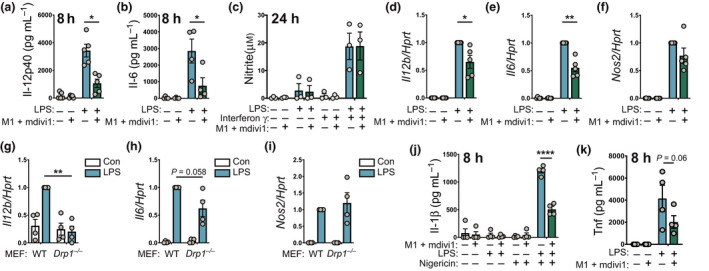
Mitochondrial fission promotes selective inflammatory responses. (a, b) BMMs were pretreated for 1 h with vehicle (DMSO) or M1 + mdivi1 and then treated with or without LPS (100 ng mL‐1) for 8 h. Supernatants were collected and assessed for levels of (a) Il‐12p40 or (b) Il‐6 [mean ± s.e.m., five independent experiments, two‐way ANOVA with Sidak’s multiple comparisons test]. (c) BMMs were stimulated with interferon‐γ for 18 h and then treated for 1 h with vehicle (DMSO) or M1 + mdivi1. Cells were then stimulated for 24 h with LPS (100 ng mL−1). Supernatants were collected and assessed for nitrite by Griess assay (mean ± s.e.m., four independent experiments, two‐way ANOVA with Sidak’s multiple comparisons test). (d–f) DMSO‐ or M1 + mdivi1‐pretreated BMMs were stimulated with LPS (100 ng mL−1) for 8 h. Total RNA was collected and mRNA levels of (d) Il12b, (e) Il6 and (f) Nos2 (relative to Hprt) were determined by real‐time‐quantitative PCR. Data (mean ± s.e.m., n = 5, two‐way ANOVA with Šidák’s multiple comparisons test) are combined from five independent experiments and are normalized to the LPS‐treated sample. (g–i) WT and Drp1 −/− MEF cells were stimulated with LPS (100 ng mL−1) for 4 h and mRNA levels of the indicated genes were assessed. Data (mean ± s.e.m., n = 4, two‐way ANOVA with Šidák’s multiple comparisons test) are combined from four independent experiments and are normalized to the WT LPS‐treated sample. (j) BMMs were treated as in a and b, but at 4 h after LPS stimulation, nigericin (10 µm) was added for another 4 h. Supernatants were collected and assessed for Il‐1 β levels (mean ± s.e.m., four independent experiments, two‐way ANOVA with Šidák’s multiple comparisons test). (k) BMMs were stimulated as in a and b and levels of secreted Tnf (mean ± s.e.m., four independent experiments, two‐way ANOVA with Šidák’s multiple comparisons test) were determined by ELISA. In all panels, *P < 0.05, **P < 0.01, ****P < 0.0001. BMMs, bone marrow‐derived macrophages; Con, control; DMSO, dimethyl sulfoxide; IL, interleukin; LPS, lipopolysaccharide; MEF, mouse embryonic fibroblast; mRNA, messenger RNA; Tnf, tumor necrosis factor; WT, wild type.
Discussion
LPS is a potent regulator of metabolism and mitochondrial functions in macrophages, 7 but only a limited number of studies have explored LPS‐regulated mitochondrial fission in this cell type. 19 , 20 , 22 , 39 Here we build on these studies, profiling LPS‐inducible fission in both primary human and mouse macrophages, examining other important parameters such as mitochondrial DNA synthesis in parallel, defining signaling components that are required for the response and using complementary genetic and pharmacological approaches to validate key findings. In so doing, we provide compelling evidence of LPS‐inducible mitochondrial fission in macrophages, delivering new insights into molecular mechanisms involved and the downstream consequences. We note, however, that although we have demonstrated that LPS increases mitochondrial fragmentation and numbers in human monocyte‐derived macrophages, we did not examine molecular mechanisms of inducible fission in these cells. Given known differences between human and murine macrophages responding to LPS, 26 , 27 such studies are clearly warranted. Furthermore, LPS‐inducible glycolysis has been reported to be dependent on Drp1 and/or fission (discussed later), yet LPS did not increase glycolysis in human macrophages. 28 Thus, the functional consequences of LPS‐inducible fission in human macrophages also require further investigation.
In investigating underlying molecular mechanisms in mouse BMMs, we found that both Tlr4 and MyD88 are required for LPS‐inducible mitochondrial fission. A previous study reported that a low dose of LPS induced mitochondrial fragmentation in BMMs via an IL‐1 receptor‐associated kinase 1‐dependent mechanism. 39 Our findings are consistent with this study, because IL‐1 receptor‐associated kinase 1 lies downstream of MyD88 in the Tlr4 pathway. 40 We also show that LPS triggers sequential post‐translational modifications on Drp1, namely transient phosphorylation on S635 and sustained dephosphorylation on S656. Unlike S635 phosphorylation, S656 dephosphorylation was dependent on MyD88, implying that this modification is likely to be particularly important for initiating LPS‐inducible mitochondrial fission. Phosphorylation of S616 on human DRP1 (corresponding to mouse S635) by either cyclin B/Cdk1 or extracellular signal‐regulated kinase 2 was linked to the recruitment of DRP1 to the mitochondria and the initiation of fission. 41 , 42 The acute LPS‐inducible phosphorylation of S635 observed here did not require MyD88, so the role of this modification in macrophage responses to LPS remains to be determined. It may be involved in the early increase in mitochondrial numbers that we observed here (1–2 h after LPS stimulation). Another possibility is that it is involved in LPS‐inducible inflammatory responses independently of fission. This may be important, given that some studies on LPS‐inducible fission have focused on regulation of S616/S635. 19 , 43 In contrast to inducible S635 phosphorylation, dephosphorylation of S656 did require MyD88, implicating this post‐translational modification in the inducible fission response. Because S656 dephosphorylation only occurred several hours after LPS stimulation, it is likely that autocrine/paracrine factors and/or regulated expression of phosphatases and/or kinases that modify this residue may be important. Calcineurin and cyclic adenosine monophosphate‐dependent protein kinase, which both modulate the phosphorylation status of S656 in other cell types, 44 are also implicated in LPS responses. 45 , 46 Furthermore, a role for calcineurin in LPS‐mediated Drp1 dephosphorylation in microglia has previously been demonstrated. 47 These therefore represent potential candidates for regulating TLR‐driven Drp1 dephosphorylation and fission in BMMs and other macrophage populations. In addition, several other unknowns around the signaling framework for LPS‐inducible fission in macrophages remain. Specifically, whether S635 phosphorylation and/or S656 dephosphorylation have causal roles in LPS‐inducible fission in macrophages is yet to be determined. This could be interrogated in the future through reconstitution of Drp1‐deficient cells with S635A and/or S656E Drp1 mutants. Roles for other TLR adaptor molecules such as TRIF and TRAM, particularly in acute S635 phosphorylation and the early fission response, also need to be addressed.
Whereas our data clearly show that LPS induces mitochondrial fission in BMMs, we found no evidence of increased mitochondrial DNA replication or biogenesis in these cells. We note that we examined the effect of LPS on mitochondrial DNA copy number over a time course in BMMs from both male and female mice. Our findings contrast with those of Zhong et al. who also studied BMMs, finding that LPS increased mitochondrial DNA content and that this primed inflammasome responses. 23 The reasons for the different observations in this study versus our own are unclear, but might be related to variation in cell culture conditions. Although both studies used similar concentrations of LPS and glucose in culture media, there were differences in other variables such as the macrophage differentiation methods. Whatever the explanation, our study clearly shows that LPS‐inducible fission can occur in the absence of inducible mitochondrial DNA synthesis in macrophages. This might also be true in human disease, as it was shown that DRP1 protein expression was elevated in tissues of critically ill patients versus those of controls, whereas there was no difference in mitochondrial DNA copy number between these groups. 48 Our findings are also somewhat consistent with a study in PC‐12 cells where rotenone promoted mitochondrial fission but actually impaired mitochondrial biogenesis via the transcription factor peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha. 49 We suggest that the LPS‐induced increase in mitochondrial numbers without a corresponding increase in mitochondrial DNA copy number might be analogous to a cell that has a greater abundance of naïve mitochondria. This occurs in oocytes, for example, where there is less dependence on oxidative phosphorylation, matched by fewer copies of the mitochondrial genome per mitochondrion. 50 , 51 In this scenario, LPS‐induced fission in macrophages might be predicted to result in a decrease in the average number of mitochondrial genomes per mitochondrion. Future studies should examine the relationship between inducible fission and mitochondrial DNA synthesis in activated macrophages in vivo in different inflammatory contexts.
In both smooth muscle cells 43 and microglia, 20 mitochondrial fission has been implicated in LPS‐inducible glycolysis. This metabolic response is known to be required for the production of a subset of inflammatory mediators, 5 thus implying that fission may promote inflammatory responses. Consistent with this, we found that genetic as well as pharmacological targeting of fission reduced the production of several proinflammatory cytokines, including Il‐12p40, Il‐6, Tnf and Il‐1β. These findings are broadly consistent with other studies undertaken in microglia. 20 , 22 Interestingly, the glycolytic enzyme pyruvate kinase 2 isoform M2 was shown to modulate mitochondrial dynamics via an interaction with mitofusin 2. 52 This interaction promoted mitochondrial fusion and controlled the metabolic switch between glycolysis and oxidative phosphorylation in cancer. 52 Pyruvate kinase 2 isoform M2 also drives LPS‐inducible Il‐1β production in murine macrophages, 53 , 54 a cytokine we found to be downstream of LPS‐induced fission. One possibility is that LPS‐induced fission inhibits the fusion‐promoting activity of pyruvate kinase 2 isoform M2, enabling it to perform its inflammatory functions. Thus, it is plausible that glycolysis drives LPS‐inducible Il‐1β production as a consequence of increased mitochondrial fission. We also observed that Il‐12p40 production was particularly dependent on the mitochondrial fission response, with LPS‐inducible Il12b mRNA expression being abrogated by Drp1 deletion in MEF cells. Consistent with our data, Gao et al. found that promoting mitochondrial fission by silencing Fam73b increased IL‐12 production in macrophages. 55 Thus, future studies on Il12b regulation are likely to provide insights into the molecular mechanisms by which Drp1 and/or mitochondrial fission promote macrophage inflammatory responses. Mitochondrial fission was recently linked to sepsis‐induced cardiomyopathy. 56 Moreover, antagonism of mitochondrial fission slowed disease progression in a mouse model of amyotrophic lateral sclerosis and abrogated inflammation associated with septic encephalopathy. 57 , 58 Further advances in our understanding of fission‐mediated inflammatory responses may therefore provide opportunities for targeting inflammation‐mediated disease in the future. In summary, LPS promotes acute S635 phosphorylation and sustained S656 dephosphorylation of Drp1, recruitment of Drp1 to mitochondria, mitochondrial fission and Drp1‐dependent inflammatory responses in macrophages (Figure 5). Future studies should address the involvement of each of these post‐translational modifications in specific macrophage inflammatory responses and the molecular mechanisms involved.
Figure 5.
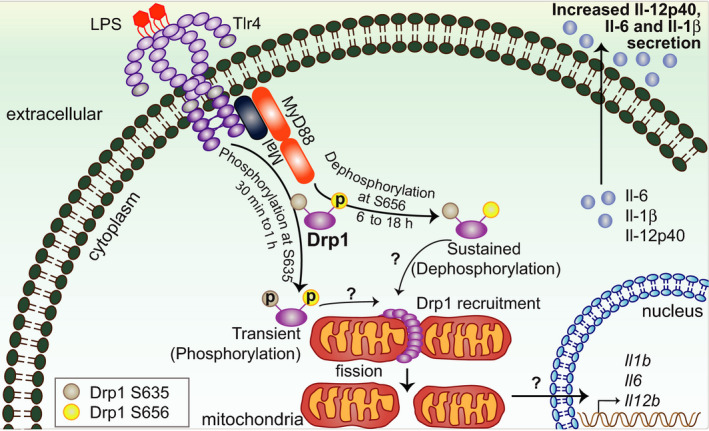
Current model of mechanisms of TLR4‐inducible mitochondrial fission and its proinflammatory effects. Upon Tlr4 stimulation, Drp1 is acutely phosphorylated at S635 independently of the adaptor molecule MyD88. By contrast, MyD88 is required for LPS‐mediated dephosphorylation of Drp1 at S656 and for mitochondrial fission. Fission enhances the expression of specific proinflammatory genes, for example, Il12b. Drp1, dynamin‐related protein 1; IL, interleukin; LPS, lipopolysaccharide; MyD88, myeloid differentiation primary response 88; TLR, Toll‐like receptor.
Methods
Ethics statement
All experiments involving primary human cells were approved by the University of Queensland Medical Research Ethics Committee (approved certificate numbers 2009001051 and 2011000826). Use of primary mouse cells was approved by The University of Queensland Animal Ethics Committee (IMB/123/18, UQDI058/19).
Reagents, animals, cell isolation and culture, quantitative PCR and quantification of mitochondrial DNA copy number and microscopy quantification
These details are provided in Supplementary material.
Gene silencing
BMMs were resuspended in media containing HEPES (10 mmol L−1; Gibco, Waltham, MA, USA). Indicated small interfering RNAs (Supplementary table 1, 1 μM final), 300 μL cell suspension and media up to a total volume of 400 μL were added to a 0.4‐cm cuvette. The cuvette was then subjected to electroporation using a Gene Pulser Xcell Electroporation System (Bio‐Rad, Hercules, CA, USA) (240 V, 1000 μF, resistance of ∞). Cells were allowed to recover at room temperature (RT) for 10–15 min and then plated at a concentration of 0.5 × 106 cells mL−1.
Microscopy
BMMs (2 × 105) or MEF (2.5 × 104) cells were plated on cover slips in 24‐well plates. For mitochondrial staining, after appropriate stimulation, media was replaced and new media containing MitoTracker Deep Red FM (Invitrogen, Waltham, MA, USA; 150 nmol L−1 for BMMs and 200 nmol L−1 for MEF cells) was added for 30 min. Cells were washed three times with phosphate‐buffered saline (PBS (Gibco); 5 min each wash) before fixing with 4% paraformaldehyde for 15 min at RT. After fixation, cells were washed three times with PBS and stained with 4′,6‐diamidino‐2‐phenylindole (20 ng mL−1) for 30 min. For immunostaining with anti‐Tom20 and anti‐Hsp60 antibodies, after fixing with 4% paraformaldehyde (Sigma, St. Louis, MO, USA), cells were permeabilized using 0.1% Triton X‐100 (Sigma) in PBS for 5 min. Next, cells were blocked in 0.5% bovine serum albuminin PBS for 1 h at RT. Cells were then incubated with primary antibodies, rabbit anti‐Tom20 or rabbit anti‐Hsp60, diluted in 0.5% bovine serum albumin in PBS for 1 h at RT, followed by incubation in a secondary antibody (goat antirabbit Alexa Fluor 488) diluted in 0.5% bovine serum albumin in PBS and 4′,6‐diamidino‐2‐phenylindole (20 ng mL−1) for 1 h at RT. The coverslips were mounted using mounting media (homemade IMBiol) and cells were imaged using a Zeiss Axiovert 200 Upright Microscope Stand with LSM 710 Meta Confocal Scanner and Spectral detection with 63× magnification (Zeiss, Oberkochen, Germany).
ELISA and nitrite production
BMMs were plated in 96‐well plates at 100 000 cells per 100 µL overnight, after which cells were pretreated with mitochondrial fission inhibitors M1 (20 µm) and mdivi1 (10 µm) for 1 h, prior to stimulation with LPS (100 ng mL−1) for the indicated time points. To induce IL‐1β secretion, BMMs were stimulated for 4 h with LPS, followed by treatment with nigericin (10 µm; Sigma, St. Louis, MO, USA) for 4 h. To assess nitric oxide production, BMMs were pretreated with IFNγ (5 ng mL−1; R&D System, Minneapolis, MN, USA) for 18 h, after which cells were stimulated with LPS for 24 h. Nitrite levels were measured by Griess assay (Promega, Madison, WI, USA) and levels of secreted Il‐12p40, Il‐6, Il‐1β and Tnf were measured by ELISA according to the manufacturer’s instructions [TNF and IL‐1β kits: BD Biosciences, Franklin Lakes, NJ, USA; IL‐6 antibodies: BD Biosciences (Capture: #554400, Detection: #554402); IL‐12p40 antibodies (Capture: #551219, Detection: #554476)].
Immunoblotting
Cells (2 × 106) were plated in six‐well plates and, after appropriate stimulations, they were lysed using radioimmunoprecipitation assay buffer (50 mmol L−1 Tris–HCl, 150 mmol L−1 NaCl, 1 mmol L−1 ethylenediaminetetraacetic acid, 1% Triton X‐100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate) supplemented with cOmplete EDTA‐free protease inhibitor cocktail (Sigma) and PhosSTOP phosphatase inhibitor tablets (Sigma). Cell lysates (10 µg protein measured by bicinchoninic acid assay; Thermo Fisher Scientific, Waltham, MA, USA) were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred on to nitrocellulose membranes (Bio‐Rad). Membranes were probed with antibodies (Supplementary table 2) and then developed by chemiluminescence using Clarity ECL (Bio‐Rad).
Statistics
Experimental data from three or more repeat experiments were combined (taking the mean of technical replicates from each experiment) and GraphPad Prism 8 (San Diego, CA, USA) was used for specific statistical analyses, as indicated in individual figure legends. Data with n ≥ 3 are represented as mean + s.e.m., where n represents number of experiments. Statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001) was determined using an unpaired t‐test, one sample t‐test, one‐way or two‐way ANOVA, depending on the type of data.
Author Contribution
Ronan Kapetanovic: Conceptualization; Funding acquisition; Investigation; Methodology; Supervision; Writing original draft. Syeda Farhana Afroz: Investigation; Methodology; Writing original draft. Divya Ramnath: Investigation; Methodology; Visualization; Writing original draft. Grace MEP Lawrence: Investigation; Methodology. Takashi Okada: Investigation; Methodology. James EB Curson: Investigation. Jost de Bruin: Investigation. David P Fairlie: Funding acquisition. Kate Schroder: Resources; Writing original draft. Justin C St. John: Investigation; Methodology; Supervision; Writing original draft. Antje Blumenthal: Resources; Writing original draft. Matthew J Sweet: Conceptualization; Funding acquisition; Methodology; Supervision; Writing original draft.
Conflict of Interest
All authors declare that they have no conflict of interest.
Supporting information
Acknowledgments
This work was supported by a National Health and Medical Research Council of Australia (NHMRC) project grant to MJS and DPF (APP1125316), an NHMRC project grant to JCSJ (APP1160106), as well as a Rebecca Cooper Foundation Research Grant to RK (PG2018095). MJS is supported by an NHMRC Senior Research Fellowship (APP1107914), DPF is supported by an NHMRC Senior Principal Research Fellowship (APP1117017) and the Australian Research Council Centre of Excellence in Advanced Molecular Imaging (CE140100011), RK was supported by an Australian Research Council Discovery Early Career Researcher Award Fellowship (DE1310470), KS is supported by an NHMRC Career Development Fellowship (APP1141131) and JdB was supported by an NL K.F. Hein Fonds Grant (1652430). We acknowledge the support of the Institute for Molecular Bioscience Dynamic Imaging Facility for Cancer Biology at The University of Queensland, established with the support of the Australian Cancer Research Foundation. We also thank the Biological Research Facility at the Translational Research Institute, Dr Nicholas Condon (Institute for Molecular Bioscience, The University of Queensland) for assistance with IMARIS, Dr Alina Zamoshnikova (The University of Queensland Diamantina Institute) for technical support, the Australian Red Cross Blood Service for providing buffy coats for the isolation of human monocytes and Professor Mike Ryan (Monash Biomedicine Discovery Institute, Monash University, Melbourne, Australia) for providing Drp1−/− and wild‐type control MEF cells. The funders had no role in study design, data collection and interpretation, or the decision to submit this work for publication.
Contributor Information
Ronan Kapetanovic, Email: r.kapetanovic@imb.uq.edu.au.
Matthew J Sweet, Email: m.sweet@imb.uq.edu.au.
References
- 1. Kawasaki T, Kawai T. Toll‐Like Receptor Signaling Pathways. Front Immunol 2014; 5: 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Luo L, Bokil NJ, Wall AA, et al SCIMP is a transmembrane non‐TIR TLR adaptor that promotes proinflammatory cytokine production from macrophages. Nat Commun 2017; 8: 14133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balka KR, De Nardo D. Understanding early TLR signaling through the Myddosome. J Leukoc Biol 2019; 105: 339–351. [DOI] [PubMed] [Google Scholar]
- 4. West AP, Brodsky IE, Rahner C, et al TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011; 472: 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ryan DG, O'Neill LAJ. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett 2017; 591: 2992–3006. [DOI] [PubMed] [Google Scholar]
- 6. Lauterbach MA, Hanke JE, Serefidou M, et al Toll‐like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP‐citrate lyase. Immunity 2019; 51: 997–1011.e7. [DOI] [PubMed] [Google Scholar]
- 7. Tannahill GM, Curtis AM, Adamik J, et al Succinate is an inflammatory signal that induces IL‐1β through HIF‐1α. Nature 2013; 496: 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 2020; 21: 204–224. [DOI] [PubMed] [Google Scholar]
- 9. Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab 2016; 27: 105–117. [DOI] [PubMed] [Google Scholar]
- 10. Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol 2007; 178: 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin‐related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 2001; 12: 2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Otera H, Wang C, Cleland MM, et al Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 2010; 191: 1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 2013; 24: 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rowland AA, Voeltz GK. Endoplasmic reticulum‐mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 2012; 13: 607–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin‐related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 2007; 282: 11521–11529. [DOI] [PubMed] [Google Scholar]
- 16. Chang CR, Blackstone C. Cyclic AMP‐dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 2007; 282: 21583–21587. [DOI] [PubMed] [Google Scholar]
- 17. Cereghetti GM, Stangherlin A, Martins de Brito O, et al Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 2008; 105: 15803–15808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu X, Jia L, Yu W, Du H. Dephosphorylation by calcineurin regulates translocation of dynamin‐related protein 1 to mitochondria in hepatic ischemia reperfusion induced hippocampus injury in young mice. Brain Res 2019; 1711: 68–76. [DOI] [PubMed] [Google Scholar]
- 19. Katoh M, Wu B, Nguyen HB, et al Polymorphic regulation of mitochondrial fission and fusion modifies phenotypes of microglia in neuroinflammation. Sci Rep 2017; 7: 4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nair S, Sobotka KS, Joshi P, et al Lipopolysaccharide‐induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo . Glia 2019; 67: 1047–1061. [DOI] [PubMed] [Google Scholar]
- 21. Park S, Won JH, Hwang I, Hong S, Lee HK, Yu JW. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci Rep 2015; 5: 15489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park J, Choi H, Min JS, et al Mitochondrial dynamics modulate the expression of pro‐inflammatory mediators in microglial cells. J Neurochem 2013; 127: 221–232. [DOI] [PubMed] [Google Scholar]
- 23. Zhong Z, Liang S, Sanchez‐Lopez E, et al New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018; 560: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Joshi AU, Minhas PS, Liddelow SA, et al Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci 2019; 22: 1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang X, Chen Z, Fan X, et al Inhibition of DNM1L and mitochondrial fission attenuates inflammatory response in fibroblast‐like synoviocytes of rheumatoid arthritis. J Cell Mol Med 2020; 24: 1516–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun J, Li N, Oh KS, et al Comprehensive RNAi‐based screening of human and mouse TLR pathways identifies species‐specific preferences in signaling protein use. Sci Signal 2016; 9: ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schroder K, Irvine KM, Taylor MS, et al Conservation and divergence in Toll‐like receptor 4‐regulated gene expression in primary human versus mouse macrophages. Proc Natl Acad Sci USA 2012; 109: E944–E953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vijayan V, Pradhan P, Braud L, et al Human and murine macrophages exhibit differential metabolic responses to lipopolysaccharide—a divergent role for glycolysis. Redox Biol 2019; 22: 101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Poltorak A, He X, Smirnova I, et al Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998; 282: 2085–2088. [DOI] [PubMed] [Google Scholar]
- 30. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science 2013; 341: 1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kayagaki N, Wong MT, Stowe IB, et al Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013; 341: 1246–1249. [DOI] [PubMed] [Google Scholar]
- 32. Wang D, Wang J, Bonamy GM, et al A small molecule promotes mitochondrial fusion in mammalian cells. Angew Chem Int Ed Engl 2012; 51: 9302–9305. [DOI] [PubMed] [Google Scholar]
- 33. Cassidy‐Stone A, Chipuk JE, Ingerman E, et al Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak‐dependent mitochondrial outer membrane permeabilization. Dev Cell 2008; 14: 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wakabayashi J, Zhang Z, Wakabayashi N, et al The dynamin‐related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol 2009; 186: 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Osellame LD, Singh AP, Stroud DA, et al Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci 2016; 129: 2170–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bordt EA, Clerc P, Roelofs BA, et al The Putative Drp1 Inhibitor mdivi‐1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev Cell 2017; 40: 583–594.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Black RA, Rauch CT, Kozlosky CJ, et al A metalloproteinase disintegrin that releases tumour‐necrosis factor‐α from cells. Nature 1997; 385: 729–733. [DOI] [PubMed] [Google Scholar]
- 38. Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin‐1 family cytokines. J Exp Med 2020; 217: e20190314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baker B, Maitra U, Geng S, Li L. Molecular and cellular mechanisms responsible for cellular stress and low‐grade inflammation induced by a super‐low dose of endotoxin. J Biol Chem 2014; 289: 16262–16269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL‐1 receptor complex. Immunity 1997; 7: 837–847. [DOI] [PubMed] [Google Scholar]
- 41. Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol 2011; 13: 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kashatus JA, Nascimento A, Myers LJ, et al Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK‐driven tumor growth. Mol Cell 2015; 57: 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang L, Ma C, Wang X, et al Lipopolysaccharide‐induced proliferation and glycolysis in airway smooth muscle cells via activation of Drp1. J Cell Physiol 2019; 234: 9255–9263. [DOI] [PubMed] [Google Scholar]
- 44. Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin‐related protein Drp1. Ann N Y Acad Sci 2010; 1201: 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suzuki J, Bayna E, Li HL, Molle ED, Lew WY. Lipopolysaccharide activates calcineurin in ventricular myocytes. J Am Coll Cardiol 2007; 49: 491–499. [DOI] [PubMed] [Google Scholar]
- 46. Wall EA, Zavzavadjian JR, Chang MS, et al Suppression of LPS‐induced TNF‐α production in macrophages by cAMP is mediated by PKA‐AKAP95‐p105. Sci Signal 2009; 2: ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Park J, Choi H, Kim B, et al Peroxiredoxin 5 (Prx5) decreases LPS‐induced microglial activation through regulation of Ca2+/calcineurin‐Drp1‐dependent mitochondrial fission. Free Radic Biol Med 2016; 99: 392–404. [DOI] [PubMed] [Google Scholar]
- 48. Vanhorebeek I, Gunst J, Derde S, et al Mitochondrial fusion, fission, and biogenesis in prolonged critically ill patients. J Clin Endocrinol Metab 2012; 97: E59–E64. [DOI] [PubMed] [Google Scholar]
- 49. Peng K, Yang L, Wang J, et al The Interaction of Mitochondrial Biogenesis and Fission/Fusion Mediated by PGC‐1α Regulates Rotenone‐Induced Dopaminergic Neurotoxicity. Mol Neurobiol 2017; 54: 3783–3797. [DOI] [PubMed] [Google Scholar]
- 50. Piko L, Matsumoto L. Number of mitochondria and some properties of mitochondrial DNA in the mouse egg. Dev Biol 1976; 49: 1–10. [DOI] [PubMed] [Google Scholar]
- 51. Piko L, Taylor KD. Amounts of mitochondrial DNA and abundance of some mitochondrial gene transcripts in early mouse embryos. Dev Biol 1987; 123: 364–374. [DOI] [PubMed] [Google Scholar]
- 52. Li T, Han J, Jia L, Hu X, Chen L, Wang Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell 2019; 10: 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Palsson‐McDermott EM, Curtis AM, Goel G, et al Pyruvate kinase M2 regulates Hif‐1α activity and IL‐1β induction and is a critical determinant of the warburg effect in LPS‐activated macrophages. Cell Metab 2015; 21: 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Das Gupta K, Shakespear MR, Curson JEB, et al Class IIa histone deacetylases drive toll‐like receptor‐inducible glycolysis and macrophage inflammatory responses via pyruvate kinase M2. Cell Rep 2020; 30: 2712–2728.e8. [DOI] [PubMed] [Google Scholar]
- 55. Gao Z, Li Y, Wang F, et al Mitochondrial dynamics controls anti‐tumour innate immunity by regulating CHIP‐IRF1 axis stability. Nat Commun 2017; 8: 1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Haileselassie B, Mukherjee R, Joshi AU, et al Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J Mol Cell Cardiol 2019; 130: 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Joshi AU, Saw NL, Vogel H, Cunnigham AD, Shamloo M, Mochly‐Rosen D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol Med 2018; 10: e8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Haileselassie B, Joshi AU, Minhas PS, Mukherjee R, Andreasson KI, Mochly‐Rosen D. Mitochondrial dysfunction mediated through dynamin‐related protein 1 (Drp1) propagates impairment in blood brain barrier in septic encephalopathy. J Neuroinflammation 2020; 17: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials