

Abstract
The display of complex proteins on the surface of cells is of great importance for protein engineering and other fields of biotechnology. Herein, we describe a modular approach, in which the membrane anchor protein Lpp‐OmpA and a protein of interest (passenger) are expressed independently as genetically fused SpyCatcher and SpyTag units and assembled in situ by post‐translational coupling. Using fluorescent proteins, we first demonstrate that this strategy allows the construct to be installed on the surface of E. coli cells. The scope of our approach was then demonstrated by using three different functional enzymes, the stereoselective ketoreductase Gre2p, the homotetrameric glucose 1‐dehydrogenase GDH, and the bulky heme‐ and diflavin‐containing cytochrome P450 BM3 (BM3). In all cases, the SpyCatcher‐SpyTag method enabled the generation of functional whole‐cell biocatalysts, even for the bulky BM3, which could not be displayed by conventional fusion with Lpp‐OmpA. Furthermore, by using a GDH variant carrying an internal SpyTag, the system could be used to display an enzyme with unmodified N‐ and C‐termini.
Keywords: biocatalysis, Lpp-OmpA, membrane proteins, self-assembly, surface display
Room for independent folding. A surface display method employs the SpyCatcher‐SpyTag system for post‐translational in situ coupling of independently expressed and properly folded passenger enzymes and membrane anchors. The efficient surface display of delicate and complex enzymes expands the toolbox for the fabrication of productive whole‐cell biocatalysts.
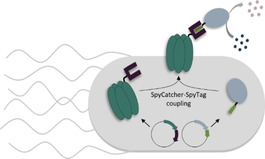
The presentation of peptides and proteins on the surface of bacterial cells is essential for their interaction with the environment.1, 2 In biotechnology, this process has been exploited for applications in protein library screening,3, 4 the production of recombinant bacterial vaccines5 or biosensors,6 and it is also used as a powerful tool for the generation of whole‐cell biocatalysts.6, 7 In the latter case, the employment of microorganisms instead of purified enzymes as biocatalysts can circumvent cost‐intensive enzyme production and purification as well as simplify production processes. Compared to conventional whole‐cell biocatalysis using intracellular enzymes, the presentation of enzymes on the cell surface can largely reduce mass transfer limitations by eliminating the need of substrates or products to cross the cell envelope. This can open up ways for processing of membrane‐impermeable small molecules, which account for a large proportion of pharmaceutically and industrially relevant compounds.
Common systems for the display of proteins on E. coli cells are based on genetic fusions of the protein of interest (passenger) to a membrane anchor that facilitates the transport across the cell envelope and anchors the passenger in the bacterial outer membrane. In the past decades a large number of membrane anchors have been developed.6, 7, 8, 9 Impressive examples of the successful surface display of demanding enzymes include the homotetrameric glucose 1‐dehydrogenase10 as well as the cofactor containing enzymes alditol oxidase11 or the bulky cytochrome P450 BM3 monooxygenase12, 13, 14 using membrane anchors such as INP15 or AIDA‐I.16 However, successful surface display of a given passenger is highly dependent on special features of the membrane anchor such as the potential fusion site with the passenger (N‐terminal, C‐terminal or internal) or the compatibility with the passenger's three‐dimensional structure within the genetic fusion. Incompatible combination of membrane anchors and passenger proteins can lead to unfavourable domain interactions, misfolding of the fusion protein, degradation or even complete loss of functionality, thus strongly effecting surface display efficiency and whole‐cell activities.17
Recently, so‐called post‐translational bioconjugation methods are being investigated for the modular assembly of protein constructs that cannot be generated directly by genetic fusion. The SpyCatcher‐SpyTag bioconjugation system, consisting of the 113 amino acid SpyCatcher (SC) protein and the 13 amino acid SpyTag (ST) peptide,18 is a powerful method that has been employed for this purpose.19, 20 SC and ST form a covalent isopeptide bond between a proximal aspartic acid and lysine residue, thereby enabling the covalent coupling of correctly folded proteins that are independently expressed as genetic fusions with the SC and ST domains. The SC‐ST coupling occurs over a wide range of physiological conditions, does not require reagents and has therefore also been applied for in vivo applications inside bacterial cells.21, 22, 23, 24, 25, 26
To extend the toolbox for surface display of complex, high molecular weight enzymes on E. coli cells, we here report on the investigation of a novel modular surface display method in which the membrane anchor and the passenger are expressed separately and assembled by post‐translational bioconjugation via SC‐ST coupling (Figure 1). As this allows independent folding of the anchor and passenger proteins, we reasoned that our approach might reduce unfavourable domain interactions and misfolding, thereby increasing the display efficiency and the whole‐cell biocatalytic activity. To demonstrate the functionality and utility of our approach we chose the well‐established Lpp‐OmpA hybrid as a model membrane anchor.27 This anchor consists of the transmembrane domain (amino acids 46‐159) from outer membrane protein A (OmpA) as well as the signal peptide and the first 9 N‐terminal amino acids of the E. coli lipoprotein (Lpp). Lpp‐OmpA has already been used for stable membrane anchoring of functional enzymes fused to its C‐terminus28, 29, 30, 31 but, like many others, seems to be limited in terms of passenger size.17
Figure 1.
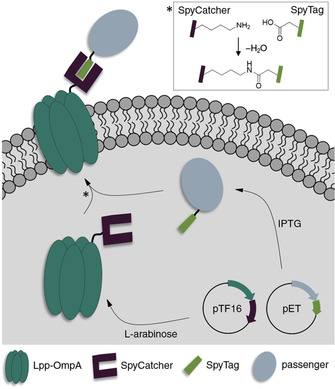
Concept of the E. coli surface display based on post‐translational SC‐ST coupling. The membrane anchor Lpp‐OmpA is genetically fused to a SC domain, while the passenger is fused to the smaller ST. Subsequent to expression from individual plasmids, the fusion proteins covalently couple through a specific SC‐ST interaction. The complex is anchored in the E. coli outer membrane by Lpp‐OmpA.
As illustrated in Figure 1, our SC‐ST display system is comprised of the membrane anchor Lpp‐OmpA genetically fused to a SC domain, while the passenger is fused to the smaller ST to minimize possible interactions during folding of complex large enzyme passengers. Expression of both components takes place separately and covalent interaction occurs by post‐translational SC‐ST isopeptide bond formation. Anchoring of the ligated complex into the E. coli outer membrane is mediated by Lpp‐OmpA to facilitate surface display of the passenger. As previous studies indicated that the expression of Lpp‐OmpA fusions from high copy number plasmids employing a leaky promoter led to slow growth, altered membrane integrity or even cell lysis,29, 32, 33 all Lpp‐OmpA fusions were expressed from a medium copy number pTF16 derived plasmid employing a p15A ori, an ampicillin resistance gene and the tight l‐arabinose‐inducible araB promoter.24, 34 A fully orthogonal pET derived plasmid with pBR322 ori, using an independent replication mechanism and employing a chloramphenicol resistance gene, was chosen as the second plasmid to ensure plasmid stability and to allow for controlled induction of the passenger‐ST via an IPTG inducible T7 promoter. A detailed presentation of the construct design is shown in Figure S1 in the Supporting Information.
To examine the functionality of the modular SC‐ST surface display, we first investigated on the formation and integration of the SC‐ST complex into the cell envelope using fluorescent anchor and passenger modules (Figure 2). To this end, ST was fused to the monomeric red fluorescent protein mRFP and the Lpp‐OmpA‐SC fusion was modified with eGFP. Functional interaction of mRFP‐ST with Lpp‐OmpA‐eGFP‐SC and subsequent translocation to the cell surface was expected to result in a co‐localization of mRFP and eGFP fluorescence signals in the cell envelope. Confocal fluorescence microscopy imaging indeed allowed to detect both fusion proteins in the outer areas of E. coli, thus indicating successful covalent interaction and translocation to the cell envelope (Figure 2A). As expected, controls in which expression of Lpp‐OmpA‐eGFP‐SC was not induced showed no such co‐localization (Figure 2B). Treatment of the cells with proteinase K, which cannot penetrate intact cells, degraded the SC‐ST complex as revealed by western blot analysis (Figure S2), thereby confirming the surface accessibility of the complex. To ensure that the surface presentation is not a result of intercellular cross‐coupling events, in which the passenger leaks into the medium and subsequently couples to surface presented Lpp‐OmpA‐SC fusions of neighbouring cells, additional experiments were performed with co‐cultured cells presenting mRFP or eGFP on their surface (Figure S3). As we could only detect cells with either mRFP or eGFP fluorescence but no cells that show the fluorescent signals of both mRFP and eGFP, we concluded that no cross‐coupling occurs and that the SC‐ST interaction takes place inside the cell before translocation of the complex to the cell surface.
Figure 2.
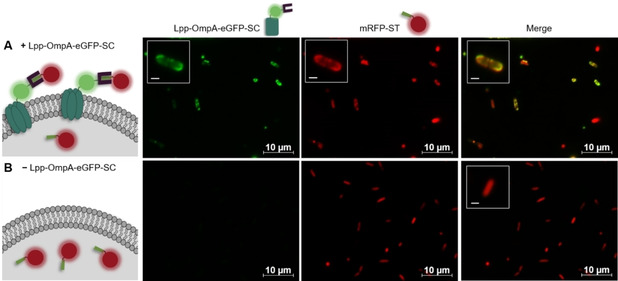
The passenger mRFP carrying a ST interacts with Lpp‐OmpA‐eGFP‐SC and is translocated to the E. coli cell envelope. E. coli cells harboring plasmids pET_mRFP‐ST and pTF16_Lpp‐OmpA‐eGFP‐SC were either A) induced with IPTG and l‐arabinose for translocation of mRFP‐ST to the cell surface after covalent interaction with Lpp‐OmpA‐eGFP‐SC or B) induced with only IPTG for cytosolic expression of mRFP‐ST. Fluorescence microscopy pictures were taken 6 h after induction. Scale bars of magnified insets: 1 μm.
To investigate the applicability of our system for the surface display of active enzymes, we chose the monomeric (S)‐selective methylglyoxal reductase Gre2p (EC 1.1.1.283) from Saccharomyces cerevisiae YJM19335 as well as the more complex homotetrameric glucose 1‐dehydrogenase (GDH) (EC 1.1.1.47) from Bacillus subtilis 36 and a variant of the bulky (119 kDa) heme‐ and diflavin‐containing cytochrome P450 BM3 monooxygenase (BM3) from Bacillus megaterium (CYP102 A1, EC 1.14.14.1) as passengers.37 Surface display of the two latter ones has so far only been reported using more recently established membrane anchors such as INP or AIDA‐I.10, 12, 13, 14 We initially demonstrated that covalent interaction of the three passenger‐ST fusions with Lpp‐OmpA‐SC can be precisely controlled by the induction of the expression of Lpp‐OmpA‐SC as verified in whole‐cell lysates using western blot analysis (Figure S4). We then confirmed the surface exposure of the functional passengers via their activity in whole‐cell biocatalytic assays. To this end, whole cells that were supposed to display the biocatalyst on their surface were incubated with substrates and cofactors, with at least one not being able to penetrate the cell membrane and the enzymatic activity was determined by fluorescence read‐outs (Figure 3, for details on the enzymatic reactions employed see Figure S5). Control cells expressing the passenger‐ST fusions intracellularly only converted the selected substrates upon permeabilization with toluene (Figure S6A), thereby confirming that the substrates were indeed not able to penetrate into intact cells. Compared to these control cells, the conversion was significantly increased, when the passenger‐ST fusions were targeted to the cell surface via the expression of Lpp‐OmpA‐SC, as indicated in Figure 3B. These results gave a clear indication that the SC‐ST surface display system can be used to effectively anchor the passenger on the cells, thereby reducing mass transfer limitations between the enzymes and the surrounding medium. The permeabilization of the cells further increased the conversion rates (Figure S6B), presumably because excess passenger‐ST fusions that were not coupled to Lpp‐OmpA‐SC (Figure S4) remain in the cytosol of the cells and can contribute to the activity of the biocatalyst after permeabilization.
Figure 3.
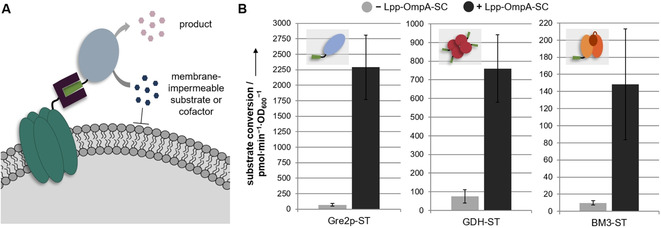
Display of ST‐modified passenger enzymes by post‐translational conjugation to Lpp‐OmpA‐SC leads to a strong increase in the conversion rates for membrane impermeable substrates or cofactors. A) Schematic representation of whole‐cell biocatalytic activity assays; details are given in Figure S5. B) Cells in which the passenger enzymes Gre2p‐ST, GDH‐ST or BM3‐ST are targeted to the cell surface by the expression of Lpp‐OmpA‐SC (+Lpp‐OmpA‐SC, black bars) show significantly increased whole‐cell biocatalytic activities, as compared to cells expressing the passenger‐ST fusions intracellularly (−Lpp‐OmpA‐SC, grey bars). Error bars were obtained from at least two independent experiments.
It was observed in previous studies that high‐level expression of Lpp‐OmpA fusions can cause substantial alterations in the E. coli outer membrane permeability.32, 33 Due to this phenomenon, and because we observed irregular cell shapes upon longer induction times (Figure S7), we suspected that the above described increase in whole‐cell biocatalytic activity might not be completely conclusive to prove the passenger localization on the cell surface. We therefore carried out additional experiments to investigate the membrane integrity. To this end, we determined the whole‐cell biocatalytic activity of a control sample overexpressing non‐coupling Lpp‐OmpA‐ST in the outer membrane and BM3‐ST in the cytosol (non‐coupling control, see Figure S8). We found modest conversion of the substrate, suggesting that the overexpression of Lpp‐OmpA fusions from the pTF16 derived vector leads to increased permeability of the E. coli outer membrane, thereby allowing the diffusion of substrates and cofactors into the cell and their subsequent conversion in the cytosol. These alterations in membrane integrity are presumably caused by the all‐or‐none response of the l‐arabinose inducible araB promoter used to control the expression of Lpp‐OmpA fusions in this study. According to Siegele et al.38 the l‐arabinose concentration in the medium influences the number of fully induced cells rather than adjusting the expression level in individual cells. This leads to a high‐level expression of Lpp‐OmpA fusions in the induced cells, which might cause an altered membrane permeability, as reported previously.32, 33 However, since permeabilization of the cells by addition of toluene led to a strong further increase in the conversion rate (Figure S8), the membrane barrier does not seem to be completely disrupted. Owing to the observed changes in membrane permeability, the whole‐cell biocatalytic activity assays could not be used as a firm proof for the surface localization of passenger enzymes. To further investigate the passenger localization at the cellular level, we therefore carried out immunofluorescence microscopy studies. To this end, cells displaying BM3‐ST modified with a C‐terminal Myc epitope tag were incubated with a fluorescently labelled anti‐Myc antibody (Figure 4). We found that cells displaying BM3 with the SC‐ST display system clearly turned fluorescent (Figure 4B). Due to the above discussed observation that the membrane integrity was compromised by the overexpression of the membrane anchor, we confirmed that the antibody could not penetrate into the cytosol of the cells by employment of a non‐coupling control with membrane integrated Lpp‐OmpA‐ST and cytosolic BM3‐ST‐Myc. As expected, almost no fluorescence was detected for these control cells (Figure 4B). Altogether, the results gave clear evidence for the surface presentation of BM3 using the SC‐ST display system. Since immunofluorescence microscopy proved to be a reliable method for the detection of surface presentation, similar experiments carried out with the Gre2p‐ST and GDH‐ST passenger enzymes demonstrated the successful surface presentation of these enzymes (Figure S9).
Figure 4.
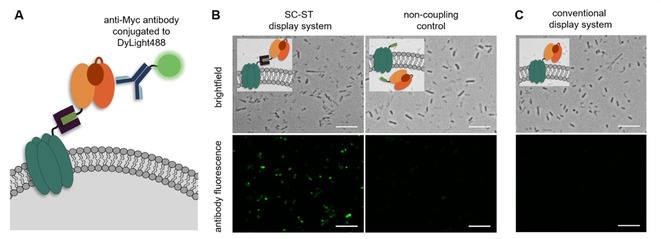
Verification of BM3 surface exposure by means of immunofluorescence microscopy. A) BM3 fusions modified with a C‐terminal Myc epitope tag were detected by using an anti‐Myc antibody conjugated to DyLight488. B) Cells presenting BM3‐ST with the SC‐ST display system were successfully stained with the antibody, whereas cells employing the corresponding non‐coupling control with membrane integrated Lpp‐OmpA‐ST and cytosolic BM3‐ST showed almost no fluorescence. C) Cells employing the conventional display system did not exhibit any antibody fluorescence. Scale bars: 10 μm.
Next, we investigated whether the SC‐ST display system offers the expected advantages over conventional display systems in terms of increased display efficiency. To this end, we constructed the direct genetic fusion Lpp‐OmpA‐BM3‐Myc. To ensure comparability regarding the expression levels of the membrane anchor, this fusion protein was also expressed from the pTF16 vector (Figure S1C). As demonstrated by immunofluorescence microscopy using the anti‐Myc antibody (Figure 4C), no fluorescent cells were detected. This observation was confirmed by flow cytometry analysis (Figure S10A), revealing only a very weak fluorescence for 2.4 % of the E. coli population expressing the Lpp‐OmpA‐BM3‐Myc fusion. In contrast the cells displaying the passenger with the SC‐ST system showed a much higher fluorescence, which was also present in a much larger proportion (12.4 %) of the population. A reduced viability was determined for cells that successfully displayed the passenger, presumably resulting from the observed alterations in membrane integrity (Figure S10B). Nevertheless, the results clearly indicate that BM3 indeed could only be displayed using Lpp‐OmpA when employing the SC‐ST display system. Based on SDS‐PAGE and western blot analyses (Figure S11A), as well as whole‐cell biocatalytic activity measurements (Figure S11B), wherein no substrate conversion was detected for the conventional display system even when the cells were permeabilized by addition of toluene, it is evident that these observations were due to absent expression of the Lpp‐OmpA‐BM3 fusion. In fact, these results are in line with previously published work suggesting that the applicability of the membrane anchor Lpp‐OmpA is limited in terms of passenger size.17 Hence, the results clearly confirm the advantages of the post‐translational assembly of separately folded membrane anchors and passengers. We hypothesize that this improvement is due to avoidance of possible unfavourable domain interactions that might occur when the bulky passenger is directly fused to Lpp‐OmpA. In contrast SC and ST are smaller and stable modifications, which allow for both the membrane anchor and the passenger to be folded in the most native way possible, thereby reducing misfolding and degradation.
As previous studies demonstrated that ST remains functional when fused to the N‐terminus, C‐terminus or even into internal loops of a protein of interest,18, 39 we reasoned that our system might also be able to display passenger enzymes carrying an internal ST. We therefore constructed a variant of the passenger enzyme GDH carrying the ST sequence inserted into an internal flexible loop (GDH(ST), Figure S12). As verified by immunofluorescence microscopy (Figure 5A), the GDH(ST) variant was successfully displayed on E. coli. Furthermore, the surface displayed GDH(ST) variant shows similar levels of substrate conversion (Figure 5B) as compared to the GDH‐ST variant carrying a C‐terminal ST (compare Figure 3B). These results clearly illustrate that the herein developed display system can also be used with an internal ST, thereby enabling the display of passenger enzymes with native N‐ and C‐termini. To the best of our knowledge, this is not possible with any other surface display system reported so far.
Figure 5.
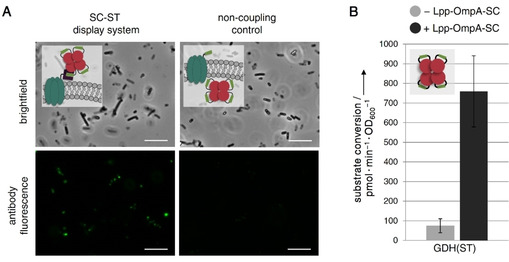
Surface display of a GDH variant carrying an internal ST. A) Successful surface display of GDH(ST) employing a His6 tag was verified by immunofluorescence microscopy using an anti‐His6 antibody conjugated to AlexaFluor488. Cells presenting the passenger on their surface employing the SC‐ST display system clearly turned fluorescent, whereas the corresponding non‐coupling control showed almost no fluorescence. Scale bars:10 μm. B) Surface display of GDH(ST) after expression of Lpp‐OmpA‐SC (+Lpp‐OmpA‐SC, black bars) significantly increased the whole‐cell biocatalytic activity, as compared to cells expressing GDH(ST) intracellularly (−Lpp‐OmpA‐SC, grey bars). Note that the GDH(ST) displaying cells showed similar levels of substrate conversion, as compared to cells displaying the GDH‐ST variant (C‐terminal tag, compare Figure 3B). The whole‐cell biocatalytic activity measurement was carried out as in Figure 3B with details given in Figure S5. Error bars were obtained from at least two independent experiments.
In conclusion we have reported on a novel approach that enables surface display of functional, complex enzymes on E. coli. As demonstrated by whole‐cell biocatalytic activity assays and immunofluorescence microscopy, the herein described post‐translational SC‐ST mediated coupling of passenger enzymes and the model membrane anchor Lpp‐OmpA allowed successful surface display of the enzymes Gre2p, the homotetrameric GDH and the highly demanding heme‐ and diflavin‐containing BM3. The resulting whole‐cell biocatalysts revealed high conversion rates for membrane impermeable substrates, thereby suggesting substantially reduced mass transfer limitations as compared to cytosolic expression. The observed alterations in outer membrane permeability and cell viability, induced by the l‐arabinose controlled overexpression of Lpp‐OmpA, might be avoided by optimizing the amount of Lpp‐OmpA in the cell membrane, for instance, by using an expression system that offers a more gradual modulation of induction. Since Lpp‐OmpA‐SC is presumably translocated to the periplasm via the Sec machinery due to the Lpp peptide,40, 41, 42, 43 but can obviously interact with the passenger‐ST fusions in the cytosol, we hypothesize that during lipid modification of the N‐terminal Lpp domain at the inner membrane41, 42, 43 the hydrophobic OmpA domain might get incorporated into the lipid bilayer with the C‐terminal SC domain being exposed towards the cytoplasm, allowing the covalent binding of the passenger‐ST fusions with subsequent translocation to the outer membrane, as reported for the native Lpp lipoprotein.41, 42, 43 Due to the fast and covalent binding of the SC‐ST system such processes could be studied in more detail especially when combined with super‐resolution microscopy. Other than the conventional direct genetic fusion to Lpp‐OmpA, which results in no detectable expression of the passenger, the successful BM3 display by aid of our SC‐ST system clearly indicates the advantages of this approach. We assume that the improved performance is due to reduced misfolding and degradation. Furthermore, the flexibility in design of the passenger‐ST fusion (N‐terminal, C‐terminal or even internal ST) allows the display system to be easily adjusted to the individual needs of the passenger. This could be particularly useful for enzymes that do not tolerate genetic fusions to specific termini. Since the system uses completely orthogonal expression plasmids for membrane anchor‐SC and passenger‐ST fusions, it also enables a combinatorial screening, without the need to generate each individual genetic fusion. Based on these arguments, we believe that our approach will expand the toolbox for efficient presentation of delicate enzymes on bacterial cell surfaces, thereby further contributing to the development of productive whole‐cell biocatalysts.
Experimental Section
Experimental Details about plasmid construction, amino acid sequences and experimental procedures are given in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Helmholtz program “Bio‐Interfaces in Technology and Medicine”. S.G. is grateful for a Kekulé fellowship by Fonds der Chemischen Industrie (FCI). We thank Alex Fenchel and Cornelia Ziegler for experimental help, Ishtiaq Ahmed for synthesis of substrates and Pia Schwitters for cloning of GDH(ST).
S. Gallus, T. Peschke, M. Paulsen, T. Burgahn, C. M. Niemeyer, K. S. Rabe, ChemBioChem 2020, 21, 2126.
References
- 1. Scott J. R., Barnett T. C., Annu. Rev. Microbiol. 2006, 60, 397–423. [DOI] [PubMed] [Google Scholar]
- 2. Bos M. P., Robert V., Tommassen J., Annu. Rev. Microbiol. 2007, 61, 191–214. [DOI] [PubMed] [Google Scholar]
- 3. van Bloois E., Winter R. T., Kolmar H., Fraaije M. W., Trends Biotechnol. 2011, 29, 79–86. [DOI] [PubMed] [Google Scholar]
- 4. Becker S., Schmoldt H.-U., Adams T. M., Wilhelm S., Kolmar H., Curr. Opin. Biotechnol. 2004, 15, 323–329. [DOI] [PubMed] [Google Scholar]
- 5. Georgiou G., Stathopoulos C., Daugherty P. S., Nayak A. R., Iverson B. L., Curtiss R., Nat. Biotechnol. 1997, 15, 29–34. [DOI] [PubMed] [Google Scholar]
- 6. Han L., Zhao Y., Cui S., Liang B., Appl. Biochem. Biotechnol. 2018, 185, 396–418. [DOI] [PubMed] [Google Scholar]
- 7. Schüürmann J., Quehl P., Festel G., Jose J., Appl. Microbiol. Biotechnol. 2014, 98, 8031–8046. [DOI] [PubMed] [Google Scholar]
- 8. Samuelson P., Gunneriusson E., Nygren P.-Å., Ståhl S., J. Biotechnol. 2002, 96, 129–154. [DOI] [PubMed] [Google Scholar]
- 9. Lee S. Y., Choi J. H., Xu Z., Trends Biotechnol. 2003, 21, 45–52. [DOI] [PubMed] [Google Scholar]
- 10. Liang B., Li L., Tang X., Lang Q., Wang H., Li F., Shi J., Shen W., Palchetti I., Mascini M., Liu A., Biosens. Bioelectron. 2013, 45, 19–24. [DOI] [PubMed] [Google Scholar]
- 11. van Bloois E., Winter R. T., Janssen D. B., Fraaije M. W., Appl. Microbiol. Biotechnol. 2009, 83, 679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yim S.-K., Kim D. H., Jung H. C., Pan J. G., Kang H. S., Ahn T., Yun C. H., J. Microbiol. Biotechnol. 2010, 20, 712–717. [DOI] [PubMed] [Google Scholar]
- 13. Park T. J., Heo N. S., Yim S. S., Park J. H., Jeong K. J., Lee S. Y., Microb. Cell Fact. 2013, 12, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Strohle F. W., Kranen E., Schrader J., Maas R., Holtmann D., Biotechnol. Bioeng. 2016, 113, 1225–1233. [DOI] [PubMed] [Google Scholar]
- 15. Jung H. C., Lebeault J. M., Pan J. G., Nat. Biotechnol. 1998, 16, 576–580. [DOI] [PubMed] [Google Scholar]
- 16. Maurer J., Jose J., Meyer T. F., J. Bacteriol. 1997, 179, 794–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wan H.-M., Chang B.-Y., Lin S.-C., Biotechnol. Bioeng. 2002, 79, 457–464. [DOI] [PubMed] [Google Scholar]
- 18. Zakeri B., Fierer J. O., Celik E., Chittock E. C., Schwarz-Linek U., Moy V. T., Howarth M., Proc. Natl. Acad. Sci. USA 2012, 109, E690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sutherland A. R., Alam M. K., Geyer C. R., ChemBioChem 2019, 20, 319–328. [DOI] [PubMed] [Google Scholar]
- 20. Reddington S. C., Howarth M., Curr. Opin. Chem. Biol. 2015, 29, 94–99. [DOI] [PubMed] [Google Scholar]
- 21. Nguyen P. Q., Botyanszki Z., Tay P. K. R., Joshi N. S., Nat. Commun. 2014, 5, 4945. [DOI] [PubMed] [Google Scholar]
- 22. Alves N. J., Turner K. B., Daniele M. A., Oh E., Medintz I. L., Walper S. A., ACS Appl. Mater. Interfaces 2015, 7, 24963–24972. [DOI] [PubMed] [Google Scholar]
- 23. Keeble A. H., Banerjee A., Ferla M. P., Reddington S. C., Anuar I. N. A. K., Howarth M., Angew. Chem. Int. Ed. Engl. 2017, 56, 16521–16525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peschke T., Rabe K. S., Niemeyer C. M., Angew. Chem. Int. Ed. Engl. 2017, 56, 2183–2186. [DOI] [PubMed] [Google Scholar]
- 25. Plavec T. V., Berlec A., Acta Chim. Slov. 2019, 66, 18–27. [PubMed] [Google Scholar]
- 26. Hatlem D., Trunk T., Linke D., Leo J. C., Int. J. Mol. Sci. 2019, 20, 2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Francisco J. A., Earhart C. F., Georgiou G., Proc. Natl. Acad. Sci. USA 1992, 89, 2713–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Francisco J. A., Stathopoulos C., Warren R. A. J., Kilburn D. G., Georgiou G., Nat. Biotechnol. 1993, 11, 491–495. [DOI] [PubMed] [Google Scholar]
- 29. Richins R. D., Kaneva I., Mulchandani A., Chen W., Nat. Biotechnol. 1997, 15, 984–987. [DOI] [PubMed] [Google Scholar]
- 30. Yang C., Zhao Q., Liu Z., Li Q., Qiao C., Mulchandani A., Chen W., Environ. Sci. Technol. 2008, 42, 6105–6110. [DOI] [PubMed] [Google Scholar]
- 31. Jo J.-H., Han C.-W., Kim S.-H., Kwon H.-J., Lee H.-H., J. Microbiol. 2014, 52, 856–862. [DOI] [PubMed] [Google Scholar]
- 32. Georgiou G., Stephens D. L., Stathopoulos C., Poetschke H. L., Mendenhall J., Earhart C. F., Protein Eng. Des. Sel. 1996, 9, 239–247. [DOI] [PubMed] [Google Scholar]
- 33. Stathopoulos C., Georgiou G., Earhart C. F., Appl. Microbiol. Biotechnol. 1996, 45, 112–119. [DOI] [PubMed] [Google Scholar]
- 34. Binder D., Probst C., Grünberger A., Hilgers F., Loeschcke A., Jaeger K.-E., Kohlheyer D., Drepper T., PLoS One 2016, 11, e0160711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Müller M., Katzberg M., Bertau M., Hummel W., Org. Biomol. Chem. 2010, 8, 1540–1550. [DOI] [PubMed] [Google Scholar]
- 36. Hilt W., Pfleiderer G., Fortnagel P., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1991, 1076, 298–304. [DOI] [PubMed] [Google Scholar]
- 37. Neufeld K., z Berstenhorst S. M., Pietruszka J., Analyt. Biochem. 2014, 456, 70–81. [DOI] [PubMed] [Google Scholar]
- 38. Siegele D. A., Hu J. C., Proc. Natl. Acad. Sci. USA 1997, 94, 8168–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bitterwolf P., Gallus S., Peschke T., Mittmann E., Oelschlaeger C., Willenbacher N., Rabe K. S., Niemeyer C. M., Chem. Sci. 2019, 118, 801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goemans C., Denoncin K., Collet J.-F., Biochim. Biophys. Acta. 2014, 1843, 1517–1528. [DOI] [PubMed] [Google Scholar]
- 41. Green E. R., Mecsas J., Microbiol. Spectr. 2016, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zückert W. R., Biochim. Biophys. Acta. 2014, 1843, 1509–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Konovalova A., Silhavy T. J., Philos. Trans. R. Soc. London Ser. A 2015, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary