

Abstract
Three molybdenum(VI) dioxido complexes [MoO2(L)2] bearing Schiff base ligands were reacted with B(C6F5)3 to afford the corresponding adducts [MoO{OB(C6F5)3}(L)2], which were fully characterized. They exhibit Frustrated Lewis‐Pairs reactivity when reacting with silanes. Especially, the [MoO{OB(C6F5)3}(L)2] complex with L=2,4‐dimethyl‐6‐((phenylimino)methyl)phenol proved to be active as catalyst for the hydroalkylation of aryl alkenes with organohalides and for the Atom‐Transfer Radical Addition (ATRA) of organohalides to aliphatic alkenes. A series of gem‐dichloride and gem‐dibromide compounds with potential for further derivatization were synthesized from simple alkenes and organohalides, like chloroform or bromoform, using low catalyst loading.

Keywords: C−C coupling, Frustrated Lewis pairs, Hydroalkylation, Molybdenum-oxido complexes, Silanes
Introduction
Since the discovery of their exceptional reactivity by Stephan and Erker,1, 2 Frustrated Lewis Pairs (FLP) became a tool of choice as catalysts for many chemical transformations, especially in the field of Small Molecules Activation.2, 3 FLP are Lewis acid‐base pairs that form no or a poor bond between them, for example by deliberately introducing steric hindrance. Among the numerous examples of activation of unreactive molecules by FLP developed over the past ten years, splitting of H2 for metal‐free hydrogenation of imines,4 carbonyls,5 alkenes6 or alkynes,7 as well as functionalization of CO2 8 are the most prominent examples. In FLP, the role of the Lewis acid is often played by tris(pentafluorophenyl)borane B(C6F5)3, due to its high electrophilicity and steric demand. Besides, B(C6F5)3 is itself a good catalyst for several reactions, especially hydrosilylation reactions.9, 10 Recently, the concept of FLP was extended to transition metal compounds, where they act as the Lewis acid or base, allowing for further reactivity not observed with main group frustrated Lewis pairs.11 In particular, transition metals complexes bearing an oxido ligand could be used as the Lewis base in combination with a Lewis acid such as B(C6F5)3, leading to new reactivity for the complexes, as reported by the group of Schrock12 and the group of Ison (Scheme 1).13, 14
Scheme 1.

Top: Lewis adduct formation and frustrated Lewis pair; bottom: Lewis adduct formation between M=O and B(C6F5)3.
Inspired by these reports, our group became interested in investigating the influence of bulky Lewis acids on oxygen activation and oxidative properties of our previously published molybdenum dioxido and oxido‐imido complexes.15, 16, 17, 18 We recently reported the synthesis of a molybdenum oxido‐imido complex that reacted with B(C6F5)3 under formation of an adduct having FLP‐like properties.19 With this molybdenum‐oxido based Lewis pair, heterolytic cleavage of silicon‐hydrogen bonds was demonstrated, leading to cationic Mo(VI) species of the formula [Mo(OSiR3)(NtBu)L2] [HB(C6F5)3] that were isolated and fully characterized. Ultimately, it was shown that such FLP‐like adducts could catalyze the hydrosilylation of benzaldehyde via insertion of the substrate into the boron‐hydrogen bond of the borate.
In this paper, we further investigate the reactivity of these FLP‐like adducts of B(C6F5)3 with molybdenum oxido complexes towards alkenes. We found that an unexpected addition of halides to the alkenes takes place in presence of catalytic amounts of the metal FLP and silanes. The addition of organohalides to alkenes is known as Atom‐Transfer Radical Addition (ATRA) or Kharasch Addition, named after Morris Kharasch, who studied the reaction of HBr, CHCl3 and CCl4 with unsymmetrical alkenes in presence of peroxides.20 The reaction starts with formation of a radical species from the organohalide, generally CX4 or CHX3, in presence of an initiator or a transition‐metal catalyst (Scheme 2). The generated .CX3 or .CHX2 radical can then react with the alkene, leading to an anti‐Markovnikov addition intermediate, which can subsequently combine with the other radical .X from the organohalide to afford the final product. Alternatively, the radical intermediate can react with one or several equivalents of the initial alkene to afford a polymer. In this case, the reaction is called Atom‐Transfer Radical Polymerization (ATRP). In a third outcome, the addition intermediate, when sufficiently reactive, can react with a hydrogen donor to lead to the corresponding hydroalkylation product. Such hydroalkylation may also be referred to as a Giese addition.21
Scheme 2.

ATRA (Kharasch addition), ATRP and hydroalkylation of alkenes with organohalides.
ATRA reactions are nowadays broadly used for the synthesis of halogenated functionalized molecules.22 Ruthenium23 or copper24 catalysts bearing ligands that could photo‐ or thermo‐initiate the formation of the radical are usually needed to perform ATRA reactions, but examples using molybdenum complexes were also reported.25, 26 The molybdenum dioxido‐B(C6F5)3 compounds presented in this paper can not only catalyze the ATRA reaction when aliphatic alkenes are used as substrates, but more importantly the hydroalkylation of aryl alkenes since these ones form more reactive intermediates. Hence, simple organohalides like chloroform or bromoform reacted with a variety of alkenes that were reduced forming new gem‐dichloride and gem‐dibromide compounds that are of interest for further reactivity and functionalization.
Results and Discussion
Synthesis of Dioxido Complexes. Three molybdenum dioxido complexes bearing iminophenolate ligands with different steric and electronic properties were synthesized in order to study their reaction with B(C6F5)3 (Scheme 3). Complex [MoO2(L1)2] (1) was synthesized following a previously published procedure16 and complexes [MoO2(L2)2] (2) and [MoO2(L3)2] (3) were synthesized using similar protocols: two equiv of ligands HL2 27 or HL3 28 were reacted with one equiv of [MoO2Cl2] in presence of excess NEt3 in an appropriate solvent using Schlenk techniques. After filtration and purification, the three molybdenum dioxido complexes were isolated as orange or yellow solids in good yields (65%–78%, Scheme 3). As reported for 1 in our previous publication,16 complex 2 exhibits a dynamic isomerism in solution, reflected by two distinct sets of resonances with different intensity in the 1H and 19F NMR spectra. Complex 3 is very poorly soluble in organic solvents and reliable NMR spectroscopy data could be only obtained using DMSO. For this complex, only one set of signals is clearly visible, while formation of a second isomer might be the cause of the other broad signals (see SI). As confirmed by X‐ray crystallography, one of the two isomers for each complex has nitrogen atoms of both ligands trans to the oxido group (N,N isomer), whereas the N,O isomer forms as well but did not crystallize in our attempts. The three complexes are slightly sensitive towards moisture and only partially soluble in acetonitrile.
Scheme 3.

Synthesis of molybdenum dioxido complexes 1–3 and molybdenum oxido Lewis adducts 4–6.
Synthesis of Molybdenum Oxido Lewis Adducts. In a similar procedure to a previous publication,19 addition of one equiv of the Lewis acid B(C6F5)3 to the yellow suspension of the molybdenum(VI) dioxido precursors 1–3 in pentane led to an immediate color change to deep red and subsequent formation of the Lewis adducts [MoO{OB(C6F5)3}(L)2] (L1–L3, 4–6) as deep red crystalline precipitates. Compounds 4–6 were isolated as red to dark red solids in very good yields after purification (Scheme 3).
Compounds 4–6 are highly sensitive to moisture and soluble in most polar organic solvents, but only sparingly soluble in benzene and toluene. Like previous observations for the related oxido imido borane adduct,19 NMR spectroscopy reveals that compounds 4–6 exist as single isomers in solution, which is in contrast to the isomeric equilibrium observed for the dioxido molybdenum complexes 1–3. The 1H NMR spectra feature two distinct signal sets for the ligands, indicating coordination at only one Mo=O moiety. The coordination of the Lewis acid is confirmed by a new set of signals corresponding to the meta, ortho and para fluorines of B(C6F5)3, observable in 19F NMR spectroscopy. Especially, the pronounced shift of the para‐F resonance (−158.8 ppm, C6D6) compared to free borane (−142.3 ppm, C6D6) is characteristic of such coordination.14, 19 The para‐F shift is less pronounced in both adducts 5 (−148.2 ppm, C6D6) and 6 (−157.6 ppm, C6D6). Due to the quadrupolar nature of 11B nucleus, 11B NMR spectroscopy gave no meaningful data for the complexes 4–6, but IR spectroscopy was used to confirm Mo=O−B coordination, with a characteristic strong signal at around 980 cm−1 for each complex.19
Molecular Structures. The molecular structures of complexes 1–3 and 4–6 were determined by single crystal X‐ray diffraction analyses. The molecular views of complexes 1–3 are shown in Figure 1 and of 4–6 in Figure 2. Selected bond lengths and angles are shown in Table 1 and full crystallographic details are provided in the supporting information. The dioxido complexes 1–3 show similar structures, as already reported for 1 16 and for other published molybdenum dioxido complexes.15, 18 As reported for similar oxido‐imido complexes forming Lewis adducts,19 the Mo=O bond that interacts with B(C6F5)3 in 4–6 becomes elongated in comparison to the parent dioxido complexes 1–3. Thus, the Mo−N bond trans to the oxido‐borane moiety is shortened for all three compounds when compared with Mo−N bond from dioxido complexes. Overall, the structures of compounds 4–6 are similar in terms of bond lengths, with the Mo1−O2 bond ranging from 1.783(2) Å in 5 to 1.7900(10) Å in 4 and the O2−B1 bond ranging from 1.530(2) Å in 4 to 1.5371(15) Å in 6. The major difference between complex 4 and 5–6 is the angle B1−O2−Mo1 being larger in the case of 4 [159.08(9)°] compared to 155.8(2)° for 5 and 153.13(8)° for 6, due to the presence of tert‐butyl and phenyl groups at the ligand.
Figure 1.

Molecular views (50% probability level) of 1–3 (from left to right); hydrogen atoms as well as solvent molecules are omitted for clarity. For disordered fragments, only atoms with the higher site occupation factors are depicted.
Figure 2.

Molecular views (50% probability level) of 4–6 (from top to bottom); hydrogen atoms as well as solvent molecules are omitted for clarity. For disordered fragments, only atoms with the higher site occupation factors are depicted.
Table 1.
Selected bond lengths [Å] and angles [°] for complexes 1–3 and 4–6.
|
|
1 |
2 |
3 |
|---|---|---|---|
|
Mo1−O1 |
1.6983(15) |
1.6989(17) |
1.705(4) |
|
Mo1−O2 |
1.6984(13) |
1.7048(17) |
1.712(3) |
|
Mo1−O11 |
1.9608(13) |
1.9404(16) |
1.950(3) |
|
Mo1−O21 |
1.9409(13) |
1.9412(16) |
1.946(4) |
|
Mo1−N17 |
2.3896(16) |
2.395(2) |
2.407(4) |
|
Mo1−N27 |
2.3898(15) |
2.358(2) |
2.382(4) |
|
O1−Mo1−O2 |
105.22(7) |
105.97(9) |
104.04(18) |
|
O11−Mo1−O21 |
152.82(5) |
154.66(7) |
154.52(14) |
|
O1−Mo1−N17 |
169.72(6) |
167.87(8) |
167.46(16) |
|
O2−Mo1−N27 |
170.77(6) |
167.74(8) |
165.53(15) |
|
N17−Mo1−N27 |
86.27(5) |
81.84(7) |
77.19(14) |
|
|
4 |
5 |
6 |
|---|---|---|---|
|
Mo1−O1 |
1.6909(11) |
1.680(2) |
1.6892(9) |
|
Mo1−O2 |
1.7900(10) |
1.783(2) |
1.7839(8) |
|
Mo1−O11 |
1.9008(10) |
1.8937(19) |
1.9121(9) |
|
Mo1−O21 |
1.9229(10) |
1.922(2) |
1.9072(9) |
|
Mo1−N17 |
2.3569(12) |
2.381(3) |
2.3950(10) |
|
Mo1−N27 |
2.2967(13) |
2.312(3) |
2.3084(10) |
|
O2−B1 |
1.530(2) |
1.535(4) |
1.5371(15) |
|
O1−Mo1−O2 |
104.47(5) |
103.85(10) |
104.37(4) |
|
O11−Mo1−O21 |
160.52(5) |
157.21(9) |
157.99(4) |
|
O1−Mo1−N17 |
163.63(5) |
165.94(10) |
159.94(4) |
|
O2−Mo1−N27 |
169.59(4) |
171.60(10) |
169.94(4) |
|
N17−Mo1−N27 |
79.02(4) |
82.21(9) |
74.80(3) |
|
B1−O2−Mo1 |
159.08(9) |
155.8(2) |
153.13(8) |
Reactivity in ATRA and Hydroalkylation of alkenes. During initial attempts to use complex 4 as catalyst in olefin hydrosilylation with phenylsilane, we noticed unexpected reactivity upon use of CHCl3 as reaction solvent. Thus, using styrene as substrate, we did not observe the hydrosilylated product, but it was rather converted to 3,3‐dichloropropyl benzene (7 a) and to 1,3,3‐trichloropropylbenzene (7 a’) (Scheme 4). This unexpected observation prompted us to investigate the generalizability and scope of this reaction in terms of catalyst, olefinic substrate, organohalide and silane. In this reaction, 1,3,3‐trichloropropylbenzene (7 a’) is formed by addition of chloroform to styrene, representing an ATRA reaction, which does typically require no silane nor borane. Such catalytic transformation was already reported to be catalyzed by molybdenum(VI) complexes in presence of triethylamine using CCl4 as organohalide.26 We envisioned that formation of 3,3‐dichloropropyl benzene (7 a) most likely occurs due to the presence of silane (vide infra).
Scheme 4.

Observed catalytic transformation of styrene into 3,3‐dichloropropyl benzene (7 a) and 1,3,3‐trichloropropylbenzene (7 a′).
With this knowledge at hand, we performed a series of experiments in order to evaluate which of the components of the reaction shown in Scheme 4 are needed for the catalytic conversion. The results are summarized in Table 2. Styrene was fully converted to 7 a in 91% yield and 7 a’ in 9% yield using 1 mol% catalyst 4 with 2 equiv PhSiH3 after 2 h at 50 °C (entry 1). Using 1 equiv PhSiH3 also afforded 7 a with almost complete conversion of the substrate, albeit slower (entry 2). Blank and control experiments revealed no conversion of styrene in absence of complex 4 (entry 3), in absence of silane or in absence of borane (using the dioxido complex 1, entry 4). The latter was corroborated using commercially available [MoO2(acac)2], with which also no reactivity was observed (entry 5). Furthermore, while the use of B(C6F5)3 did lead to a partial consumption of styrene, no formation of the chlorinated products was observed. Instead, using B(C6F5)3 led to a mixture of unidentified products accompanied by small quantities of the hydrosilylation product (entry 6). It is also interesting to note that the use of the two other borane adducts 5 or 6, or the presence of [MoO2(acac)2] and B(C6F5)3 together in the reaction mixture, although showing slow conversion of styrene, lead to no formation or in a small amount of chlorinated products (entry 7 to 9).
Table 2.
Control experiments for the addition of CHCl3 to styrene in presence of silane.
|
| ||||||
|---|---|---|---|---|---|---|
|
Entry |
Catalyst (1 mol%) |
PhSiH3 (equiv) |
Reaction time |
Conversion (%) |
Selectivity for 7 a (%) |
Selectivity for 7 a’ (%) |
|
1 |
4 |
2 |
2 h |
>98 |
91 |
9 |
|
2 |
4 |
1 |
2 h/5 h |
95/>98 |
77/80 |
23/16 |
|
3 |
none |
2 |
20 h |
0 |
0 |
0 |
|
4 |
1 |
2 |
20 h |
0 |
0 |
0 |
|
5 |
[MoO2(acac)2] |
2 |
20 h |
0 |
0 |
0 |
|
6 |
B(C6F5)3 |
2 |
20 h |
5 |
0 |
0 |
|
7 |
5 |
2 |
2 h/20 h |
56/95 |
0/7 |
0/2 |
|
8 |
6 |
2 |
2 h/20 h |
11/61 |
0/0 |
0/0 |
|
9 |
[MoO2(acac)2]+B(C6F5)3 |
2 |
20 h |
83 |
0 |
0 |

General conditions: 1 or 2 equiv PhSiH3, 50 °C, 0.5 mL CHCl3 as solvent. Conversion of styrene as determined by GC‐MS and selectivity as determined by 1H NMR spectroscopy.
Thus, exclusively the combination of the oxido‐adduct 4 with silane as shown in Scheme 4 leads to catalytic hydroalkylation of styrene.
With catalyst 4, reaction optimization in toluene with varying amounts of CHCl3 was performed revealing optimal conversion and selectivity for 7 a with 5 equiv of chloroform (Table 3, entries 2–4). Solvent screening with 5 equiv of CHCl3 showed 93% conversion of styrene in chlorobenzene were after 2 h (entry 5), while no consumption of styrene took place in acetonitrile as it coordinates to B(C6F5)3 (entry 6).
Table 3.
Solvent screening for the addition of CHCl3 to styrene in presence of silane.
|
| |||||
|---|---|---|---|---|---|
|
Entry |
Solvent |
Equiv CHCl3 |
Conv. (%) |
Select. for 7 a (%) |
Select.for 7 a’ (%) |
|
1 |
CHCl3 |
– |
>98 |
91 |
9 |
|
2 |
Tol. |
1 |
0 |
0 |
0 |
|
3 |
Tol. |
2 |
60 |
73 |
0 |
|
4 |
Tol. |
5 |
77 |
75 |
7 |
|
5 |
PhCl |
5 |
93 |
92 |
8 |
|
6 |
MeCN |
5 |
0 |
0 |
0 |

General conditions: 1 mol% cat 4, 2 equiv PhSiH3, 50 °C, 2 h. Conversion of styrene as determined by GC‐MS and selectivity as determined by 1H NMR spectroscopy.
As the selectivity toward formation of 3,3‐dichloropropyl benzene is very high in chlorobenzene, where only the ATRA product was observed as a side product, the latter was used as preferred solvent for the reaction.
In order to evaluate the scope of applicable chlorinated substrates, styrene was reacted with various organochlorides and organobromides. Results are summarized in Table 4. Chloroform and carbon tetrachloride react straightforward with styrene using 1 mol% of 4, affording the corresponding hydroalkylation products with good selectivity (Table 4, entry 1 and 2).
Table 4.
Hydroalkylation of styrene with different organohalides catalyzed by 4 in presence of phenylsilane.
|
| ||||||
|---|---|---|---|---|---|---|
|
Entry |
R−CXxHy |
solvent |
Time (h) |
Cat. loading (mol%) |
Conversion (%) |
Selectivity for hydroalkylation product (%) |
|
1 |
CHCl3 |
– |
2 |
1 |
>98 |
91 |
|
2 |
CCl4 |
PhCl |
20 |
1 |
100 |
70 |
|
3 |
CH2Cl2 |
– |
20 |
1 |
0 |
0 |
|
4 |
CHBr3 |
PhCl |
1 |
1 |
100 |
>98 |
|
5 |
CBr4 |
PhCl |
3 |
1 |
100 |
25 |
|
6 |
CHI3 |
PhCl |
20 |
1 |
0 |
0 |
|
7 |
MeCCl3 |
PhCl |
20 |
2 |
0 |
0 |
|
8 |
PhCCl3 |
PhCl |
20 |
2 |
0 |
0 |
|
9 |
NCCCl3 |
toluene |
20 |
1 |
0 |
0 |
|
10 |
(EtO2C)2CHBr |
PhCl |
20 |
2 |
0 |
0 |

General conditions: 2 equiv PhSiH3, 50 °C, 0.5 mL solvent. Conversion of styrene as determined by GC‐MS and selectivity as determined by 1H NMR spectroscopy.
It is noteworthy that the main side product in these reactions is the ATRA product, where the vicinal carbon of the aryl group is substituted by a chlorine atom. Bromoform is the most reactive organohalide, leading to 100% conversion and almost selective formation of the hydroalkylation product after only 1 hour (Table 4, entry 4). Despite full conversion of styrene after 3 h, the reaction with carbon tetrabromide resulted in only a small amount of brominated product, possibly due to the instability of CBr4 (Table 4, entry 5). Bulkier CHI3 or substituted organohalides such as MeCCl3, PhCCl3, NCCCl3 or diethyl bromomalonate did not react under the reaction conditions (entry 6 to 10). Dichloromethane proved unreactive as well (entry 3).
The fact that the reaction with CCl4 lead mainly to formation of the hydroalkylation product is a proof that the silane is the source of the hydrogen atom added to the double bond. In order to confirm this, an experiment using CDCl3 as the organohalide in the hydroalkylation of styrene was performed, then the 1H NMR spectrum in CDCl3 of the obtained product was compared to the one when CHCl3 is used in C6D6 (Figure 3).
Figure 3.

1H NMR spectra of hydroalkylation of styrene using CDCl3 (top) or CHCl3 in C6D6 (bottom) as the organohalide.
Disappearance of the triplet signal at 5.2 ppm could be observed and the integration for the two signals corresponding to the two CH2 groups remained the same, indicating that the CDCl2 moiety is incorporated to the product and no C−H bond from the organohalide is cleaved during the reaction. This 1H NMR spectroscopy study of the reaction of styrene with CHCl3 in C6D6 in presence of PhSiH3 and 4 confirmed the formation of PhSiH2Cl and PhSiHCl2 as side‐products from the reaction. Furthermore, the hydroalkylation of styrene takes place and affords 7 a not only with phenylsilane but also with secondary and tertiary silanes, albeit slower (Table 5).
Table 5.
Silane screening for the addition of CHCl3 to styrene in toluene.
|
| |||||
|---|---|---|---|---|---|
|
|
Silane |
Time |
Conv. (%) |
Select. for 7 a (%) |
Select.for 7 a′ (%) |
|
1 |
PhSiH3 |
2 h |
77 |
96/>98 |
4/0 |
|
2 |
PhMeSiH2 |
2 h/20 h |
72/>98 |
76/>98 |
0/0 |
|
3 |
Et3SiH |
2 h/20 h |
0/51 |
0/88 |
0/0 |
|
4 |
PhMe2SiH |
20 h |
44 |
0 |
0 |

General conditions: 1 mol% cat 4, 2 equiv Silane, 5 equiv CHCl3, 50 °C in toluene. Conversion of styrene as determined by GC‐MS and selectivity as determined by 1H NMR spectroscopy.
The observation of two different products in the here described system is in contrast to a classical ATRA, where the halide of the organohalide is incorporated to the alkene substrate. Since the ATRA and the hydroalkylation reactions involve formation of radical species, we investigated if indeed our catalytic reaction proceeds in a similar fashion. Addition of the free radical‐scavenger galvinoxyl to the typical reaction mixture for the hydroalkylation of styrene (1 mol% 4, 2 equiv PhSiH3, 5 equiv CHBr3) shut down the reactivity and styrene remained intact, providing evidence that the organohalide is added to the alkene via formation of radical species. Furthermore, a 1H NMR spectroscopy study of the reaction of PhSiH3 with CHCl3 in C6D6 in presence of 4, without substrate, confirmed the formation of radical species (Figure 4). In these conditions, PhSiH3 reacts with CHCl3 to form a mixture of PhSiH2Cl, PhSiHCl2, CH2Cl2 and CH3Cl, similar to the chlorination of hydrosilanes reported by Chulsky and Dobrovetsky.10
Figure 4.

1H NMR spectrum in C6D6 of the chlorination of PhSiH3 with CHCl3 in presence of 4 without radical scavenger.
When the reaction is run with a stoichiometric amount of galvinoxyl, the 1H NMR spectrum showed no exchange between PhSiH3 and CHCl3. This indicates that PhSiH3 reacts with the organohalide in presence of borane adduct 4 generating a radical species. This exchange reaction with PhSiH3 is very fast for CHBr3, but surprisingly does not proceed when using CHI3.
Besides, an EPR measurement of the reaction of complex 4 with 1 equiv PhSiH3 in C6D6 without substrate or organohalide was performed and showed that the reaction led to formation of a Mo(V) intermediate (see Supporting Information). Although this intermediate might not be the catalytically active species, this result shows that reduction of the molybdenum center occurs during the reaction. The reactivity of complexes 5 and 6 was also tested without substrate to see if this chlorination of PhSiH3 takes place. We observed the same formation of PhSiH2Cl and CH2Cl2, but only in trace amounts and the reaction is slower than for 4. This lack of reactivity of PhSiH3 with CHCl3, after reacting with the FLP adducts 5 or 6, could be explained by the presence of electron‐withdrawing substituents at the ligands. The Mo(V) intermediate arising from the reaction of PhSiH3 with 5 or 6 would be stable enough to react with the radical species or other molecules present in the mixture, leading to deactivation of the catalyst, while for complex 4, the Mo(V) intermediate is probably a transient species, allowing reaction of H2PhSi. with CHCl3. These results led us to consider a plausible mechanism for the reaction similar to the mechanism reported by Hell et al. for the silyl radical‐mediated activation of sulfamoyl chlorides (Scheme 5).21d
Scheme 5.

Plausible mechanism for the hydroalkylation or ATRA of an alkene with an organohalide in presence of 4 and PhSiH3.
In the proposed mechanism, PhSiH3 reacts at first with the FLP complex 4 to form the corresponding Lewis Pair I, that undergoes Single Electron‐Transfer (SET) to activate another PhSiH3 molecule, leading to the radical H2PhSi. II, a Mo(V) intermediate III and a free proton. H2PhSi. then abstracts a halogen atom of the organohalide, forming the side‐product PhSiH2X and the radical .CHX2 IV. This radical adds to the alkene, forming a new reactive radical intermediate V. The catalytic cycle is closed by recombination of this radical with HB(C6F5), affording the final product VI, and a SET regenerating the Mo(VI) complex 4. In the case of aliphatic alkenes, the radical intermediate V is less reactive and has time to react with the organohalide, leading to ATRA product VII.
As reactions with bromoform lead exclusively to the hydroalkylation product, the experiments to extent the scope of the olefinic substrates as summarized in Table 6 were performed using CHBr3 as the organohalide. The most striking observation concerns the substrate structure, where a phenyl group adjacent to the double bond led to quick conversion into the hydroalkylation product (3,3‐dibromo derivatives). Aliphatic substrates with terminal double bonds reacted much slower and formed the ATRA product (1,3,3‐tribromo derivatives) exclusively (Scheme 6). The reaction of cyclooctene with CHBr3, although showing conversion of the substrate, did not lead to formation of brominated product. However, reaction of cyclooctene with CCl4 in chlorobenzene using 1 mol% of catalyst 4 shows full conversion of the substrate to a mixture of 1,2‐ and 1,4‐addition products, as reported in previous publications (Table 6, entry 3).29
Table 6.
Products scope of the reaction of various alkenes with organohalides in presence of catalyst 4 and phenylsilane.
|
| |||||||
|---|---|---|---|---|---|---|---|
|
Entry |
Substrate/Conversion (%) |
Organohalide |
Product/Isolated yield (%) |
Catalyst 4 loading (mol%) |
Time (h) |
||
|
1 |
|
>98 |
CHCl3 |
|
7 a, 86 (GC) |
1 |
2 |
|
2 |
|
>98 |
CHBr3 |
|
7 b, 86 |
1 |
3 |
|
3 |
|
100 |
CCl4 |
|
1 |
2 |
|
|
4 |
|
79 |
CHCl3 |
|
7 a, 79 |
1 |
20 |
|
5 |
|
100 |
CHBr3 |
|
7 c, 90 |
1 |
1 |
|
6 |
|
100 |
CHBr3 |
|
7 d, 96 |
2 |
1 |
|
7 |
|
57 |
CHBr3 |
|
7 e, 42 |
2 |
24 |
|
8 |
|
46 |
CHBr3 |
|
7 f, 41 |
2 |
24 |
|
9 |
|
95 |
CHBr3 |
|
7 g, 76 |
3 |
24 |
|
10 |
|
36 |
CHBr3 |
|
7 h, 18 |
2 |
24 |
|
11 |
|
71 |
CHBr3 |
|
7 i, 46 |
1 |
24 |
|
12 |
|
100 |
CHBr3 |
|
7 j, 90 |
1 |
3 |
|
13 |
|
100 |
CHBr3 |
|
7 k, 90 |
1 |
3 |
|
14 |
|
100 |
CHBr3 |
|
7 l, 18 |
2 |
5 |
|
15 |
|
100 |
CHBr3 |
unidentified |
|
1 |
24 |
|
16 |
|
0 |
CHBr3 |
– |
|
1 |
24 |
























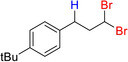






General conditions: 1 mmol substrate, 2 equiv PhSiH3, 5 equiv organohalide, 50 °C in chlorobenzene. Conversion of substrate as determined by GC‐MS.
Scheme 6.

Different outcome of the reaction of various alkenes with CHBr3 in presence of catalyst 4 and phenylsilane.
Formation of the ATRA product as side product was also observed for styrene when using CCl4 or CHCl3, but it is interesting to see that these ATRA products can react in the reaction conditions to form the hydroalkylation product (entry 4). Furthermore, alkenes with a phenyl substituent proved to be significantly more reactive if terminal rather than internal. Hence, aryl‐substituted alkenes such as styrene, 1,1‐diphenylethene or α‐methylstyrene react quickly with CHBr3 with low catalyst loading to afford the corresponding hydroalkylation products 7 a–d in excellent isolated yields (entry 2, 5 and 6). On the other hand, reaction of 4‐phenyl‐1‐butene or 1‐octene with bromoform using 2 mol% of complex 4 in presence of phenylsilane lead to the isolation of Kharasch addition products (3,5,5‐tribromopentyl)benzene 7 e and 1,3,3‐tribromononane 7 f in 42% and 41% yield, respectively (entry 7 and 8). Substrates that possess an aryl group adjacent to an internal double bond, such as prop‐1‐en‐1‐ylbenzene react with CHBr3 to afford the hydroalkylation products 7 g–i, albeit requiring longer reaction time and higher catalyst loading (entry 9–11). We also investigated the influence of substituents at the aryl group onto the activity of the catalyst. Using 1‐bromo‐4‐vinylbenzene or 1‐(tert‐butyl)‐4‐vinylbenzene as substrate lead to excellent isolated yields, 90% for both products 7 j and 7 k, after 3 h using 1 mol% catalyst (entry 12–13). Having a methoxy group trans to the vinyl functionality lead to formation of numerous side products by reaction with the silane. No halogenation product was observed in the case of 1‐methoxy‐4‐vinylbenzene, and the hydroalkylation product 7 l of the reaction with trans‐anethole appears to involve etherification of the anethole (entry 14 and 15). However, reaction with 4‐vinylbenzonitrile or 2‐vinylpyridine did not lead to conversion of the substrate, possibly because of the interaction of the donor atom with the borane moiety (entry 16).
Conclusion
In summary, we synthesized and characterized a molybdenum‐oxido Frustrated Lewis Pair adduct [MoO{OB(C6F5)3}(L)2] (L=2,4‐dimethyl‐6‐((phenylimino)methyl)phenol) which is able to react with phenylsilane to catalyze the hydroalkylation of various aryl alkenes with organohalides and the ATRA of organohalides to aliphatic alkenes. Examples of such catalytic hydroalkylation of alkene using simple chlorine and bromine derivatives, like bromoform, are scarce. This catalytic system leads to formation of gem‐dichloride and gem‐dibromide derivatives in very good yields using low catalyst loadings.
Experimental Section
General Informations. If not otherwise noted, reactions were carried out under N2 atmosphere, using standard Schlenk‐techniques or a N2‐filled glovebox. The substrates were purchased from commercial sources and used as received. Solvents were purified via a Pure‐Solv MD‐4‐EN solvent purification system from Innovative Technology, Inc. CHCl3, CCl4 and chlorobenzene were purchased from commercial sources and distilled prior to use. The complex [MoO2(L1)2],16 the Schiff base ligands HL2 27 and HL3 28 as well as B(C6F5)3 30 were synthesized according to previously published literature. The 1H, 11B, 13C and 19F NMR spectra were recorded on a Bruker Optics instrument at 300/96/75/282 MHz. Peaks are denoted as singlet (s) doublet (d), doublet of doublets (dd), triplet (t), quartet (q) and multiplet (m), broad peaks are denoted (br) and all peaks are referenced to the solvent residual signal. Shifts in 11B and 19F NMR spectra are referenced to external standards (BF3 ⋅ Et2O and CFCl3, respectively). Used solvents and peak assignment are mentioned at the specific data sets. GC‐MS analyses were performed with an Agilent 7890A GC system with an Agilent 19091J‐433 column coupled to a 5975C inert XL EI/CI mass selective detector (MSD). IR spectra were measured as solid samples on a Bruker Alpha‐P Diamond FTIR‐ATR spectrometer. Elemental analyses were carried out using a Heraeus Vario Elementar automatic analyzer at the Institute of Inorganic Chemistry at the Graz University of Technology.
Synthesis of Complex 2 [MoO2(L2)2]. [MoO2Cl2] (1 equiv, 0.12 g, 0.60 mmol) was added to a solution of HL2 (2.1 equiv, 0.56 g, 1.26 mmol) and NEt3 (2.4 equiv, 0.2 mL, 1.43 mmol) in acetonitrile (5 mL) under stirring. The addition was accompanied by a color change from bright yellow to orange‐red. The reaction mixture was subsequently stirred overnight at room temperature, whereupon a yellow precipitate had formed. The precipitate was filtered off, washed with cold acetonitrile (3×2 mL) and dried in vacuo to obtain complex 2 as bright yellow solid (0.394 g, 65%).1H NMR (300 MHz, C6D6): δ=7.61 (d, 2H, ArH), 7.60 (s, 2H, CH=N), 7.53 (br s, 2H, Ph), 7.34 (br s, 4H, ArH), 7.06 (d, 2H, ArH), 1.31 (s, 18H, tert‐Bu), 1.07 (s, 18H, tert‐Bu). 13C NMR (75 MHz, C6D6): δ=171.30 (C=N), 161.08 (Ar−O), 154.53, 143.47, 139.85 (q‐C), 133.01, 132.57 (CF3), 130.54 (ArH), 125.14 (q‐C), 123.50 (br, 2x Ph), 121.52 (br, Ph), 121.06 (q‐C), 35.20, 34.47, 31.35, 29.24 (tert‐Bu). 19F NMR (282 MHz, C6D6): δ=−62.75 (s, 6F, CF3). IR (ATR, cm−1): υ=2960 (m), 1598 (s, C=N), 1475 (s), 1364 (s), 1276 (s), 1179 (s), 1134 (s), 935 (m), 916 (m), 893 (s, Mo=O), 847 (s), 682 (m), 555 (m, Mo−O). Anal. calcd for C46H48F12MoN2O4: C, 54.34; H, 4.76; N, 2.76; Found: C, 54.21; H, 4.97; N, 2.72.
Synthesis of Complex 3 [MoO2(L3)2]. [MoO2Cl2] (1 equiv, 0.52 g, 2.63 mmol) was suspended in acetonitrile (20 mL), whereupon HL3 (2.1 equiv, 1.40 g, 5.26 mmol) and NEt3 (2.4 equiv, 0.88 mL, 6.31 mmol) were added under stirring. The addition was accompanied by a color change from bright yellow to deep red. The reaction mixture was subsequently stirred overnight at room temperature, whereupon a beige precipitate had formed. The precipitate was filtered off, washed with cold acetonitrile (3×15 mL) and pentane (2×10 mL) and dried in vacuo to obtain 3 as bright yellow solid (1.34 g, 78%). Single crystals of 3 suitable for X‐ray diffraction analysis were obtained via slow evaporation from a concentrated solution of 3 in dichloromethane layered with n‐heptane. 1H NMR (300 MHz, (CD3)2SO): δ=8.51 (s, 2H, CH=N), 7.88–7.11 (m, 14H, ArH+Ph). 13C NMR (300 MHz, (CD3)2SO): δ=167.41, 152.88, 150.33, 134.10, 132.71, 132.26, 130.72, 129.65, 128.81, 128.32, 126.66, 124.70, 123.90, 123.32, 123.06, 122.58, 121.53. IR (ATR, cm−1): υ=1613 (s, C=N), 1444 (s), 1375 (m), 1279 (s), 1177 (s), 916 (s), 900 (s, Mo=O), 873 (s), 857 (s), 783 (s), 733 (s), 699 (s), 608 (s), 543 (s, Mo−O), 509 (s), 483 (s), 463 (s). Anal. calcd for C26H16Cl4MoN2O4 ⋅ 0.2 C5H12: C, 48.21; H, 2.76; N, 4.16; Found: C, 48.48; H, 2.80; N, 4.16.
Synthesis of Complex 4 [MoO{OB(C6F5)3}(L1)2]. A solution of B(C6F5)3 (1 equiv, 0.068 g, 0.13 mmol) in dry pentane (2 mL) was added to a suspension of complex 1 (1 equiv, 0.1 g, 0.13 mmol) in the same solvent (3 mL). The addition was accompanied by an immediate color change of the suspension from yellow to dark red. The reaction mixture was subsequently stirred at room temperature for 6 h, whereupon a large quantity of a dark red precipitate had formed. The precipitate was subsequently filtered off, washed thoroughly with cold pentane (3×5 mL) and dried in vacuo to yield complex 4 as a dark red solid (0.153 g, 91%). Single crystals suitable for X‐ray diffraction analysis were obtained via vapor diffusion of pentane into a saturated toluene solution of 4 at room temperature. 1H NMR (300 MHz, C6D6): δ=7.76 (s, 1H, CH=N), 7.56 (d+s, 2H, ArH+CH=N), 7.44 (d, 1H, ArH), 6.95–6.82 (m, 5H, Ph), 6.78 (d, 1H, ArH), 6.75 (d, 1H, ArH), 6.62–6.50 (m, 5H, Ph), 1.23 (s, 9H, tert‐Bu), 1.10 (s, 9H, tert‐Bu), 1.06 (s, 9H, tert‐Bu), 1.03 (s, 9H, tert‐Bu). 13C NMR (75 MHz, C6D6): δ=171.64, 169.14 (C=N), 159.36, 155.36 (Ar−O), 152.46, 151.20 (q‐C), 149.97, 146.81 (C6F5), 146.48, 145.18 (q‐C), 141.84 (C6F5), 139.35, 139.17 (q‐C), 135.74 (C6F5), 133.06, 132.92 130.48, 129.89 (ArH), 128.84, 128.73, 127.36, 127.24, 124.04, 123.65 (Ph), 123.46, 121.69 (q‐C), 35.46, 35.22, 34.50, 34.36 (q‐tert‐Bu), 31.21, 31.16, 30.74, 29.56 (tert‐Bu). 19F NMR (282 MHz, C6D6): δ=−130.25 (dd, 6F, o‐F), −158.78 (t, 3F, p‐F), ‐165.03 (m, 6F, m‐F). IR (ATR, cm−1): υ=2962 (m, C−H), 1606 (w, C=N), 1514 (m), 1467 (s), 1235 (m), 1094 (s), 977 (s), 880 (s), 843 (s), 765 (s), 555 (s, Mo−O). Anal. calcd for C60H52BF15MoN2O4: C, 57.34; H, 4.17; N, 2.23; Found: C, 57.14; H, 4.08; N, 2.23.
Synthesis of Complex 5 [Mo{OB(C6F5)3}O(L2)2]. A solution of B(C6F5)3 (1 equiv, 0.025 g, 0.05 mmol) in dry pentane (1 mL) was added to a suspension of complex 2 (1 equiv, 0.05 g, 0.05 mmol) in the same solvent (2 mL). The addition was accompanied by an immediate color change of the suspension from yellow to deep red and by the formation of a dark red precipitate. The reaction mixture was subsequently stirred at room temperature for 6 h, the precipitate was filtered off, washed thoroughly with cold pentane (3×5 mL) and dried in vacuo to yield complex 5 as a dark red‐brownish solid (0.063 g, 84%). Single crystals suitable for X‐ray diffraction analysis were obtained from a saturated pentane solution of 5 at −35 °C. 1H NMR (300 MHz, C6D6): δ =7.58 (d, 2H, ArH), 7.54 s, 2H, CH=N), 7.51 (br s, 2H, Ph), 7.26 (br s, 4H, Ph), 7.03 (d, 2H, ArH), 1.26 (s, 18H, tert‐Bu), 0.91 (s, 18H, tert‐Bu). 13C NMR (75 MHz, C6D6, C6F5 obscured): δ=171.99 (C=N), 159.22 (Ar−O), 153.85, 145.45, 139.63 (q‐C), 134.05 (ArH), 133.07 (q, 1JC‐F=33.9 Hz, CF3), 130.98 (ArH), 124.90 (q‐C), 123.53 (br, 2 Ph), 121.28 (q‐C), 121.16 (Ph), 35.09, 34.55 (q‐tert‐Bu), 31.13, 29.33 (tert‐Bu). 19F NMR (282 MHz, C6D6): δ=−62.81 (s, 12F, CF3), −132.31 (d, 6F, o‐F), −148.23 (br s, 3F, p‐F), −161.03 (m, 6F, m‐F). IR (ATR, cm−1): υ=2962 (m, C−H), 1596 (w, C=N), 1517 (m), 1467 (s), 1365 (s), 1278 (m), 1176 (m), 1141 (s), 1094 (s), 978 (s), 880 (s), 847 (s), 762 (m), 682 (s), 560 (s, Mo−O). Anal. calcd for C64H48BF27MoN2O4 ⋅ 0.5 C5H12: C, 51.04; H, 3.48; N, 1.79; Found: C, 50.99; H, 3.84; N, 1.75.
Synthesis of Complex 6 [Mo{OB(C6F5)3}O(L3)2]. A solution of B(C6F5)3 (1 equiv, 0.2 g, 0.39 mmol) in dry pentane (10 mL) was added to a suspension of complex 3 (1 equiv, 0.26 g, 0.39 mmol) in the same solvent (10 mL). The addition was accompanied by an immediate color change from yellow to deep red and by the formation of a red precipitate. The reaction mixture was subsequently stirred at room temperature overnight, the precipitate was filtered off, washed thrice with cold pentane (3×10 mL) and dried in vacuo to yield 6 as a brick red solid (0.35 g, 76%). Single crystals suitable for X‐ray diffraction analysis were obtained from a concentrated benzene solution of 6 at room temperature or a concentrated toluene solution at −35 °C. 1H NMR (300 MHz, CD2Cl2): δ=8.24 (s, 1H, CH=N), 8.09 (s, 1H, CH=N), 7.40–7.35 (m, 2H, ArH), 7.27–7.07 (br m, 10H, Ph), 6.72–6.69 (m, 2H, ArH). 13C NMR (75 MHz, CD2Cl2): δ=166.71, 166.46, 153.10, 152.40, 150.38, 150.06, 146.77, 141.95, 139.32, 138.47, 137.54, 136.08, 135.88, 135.57, 132.57, 132.29, 129.76, 129.52, 128.90, 128.50, 128.04, 126.15, 125.98, 123.83, 123.41, 123.02, 122.38. 19F NMR (282 MHz, CD2Cl2): δ=−132.0 (d, 6F, o‐F), −158.97 (t, 3F, p‐F), −164.93 (m, 6F, m‐F). IR (ATR, cm−1): υ=1610 (m, C=N), 1546 (m), 1465 (s), 1446 (s), 1269 (s), 1093 (s), 976 (s), 882 (s), 790 (s), 732 (m), 701 (m), 671 (m), 609(m), 551 (s, Mo−O), 520 (m). Anal. calcd for C44H16BCl4F15MoN2O4: C, 45.16; H, 1.38; N, 2.39; Found: C, 45.07; H, 1.42; N, 2.58.
Procedure for Catalytic Runs. All catalytic experiments for determination of conversion using Gas Chromatography were performed under inert conditions (N2 atmosphere, exclusion of moisture) in Mininert® reaction vessels. In a typical experiment, an aliquot of a chlorobenzene stock solution of the respective catalyst was added to 0.5 mL of chlorobenzene containing 0.1 mmol of the substrate, two equivalents of silane and five equivalents of the respective halide. 0.1 mmol of mesitylene was used as internal standard. Samples for GC‐MS measurements were withdrawn at given time intervals with a microliter syringe (10 μL), quenched with Na2CO3 and diluted by a factor of 50 with HPLC grade ethyl acetate. A 0 h sample was withdrawn before addition of the silane. All catalytic experiments for determination of isolated yields of products 7 a–7 l were performed under inert conditions using Schlenk techniques. 0.01–0.03 mmol of the catalyst was weighted in the glovebox in a Schlenk flask and dissolved in 5 mL chlorobenzene, then 1 mmol of the substrate, 2 mmol PhSiH3 and 5 mmol of the respective halide were added. After stirring at 50 °C for the corresponding reaction time, the reaction was quenched with aqueous sodium carbonate, the solvent and volatiles were removed under vacuum, the residue re‐dissolved in dichloromethane and filtered over a plug of silica. Isolated yields were obtained by purifying the products using column chromatography over silica with a Biotage Isolera Four equipment, using cyclohexane/ethyl acetate mixtures (10:1) as the eluent.
Analytical data for (3,3‐dichloropropyl)benzene 7 a. Using 0.1 g 7 a’ as substrate, 7 a was isolated in 79% yield (0.067 g). NMR data are supported by previous publication.31
1H NMR (300 MHz, CDCl3): δ=7.34–7.20 (m, 5H, ArH), 5.66 (t, 1H, 1JC‐H=6.1 Hz, −CHCl2), 2.91–2.86 (m, 2H, Ar−CH2), 2.55–2.48 (m, 2H, −CH2).
Analytical data for (3,3‐dibromopropyl)benzene 7 b. Using 0.1 g styrene as substrate, 7 b was isolated in 86% yield (0.23 g). NMR data are supported by previous publication.32 1H NMR (300 MHz, CDCl3): δ=7.35–7.20 (m, 5H, ArH), 5.60 (t, 1H, 1JC‐H=6.3 Hz, −CHBr2), 2.89–2.85 (m, 2H, Ar−CH2), 2.74–2.67 (m, 2H, −CH2).
Analytical data for (3,3‐dibromopropane‐1,1‐diyl)dibenzene 7 c. Using 0.18 g 1,1‐diphenylethylene as substrate, 7 c was isolated in 90% yield (0.318 g). 1H NMR (300 MHz, CDCl3): δ=7.36–7.22 (m, 10H, ArH), 5.30 (t, 1H, 1JC‐H=6.9 Hz, −CHBr2), 4.28 (t, 1H, 1JC‐H=7.7 Hz, Ar2CH), 3.16–3.11 (dd, 2H, 1JC‐H=6.9 Hz, 1JC‐H=7.7 Hz, −CH2). 13C NMR (75 MHz, CDCl3): δ=142.20, 129.00, 127.91, 127.07, 51.05, 49.95, 44.38.
Analytical data for (4,4‐dibromobutan‐2‐yl)benzene 7 d. Using 0.118 g α‐methylstyrene as substrate, 7 d was isolated in 96% yield (0.278 g). 1H NMR (300 MHz, CDCl3): δ=7.35–7.20 (m, 5H, ArH), 5.27–5.22 (m, 1H, −CHBr2), 3.11–2.99 (m, 1H, ArCHMe), 2.69–2.63 (m, 2H, −CH2), 1.31 (d, 1H, 1JC‐H=7.0 Hz, −CH3). 13C NMR (75 MHz, CDCl3): 144.05, 128.98, 127.13, 126.99, 53.72, 44.72, 39.29, 21.80.
Analytical data for (3,5,5‐tribromopentyl)benzene 7 e. Using 0.132 g 4‐phenyl‐1‐butene as substrate, 7 e was isolated in 42% yield (0.16 g). 1H NMR (300 MHz, CDCl3): δ=7.34–7.21 (m, 5H, ArH), 5.93–5.89 (m, 1H, −CHBr2), 4.16–4.07 (m, 1H, CHBr), 2.98–2.74 (m, 4H, −CH2), 2.22–2.13 (m, 2H, −CH2). 13C NMR (75 MHz, CDCl3): 140.34, 128.75, 128.59, 126.48, 53.69, 53.52, 43.35, 40.21, 33.55.
Analytical data for 1,1,3‐tribromononane 7 f. Using 0.112 g 1‐octene as substrate, 7 f was isolated in 41% yield (0.149 g). NMR data are supported by previous publication.33 1H NMR (300 MHz, CDCl3): δ=5.92–5.88 (m, 1H, −CHBr2), 4.16–4.08 (m, 1H, CHBr), 2.84–2.76 (m, 2H, −CH2), 1.89–1.84 (m, 2H,−CH2), 1.30 (bs, 8H, −CH2), 0.89 (t, 3H, 1JC‐H=6.6 Hz, −CH3).
Analytical data for (3,3‐dibromo‐2‐methylpropyl)benzene 7 g. Using 0.118 g trans‐β‐methylstyrene as substrate, 7 g was isolated in 76% yield (0.22 g). 1H NMR (300 MHz, CDCl3): δ=7.36–7.20 (m, 5H, ArH), 5.73 (d, 1H, 1JC‐H=2.6 Hz, −CHBr2), 2.88–2.81 (dd, 1H, 1JC‐H=13.7, 7.3 Hz, −CH2), 2.63 (dd, 1H, 1JC‐H=13.7, 7.3 Hz, −CH2), 2.44–2.32 (m, 1H, −CHMe), 1.20 (d, 3H, 1JC‐H=6.5 Hz, −CH3). 13C NMR (75 MHz, CDCl3): δ=138.85, 129.17, 128.78, 126.73, 54.53, 46.83, 40.62, 16.58.
Analytical data for (3,3‐dibromopropane‐1,2‐diyl)dibenzene 7 h. Using 0.18 g cis‐stilbene as substrate, 7 h was isolated in 18% yield (0.064 g). 1H NMR (300 MHz, CDCl3): δ=7.41–7.10 (m, 10H, ArH), 5.85 (d, 1H, 1JC‐H=4.5 Hz, −CHBr2), 3.60–3.56 (m, 1H, −CHPh), 3.48–3.41 (m, 1H, −CH2), 3.17–3.10 (m, 1H, −CH2). 13C NMR (75 MHz, CDCl3): δ=138.61, 138.58, 129.40, 129.16, 128.61, 128.31, 128.01, 126.65, 58.14, 51.64, 38.87.
Analytical data for 2‐(dibromomethyl)‐1,2,3,4‐tetrahydronaphthalene 7 i. Using 0.13 g 1,2‐dihydronaphtalene as substrate, 7 i was isolated in 46% yield (0.14 g). 1H NMR (300 MHz, CDCl3): δ=7.14–7.12 (m, 4H, ArH), 5.87 (d, 1H, 1JC‐H=4.2 Hz, −CHBr2), 3.12–2.82 (m, 4H, −CH2), 2.45–2.32 (m, 1H, CH2), 2.21–2.12 (m, 1H, CH2), 1.80–1.65 (m, 1H, CH). 13C NMR (75 MHz, CDCl3): δ=135.87, 134.79, 129.38, 128.83, 126.16, 126.09, 53.12, 46.18, 32.97, 28.92, 27.60.
Analytical data for 1‐(tert‐butyl)‐4‐(3,3‐dibromopropyl)benzene 7 j. Using 0.16 g 4‐tert‐butylstyrene as substrate, 7 j was isolated in 90% yield (0.3 g). 1H NMR (300 MHz, CDCl3): δ=7.35–7.32 (m, 2H, Ar), 7.16–7.13 (m, 2H, Ar), 5.60 (t, 1H, 1JC‐H=6.4 Hz, −CHBr2), 2.86–2.81 (m, 2H, CH2), 2.73–2.66 (m, 2H, CH2), 1.32 (s, 9H, tert‐Bu). 13C NMR (75 MHz, CDCl3): δ=149.56, 136.11, 128.35, 125.73, 46.90, 45.57, 34.58, 33.70, 31.51.
Analytical data for 1‐bromo‐4‐(3,3‐dibromopropyl)benzene 7 k. Using 0.183 g 4‐bromostyrene as substrate, 7 k was isolated in 90% yield (0.32 g). 1H NMR (300 MHz, CDCl3): δ=7.45–7.42 (m, 2H, Ar), 7.10–7.07 (m, 2H, Ar), 5.58 (t, 1H, 1JC‐H=6.2 Hz, −CHBr2), 2.85–2.80 (m, 2H, CH2), 2.70–2.63 (m, 2H, CH2). 13C NMR (75 MHz, CDCl3): δ=138.18, 131.92, 130.42, 120.52, 46.55, 44.82, 33.64.
Analytical data for 4,4′‐oxybis((3,3‐dibromo‐2‐methylpropyl)benzene) 7 l. Using 0.148 g anethole as substrate, 7 l was isolated in 18% yield (0.107 g). 1H NMR (300 MHz, CDCl3): δ=7.08–7.05 (m, 4H, ArH), 6.80–6.77 (m, 4H, ArH), 5.71 (d, 2H, 1JC‐H=2.6 Hz, −CHBr2), 2.77–2.70 (dd, 2H, 1JC‐H=13.8, 7.3 Hz, −CH2), 2.58–2.51 (dd, 2H, 1JC‐H=13.8, 7.3 Hz, −CH2), 2.35–2.23 (m, 2H, −CHMe), 1.17 (d, 6H, 1JC‐H=6.5 Hz, −CH3). 13C NMR (75 MHz, CDCl3): 154.30, 131.01, 130.32, 115.60, 54.60, 46.97, 39.76, 16.48. ESI‐MS (135 V): m/z=592.8 [M−H]−.
Crystallographic Data for Complexes 1–6. The X‐ray data collections were performed with a Bruker AXS SMART APEX‐II CCD diffractometer at 100 K with Mo‐K α radiation (λ=0.71073 Å) from an Incoatec microfocus sealed tube equipped with a multilayer monochromator. Absorption corrections were made semi‐empirically from equivalents. The structures were solved by direct methods (SHELXS‐97) and refined by full‐matrix least‐squares techniques against F 2 (SHELXL‐2014/6). A weighting scheme of w=1/[σ2(Fo 2)+(aP)2+bP] where P=(Fo 2+2Fc 2)/3 was used. The absolute configuration of 3 was established by anomalous dispersion effects in the diffraction measurements of the crystal. The non‐hydrogen atoms of 1, 3, 4, and 6 were refined with anisotropic displacement parameters without any constraints. The H atoms of the phenyl rings including any adjacent CH=N groups were put at the external bisectors of the C−C−C angles at C−H distances of 0.95 Å and common isotropic displacement parameters were refined for the H atoms of the same ring. The H atoms of the tert‐butyl groups were refined with common isotropic displacement parameters for the H atoms of the same group and idealized geometries with tetrahedral angles, enabling rotations around the C−C bonds, and C−H distances of 0.98 Å. Crystallographic data for the structures of compounds 1–6 have been deposited with the Cambridge Crystallographic Data Center (CCDC 1940999 to CCDC 1941004 for 1 to 6).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the Austrian Science Fund (FWF, grant number P26264) and NAWI Graz is greatly acknowledged.
N. Zwettler, A. Dupé, S. Klokić, A. Milinković, D. Rodić, S. Walg, D. Neshchadin, F. Belaj, N. C. Mösch-Zanetti, Adv. Synth. Catal. 2020, 362, 3170.
References
- 1.
- 1a. Stephan D. W., Dalton Trans. 2009, 3129–3136; [DOI] [PubMed] [Google Scholar]
- 1b. Stephan D. W., Erker G., Angew. Chem. Int. Ed. 2010, 49, 46–76; [DOI] [PubMed] [Google Scholar]
- 1c. Spies P., Erker G., Kehr G., Bergander K., Fröhlich R., Grimme S., Stephan D. W., Chem. Commun. 2007, 5072–5074. [DOI] [PubMed] [Google Scholar]
- 2. Welch G. C., San Juan R. R., Masuda J. D., Stephan D. W., Science 2006, 314, 1124–1126. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Simonneau A., Turrel R., Vendier L., Etienne M., Angew. Chem. Int. Ed. 2017, 56, 12268–12272; [DOI] [PubMed] [Google Scholar]
- 3b. Melen R. L., Angew. Chem. Int. Ed. 2018, 57, 880–882; [DOI] [PubMed] [Google Scholar]
- 3c. Stephan D. W., Erker G., Chem. Sci. 2014, 5, 2625–2641; [Google Scholar]
- 3d. Stephan D. W., Science 2016, 354, aaf7229. [Google Scholar]
- 4. Chase P. A., Welch G. C., Jurca T., Stephan D. W., Angew. Chem. Int. Ed. 2007, 46, 8050–8053. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Mahdi T., Stephan D. W., J. Am. Chem. Soc. 2014, 136, 15809–15812; [DOI] [PubMed] [Google Scholar]
- 5b. Scott D. J., Fuchter M. J., Ashley A. E., J. Am. Chem. Soc. 2014, 136, 15813–15816. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Paradies J., Angew. Chem. Int. Ed. 2014, 53, 3552–3557; [DOI] [PubMed] [Google Scholar]
- 6b. Greb L., Oña-Burgos P., Schirmer B., Grimme S., Stephan D. W., Paradies J., Angew. Chem. Int. Ed. 2012, 51, 10164–10168. [DOI] [PubMed] [Google Scholar]
- 7. Chernichenko K., Madarász Á., Pápai I., Nieger M., Leskelä M., Repo T., Nat. Chem. 2013, 5, 718. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Travis A. L., Binding S. C., Zaher H., Arnold T. A. Q., Buffet J.-C., O′Hare D., Dalton Trans. 2013, 42, 2431–2437; [DOI] [PubMed] [Google Scholar]
- 8b. Ashley A. E., Thompson A. L., O′Hare D., Angew. Chem. Int. Ed. 2009, 121, 10023–10027; [Google Scholar]
- 8c. Bontemps S., Coord. Chem. Rev. 2016, 308, 117–130. [Google Scholar]
- 9.
- 9a. Parks D. J., Piers W. E., J. Am. Chem. Soc. 1996, 118, 9440–9441; [Google Scholar]
- 9b. Rubin M., Schwier T., Gevorgyan V., J. Org. Chem. 2002, 67, 1936–1940; [DOI] [PubMed] [Google Scholar]
- 9c. Lawson J. R., Melen R. L., Inorg. Chem. 2017, 56, 8627–8643; [DOI] [PubMed] [Google Scholar]
- 9d. Hackel T., McGrath N. A., Molecules 2019, 24, 432; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. Oestreich M., Hermeke J., Mohr J. A., Chem. Soc. Rev. 2015, 44, 2202–2220; [DOI] [PubMed] [Google Scholar]
- 9f. Lipke M. C., Liberman-Martin A. L., Tilley T. D., Angew. Chem. Int. Ed. 2017, 56, 2260–2294. [DOI] [PubMed] [Google Scholar]
- 10. Chulsky K., Dobrovetsky R., Angew. Chem. Int. Ed. 2017, 56, 4744–4748. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Berkefeld A., Piers W. E., Parvez M., Castro L., Maron L., Eisenstein O., Chem. Sci. 2013, 4, 2152–2162; [Google Scholar]
- 11b. Flynn S. R., Wass D. F., ACS Catal. 2013, 3, 2574–2581; [Google Scholar]
- 11c. Zhang S., Appel A. M., Bullock R. M., J. Am. Chem. Soc. 2017, 139, 7376–7387; [DOI] [PubMed] [Google Scholar]
- 11d. Campos J., J. Am. Chem. Soc. 2017, 139, 2944–2947; [DOI] [PubMed] [Google Scholar]
- 11e. Owen G. R., Chem. Commun. 2016, 52, 10712–10726. [DOI] [PubMed] [Google Scholar]
- 12. Peryshkov D. V., Forrest W. P., Schrock R. R., Smith S. J., Müller P., Organometallics 2013, 32, 5256–5259. [Google Scholar]
- 13.
- 13a. Lambic N. S., Brown C. A., Sommer R. D., Ison E. A., Organometallics 2017, 36, 2042–2051; [Google Scholar]
- 13b. Lambic N. S., Lilly C. P., Robbins L. K., Sommer R. D., Ison E. A., Organometallics 2016, 35, 2822–2829; [Google Scholar]
- 13c. Lambic N. S., Sommer R. D., Ison E. A., J. Am. Chem. Soc. 2016, 138, 4832–4842; [DOI] [PubMed] [Google Scholar]
- 13d. Lambic N. S., Sommer R. D., Ison E. A., ACS Catal. 2017, 7, 1170–1180. [Google Scholar]
- 14. Smeltz J. L., Lilly C. P., Boyle P. D., Ison E. A., J. Am. Chem. Soc. 2013, 135, 9433–9441. [DOI] [PubMed] [Google Scholar]
- 15. Dupé A., Judmaier M. E., Belaj F., Zangger K., Mösch-Zanetti N. C., Dalton Trans. 2015, 44, 20514–20522. [DOI] [PubMed] [Google Scholar]
- 16. Zwettler N., Judmaier M. E., Strohmeier L., Belaj F., Mösch-Zanetti N. C., Dalton Trans. 2016, 45, 14549–14560. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Zwettler N., Grover N., Belaj F., Kirchner K., Mösch-Zanetti N. C., Inorg. Chem. 2017, 56, 10147–10150; [DOI] [PubMed] [Google Scholar]
- 17b. Zwettler N., Mösch-Zanetti N. C., Chem. Eur. J. 2019, 25, 6064–6076. [DOI] [PubMed] [Google Scholar]
- 18. Zwettler N., Ehweiner M. A., Schachner J. A., Dupé A., Belaj F., Mösch-Zanetti N. C., Molecules 2019, 24, 1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zwettler N., Walg S. P., Belaj F., Mösch-Zanetti N. C., Chem. Eur. J. 2018, 24, 7149–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Kharasch M. S., Engelmann H., Mayo F. R., J. Org. Chem. 1937, 02, 288–302; [Google Scholar]
- 20b. Kharasch M. S., Jensen E. V., Urry W. H., Science 1945, 102, 128. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Giese B., Angew. Chem. Int. Ed. Engl. 1983, 22, 753–764; [Google Scholar]
- 21b. Courant T., Masson G., J. Org. Chem. 2016, 81, 6945–6952; [DOI] [PubMed] [Google Scholar]
- 21c. Mizuta S., Verhoog S., Engle K. M., Khotavivattana T., O′Duill M., Wheelhouse K., Rassias G., Médebielle M., Gouverneur V., J. Am. Chem. Soc. 2013, 135, 2505–2508; [DOI] [PubMed] [Google Scholar]
- 21d. Hell S. M., Meyer C. F., Laudadio G., Misale A., Willis M. C., Noël T., Trabanco A. A., Gouverneur V., J. Am. Chem. Soc. 2020, 142, 720–725. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Muñoz-Molina J. M., Belderrain T. R., Pérez P. J., Eur. J. Inorg. Chem. 2011, 3155–3164; [Google Scholar]
- 22b. Muñoz-Molina J. M., Belderrain T. R. in Science of Synthesis: C-1 Building Blocks in Organic Synthesis, vol. 2 (Ed. P. W. N. M. Van Leuween), Thieme, 2014. [Google Scholar]
- 23.
- 23a. Severin K., Chimia 2012, 66, 386–388; [DOI] [PubMed] [Google Scholar]
- 23b. Wallentin C.-J., Nguyen J. D., Finkbeiner P., Stephenson C. R. J., J. Am. Chem. Soc. 2012, 134, 8875–8884; [DOI] [PubMed] [Google Scholar]
- 23c. Lee J., Grandner J. M., Engle K. M., Houk K. N., Grubbs R. H., J. Am. Chem. Soc. 2016, 138, 7171–7177; [DOI] [PubMed] [Google Scholar]
- 23d. Chotard F., Malacea-Kabbara R., Balan C., Bodio E., Picquet M., Richard P., Ponce-Vargas M., Fleurat-Lessard P., Le Gendre P., Organometallics 2018, 37, 812–820; [Google Scholar]
- 23e. Das K., Dutta M., Das B., Kumar Srivastava H., Kumar A., Adv. Synth. Catal. 2019, 361, 2965–2980. [Google Scholar]
- 24.
- 24a. Matsuo K., Yamaguchi E., Itoh A., Asian J. Org. Chem. 2018, 7, 2435–2438; [Google Scholar]
- 24b. Reiser O., Acc. Chem. Res. 2016, 49, 1990–1996. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Shvo Y., Green R., J. Organomet. Chem. 2003, 675, 77–83; [Google Scholar]
- 25b. Davis R., Groves I. F., J. Chem. Soc. Dalton Trans. 1982, 2281–2287. [Google Scholar]
- 26. Boualy B., Harrad M. A., El Firdoussi L., Ait Ali M., El Houssame S., Karim A., Catal. Commun. 2011, 12, 1295–1297. [Google Scholar]
- 27. Cameron P. A., Gibson V. C., Redshaw C., Segal J. A., Solan G. A., White A. J. P., Williams D. J., J. Chem. Soc. Dalton Trans. 2001, 1472–1476. [Google Scholar]
- 28. Carril M., Altmann P., Drees M., Bonrath W., Netscher T., Schütz J., Kühn F. E., J. Catal. 2011, 283, 55–67. [Google Scholar]
- 29. Dneprovskii A. S., Kasatochkin A. N., Boyarskii V. P., Ermoshkin A. A., Yakovlev A. A., Russ. J. Org. Chem. 2006, 42, 1120–1130. [Google Scholar]
- 30. Lancaster S., ChemSpider SyntheticPages 2003. [Google Scholar]
- 31. Aghapour G., Afzali A., Synth. Commun. 2008, 38, 4023–4035. [Google Scholar]
- 32. King S. M., Ma X., Herzon S. B., J. Am. Chem. Soc. 2014, 136, 6884–6887. [DOI] [PubMed] [Google Scholar]
- 33. Mitsuo N., Kunieda T., Takizawa T., J. Org. Chem. 1973, 2255–2257. [Google Scholar]
- 34. Sheldrick G. M., Acta Crystallogr. Sect. A 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- 35. Sheldrick G. M., Acta Crystallogr. Sect. C 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary