

Abstract
Therapeutic vaccination offers great promise as an intervention for a diversity of infectious and non‐infectious conditions. Given that most chronic health conditions are thought to have an immune component, vaccination can at least in principle be proposed as a therapeutic strategy. Understanding the nature of protective immunity is of vital importance, and the progress made in recent years in defining the nature of pathological and protective immunity for a range of diseases has provided an impetus to devise strategies to promote such responses in a targeted manner. However, in many cases, limited progress has been made in clinical adoption of such approaches. This in part results from a lack of safe and effective vaccine adjuvants that can be used to promote protective immunity and/or reduce deleterious immune responses. Although somewhat simplistic, it is possible to divide therapeutic vaccine approaches into those targeting conditions where antibody responses can mediate protection and those where the principal focus is the promotion of effector and memory cellular immunity or the reduction of damaging cellular immune responses as in the case of autoimmune diseases. Clearly, in all cases of antigen‐specific immunotherapy, the identification of protective antigens is a vital first step. There are many challenges to developing therapeutic vaccines beyond those associated with prophylactic diseases including the ongoing immune responses in patients, patient heterogeneity, and diversity in the type and stage of disease. If reproducible biomarkers can be defined, these could allow earlier diagnosis and intervention and likely increase therapeutic vaccine efficacy. Current immunomodulatory approaches related to adoptive cell transfers or passive antibody therapy are showing great promise, but these are outside the scope of this review which will focus on the potential for adjuvanted therapeutic active vaccination strategies.
Keywords: adjuvant, autoimmunity, cancer, cellular immunity, therapeutic, vaccine
1. INTRODUCTION
Vaccines have made an enormous contribution to the reduction of morbidity and mortality across the globe. The expression “therapeutic vaccine” may sound counter intuitive since “vaccines” are traditionally used as prophylactic medicines with the aim to prevent, rather than to treat, diseases, mostly viral, or bacterial infections. The terms “therapeutic immunization” or “therapeutic vaccines” used in this paper define vaccines, which are used therapeutically to treat a medical condition, such as an ongoing, chronic, often debilitating health problem, or an unwanted biological response. Therapeutic vaccines aim to reprogram the immune system of the patient in order to better recognize and neutralize specific deleterious molecular targets or immune cells. 1 Hence, the vaccine could target antigens associated with an infectious (chronic) disease or non‐infectious diseases such as cancer, allergy, drug (eg, nicotine) addiction, or self‐molecules associated with autoimmune/autoinflammatory situations (eg, hypertension, neurological disorders, atherosclerosis, diabetes). 2 A therapeutic vaccine contains a carefully chosen molecular entity, the vaccine antigen(s), which is the target of the vaccination‐induced immune response. In addition, the therapeutic vaccine generally requires a suitable adjuvant or immune modulator to induce and direct the desired type of therapeutic immune reaction. This article provides an overview of therapeutic vaccines in clinical, preclinical, and experimental use, their molecular and/or cellular targets, and the key role of adjuvants in promoting effective immune responses.
Given the breadth of the targeted medical conditions, therapeutic vaccines may come in many forms. Figure 1 presents an overview of the contexts where therapeutic vaccines exist or are being developed.
FIGURE 1.
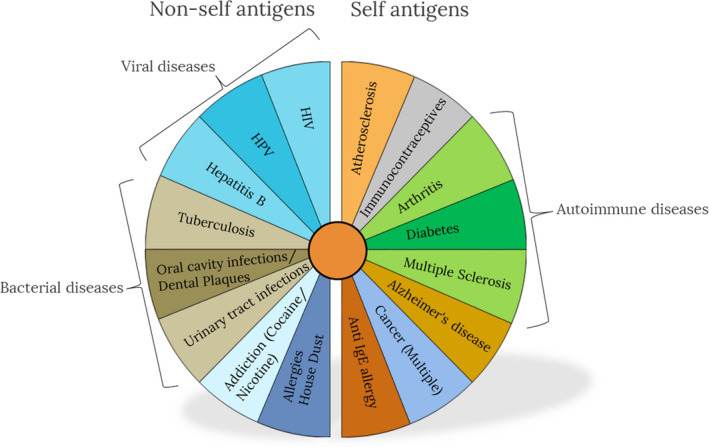
Indications for therapeutic vaccines
Since therapeutic vaccines can be designed for many indications including chronic infectious disease, allergies, addictive drugs, cancer cells, and even chronic non‐infectious disease such as atherosclerosis, a range of different vaccine approaches is required. Rational vaccine design, for both prophylactic and therapeutic vaccines, first requires elucidation of the nature of protective or beneficial immunity for a specific condition. The first step in rational therapeutic vaccine design therefore involves identification and selection of antigens, which require knowledge of the pathogenesis of the condition and accessible, and ideally stably expressed, key molecular targets. For example, an infectious target with an extracellular lifecycle (or stage) can be targeted with specific antibodies elicited by immunization, while an intracellular target (eg, a pathogen with an intracellular life cycle) needs to be targeted with a cell‐mediated immune reaction, which eventually eliminates (sacrifices) the target cell. Similarly, tumor cell targets expressed on the membrane of the tumor are accessible by antibody‐mediated cytotoxicity (ADCC), while intracellular tumor antigens need to be targeted by cellular immune responses.
The next challenge is how to generate the desired immune response against the chosen antigen, ideally with long‐term immunological memory. This requires the choice of a suitable immunostimulator that is able to induce, prolong, magnify, and steer the required immune response. This is the role for vaccine adjuvants, which with the increasing knowledge in innate immune mechanisms has moved from being “immunologists dirty little secret” to vaccinologists requisite for success. The two main objectives for the vaccine adjuvants are to (a) ensure that the antigens are delivered to those cells of the innate immune system specialized in inducing the desired immune response and (b) activate the innate immune system to direct the inducible adaptive immunity in the required direction. Modern adjuvants thus typically consist of both a delivery system, that can be composed of emulsions, liposomes, polymeric nanoparticles etc and immunostimulators that are typically ligands for so‐called pattern recognition receptors (PRRs) on the innate immune cells. These ligands are typically mimicking pathogen or damage‐associated molecular patterns (PAMPs/DAMPs), including Toll‐like receptor (TLR) agonists, C‐type lectin receptor (CLR) agonists, Nod‐like receptor (NLR) agonists, and RIG‐like receptor (RLR) agonists. Only a few adjuvants have been approved in vaccines for human use, including aluminum salts, Virosomes, MF59™, AS01™, AS03™, AS04™, and CpG, but many more are in clinical development and the repertoire will expand during the next decade. This subject has recently been reviewed in details in the book “Immunopotentiators in Modern Vaccines” which provides in‐depth insights and overviews of the most successful adjuvants, including those that have been included in licensed products and the most promising emerging technologies. 3
Other key issues at this stage are formulation safety, stability, capacity for sterile filtration, consistency of component availability, and cost.
1.1. Antibody‐based immune responses against extracellular targets
Many molecules that are targets of monoclonal antibody therapies may also be candidates for actively elicited antibodies, triggered by therapeutic immunization. Hence, neutralizing antibodies induced by vaccination may be alternatives to, often very expensive, passively administered monoclonal antibodies. However, the duration of such active circulating antibody responses vs declining concentrations of passively administered monoclonals needs to be considered for safety reasons. In case of drug addictions, neutralizing antibodies induced by vaccines against cocaine or nicotine may be ideal; however, when acute reversibility or temporary inhibition of responses is the aim, passive delivery of monoclonal antibodies may be preferred over actively generated antibodies.
One of the main effector processes triggered by a vaccine is the production of antibodies by antigen‐specific B lymphocytes. Soluble antibody molecules can specifically detect and bind their antigen target, promoting its neutralization and opsonization by phagocytes (macrophages and neutrophils). Antibodies are also able to (a) block the activity of certain toxins, (b) prevent the spread of harmful infectious agents, (c) keep viruses from entering healthy cells, and (d) activate the complement cascade, which helps in microbe clearance. B cells can directly recognize antigenic moieties of extracellular pathogens through the immunoglobulins present in their outer membrane, the so‐called B cell receptors (BCR). Upon encountering the antigen, B cells become activated and differentiate into plasma cells, which produce and secrete specific IgM antibodies (without class switching) in a process known as T cell–independent activation. 4 Activation of CD4+ T lymphocytes is essential for optimal B cell priming by enhancing antigen presentation. 5 Detailed stages and processes of the germinal center and extrafollicular B cell immune response in the context of an innate immune response are recently described in detail by Shlomchik et al. 6 T cells are activated by antigen‐presenting cells (APCs), particularly dendritic cells (DCs). Upon encountering pathogen‐associated molecular patterns (PAMPs) or danger‐associated molecular patterns (DAMPs), DCs undergo rapid maturation, modulate their surface receptors, and migrate to secondary lymphoid organs, such as lymph nodes, where B and T cell–mediated adaptive responses are initiated. 7 In the case of extracellular pathogens and many vaccines, DCs patrolling the body can recognize and take up antigens. Depending on the nature of the antigen (bacteria, fungus, virus, etc) and immunostimulatory adjuvant moieties present, the resulting epitopes are loaded onto MHC class I or class II molecules. Epitopes derived from pathogens that enter the endosomal pathway are mainly presented by MHC class II molecules. 8 Maturation signals induced by adjuvant molecules facilitate transport of antigen‐loaded MHC class II molecules onto the plasma membrane, where interaction with TCRs and costimulatory molecules results in activation of CD4+ T‐helper cells. 9 T‐helper lymphocytes were initially subdivided into two main groups with counter‐regulatory functions depending on the cytokine pattern: a Th1 subset, which participates in cell‐mediated immunity and is associated with IFN‐γ secretion; and a Th2 subset, which was associated with enhanced proliferation of activated B cells, promoting differentiation toward plasma cells, enhanced expression of MHC class II molecules, and isotype class switching by secreting several cytokines such as IL‐4 and IL‐13. 10 This dichotomy became outdated following the description of other CD4+ T‐helper populations including Th9, Th17, and Th22 cells, regulatory T cells (TREG), and T follicular helper (TFH) cells. 11
In terms of therapeutic vaccines for treating autoimmune and inflammatory conditions and maintaining homeostasis and self‐tolerance, TREG play a central role. 12 , 13 , 14 TREG constitute 5% of circulating T cells and are characterized by expression of the transcription factor Foxp3. 15 In addition to these “natural” TREG, a population of inducible type 1 TREG cells (Tr1 cells) is characterized by the secretion of IL‐10 and expression of CD49b and lymphocyte activation gene 3 protein (LAG3). 16
Of particular relevance to vaccine‐induced antibody responses, are the follicular TFH cells, which are specialized in providing T cell help to B cells. 17 , 18 Their specific location in the lymph node B cell follicle and cellular interactions allows them to play a key role in the induction and regulation of antibody production 19 and B cell memory responses. This T cell–dependent B cell activation enables antibody class switching and elicits more robust responses and higher‐affinity antibodies than the T‐independent activation, highlighting the importance of optimal targeting of DCs and the subsequent instruction of CD4+ T cells in the adaptive response to a vaccine. These interactions and potential adjuvant interventions are highlighted in Figure 2. Differentiation of TFH cells is mediated by STAT‐3 (signal transducer and activator of transcription 3), activating cytokines, secreted from DCs (IL‐6, IL‐12 and IL‐27), B cells (IL‐6 and probably IL‐27), and CD4 + T cells (IL‐21) although there are significant differences in cytokine requirements between mice and humans. 17 The cytokines function alone or together to induce or increase the expression of transcription factors BCL 6, MAF, BATF (basic leucine zipper transcriptional factor ATF‐like), and IRF4 (interferon‐regulatory factor 4), which then causes transcription of CXCR5, ICOS (inducible T cell costimulator), IL21, and PD‐1 (programmed cell death protein 1). CD28‐CD86, CD40 ligand (CD40L)‐CD40, and ICOS‐ICOS ligand (ICOSL) interactions are central to differentiation of TFH cells.
FIGURE 2.
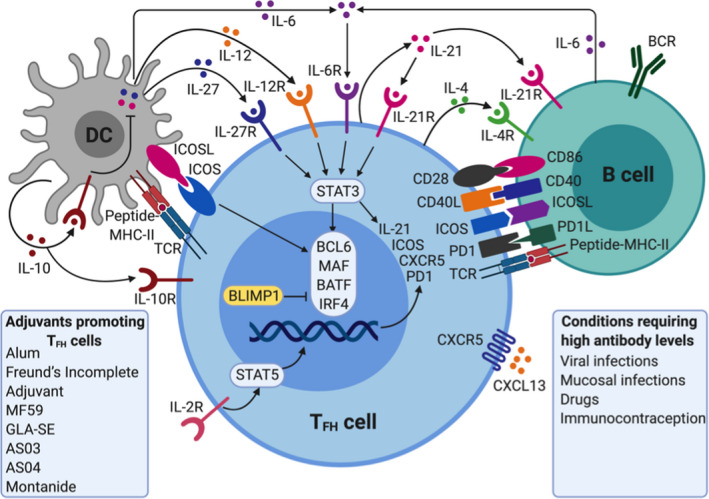
Adjuvants activate dendritic cells (DCs) to enhance generation of TFH cells
Vaccines composed of molecules with highly repetitive domains, such as polysaccharides, predominantly elicit T‐independent B cell responses, while peptide in protein‐based antigens are necessary to provide the T cell epitopes in T‐dependent B cell responses. With a view toward rational vaccine design, it is important to note that pathogen‐derived antigens often induce both pathways and these can be complementary. In addition, whereas whole pathogens possess a mixture of PAMPs that serve as adjuvants and promote DC and B cell activation, immunostimulatory adjuvants are essential to facilitate the induction of effector responses in the case of subunit vaccines. 20 Although an increasing number of different antibody‐inducing adjuvants are included in prophylactic vaccines, including mainly aluminum salts (eg, Alhydrogel®, AS04™, and Adjuphos®) and squalene emulsions (MF59™ and AS03™), only aluminum salts are currently used in licensed therapeutic vaccines. Aluminum hydroxide is thus added to several allergy vaccines, including various pollens, animal hair/dandruff, insect bites, and house dustmites, to increase antibody responses.
2. THERAPEUTIC VACCINES MAINLY INDUCING ANTIBODY‐BASED IMMUNE RESPONSES
2.1. Allergy vaccines
Allergen immunotherapy (AIT) can provide a safe and effective treatment for allergic diseases. There are currently two types of AIT in clinical practice: subcutaneous (SCIT) and sublingual (SLIT) immunotherapy. The clinical effect of both SCIT and SLIT has been well demonstrated in several randomized, double‐blind, placebo‐controlled clinical trials, showing significant reductions in symptoms and medication scores. 21 The principle behind AIT is to expose the allergic individual to the specific allergen(s), leading to immunological tolerance or altered antibody responses with a lasting effect, even after termination of the treatment. 22 , 23
Adjuvants and adjunct therapies have, in the context of AIT, been investigated since the 1990s, with the aim to either increase the efficacy, lower the adverse events, or shorten the treatment. So far, we have not seen a major breakthrough, but several clinical trials are still aiming to change this picture (Table 1). As previously mentioned, aluminum hydroxide (alum) has been the adjuvant of choice in SCIT. The effect of alum can partly be attributed to its adsorptive properties, which leads to increased immunogenicity due to the slow and sustained allergen release from the alum particles 24 (depot effect). Importantly, allergens strongly adsorbed to alum particles are less accessible to the immune system, which has been shown to decrease local side effects. 25 A number of pathways have been implicated in alum adjuvanticity, including DNA release, 26 prostaglandin E2, and cholesterol interaction. 27 The mineral salt, calcium phosphate, and the natural amino acid L‐tyrosine are also used as depot adjuvants in SCIT. Both of these adjuvants enhance IgG production with a limited increase of IgE. 28
TABLE 1.
Adjuvants used in clinical trials against allergy
| Adjuvant | Delivery system | Immunostimulator | Receptor | Disease | NCT number |
|---|---|---|---|---|---|
| Al(OH)3 | ‐ | Multiple theories | ‐ | Grass pollen allergy | 01538979 |
| MPLA | ‐ | MPLA | TLR4 | Tree pollen allergy | 00118625 |
| GLA | ‐ | TLR4 agonist | TLR4 | peanut | 03463135 |
The table displays adjuvants, which have been or are currently used in clinical trials of allergy. Clinical trials in which the adjuvants have been used are identified by the Clinical Trials gov. identifier number (NCT). Numbers are examples, and only one number is given although some adjuvants are used in several clinical trials.
More recently, Toll‐like receptor (TLR) ligands have been applied as adjuvants in SCIT. One example is monophosphoryl lipid A (MPL), which is a non‐toxic derivative of lipopolysaccharide (LPS) which interacts with TLR4 on innate immune cells and can promote Th1 responses. 29 In combination with L‐tyrosine, MPL is being investigated in phase III clinical studies as an adjuvant in short‐course SCIT treatment of pollen‐induced allergies. However, the most recent of these studies, treating birch pollen allergics, failed to meet the primary endpoint of reducing the combined symptom and medication score (Study B301). Glucopyranosyl lipid A (GLA) is similarly a TLR4 ligand, inducing a Th1 response by activating dendritic cells. 30 This adjuvant is currently being evaluated in a clinical study with SLIT treatment of peanut allergic patients (NCT03463135). Attempts have been made to improve AIT using adjuvants targeting TLRs other than TLR4. CpG oligodeoxynucleotides (CpG‐ODN) resemble bacterial DNA and bind to the endosomal receptor TLR9. In animal models of asthma, it has been shown that CpG‐ODN downregulate the allergic Th2 response and induce a Th1 and T‐regulatory response. 31 This concept was tested by SCIT treatment of ragweed allergic patients where the antigen Amb a 1 was conjugated to a CpG‐ODN. Initial results from a phase II study showed promising results, but in a later phase IIb study, the effect was not different from placebo. 28
Nanoscale adjuvants, including virus‐like particles (VLP), have also been tested for efficacy in SCIT. VLPs are virus‐shaped particles made of coat proteins or capsids from viruses or bacteriophages. These particles are highly immunogenic and linking allergens to their surface enhances their immunogenicity. However, it has been demonstrated that VLP bound allergen has a strongly impaired ability to bind to surface bound IgE and to induce mast cell degranulation. 32 The enhanced immunogenicity was demonstrated in a small clinical trial, where high titers of specific antibodies were observed after immunization with the antigen Der p 1 conjugated to VLP. 33 Furthermore, VLP combined with CpG‐ODN has been tested as a SCIT adjuvant in clinical trials, where it showed reduction of clinical symptoms in house‐dust mite allergic patients. 34 Interestingly, this reduction could be seen both with and without coupling of allergen to the VLPs. 35
An alternative to adjuvants is co‐administration of adjunct biopharmaceuticals. In several studies, AIT has been combined with omalizumab (anti‐IgE antibody) for the treatment of allergic rhinitis or asthma, resulting in fewer side effects compared to AIT alone. 36 In food allergy, it has been shown that addition of omalizumab to oral immunotherapy was beneficial effect in terms of adverse events, but had no effect on tolerance induction or sustained responses. 36 Dupilumab, an anti‐IL‐4 receptor alpha‐specific antibody, is currently being investigated as adjunct therapy to peanut AIT. Mouse studies have indicated that peanut AIT and anti‐IL‐4R antibody treatment show a synergistic treatment effect.
2.2. Vaccines for substance use disorders
Addictive drugs are a chemically heterogeneous group of small molecules that are able to pass the blood‐brain barrier (BBB) and target the distinct neurotransmitter systems in the brain. Vaccines have the potential to induce anti‐drug antibodies that cannot cross the BBB but can potentially bind to the drug and inhibit its transport to the brain, without altering brain function. Because the addictive drugs are small non‐immunogenic molecules, a hapten‐carrier approach is used which requires conjugation to a carrier protein to enhance the induction of drug‐specific antibodies. For example, the key components of a conjugate nicotine vaccine are: a B cell epitope, in this case nicotine; a T cell epitope, provided by the carrier protein as used in polysaccharide conjugate vaccines for capsulated bacterial pathogens and an adjuvant that enhances vaccine immunogenicity. 37 Although the vaccine principle is the same for all drugs, when designing the hapten vaccine, it is important to consider drug properties such as size, chemical structure, metabolism, and biodistribution. 38 The efficacy of induced antibodies will depend on a number of variables including the carrier, hapten density, aggregates and adducts, and a choice of adjuvant. 38 , 39 , 40 , 41 , 42 , 43
Nicotine, a psychostimulant, is the main addictive component in tobacco products. Nicotine‐targeting vaccines have been the best studied of all addictive drug vaccines, with six candidates reaching clinical trials. 44 NicVax is the first‐generation anti‐nicotine vaccine that has demonstrated promising results in phase 2 trials, but has failed to show improvement in smoking cessation compared to placebo in phase III trials. 45 , 46 , 47 Esterlis et al 48 have shown that the vaccine resulted in only a 12.5% decrease of nicotine concentration in the brain. NicVAX consists of a hapten, 3’aminomethylnicotine, conjugated to the exoprotein A from Pseudomonas aeruginosa and alum as an adjuvant. 44
Improved responses have been seen with new hapten designs involving conjugation to cross‐reactive material 197 (CRM197), a non‐toxic derivative of diphtheria toxin (DT), and addition of CpG adjuvant (TLR agonist) in addition to alum. 41 , 49 , 50 , 51 Indeed, this formulation induced higher titers of nicotine‐binding antibodies in rats and non‐human primates (NHPs), and the study showed that a combination of alum and CpG adjuvants can enhance both the antibody titer and affinity. 49 Because of the positive preclinical results, the vaccine (NIC7‐001) is currently being tested in a phase I clinical study; however, the results are not yet available. The N4N vaccine is another second‐generation vaccine that has shown promise for nicotine vaccination. 52 The N4N hapten is a covalent modification of pyridine and has much higher nicotine affinity than 3’aminomethylnicotine from the NicVax vaccine. The N4N hapten is conjugated to flagellin but has not yet been tested clinically.
A different vaccine approach for inducing drug‐specific antibody responses involves particle‐based vaccines, which are built from either polymers, liposomes, peptides, virus‐like particles, or other combinations. 53 , 54 , 55 These self‐assembling particle vaccines are anticipated to enhance the activation of antigen‐presenting cells (APC), to promote stronger T‐helper cell responses, and to stimulate the differentiation of memory B cells. 56 , 57 Additionally, the hapten load can be controlled and the delivery of adjuvants and other immunomodulators to APCs made more efficient. 42 The nanoparticle‐based vaccine SEL‐068 from Selecta Bioscience consists of nicotine bound to the surface of polymers, a synthetic TLR ligand, and a T‐cell helper peptide. In preclinical studies in non‐human primates, the vaccine blocked the development of nicotine discrimination, a behavioral experimental procedure to test the effect of nicotine. 58 The Selecta group showed that codelivery of an antigen with a TLR7/8 or TLR9 agonist in synthetic polymer nanoparticles increased drug immunogenicity with minimal systemic production of inflammatory cytokines. 59 SEL‐068 is currently being evaluated in phase 1 clinical trials.
Another particle‐like vaccine in preclinical studies incorporates a synthesized short trimeric coiled‐coil peptide (TCC) that creates a series of B and T cell epitopes with uniform stoichiometry and high density. 60 Vaccination with this antigen and alum and a TLR4 agonist (GLA‐SE) could prevent 90% of a nicotine dose equivalent to three smoked cigarettes from reaching the brain. The TLR4‐based adjuvant, as a potent stimulator of T cell–mediated antibody responses, has shown superiority compared to alum, with higher antibody titers and improved antibody affinities.
More recently, a hybrid nanoparticle‐based nicotine vaccine (NanoNiccine) has been developed with an aim to improve specificity and induce more sustained responses. 61 NanoNiccine is composed of a poly(lactide‐co‐glycolide) acid (PLGA) core, keyhole limpet hemocyanin (KLH) as an adjuvant protein enclosed within the PLGA core, a lipid layer, and nicotine haptens conjugated to the outer surface of the lipid layer. The vaccine showed superior immunogenicity compared to traditional nicotine‐protein conjugate vaccines. The particles were efficiently taken up by dendritic cells, and the principal adaptive immune response detected was the induction of antigen‐specific IgG antibodies. The same group have demonstrated that the immunogenicity of the NanoNiccine vaccine can be further improved by modulating factors such as particle size, 62 , 63 hapten localization, and density, 62 , 63 combinations of adjuvants, 64 , 65 conjugation of potent carrier proteins, 64 , 65 and degree of pegylation. 66 In addition to the great potential of novel adjuvant approaches to enhance the magnitude and quality of the anti‐drug antibody response, the nature of the hapten used and the degree to which it can trigger high affinity anti‐drug antibodies is pivotal. However, detailed discussion of this topic is outside the scope of this review.
2.3. Vaccines against chemical hazards
DDT (1,1,1‐trichloro‐2,2‐bis (p‐chlorophenyl) ethane) has been used widely as a pest control agent but was banned in 1970 because of its harmful effects on wildlife and human health. 67 This compound is therefore a chemical hazard for both human health and the environment. The biodegradation of DDT is very slow, 68 and in animals and humans, the DDT accumulates and is stored in adipose tissue. 69 When an organism is a part of the food chain, a biomagnification occurs, where the hazard is accumulated. 70 Although the liver is able to transform some of the DDT to DDE or DDD, 71 these degradation compounds are not eliminated, but are stored even more avidly. 72
Research has been conducted into the potential of the immune system to eliminate DDT following vaccination. The toxin DDT was made immunogenic by conjugating it with keyhole limpet hemocyanine (DDT‐KLH). Mice were immunized subcutaneously using aluminum hydroxide adsorbed DDT‐KLH conjugate, where the second group only received KLH adsorbed to aluminum hydroxide, and the third group was used as a control to provide information on DDT levels in serum in untreated animals. The mice were then fed with chow containing 40 mg/kg of DDT for 45 days. 73 The concentration of DDT and its metabolites (DDE and DDD) was analyzed in various tissues, and DDT‐specific antibody titers were determined. Higher antibody responses were detected in mice vaccinated with the DDT‐KLH conjugate, and DDT, DDE, and DDD levels in adipose tissue, blood, brain, and spleen were significantly reduced, compared to the group that received native unconjugated DDT. 73 This demonstrates that immunization against a chemical hazard may be used to treat animals or humans exposed to high levels of such toxic compounds, as well as prevent them from having these compounds accumulating in their body.
2.4. Alzheimer's disease vaccines
Alzheimer's disease (AD) is a progressive neurodegenerative disorder, characterized by plaques of misfolded amyloid β (Aβ) and tau protein aggregates in neural tissue, and cerebrovascular dysfunction resulting from damaged small blood vessels in the brain, 74 which eventually leads to dementia in the elderly population. Currently, it is considered an incurable disorder with limited treatment options. The mechanism(s) underlying the cognitive decline in Alzheimer's disease still has not been clearly unraveled, which makes it difficult to judge whether current therapies target the symptoms, rather than the key molecule(s) causing the pathology. In AD transgenic animal models, immunotherapeutic targeting of B cell epitopes of (misfolded) Aβ and/or tau seemed a most logical strategy to target neural plaques or vascular aggregates. Indeed, both vaccines, and passive antibody‐based interventions, resulted in a clear reduction of Aβ pathology and cognitive benefits, with limited local inflammatory adverse effects. However, despite these promising preclinical results, such approaches in human studies showed limited evidence of significant clinical benefits, even despite the postmortem observed clearance of amyloid pathology, and acceptable tolerability. 75 , 76 , 77
Several redesigned vaccines against the (modified) variants of soluble or aggregated Aβ and tau protein (with or without protein carrier Qβ or KLH, lacking common T cell epitopes) formulated with saponin QS‐21, liposomes plus TLR4 agonist, or KLH‐alum, as immune modulating adjuvants, are being evaluated in early‐stage trials.
2.5. Atherosclerosis vaccines
Accumulation of leukocytes in the intima of the arterial wall and subsequent uptake of lipids in the form of low‐density lipoprotein (LDL) by macrophages initiate the formation of atherosclerotic lesions in large and medium sized arteries. As lesions grow, they become increasingly unstable, ultimately causing them to rupture, which can result in thrombotic occlusion of blood vessels. As such, atherosclerosis is the major underlying pathology of cardiovascular events like myocardial infarction or stroke and is the leading cause of death in the Western world. Although current treatment strategies focus on controlling LDL levels (eg, by statins, PCSK9 inhibitors), immunomodulatory interventions, including vaccination, are currently in preclinical and clinical investigation.
Development of atherosclerotic plaques is driven by an inability to clear LDL from the lesion, resulting in a chronic inflammatory reaction involving both innate and adaptive immunity, and therefore, LDL has been the most intensively studied antigen. Indeed, LDL‐specific T cells have been identified in atherosclerotic lesions 78 and circulating auto‐antibodies against LDL or modified LDL can be readily detected in most individuals. 79 Interestingly, induction of anti‐LDL antibodies by immunizing rabbits with chemically modified LDL particles suspended in the water‐in‐oil emulsion, Complete Freund's adjuvant (CFA), or AdjuPrime had a protective effect with less prominent atherosclerotic lesions in the vaccinated animals. 80 , 81 Similarly, vaccination with peptides derived from the largest protein in LDL, ApoB100, formulated with aluminum phosphate and cationized BSA, or CFA, reduced atherosclerosis in mice. 82 , 83
2.6. Cancer vaccines against tumor‐associated antigens (TAAs)
Antibodies targeting tumor‐associated cell surface antigens have been shown to be able to eliminate circulating tumor cells, 84 and vaccine‐induced antibodies against these antigens, especially carbohydrates, have correlated with improved prognosis in clinical settings. 85 Thus, tumor‐associated carbohydrate antigens, (TACAs), which are aberrantly expressed at the surface of tumor cells compared with normal cells, either as glycolipids (GM2, GD2, GD3, Globo H) or as mucin glycoproteins (Tn, TF, STn, MUC1), 86 have become important targets for antibody recognition and immune attack against cancers (Figure 3, Table 2). A relationship exists between tumor type and expression of these abnormal TACAs. 87 Since the first pioneering hypotheses on the cell biological significance of aberrant glycosylation appeared in 1985, 88 efforts focused on the development of anti‐TACA‐based cancer vaccines have expanded. 85 , 89 , 90 , 91 Despite these efforts, however, no anti‐TACA cancer vaccine has been approved by the FDA. The failures experienced by the TACA‐based vaccines that entered clinical trials can be ascribed to several factors, being the most compelling one that carbohydrate antigens are strictly B cell epitopes and induce a T cell–independent humoral response. Moreover, it should be considered that targeting TAAs with antibody‐based immune responses may not target variant tumor cells expressing different TAAs in case of tumor heterogeneity, resulting in tumor immune escape from vaccine‐induced immune reactions. 92
FIGURE 3.
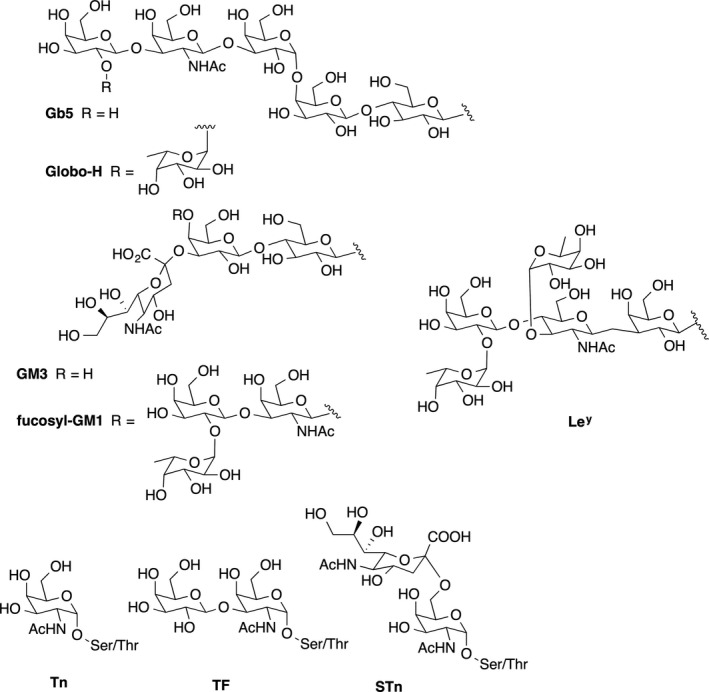
Examples of major Tumor‐associated carbohydrate antigens (TACAs)
TABLE 2.
Antigens targeted in therapeutic antibody‐inducing cancer vaccines
| Antigens | Tumors |
|---|---|
| GM2, globo H, sTn, TF, Ley | Ovary |
| GM2, Tn, sTn, TF, Ley | Prostate |
| GM2, globo H, Tn, sTn, TF, Ley | Breast |
| GM2, fucosyl GM1, polysialic acid, globo H, sialyl Lea | Small‐cell lung cancer |
| GM2, GD2, GD3 | Sarcoma |
| GM2, GD2, GD3, polysialic acid | Neuroblastoma |
| GM2, GD2, GD3 | Melanoma |
Vaccine‐elicited antibodies against TAAs can mediate elimination of tumor cells by complement‐dependent cytotoxicity (CDC) and/or antibody‐dependent cellular cytotoxicity (ADCC). Another important function of these antibodies is opsonization of tumor cells to promote their uptake by antigen‐presenting cells. The basis for vaccines including this type of TAA is the correlation between overall antibody titer against tumor cells and the CDC or ADCC effector mechanisms. Antibody‐inducing vaccines in mice and cancer patients have included cell surface carbohydrate antigens conjugated to carrier proteins such as KLH or bovine serum albumin (BSA), co‐administered with a variety of distinct types of adjuvant, for example the natural saponin adjuvant QS‐21, the semisynthetic saponin adjuvant mixture GPI‐0100, β‐glucan, or the nanoparticulate product GlycoVaxGM3 with Montanide ISA 51 (Table 3). 93 Some other glycoprotein antigens such as MUC1 and KSA in epithelial cancers, PSMA in prostate cancers, and MUC16 in ovarian cancer are also abundantly expressed on other types of cancers and have also been a target for antibody‐inducing vaccines.
TABLE 3.
Adjuvants used in cancer vaccines
| Adjuvant | Delivery system | Immuno‐stimulator | Receptor | Type of cancer | Antigen a | NCT number b |
|---|---|---|---|---|---|---|
| Montanide ISA51 | W/O emulsion | ‐ | ‐ |
1.CNS tumors 2.Breast 3.Leukemia |
1.TAA peptide mix 2.P10s‐PADRE 3.PR1 peptide |
1.00935545 2.01390064 3.00004918 |
| Hiltonol | ‐ | Poly:IC‐LC |
TLR3/ RIG‐I |
1.Solid tumors 2.Lung 3.Melanoma |
1.Neo‐antigen 2.MUC‐1 3.pBCAR3 peptide |
1.02721043 2.01720836 3.01846143 |
| AS15 | Neutral liposomes | CpG/MPLA/QS‐21 | TLR9/4 |
1.Melanoma 2.Breast 3.Lung |
1.MAGE‐A3 2,MAG‐Tn3 3.PRAME |
1.01425749 2.02364492 3.01853878 |
| CAF09b | Cationic liposomes |
MMG/ Poly:IC |
Mincle/ TLR3/RIG‐I |
1.Solid tumors 2.Prostate |
1.Neo‐antigen 2.Bcl‐XL |
1.03715985 2.03412786 |
| ISA51 + Hiltonol | W/O emulsion | Poly:IC‐LC |
TLR3/ RIG‐I |
1.Melanoma 2.AML 3.Glioma |
1.NY‐ESO‐1 2.WT1 peptide 3.GAA/TT‐peptide |
1.01079741 2.01842139 3.00795457 |
| ISA51 + CpG7909 | W/O emulsion | CpG | TLR9 |
1:Lung 2:esophageal 3:Melonma |
1:NY‐ESO‐1b 2:URLC10‐177 3: MAGE‐3.1 |
1:00199836 2:00669292 3:00085189 |
| ISA51 + Aldara | W/O emulsion | Imiquimod | TLR7/8 |
1:Melanoma 2:Prostate |
1:gp100 2:Pros.spec. peptide |
1:00273910 2:02452307 |
| Detox B | Oil droplet emulsion | MPL/mb. cell wall | TLR4+? |
1:Solid tumors 2:Breast |
1:ras peptide 2:THERATOPE |
1:00019006 2:00003638 |
| QS‐21 | ‐ | QS‐21 | Unknown |
1:Ovarian 2:Breast 3:Prostate |
1:Globo H‐GM2 2:sialyl Lewisª 3:MUC‐2‐KLH |
1:01248273 2:00470574 3:00004929 |
| Iscomatrix | ISCOMs | QuilA | Unknown | Melanoma | NY‐ESO‐1 | 00199901 |
| Resiquimod | ‐ | Resiquimod | TLR7/8 |
1:Melanoma 2:Bladder 3:Glioma |
1:CDX‐1401 2:CDX‐1307 3:Tumor lysate |
1:00948961 2:01094496 3:01204684 |
| Resiquimod + Hiltonol | ‐ |
Resiquimod/ Poly:IC‐LC |
TLR7/8/ TLR3/RIG‐I |
Melanoma | CDX‐1401 | 00948961 |
| Sargramostim | ‐ | GM‐CSF | GM‐CSFR |
1:Breast 2:Lung 3:Ovarian |
1:HER2 Peptide 2: ras peptide 3:ALVAC‐NY‐ESO‐1 |
1:00003002 2:00005630 3:00803569 |
| GLA‐SE | ‐ | GLA | TLR4 |
1:Melanoma 2:Ovarian 3:Lung |
1:MART‐1 2:IDC‐G305 3:CMB305 |
1:02320305 2:02015416 3:02387125 |
| ISA51 + GM‐CSF | W/O emulsion | GM‐CSF | GM‐CSFR |
1:Multiple Mye. 2:Leukemia 3:Melanoma |
1:SVN53‐67‐KLH 2:PR1 peptide 3:MART‐1 |
1:02334865 2:00004918 3:00031733 |
| Alhydrogel | ‐ | ‐ | ‐ | Prostate cancer | rsPSMA | 00705835 |
| Allostatine | ‐ | ? | ? | Various | ‐ | ‐ |
The table displays an overview of adjuvants, which have been or are currently used in clinical trials of cancer vaccines. The type of cancer, antigen type, and Clinical Trials gov. identifier number (NCT) is given.
AML, Acute Myeloid Leukemia.
For therapeutic vaccines containing multiple antigens, only one is displayed in the table.
Note that although some adjuvants are used in trials against several cancer types, only up to three trial (NCT) numbers are displayed.
Despite some promise, the clinical anti‐tumor effects of the antibodies induced by early formulations of carbohydrate‐protein conjugate vaccines were not significant, likely due to the advanced stages of disease and tumor immune escape. 94 These disappointing outcomes in terms of overall survival have driven the development of alternative synthetic carbohydrate vaccines, which have been shown to be immunogenic without the need for a conjugated protein carrier or an external adjuvant. Some of these new vaccine constructs have focused on glycosylated MUC1 B‐cell epitopes, which have elicited robust IgG antibody responses in MUC1.Tg mice that were able to kill MUC1‐bearing tumor cells by CDC and ADCC. 95 , 96 The antibodies showed selectivity toward human breast cancer tissues, and the vaccine candidates exhibited therapeutic anti‐tumor effects, paving the way for potential translation into the clinic.
In recent times, the development of TACA‐based vaccines has placed special emphasis on the impact of several factors on the immune response: that is, (a) antigen modification and density, (b) the carrier, and (c) the selected adjuvant. Different strategies have thus been proposed to induce improved anti‐TACA immune responses, spanning from synthetic modification of antigens to multiantigen vaccine constructs, relying on nanosized, peptidic, or protein carriers. 91 In Box 1, we summarize a number of cancer vaccine prototypes where the objective is targeting of TACAs with antibodies.
BOX 1. Cancer vaccine prototypes based on TACAs targeted by antibody induction.
Ley AND SLea‐BASED VACCINES
A Ley‐based vaccine, effective in preclinical trails, was generated by conjugating the synthetic pentasaccharide Ley to KLH and injected with QS‐21 as an adjuvant. Vaccination triggered IgM and, to a lesser extent, IgG antibodies and proved to be selectively toxic to Ley‐positive cells. 97 This potential vaccine entered a phase I clinical trial against ovarian cancer, but was discontinued because the antibodies induced were low affinity IgM molecules. The hexasaccharide SLea (CA19‐9) was also used as a TACA linked to KLH and co‐administered with QS‐21 or with the semisynthetic saponin adjuvant mixture GPI‐0100. In animal tests, vaccination with SLea‐KLH alone induced moderate antibody titers, which were increased by using GPI‐0100 as an adjuvant. The induced IgM and IgG antibodies mediated cytotoxicity against the SLea‐positive human adenocarcinoma cell line SW626. No cross‐reactivity to other or similar carbohydrate antigens (SLex, Lea or Ley) was detected. 98
GLOBO H‐BASED VACCINES
The first total synthesis of the Globo H hexasaccharide was reported in 1995. 99 The enzymatic synthesis of Globo H by large scale manufacturing of this demanding TACA enabled the development of Globo H‐based vaccines. A number of different carriers and adjuvants have been tested to enhance Globo H‐specific immune responses. Vaccination with the resulting Globo H‐KLH conjugate in combination with QS‐21 in a phase I clinical trial for breast cancer was well‐tolerated, and increased CDC and ADCC activity was observed in several patients. In a second‐generation vaccine in which Globo H was linked to the diphtheria toxin mutant CRM197 and used with the glycolipid adjuvant C34, a synthetic ligand for CD1d, improved immune responses were obtained. 100
GD2‐, GD3‐, GM3‐, AND FUC‐GM1‐BASED VACCINES
GD2 and GD3 are neuroblastoma TACAs that were used in a vaccine that entered a phase I clinical trial. 101 The two disialogangliosides, extracted from natural sources (rabbit brain and bovine buttermilk, respectively), were linked to KLH and administered with the QS‐21‐related saponin OPT‐821 or β‐glucan as adjuvants. The bivalent GD2/GD3 antigen/β‐glucan vaccine induced anti‐GD2 and anti‐GD3 antibody responses and was well tolerated, without major toxicity signs or induction of neuropathic pain.
The monosialoganglioside GM3 has also been considered as a TACA. However, a lactone metabolite of GM3 has been demonstrated to be a more discriminating antigen on cancer cells. Under physiological conditions, the expression of this lactone is below the recognition threshold. 102 Thus, GM3 has been the subject of extensive synthetic studies, and structural analogues have been proposed for the development of cancer vaccines. 103 , 104 , 105
The NeuGcGM3 ganglioside is a natural, GM3‐containing glycoantigen overexpressed on melanoma cells and found in other carcinomas, but poorly expressed in most normal human tissues. NeuGCGM3 was included in a proteoliposome of Neisseria meningitidis and was tested as a nanoparticulate product (GlycoVaxGM3) with Montanide ISA 51 as an adjuvant. Patients with melanoma 106 or breast cancer 107 treated with GlycoVaxGM3 developed IgM and IgG antibodies to NeuGcGM3 that could mediate complement‐dependent cytotoxicity (CDC) against P3X63 myeloma cell lines. Clinical trials with GlycoVaxGM3 are ongoing. 108
The fucosyl‐GM1 hexasaccharide is expressed on many small‐cell lung cancers but not on normal cells. The synthetic fucosyl‐GM1 linked to KLH was employed in combination with QS‐21 as an adjuvant. 109 Upon vaccination, an IgM antibody response against fucosyl‐GM1 and against tumor cells expressing fucosyl‐GM1 was elicited.
Tn‐, STn‐, TF‐, AND STF‐BASED VACCINES
The MUC1‐related Tn, TF, STn, and STF TACAs, expressed in more than 90% of primary adenocarcinomas, are considered pancarcinoma antigens and have been widely used to design cancer vaccines. They can be found attached through O‐glycosidic linkages to serine or threonine residues of tandem repeat peptide sequences. A synthetic, Tn/TF‐containing MUC1 peptide sequence conjugated to bovine serum albumin (BSA) has been developed as a vaccine candidate. A higher antibody response was obtained when a Tn or a TF residue was linked to a threonine residue (19). The same authors also synthesized the MUC1 tandem peptide glycosylated with STn and 2,6‐STF antigens at a serine residue (S15), maintaining Tn or TF antigens linked to threonine (T9). After conjugation to BSA, the vaccine candidates were co‐administered with complete Freund's adjuvant for the first inoculation and with incomplete Freund's adjuvant for each subsequent immunization. 110 These vaccines generated a strong antibody response, with elicitation of IgM antibodies and IgG1 as the predominant antibody isotype.
A tricomponent vaccine comprising the TLR2 ligand Pam3CysSK4 as an adjuvant, a T‐helper cell epitope peptide, and a Tn glycosylated MUC1‐derived peptide induced a potent humoral and cellular response, including CTL and ADCC‐mediating antibodies. This vaccine was able to generate a therapeutic response due to specific immunity against MUC1 and to a non‐specific anti‐tumor response elicited by the adjuvant. 95
Another example of self‐adjuvating potential cancer vaccine was developed by covalent conjugation of α‐galactosylceramide (αGalCer), a CD1d and invariant natural killer T (iNKT) cell ligand, with sialyl Tn (STn) TACA. This vaccine showed remarkable efficacy in inducing a strong STn‐specific IgG response. 111
In an effort to improve the multivalent presentation of TACAs as well as to improve anti‐glycan immunity, the formulation of Tn antigen conjugated to αGalCer into liposomes was recently published. 112 Liposomes containing 1,2‐diasteraroyl‐sn‐glycero‐3‐phoshpocholine (DSPC) and cholesterol were prepared by lipid extrusion and co‐formulated with Tn‐αGalCer glycoconjugate and αGalCer. The combination of DSPC and cholesterol for liposomal vaccines was known to induce a strong antibody (particularly IgG1) response in mice. 113
In another vaccine design, the Tn antigen was linked to virus‐like particles (VLP) of the bacteriophage Qβ, eliciting higher and more diverse antibody responses. 114 A four‐component self‐adjuvating multivalent cancer vaccine prototype was also proposed, relying on a cyclic peptide scaffold presenting four residues of Tn antigen and decorated with a B‐cell epitope. The multivalent scaffold also incorporated the peptidic Th epitope (PADRE), a CTL epitope from OVA, and the TLR2 ligand palmitic acid as an adjuvant. In mouse models, vaccination with this construct elicited IgG antibodies and offered protection against tumor growth. 115 A STn‐KLH glycoconjugate vaccine, THERATOPE®, reached phase III clinical trials, although with a poor clinical outcome. In the pilot study, the STn‐KLH vaccine was administered to patients with metastatic breast cancer pretreated with cyclophosphamide. All patients developed IgM and IgG antibodies that recognized both the synthetic and natural STn disaccharide. 116
Given the ability of certain zwitterionic polysaccharides (ZPSs) to evoke an MHC class‐II‐mediated T‐cell response, a new protein‐free, saccharide‐based vaccine construct was proposed consisting of Tn antigen chemically conjugated to PSA1 ZPS from Bacteroides fragilis. 117 This Tn‐PSA1 vaccine was administered to C57BL/6 mice in combination with TiterMaxGold as an adjuvant, generating high antibody titers specific to the Tn moiety. Subsequently, the same authors developed a related STn‐PSA1 vaccine candidate. In mice co‐administered with MPLA as an adjuvant, this vaccine elicited strong and functional humoral and cellular responses, with high titers of antibodies able to bind to and mediate CDC against STn‐expressing cancer cell lines. 118
Currently, the most intensively studied TACAs for the development of cancer vaccines are Lewis determinants (Lea, Ley, SLea) and glycans of the Globo class (Gb5, Globo H), which are overexpressed in a range of tumors, as well as carbohydrates present in gangliosides (GM2, GM3, GD2, fucosyl GM1) overexpressed in melanomas, lung, colon, prostate cancer, and mucin‐related TACAs (Tn TF, STn, STF) characterized by their rather simple structures and largely expressed in adenocarcinomas. Expression of these TACAs correlates with tumor malignancy.
3. THERAPEUTIC VACCINES MAINLY INDUCING ENHANCED OR MODIFIED CELLULAR IMMUNE RESPONSES
3.1. Cell‐based immune responses to intracellular targets
Cytotoxic CD8+ T lymphocytes (CTLs) play a crucial role in tumor control and chronic infections by intracellular pathogens. Indeed, they display a large spectrum of cytotoxic mediators in response to the specific recognition of tumor antigens presented in the context of the MHC class I complex on the surface of the target cell. CTLs mediate tumor or virus‐infected cell death by direct or indirect processes: (a) the release of lytic molecules, (b) the binding to pro‐apoptotic receptors expressed by tumor cells, (c) the secondary recruitment of effector cells, and (d) the increase of tumor cell recognition via the induction of MHC‐I molecule expression. Such highly cytotoxic capacities make their induction and infiltration into the tumor an attractive therapeutic strategy.
3.2. Therapeutic cancer vaccines
Anti‐tumor therapeutic vaccination should generate not only tumor‐specific CD8+ T cells but also tumor‐specific tissue‐resident memory T cells (Trm) at the tumor site. 119 Indeed, Trm are emerging as an essential actor in tissue immunosurveillance due to their specific expression of defined adhesion molecules (CD103 or CD49a integrins, chemokine receptors) facilitating their retention in tissues and consequently into the tumor. Their well‐positioned location in close contact with tumor cells and their higher cytotoxic capacities explain why their tumor infiltration is correlated with good clinical outcomes in many cancers. Accordingly, it appears that eliciting Trm represents a key target for the success of cancer vaccines.
The phenomenon of antigen spreading, which corresponds to the secondary CD8+ T cell response against antigen epitopes that are not present in the vaccine but likely result from local release after the first wave of tumor‐specific CD8+ T cells, provides additional evidence for the important role of these effector cells. 120 Indeed, in human clinical case studies, it was reported that tumor regression may be dependent on CD8+ T cells directed against tumor antigens not present in the vaccine, and thus, the induction of CD8+ T cells might be a suitable surrogate biomarker based on immune‐related response criteria to evaluate therapeutic vaccines. 121
One of the most critical issues for efficient priming of naïve CD8+ T cells relates to their contact with mature antigen‐presenting DCs. Some vaccine formulations including DNA‐, RNA‐, or DC‐based vaccines exhibit direct immunostimulatory properties and have been shown to elicit CD8+ T cells in mice and in humans. 122 However, some discrepancies between results obtained in mice and humans, especially for DNA vaccines, have to be addressed. 123 The plasmid‐based DNA vaccines were thus very efficient in many species including mice and non‐human primates, but early human studies failed due to low efficacy. The plasmid technology has greatly improved over the years, both when it comes to codon optimization increasing expression of the antigens, and plasmid delivery within the body. So, it will be interesting to follow these techniques in the future.
Immunostimulatory adjuvants such as TLR ligands, CD40 agonists, cytokines, or activators of stimulator of IFN genes (STING) are indicated to potentiate vaccine immunogenicity and to break self‐TAA tolerance. 124 Interestingly, some adjuvants can also preferentially polarize the immune cells to a Tc1‐type CD8+ T cell response. For example, vaccination with CpG and Poly I:C increased the ratio between effector CD8+ T cells and TREG. 125 , 126 Activators of STING delivered via a particle platform to reach the cytosol also favor the induction of CD8+ T cells. 127 To date, relatively few immunostimulants have been approved for human use, although several are under evaluation in clinical trials. 128
One of the most promising strategies for development of therapeutic vaccines against tumor targets uses recombinant, attenuated live vectors derived from viruses or bacteria as a vehicle for the tumor target antigen. Because of their intrinsic capacity to reach the cytosol of the cells, these recombinant pathogen‐derived delivery systems favor MHC class I‐restricted peptide presentation and generally induce a better CD8+ T cell response than free proteins or peptide vaccines. 129 In addition, they are highly immunogenic as they express PAMPs and can be engineered to also express activating molecules like cytokines (IL‐2) or immunomodulatory molecules (B7.1; or a triade of costimulatory molecules called TRICOM: B7.1, ICAM‐1, and LFA‐3) to further amplify the vaccine response. 130 However, it has to be considered that the efficacy of live vectors can be reduced after repeated vaccinations due to anti‐vector antibody responses. A diversified, prime‐pull strategy using distinct attenuated pathogens is required to overcome the neutralization of the vector bacteria or virus by host immunity. A recently described deep learning approach to neoantigen identification 131 offers promise for more rapid and targeted tumor antigen discovery, and novel adjuvant and delivery strategies are being evaluated. Several non‐replicating live vectors have been tested in clinical trials with minimal toxicity and the ability to generate antigen‐specific CD8+ T cells. 132 Adjuvant systems such as ISCOMS, saponins, emulsions, liposomes, virosomes, or nanoparticles (Table 3) can also favor cross‐presentation to CD8+ T cells in parallel with delivering immune signals. Allostatine is a short peptide derived from Alloferons, a group of naturally occurring peptides isolated from bacteria‐challenged larvae of the blow fly Calliphora vicina. These larvae are well known for medical use in wound healing: Napoleon's army was using them on wounded soldiers (a.k.a. “Surgical maggots”). Allostatine is a synthetic linear peptide having amino acid sequence HGVSGWGQHGTHG. It demonstrated strong adjuvant properties in a mouse P388/DBA2 tumor transplantation model when combined with a vaccine consisting of X‐ray inactivated tumor cells. 133 Although the vaccine alone demonstrated only weak tumoristatic effect in a quarter of the recipients, its combination with allostatine caused a tumoristatic effect in 65% of recipients and prevented tumor appearance in another 30% (the overall positive effect was 95%). The anti‐tumor effect of allostatine "alone" (tumor‐free animals two months after tumor implantation) has been detected in only 9% of the animals. Alloferons are antiviral and anti‐tumor peptides isolated from insects, exhibiting stimulatory effect on the cytotoxic activity of human and mouse NK cells. 134 , 135 The Ig site is an evolutionarily conserved pattern in all human and mouse immunoglobulins. Even very low concentration of allostatine in culture medium (at ng/ml scale) causes rearrangement of NK and T cell receptors, stimulation of NK cell cytotoxic activity against cancer cells, and an increased number of IFN‐γ and IL‐2‐producing cells. Allostatine is non‐toxic to immune cells, even at high dose, water soluble, stable in aqueous solutions and can be produced in large quantities at relatively low cost.
Short CD8+ peptides can bypass the need for cross‐presentation by directly binding to MHC‐I molecules on the surface of the APC, but they show low efficiency due to the lack of CD4+ T cell help, illustrating the importance of cross‐presentation. It is well‐established that CD4+ T cells and especially Th1 cells play a key role in promoting anti‐tumor cellular immunity, mediated via their “helper” function. 136 Activation of CD4+ T cells is essential for optimal CD8+ T cell priming, by enhancing antigen presentation and favoring cross‐priming of tumor antigen by DC 137 (see also Figure 4). CD4+ T cells also support recruitment of CD8+ T cells to the tumor site, maintenance, and expansion of a CD8+ T cell memory response. 138 Besides their “help” to CD8+ T cell responses, CD4+ T cells can mediate other anti‐tumor effects such as direct killing of tumor cells, recruitment toward and activation of innate immune cells (eg, NK cells or M1 macrophages) at the tumor site, and modulation of the tumor microenvironment by anti‐angiogenic effects. 139 , 140 CD4+ Th1 cells also promote vessel normalization leading to better intratumoral CD8+ T cell infiltration. 141
FIGURE 4.
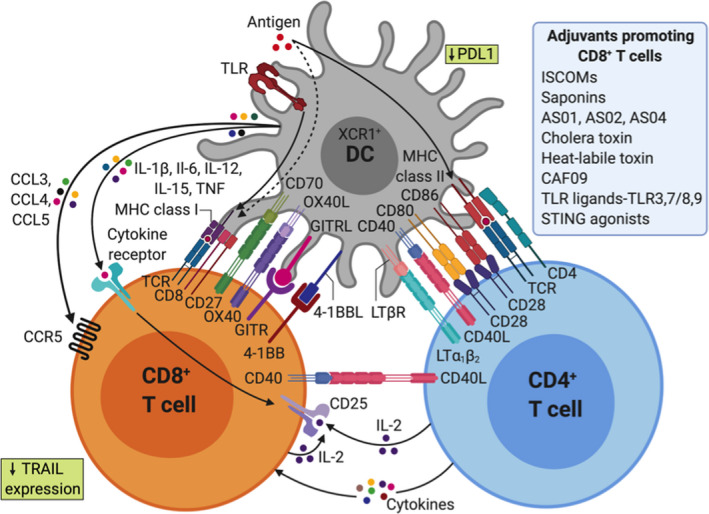
Adjuvants shape CD4+ and CD8+ T cell immunity through direct and indirect effects on dendritic cells (DCs)
Certain DC subpopulations, especially human CD141+ DC designated as cDC1, are thought to be specialized for cross‐presentation. 142 Recent studies revealed that antibody‐based targeting of antigens to specific DC receptors can enhance antigen uptake and anti‐tumor vaccine efficiency. 143 Alternative targeting strategies are also possible based on the fusion of tumor antigen(s) to a protein that can engage with receptors on the DC membrane in situ. For example, the group of Eric Tartour demonstrated that targeting DC in vivo with the B subunit of Shiga toxin (STxB), a non‐toxic vector, which binds to Gb3, preferentially expressed by DCs, significantly increased antigen‐specific CD8+ T cell responses when coupled to various antigens. 144 From this pioneering study, a number of cancer vaccines have been developed based on the targeting of surface molecules preferentially expressed on DC (DEC‐205, Clec9a/DNGR, XCR1, CD11c). 145 Antibodies against human DC receptors such as DEC‐205 and Clec9a coupled with model antigens reported an increase of cross‐presentation, resulting in enhanced induction of antigen‐specific CD8+ T cells. 146 Alternatively, tumor antigen may be formulated and administered ex vivo to DCs in the form of a cell therapy product. 147 However, the choice of the subpopulation of DC to be selected, the maturation signal to be used and the optimal formulation of antigen remain a matter of debate. 148 While this type of cancer vaccine has been shown to induce specific CD8+ T cells, it has had variable clinical impact, 149 although the approach remains under evaluation. In this context, specific adjuvants may have potential to optimally activate DC in vitro before adoptive transfer into patients.
The ideal setting for treatment with a therapeutic anti‐cancer vaccine is after surgical resection and/or chemo/radiotherapy, where the induction of anti‐tumor immunity can contribute to eliminate residual circulating cancer cells and micrometastasis. However, despite extensive efforts and promising preclinical studies, therapeutic cancer vaccines have shown limited objective tumor regression and therefore have mostly proven unsuccessful in the clinic. This may be explained by the state of immune suppression after chemotherapy and radiotherapy in the patient and the escape from immune surveillance of newly mutated tumor cells. These anti‐cancer vaccines have been hampered by poor immunogenicity and limited triggering of rare tumor‐specific immune cells, which makes the elicitation of both humoral and cellular immunity challenging and in most cases unsuccessful. 150
The range of tumor‐associated antigens recognized by T cells provides a variety of potential targets for cancer immunotherapy. In the case of TAAs targeted via cell‐mediated immunity, the activated T cells induced by vaccination recognize the MHC‐peptide complexes of the tumor antigen on the cell surface and not the surface protein itself. Thus, the optimal tumor‐associated antigens for triggering cellular immune responses are not necessarily cell surface proteins and can be classified in several categories 151 :
Cancer‐testis antigens—normally expressed in germ cells but aberrantly expressed in tumor cells: BAGE, GAGE, MAGE (in melanoma, bladder cancers), and NY‐ESO1 (in melanomas and ovarian cancers; antibody and T‐cell responses have been observed in patients).
Differentiation antigens—expressed on tumor cells and normal cells from the same tissue: Melan‐A/Mart‐1, gp100, tyrosinase (in melanomas), PSA (in prostate cancer), and CEA (colon).
Overexpressed antigens—expressed on normal cells at low levels, but at higher levels on tumor cells: HER2 (breast), hTERT, p53, survivin (melanoma), and MUC1 (breast, prostate, colon).
Tumor‐associated peptides antigens, in particular MHC class I‐restricted peptides derived from cancer‐testis and differentiation antigens, have been used in cancer vaccine clinical trials as a means to generate anti‐tumor T‐lymphocyte responses. These vaccines have generated some cytotoxic T‐lymphocyte (CTL) activity; however, the frequency and duration of these responses have been uniformly low and the clinical outcomes have been limited. 150 Accordingly, the design of therapeutic vaccines against cancer, especially for treating early disease, should not focus only on skewing a response to a single T‐helper cell type (for instance, Th1) or one effector mechanism (for instance, CTL). Rather, the ideal cancer vaccine should seek the simultaneous activation and synergy of both arms of the immune system to generate as many effector cells as possible to eradicate the tumor. Instead of using single peptide, protein, or carbohydrate antigens as TAA for cancer vaccines, cancer antigens prepared from surgically removed autologous or allogeneic tumor tissue may provide a source of TAAs with all, or many, of the required antigens in one preparation. 152 , 153
Despite extensive efforts and clinical trials with vaccines targeting TAAs, the clinical success of anti‐cancer vaccine therapies remains limited, with the DC‐based prostate cancer vaccine Sipuleucel‐T being the first and only human therapeutic cancer vaccine approved by the US Food and Drug Administration (FDA). 154 To advance clinical development of cancer vaccines, further efforts are required, not only to optimize and discover new tumor‐associated antigens, but also to identify and develop improved vaccine adjuvants, delivery systems, and formulations, building upon an increased understanding of the mechanisms underlying the induction of the required immune responses. Moreover, the recent success of checkpoint inhibitors has set the stage for combination strategies and opened the door to revisit cancer vaccine design. Taken together, progress on this front will enable realization of the full potential of a patient's immune response to target and combat cancer.
3.3. Atherosclerosis vaccines
Although promising results, as mentioned earlier in this paper, suggest an important role for LDL‐specific antibodies in clearing modified LDL particles and reducing atherosclerosis development; not all ApoB100 peptides elevate LDL‐specific antibody titers after vaccination. In fact, an increasing body of evidence points to a role for T cells in mediating the protective effect of LDL vaccination. Transfer of CD8+ T cells from alum‐ApoB100 peptide vaccinated mice reduced atherosclerosis in the recipient mice, suggesting that CD8+ T cells play a protective role, potentially by cytolysis of antigen‐presenting cells. However, more literature points to a crucial role of regulatory T cells and the induction of immunological tolerance toward LDL. Continuous exposure to ApoB100 peptides with an osmotic pump, mucosal vaccination with ApoB100 peptides covalently linked to cholera toxin B subunit, or oral vaccination with oxidized LDL induced IL‐10 producing regulatory T cells, which have previously been reported to be atheroprotective. These results strongly suggest that vaccine strategies should avoid overt inflammation and should aim at induction of regulatory T cells. Kobiyama and colleagues showed that the adjuvant, AddaVax (a squalene emulsion‐based adjuvant), can replace CFA and reduce atherosclerosis and that production of IL‐10 may be achieved by appropriate adjuvant selection. Specifically aiming at induction of regulatory T cells, Benne and colleagues showed that vaccination with tolerogenic liposomes containing ApoB100 peptides can reduce atherosclerosis and inflammation inside the lesion. Hence, currently it is expected that further development of adjuvants specifically designed for induction of tolerance or regulatory T cells may greatly benefit vaccination against atherosclerosis.
4. THERAPEUTIC VACCINES TO MODULATE CELLULAR IMMUNE RESPONSES FOR INFECTIOUS DISEASES
4.1. Therapeutic vaccines targeting human immunodeficiency virus
Human immunodeficiency virus (HIV) therapeutic vaccine candidates are designed to induce protective immune responses that control viral replication in the presence or absence of anti‐retroviral therapy (ART). To date, no candidate has proven successful in a controlled randomized trial to achieve long‐term HIV remission. 155 The low efficacy of many documented strategies appears to result from their inability to induce a broad immune response which suppresses the diverse escape variants that emerge during viral rebound and to purge the HIV reservoir. However, recently published combinatorial approaches demonstrated a high degree of efficacy following anti‐retroviral therapy interruption (ATI) with highly potent antiviral activity demonstrated in non‐human primates (NHP). 156 , 157
Most of the unsuccessful candidates were designed to stimulate HIV‐1 gag‐specific CD8+ T cell responses as this has been associated with virologic control in preclinical models and in long‐term non‐progressors (LTNP). For example, the use of a recombinant adenovirus serotype 5 (Ad5) expressing the Gag antigen in HIV‐positive patients under ART was unable to induce a strong T cell response and to control viral rebound after ATI (anti‐retroviral therapy interruption). 158 Also, the use of a MVA poxvirus (MVA.HIVconsv) expressing 14 conserved regions of HIV proved poorly immunogenic and did not impact the viral reservoir. 159 The use of such therapeutic vaccines may be more efficient when combined with drugs able to reactivate the HIV reservoir. Similarly, candidate vaccines designed to induce neutralizing Ab response have not been successful. In the TUTI‐16 trial, a therapeutic vaccine targeting a conserved Tat B‐cell epitope proved unable to block viral rebound after ATI. 160
Current therapeutic approaches aim to elicit a broader immune response to target escape variants which emerge during viral rebound. The use of autologous dendritic cells pulsed with heat‐inactivated HIV (DC‐HIV) in viremic untreated HIV+ patients induces broad T‐cell responses, resulting in a significant decrease in viral load. 161 Combination of the poxvirus‐based ALVAC‐HIV (expressing Env, Gag, Pol, and Nef) with Lipo‐6T (Nef, Gag, and Pol peptides combined with a tetanus toxoid peptide and a lipid tail), followed by interleukin‐2, significantly decreased viral rebound after ATI. 162 Polyfunctional T cell responses were identified as correlates of efficacy. Very recently, it has been confirmed that the addition of IL‐2 with a therapeutic vaccine could reverse T‐cell anergy and increase Ag‐specific T cell responses. 163 Combination of the DNA‐based vaccine GTUMultiHIV B‐clade with the lipopeptide vaccine Lipo‐5 as well as an MVA‐based vaccine is currently under investigation at the French National Institute for Health and Medical Research‐French National Agency for Research on AIDS and Viral Hepatitis (Inserm‐ANRS). 164
Hence, from the results obtained in the different randomized clinical trials, a broad immune response seems to be required to have an effect on viral variants appearing during the viral rebound. A simian immunodeficiency (SIV) vaccine, based on a rhesus cytomegalovirus vector (rhCMV) expressing Gag, has been described by the group of Picker. 165 This vaccine induced an effector memory T cell response that cured 50% of chronically infected monkeys. Follow‐up studies showed that rhCMV elicited an unexpectedly broad T cell response. 166 Unconventional T cell responses could also contribute to the therapeutic efficacy of rhCMV, with SIV‐specific CD8 T cells recognizing peptides in the context of non‐classical MHC‐E (HLA‐E) molecules. Despite the high efficacy of this strategy, moving this approach to a human trial remains a challenge as human CMV is pathogenic and also because it is unknown whether such non‐classical HIV‐specific CD8 T‐cell responses could be induced in humans. 167
Recent promising studies, described in non‐human primates on ART, also demonstrated the need for a combination of broad therapeutic vaccines with the activation of innate immunity. A combined approach with an adenovirus serotype 26 (Ad26) and MVA vectors (both expressing Env, Gag, and Pol), with or without a toll‐like receptor 7 (TLR7) agonist, in SIV‐infected monkeys on ART proved to be very efficient. 168 The Ad26/MVA therapeutic vaccine regimen strongly increased both the magnitude and the breadth of T and B cell response. Interestingly, only the combination of Ad26/MVA with a TLR7 agonist exhibited significant reduction in viral load and a delay of viral rebound. Notably, 33% of the monkeys showed a sustained undetectable viral load following ATI. Ad26/MVA vaccine is currently being tested in HIV+ patients who initiated ART during acute HIV infection in Thailand and in the setting of chronic HIV infection at the Beth Israel Deaconnes Medical center (BIDMC). The combined approach with the TLR7 agonist will be also tested in clinical trials. Alternatively, the combination of therapeutic vaccines with existing check point inhibitors, blocking for example PD1 or CTL4, could be very potent approaches. Currently, many promising randomized clinical trials have been designed on these concepts and are under investigation during ATI phases. Thus, therapeutic vaccines able to induce broad immune responses (polyfunctional T cells and broad neutralizing antibodies) carry the potential to strongly impact the viral rebound during ATI.
4.2. Therapeutic vaccines against tuberculosis
The only licensed vaccine for tuberculosis (TB) today is a prophylactic vaccine. Bacille Calmette Guérin (BCG), an attenuated strain of Mycobacterium bovis, is moderately effective in preventing TB in infants/young children, while being unreliable in preventing pulmonary TB in adolescents and adults. As of early 2018, there were 13 candidates in the clinical vaccine development pipeline, which can be classified into the following categories: (a) prophylactic pre‐exposure (priming) vaccines, administered to neonates prior to first exposure to M tuberculosis (Mtb); (b) prophylactic postexposure vaccines, given to adolescents/adults with latent TB infection having been previously immunized with BCG, and (c) therapeutic vaccines, intended for administration as an adjunct to conventional TB therapy, to a population at higher risk of developing recurrent disease. 169 The major goal of therapeutic TB vaccines is to boost the cellular immune response (Th1 type) against Mtb, to shorten the treatment and improve outcomes, such as reducing treatment failure and preventing recurrence, particularly in multi‐drug resistant TB. Several vaccine candidates are currently under development as therapeutic vaccines, including inactivated whole‐cell vaccines, which do not require co‐administration with an adjuvant, while the recently emerging subunit vaccines capable of acting as prophylactic as well as therapeutic vaccines are usually adjuvanted.
A heat‐inactivated whole‐cell vaccine derived from M vaccae (named Vaccae), in an injectable form, has already been approved in China for the adjunctive treatment of TB. 170 It should be noted that a comprehensive review of results obtained in phase II and III trials also supports the use of multiple oral doses of Vaccae (formulated as a tablet) as an adjunct to conventional TB therapy. It not only boosted the Th1 response but also inhibited the Th2 response, thus enhancing host defense against Mtb and demonstrating a marked therapeutic potential. 171 , 172 , 173 The TB vaccine, named RUTI, on the other hand, needs to be administered following previous anti‐TB drug therapy, as it is not able to directly reduce the bacterial load. 174 Interestingly, RUTI was the first therapeutic vaccine designed to target, and subsequently destroy, the non‐replicating Mtb bacilli via the induction of a Th1 poly‐antigenic cellular response. It is composed of detoxified liposomal cellular fragments of Mtb cultured under stress conditions mimicking those that exist inside granulomas. Under these conditions, latency antigens are induced, which are normally concealed from the immune system, thus facilitating the generation of immune responses against both non‐replicating and replicating Mtb. The results obtained in phase I and II clinical studies in HIV‐positive volunteers with latent infection demonstrated safety and immunogenicity, respectively. 175 Lastly, another inactivated non‐tuberculous mycobacterial vaccine based on M indicus pranii has also been studied in a phase II trial as an adjunct to TB therapy capable of inducing a Th1 response. 176
Subunit prophylactic/therapeutic vaccine candidates either contain plasmid DNA or fusion proteins presenting a combination of antigens covering both active and latent TB stages or usually require formulation with an adjuvant. 169 ID93/GLA‐SE is a vaccine candidate containing a fusion protein, specifically a recombinant antigen called ID93, integrating virulence‐associated proteins Rv2608, Rv3619, and Rv360 or latency‐associated protein Rv1813. This recombinant antigen is formulated with the synthetic Toll‐like receptor 4 (TLR‐4) agonist GLA, a glucopyranosyl lipid A in a stable nanoemulsion (GLA‐SE). 177 , 178 The latter allows for the induction of a robust Th1 response and has shown promising preclinical results, since it significantly enhanced the efficacy of anti‐tuberculous drugs in a mouse model. 179 , 180
A plasmid DNA vaccine encoding mycobacterial heat shock protein 65 (Hsp65) and interleukin‐12 (IL‐12), delivered by the hemagglutinating virus of Japan (HVJ) envelope (E) in liposomes, 181 induced protective as well as therapeutic efficacy against drug resistant TB in murine models in combination with anti‐tuberculous drugs. 182 , 183 The efficacy of this Hsp65 vaccine was further enhanced with other plasmid DNA‐based vaccines encoding the granulysin or Ksp37. 182 Ag85 complex proteins possessing mycolyl transferase activity are highly immunogenic and are secreted by all mycobacterium species. A vaccine candidate consisting of Ag85AB protein complex emulsified and adjuvanted with a TLR9 agonist (CpG‐ODN) and dimethyldioctadecylammonium bromide elicited a strong Th1 response when co‐administered with anti‐tuberculous drugs. 184 A similar therapeutic efficacy has been achieved by DNA vaccine candidates consisting of plasmid DNA encoding either Ag85A or Ag85B which enhanced the efficacy of concomitantly administered anti‐tuberculous drugs via the augmented secretion of Th1‐type cytokines and a suppressed release of Th2‐type cytokines in preclinical trials. 185 , 186 , 187 Recent data demonstrate that the adjuvanted subunit tuberculosis vaccine M72/AS01E is immunogenic in TB‐infected patients 188 offering promise for the development of a more effective therapeutic TB vaccine.
5. THERAPEUTIC VETERINARY VACCINES
While veterinary therapeutic vaccines have a rather long history, relatively few approaches have resulted in commercial products, and most of them originated from human medical platforms. Initial attempts focused on oncological diseases in production animals. In 1958, an autologous vaccine was developed for bovine papillomatosis, a viral disease of cattle characterized by the presence of mucosal and cutaneous neoplastic lesions. It was synthesized from formalin‐inactivated supernatants of affected tissue. 196 The autologous vaccination procedure reportedly induced remission of the skin lesions in diseased cattle. 196 , 197 However, the results were based on clinical observations, and experimental data are lacking from the literature. Recent publications showed high recovery rates, using therapeutic schemes related to human papilloma virus vaccines, but none of them reached the market (reviewed by 198).
The causative agent of bovine papillomatosis, the bovine papilloma virus, is also involved in the pathogenesis of equine sarcoid, the most frequent skin tumor in horses. 199 Several research groups investigated the efficacy of autologous vaccines against equine sarcoids. 200 , 201 , 202 According to these publications, complete tumor regression was noted in a high percentage of affected horses. However, the number of studied cases was limited, while other strategies, such as chemotherapy or surgical removal of the lesions, have been suggested as alternative treatments. 203 The current absence of commercially registered therapeutic tools favors the development of small private laboratories, which are specialized in the production of autologous vaccines, not only for equine sarcoids but also for other malignancies of companion animals.
In contrast to the human medical sector, the pharmaceutical industry seems hesitant to heavily invest in veterinary anti‐cancer immunotherapeutics, with the exception of the canine oral melanoma vaccine, Oncept (Merial, Duluth, GA, USA). Oncept, primarily based on studies for human vaccines, activates an immune response to tyrosinase, an enzyme overexpressed in melanoma tumor cells, which is usually ignored by the dog's immune system. Vaccination with Oncept triggers the immune responses toward a protein expressed by a xenogenic human tyrosinase gene, incorporated into a plasmid vector. 204 Clinical trials in dogs with surgically resected tumors revealed significantly prolonged survival times, 204 , 205 although in later studies a clear correlation between immunological parameters and loco‐regional cancer control could not be established. 206
The recently licensed Cytopoint (Cytopoint, Zoetis, USA), a monoclonal antibody‐based immunotherapeutic against atopic dermatitis in dogs, illustrates the growing demand for novel products in the field of companion animal welfare. Apart from animal health‐specific issues, effective therapeutic veterinary vaccines will have a strong impact on public health through reductions in the transmission rates of potential zoonoses. For example, a vaccine against dermatophytosis (Insol Dermatophyton, Boehringer Ingelheim, Germany) also known as ringworm, a disease that can be transmitted between animals and humans, has been approved in many European countries for therapeutic or prophylactic use in dogs, cats, and horses. Insol dermatophyton is reported to accelerate the healing of skin damage caused by the fungus. A similar therapeutic effect was demonstrated particularly for cats, after administration of an inactivated vaccine containing strains of Microsporum canis, Microsporum gypseum, and Trichophyton mentagrophytes. 207
Since the field of animal vaccinology has more flexible regulatory and preclinical trial requirements, significant knowledge regarding the safety and the effectiveness of the vaccine in the target animal can be achieved relatively quickly. In addition, insights into adjuvant mechanism of action in this context could then inform studies on other vaccines with benefits for both animal and human indications. However currently, details regarding the immunomodulatory adjuvant components in these vaccines are often not publicly available, being protected as proprietary information by the vaccine manufacturer.
6. FUTURE PERSPECTIVES
Since a majority of chronic diseases affecting humans and animals have an underlying immunological basis, therapeutic vaccine approaches can be envisaged to intervene, to eliminate or reduce disease symptoms. As a result, therapeutic vaccination has great potential to address a range of infectious and non‐infectious conditions. However, there are many challenges in comparison with prophylactic vaccination including disease heterogeneity, a requirement to block or divert ongoing immune responses, and a lack of clarity on the optimal antigens.
Progress in this field will depend on developing novel adjuvant approaches and appropriate tailoring of adjuvants to achieve a specific goal. Given the enormous diversity in requirements from driving potent tumor‐specific T cells in the case of cancer to a dominant Treg response for autoimmune diseases, the types of adjuvant required will be diverse and it would be a major step forward if a panel of clinically acceptable adjuvants that can achieve these diverse outcomes were made available. While there are very promising advances in antigen discovery, exciting recent developments, particularly in situ vaccination for cancer, 208 offer the potential for adjuvant strategies to act as therapeutics without the challenge of identifying and incorporating specific antigens. Related to this, innate immune training presents strategies to modulate innate immunity to drive effector responses or reduce inflammation in an antigen‐independent manner and strategies toward identifying the optimal adjuvants to achieve this are being evaluated. 209
In the case of cancer (and autoimmune diseases), it is likely that vaccination approaches will be used in parallel with other therapeutic strategies. In particular, for cancer vaccines, optimal combinations of therapeutic vaccines and checkpoint inhibitors are likely to be more effective than the vaccines alone. In the case of autoimmune diseases, therapeutic vaccines that reduce inflammatory T cell responses could be combined with anti‐cytokine therapies to bolster anti‐inflammatory responses and reduce pathology. Overall, although this is a challenging field, the future appears bright and there will be a major role for adjuvant researchers to facilitate the generation of innovative new therapeutic vaccine strategies.
CONFLICT OF INTEREST
Jens Brimnes is an employee at ALK‐Abello A/S, a company that manufactures and sells allergy immunotherapy (AIT) products. Sergey Chernish is the owner of Alopharm, a company producing Allostatines. Virgil Schijns is shareholder and Chief Scientific Officer at Epitopoietic Research Corporation (ERC), a company producing an immunotherapy against glioma brain cancer. All other authors declare no competing interest.
ACKNOWLEDGEMENT
This article/publication is based upon work from COST Action CA16231 ENOVA (European Network of Vaccine Adjuvants), supported by COST (European Cooperation in Science and Technology—www.cost.eu).
Schijns V, Fernández‐Tejada A, Barjaktarović Ž, et al. Modulation of immune responses using adjuvants to facilitate therapeutic vaccination. Immunol Rev. 2020;296:169–190. 10.1111/imr.12889
This article is part of a series of reviews covering B and Th cell response to Ag in vivo: implications for vaccine development and diseases appearing in Volume 296 of Immunological Reviews
Contributor Information
Virgil Schijns, Email: Schijns.virgil@gmail.com.
Ed C. Lavelle, Email: lavellee@tcd.ie.
REFERENCES
- 1. Spohn G, Bachmann MF. Therapeutic vaccination to block receptor‐ligand interactions. Expert Opin Biol Ther. 2003;3:469‐476. [DOI] [PubMed] [Google Scholar]
- 2. Bachmann MF, Dyer MR. Therapeutic vaccination for chronic diseases: a new class of drugs in sight. Nat Rev Drug Discov. 2004;3:81‐88. [DOI] [PubMed] [Google Scholar]
- 3. Schijns V, O'Hagan D (Eds.). Immunopotentiators in Modern Vaccines, 2nd edn 2017:1–508. London: Academic Press Elsevier. [Google Scholar]
- 4. Vos Q, Lees A, Wu ZQ, Snapper CM, Mond JJ. B‐cell activation by T‐cell‐independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol Rev. 2000;176:154‐170. [DOI] [PubMed] [Google Scholar]
- 5. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. 2015;15:149‐159. [DOI] [PubMed] [Google Scholar]
- 6. Shlomchik MJ, Weisel F. B cell primary immune responses. Immunol Rev. 2019;288:5‐9. [DOI] [PubMed] [Google Scholar]
- 7. Storni T, Kündig TM, Senti G, Johansen P. Immunity in response to particulate antigen‐delivery systems. Adv Drug Deliv Rev. 2005;57:333‐355. [DOI] [PubMed] [Google Scholar]
- 8. Weenink SM, Gautam AM. Antigen presentation by MHC class II molecules. Immunol Cell Biol. 1997;75:69‐81. [DOI] [PubMed] [Google Scholar]
- 9. Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11:823‐836. [DOI] [PubMed] [Google Scholar]
- 10. Romagnani S. Type 1 T helper and type 2 T helper cells: functions, regulation and role in protection and disease. Int J Clin Lab Res. 1991;21:152‐158. [DOI] [PubMed] [Google Scholar]
- 11. Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. 2019;16:634‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dominguez‐Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. 2018;19:665‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression ‐ implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16:356‐371. [DOI] [PubMed] [Google Scholar]
- 14. Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol. 2007;7:875‐888. [DOI] [PubMed] [Google Scholar]
- 15. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next‐generation regulatory T cell therapy. Nat Rev Drug Discov. 2019;18:749‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gagliani N, Magnani CF, Huber S, et al. Coexpression of CD49b and LAG‐3 identifies human and mouse T regulatory type 1 cells. Nat Med. 2013;19:739‐746. [DOI] [PubMed] [Google Scholar]
- 17. Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. 2019;50:1132‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song W, Craft J. T follicular helper cell heterogeneity: time, space, and function. Immunol Rev. 2019;288:85‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mastelic Gavillet B, Eberhardt CS, Auderset F, et al. MF59 mediates its B cell adjuvanticity by promoting T follicular helper cells and thus germinal center responses in adult and early life. J Immunol. 2015;194:4836‐4845. [DOI] [PubMed] [Google Scholar]
- 20. Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med. 2013;19:1597‐1608. [DOI] [PubMed] [Google Scholar]
- 21. Bousquet J, Lockey R, Malling HJ. Allergen immunotherapy: therapeutic vaccines for allergic diseases. A WHO position paper. J Allergy Clin Immunol. 1998;102:558‐562. [DOI] [PubMed] [Google Scholar]
- 22. Durham SR, Emminger W, Kapp A, et al. SQ‐standardized sublingual grass immunotherapy: confirmation of disease modification 2 years after 3 years of treatment in a randomized trial. J Allergy Clin Immunol. 2012;129:717‐725. e715. [DOI] [PubMed] [Google Scholar]
- 23. Walker SM, Varney VA, Gaga M, Jacobson MR, Durham SR. Grass pollen immunotherapy: efficacy and safety during a 4‐year follow‐up study. Allergy. 1995;50:405‐413. [DOI] [PubMed] [Google Scholar]
- 24. Kool M, Fierens K, Lambrecht BN. Alum adjuvant: some of the tricks of the oldest adjuvant. J Med Microbiol. 2012;61:927‐934. [DOI] [PubMed] [Google Scholar]
- 25. Jensen‐Jarolim E. Aluminium in Allergies and Allergen immunotherapy. World Allergy Organ J. 2015;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marichal T, Ohata K, Bedoret D, et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med. 2011;17:996‐1002. [DOI] [PubMed] [Google Scholar]
- 27. Oleszycka E, Lavelle EC. Immunomodulatory properties of the vaccine adjuvant alum. Curr Opin Immunol. 2014;28:1‐5. [DOI] [PubMed] [Google Scholar]
- 28. Zubeldia JM, Ferrer M, Dávila I, Justicia JL. Adjuvants in allergen‐specific immunotherapy: modulating and enhancing the immune response. J Investig Allergol Clin Immunol. 2019;29:103‐111. [DOI] [PubMed] [Google Scholar]
- 29. Puggioni F, Durham SR, Francis JN. Monophosphoryl lipid A (MPL) promotes allergen‐induced immune deviation in favour of Th1 responses. Allergy. 2005;60:678‐684. [DOI] [PubMed] [Google Scholar]
- 30. Pantel A, Cheong C, Dandamudi D, et al. A new synthetic TLR4 agonist, GLA, allows dendritic cells targeted with antigen to elicit Th1 T‐cell immunity in vivo. Eur J Immunol. 2012;42:101‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fonseca DE, Kline JN. Use of CpG oligonucleotides in treatment of asthma and allergic disease. Adv Drug Deliv Rev. 2009;61:256‐262. [DOI] [PubMed] [Google Scholar]
- 32. Klimek L, Kündig T, Kramer MF, et al. Virus‐like particles (VLP) in prophylaxis and immunotherapy of allergic diseases. Aller J Int. 2018;27:245‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kündig TM, Senti G, Schnetzler G, et al. Der p 1 peptide on virus‐like particles is safe and highly immunogenic in healthy adults. J Allergy Clin Immunol. 2006;117:1470‐1476. [DOI] [PubMed] [Google Scholar]
- 34. Senti G, Johansen P, Haug S, et al. Use of A‐type CpG oligodeoxynucleotides as an adjuvant in allergen‐specific immunotherapy in humans: a phase I/IIa clinical trial. Clin Exp Allergy. 2009;39:562‐570. [DOI] [PubMed] [Google Scholar]
- 35. Klimek L, Willers J, Hammann‐Haenni A, et al. Assessment of clinical efficacy of CYT003‐QbG10 in patients with allergic rhinoconjunctivitis: a phase IIb study: Treatment of allergy with novel immunotherapeutic. Clin Exp Allergy. 2011;41:1305‐1312. [DOI] [PubMed] [Google Scholar]
- 36. Dantzer JA, Wood RA. The use of omalizumab in allergen immunotherapy. Clin Exp Allergy. 2018;48:232‐240. [DOI] [PubMed] [Google Scholar]
- 37. Pentel PR, LeSage MG. New directions in nicotine vaccine design and use. Adv Pharmacol. 2014;69:553‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bremer PT, Janda KD. Conjugate vaccine immunotherapy for substance use disorder. Pharmacol Rev. 2017;69:298‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alving CR, Matyas GR, Torres O, Jalah R, Beck Z. Adjuvants for vaccines to drugs of abuse and addiction. Vaccine. 2014;32:5382‐5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Janda KD, Treweek JB. Vaccines targeting drugs of abuse: is the glass half‐empty or half‐full? Nat Rev Immunol. 2011;12:67‐72. [DOI] [PubMed] [Google Scholar]
- 41. McCluskie MJ, Thorn J, Mehelic PR, et al. Molecular attributes of conjugate antigen influence function of antibodies induced by anti‐nicotine vaccine in mice and non‐human primates. Int Immunopharmacol. 2015;25:518‐527. [DOI] [PubMed] [Google Scholar]
- 42. Pravetoni M. Biologics to treat substance use disorders: current status and new directions. Hum Vaccin Immunother. 2016;12:3005‐3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pryde DC, Jones LH, Gervais DP, et al. Selection of a novel anti‐nicotine vaccine: influence of antigen design on antibody function in mice. PLoS One. 2013;8:e76557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heekin RD, Shorter D, Kosten TR. Current status and future prospects for the development of substance abuse vaccines. Exp Rev Vaccin. 2017;16:1067‐1077. [DOI] [PubMed] [Google Scholar]
- 45. Hatsukami DK, Jorenby DE, Gonzales D, et al. Immunogenicity and smoking‐cessation outcomes for a novel nicotine immunotherapeutic. Clin Pharmacol Ther. 2011;89:392‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hatsukami DK, Rennard S, Jorenby D, et al. Safety and immunogenicity of a nicotine conjugate vaccine in current smokers. Clin Pharmacol Ther. 2005;78:456‐467. [DOI] [PubMed] [Google Scholar]
- 47. Hoogsteder PHJ, Kotz D, van Spiegel PI, Viechtbauer W, van Schayck OCP. Efficacy of the nicotine vaccine 3'‐AmNic‐rEPA (NicVAX) co‐administered with varenicline and counselling for smoking cessation: a randomized placebo‐controlled trial. Addiction. 2014;109:1252‐1259. [DOI] [PubMed] [Google Scholar]
- 48. Esterlis I, Hannestad JO, Perkins E, et al. Effect of a nicotine vaccine on nicotine binding to β2*‐nicotinic acetylcholine receptors in vivo in human tobacco smokers. Am J Psychiatry. 2013;170:399‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McCluskie MJ, Pryde DC, Gervais DP, et al. Enhancing immunogenicity of a 3'aminomethylnicotine‐DT‐conjugate anti‐nicotine vaccine with CpG adjuvant in mice and non‐human primates. Int Immunopharmacol. 2013;16:50‐56. [DOI] [PubMed] [Google Scholar]
- 50. McCluskie MJ, Thorn J, Gervais DP, et al. Anti‐nicotine vaccines: Comparison of adjuvanted CRM197 and Qb‐VLP conjugate formulations for immunogenicity and function in non‐human primates. Int Immunopharmacol. 2015;29:663‐671. [DOI] [PubMed] [Google Scholar]
- 51. Thorn JM, Bhattacharya K, Crutcher R, et al. The effect of physicochemical modification on the function of antibodies induced by anti‐nicotine vaccine in mice. Vaccines (Basel). 2017;5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jacob NT,Lockner JW, Schlosburg JE, et al. Investigations of enantiopure nicotine haptens using an adjuvanting carrier in anti‐nicotine vaccine development. J Med Chem. 2016;59:2523‐2529. [DOI] [PubMed] [Google Scholar]
- 53. Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic nanoparticles for vaccines and immunotherapy. Chem Rev. 2015;115:11109‐11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moon JJ, Huang B, Irvine DJ. Engineering nano‐ and microparticles to tune immunity. Adv Mater Weinheim. 2012;24:3724‐3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singha S, Shao K, Ellestad KK, Yang Y, Santamaria P. Nanoparticles for immune stimulation against infection, cancer, and autoimmunity. ACS Nano. 2018;12:10621‐10635. [DOI] [PubMed] [Google Scholar]
- 56. Braun M, Jandus C, Maurer P, et al. Virus‐like particles induce robust human T‐helper cell responses. Eur J Immunol. 2012;42:330‐340. [DOI] [PubMed] [Google Scholar]
- 57. Zabel F, Mohanan D, Bessa J, et al. Viral particles drive rapid differentiation of memory B cells into secondary plasma cells producing increased levels of antibodies. J Immunol. 2014;192:5499‐5508. [DOI] [PubMed] [Google Scholar]
- 58. Desai RI, Bergman J. Effects of the nanoparticle‐based vaccine, SEL‐068, on nicotine discrimination in squirrel monkeys. Neuropsychopharmacology. 2015;40:2207‐2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ilyinskii PO, Johnston LPM. Nanoparticle‐based nicotine vaccine In: Montoya ID, ed. Biologics to Treat Substance Use Disorders: Vaccines, Monoclonal Antibodies, and Enzymes. Cham, Switzerland: Springer International Publishing; 2016:249‐278. [Google Scholar]
- 60. Miller KD, Roque R, Clegg CH. Novel anti‐nicotine vaccine using a trimeric coiled‐coil hapten carrier. PLoS One. 2014;9:e114366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hu Y, Smith D, Frazier E, Hoerle R, Ehrich M, Zhang C. The next‐generation nicotine vaccine: a novel and potent hybrid nanoparticle‐based nicotine vaccine. Biomaterials. 2016;106:228‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhao Z, Hu Y, Harmon T, Pentel P, Ehrich M, Zhang C. Rationalization of a nanoparticle‐based nicotine nanovaccine as an effective next‐generation nicotine vaccine: a focus on hapten localization. Biomaterials. 2017;138:46‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhao Z, Powers K, Hu Y, Raleigh M, Pentel P, Zhang C. Engineering of a hybrid nanoparticle‐based nicotine nanovaccine as a next‐generation immunotherapeutic strategy against nicotine addiction: a focus on hapten density. Biomaterials. 2017;123:107‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhao Z, Harris B, Hu Y, Harmon T, Pentel PR, Ehrich M, Zhang C. Rational incorporation of molecular adjuvants into a hybrid nanoparticle‐based nicotine vaccine for immunotherapy against nicotine addiction. Biomaterials. 2018;155:165‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhao Z, Hu Y, Harmon T, Pentel PR, Ehrich M, Zhang C. Hybrid nanoparticle‐based nicotine nanovaccines: Boosting the immunological efficacy by conjugation of potent carrier proteins. Nanomedicine. 2018;14:1655‐1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hu Y, Zhao Z, Harmon T, Pentel PR, Ehrich M, Zhang C. Paradox of PEGylation in fabricating hybrid nanoparticle‐based nicotine vaccines. Biomaterials. 2018;182:72‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Leber ER, Benya TJ. Chlorinated hydrocarbon insecticides In: Clayton GD, Clayton FE, eds. Patty's Industrial Hygiene and Toxicology, vol. 2 (Part B). New York, NY: John Wiley and Sons Inc; 1994:1503‐1566. [Google Scholar]
- 68. Klaassen CD. Toxicology In: Hardman JG, Gilman AG, Limbird LE, eds. Goodman and Gilman's The Pharmacological Basis of Therapeutics, 9th edn New York, NY: Pergamon; 1996:1684‐1688. [Google Scholar]
- 69. World Health Organization , WHO Task Group on Environmental Health Criteria for DDT and its Derivatives . DDT and its derivatives/published under the joint sponsorship of the United Nations Environment Programme and the World Health Organization. Albany, NY, Geneva: World Health Organization;1979. https://apps.who.int/iris/handle/10665/39562 [Google Scholar]
- 70. Smith AG. Chlorinated hydrocarbon insecticides In: Hayes WJ, Laws ER, eds. Handbook of Pesticides Toxicology. San Diego, CA: Academic Press Inc; 1991:731‐915. [Google Scholar]
- 71. Nims RW, Jones SD, Reddy PE, Kocarek TA. Comparative pharmacodynamics of CYP2B induction by DDT, DDE, and DDD in male rat liver and cultured rat hepatocytes. J Toxicol Environ Health Part A. 1998;53:455‐477. [DOI] [PubMed] [Google Scholar]
- 72. Wallcave L, Bronczyk S, Gingell R. Excreted metabolites of 1,1,1‐trichloro‐2,2‐bis(p‐chlorophenyl)ethane in the mouse and hamster. J Agric Food Chem. 1974;22:904‐908. [DOI] [PubMed] [Google Scholar]
- 73. Hrafnkelsdottir K, Valgeirsson J, Bjarnadottir S, et al. Immunization prevents DDT buildup in mouse tissues. Int Immunopharmacol. 2007;7:1179‐1184. [DOI] [PubMed] [Google Scholar]
- 74. Marchesi VT. Alzheimer's dementia begins as a disease of small blood vessels, damaged by oxidative‐induced inflammation and dysregulated amyloid metabolism: implications for early detection and therapy. FASEB J. 2011;25:5‐13. [DOI] [PubMed] [Google Scholar]
- 75. Herline K, Drummond E, Wisniewski T. Recent advancements toward therapeutic vaccines against Alzheimer's disease. Exp Rev Vaccin. 2018;17:707‐721. [DOI] [PubMed] [Google Scholar]
- 76. Holmes C,Boche D, Wilkinson D, et al. Long‐term effects of Abeta42 immunisation in Alzheimer's disease: follow‐up of a randomised, placebo‐controlled phase I trial. Lancet. 2008;372:216‐223. [DOI] [PubMed] [Google Scholar]
- 77. Wisniewski T, Drummond E. Developing therapeutic vaccines against Alzheimer's disease. Exp Rev Vaccin. 2016;15:401‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Stemme S, Faber B, Holm J, Wiklund O, Witztum JL, Hansson GK. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1995;92:3893‐3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Investig. 1991;88:1785‐1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ameli S, Hultgårdh‐Nilsson A,Regnström J, et al. Effect of immunization with homologous LDL and oxidized LDL on early atherosclerosis in hypercholesterolemic rabbits. Arterioscler Thromb Vasc Biol. 1996;16:1074‐1079. [DOI] [PubMed] [Google Scholar]
- 81. Palinski W, Miller E, Witztum JL. Immunization of low density lipoprotein (LDL) receptor‐deficient rabbits with homologous malondialdehyde‐modified LDL reduces atherogenesis. Proc Natl Acad Sci USA. 1995;92:821‐825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fredrikson GN, Söderberg I, Lindholm M, et al. Inhibition of atherosclerosis in apoE‐null mice by immunization with apoB‐100 peptide sequences. Arterioscler Thromb Vasc Biol. 2003;23:879‐884. [DOI] [PubMed] [Google Scholar]
- 83. Gisterå A, Hermansson A, Strodthoff D, et al. Vaccination against T‐cell epitopes of native ApoB100 reduces vascular inflammation and disease in a humanized mouse model of atherosclerosis. J Intern Med. 2017;281:383‐397. [DOI] [PubMed] [Google Scholar]
- 84. Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3:630‐641. [DOI] [PubMed] [Google Scholar]
- 85. Livingston PO, Wong GY, Adluri S, et al. Improved survival in stage III melanoma patients with GM2 antibodies: a randomized trial of adjuvant vaccination with GM2 ganglioside. J Clin Oncol. 1994;12:1036‐1044. [DOI] [PubMed] [Google Scholar]
- 86. Feizi T. Demonstration by monoclonal antibodies that carbohydrate structures of glycoproteins and glycolipids are onco‐developmental antigens. Nature. 1985;314:53‐57. [DOI] [PubMed] [Google Scholar]
- 87. Wilson RM, Danishefsky SJ. A vision for vaccines built from fully synthetic tumor‐associated antigens: from the laboratory to the clinic. J Am Chem Soc. 2013;135:14462‐14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hakomori S. Aberrant glycosylation in cancer cell membranes as focused on glycolipids: overview and perspectives. Cancer Res. 1985;45:2405‐2414. [PubMed] [Google Scholar]
- 89. Hakomori S. Tumor‐associated carbohydrate antigens defining tumor malignancy: basis for development of anti‐cancer vaccines. Adv Exp Med Biol. 2001;491:369‐402. [DOI] [PubMed] [Google Scholar]
- 90. Lloyd KO. Humoral immune responses to tumor‐associated carbohydrate antigens. Semin Cancer Biol. 1991;2:421‐431. [PubMed] [Google Scholar]
- 91. Nativi C, Renaudet O. Recent progress in antitumoral synthetic vaccines. ACS Med Chem Lett. 2014;5:1176‐1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991‐998. [DOI] [PubMed] [Google Scholar]
- 93. Morse MA, Clay TM, Kim Lyerly H (Eds.) Handbook of Cancer Vaccines. Totowa, NJ: Humana Press; 2004. [Google Scholar]
- 94. Livingston PO, Natoli EJ, Calves MJ, Stockert E, Oettgen HF, Old LJ. Vaccines containing purified GM2 ganglioside elicit GM2 antibodies in melanoma patients. Proc Natl Acad Sci USA. 1987;84:2911‐2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lakshminarayanan V, Thompson P, Wolfert MA, et al. Immune recognition of tumor‐associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proc Natl Acad Sci USA. 2012;109:261‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wu X, Yin Z, McKay C, et al. Protective epitope discovery and design of MUC1‐based vaccine for effective tumor protections in immunotolerant mice. J Am Chem Soc. 2018;140:16596‐16609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kudryashov V, Kim HM, Ragupathi G, Danishefsky SJ, Livingston PO, Lloyd KO. Immunogenicity of synthetic conjugates of Lewis(y) oligosaccharide with proteins in mice: towards the design of anticancer vaccines. Cancer Immunol Immunother. 1998;45:281‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ragupathi G, Damani P, Srivastava G, et al. Synthesis of sialyl Lewis(a) (sLe (a), CA19‐9) and construction of an immunogenic sLe(a) vaccine. Cancer Immunol. Immunother. 2009;58:1397‐1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bilodeau MT, Park TK, Hu S, et al. Total synthesis of a human breast tumor associated antigen. J Am Chem Soc. 1995;117:7840‐7841. [Google Scholar]
- 100. Danishefsky SJ, Shue Y‐K, Chang MN, Wong C‐H. Development of Globo‐H cancer vaccine. Acc Chem Res. 2015;48:643‐652. [DOI] [PubMed] [Google Scholar]
- 101. Kushner BH, Cheung IY, Modak S, Kramer K, Ragupathi G, Cheung NK. Phase I trial of a bivalent gangliosides vaccine in combination with β‐glucan for high‐risk neuroblastoma in second or later remission. Clin Cancer Res. 2014;20:1375‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nores GA, Dohi T, Taniguchi M, Hakomori S. Density‐dependent recognition of cell surface GM3 by a certain anti‐melanoma antibody, and GM3 lactone as a possible immunogen: requirements for tumor‐associated antigen and immunogen. J Immunol. 1987;139:3171‐3176. [PubMed] [Google Scholar]
- 103. Arcangeli A, Toma L, Contiero L, et al. Stable GM3 lactone mimetic raises antibodies specific for the antigens expressed on melanoma cells. Bioconjug. Chem. 2010;21:1432‐1438. [DOI] [PubMed] [Google Scholar]
- 104. Tietze LF, Keim H, Janssen CO, Tappertzhofen C, Olschimke J. Synthesis of a novel ether‐bridged GM3‐lactone analogue as a target for an antibody‐based cancer therapy. Chemistry. 2000;6:2801‐2808. [DOI] [PubMed] [Google Scholar]
- 105. Wilstermann M, Kononov LO, Nilsson U, Ray AK, Magnusson G. Synthesis of ganglioside lactams corresponding to GM1‐, GM2‐, GM3‐, and GM4‐ganglioside lactones. J Am Chem Soc. 1995;117:4742‐4754. [Google Scholar]
- 106. Pérez K, Osorio M, Hernández J, Carr A, Fernández LE. NGcGM3/VSSP vaccine as treatment for melanoma patients. Hum Vaccin Immunother. 2013;9:1237‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mulens V, de la Torre A, Marinello P, et al. Immunogenicity and safety of a NeuGcGM3 based cancer vaccine: results from a controlled study in metastatic breast cancer patients. Hum Vaccin. 2010;6:736–744. [DOI] [PubMed] [Google Scholar]
- 108. Labrada M, Dorvignit D, Hevia G, et al. GM3(Neu5Gc) ganglioside: an evolution fixed neoantigen for cancer immunotherapy. Semin Oncol. 2018;45:41‐51. [DOI] [PubMed] [Google Scholar]
- 109. Krug LM, Ragupathi G, Hood C, et al. Vaccination of patients with small‐cell lung cancer with synthetic fucosyl GM‐1 conjugated to keyhole limpet hemocyanin. Clin Cancer Res. 2004;10:6094‐6100. [DOI] [PubMed] [Google Scholar]
- 110. Cai H, Huang ZH, Shi L, et al. Variation of the glycosylation pattern in MUC1 glycopeptide BSA vaccines and its influence on the immune response. Angew Chem Int Ed Engl. 2012;51:1719‐1723. [DOI] [PubMed] [Google Scholar]
- 111. Yin X‐G, Chen XZ, Sun WM, et al. IgG antibody response elicited by a fully synthetic two‐component carbohydrate‐based cancer vaccine candidate with α‐galactosylceramide as built‐in adjuvant. Org Lett. 2017;19:456‐459. [DOI] [PubMed] [Google Scholar]
- 112. Broecker F, Götze S, Hudon J, et al. Synthesis, liposomal formulation, and immunological evaluation of a minimalistic carbohydrate‐α‐GalCer vaccine candidate. J Med Chem. 2018;61:4918‐4927. [DOI] [PubMed] [Google Scholar]
- 113. Watson DS, Endsley AN, Huang L. Design considerations for liposomal vaccines: influence of formulation parameters on antibody and cell‐mediated immune responses to liposome associated antigens. Vaccine. 2012;30:2256‐2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Yin Z, Chowdhury S, McKay C, et al. Significant impact of immunogen design on the diversity of antibodies generated by carbohydrate‐based anticancer vaccine. ACS Chem Biol. 2015;10:2364‐2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Renaudet O, BenMohamed L, Dasgupta G, Bettahi I, Dumy P. Towards a self‐adjuvanting multivalent B and T cell epitope containing synthetic glycolipopeptide cancer vaccine. ChemMedChem. 2008;3:737‐741. [DOI] [PubMed] [Google Scholar]
- 116. Ibrahim NK, Murray JL. Clinical development of the STn‐KLH vaccine (Theratope). Clin Breast Cancer. 2003;3(Suppl 4):S139‐143. [DOI] [PubMed] [Google Scholar]
- 117. De Silva RA, Wang Q, Chidley T, Appulage DK, Andreana PR. Immunological response from an entirely carbohydrate antigen: design of synthetic vaccines based on Tn‐PS A1 conjugates. J Am Chem Soc. 2009;131:9622‐9623. [DOI] [PubMed] [Google Scholar]
- 118. Shi M, Kleski KA, Trabbic KR, Bourgault J‐P, Andreana PR. Sialyl‐Tn polysaccharide A1 as an entirely carbohydrate immunogen: synthesis and immunological evaluation. J Am Chem Soc. 2016;138:14264‐14272. [DOI] [PubMed] [Google Scholar]
- 119. Granier C, Blanc C, Karaki S, Tran T, Roussel H, Tartour E. Tissue‐resident memory T cells play a key role in the efficacy of cancer vaccines. Oncoimmunology. 2017;6:e1358841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Corbière V, Chapiro J, Stroobant V, et al. Antigen spreading contributes to MAGE vaccination‐induced regression of melanoma metastases. Cancer Res. 2011;71:1253‐1262. [DOI] [PubMed] [Google Scholar]
- 121. Bilusic M, Gulley JL. Editorial: local immunotherapy: a way to convert tumors from "cold" to "hot". J Natl Cancer Inst. 2017;109:djx132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355‐1360. [DOI] [PubMed] [Google Scholar]
- 123. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour‐specific, therapeutic vaccines for cancer. Nat Rev Immunol. 2018;18:168‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Banday AH, Jeelani S, Hruby VJ. Cancer vaccine adjuvants–recent clinical progress and future perspectives. Immunopharmacol Immunotoxicol. 2015;37:1‐11. [DOI] [PubMed] [Google Scholar]
- 125. Perret R, Sierro SR, Botelho NK, et al. Adjuvants that improve the ratio of antigen‐specific effector to regulatory T cells enhance tumor immunity. Cancer Res. 2013;73:6597‐6608. [DOI] [PubMed] [Google Scholar]
- 126. Speiser DE, Liénard D, Rufer N, et al. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Investig. 2005;115:739‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Junkins RD, Gallovic MD, Johnson BM, et al. A robust microparticle platform for a STING‐targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J Controlled Release. 2018;270:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Gutjahr A, Tiraby G, Perouzel E, Verrier B, Paul S. Triggering intracellular receptors for vaccine adjuvantation. Trends Immunol. 2016;37:716. [DOI] [PubMed] [Google Scholar]
- 129. Cawood R,Hills T, Wong SL, et al. Recombinant viral vaccines for cancer. Trends Mol Med. 2012;18:564‐574. [DOI] [PubMed] [Google Scholar]
- 130. Yang S, Hodge JW, Grosenbach DW, Schlom J. Vaccines with enhanced costimulation maintain high avidity memory CTL. J Immunol. 2005;175:3715‐3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Bulik‐Sullivan B, Busby J, Palmer CD, et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2019;37(1):55–63. [DOI] [PubMed] [Google Scholar]
- 132. Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral‐based PSA‐targeted immunotherapy in metastatic castration‐resistant prostate cancer. J Clin Oncol. 2010;28:1099‐1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chernysh S, Kozuharova I. Anti‐tumor activity of a peptide combining patterns of insect alloferons and mammalian immunoglobulins in naïve and tumor antigen vaccinated mice. Int Immunopharmacol. 2013;17:1090‐1093. [DOI] [PubMed] [Google Scholar]
- 134. Chernysh S, Irina K, Irina A. Anti‐tumor activity of immunomodulatory peptide alloferon‐1 in mouse tumor transplantation model. Int. Immunopharmacol. 2012;12:312‐314. [DOI] [PubMed] [Google Scholar]
- 135. Chernysh S, Kim SI, Bekker G, et al. Antiviral and antitumor peptides from insects. Proc Natl Acad Sci USA. 2002;99:12628‐12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Smith KD, Andersen‐Nissen E, Hayashi F, et al. Toll‐like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nat Immunol. 2003;4:1247‐1253. [DOI] [PubMed] [Google Scholar]
- 137. Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635‐647. [DOI] [PubMed] [Google Scholar]
- 138. Bos R, Sherman LA. CD4+ T‐cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70:8368‐8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Galaine J, Borg C, Godet Y, Adotévi O. Interest of tumor‐specific CD4 T helper 1 cells for therapeutic anticancer vaccine. Vaccines (Basel). 2015;3:490‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Qin Z, Blankenstein T. CD4+ T cell–mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity. 2000;12:677‐686. [DOI] [PubMed] [Google Scholar]
- 141. Tian L, Goldstein A, Wang H, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature. 2017;544:250‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Segura E, Amigorena S. Cross‐presentation in mouse and human dendritic cells. Adv Immunol. 2015;127:1‐31. [DOI] [PubMed] [Google Scholar]
- 143. Macri C, Dumont C, Johnston AP, Mintern JD. Targeting dendritic cells: a promising strategy to improve vaccine effectiveness. Clin Transl Immunology. 2016;5:e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Adotevi O, Vingert B, Freyburger L, et al. B subunit of Shiga toxin‐based vaccines synergize with alpha‐galactosylceramide to break tolerance against self antigen and elicit antiviral immunity. J Immunol. 2007;179:3371‐3379. [DOI] [PubMed] [Google Scholar]
- 145. Terhorst D, Fossum E, Baranska A, et al. Laser‐assisted intradermal delivery of adjuvant‐free vaccines targeting XCR1+ dendritic cells induces potent antitumoral responses. J Immunol. 2015;194:5895‐5902. [DOI] [PubMed] [Google Scholar]
- 146. Tullett KM, Leal Rojas IM, Minoda Y, et al. Targeting CLEC9A delivers antigen to human CD141+ DC for CD4+ and CD8+T cell recognition. JCI Insight. 2016;1:e87102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Tanyi JL, Bobisse S, Ophir E, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med. 2018;10:eaao5931. [DOI] [PubMed] [Google Scholar]
- 148. Strioga M, Schijns V, Powell DJ, Pasukoniene V, Dobrovolskiene N, Michalek J. Dendritic cells and their role in tumor immunosurveillance. Innate Immun. 2013;19:98‐111. [DOI] [PubMed] [Google Scholar]
- 149. Schuler PJ, Harasymczuk M, Visus C, et al. Phase I dendritic cell p53 peptide vaccine for head and neck cancer. Clin Cancer Res. 2014;20:2433‐2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Buonaguro L, Petrizzo A, Tornesello ML, Buonaguro FM. Translating tumor antigens into cancer vaccines. Clin Vaccine Immunol. 2011;18:23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Melero I, Gaudernack G, Gerritsen W, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11:509‐524. [DOI] [PubMed] [Google Scholar]
- 152. Schijns VEJC, Pretto C, Devillers L, et al. First clinical results of a personalized immunotherapeutic vaccine against recurrent, incompletely resected, treatment‐resistant glioblastoma multiforme (GBM) tumors, based on combined allo‐ and auto‐immune tumor reactivity. Vaccine. 2015;33:2690‐2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Schijns VEJC, Pretto C, Strik A, et al. Therapeutic immunization against glioblastoma. Int J Mol Sci. 2018;19:2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Cheever MA, Higano CS. PROVENGE (Sipuleucel‐T) in prostate cancer: the first FDA‐approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17:3520‐3526. [DOI] [PubMed] [Google Scholar]
- 155. Gray GE, Laher F, Lazarus E, Ensoli B, Corey L. Approaches to preventative and therapeutic HIV vaccines. Curr Opin Virol. 2016;17:104‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Seddiki N, Lévy Y. Therapeutic HIV‐1 vaccine: time for immunomodulation and combinatorial strategies. Curr Opin HIV AIDS. 2018;13:119‐127. [DOI] [PubMed] [Google Scholar]
- 157. Stephenson KE. Therapeutic vaccination for HIV: hopes and challenges. Curr Opin HIV AIDS. 2018;13:408‐415. [DOI] [PubMed] [Google Scholar]
- 158. Li JZ,AIDS Clinical Trials Group A5197 Study Team , Brumme ZL, et al. Factors associated with viral rebound in HIV‐1‐infected individuals enrolled in a therapeutic HIV‐1 gag vaccine trial. J Infect Dis. 2011;203:976‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hancock G, Morón‐López S, Kopycinski J, et al. Evaluation of the immunogenicity and impact on the latent HIV‐1 reservoir of a conserved region vaccine, MVA.HIVconsv, in antiretroviral therapy‐treated subjects. J Int AIDS Soc. 2017;20:21171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Goldstein G, Damiano E, Donikyan M, Pasha M, Beckwith E, Chicca J. HIV‐1 Tat B‐cell epitope vaccination was ineffectual in preventing viral rebound after ART cessation: HIV rebound with current ART appears to be due to infection with new endogenous founder virus and not to resurgence of pre‐existing Tat‐dependent viremia. Hum Vaccin Immunother. 2012;8:1425‐1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. García F, Climent N, Assoumou L, et al. A therapeutic dendritic cell‐based vaccine for HIV‐1 infection. J Infect Dis. 2011;203:473‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Lévy Y, Gahéry‐Ségard H, Durier C, et al. Immunological and virological efficacy of a therapeutic immunization combined with interleukin‐2 in chronically HIV‐1 infected patients. AIDS. 2005;19:279‐286. [PubMed] [Google Scholar]
- 163. Brezar V, Hani L, Surenaud M, et al. Negative modulation of suppressive HIV‐specific regulatory T cells by IL‐2 adjuvanted therapeutic vaccine. PLoS Pathog. 2017;13:e1006489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Nct . Lipopeptide Immunisation With GTU‐multiHIV Trial. clinicaltrials.gov. Accessed July 5, 2019.
- 165. Hansen SG, Sacha JB, Hughes CM, et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science. 2013;340:1237874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Hansen SG, Wu HL, Burwitz BJ, et al. Broadly targeted CD8⁺ T cell responses restricted by major histocompatibility complex E. Science. 2016;351:714‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Früh K, Picker L. CD8+ T cell programming by cytomegalovirus vectors: applications in prophylactic and therapeutic vaccination. Curr Opin Immunol. 2017;47:52‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Borducchi EN, Cabral C, Stephenson KE, et al. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV‐infected rhesus monkeys. Nature. 2016;540:284‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Kaufmann SHE, Weiner J, von Reyn CF. Novel approaches to tuberculosis vaccine development. Int J Infect Dis. 2017;56:263‐267. [DOI] [PubMed] [Google Scholar]
- 170. Weng H, Huang J‐Y, Meng X‐Y, Li S, Zhang G‐Q. Adjunctive therapy of Mycobacterium vaccae vaccine in the treatment of multidrug‐resistant tuberculosis: a systematic review and meta‐analysis. Biomed Rep. 2016;4:595‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Butov DA, Efremenko YV, Prihoda ND, et al. Adjunct immune therapy of first‐diagnosed TB, relapsed TB, treatment‐failed TB, multidrug‐resistant TB and TB/HIV. Immunotherapy. 2012;4:687‐695. [DOI] [PubMed] [Google Scholar]
- 172. Efremenko YV, Butov DA, Prihoda ND, et al. Randomized, placebo‐controlled phase II trial of heat‐killed Mycobacterium vaccae (Longcom batch) formulated as an oral pill (V7). Hum Vaccin Immunother. 2013;9:1852‐1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Gröschel MI, Prabowo SA, Cardona P‐J, Stanford JL, van der Werf TS. Therapeutic vaccines for tuberculosis–a systematic review. Vaccine. 2014;32:3162‐3168. [DOI] [PubMed] [Google Scholar]
- 174. Cardona P‐J, Amat I, Gordillo S, et al. Immunotherapy with fragmented Mycobacterium tuberculosis cells increases the effectiveness of chemotherapy against a chronical infection in a murine model of tuberculosis. Vaccine. 2005;23:1393‐1398. [DOI] [PubMed] [Google Scholar]
- 175. Nell AS, D’lom E, Bouic P et al. Safety, tolerability, and immunogenicity of the novel antituberculous vaccine RUTI: randomized, placebo‐controlled phase II clinical trial in patients with latent tuberculosis infection. PLoS One. 2014;9:e89612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kamal R, Pathak V, Kumari A, Natrajan M, Katoch K, Kar HK. Addition of Mycobacterium indicus pranii vaccine as an immunotherapeutic to standard chemotherapy in borderline leprosy: a double‐blind study to assess clinical improvement (preliminary report). Br J Dermatol. 2017;176:1388‐1389. [DOI] [PubMed] [Google Scholar]
- 177. Bertholet S,Ireton GC, Ordway DJ, et al. A defined tuberculosis vaccine candidate boosts BCG and protects against multidrug‐resistant Mycobacterium tuberculosis. Sci Transl Med. 2010;2:53ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Coler RN, Bertholet S, Pine SO, et al. Therapeutic immunization against Mycobacterium tuberculosis is an effective adjunct to antibiotic treatment. J Infect Dis. 2013;207:1242‐1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Larsen SE, Baldwin S, Orr M, et al. Enhanced anti‐mycobacterium tuberculosis immunity over time with combined drug and immunotherapy treatment. Vaccines (Basel). 2018;6(2):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Penn‐Nicholson A, Tameris M, Smit E, et al. Safety and immunogenicity of the novel tuberculosis vaccine ID93 + GLA‐SE in BCG‐vaccinated healthy adults in South Africa: a randomised, double‐blind, placebo‐controlled phase 1 trial. Lancet Respir Med. 2018;6:287‐298. [DOI] [PubMed] [Google Scholar]
- 181. Okada M, Kita Y, Nakajima T, et al. Novel prophylactic and therapeutic vaccine against tuberculosis. Vaccine. 2009;27:3267‐3270. [DOI] [PubMed] [Google Scholar]
- 182. Kita Y, Hashimoto S, Nakajima T, et al. Novel therapeutic vaccines [(HSP65 + IL‐12)DNA‐, granulysin‐ and Ksp37‐vaccine] against tuberculosis and synergistic effects in the combination with chemotherapy. Hum Vaccin Immunother. 2013;9:526‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Okada M, Kita Y, Hashimoto S, et al. Preclinical study and clinical trial of a novel therapeutic vaccine against multi‐drug resistant tuberculosis. Hum Vaccin Immunother. 2017;13:298‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Giri PK, Verma I, Khuller GK. Adjunct immunotherapy with Ag85 complex proteins based subunit vaccine in a murine model of Mycobacterium tuberculosis infection. Immunotherapy. 2009;1:31‐37. [DOI] [PubMed] [Google Scholar]
- 185. Liang Y, Wu X, Zhang J, et al. The treatment of mice infected with multi‐drug‐resistant Mycobacterium tuberculosis using DNA vaccines or in combination with rifampin. Vaccine. 2008;26:4536‐4540. [DOI] [PubMed] [Google Scholar]
- 186. Liang Y, Wu X, Zhang J, et al. Treatment of multi‐drug‐resistant tuberculosis in mice with DNA vaccines alone or in combination with chemotherapeutic drugs. Scand J Immunol. 2011;74:42‐46. [DOI] [PubMed] [Google Scholar]
- 187. Sheikh JA, Khuller GK, Verma I. Immunotherapeutic role of Ag85B as an adjunct to antituberculous chemotherapy. J Immune Based Ther Vaccines. 2011;9:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Gillard P, Yang P‐C, Danilovits M, et al. Safety and immunogenicity of the M72/AS01E candidate tuberculosis vaccine in adults with tuberculosis: A phase II randomised study. Tuberculosis. 2016;100:118‐127. [DOI] [PubMed] [Google Scholar]
- 189. Chyu K‐Y, Zhao X, Dimayuga PC, et al. CD8+ T cells mediate the athero‐protective effect of immunization with an ApoB‐100 peptide. PLoS One. 2012;7:e30780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Herbin O, Ait‐Oufella H, Yu W, et al. Regulatory T‐cell response to apolipoprotein B100‐derived peptides reduces the development and progression of atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2012;32:605‐612. [DOI] [PubMed] [Google Scholar]
- 191. Klingenberg R, Lebens M, Hermansson A, et al. Intranasal immunization with an apolipoprotein B‐100 fusion protein induces antigen‐specific regulatory T cells and reduces atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:946‐952. [DOI] [PubMed] [Google Scholar]
- 192. van Puijvelde GHM, Hauer AD, de Vos P, et al. Induction of oral tolerance to oxidized low‐density lipoprotein ameliorates atherosclerosis. Circulation. 2006;114:1968‐1976. [DOI] [PubMed] [Google Scholar]
- 193. Foks AC, Frodermann V, ter Borg M, et al. Differential effects of regulatory T cells on the initiation and regression of atherosclerosis. Atherosclerosis. 2011;218:53‐60. [DOI] [PubMed] [Google Scholar]
- 194. Kobiyama K, Vassallo M, Mitzi J, et al. A clinically applicable adjuvant for an atherosclerosis vaccine in mice. Eur J Immunol. 2018;48:1580‐1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Benne N, van Duijn J, Lozano Vigario F, et al. Anionic 1,2‐distearoyl‐sn‐glycero‐3‐phosphoglycerol (DSPG) liposomes induce antigen‐specific regulatory T cells and prevent atherosclerosis in mice. J Control Release. 2018;291:135‐146. [DOI] [PubMed] [Google Scholar]
- 196. Pearson JKL, Kerr WR, McCartney WDJ. Tissue vaccine in the treatment of bovine papillomas. Vet Rec. 1958;70:971‐973. [Google Scholar]
- 197. Amstutz HE. Bovine skin conditions: a brief review. Mod Vet Pract. 1979;60821–824. [PubMed] [Google Scholar]
- 198. Araldi RP, Assaf SM, Carvalho RF, et al. Papillomaviruses: a systematic review. Genet Mol Biol. 2017;40:1‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Nasir L, Campo MS. Bovine papillomaviruses: their role in the aetiology of cutaneous tumours of bovids and equids. Vet Dermatol. 2008;19:243‐254. [DOI] [PubMed] [Google Scholar]
- 200. Epsy BK. How to treat equine sarcoids by autologous implantation. Proc Am Assoc Equine Pract. 2008;54:68–73. [Google Scholar]
- 201. Kinnunen RE, Tallberg T, Stenbäck H, Sarna S. Equine sarcoid tumour treated by autogenous tumour vaccine. Anticancer Res. 1999;19:3367‐3374. [PubMed] [Google Scholar]
- 202. Rothacker CC, Boyle AG, Levine DG. Autologous vaccination for the treatment of equine sarcoids: 18 cases (2009–2014). Can Vet J. 2015;56:709‐714. [PMC free article] [PubMed] [Google Scholar]
- 203. Taylor S, Haldorson G. A review of equine sarcoid: Equine cutaneous tumours. Equine Vet Educ. 2013;25:210‐216. [Google Scholar]
- 204. Bergman PJ, Camps‐Palau MA, McKnight JA, et al. Development of a xenogeneic DNA vaccine program for canine malignant melanoma at the Animal Medical Center. Vaccine. 2006;24:4582‐4585. [DOI] [PubMed] [Google Scholar]
- 205. Grosenbaugh DA, Leard AT, Bergman PJ, et al. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am J Vet Res. 2011;72:1631‐1638. [DOI] [PubMed] [Google Scholar]
- 206. Ottnod JM, Smedley RC, Walshaw R, Hauptman JG, Kiupel M, Obradovich JE. A retrospective analysis of the efficacy of Oncept vaccine for the adjunct treatment of canine oral malignant melanoma. Vet Comp Oncol. 2013;11:219‐229. [DOI] [PubMed] [Google Scholar]
- 207. Westhoff DK, Kloes MC, Orveillon FX, Farnow D, Elbers K, Mueller RS. Treatment of feline dermatophytosis with an inactivated fungal vaccine. Open Mycol J. 2010;4:10‐17. [Google Scholar]
- 208. Sagiv‐Barfi I,Czerwinski DK, Levy S, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med. 2018;10:eaan4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Mulder WJM, Ochando J, Joosten LAB, Fayad ZA, Netea MG. Therapeutic targeting of trained immunity. Nat Rev Drug Discov. 2019;18:553‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]