

Summary
The Honghe Hani rice terraces system (HHRTS) is a traditional rice cultivation system where Hani people cultivate remarkably diverse rice varieties. Recent introductions of modern rice varieties to the HHRTS have significantly increased the severity of rice diseases within the terraces. Here, we determine the impacts of these recent introductions on the composition of the rice‐associated microbial communities. We confirm that the HHRTS contains a range of both traditional HHRTS landraces and introduced modern rice varieties and find differences between the microbial communities of these two groups. However, this introduction of modern rice varieties has not strongly impacted the overall diversity of the HHRTS rice microbial community. Furthermore, we find that the rice varieties (i.e. groups of closely related genotypes) have significantly structured the rice microbial community composition (accounting for 15%–22% of the variance) and that the core microbial community of HHRTS rice plants represents less than 3.3% of all the microbial taxa identified. Collectively, our study suggests a highly diverse HHRTS rice holobiont (host with its associated microbes) where the diversity of rice hosts mirrors the diversity of their microbial communities. Further studies will be needed to better determine how such changes might impact the sustainability of the HHRTS.
Introduction
During and since the Green Revolution, governments and agricultural stakeholders all around the world have promoted the widespread introduction to cropping systems of modern high‐yielding crop varieties. By contributing to the simplification and homogenization of cropping systems this trend has likely also inadvertently contributed to increases in the occurrence of various crop diseases (Keesing et al., 2006; Stuckenbrock and McDonald, 2008; Roossinck and Garcia‐Arenal, 2015; Bernardo et al., 2018). Nevertheless, there are a number of Globally Important Agricultural Heritage Systems (GIAHS) within which landraces (traditional varieties) are still cultivated (FAO, http://www.fao.org/giahs/en/). Although many of these GIAHSs were left largely unchanged by the Green Revolution, some, such as the Honghe Hani rice terraces system (HHRTS), have more recently experienced both the introduction of modern high‐yielding rice varieties and an increase in chemical usage (Yang et al., 2017; Dedeurwaerdere and Hannachi, 2019).
The HHRTS, which was recently listed as a World Cultural Heritage Site (UNESCO, 2013), is a renowned rice terrace landscape where remarkably diverse rice varieties have been cultivated for over 1300 years by the Hani people (Jiao et al., 2012). The HHRTS consists of a collection of unique man‐made vertically structured ecological landscapes comprising cascading terraced rice fields sandwiched between forests and villages at the top, and a river at the bottom (Cui et al., 2008; Yang et al., 2017). Hundreds of years of rice varietal selection within the HHRTS has yielded at least 195 local rice landraces (Oryza sativa) and 47 wild rice landraces (Oryza rufipogon and Oryza nivara) (Jiao et al., 2012). These diverse rice landraces are grown by each Hani household in complex heterogeneous mosaics, in narrow paddy fields averaging ~150 m2 within each of which only one rice landrace is cultivated.
While increased diversification of rice varieties has likely reduced the severity of rice‐associated diseases (Zhu et al., 2000), it is usually hypothesized that the structure of the HHRTS has likely also limited the occurrence and spread of diseases within the system. For example, the spread of Pyricularia oryzae, the causal agent of rice blast, is apparently hampered by the high diversity of basal and effector‐triggered immune responses that are displayed by the japonica and indica rice varieties commonly cultivated within the HHRTS (Liao et al., 2016).
However, a recent socio‐economic survey has revealed a significant increase in the severity of diseases occurring within HHRTS fields where the government‐promoted ‘HongYang’ improved rice variety has been cultivated (Dedeurwaerdere and Hannachi, 2019). Increased use of this improved variety since 2010 may, for example, account for increases over the past decade in the prevalence within the HHRTS of Southern rice black‐streaked dwarf virus (SRBSDV) (Alonso et al., 2019).
Besides the role played in plant health by intrinsic plant mechanisms, plant‐associated microbial communities are also likely to be directly or indirectly involved in plant health and plant development (Berendsen et al., 2012; Hacquard et al., 2017; Hassani et al., 2019; Vannier et al., 2019). It is now widely accepted that the structuring of plant microbiota is controlled by plant‐specific factors (Vorholt, 2012; Bulgarelli et al., 2013; Reinhold‐Hurek et al., 2015; Hamonts et al., 2018) and that plant genotype may further impact microbial communities (Bulgarelli et al., 2015; Sapkota et al., 2015; Wagner et al., 2016). Specifically, plant domestication or selection has possibly contributed to a small but significant change in plant microbial communities (Redford et al., 2010; Peiffer and Ley, 2013; Bouffaud et al., 2014; Ofek‐Lalzar et al., 2014; Bulgarelli et al., 2015; Edwards et al., 2015). The HHRTS is therefore a good potential candidate for determining the impacts of cultivating modern rice varieties on aspects of traditional rice cultivation systems: impacts such as the composition of their associated microbial communities. Overall, modern varieties have low genetic diversity due to the recurrent selection for traits contributing to, among other things, plant yield, rice quality, and resistance to biotic and abiotic stresses (Meyer and Purugganan, 2013). This genetic simplification has often been implicated in reducing the bacterial and fungal diversity in the rhizosphere and phyllosphere of modern cultivated crop varieties relative to that found in the rhizosphere and phyllosphere of wild species and landraces (reviewed in Cordovez et al., 2019). For example, modern cultivated pea and broad bean varieties have less promiscuous interactions with symbionts than do their wild relatives (Mutch and Young, 2004). Similarly, the bacterial and fungal diversity in the rhizosphere and phyllosphere of uncultivated agave species is higher than that of cultivated agave (Coleman‐Derr et al., 2016). Small but significant domestication effects have also been noted when comparing the root microbial communities of wild and modern varieties of barley (Bulgarelli et al., 2015; Szoboszlay et al., 2015). Two potential exceptions to this trend are modern lettuce and sunflower varieties, which harbour degrees of rhizobacterial diversity that are similar to, or in some cases higher than, those of their uncultivated relatives (Cardinale et al., 2015; Leff et al., 2017).
The microbial community associated with the rice rhizosphere has been the most intensively investigated (Edwards et al., 2015; Wang et al., 2016; Edwards et al., 2018; Moronta‐Barrios et al., 2018; Ding et al., 2019). Specifically, several studies have focused on the comparison of the rhizosphere or seed‐associated microbial communities of wild Oryza, rice landraces and modern rice varieties (Shenton et al., 2016; Shi et al., 2019; Kim et al., 2020). For instance, Shenton and colleagues (2016) have shown that the rhizosphere bacterial community of wild Oryza differed substantially from that of cultivated rice in terms of both species richness and composition (Shenton et al., 2016). Interestingly, this study also revealed that landraces had a rhizosphere bacterial community composition that was intermediate between that of wild Oryza and cultivated rice varieties (Shenton et al., 2016). It is also apparent that fungal communities differ more substantially than bacterial communities between the seed or rhizosphere microbial communities of cultivated varieties and wild Oryza (Shi et al., 2019; Kim et al., 2020).
Here, we hypothesized that the introduction of modern rice varieties to the HHRTS may have impacted rice plant microbial communities within the HHRTS. We initially determined the degree of rice genetic diversity within the paddy fields of a single HHRTS village, Malizhai that has adopted a mixed landrace/modern variety system. Then, we compared the diversity and composition of the microbial communities of introduced modern rice varieties and HHRTS landraces.
Results and discussion
Both landraces and modern rice varieties are grown in the Malizhai HHRTS
Malizhai is a village broadly representative of the HHRTS throughout the Hani region (Jiao et al., 2012). To obtain a detailed understanding of rice genetic diversity within the Malizhai HHRTS, we sampled 19 rice paddies, each in a 2 km2 area of Malizhai. While 11 of the rice paddies from which samples were collected were referred to as ‘traditional varieties’ by the Malizhai farmers, eight were referred to as ‘modern varieties’. We performed a genotyping by sequencing (GBS) analysis of the 19 sampled rice paddies (hereafter referred to as our Malizhai GBS dataset) and compared these to a reference dataset containing whole‐genome re‐sequencing data produced by the 3000 Rice Genomes Project (Wang et al., 2018) from nine rice accessions randomly selected from among four representative subpopulations of the indica rice subspecies (subpopulations XI‐1A, XI‐1B, XI‐2 and XI‐3).
This GBS comparison revealed that only one of the groups of Malizhai rice genotypes belonged to the japonica rice subspecies with the remaining 18 group of rice genotypes belonging to the indica rice subspecies (Fig. 1A, B). Whereas 10 of the 18 group of Malizhai indica genotypes were identified by farmers as being ‘traditional varieties’ and will hereafter be referred to as the ‘group of HHRTS landraces’, the other eight genotypes were identified by farmers as being ‘modern varieties’ and will hereafter be referred to as the ‘group of modern rice’. Our inference of population subdivision by partitioning genotypes into K ancestral populations, based both on the cross‐entropy criterion (Fig. S1) and visual analysis of clustering patterns (Fig. 1B), revealed that the model with K = 4 clusters captured most of the structure in the data. Hence, at K = 4, besides the indica rice genotypes from the 3000 Rice Genomes Project (coloured in blue, Fig. 1B), three groups from the Malizhai HHRTS were distinguishable, including a group of japonica genotypes (coloured in orange, Fig. 1B), the group of HHRTS landraces (coloured in green, Fig. 1B), and the group of modern varieties (coloured in brown, Fig. 1B). Our GBS analysis confirmed that the rice genotypes are heterogeneously distributed in the Malizhai HHRTS forming a mosaic landscape of modern genotypes and landraces (Fig. 1C). The eight groups of genotypes representing the group of modern varieties included commercial modern hybrid F1 varieties that are widely and intensively cultivated in the rice‐growing plains of China (Table 1). Nucleotide diversity was slightly lower within the group of modern varieties (π = 8.05 × 10−4) than that of the HHRTS landraces group (π = 9.43 × 10−4).
Fig 1.
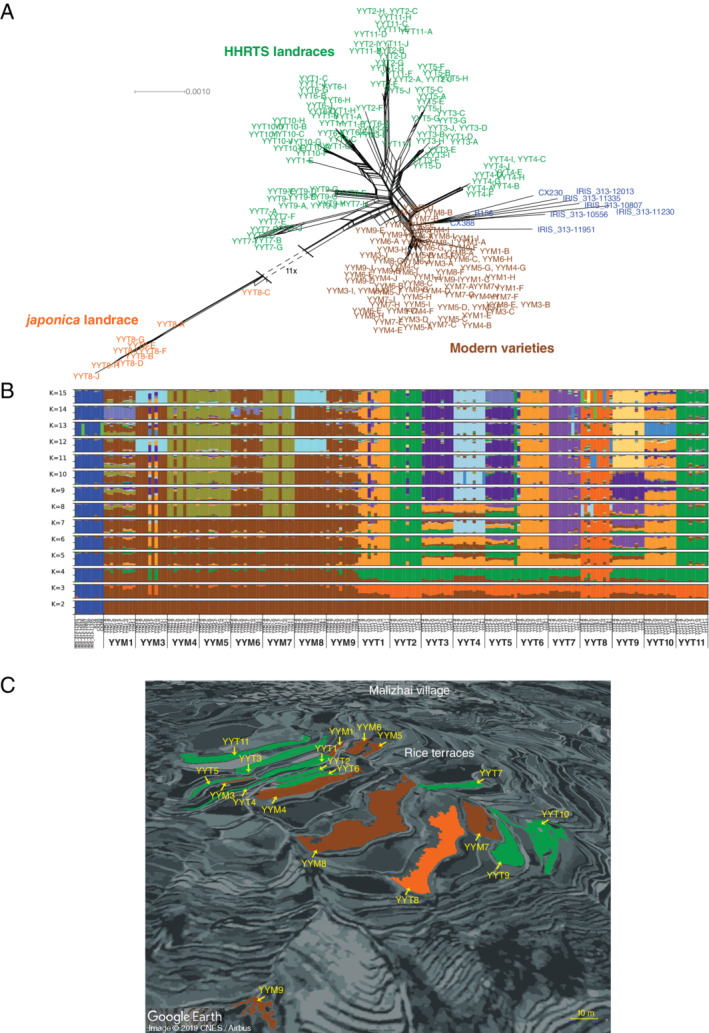
A. Neighbour‐Net split decomposition network indicating the relationships between rice accessions based on 10 028 analysed SNPs. Plant samples assigned to the ‘HHRTS landraces’, the ‘modern rice varieties’, the japonica subspecies and representative subset of indica subgroups from the 3000 Rice Genomes Project are labelled in green, brown, orange and blue respectively. B. Ancestry proportions inferred using sNMF for models with K = 2 to K = 15 ancestral populations. Each accession is represented by a vertical bar divided into K segments of different colours, representing proportions of ancestry in K ancestral populations for a single accession, with colours corresponding to ancestral populations. C. Map of the Malizhai rice terraces and location of the rice fields that were collected in 2016. Terraces were HHRTS landraces, modern rice varieties and japonica varieties, which are coloured in green, brown and orange respectively.
Table 1.
Genotyping of rice varieties from the Yuanyang rice terraces of China and assignment of rice varieties to three rice genetic groups (‘HHRTS landraces group’, ‘Modern rice group’ and Oryza sativa subspecies japonica).
| Sampling field | Rice genetic group | Rice variety name(given by the farmers) | Number of samples |
|---|---|---|---|
| YYT1 | HHRTS landraces group (HongYang) | Chepugu | 9 |
| YYT2 | HHRTS landraces group | Lubaigu | 10 |
| YYT3 | HHRTS landraces group | Zaogu | 10 |
| YYT4 | HHRTS landraces group | Nuogu | 10 |
| YYT5 | HHRTS landraces group | Epugu | 10 |
| YYT6 | HHRTS landraces group (HongYang) | Chepugu | 9 |
| YYT7 | HHRTS landraces group | Jianshuigu | 10 |
| YYT8 | Oryza sativa ssp. japonica | Honglueduolu | 10 |
| YYT9 | HHRTS landraces group | Nuogu | 10 |
| YYT10 | HHRTS landraces group (HongYang) | Jinpinggu | 10 |
| YYT11 | HHRTS landraces group | Luhonggu | 10 |
| YYM1 | Modern rice group | Mingliangyou 527 | 10 |
| YYM3 | Modern rice group | Mingliangyou 528 | 8 |
| YYM4 | Modern rice group | Hefeng 177 | 10 |
| YYM5 | Modern rice group | Zhongyou 177 | 10 |
| YYM6 | Modern rice group | Liangyou 2186 | 10 |
| YYM7 | Modern rice group | Guofeng 1 | 10 |
| YYM8 | Modern rice group | Liangyou 725 | 10 |
| YYM9 | Modern rice group | Liangyou 2161 | 10 |
The model with K = 8 clusters also revealed patterns of clustering that seem biologically sensible. At K = 8, the group of modern varieties could be further split into two subgroups, referred to as the modern rice subgroup1 (samples from fields YYM1, YYM3, YYM6, YYM8 and YYM9, Fig. 1B) and the modern rice subgroup2 (samples from fields YYM4, YYM5 and YYM7, Fig. 1B). Unexpectedly, but in accordance with farmer assignments, the government‐promoted HongYang varieties grown in three fields (YYT1, YYT6 and YYT10; Table 1) that were believed to be ‘modern improved varieties’ clearly share a recent ancestry with the HHRTS landraces (Fig. 1A). This implies that HongYang is based on a traditional landrace that likely originated from the group of HHRTS landraces (Dedeurwaerdere and Hannachi, 2019). Clustering patterns at K = 8 also revealed that the group of HHRTS indica landraces can be further subdivided into four subgroups, which are referred to as HHRTS landraces subgroup1 (YYT1, YYT6 and YYT10, i.e. HongYang varieties), HHRTS landraces subgroup2 (YYT2, YYT11), HHRTS landraces subgroup3 (YYT3, YYT5) and HHRTS landraces subgroup4 (YYT7, YYT9) (Fig. 1B).
We have thus found that rice varieties grown in the Malizhai HHRTS fall into two main genetic groups; one including newly introduced commercial Hybrid F1 modern varieties and the other including several traditional or improved (HongYang) varieties. This confirms that changes in cultural practices in the Malizhai HHRTS, and likely in other villages as well, have resulted in a shift in the genetic makeup of cultivated rice in the terraces. We also find evidence that further replacement of HHRTS landraces by modern varieties could slightly reduce the over‐all genetic diversity of rice within the HHRTS.
The microbial communities of rice roots and stems are highly divergent
Having characterized the genetic diversity of rice grown in the Malizhai HHRTS, we next sought to characterize the bacterial and fungal components of the microbial communities of sampled rice plants. Our samples included the microbes living at the surface and within plant tissue. Based on the analysis of GBS and whole‐genome resequencing data, we split the sampled indica rice plants into a group of modern varieties (including 78 plants from eight different modern varieties) and a group of HHRTS landraces (including 98 plants from 10 different traditional or improved HHRTS varieties). For consistency, we excluded plants of the YYT8 japonica rice subspecies (Table 1) from this analysis. We then examined the stem and root microbial communities of plants belonging to the HHRTS landrace and modern variety groups.
Metabarcoding of bacterial communities produced after the bioinformatics processing 120 666 high‐quality reads from the root compartment and 92 400 from the stem compartment respectively corresponding to 325 and 91 Operational taxonomic units (OTUs) with each OTU representing over 50% prevalence per rice variety (Table S1). For the fungal communities, we obtained 30 858 high‐quality reads for the root compartment and 21 750 high‐quality reads for the stem for the root compartment respectively representing 110 and 105 OTUs with each OTU representing over 50% prevalence per rice variety (Table S1). Details of the number of sequences and OTUs recovered after each step of the bioinformatics processing are available in Table S1. Given that too few samples from the field YYT4 yielded enough high‐quality reads to reach the rarefaction thresholds, this variety was further excluded from the group of HHRTS landraces in all subsequent analyses. Data for the group of HHRTS landraces were drawn from 88 plant samples belonging to nine different rice traditional varieties. The OTUs tables are available in Tables S2 and S3.
Root bacterial communities were significantly richer than those of the stem (Fig. S2A) and the structure and composition of these bacterial communities were also significantly different (Fig. S2C). Additionally, while the α diversity indices (measured by the richness index and Shannon index) of fungal root and stem communities were not significantly different (Fig. S2B), the fungal community compositions were significantly different between the stem and root compartments, albeit to a lower degree than that observed for the bacterial communities (Fig. S2C/D). These results remain consistent with previous observations in other species that the strongest determinant of the microbial community compositions is the compartment from which these are drawn (Knief et al., 2012; Lundberg et al., 2012; Coleman‐Derr et al., 2016).
Wide inter‐individual variation in α diversity of microbial communities associated with HHRTS rice plants
The root and stem microbial community of HHRTS rice plants were subsequently studied separately to detect potential impacts on these of the shifting composition of rice plant populations that is currently occurring in the HHRTS (landraces vs. modern varieties). Alpha diversity indices were not significantly different between modern rice varieties and HHRTS landraces (Fig. 2) when considering either bacterial or fungal communities in either the stem or root samples. Additionally, the six‐subgroups partitioning revealed a low variability in alpha diversity indices among HHRTS rice subgroups even if the indices of α diversity between landraces subgroups 3 and 4 were significantly different (Fig. S3). It should be stressed, however, that the α diversity indices estimated across landraces and modern varieties, or for each of the six subgroups of landraces and modern varieties were highly variable (Fig. 2 and Fig. S3), suggesting that factors other than rice genotype may be shaping the richness of microbial communities at the individual rice plant level. Such wide inter‐individual variations in α diversity are reminiscent of the inter‐individual variability of microbial communities observed previously in human saliva. This variability in saliva has been interpreted as a possible consequence of the human mouth being exposed to highly variable external (food) and internal (digestion exchanges) environments, and being subjected to a wide variety of oral hygiene regimes (Hall et al., 2017; Verma et al., 2018). Accordingly, our results suggest that individual rice plants within a field may have encountered during their lifetimes a variety of physical and chemical environmental conditions that have left an imprint on the composition of their associated microbial communities. The impacts of heterogeneous micro‐environments on rice‐associated microbial community structures may be particularly pertinent to flooded rice systems in general (Fernández‐Valiente and Quesada, 2004; Cui et al., 2008; Xie et al., 2011; Jiao et al., 2012), and specifically to the HHRTS flooded rice system, where micro‐environment variability could conceivably arise for a multitude of different reasons including (i) the high densities at which rice is planted; (ii) the compactness of the terraces; (iii) the low depth of the floodwaters within the terraces; (iv) the variability of ancillary food production systems that are co‐located in the terraces which can include ducks, fish, frogs, and snails; and (v) erratic increases in the levels of suspended soil particles in the floodwaters arising as a consequence of periodic disturbances of silt by labourers and water buffalos within the paddies.
Fig 2.
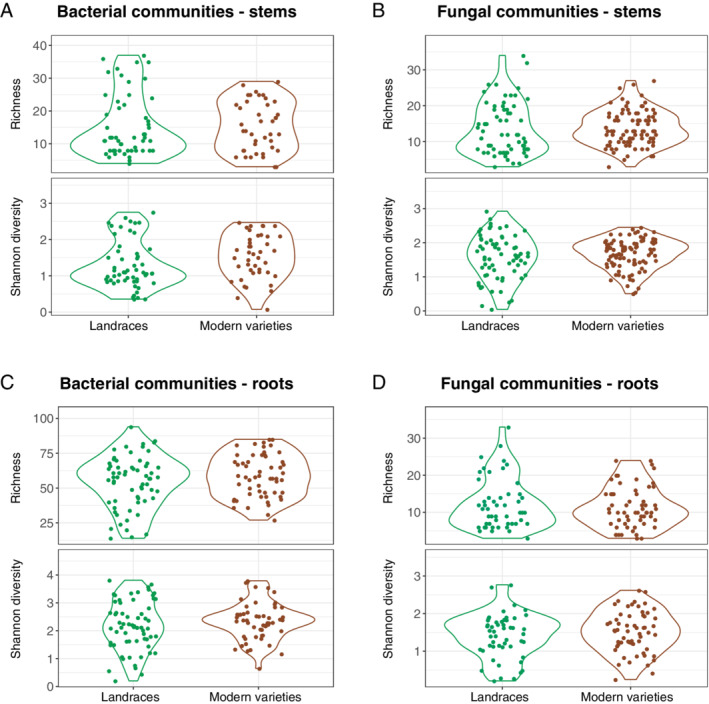
Violin plots of rice microbial α diversity (richness and Shannon diversity indexes) across HHRTS landraces and modern rice varieties for (A) the rice stem bacterial communities and (B) the rice stem fungal communities as well as (C) the rice root bacterial communities and (D) the rice root fungal communities. P‐values of Tukey HSD tests were all >0.05 between landrace and modern communities whatever the community (bacteria or fungi) and the diversity indexes.
Microbial community structure of modern rice varieties and HHRTS landraces are slightly different
We further examined differences between the microbial community compositions associated with modern rice varieties and those associated with HHRTS landraces. We first calculated the ß‐diversity and used a permutational multivariate analysis of variance (PERMANOVA) of microbial communities between modern rice varieties and HHRTS landraces for both stem and root samples using phylogeny‐based UniFrac distances either weighted or unweighted by the abundance of OTUs. These PERMANOVA analyses revealed a slight (R 2 values ranged from 0.03 to 0.06), but significant effect of variety type for both stem and root bacterial and fungal communities using the unweighted UniFrac distances (Fig. 3). This finding supports the hypothesis that the introduction of modern rice varieties in the HHRTS could have caused a slight modification in the structure of rice‐associated microbial communities. This slight effect is consistent with findings from recent studies on other plant species (Emmett et al., 2017; Hamonts et al., 2018; Compant et al., 2019). In contrast, the quantitative weighted UniFrac distances did not support a difference between the structures of HHRTS landrace microbial communities and the modern variety microbial communities (Fig. S4). This result suggests that the slight microbial variations observed between both rice groups in the Malizhai HHRTS are primarily driven by differences in the distributions of rare taxa between the microbial community associated with the HHRTS landrace and modern variety groups.
Fig 3.
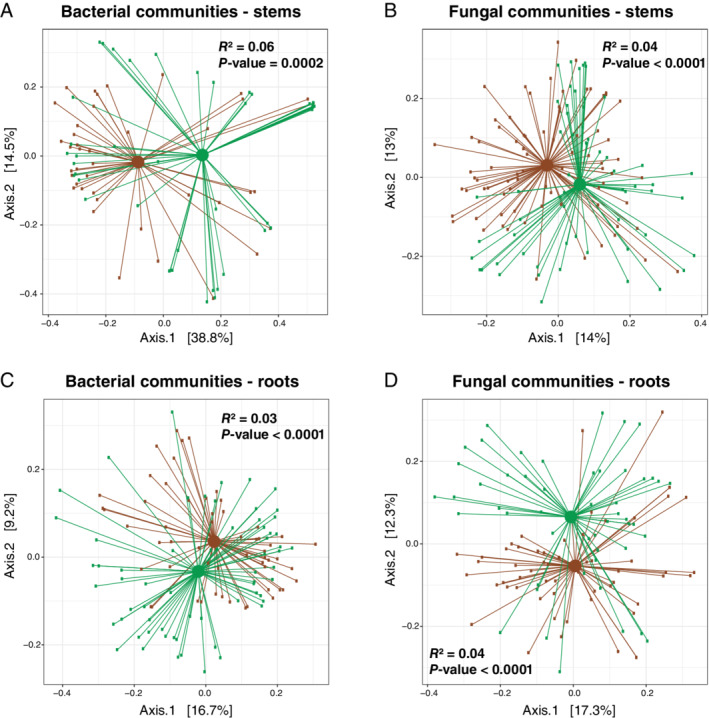
Principal coordinates analysis (PCoA) plots based on unweighted UniFrac distances of (A) rice stem bacterial communities and (B) rice stem fungal communities as well as (C) rice root bacterial communities and (D) rice root fungal communities. Communities from the HHRTS landraces and from the modern rice varieties are labelled in green and brown respectively. Axes represent the two dimensions explaining the greatest proportion of variance in the communities for each analysis. Permutational multivariate analysis of variance (PERMANOVA) results are indicated (R 2 and the P‐value).
Rice variety is a key factor of the structure of the microbial community in Malizhai HHRTS
We examined differences between the microbial community compositions associated with the six rice subgroups of landraces and modern varieties that were sampled in 2016 in the Malizhai HHRTS and identified using clustering algorithms assuming K = 8 clusters (Fig. 4). The PERMANOVA analysis revealed a significant effect of the rice subgroup (R 2 values ranged from 0.14 to 0.22, P < 0.0001) for both stem and root bacterial and fungal communities using the unweighted UniFrac distances (Fig. 4). This result suggests that rice subgroups, each composed of related rice varieties, play a role in structuring the HHRTS microbial communities. This finding is in line with results from another study that revealed that genotypic differences in rice had a significant effect on root‐associated microbial communities (Edwards et al., 2015). Principal Coordinate Analyses (PCoA) based on unweighted UniFrac distances showed that bacterial communities from both stems and roots, and fungal communities from the stems of the modern subgroups and landrace subgroup1 (corresponding to the governmental‐promoted HongYang varieties) were more similar to each other and farther from the three other landraces subgroups (Fig. 4). The pairwise PERMANOVA analysis using the unweighted UniFrac distances further confirmed that the bacterial communities from roots were not significantly different between both modern rice subgroups and between the modern rice subgroup2 and the landrace subgroup1 (Table S4). This result suggests that varietal improvement for higher yields or pest resistance that was derived either from improved local landraces (HongYang varieties, landrace subgroup1) or from exogenous rice varieties (modern high‐yielding rice hybrids) has led to a partial homogenization of microbial communities among the improved varieties grown in the Malizhai HHRTS. This convergence in microbial communities can be explained by shared selected features of improved rice varieties such as their root morphology or their root exudation (or other traits) that have induced the assembly of similar microbial communities in the Malizhai HHRTS context (Szoboszlay et al., 2015; Perez‐Jaramillo et al., 2018; Cordovez et al., 2019). Based on these results, we tested whether partitioning the Malizhai HHRTS varieties into ‘improved varieties’ (including modern and HongYang varieties) and ‘traditional landraces’ (including landraces subgroups 2, 3 and 4) would have revealed a higher degree of population structuring in the Malizhai HHRTS microbial communities. The PERMANOVA analysis revealed a slight (R 2 values ranged from 0.04 to 0.05), but significant effect of variety type for both stem and root bacterial and fungal communities using the unweighted UniFrac distances, suggesting that the ‘varietal improvement’ factor explain less the structure of microbial communities than the ‘rice genotype’ factor.
Fig 4.
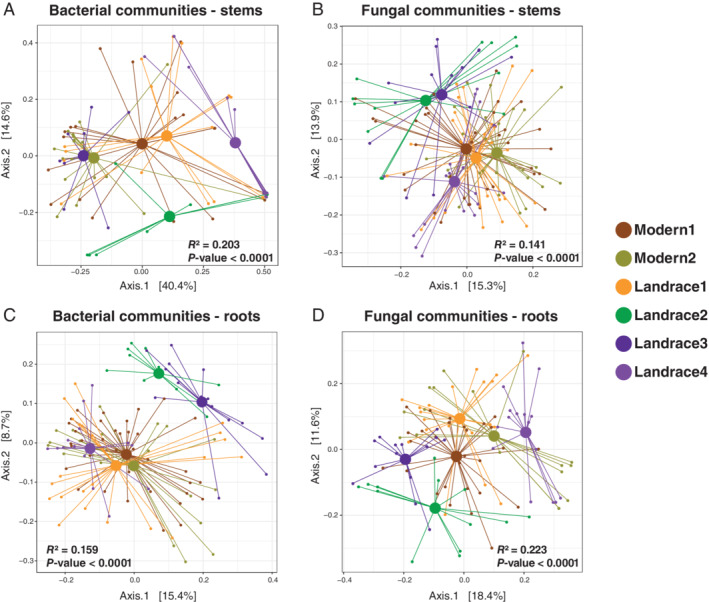
Principal coordinate analysis (PCoA) plots based on unweighted UniFrac distances of (A) rice stem bacterial communities, (B) rice stem fungal communities, (C) rice root bacterial communities and (D) rice root fungal communities. Communities associated with the six subgroups of HHRTS rice varieties are labelled in orange (HHRTS landraces subgroup1), green (HHRTS landraces subgroup2), blue (HHRTS landraces subgroup3), purple (HHRTS landraces subgroup4), brown (modern subgroup1) and olive green (modern subgroup2) respectively. Axes represent the two dimensions explaining the greatest proportion of variance in the communities for each analysis. Permutational multivariate analysis of variance (PERMANOVA) results are indicated (R 2 and the P‐values).
We further compared the relative abundance of individual OTUs between the six rice subgroups of landraces and modern varieties using Wilcoxon signed‐rank tests. We then used the Metacoder approach, which relies on a taxonomic tree‐based visualization (called a heat tree), to illustrate whether the relative abundance of microbial OTUs differed significantly between the six rice subgroups (Figs [Link], [Link], [Link] and S8). These analyses highlighted 243 OTUs with significantly different (with a P‐value threshold <0.05) degrees of relative abundance between the six rice subgroups (Tables S5 and S6). Only two fungal OTUs of the 243 OTUs were more abundant in stems of all four rice subgroups of traditional and improved HHRTS varieties than in both modern rice subgroups (Table S6), suggesting a reduced core microbial community of rice varieties cultivated in the Malizhai HHRTS.
Phylogenetic distances between the microbial community and rice genotypes were correlated
Given that we found differences between the microbial communities of modern rice varieties and HHRTS landraces, we investigated whether more closely related genotypes tend to have more similar microbial communities. We conducted Mantel tests using the unweighted UniFrac distances of OTUs and genetic distances between rice genotypes. All four of the unweighted UniFrac distance matrices of microbial communities were significantly correlated with the genetic distances between the rice genotypes they are associated with (Table S7). This result is consistent with previous studies focusing on wild and modern rice varieties (Shenton et al., 2016; Kim et al., 2020), maize and other Poaceae (Bouffaud et al., 2014) suggesting that the evolutionary history of poaceous crop plants has had an impact on the evolutionary history of their microbial communities.
The core microbial community associated with HHRTS rice plants encompassed a small number of taxa
We hypothesized that the ‘core’ microbial community in the stem and root of Malizhai HHRTS rice plants is limited in size. To test this hypothesis, we considered OTUs only present in at least 80% of the stem and root samples from plants belonging to the 18 indica varieties (including all the modern varieties and HHRTS landraces), with no consideration of the relative abundance of the taxa. The core stem microbial community consisted of just two fungal OTUs and one bacterial OTU and the core root microbial community consisted of just two fungal OTUs and three bacterial OTUs (Table 2). We further considered separately the stem and root core microbial communities of the modern rice varieties and HHRTS landraces. The core stem microbial community of modern rice varieties consisted of two fungal OTUs and the HHRTS landraces core stem microbial community included these two‐same fungal OTUs and one additional bacterial OTU (Table 2 and Table S8). Whereas the core root microbial community of the modern rice varieties consisted of six bacterial OTUs and one fungal OTU that of the HHRTS landraces consisted of two bacterial OTUs and one fungal OTU (Table 2 and Table S8). Consequently, the core microbial community defined by our criteria ranged from 0.7% to 3.3% of the total number of taxa identified within and among the HHRTS landraces and modern rice varieties (Table 2). These low percentages are consistent with another study that identified a core microbial community constituted of 1.5% of the total OTUs associated with rice plants sampled from three Californian rice fields (Edwards et al., 2015). The relative abundances of the ‘core’ OTUs associated with the Malizhai HHRTS rice varieties accounted for between 0.7% and 26.4% of the total number of assigned sequencing reads (see relative abundances in Table S8). The core microbial community, therefore, consisted of a combination of rare and abundant OTUs (Table S8). Besides revealing a high heterogeneity of HHRTS microbial community, our study also indicates that the HHRTS rice holobiont [host with its associated microbes that can potentially affect the phenotypes of rice plants (Vandenkoornhuyse et al., 2015; Theis et al., 2016)] is highly diverse with microbial communities mirroring the diversity their rice hosts.
Table 2.
The proportion of core reads and core OTUs within and among the rice genetic groups.
| Community | Plant compartment | Genotype group | Number of core reads | Total number of reads | Proportion of core reads | Number of core OTUs | Total number of OTUs | Proportion of core OTUs |
|---|---|---|---|---|---|---|---|---|
| Bacterial | Stem | Modern + HHRTS landraces | 18 703 | 101 850 | 18.3 | 1 | 91 | 1.0 |
| Modern | 0 | 44 100 | 0.0 | 0 | 63 | 0.0 | ||
| HHRTS landraces | 11 233 | 57 750 | 19.5 | 1 | 81 | 1.2 | ||
| Root | Modern + HHRTS landraces | 41 838 | 120 666 | 34.7 | 3 | 325 | 0.9 | |
| Modern | 28 417 | 56 784 | 50.0 | 6 | 240 | 2.5 | ||
| HHRTS landraces | 23 336 | 63 882 | 36.5 | 2 | 288 | 0.7 | ||
| Fungal | Stem | Modern + HHRTS landraces | 13 737 | 45 240 | 30.4 | 2 | 110 | 1.8 |
| Modern | 8185 | 20 590 | 39.8 | 2 | 60 | 3.3 | ||
| HHRTS landraces | 5501 | 24 650 | 22.3 | 2 | 91 | 2.2 | ||
| Root | Modern + HHRTS landraces | 6534 | 30 858 | 21.2 | 2 | 105 | 1.9 | |
| Modern | 2729 | 13 344 | 20.4 | 1 | 71 | 1.4 | ||
| HHRTS landraces | 1898 | 17 514 | 10.8 | 1 | 79 | 1.3 |
Towards disentangling the biotic and abiotic factors affecting the Malizhai HHRTS rice microbial community
Our results show that the population structure of Malizhai HHRTS rice varieties is one of the factors that impact the community structure of associated microbes. However, even if the genetic makeup of rice hosts explained the largest proportion of variance in the composition of microbial communities in Malizhai HHRTS (15%–22%), other factors not covered by our study may also explain the observed patterns of microbial diversity. Indeed, the small sampling area selected (<2 km2) was more environmentally heterogeneous than initially envisioned. It is known that plant root microbial communities are influenced by soil cultivation histories and agricultural practices (Peiffer et al., 2013; Li et al., 2019). Hence, the turnover of rice varieties in HHRTS fields and other characteristics of the flooded terraces—such as the use of living animal fertilizers (ducks, snails, fish and cattle)—may also explain the heterogeneity of the microbial communities of different rice plants (Xie et al., 2011; Jiao et al., 2012). Precise information pertaining to the soil compositions or fertilizer usage history of our sampling sites is not available. However, previous surveys of farming practices in the portion of the HHRTS that we studied revealed that farmers employ a variety of cropping practices and frequently alter the diverse set of rice varieties that they use (Dedeurwaerdere and Hannachi, 2019).
Another factor that could have contributed to environmental heterogeneity is the presence of rice pathogens. Several recent studies have indicated both that the impact of microbial communities on the course and outcome of host diseases can be substantial, and that host diseases can reciprocally have a large impact on microbial community compositions (Berendsen et al., 2012; Ritpitakphong et al., 2016; Koskella et al., 2017; Zhang et al., 2018; Vannier et al., 2019). We have recently shown that the SRBSDV was prevalent in 2016 in the Malizhai HHRTS and that 23 of the 166 rice plants examined in the present study were infected by SRBSDV (Alonso et al., 2019). We, therefore, examined the impact of SRBSDV infections on the compositions of microbial communities but no significant difference was observed between the microbial communities of SRBSDV‐infected rice plants and those of SRBSDV‐uninfected plants (Table S9). Interestingly, several of the HHRTS landraces and modern rice varieties had no SRBSDV infected plants. If these landraces and varieties are either resistant or tolerant to SRBSDV (Alonso et al., 2019), it is most probable that this resistance would be attributable to host genetic factors, i.e. host resistance, rather than to their respective microbial communities. SRBSDV is transmitted by a flying insect such that root microbial communities are less likely to play a significant role in SRBSDV transmission than in the transmission of soil‐borne rice pathogens.
Finally, in addition to deterministic factors, like host genotype that did explain the largest proportion of variance in the compositions of rice microbial communities, and other factors not covered by this study, we cannot rule out that stochastic processes could have also influenced microbial community compositions. Indeed, stochastic processes might account for the high variability in microbial community compositions that were observed both between plants belonging to the same rice genotypes and between plants sampled from the same HHRTS paddy fields (Zhou and Ning, 2017). Stochasticity could for instance arise during the colonization of plants by microbes wherever early arriving taxa modify the surface or within‐plant niches, making these niches more or less suitable for later‐arriving species (Maignien et al., 2014). Therefore, besides better disentangling the roles that biotic and abiotic factors play in modifying the microbial community of HHRTS rice plants, further studies will be needed to determine the impacts over time of stochastic processes on dynamic changes in rice microbial community compositions.
Materials and methods
Study area and sampling site
The village of Malizhai is located near the town of Xinjie in the Honghe Hani Yi Autonomous Prefecture (Yunnan province, China). This village has recently adopted a mixed landrace/modern variety system but, prior to 2010 was representative of the cultural landscape of the Hani rice terraces (Jiao et al., 2012). The sampling site in Malizhai covered an area of approximately 2 km2 (N23°07′55.04″ E102°46′03.95″) at an altitude ranging between 1570 and 1608 m.
Plant sampling
Nineteen small Malizhai paddy fields were sampled in July 2016, including 11 fields cultivated with HHRTS landraces and eight fields cultivated with “modern rice varieties. Ten plants were collected per field, regardless of the presence of disease symptoms. It is noteworthy that at the time of the sampling survey disease pressure was very low and a very few symptoms were visible on rice plants throughout the Malizhai terraces. Roots, stems and leaves were separately collected and rinsed with water in order to remove the attached soil. All samples were individually stored at 4°C in a mobile fridge. Within 24 h, all samples were dried in the presence of CaCl2 until DNA extraction.
Plant and microbial DNA extractions
Plant DNA extractions and genotyping‐by‐sequencing GBS were carried out on 30 mg of dried individual leaf samples as described previously (Alonso et al., 2019). We performed microbial DNA extractions on each stem sample with both epiphyte and endophyte microorganisms and on each root sample with its rhizoplane (root surface) and endophyte microorganisms. In total, 30 mg of the 190 sample rice stems and the 190 sample rice roots were individually frozen in liquid nitrogen then ground with bead beating (two steel beads, diameter 0.1 cm and one ceramic bead, diameter 0.5 cm) using a FastPrep‐24 5G System (MP Biomedicals ‐ Fisher Scientific) for 2 × 30 s at 6 ms−1. Total genomic DNA was extracted from the resulting powder using the NucleoMag Plant Kit (Macherey–Nagel, Germany) and KingFisher Flex Purification System (ThermoFisher Scientific, MA, USA), following the manufacturer's instructions.
Analysis of rice genotyping‐by‐sequencing data
GBS reads for rice landraces and whole‐genome re‐sequencing data for nine randomly selected individuals from four representative subpopulations of indica (XI‐1A, XI‐1B, XI‐2 and XI‐3) described in the 3000 Rice Genomes Project (Wang et al., 2018) were aligned against the Nipponbare rice reference genome (MSU7) using Bowtie2.3.5 (Langmead et al., 2009; Langmead and Salzberg, 2012; Wang et al., 2018; Langmead et al., 2019). The GBS raw sequence read data are available from the NCBI Sequence Read Archive, under the BioProject ID number PRJNA573048. SNP‐calling using bcftools mpileup (options ‐‐max‐depth 500 ‐a DP) was carried out independently for the 3000 Rice Genomes Project dataset and the GBS dataset (https://www.sanger.ac.uk/science/tools/samtools-bcftools-htslib). For the GBS dataset, sites with either AN ≤ 300 or MQ ≤ 20 or which were tagged as ‘LowQual’ were removed using bcftools filter (INFO/AN ≤ 300; INFO/MQ ≤ 20) and bash (grep ‐v ‘LowQual’). Genotypes with DP ≤ 5 were converted to missing data using vcftools (‐‐minDP 5). For the 3000 Rice Genomes Project dataset, sites with DP ≤ 5 or MQ ≤ 20 were filtered out using bcftools filter (INFO/DP ≤ 5; INFO/MQ ≤ 20). The VCF files for the 3000 Rice Genomes Project and GBS accessions were merged using bcftools merge. The merged dataset included 143 611 sites with ≤50% missing data and 10 028 sites with ≤10% missing data. We used the dataset with ≤10% missing data for analyses of genealogical relationships among genotypes.
We constructed a neighbour‐network using SplitsTree 4.13, to visualize evolutionary relationships between the indica rice genotypes while taking the possibility of recombination or incomplete lineage sorting into account (Huson and Bryant, 2006). We used the program sNMF (http://membres-timc.imag.fr/Olivier.Francois/snmf/index.htm) to test the dataset with ≤50% missing data for evidence of population subdivision by partitioning genotypes into K ancestral populations and estimating individual ancestry coefficients in the K populations (Frichot et al., 2014). We ran sNMF for a number of clusters K‐values ranging from 1 to 15, and for each K we performed 10 replicates. We used the Greedy algorithm in CLUMPP 1.1.2 (http://web.stanford.edu/group/rosenberglab/clumpp.html) (Jakobsson and Rosenberg, 2007) to identify runs representing the same clustering solution (i.e. same mode). We randomly selected one representative of runs belonging to the major mode for graphical representation as a stacked barplot using the Matplotlib package in Python. Nucleotide diversity (π) was estimated using the scikit‐allel in Python (https://github.com/cggh/scikit-allel).
PCR amplification and sequencing
The composition and diversity of rice‐associated microbial communities were characterized by applying a high‐throughput sequencing‐based protocol that targets PCR‐generated amplicons. Bacterial communities were characterized from the variable region, V3‐V4, of the 16S rRNA gene using the primers 341‐F (5′CTACGGGNGGCWGCAG 3′) and 785‐R (5′GACTACHVGGGTATCTAATCC3′) as universal primers to maximize bacterial taxonomic assignment (Thijs et al., 2017). We used ITS86‐F (5′GTGAATCATCGAATCTTTGAA3′) and ITS4‐R (5′ TCCTCCGCTTATTGATATGC3′) as universal primers for amplification of the fungal ITS1 region (Op De Beeck et al., 2014).
DNA amplification was performed by PCR in a total volume of 25 μl containing 1× GoTaq G2 DNA polymerase buffer (Promega Corporation, Madison, USA), 0.5 μM of each primer, and 0.2 μM dNTPs and 1 μl of genomic DNA. We used peptide nucleotide acid (PNA) clamps, which specifically bind to mitochondrial or chloroplast sequences to block the amplification of plant derived‐DNA. We added to the PCR mix PNA blocker oligos (PNA Bio, Thousand Oaks, CA, USA) at 0.5 μM targeting the 16S rRNA gene of plant mitochondria (PNAm: GGCAAGTGTTCTTCGGA), and chloroplasts (PNSp: GGCTCAACCCTGGACAG) (Jackrel et al., 2017). All amplifications were performed in a thermal cycler (Biometra, Gottingen, Germany) under the following conditions for 16S rRNA gene amplifications: an initial denaturation at 98°C for 3 min followed by 30 cycles of denaturation at 98°C for 15 s, PNA annealing at 75°C for 10 s, primers annealing at 52°C for 10 s, extension at 72°C for 30 s, and a final extension at 72°C for 10 min. ITS amplifications were performed under the following conditions: an initial denaturation at 94°C for 3 min followed by 30 cycles of denaturation at 94°C for 45 s, primers annealing at 55°C for 45 s, extension at 72°C for 2 min, and a final extension at 72°C for 10 min. Each PCR product was tagged with a combination of two different barcodes designed by a genomic platform (GenSeq, University of Sciences, UMII, Montpellier, France) that allows for the identification of 384 different PCR products loaded onto the same MiSeq flow cell. Negative controls from the extraction step and PCR reaction were sequenced with the plant samples to evaluate and exclude contaminant reads from the sample data set. All PCR products were pooled and purified, and the library was constructed and sequenced using a GenSeq platform with Illumina paired‐end 2 × 250‐bp technology and V2 chemistry.
Sequence processing, OTU clustering, and OTU filtering
Base calling and demultiplexing of Illumina sequences were carried out using RTA v1.18.54, MCS 2.6 and bcl2fastq2.17. Paired Illumina MiSeq reads were assembled with VSEARCH v2.11.0 (Rognes et al., 2016) using the command fastq_mergepairs and the option fastq_allowmergestagger. Primer clipping was performed with cutadapt v1.9 (Martin, 2011) allowing a 2/3‐length partial match for forward and reverse primers. Only reads containing both primers were retained. The expected error per read was estimated with the VSEARCH command fastq_filter and the option eeout. Each sample was then dereplicated by merging identical reads using vsearch's command, derep_fulllength, and converted to FASTA format. To prepare for clustering, the samples were pooled and further dereplicated with VSEARCH. Files containing per‐read expected error values were also dereplicated to retain only the lowest expected error for each unique sequence. Clustering was performed with Swarm v3.0.0 (Mahe et al., 2015), using a local threshold of one difference and the fastidious option.
OTU representative sequences were then searched for evidence of chimeras with the VSEARCH command, uchime_denovo (Edgar et al., 2011). In parallel, representative sequences were assigned to taxa using the stampa pipeline (https://github.com/frederic-mahe/stampa/) and the ribosomal database SILVA v138 (https://www.arb-silva.de/) (Quast et al., 2013) for the bacterial community, and a custom version of the ITS database UNITE v8 (https://unite.ut.ee/) (Abarenkov et al., 2010; Koljalg et al., 2013) for the fungal community.
Clustering results, expected error values, taxonomic assignments and chimera detection results were used to build a raw OTU table. Up to that point, reads without primers, reads shorter than 32 nucleotides and reads with uncalled bases (‘N’) had been eliminated. To create the ‘cleaned’ OTU table, additional filters were applied to retain only non‐chimeric OTUs, OTUs with an expected error per nucleotide below 0.0002, OTUs containing more than three reads or which were found two or more samples. ITS and 16S OTUs tables obtained after the initial cleaning step (chimeric OTUs removal) can be found at https://github.com/P‐alonso/HHRTS_microbial_diversit (Yunnan_Rice_2016_16S_roots_and_stems_384_samples.OTU.filtered.table, Yunnan_Rice_2016_ITS2_roots_and_stems_384_samples.OTU.filtered.table). All 16S and ITS sequences with a higher abundance in at least one negative control than the rice samples were excluded from the final dataset. All 16S OTUs assigned to chloroplast or mitochondrial sequences were excluded. Similarly, ITS OTUs not assigned to fungal reference sequences were excluded. All codes and representative OTU sequences can be found in HTML format in Supporting information file S1. The raw data are available from the NCBI Sequence Read Archive (SRA) under BioProject ID number PRJNA573048.
Statistical analyses of microbial community data
All statistical analyses were performed in R (http://www.R-project.org) with the Phyloseq (McMurdie and Holmes, 2013), Vegan (Oksanen et al., 2018), pairwiseAdonis (Martinez Arbizu, 2020) and Metacoder packages. Biological replicates corresponding to 10 plants per field were used to fix a biological reproducibility threshold in order to perform the sequencing de‐noising. Only OTUs with over 50% prevalence per paddy field were considered. Samples with less than 1000 reads for bacterial communities and less than 5000 reads for fungal communities were discarded following which the OTUs abundances were rarefied to homogenize sequencing depth.
We estimated the microbial diversity using richness and Shannon's diversity indices for α‐diversity calculations and UniFrac distances for β‐diversity calculations. To assess the relationships between plant genotypes (HHRTS landraces vs. modern rice varieties) and plant‐associated microbial community, a Tukey HSD test and a PERMANOVA were performed for the α‐diversity and the β‐diversity respectively. Differences in community composition between groups of samples were statistically evaluated by PERMANOVA using the UniFrac distance matrices. The adonis() function was used to calculate PERMANOVA with 10 000 permutations between HHRTS and modern varieties. A pairwise post hoc comparison was performed to evaluate the difference in community compositions across the six rice subgroups (HHRTS landraces subgroup1, HHRTS landraces subgroup2, HHRTS landraces subgroup3, HHRTS landraces subgroup4, modern subgroup1 and modern subgroup2) using the pairwise.adonis() function. Differences in community composition were assessed by PCoA based on weighted UniFrac and unweighted UniFrac distances. PCoA is an ordination method that represents pairwise (dis)similarities between samples in a low‐dimensional space, so that samples placed closer in the graph are more similar than those placed further apart (McMurdie and Holmes, 2013). R codes used in statistical analyses are provided at https://github.com/P-alonso/HHRTS_microbial_diversit/.
The Metacoder package was used to visualize differential abundances in taxa between the modern varieties and HHRTS landrace groups using the function compare_groups() among plant microbial communities on a differential heat tree. The ratio of the mean OTU abundance between landraces and modern rice varieties was calculated. For each taxon, a Wilcoxon Rank Sum test was used to test for differences between the median abundances of samples between landraces and modern rice varieties. The taxon abundance of bacterial and fungal communities was plotted on a taxonomic tree and the result of the Wilcoxon Rank Sum test with a fold‐change cutoff of 1.5 and a P‐value cutoff of 0.05 were used to highlight the differences in taxon abundance between HHRTS landraces and modern rice varieties. To test for significant associations between microbial community dissimilarities and the phylogenetic distances between rice plants that hosted these communities, we conducted partial Mantel tests, as implemented in the vegan package in R (Oksanen et al., 2018) between the unweighted UniFrac distances of microbial communities and the patristic distances of rice plants calculated with the cophenetic() function.
The core microbial taxa within the stem and root microbial communities were identified using the Microbiome R package based on a criterion of prevalence in at least 80% of the samples from the 18 indica varieties (including all the modern varieties and HHRTS landraces) with no criterion related to the relative abundance of the taxa, in order to consider rare but prevalent microbial taxa. A second analysis focused on stem and root microbial taxa identified from each group of rice genotypes using the same criterion. Based on this criterion, a list of core taxa was identified and their relative abundance was calculated.
Supporting information
File S1 Codes and bioinformatics methods used to produce OTU tables after the initial cleaning (chimeric OTUs removal) (HTML format).
Fig. S1 Relationship between minimal cross‐entropy and number of ancestral populations (K) modelled in the sNMF analysis of population subdivision.
Fig. S2 Violin plots of rice microbial alpha diversity (richness and Shannon diversity indexes) of (A) root and stem bacterial communities and (B) roots and stems fungal communities. P‐values of Tukey HSD tests are shown with ***P < 0.001 or not shown if P > 0.05. PCoA plots based on unweighted UniFrac distances of (C) root and stem bacterial communities and (D) root and stem fungal communities. Communities identified from stems and roots are coloured in blue and red respectively. Axes represent the two dimensions explaining the greatest proportion of variances in the communities for each analysis. Permutational multivariate analysis of variance (PERMANOVA) results are indicated (R2 and the P‐value).
Fig. S3 Violin plots of rice microbial α diversity (richness and Shannon diversity indices) across the six subgroups of rice sampled in the HHRTS (HHRTS landraces subgroup1 (L1), HHRTS landraces subgroup2 (L2), HHRTS landraces subgroup3 (L3), HHRTS landraces subgroup4 (L4), modern subgroup1 (M1) and modern subgroup2 (M2)) for (A) the rice stem bacterial communities, (B) the rice stem fungal communities, (C) the rice root bacterial communities, and (D) the rice root fungal communities. Results from Tukey HSD tests are presented as letters denoting groups that are significantly different (P‐values <0.05).
Fig. S4 PCoA plots based on weighted UniFrac distances of (A) rice stem bacterial communities and (C) rice stem fungal communities as well as (B) rice roots bacterial communities and (D) rice roots fungal communities. Communities from the HHRTS landraces and from the modern rice varieties are labelled in green and brown respectively. Axes represent the two dimensions explaining the greatest proportion of variances in the communities for each analysis. Results of a permutational multivariate analysis of variance (PERMANOVA) are indicated (R2 and the P‐value).
Fig. S5 Pairwise comparison of number of reads of rice stem bacterial communities assigned to bacterial species among the six HHRTS rice subgroups. The node width indicates the number of reads assigned to each taxonomic rank level along the tree and the colour indicates the statistically significant differences in relative bacterial taxa abundance. A taxon coloured brown is more abundant in the communities in the column and a taxon coloured green is more abundant in the communities in the row.
Fig. S6 Pairwise comparison of number of reads of rice root bacterial communities assigned to bacterial species among the six HHRTS rice subgroups. The node width indicates the number of reads assigned to each taxonomic rank level along the tree and the colour indicates the statistically significant differences in relative bacterial taxa abundance. A taxon coloured brown is more abundant in the communities in the column and a taxon coloured green is more abundant in the communities in the row.
Fig. S7 Pairwise comparison of number of reads of rice stem fungal communities assigned to fungal species among the six HHRTS rice subgroups. The node width indicates the number of reads assigned to each taxonomic rank level along the tree and the colour indicates the statistically significant differences in relative fungal taxa abundance. A taxon coloured brown is more abundant in the communities in the column and a taxon coloured green is more abundant in the communities in the row.
Fig. S8 Pairwise comparison of number of reads of rice root fungal communities assigned to fungal species among the six HHRTS rice subgroups. The node width indicates the number of reads assigned to each taxonomic rank level along the tree and the colour indicates the statistically significant differences in relative fungal taxa abundance. A taxon coloured brown is more abundant in the communities in the column and a taxon coloured green is more abundant in the communities in the row.
Table S1 Abundance of reads and OTUs in bacterial and fungal community data sets for stem and root samples through the different steps of the bio‐informatics treatments.
Table S2 Abundance of bacterial OTUs in the rice communities of modern varieties (YYM) and HHRTS landraces (YYT) obtained from root (R as last letter) or stem (T as last letter) and taxonomic assignation. Different numbers indicate different rice paddies. Cleaned data obtained after the rarefaction process.
Table S3 Abundance of fungal OTUs in the rice communities of modern varieties (YYM) and HHRTS landraces (YYT) obtained from root (R as last letter) or stem (T as last letter) and taxonomic assignation. Different numbers indicate different rice paddies. Cleaned data obtained after the rarefaction process.
Table S4 Pairwise permutational multivariate analysis of variance (PERMANOVA) results for bacterial and fungal communities for both stem and root samples using unweighted UniFrac distance values (10,000 permutations), R2 denotes the proportion of variance that could be explained by the grouping. P. adjusted corresponds to the Bonferroni correction applied to adjust the P‐value for multiple comparisons within each group. Sig significance level *P. adjusted <0.05, **P. adjusted <0.01 and NS not significant.
Table S5 Bacterial taxa from stems and roots that are significantly differentially abundant between the six rice subgroups using Wilcoxon signed rank tests. Only, significant P‐values <0.05 are indicated.
Table S6 Fungal taxa from stems and roots that are significantly differentially abundant between the six rice subgroups using Wilcoxon signed rank tests. Only, significant P‐values <0.05 are indicated.
Table S7 Detection of phylogenetic signal in root‐associated and stem‐associated microbial communities. Mantel statistic based on Pearson's product–moment correlation to assess the correlation between unweighted UniFrac distances matrix of microbial community dissimilarities and the rice genetic distances (10,000 permutations), R2 denotes the proportions of variances that could be explained by the grouping.
Table S8 List of the taxa that were present in more than 80% of the plant samples of each rice genetic group.
Table S9 Permutational multivariate analysis of variance (PERMANOVA) results for the influence of infection of SRBSDV on bacterial and fungal composition using UniFrac distance values (10,000 permutations), R2 denotes the proportions of variances that could be explained by the grouping.
Acknowledgements
This study was funded by the French International and Agricultural Research Agency (CIRAD), the Agropolis Fondation (E‐Space Flagship Program, grant number 1504‐004), CGIAR Research Program (CRP) on rice agri‐food systems (RICE, 2017‐2022) and the French National Research Agency (Next Generation Biomonitoring Project, grant number ANR‐17‐CE32‐0011). We thank the Yunnan Agricultural University and the International Associated Laboratory Plantomix (INRAE/Yunnan Agricultural University) for their technical logistics and support. D.P.M. has received a research grant from the National Research Foundation of South Africa.
References
- Abarenkov, K. , Henrik Nilsson, R. , Larsson, K.‐H. , Alexander, I.J. , Eberhardt, U. , Erland, S. , et al (2010) The UNITE database for molecular identification of fungi – recent updates and future perspectives. New Phytol 186: 281–285. [DOI] [PubMed] [Google Scholar]
- Alonso, P. , Gladieux, P. , Moubset, O. , Shih, P.J. , Mournet, P. , Frouin, J. , et al (2019) Emergence of southern rice black‐streaked dwarf virus in the centuries‐old Chinese Yuanyang Agrosystem of rice landraces. Viruses 11: v11110985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen, R.L. , Pieterse, C.M. , and Bakker, P.A. (2012) The rhizosphere microbiome and plant health. Trends Plant Sci 17: 478–486. [DOI] [PubMed] [Google Scholar]
- Bernardo, P. , Charles‐Dominique, T. , Barakat, M. , Ortet, P. , Fernandez, E. , Filloux, D. , et al (2018) Geometagenomics illuminates the impact of agriculture on the distribution and prevalence of plant viruses at the ecosystem scale. ISME J 12: 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouffaud, M.L. , Poirier, M.A. , Muller, D. , and Moenne‐Loccoz, Y. (2014) Root microbiome relates to plant host evolution in maize and other Poaceae . Environ Microbiol 16: 2804–2814. [DOI] [PubMed] [Google Scholar]
- Bulgarelli, D. , Garrido‐Oter, R. , Munch, P.C. , Weiman, A. , Droge, J. , Pan, Y. , et al (2015) Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17: 392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulgarelli, D. , Schlaeppi, K. , Spaepen, S. , Ver Loren van Themaat, E. , and Schulze‐Lefert, P. (2013) Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol 64: 807–838. [DOI] [PubMed] [Google Scholar]
- Cardinale, M. , Grube, M. , Erlacher, A. , Quehenberger, J. , and Berg, G. (2015) Bacterial networks and co‐occurrence relationships in the lettuce root microbiota. Environ Microbiol 17: 239–252. [DOI] [PubMed] [Google Scholar]
- Coleman‐Derr, D. , Desgarennes, D. , Fonseca‐Garcia, C. , Gross, S. , Clingenpeel, S. , Woyke, T. , et al (2016) Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol 209: 798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compant, S. , Samad, A. , Faist, H. , and Sessitsch, A. (2019) A review on the plant microbiome: ecology, functions, and emerging trends in microbial application. J Adv Res 19: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordovez, V. , Dini‐Andreote, F. , Carrion, V.J. , and Raaijmakers, J.M. (2019) Ecology and evolution of plant microbiomes. Annu Rev Microbiol 73: 69–88. [DOI] [PubMed] [Google Scholar]
- Cui, B. , You, Z. , and Yao, M. (2008) Vertical characteristics of the Hani terraces paddyfield ecosystem in Yunnan, China. Front Biol China 3: 351–359. [Google Scholar]
- Dedeurwaerdere, T. , and Hannachi, M. (2019) Socio‐economic drivers of coexistence of landraces and modern crop varieties in agro‐biodiversity rich Yunnan rice fields. Ecol Econ 159: 177–188. [Google Scholar]
- Ding, L.J. , Cui, H.L. , Nie, S.A. , Long, X.E. , Duan, G.L. , and Zhu, Y.G. (2019) Microbiomes inhabiting rice roots and rhizosphere. FEMS Microbiol Ecol 95: fiz040. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. , Haas, B.J. , Clemente, J.C. , Quince, C. , and Knight, R. (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, J. , Johnson, C. , Santos‐Medellin, C. , Lurie, E. , Podishetty, N.K. , Bhatnagar, S. , et al (2015) Structure, variation, and assembly of the root‐associated microbiomes of rice. Proc Natl Acad Sci U S A 112: E911–E920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, J.A. , Santos‐Medellin, C.M. , Liechty, Z.S. , Nguyen, B. , Lurie, E. , Eason, S. , et al (2018) Compositional shifts in root‐associated bacterial and archaeal microbiota track the plant life cycle in field‐grown rice. PLoS Biol 16: e2003862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmett, B.D. , Youngblut, N.D. , Buckley, D.H. , and Drinkwater, L.E. (2017) Plant phylogeny and life history shape rhizosphere bacterial microbiome of summer annuals in an agricultural field. Front Microbiol 8: 2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Valiente, E. , and Quesada, A. (2004) A shallow water ecosystem: rice‐fields. The relevance of cyanobacteria in the ecosystem. Limnetica 23: 95–107. [Google Scholar]
- Frichot, E. , Mathieu, F. , Trouillon, T. , Bouchard, G. , and Francois, O. (2014) Fast and efficient estimation of individual ancestry coefficients. Genetics 196: 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacquard, S. , Spaepen, S. , Garrido‐Oter, R. , and Schulze‐Lefert, P. (2017) Interplay between innate immunity and the plant microbiota. Annu Rev Phytopathol 55: 565–589. [DOI] [PubMed] [Google Scholar]
- Hall, M.W. , Singh, N. , Ng, K.F. , Lam, D.K. , Goldberg, M.B. , Tenenbaum, H.C. , et al (2017) Inter‐personal diversity and temporal dynamics of dental, tongue, and salivary microbiota in the healthy oral cavity. NPJ Biofilms Microbiomes 3: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamonts, K. , Trivedi, P. , Garg, A. , Janitz, C. , Grinyer, J. , Holford, P. , et al (2018) Field study reveals core plant microbiota and relative importance of their drivers. Environ Microbiol 20: 124–140. [DOI] [PubMed] [Google Scholar]
- Hassani, M.A. , Özkurt, E. , Seybold, H. , Dagan, T. , and Stukenbrock, E.H. (2019) Interactions and coadaptation in plant metaorganisms. Annu Rev Phytopathol 57: 483–503. [DOI] [PubMed] [Google Scholar]
- Huson, D.H. , and Bryant, D. (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23: 254–267. [DOI] [PubMed] [Google Scholar]
- Jackrel, S.L. , Owens, S.M. , Gilbert, J.A. , and Pfister, C.A. (2017) Identifying the plant‐associated microbiome across aquatic and terrestrial environments: the effects of amplification method on taxa discovery. Mol Ecol Resour 17: 931–942. [DOI] [PubMed] [Google Scholar]
- Jakobsson, M. , and Rosenberg, N.A. (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806. [DOI] [PubMed] [Google Scholar]
- Jiao, Y. , Li, X. , Liang, L. , Takeuchi, K. , Okuro, T. , Zhang, D. , and Sun, L. (2012) Indigenous ecological knowledge and natural resource management in the cultural landscape of China's Hani terraces. Ecol Res 27: 247–263. [Google Scholar]
- Keesing, F. , Holt, R.D. , and Ostfeld, R.S. (2006) Effects of species diversity on disease risk. Ecol Lett 9: 485–498. [DOI] [PubMed] [Google Scholar]
- Kim, H. , Lee, K.K. , Jeon, J. , Harris, W.A. , and Lee, Y.‐H. (2020) Domestication of Oryza species eco‐evolutionarily shapes bacterial and fungal communities in rice seed. Microbiome 8: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knief, C. , Delmotte, N. , Chaffron, S. , Stark, M. , Innerebner, G. , Wassmann, R. , et al (2012) Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J 6: 1378–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koljalg, U. , Nilsson, R.H. , Abarenkov, K. , Tedersoo, L. , Taylor, A.F. , Bahram, M. , et al (2013) Towards a unified paradigm for sequence‐based identification of fungi. Mol Ecol 22: 5271–5277. [DOI] [PubMed] [Google Scholar]
- Koskella, B. , Meaden, S. , Crowther, W.J. , Leimu, R. , and Metcalf, C.J.E. (2017) A signature of tree health? Shifts in the microbiome and the ecological drivers of horse chestnut bleeding canker disease. New Phytol 215: 737–746. [DOI] [PubMed] [Google Scholar]
- Langmead, B. , and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , Trapnell, C. , Pop, M. , and Salzberg, S.L. (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , Wilks, C. , Antonescu, V. , and Charles, R. (2019) Scaling read aligners to hundreds of threads on general‐purpose processors. Bioinformatics 35: 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leff, J.W. , Lynch, R.C. , Kane, N.C. , and Fierer, N. (2017) Plant domestication and the assembly of bacterial and fungal communities associated with strains of the common sunflower, Helianthus annuus . New Phytol 214: 412–423. [DOI] [PubMed] [Google Scholar]
- Li, X. , Jousset, A. , de Boer, W. , Carrion, V.J. , Zhang, T. , Wang, X. , and Kuramae, E.E. (2019) Legacy of land use history determines reprogramming of plant physiology by soil microbiome. ISME J 13: 738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, J. , Huang, H. , Meusnier, I. , Adreit, H. , Ducasse, A. , Bonnot, F. , et al (2016) Pathogen effectors and plant immunity determine specialization of the blast fungus to rice subspecies. Elife 5: e19377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg, D.S. , Lebeis, S.L. , Paredes, S.H. , Yourstone, S. , Gehring, J. , Malfatti, S. , et al (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488: 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahe, F. , Rognes, T. , Quince, C. , de Vargas, C. , and Dunthorn, M. (2015) Swarm v2: highly‐scalable and high‐resolution amplicon clustering. PeerJ 3: e1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maignien, L. , DeForce, E.A. , Chafee, M.E. , Eren, A.M. , and Simmons, S.L. (2014) Ecological succession and stochastic variation in the assembly of Arabidopsis thaliana phyllosphere communities. mBio 5: e00682‐00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M.P. (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal 17: 200. [Google Scholar]
- Martinez Arbizu, P. (2020) pairwiseAdonis: Pairwise multilevel comparison using adonis. R package version 0.4.
- McMurdie, P.J. , and Holmes, S. (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8: e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, R.S. , and Purugganan, M.D. (2013) Evolution of crop species: genetics of domestication and diversification. Nat Rev Genet 14: 840–852. [DOI] [PubMed] [Google Scholar]
- Moronta‐Barrios, F. , Gionechetti, F. , Pallavicini, A. , Marys, E. , and Venturi, V. (2018) Bacterial microbiota of rice roots: 16S‐based taxonomic profiling of endophytic and rhizospheric diversity, endophytes isolation and simplified endophytic community. Microorganisms 6: 6010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch, L.A. , and Young, J.P. (2004) Diversity and specificity of Rhizobium leguminosarum biovar viciae on wild and cultivated legumes. Mol Ecol 13: 2435–2444. [DOI] [PubMed] [Google Scholar]
- Ofek‐Lalzar, M. , Sela, N. , Goldman‐Voronov, M. , Green, S.J. , Hadar, Y. , and Minz, D. (2014) Niche and host‐associated functional signatures of the root surface microbiome. Nat Commun 5: 4950. [DOI] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F.G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. et al. (2018) Vegan: Community Ecology Package. R package version 2.5.3.
- Op De Beeck, M. , Lievens, B. , Busschaert, P. , Declerck, S. , Vangronsveld, J. , and Colpaert, J.V. (2014) Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS One 9: e97629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer, J.A. , and Ley, R.E. (2013) Exploring the maize rhizosphere microbiome in the field: a glimpse into a highly complex system. Commun Integr Biol 6: e25177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer, J.A. , Spor, A. , Koren, O. , Jin, Z. , Tringe, S.G. , Dangl, J.L. , et al (2013) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci U S A 110: 6548–6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Jaramillo, J.E. , Carrion, V.J. , de Hollander, M. , and Raaijmakers, J.M. (2018) The wild side of plant microbiomes. Microbiome 6: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , and Yarza, P. (2013) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redford, A.J. , Bowers, R.M. , Knight, R. , Linhart, Y. , and Fierer, N. (2010) The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ Microbiol 12: 2885–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold‐Hurek, B. , Bunger, W. , Burbano, C.S. , Sabale, M. , and Hurek, T. (2015) Roots shaping their microbiome: global hotspots for microbial activity. Annu Rev Phytopathol 53: 403–424. 10.1146/annurev-phyto-082712-102342. [DOI] [PubMed] [Google Scholar]
- Ritpitakphong, U. , Falquet, L. , Vimoltust, A. , Berger, A. , Metraux, J.P. , and L'Haridon, F. (2016) The microbiome of the leaf surface of Arabidopsis protects against a fungal pathogen. New Phytol 210: 1033–1043. [DOI] [PubMed] [Google Scholar]
- Rognes, T. , Flouri, T. , Nichols, B. , Quince, C. , and Mahe, F. (2016) VSEARCH: a versatile open source tool for metagenomics. PeerJ 4: e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roossinck, M.J. , and Garcia‐Arenal, F. (2015) Ecosystem simplification, biodiversity loss and plant virus emergence. Curr Opin Virol 10: 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota, R. , Knorr, K. , Jorgensen, L.N. , O'Hanlon, K.A. , and Nicolaisen, M. (2015) Host genotype is an important determinant of the cereal phyllosphere mycobiome. New Phytol 207: 1134–1144. [DOI] [PubMed] [Google Scholar]
- Shenton, M. , Iwamoto, C. , Kurata, N. , and Ikeo, K. (2016) Effect of wild and cultivated rice genotypes on rhizosphere bacterial community composition. Rice 9: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, S. , Chang, J. , Tian, L. , Nasir, F. , Ji, L. , Li, X. , and Tian, C. (2019) Comparative analysis of the rhizomicrobiome of the wild versus cultivated crop: insights from rice and soybean. Arch Microbiol 201: 879–888. [DOI] [PubMed] [Google Scholar]
- Stuckenbrock, E.H. , and McDonald, B.A. (2008) The origins of plant pathogens in agro‐ecosystems. Annu Rev Phytopathol 46: 75–100. [DOI] [PubMed] [Google Scholar]
- Szoboszlay, M. , Lambers, J. , Chappell, J. , Kupper, J.V. , Moe, L.A. , and McNear, D.H. (2015) Comparison of root system architecture and rhizosphere microbial communities of Balsas teosinte and domesticated corn cultivars. Soil Biol Biochem 80: 34–44. [Google Scholar]
- Theis, K.R. , Dheilly, N.M. , Klassen, J.L. , Brucker, R.M. , Baines, J.F. , Bosch, T.C. , et al (2016) Getting the hologenome concept right: an eco‐evolutionary framework for hosts and their microbiomes. mSystems 1: 00028‐00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijs, S. , Op De Beeck, M. , Beckers, B. , Truyens, S. , Stevens, V. , Van Hamme, J.D. , et al (2017) Comparative evaluation of four bacteria‐specific primer pairs for 16S rRNA gene surveys. Front Microbiol 8: 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UNESCO . (2013) Convention concerning the protection of the world cultural and natural heritage In WHC‐13/37COM/20. Phnom Penh, Cambodia: UNESCO, p. 248. [Google Scholar]
- Vandenkoornhuyse, P. , Quaiser, A. , Duhamel, M. , Le Van, A. , and Dufresne, A. (2015) The importance of the microbiome of the plant holobiont. New Phytol 206: 1196–1206. [DOI] [PubMed] [Google Scholar]
- Vannier, N. , Agler, M. , and Hacquard, S. (2019) Microbiota‐mediated disease resistance in plants. PLoS Pathog 15: e1007740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma, D. , Garg, P.K. , and Dubey, A.K. (2018) Insights into the human oral microbiome. Arch Microbiol 200: 525–540. [DOI] [PubMed] [Google Scholar]
- Vorholt, J.A. (2012) Microbial life in the phyllosphere. Nat Rev Microbiol 10: 828–840. [DOI] [PubMed] [Google Scholar]
- Wagner, M.R. , Lundberg, D.S. , Del Rio, T.G. , Tringe, S.G. , Dangl, J.L. , and Mitchell‐Olds, T. (2016) Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat Commun 7: 12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Mauleon, R. , Hu, Z. , Chebotarov, D. , Tai, S. , Wu, Z. , et al (2018) Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 557: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Zhai, Y. , Cao, L. , Tan, H. , and Zhang, R. (2016) Endophytic bacterial and fungal microbiota in sprouts, roots and stems of rice (Oryza sativa L.). Microbiol Res 188‐189: 1–8. [DOI] [PubMed] [Google Scholar]
- Xie, J. , Hu, L. , Tang, J. , Wu, X. , Li, N. , Yuan, Y. , et al (2011) Ecological mechanisms underlying the sustainability of the agricultural heritage rice‐fish coculture system. Proc Natl Acad Sci U S A 108: E1381–E1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L. , Liu, M. , Lun, F. , Yuan, Z. , Zhang, Y. , and Min, Q. (2017) An analysis on crops choice and its driving factors in agricultural heritage systems—a case of Honghe Hani Rice terraces system. Sustainability 9: 1162. [Google Scholar]
- Zhang, Z. , Luo, L. , Tan, X. , Kong, X. , Yang, J. , Wang, D. , et al (2018) Pumpkin powdery mildew disease severity influences the fungal diversity of the phyllosphere. PeerJ 6: e4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , and Ning, D. (2017) Stochastic community assembly: does it matter in microbial ecology? Microbiol Mol Biol Rev 81: e00002‐00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y. , Chen, H. , Fan, J. , Wang, Y. , Li, Y. , Chen, J. , et al (2000) Genetic diversity and disease control in rice. Nature 406: 718–722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1 Codes and bioinformatics methods used to produce OTU tables after the initial cleaning (chimeric OTUs removal) (HTML format).
Fig. S1 Relationship between minimal cross‐entropy and number of ancestral populations (K) modelled in the sNMF analysis of population subdivision.
Fig. S2 Violin plots of rice microbial alpha diversity (richness and Shannon diversity indexes) of (A) root and stem bacterial communities and (B) roots and stems fungal communities. P‐values of Tukey HSD tests are shown with ***P < 0.001 or not shown if P > 0.05. PCoA plots based on unweighted UniFrac distances of (C) root and stem bacterial communities and (D) root and stem fungal communities. Communities identified from stems and roots are coloured in blue and red respectively. Axes represent the two dimensions explaining the greatest proportion of variances in the communities for each analysis. Permutational multivariate analysis of variance (PERMANOVA) results are indicated (R2 and the P‐value).
Fig. S3 Violin plots of rice microbial α diversity (richness and Shannon diversity indices) across the six subgroups of rice sampled in the HHRTS (HHRTS landraces subgroup1 (L1), HHRTS landraces subgroup2 (L2), HHRTS landraces subgroup3 (L3), HHRTS landraces subgroup4 (L4), modern subgroup1 (M1) and modern subgroup2 (M2)) for (A) the rice stem bacterial communities, (B) the rice stem fungal communities, (C) the rice root bacterial communities, and (D) the rice root fungal communities. Results from Tukey HSD tests are presented as letters denoting groups that are significantly different (P‐values <0.05).
Fig. S4 PCoA plots based on weighted UniFrac distances of (A) rice stem bacterial communities and (C) rice stem fungal communities as well as (B) rice roots bacterial communities and (D) rice roots fungal communities. Communities from the HHRTS landraces and from the modern rice varieties are labelled in green and brown respectively. Axes represent the two dimensions explaining the greatest proportion of variances in the communities for each analysis. Results of a permutational multivariate analysis of variance (PERMANOVA) are indicated (R2 and the P‐value).
Fig. S5 Pairwise comparison of number of reads of rice stem bacterial communities assigned to bacterial species among the six HHRTS rice subgroups. The node width indicates the number of reads assigned to each taxonomic rank level along the tree and the colour indicates the statistically significant differences in relative bacterial taxa abundance. A taxon coloured brown is more abundant in the communities in the column and a taxon coloured green is more abundant in the communities in the row.
Fig. S6 Pairwise comparison of number of reads of rice root bacterial communities assigned to bacterial species among the six HHRTS rice subgroups. The node width indicates the number of reads assigned to each taxonomic rank level along the tree and the colour indicates the statistically significant differences in relative bacterial taxa abundance. A taxon coloured brown is more abundant in the communities in the column and a taxon coloured green is more abundant in the communities in the row.
Fig. S7 Pairwise comparison of number of reads of rice stem fungal communities assigned to fungal species among the six HHRTS rice subgroups. The node width indicates the number of reads assigned to each taxonomic rank level along the tree and the colour indicates the statistically significant differences in relative fungal taxa abundance. A taxon coloured brown is more abundant in the communities in the column and a taxon coloured green is more abundant in the communities in the row.
Fig. S8 Pairwise comparison of number of reads of rice root fungal communities assigned to fungal species among the six HHRTS rice subgroups. The node width indicates the number of reads assigned to each taxonomic rank level along the tree and the colour indicates the statistically significant differences in relative fungal taxa abundance. A taxon coloured brown is more abundant in the communities in the column and a taxon coloured green is more abundant in the communities in the row.
Table S1 Abundance of reads and OTUs in bacterial and fungal community data sets for stem and root samples through the different steps of the bio‐informatics treatments.
Table S2 Abundance of bacterial OTUs in the rice communities of modern varieties (YYM) and HHRTS landraces (YYT) obtained from root (R as last letter) or stem (T as last letter) and taxonomic assignation. Different numbers indicate different rice paddies. Cleaned data obtained after the rarefaction process.
Table S3 Abundance of fungal OTUs in the rice communities of modern varieties (YYM) and HHRTS landraces (YYT) obtained from root (R as last letter) or stem (T as last letter) and taxonomic assignation. Different numbers indicate different rice paddies. Cleaned data obtained after the rarefaction process.
Table S4 Pairwise permutational multivariate analysis of variance (PERMANOVA) results for bacterial and fungal communities for both stem and root samples using unweighted UniFrac distance values (10,000 permutations), R2 denotes the proportion of variance that could be explained by the grouping. P. adjusted corresponds to the Bonferroni correction applied to adjust the P‐value for multiple comparisons within each group. Sig significance level *P. adjusted <0.05, **P. adjusted <0.01 and NS not significant.
Table S5 Bacterial taxa from stems and roots that are significantly differentially abundant between the six rice subgroups using Wilcoxon signed rank tests. Only, significant P‐values <0.05 are indicated.
Table S6 Fungal taxa from stems and roots that are significantly differentially abundant between the six rice subgroups using Wilcoxon signed rank tests. Only, significant P‐values <0.05 are indicated.
Table S7 Detection of phylogenetic signal in root‐associated and stem‐associated microbial communities. Mantel statistic based on Pearson's product–moment correlation to assess the correlation between unweighted UniFrac distances matrix of microbial community dissimilarities and the rice genetic distances (10,000 permutations), R2 denotes the proportions of variances that could be explained by the grouping.
Table S8 List of the taxa that were present in more than 80% of the plant samples of each rice genetic group.
Table S9 Permutational multivariate analysis of variance (PERMANOVA) results for the influence of infection of SRBSDV on bacterial and fungal composition using UniFrac distance values (10,000 permutations), R2 denotes the proportions of variances that could be explained by the grouping.