

Background
Copy number variants (CNVs) are genetic rearrangements, such as deletions and duplications, which result in a deviation from the normal number of copies of a given gene segment. CNVs are implicated in many neuropsychiatric disorders. Deletions of the human chromosomal region 16p11.2 are one of the most common genetic linkages to autism spectrum disorders (ASD). However, ASD is not the only presenting feature, and many patients with 16p11.2 deletions present with a variable clinical spectrum.
Methods
To better understand the nature and presentation of the syndrome throughout development, we present three different, unrelated clinical cases of children with 16p11.2 deletion and provide a detailed description of their clinical manifestations.
Results
Cognitive and motor impairments were characteristic of all three patients with 16p11.2 deletion, despite the differences in the extent and clinical presentation of impairment. Two patients had a clinical diagnosis of ASD and one showed several ASD traits. In addition, two patients also had severe speech and language impairments, which is in line with previous reports on 16p11.2 phenotypes. Although epilepsy and obesity have been frequently associated with 16p11.2 deletion, only one patient had a diagnosis of epilepsy and none of the three cases were obese.
Conclusion
This variation in clinical phenotype renders correct clinical interpretation and diagnosis challenging. Therefore, it is critical to elucidate the variable clinical phenotypes of rare CNVs, including 16p11.2 deletions, to help guide clinical monitoring and counselling of patients and families.
Keywords: autism spectrum disorder, case reports, DNA copy number variations, 16p11.2 deletion syndrome
Introduction
Copy number variants (CNVs) represent recurrent chromosomal abnormalities and are associated with various phenotypic features (Lupski, 1998; Shinawi et al., 2010; Jensen and Girirajan, 2019). Chromosome 16 is particularly rich in highly homologous low-copy repeats (LCRs), that mediate such genetic rearrangements via LCR-mediated nonallelic homologous recombination events during meiosis (Loftus et al., 1999; Stankiewicz and Lupski, 2002; Martin et al., 2004). Deletions of the human chromosomal region 16p11.2 are one of the most common genetic linkages to autism spectrum disorders (ASD) and account for approximately 1% of the cases (Kumar et al., 2008; Weiss et al., 2008; Fernandez et al., 2010). Indeed patients with duplications of 16p11.2 appear to have a similar odds ratio to developing ASD as 16p11.2 deletions but interestingly the duplication carriers have a higher odds ratio of developing attention deficit hyperactivity disorder and a higher frequency of developing overall psychiatric disorders and psychosis (Niarchou et al., 2019). However, ASD is not the only presenting feature in many patients with 16p11.2 deletion and only 25% of individuals with the deletion exhibit an autism phenotype (Hanson et al.; Jensen and Girirajan, 2019). 16p11.2 deletions are associated with a variable clinical spectrum of neurocognitive phenotypes and many other patients may manifest intellectual disability, morbid obesity, large head circumference, or epilepsy at varying degrees of penetrance (Bijlsma et al., 2009; Bochukova et al., 2010; Shinawi et al., 2010). Even unaffected carriers of these CNVs are associated with cognitive deficits which may be subtle but confer significant disadvantages in educational attainment and ability to earn income in adulthood (Kendall et al., 2019) as well as resulting in profound effects on physical health and mortality, even in those who have less profound early neurodevelopmental consequences (Crawford et al., 2019). Early identification is therefore vital to allow appropriate medical and neuropsychiatric screening, support and treatment throughout the lifespan.
As with other microdeletion syndromes, the clinical heterogeneity and incomplete penetrance in 16p11.2 patients are quite remarkable which renders correct clinical interpretation and diagnosis challenging. To provide patients with a better clinical experience and ensure an enhanced developmental outcome, 16p11.2 deletion cases should be thoroughly phenotyped clinically. Several studies have presented a detailed phenotypic characterisation of individuals with 16p11.2 imbalances at the time of diagnosis revealing a variable clinical spectrum of neurocognitive phenotypes which included macrocephaly, developmental and language delay, cognitive impairment, as well as seizures (Fernandez et al., 2010; Hanson et al., 2010; Shinawi et al., 2010). Another study has provided an elaborate description of the neurologic phenotypes of 16p11.2 deletion cases via comprehensive neurologic evaluation showing that despite the overlap with other neurodevelopmental disorders, speech and motor impairments, growth abnormalities, and tremors are striking features of 16p11.2 deletions that clinicians should be aware of (Steinman et al., 2016). Here, we present three different, unrelated clinical cases of children with 16p11.2 deletion and provide a detailed description of their clinical manifestations from birth and throughout development. Despite the limited number of cases described here, presenting the complete developmental history and progression of the symptoms will help better understand the nature and presentation of the syndrome throughout development and thereby assist in accurate clinical diagnosis and early interventions.
Methods
This study was approved by the School of Philosophy, Psychology and Language Sciences (PPLS) Research Ethics Committee of the University of Edinburgh (reference number 286-1819/2). Participants were recruited through online platforms, such as Simons Variation in Individuals Project (Simons VIP) connect social group. A thorough explanation of the study was provided to the guardians of the three children, and they gave their informed consent to participate. Parents were then interviewed in person or through video chat (example: Skype) if they were not residents of the UK, and were asked questions on (1) the clinical symptoms of their children throughout development, (2) any medications being taken, (3) any relevant family history and finally (4) social/clinical measures taken by parent/guardian since diagnosis with 16p11.2 deletion.
Results
Case 1
An 11-year-old girl presented with a diagnosis of hypermobility, memory retention difficulties (both short-term and long-term), social difficulties and epilepsy. At birth, she was reported to have a large head circumference. At 6 months of age, she had her first epileptic seizure and continued to have episodes every other month until she was diagnosed with generalised idiopathic tonic-clonic epilepsy. At 13 months, she was administered sodium valproate (Epilim) for the treatment of epilepsy. At 16 months, her parent started noticing signs of impaired mobility, such as lack of balance and frequent falling. By 3 years of age, her valproate medication was stopped because she had stopped having seizures but this resulted in a longer seizure that kept reoccurring over a period of 7 h. She was treated with phenytoin and put back on her former medication.
At school, this patient started showing signs of delayed learning and impaired memory retention as well as weight fluctuations. Symptoms of impaired mobility (imbalance, inward toeing while walking and continuous falling) still persisted and at 8 years old, she was diagnosed with hypermobility. By the age of 9, she was referred to an endocrinologist who offered genetic testing. Array comparative genomic hybridisation revealed a deletion in the proximal region of the genomic locus 16p11.2 (29 673 953–30 198 600), spanning approximately 524 kb. At this time, the patient continues to have moderate–severe learning and memory impairments. She is also presented with several ASD traits, although no official ASD diagnosis has been made. For example, she struggles with change of routine and gets emotionally attached to objects. Moreover, she has sensory difficulties. She dislikes loud noises, music or over-populated areas. The patient also struggles with social nuance and social cues. According to her parent, she has a very low opinion of herself and struggles to regulate her emotions. Her parent also continues to notice absence seizures, but no official diagnosis has been made.
Case 2
A 5-year-old girl presented with a diagnosis of ASD, developmental delay, speech delay and anxiety. The mother was reported to have complications during pregnancy. She haemorrhaged the night before her due date and the following afternoon she was induced. At birth, the child had low birth weight (2.88 kg) and was supported in a temperature-controlled incubator. At the age of 3, her parents started noticing signs of speech and motor delay; however, a diagnosis of ASD was not met at this stage. She was then supported by an occupational therapist, to develop and strengthen fine and gross motor skills. However, at 4 years, she was diagnosed with ASD, global delay and anxiety and was offered genetic testing by her paediatrician. Molecular karyotyping results revealed a deletion in chromosome 16, region 16p11.2 (29 634 212–30 199 805). This spans approximately 565 kb. Currently, she is not on any medications. Her parents have taken environmental measures to ensure appropriate lighting and noise and provided her with weighted and sensory toys. The patient continues to show speech and motor impairments, with no reports of seizures.
Case 3
An 8-year-old boy presented with a diagnosis of ASD together with childhood apraxia of speech. He was born with low birth weight (2.60 kg) and slight macrocephaly. From 3 to 6 months, he kept losing weight. Between 6 and 10 months, he was diagnosed with rickets and was given vitamin D supplements. By the age of 2, the patient showed symptoms of social and emotional delay and was offered occupational therapy, physical therapy and speech therapy. He was then referred to a neurologist who diagnosed him with ASD around the age of 3. The patient also showed severe language impairments, whereby the age of 3 he could only speak three words and communicated with his parents through sign language for 2 years. He was also reported to have a few episodes of febrile seizures; however, EEG tests showed no signs of epilepsy and seizures resolved during childhood. Following that, he was offered genetic testing by his paediatrician. Chromosomal microarray analysis revealed a deletion in the short arm of chromosome 16; particularly at the region 16p11.2 (29 567 295–30 177 916) which spans approximately 611 kb.
Between 4 and 5 years, the patient started exhibiting repetitive movements as well as verbal/phonetic tics triggered by calling his name. This included foot rubbing to full leg rubbing, refusing to walk on lines and not being able to walk through light beams shining through a window. Currently, he is not on any medications. He has a poor muscle tone, slight scoliosis and continues to show speech and language impairments.
Discussion
Recurrent 16p11.2 deletions are characterised by a spectrum of neurocognitive phenotypes that are variably expressed amongst patients. We describe herein the phenotypes of three subjects with the deletion of the genomic region within chromosome 16p11.2. Table 1 provide a summary of the different clinical manifestations and clinical data of the patients together with the genetic locus of the deletions. The effects of 16p11.2 deletion on mean intelligence quotient have been shown to be a decrease of approximately 2 SDs (D’Angelo et al., 2016; Hippolyte et al., 2016) and intellectual disability is more frequent amongst deletion carriers (Niarchou et al., 2019). Similarly, 16p11.2 deletion carriers showed the worst performance in a series of cognitive tests and were associated with statistically significant reductions in the number of offspring, suggesting deficits in socialising and forming families, as a consequence of cognitive, medical and behavioural problems (Kendall et al., 2019). Cognitive and motor impairments were characteristic of all three patients with 16p11.2 deletion, despite the differences in extent and clinical presentation of impairment. This is in line with the clinical findings of other studies on 16p11.2 deletion carriers (Hanson et al., 2010, 2015; Shinawi et al., 2010; Zufferey et al., 2012). The 16p11.2 deletion has been repeatedly associated with ASD, and accounts for approximately 1% of ASD cases (Weiss et al., 2008; Kumar et al., 2008; Fernandez et al., 2010). Moreover, ASD has been shown to be the second most prevalent diagnosis in 16p11.2 deletion carriers (Niarchou et al., 2019). In this study, two patients had a clinical diagnosis of ASD and one showed several ASD traits. Two of the patients also had severe speech and language impairments, which have been reported in previous phenotypic characterisation of patients with 16p11.2 deletions (Shimojima et al., 2009; Hanson et al., 2010; Shinawi et al., 2010; Schaaf et al., 2011; Zufferey et al., 2012). The 16p11.2 deletion region has also been associated with obesity (Kumar et al., 2008; Bochukova et al., 2010; Walters et al., 2010), but none of the three cases described here were obese. On the other hand, two patients were reported to be born with very low birthweights and weight fluctuations during early childhood. Although epilepsy is the most frequent neurological disorder observed in 16p11.2 deletion carriers (Shimojima et al., 2009; Hanson et al., 2010; Shinawi et al., 2010; Schaaf et al., 2011; Zufferey et al., 2012), only one patient had a diagnosis of epilepsy whereas the other two had no reports of seizures or seizures had resolved with age. Similarly, previous reports have shown that congenital anomalies, including vertebral and spinal-related anomalies (Zufferey et al., 2012), to be strongly associated with deletion carriers, scoliosis was reported in only one patient.
Table 1.
Summary of clinical data of the three cases throughout development
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Premature birth | No (1 day before due date) | No (1 day after due date) | No |
| Birth weight | Average (3.30 kg) | Low (2.88 kg) | Low (2.60 kg) |
| Head circumference at birth | N/Aa | 33.3 cm | N/Aa |
| Cognitive impairment | Yes: moderate-severe learning and memory impairments | Yes: global developmental delay, anxiety | Yes: social and emotional delay |
| Motor impairments | Yes: hypermobility, imbalance, inward-towing | Yes: motor delay | Yes: poor muscle tone |
| Language impairments | None | Yes: speech delay | Yes: childhood apraxia of speech |
| Childhood illnesses diagnosed | Hypermobility, memory retention and social difficulties, generalised idiopathic tonic-clonic epilepsy | ASD, developmental delay, speech delay and anxiety | ASD, childhood apraxia of speech, rickets |
| ASD diagnosis | No (but shows some traits) | Yes | Yes |
| Seizures | Yes: generalised idiopathic tonic-clonic epilepsy | None | Resolved: Febrile seizures |
| Medications | Sodium valproate (Epilim) | None | None |
| Age at genetic testing | 9 years | 4 years | 3 year |
| Deletion locus | 16p11.2 (29 673 953–30 198 600) | 16p11.2 (29 634 212–30 199 805) | 16p11.2 (29 567 295–30 177 916) |
N/A = measurements unavailable.
All three patients had deletion in the proximal region of the 16p11.2 locus (Fig. 1). This genomic region spans around 600 kb and contains 47 genes, 28 of which are protein-coding. The flanking LCRs each span 147 kb and contain six duplicated genes, five of which are annotated as protein-coding (Weiss et al., 2008; Jacquemont et al., 2011; Tai et al., 2016). NAHR-mediated CNV formation in vivo involves the mispairing of the flanking LCRs, which can result in either the loss or gain of a 740-kb segment equivalent to one copy of the 593-kb segment and one copy-equivalent of the LCR (Walters et al., 2010). Several studies have demonstrated a dosage effect of 16p11.2 copy number on the various clinical findings, suggesting the presence of dosage-sensitive genes within the region (Weiss et al., 2008; Shinawi et al., 2010; Blumenthal et al., 2014). Moreover, several genes within the 16p11.2 are promising candidates for the variable phenotypes reported in patients with the deletion. For example, MAPK3 is a synaptic signalling component which is reported to be necessary for several forms of learning (Mazzucchelli et al., 2002; Pucilowska et al., 2015) and quinolinate phosphoribosyltransferase encodes a key enzyme in catabolism of quinolinate, a potent endogenous exitotoxin to neurons, the elevation of which has been linked to the pathogenesis of epilepsy (Feldblum et al., 1988; Haslinger et al., 2018). Polymorphisms in the TBX6 gene were associated with congenital scoliosis in the Han population (Wu et al., 2015). Mice homozygous for a TBX6 mutation were also reported to show rib and vertebral body anomalies (Watabe-Rudolph et al., 2002), suggesting that TBX6 is a candidate gene for vertebral malformations. On a molecular level, knockdown and overexpression studies revealed the implications of several genes like MAPK3, KCTD13, MVP and TAOK2 that play critical roles in the growth and proliferation of progenitor cells as well as neurite morphogenesis in the pathology of ASD (Golzio et al., 2012; de Anda et al., 2012; Pucilowska et al., 2015). Although the functions of other genes in this CNV remain poorly defined, these studies suggest that the dysregulation of cell cycle, neuronal migration and cortical lamination is fundamental for the pathogenesis of neurodevelopmental disorders, including ASD, in the developing brain (Hanson et al., 2010; de Anda et al., 2012; Packer, 2016; Casanova, 2014). Recently, it has been shown by modelling the 16p11.2 deletion in Drosophila melanogaster that a complex interaction of CNV genes operates in conserved pathways to modulate expression of the phenotype, many suppressing or enhancing cell proliferation pathways and are enriched in a human brain-specific network, providing translational relevance in humans (Iyer et al., 2018). It is suggested that this CNV has pleiotropic effects, given that it intersects multiple genes. Recently, significantly associated phenotypes and medical consequences to 16p11.2 deletion in adult carriers included a high incidence of diabetes, osteoarthritis, hypertension, as well as asthma, anaemia and renal problems, indicating the need for regular medical monitoring (Crawford et al., 2019).
Fig. 1.
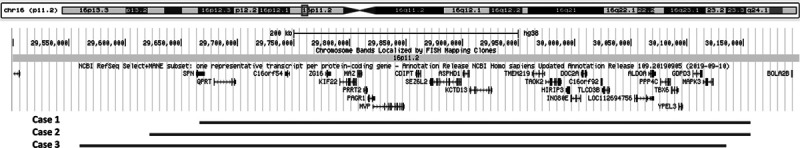
16p11.2 genomic locus. Red bars mark the approximate location of the deleted region of each case. Genes encompassed by the genomic are shown. Genomic positions are given according to the human genome build (GRCh38/hg38) assembly from the UCSC genome browser www.genome-euro.ucsc.edu.
In conclusion, it is clear that 16p11.2 deletions are manifested in a wide range of clinical symptoms which generally include developmental and language delay, cognitive impairment, seizures and ASD. The variations in clinical phenotypes in patients with 16p11.2 deletion render correct clinical interpretation challenging and frequently results in delayed diagnosis. All three cases reported here were at least 3 years of age before they received genetic testing, which highlights the importance of establishing and implementing early routine diagnostic testing so that functional and physical health outcomes can be addressed and treated at the earliest opportunity to reduce the likelihood of longer-term sequalae. As 16p11.2 deletion cannot be identified solely on the basis of clinical history, genetic testing should be a routine test in patients with neurodevelopmental disorders showing symptoms of speech and language delay, intellectual disability, or a diagnosis of ASD. Therefore, elucidating the variable clinical phenotypes of rare CNVs, including 16p11.2 deletions, remains critical to help guide clinical monitoring and counselling of patients and families.
Acknowledgements
We would like to thank Simons VIP connect social group; an online community, funded by Simons foundation, that supports families with rare genetic changes associated with autism and developmental delay that allowed for the recruitment of participants for the study. We would like to specially thank the parents and children who agreed to take part in this study.
This work was funded by the Wellcome Trust (grant number: 628TRN G32492). The funder was not involved in conducting the research or preparation/submission of the manuscript.
Conflicts of interest
There are no conflicts of interest.
References
- Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, Anderlid BM, et al. Extending the phenotype of recurrent rearrangements of 16p11.2: deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet. 2009; 52:77–87 [DOI] [PubMed] [Google Scholar]
- Blumenthal I, Ragavendran A, Erdin S, Klei L, Sugathan A, Guide JR, et al. Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am J Hum Genet. 2014; 94:870–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochukova EG, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010; 463:666–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova MF. Autism as a sequence: from heterochronic germinal cell divisions to abnormalities of cell migration and cortical dysplasias. Med Hypotheses. 2014; 83:32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K, Bracher-Smith M, Owen D, Kendall KM, Rees E, Pardiñas AF, et al. Medical consequences of pathogenic CNVs in adults: analysis of the UK Biobank. J Med Genet. 2019; 56:131–138 [DOI] [PubMed] [Google Scholar]
- D’Angelo D, Lebon S, Chen Q, Martin-Brevet S, Snyder LG, Hippolyte L, et al. ; Cardiff University Experiences of Children With Copy Number Variants (ECHO) Study; 16p11.2 European Consortium; Simons Variation in Individuals Project (VIP) Consortium. Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry. 2016; 73:20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Anda FC, Rosario AL, Durak O, Tran T, Gräff J, Meletis K, et al. Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat Neurosci. 2012; 15:1022–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldblum S, Rougier A, Loiseau H, Loiseau P, Cohadon F, Morselli PL, Lloyd KG. Quinolinic-phosphoribosyl transferase activity is decreased in epileptic human brain tissue. Epilepsia. 1988; 29:523–529 [DOI] [PubMed] [Google Scholar]
- Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet. 2010; 47:195–203 [DOI] [PubMed] [Google Scholar]
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012; 485:363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, et al. ; Simons Variation in Individuals Project Consortium. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol Psychiatry. 2015; 77:785–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson E, Nasir RH, Fong A, Lian A, Hundley R, Shen Y, et al. Cognitive and behavioral characterization of 16p11. 2 deletion syndrome. J Dev Behav Pediatr. 2010; 31:649–657 [DOI] [PubMed] [Google Scholar]
- Haslinger D, Waltes R, Yousaf A, Lindlar S, Schneider I, Lim CK, et al. Loss of the Chr16p11.2 ASD candidate gene QPRT leads to aberrant neuronal differentiation in the SH-SY5Y neuronal cell model. Mol Autism. 2018; 9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippolyte L, Maillard AM, Rodriguez-Herreros B, Pain A, Martin-Brevet S, Ferrari C, et al. ; 16p11.2 European Consortium, Simons Variation in Individuals Project Consortium. The number of genomic copies at the 16p11.2 locus modulates language, verbal memory, and inhibition. Biol Psychiatry. 2016; 80:129–139 [DOI] [PubMed] [Google Scholar]
- Iyer J, Singh MD, Jensen M, Patel P, Pizzo L, Huber E, et al. Pervasive genetic interactions modulate neurodevelopmental defects of the autism-associated 16p11.2 deletion in Drosophila melanogaster. Nat Commun. 2018; 9:2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Reymond A, Zufferey F, Harewood L, Walters RG, Kutalik Z, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011; 478:97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M, Girirajan S. An interaction-based model for neuropsychiatric features of copy-number variants. Plos Genet. 2019; 15:e1007879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall KM, Bracher-Smith M, Fitzpatrick H, Lynham A, Rees E, Escott-Price V, et al. Cognitive performance and functional outcomes of carriers of pathogenic copy number variants: analysis of the UK Biobank. Br J Psychiatry. 2019; 214:297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008; 17:628–638 [DOI] [PubMed] [Google Scholar]
- Loftus BJ, Kim UJ, Sneddon VP, Kalush F, Brandon R, Fuhrmann J, et al. Genome duplications and other features in 12 Mb of DNA sequence from human chromosome 16p and 16q. Genomics. 1999; 60:295–308 [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998; 14:417–422 [DOI] [PubMed] [Google Scholar]
- Martin J, Han C, Gordon LA, Terry A, Prabhakar S, She X, et al. The sequence and analysis of duplication-rich human chromosome 16. Nature. 2004; 432:988–994 [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, et al. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002; 34:807–820 [DOI] [PubMed] [Google Scholar]
- Niarchou M, Chawner SJRA, Doherty JL, Maillard AM, Jacquemont S, Chung WK, et al. Correction: psychiatric disorders in children with 16p11.2 deletion and duplication. Transl Psychiatry. 2019; 9:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer A. Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci Biobehav Rev. 2016; 64:185–195 [DOI] [PubMed] [Google Scholar]
- Pucilowska J, Vithayathil J, Tavares EJ, Kelly C, Karlo JC, Landreth GE. The 16p11. 2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J Neurosci. 2015; 35:3190–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Goin-Kochel RP, Nowell KP, Hunter JV, Aleck KA, Cox S. Expanding the clinical spectrum of the 16p11. 2 chromosomal rearrangements: three patients with syringomyelia. Eur J Hum Genet. 2011; 19:152–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima K, Inoue T, Fujii Y, Ohno K, Yamamoto T. A familial 593-kb microdeletion of 16p11.2 associated with mental retardation and hemivertebrae. Eur J Med Genet. 2009; 52:433–435 [DOI] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010; 47:332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002; 18:74–82 [DOI] [PubMed] [Google Scholar]
- Steinman KJ, Spence SJ, Ramocki MB, Proud MB, Kessler SK, Marco EJ, et al. 16p11. 2 deletion and duplication: characterizing neurologic phenotypes in a large clinically ascertained cohort. Am J Med Genet A. 2016; 170:2943–2955 [DOI] [PubMed] [Google Scholar]
- Tai DJ, Ragavendran A, Manavalan P, Stortchevoi A, Seabra CM, Erdin S, et al. Engineering microdeletions and microduplications by targeting segmental duplications with CRISPR. Nat Neurosci. 2016; 19:517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010; 463:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Rudolph M, Schlautmann N, Papaioannou VE, Gossler A. The mouse rib-vertebrae mutation is a hypomorphic Tbx6 allele. Mech Dev. 2002; 119:251–256 [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11. 2 and autism. New Engl J Med. 2008; 358:667–675 [DOI] [PubMed] [Google Scholar]
- Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. 2015; 372:341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, et al. ; Simons VIP Consortium; 16p11.2 European Consortium. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet. 2012; 49:660–668 [DOI] [PMC free article] [PubMed] [Google Scholar]