

Abstract
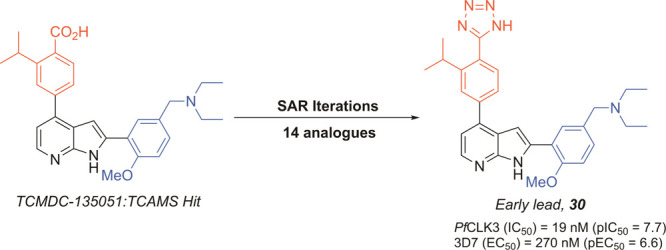
The protein kinase PfCLK3 plays a critical role in the regulation of malarial parasite RNA splicing and is essential for the survival of blood stage Plasmodium falciparum. We recently validated PfCLK3 as a drug target in malaria that offers prophylactic, transmission blocking, and curative potential. Herein, we describe the synthesis of our initial hit TCMDC-135051 (1) and efforts to establish a structure–activity relationship with a 7-azaindole-based series. A total of 14 analogues were assessed in a time-resolved fluorescence energy transfer assay against the full-length recombinant protein kinase PfCLK3, and 11 analogues were further assessed in asexual 3D7 (chloroquine-sensitive) strains of P. falciparum parasites. SAR relating to rings A and B was established. These data together with analysis of activity against parasites collected from patients in the field suggest that TCMDC-135051 (1) is a promising lead compound for the development of new antimalarials with a novel mechanism of action targeting PfCLK3.
Introduction
In the global fight against malaria, the distribution of over 0.5 billion insecticide impregnated bed nets since 2015, together with artemisinin-based combination therapies, has contributed to 20 million fewer cases of malaria being reported in 2017 compared to 2010.1 Despite this success, the rate of reduction in malaria has stalled with no significant reduction seen over the last 3 years.1 This, together with evidence of the emergence of resistance to both artemisinin2,3 and partner drugs including piperaquine and mefloquine,4,5 means that there is a danger that the progress made in the reduction of malaria will be reversed unless new medicines with novel mechanisms of action are discovered. To address this, we have been testing the notion that targeting malaria parasite protein kinases, known to be essential for the survival of the parasite, might offer a novel strategy for the development of next-generation antimalarials.6,7
In 2011, we determined that 36 of the 65 eukaryotic protein kinases are essential for the survival of the blood stage of the most virulent species of human malaria, Plasmodium falciparum.8 Among these protein kinases was PfCLK3 (PF3D7_1114700), one of the four members of the cyclin-dependent like protein kinase family (PfCLK1-4). The plasmodium CLK-family are closely related to the mammalian CLK family and the serine–arginine-rich protein kinase (SRPK) family,9 both of which are crucial mediators of multiple phosphorylation events on splicing factors necessary for the correct assembly and catalytic activity of spliceosomes.10In vitro studies showed that PfCLK3 can phosphorylate parasite SR proteins,11 indicating that PfCLK3, together with other members of the PfCLK family,12 plays a role in the processing of parasite RNA.7 We reasoned therefore that inhibitors of PfCLK3 might have parasiticidal activity at any stage of the parasite life cycle where RNA splicing played an essential role.
Screening of ∼25,000 compounds, including all 13,533 compounds of the Tres cantos antimalarial set, resulted in the discovery of TCMDC-135051 (1, Figure 1), a compound with nanomolar (nM) activity against PfCLK3 in vitro kinase assays and submicromolar (μM) parasiticidal activity in asexual blood stage P. falciparum parasites (Figure 1).13 Subsequent studies revealed that TCMDC-135051 rapidly killed P. falciparum at the trophozoite to schizont stages as well as preventing the development of stage V gametocytes and inhibiting the development of liver stage parasites. TCMDC-135051 also showed equivalent in vitro kinase activity at CLK3 from P. falciparum, Plasmodium berghei (mouse malaria), Plasmodium vivax (human malaria), and Plasmodium knowlesi (monkey and human malaria).13 Our recent studies have therefore validated PfCLK3 as a target with the potential to deliver a curative treatment, be transmission blocking, and act as a prophylactic target.13
Figure 1.

Structure and biological profile of hit PfCLK3 inhibitor TCMDC-135051, 1.
Key structural features of 1 include a core 7-azaindole scaffold with aromatic rings in the 2- and 4-positions (ring A and B, respectively). These aromatic rings are further substituted with various functional groups, including a tertiary amine (ring A) and carboxylic acid (ring B), resulting in a zwitterionic compound at physiological pH.
7-Azaindoles are a widely studied pharmacophore incorporated into several therapeutic agents.14 Kinases are the predominate target with the 7-azaindole moiety generally interacting at the ATP binding site within the kinase hinge region.15,16 An interaction occurs between the kinase hinge region peptide backbone and the azaindole via two H-bonds; the first involving the pyridine nitrogen lone pair and peptide backbone NH, and the second between the pyrrole NH and peptide backbone carbonyl. The resulting H-bond acceptor and donor bidentate 7-azaindole interaction with the hinge region of the kinase can occur in the more common “normal” orientation or the “flipped” orientation with the 2-position of the 7-azaindole projected out of the hinge region into the solvent-exposed space.17,18
A number of small molecule kinase 7-azaindole inhibitors have progressed to different stages of clinical trials.19 A potential drug candidate GSK1070916 is being developed as an aurora kinase (Ser/Thr protein kinases family) inhibitor and has reached human clinical trials (Figure 2A).20 The core scaffold is a 7-azaindole with aromatic substituents in the 2- and 4-positions. An X-ray crystal structure of the molecule:aurora kinase complex revealed a flipped hinge region binding mechanism, with 2-aryl projecting out of the hinge region into solvent and 4-aryl bound within the ribose pocket.20
Figure 2.
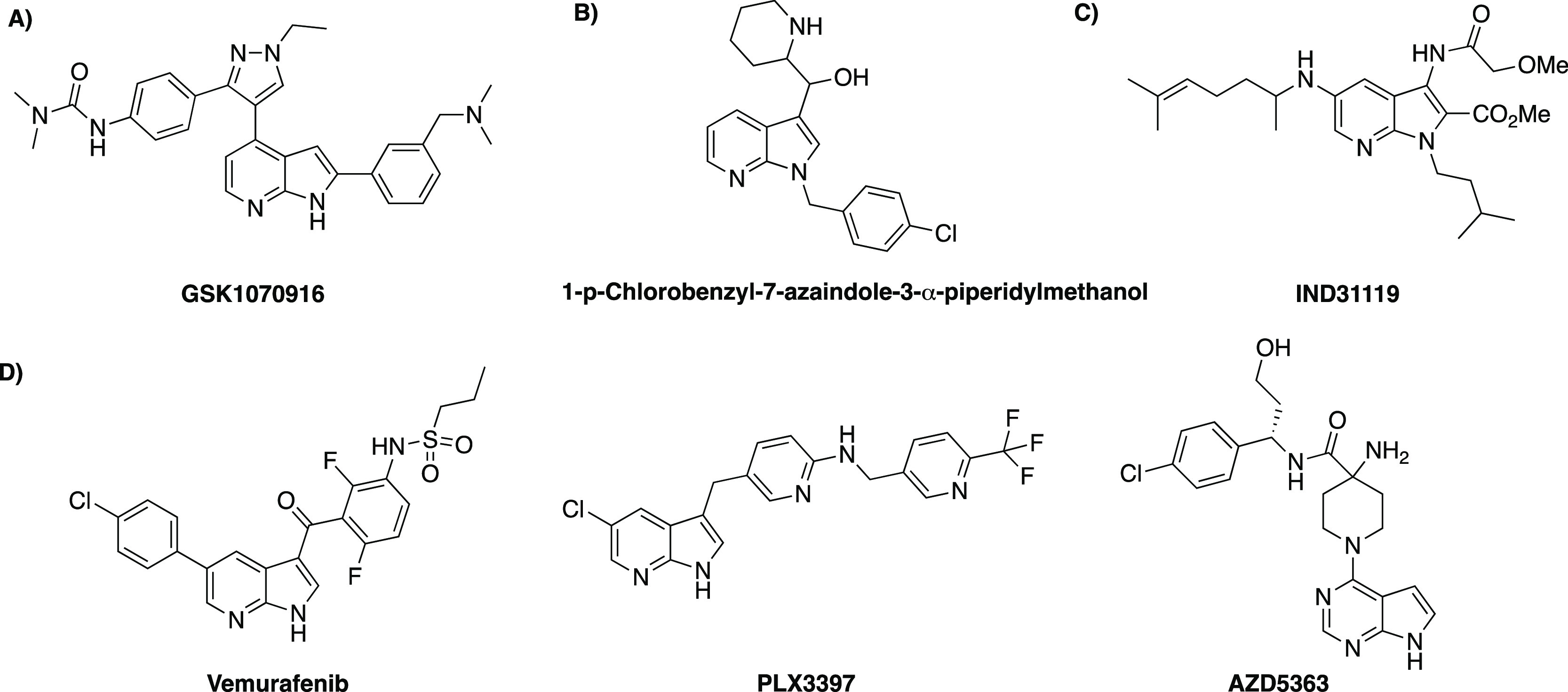
(A) Aurora kinase inhibitor GSK1070916, (B) 1-p-chlorobenzyl-7-azaindole-3-α-piperidylmethanol has in vivo activity against P. berghei, (C) PfHsp90 inhibitor IND31119, and (D) drug candidates incorporating a core 7-azaindole scaffold, vemurafenib, PLX3397, and AZD5363.
Substructure analysis using Scifinder revealed several 7-azaindole analogues described as antimalarials. In 1972, Verbiscar reported the first example of a 7-azaindole with antimalarial activity.21 1-p-Chlorobenzyl-7-azaindole-3-α-piperidylmethanol displayed antimalarial activity in mice against P. berghei(Figure 2B). More recently, Picard et al. reported 7-azaindole compounds that were identified from an in silico structure-based drug screen (1,013,483 compounds, Chembridge library).22 Compound IND31119 (Figure 2C) binds to the recombinant N-terminal domain of PfHsp90 and shows selectivity over human Hsp90 and PfHsp90 mutants.22
In addition to antimalarial activity, the 7-azaindole scaffold has been identified as having a wide variety of biological applications, including antitumor activity, and acts as an HIV-1 inhibitor in infected patients.23,24 The 7-azaindole motif can be regarded as a privileged scaffold in medicinal chemistry as it is found in three clinical candidates (vemurafenib, PLX3397, and AZD5363) (Figure 2D), which suggests its usefulness for developing novel pharmaceuticals.25−27
To investigate the structure–activity relationship (SAR) of hit compound 1, a series of analogues were prepared through varying different substituents on ring A and ring B (Figure 1). The N-diethyl amine group (ring A) was initially replaced with different amine substituents to investigate lipophilicity and solubility (Figure 1, SAR1). Next, we replaced the methoxy moiety (ring A) with hydroxyl or hydrogen to modify polarity and to explore the role of the methoxy group on activity (Figure 1, SAR2). The isopropyl substituent (ring B) was replaced with other small hydrophobic substituents to probe noncoplanar conformations that could potentially lower the energy of crystal packing and consequently improve aqueous solubility and log P (Figure 1, SAR3). Finally, we exchanged the carboxylic acid group (ring B) with other substituents to investigate its role in binding, to potentially improve metabolic stability, and to explore the effect of increased lipophilic character (Figure 1, SAR4).
TCMDC-135051 is a promising hit compound for a medicinal chemistry program to develop as a preclinical lead that meets many of the criteria set by the Medicines for Malaria Venture (capable of rapidly clearing the parasite, has multistage potency, and killing multiple parasite species with action as a transmission blocker).28,29 Here, we describe the synthetic route to TCMDC-135051 and determine a SAR that will be key for lead development.
Chemistry
To investigate the effect of the N-diethyl group of ring A on antimalarial activity, analogues of 4-(2-(5-((diethylamino)methyl)-2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-isopropylbenzoic acid (TCMDC-135051) 1, 8a–c were prepared, as shown in Scheme 1. Protection of 4-bromo-7-azaindole 2 was achieved using p-toluenesulfonyl chloride under basic conditions to provide N-tosyl-7-azaindole 3. Regioselective iodination of 3 using lithium diisopropylamide (LDA) and iodine in tetrahydrofuran (THF) at −78 °C provided 2-iodoazaindole 4, which was subsequently subjected to Suzuki coupling with (5-formyl-2-methoxyphenyl)boronic acid to give 2-aryl substituted azaindole 5. Reductive amination of the aldehyde functionality of 5 was performed with various amines in the presence of sodium triacetoxyborohydride to give amines 6a–d in excellent yields. Next, the N-tosyl protecting group was removed under basic conditions at reflux for 18 h to yield 7a–d. Finally, the desired analogues 1 and 8a–c were obtained by Suzuki cross-coupling with 2-isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid (see the Supporting Information for boronate ester synthesis).
Scheme 1. Synthesis of TCMDC-135051 1 and Analogues 8a–c & 9.
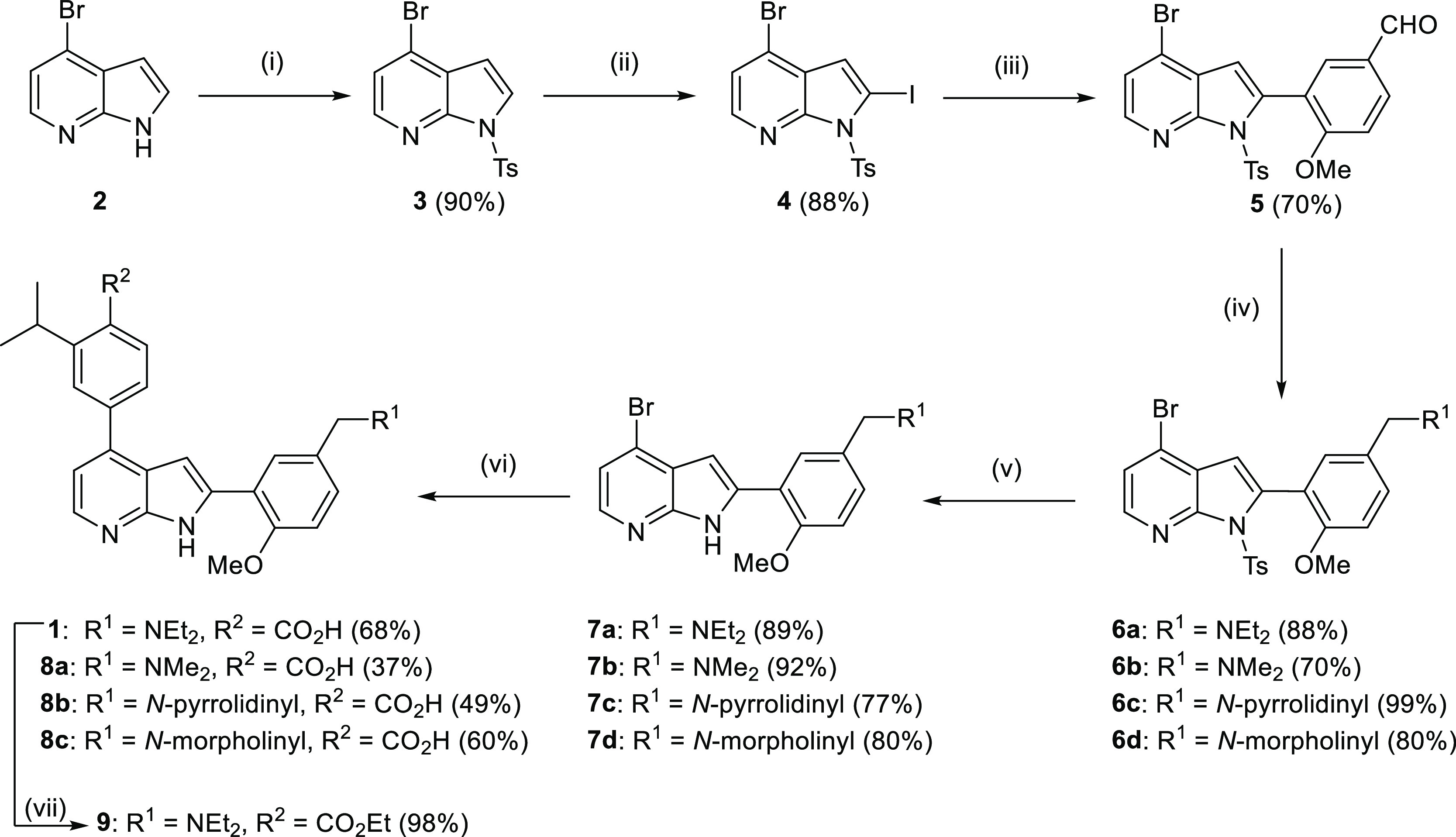
Reagents and conditions: (i) TsCl, NaH, THF, 0 °C, 2 h; (ii) LDA, I2, THF, −78 °C, 3 h; (iii) (5-formyl-2-methoxyphenyl)boronic acid, Pd(PPh3)4, Na2CO3, 1,4-dioxane, 110 °C, 12 h; (iv) amine, NaBH(AcO)3, 1,4-dioxane, 20 °C, 12 h; (v) CH3OH, K2CO3, 55 °C, 18 h; (vi) 2-isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid, Pd(dppf)Cl2·CH2Cl2, Na2CO3, 1,4-dioxane, 110 °C, 0.5 h, MW; and (vii) SOCl2, CH3CH2OH, reflux, 18 h.
Compound 9 was prepared, as described in Scheme 1. The carboxylic acid of 1 was converted to its corresponding ethyl ester by refluxing in thionyl chloride and ethanol for 18 h.
The synthetic route outlined in Scheme 1 was modified to prepare primary amine 12. The first step of this synthetic route involved coupling 2-iodo-azaindole 4 with the appropriate boronate ester under Suzuki conditions to yield nitrile 10 in 87% yield (Scheme 2). Tosyl deprotection was then achieved using K2CO3, followed by a Suzuki coupling at the 4-bromo-azaindole scaffold with the appropriate pinacol boronate ester. The crude material was reduced in situ using cobalt(II) chloride hexahydrate and sodium borohydride to provide the corresponding amine 12 (Scheme 2).
Scheme 2. Synthesis of 4-(2-(5-(Aminomethyl)-2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-isopropylbenzoic Acid 12.

Reagents and conditions: (i) 4-methoxy-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile, Pd(PPh3)4, Na2CO3, 1,4-dioxane, 110 °C, 18 h; (ii) CH3OH, K2CO3, 55 °C, 18 h; (iii) 2-isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid, Pd(dppf)Cl2.CH2Cl2, Na2CO3, 1,4-dioxane, 110 °C, 0.5 h, MW; and (iv) CoCl2·6H2O, NaBH4, CH3OH.
Similarly, analogue 15 was synthesized via Suzuki coupling of 4 with 2-methoxyphenyl boronic acid, followed by tosyl deprotection and Suzuki coupling (Scheme 3).
Scheme 3. Synthesis of 2-Isopropyl-4-(2-(2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)benzoic Acid 15, 4-(2-(5-((Diethylamino)methyl)-2-hydroxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-isopropylbenzoic Acid 19, 4-(2-(3-((Diethylamino)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-isopropylbenzoic Acid 23 and [4-(2-(3-((Diethylamino)methyl)-2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-isopropylbenzoic Acid] 27.
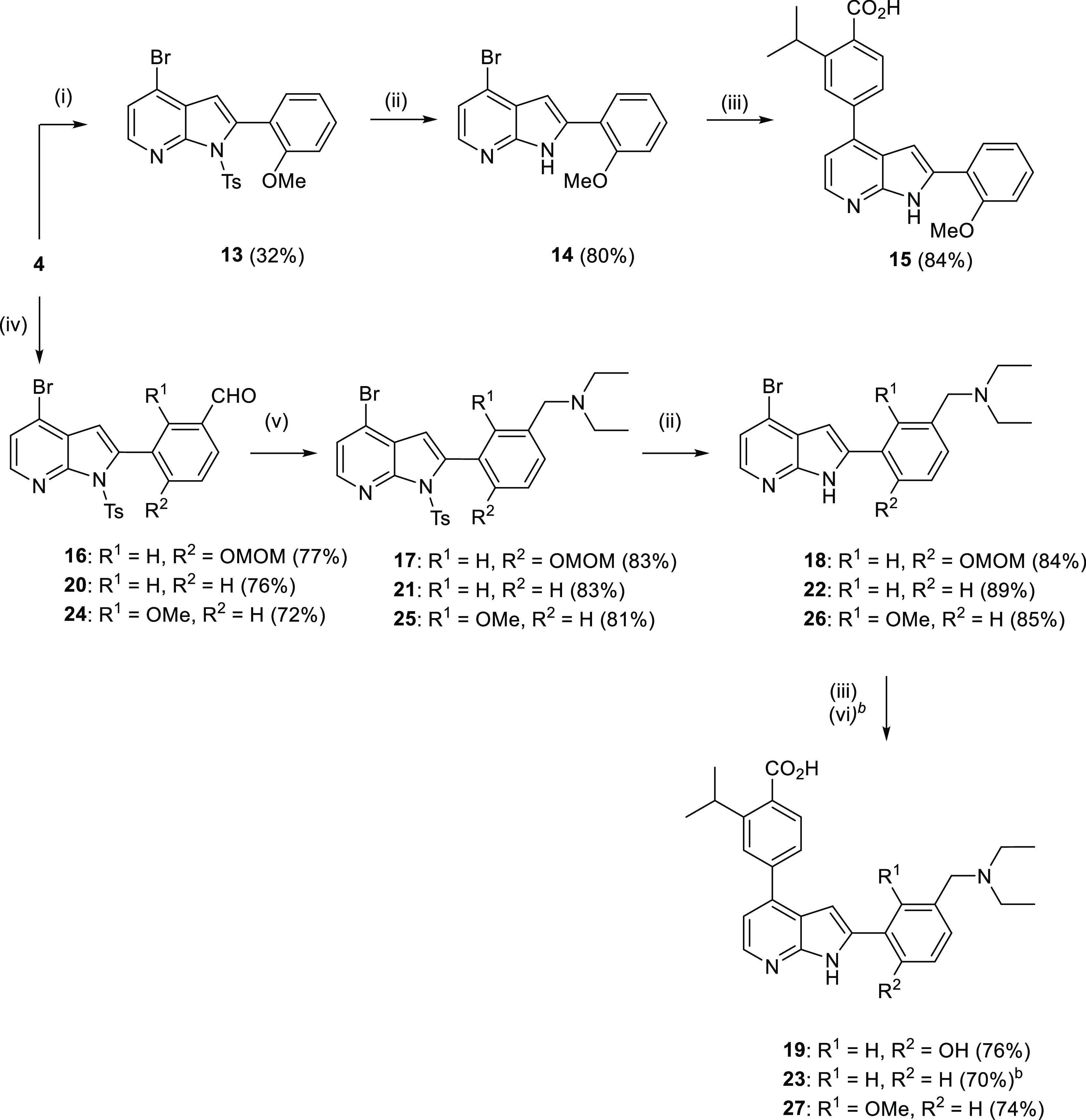
Reagents and conditions: (i) (2-methoxyphenyl)boronic acid, Pd(PPh3)4, Na2CO3, 1,4-dioxane, 110 °C, 12 h; (ii) CH3OH, K2CO3, 55 °C, 18 h; (iii) 2-isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid, Pd(dppf)Cl2·CH2Cl2, Na2CO3, 1,4-dioxane, 110 °C, 0.5 h, MW; (iv) 4-(methoxymethoxy)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde or 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde, Pd(PPh3)4, Na2CO3, 1,4-dioxane, 110 °C, 18 h; (v) Et2NH, NaBH(AcO)3, 1,4-dioxane, rt, 18 h; and (vi) HCl, MeCN/H2O 3:1.
To investigate the precise role of the methoxy group of ring A on activity, we prepared analogues 19 and 23 in which the OMe is replaced by a hydroxy group (19) and OMe is removed (23) (Scheme 3). The synthetic route toward 19 starts with Suzuki coupling of 4 with methoxymethyl ether (MOM)-protected boronate ester (see the Supporting Information for boronate ester synthesis) to yield 16. Reductive amination of 16 followed by removal of the tosyl group gave 18. Finally, 18 was coupled with boronate ester under Suzuki conditions, and the MOM protecting group was removed under acidic conditions to provide the desired analogue 19 in excellent yields. Analogue 23 was synthesized in four steps using the same methods.
To investigate whether the substitution pattern on ring A is important for antimalarial activity, we produced analogue 27 in which the methoxy group is moved from para to the ortho position relative to the methylene N-diethyl functionality (Scheme 3).
To investigate the effect of the isopropyl substituent (ring B) on efficacy, we prepared analogues 28–29, as shown in Scheme 4. 4-Bromo-7-azaindole 7a was coupled with various boronate esters under Suzuki conditions to yield the desired 4-aryl-7-azaindole analogues 28–29.
Scheme 4. Synthesis of 4-Aryl-7-azaindole Analogues, 28–32.

Reagents and conditions: (i) boronate esters, Pd(dppf)Cl2.CH2Cl2, Na2CO3, 1-4-dioxane, 110 °C, 0.5 h, MW.
To investigate the role of the carboxylic acid substituent of ring B on antimalarial activity, several analogues 30–32 were synthesized, as depicted in Scheme 4. Synthesis of analogues 30–32 was achieved by Suzuki coupling 7a with various boronate esters (R2 = 1H-tetrazole or H) to yield target compounds 30–31 in good yields.
An isomer, in which the isopropyl and carboxylic acids were switched, 32 was synthesized to investigate the role of these functional groups on antimalarial activity (Scheme 4).
Results and Discussion
In this work, we have maintained the core 7-azaindole molecular scaffold of TCMDC-135051 and focused on the SARs of the substitutes on ring A and -B to assess the requirement for this functionality. All synthesized analogues were assessed in a time-resolved fluorescence energy transfer (TR-FRET) PfCLK3 in vitro kinase assay against the full recombinant protein kinase (Tables 1 and 2). Analogues which gave low nanomolar activity were then further assessed in live parasite viability (parasiticidal) assays using laboratory strain 3D7 (chloroquine-sensitive) P. falciparum (Tables 1 and 2). The synthesized analogues were evaluated for log D7.4 (distribution co-efficient) and metabolic stability, and the results are shown in Tables 1 and 2.
Table 1. Physicochemical Properties and Activity Data of TCMDC-135051 Ring A Analogues.

|
PfCLK3 |
3D7 |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| analogue | R1 | R2 | R3 | IC50 (nM)a | pIC50 | EC50 (nM)b | pEC50 | log D7.4c | clint (mL/min/g liver)d |
| 1 | NEt2 | OMe | H | 40 | 7.4 (±0.221) | 180 | 6.7 (±0.126) | 0.93 | 1.12 |
| 8a | NMe2 | OMe | H | 29 | 7.5 (±0.224) | 457 | 6.3 (±0.129) | 0.85 | 2.53 |
| 8b | N-pyrrolidinyl | OMe | H | 38 | 7.4 (±0.113) | 382 | 6.4 (±0.081) | 2.43 | 1.94 |
| 8c | N-morpholinyl | OMe | H | 9 | 8.0 (±0.191) | 1339 | 5.9 (±0.118) | 1.20 | 1.60 |
| 12 | NH2 | OMe | H | 76 | 7.1 (±0.142) | 2801 | 5.6 (±0.104) | 0.61 | 2.92 |
| 15 | H | OMe | H | 79 | 7.1 (±0.132) | 1456 | 5.8 (±0.152) | 2.45 | 2.54 |
| 19 | NEt2 | OH | H | 22 | 7.7 (±0.115) | 3529 | 5.5 (±0.133) | 0.59 | 1.94 |
| 23 | NEt2 | H | H | 25 | 7.6 (±0.089) | 309 | 6.5 (±0.114) | 0.80 | 0.85 |
| 27 | NEt2 | H | OMe | 17 | 7.7 (±0.116) | 3167 | 5.6 (±0.109) | 0.74 | 1.65 |
IC50 (the concentration of an inhibitor where the response is reduced by half).
EC50 (the concentration of a drug that gives half-maximal response). IC50 and EC50 values are means ± SEM of three independent experiments run in triplicates (n = 3).
log D7.4 (distribution co-efficient) was estimated using HPLC chromatography.
In vitro intrinsic clearance in mouse liver microsomes.
Table 2. Physicochemical Properties and Activity Data of TCMDC-135051 Ring B Analogues.

|
PfCLK3 |
3D7 |
|||||||
|---|---|---|---|---|---|---|---|---|
| analogue | R1 | R2 | IC50 (nM)a | pIC50 | ED50 (nM)b | pEC50 | log D7.4c | Clint (mL/min/g liver)d |
| 28 | CH3 | CO2H | 24 | 7.6 (±0.10) | 1185 | 5.6 (±0.097) | 0.66 | 1.33 |
| 29 | H | CO2H | 34 | 7.5 (±0.089) | 3272 | 5.5 (±0.124) | 0.65 | 11.01 |
| 30 | CH(CH3)2 | 1H-tetrazole | 19 | 7.7 (±0.089) | 270 | 6.6 (±0.158) | 0.93 | 2.32 |
| 31 | CH(CH3)2 | H | 1300 | 6.0 (±0.091) | NDe | NDe | 4.45 | 9.54 |
| 9 | CH(CH3)2 | CO2CH2CH3 | 390 | 6.4 (±0.087) | NDe | NDe | 2.45 | 16.06 |
| 32 | CO2H | CH(CH3)2 | 1385 | 4.9 (±0.093) | NDe | NDe | NDe | NDe |
IC50 (the concentration of an inhibitor where the response is reduced by half).
EC50 (the concentration of a drug that gives half-maximal response). IC50 and EC50 values are means ± SEM of three independent experiments run in triplicates (n = 3).
log D7.4 (distribution co-efficient) was estimated using HPLC chromatography.
In vitro intrinsic clearance in mouse liver microsomes.
ND, not determined.
SAR1 corresponding to analogues 8a–c, 12, and 15 were designed to examine the effect of the N-diethyl group of ring A on antimalarial activity. In analogue 8a, the N-diethyl group was replaced with an N-dimethyl group to investigate the effect of alkyl group size and molecular lipophilicity (log D7.4 = 0.85). For in vitro kinase activity of 8a, the half maximal inhibitory activity (IC50) IC50 = 29 nM (pIC50 = 7.5 ± 0.224) remains the same in recombinant PfCLK3 kinase, whereas a 2-fold decrease in the half maximal parasite growth inhibition effect (EC50) EC50 = 457 nM (pEC50 = 6.3 ± 0.129) was observed (Table 1). To further investigate the steric requirements of this functionality and polarity, pyrrolidine (8b) and morpholino groups (8c) were introduced. Analogue 8b shows comparable in vitro kinase activity, IC50 = 38 nM (pIC50 = 7.4 ± 0.113), and a 2-fold decrease in parasite growth inhibition 8b, EC50 = 382 nM (pEC50 = 6.4 ± 0.081), was observed. However, the more polar 8c shows a slight improvement in vitro kinase activity of IC50 = 9 nM (pIC50 = 8.0 ± 0.191), a 7-fold decrease in parasite growth inhibition was observed 8c, EC50 = 1339 nM (pEC50 = 5.9 ± 0.118) (Table 1). To further investigate the polarity of this moiety, we replaced the N-diethyl functionality with a more polar primary amine (log D7.4 = 0.61). Analogue 12 incorporating a primary amine gave an IC50 = 76 nM (pIC50 = 7.1 ± 0.142), thus was 2-fold less potent in vitro and showed a dramatic loss of efficacy, EC50 = 2801 nM (pEC50 = 5.6 ± 0.104), in parasites, indicating the need to decrease the polarity of the amine group for optimal parasite growth inhibition (Table 1). Analogue 15, with the alkyl amine group removed (log D7.4 = 2.45) was less potent in vitro, IC50 = 79.0 nM (pIC50 = 7.1 ± 0.132) against recombinant PfCLK3 protein and in parasites, EC50 = 1456 nM (pEC50 = 5.8 ± 0.152). There was a 7-fold drop in the parasite growth inhibition, suggesting that the alkyl amine on ring A is important for antimalarial activity (Table 1).
To explore the importance of the methoxy group (OMe) on ring A, two analogues, hydroxyl 19 and 23, with methoxy removed, were prepared. We first replaced the OMe group with a hydroxyl to investigate the role of polarity on activity (log D7.4 = 0.59). Next, we replaced the OMe group with hydrogen (23) to investigate the importance of this functionality for activity. For both compounds 19 and 23, in vitro kinase potency was comparable, IC50 = 22 nM (pIC50 = 7.7 ± 0.115) and IC50 = 25 nM (pIC50 = 7.6 ± 0.089), respectively (Table 1). When tested in parasites, 19 shows significant loss of activity, EC50 = 3529 nM (pEC50 = 5.5 ± 0.133). Interestingly, replacing the OMe group with a hydrogen, 23 also shows a 1.5-fold decrease in potency, EC50 = 309 nM (pEC50 = 6.5 ± 0.114) against parasites. These data demonstrated that the OMe group on ring A is required for in vitro kinase and parasite growth inhibition. Overall, analogues (8a–c, 12, 15, 19, and 23) showed good metabolic stability in mouse liver microsomes (Table 1).
Next, we turned our attention to study the SAR of various functional groups on ring B. First, we examined the role of binding through van der Waals interactions in vitro and lipophilicity in parasites, imparted by the ring B isopropyl group, on antimalarial activity. In analogue 28 (log D7.4 = 0.66), the isopropyl group was replaced with methyl, whereas in analogue 29 (log D7.4 = 0.65), the isopropyl was removed and replaced with hydrogen. Analogues 28 and 29 had a modest increase on in vitro kinase activity, IC50 = 24 nM (pIC50 = 7.6 ± 0.10) and IC50 = 34 nM (pIC50 = 7.5 ± 0.089), respectively. However, a dramatic loss of potency was observed for both compounds 28 and 29 with EC50 = 1185 nM (pEC50 = 5.6 ± 0.097) and EC50 = 3272 nM (pEC50 = 5.5 ± 0.124), respectively, representing a 6-fold and 16-fold decrease in parasite growth inhibition (Table 2). Therefore, this group is not required for binding in vitro; however, the effect of lipophilicity appears to be very important for parasite growth inhibition. Analogues 28 and 29 were then evaluated for intrinsic clearance in mouse microsome. Analogue 28 (Clint = 1.33 mL/min/g liver) showed a similar clearance value to that of 1 (TCMDC-051) in mouse liver microsomes, whereas 29 had a higher clearance under the same conditions (Clint = 11.01 mL/min/g liver) indicating the importance of the isopropyl group.
At this point, we decided to increase the lipophilicity and replace the isopropyl group to the larger tert-butyl group. However, due to a restriction of the chemistry of boronate esters (instability of tert-butyl anion required for SNAr reaction), we could not synthesize the proposed molecule. From these data, the alkyl substituent on ring B appears not to be particularly important in the molecular recognition event with PfCLK3, however is key for overall molecular lipophilicity that contributes to parasite growth inhibition.
To investigate the requirements (i.e., ionic and H-bonding) of the carboxylic acid (ring B) for antimalarial activity, we employed a series of structural changes. The presence of carboxylic acid functionality in a drug molecule has several potential drawbacks, including limited permeability across biological membranes, metabolic instability, and potential idiosyncratic toxicities. A common isostere of a carboxylic acid, that overcomes many of these physicochemical limitations, is a tetrazole. We therefore designed tetrazole analogue 30. This change was orchestrated to increase the lipophilicity of the molecule, retain H-bonding capability, and investigate the role of potential ionic interactions with the enzyme. The tetrazole analogue 30 shows improved in vitro kinase potency with an IC50 = 19 nM (pIC50 = 7.7 ± 0.089), with comparable parasite growth inhibition, EC50 = 270 nM (pEC50 = 6.6 ± 0.158) (Table 2). The potency of analogue 30 was tested against resistant mutant PfCLK3 (PfCLK3_G449P) 3D7 parasites.13 The parasiticidal activity of TCMDC-135051 1 against asexual 3D7 parasites was 180 nM (pEC50 = 6.7), and 1806 nM (pEC50 = 5.74) against mutant G449P. A 15-fold shift in sensitivity to the inhibitor. The tetrazole 30, with an EC50 = 270 nM (pEC50 = 6.6) in wild type parasites showed a significant reduction is potency against the mutant G449P parasites, EC50 = 3494 nM (pEC50 = 5.4). This represents a 13-fold shift in potency.
Analogue 31 where the carboxylic acid group was replaced with a hydrogen shows significant loss of in vitro kinase activity, IC50 = 1300 nM (pIC50 = 6.0 ± 0.091). Analogue 31 has high lipophilicity (log D7.4 = 4.45) and in vitro high intrinsic clearance (Clint = 9.54 mL/min/g liver) demonstrating the importance of the carboxylic acid group (Table 2).
Analogue 9 has improved lipophilicity (log D7.4 = 2.45), yet the in vitro kinase potency was low, IC50 = 390 nM (pIC50 = 6.4 ± 0.087), indicating that the presence of a functional group capable of donating a hydrogen bond is important for activity (Table 2). This analogue was not tested in parasites because of the low potency against recombinant PfCLK3 kinase.
The final part of our SAR assessment was dedicated to exploring the possibility of orientation of key substituents. Varying the position of the OMe group from para to the ortho position 27 resulted in a 2-fold increase in in vitro kinase activity, IC50 = 17 nM (pIC50 = 7.7 ± 0.116). However, when 27 was tested in parasites, the activity was lowered by 15-fold, EC50 = 3167 nM (pEC50 = 5.6 ± 0.109) (Table 1).
We next investigated the position of the isopropyl and carboxylic acid substituents of ring B 32 on antimalarial activity. The change of orientation of the substituent is detrimental for the in vitro kinase activity, IC50 = 1385 nM (pIC50 = 4.9 ± 0.093), and therefore was not tested in parasites. These data suggested that the positioning of the isopropyl and carboxylic acid group was essential for binding to its cellular target.
No X-ray crystal structure has been reported for PfCLK3 and so a homology model of PfCLK3 was created using SWISS-MODEL to provide evidence of our proposed binding mechanism (i.e., hinge binder), which we hypothesized based on other literature examples of 7-azaindole scaffolds binding at the hinge region of the kinase domain. The structure of the human PRPF4B kinase domain was selected as the template as the closest homologue [PDB 6CNH (sequence identity 53.2%)].30 Model accuracy was determined to be reasonable using SWISS-MODEL with QMEAN (qualitative model energy analysis) score = −2.26 and GMQE (global model quality estimation) = 0.77. Overlay with Human Jnk1alpha kinase with 4-phenyl-7-azaindole IKK2 inhibitor bound (PDB 4AWI) facilitated identification of the proposed binding pocket.31 Based on this model, we propose that the 7-aza-indole scaffold interacts with the hinge region in the flipped conformation, that is, H-bonding to the peptide backbone of the hinge region (Figure 3). The benzoic acid on ring B appears to occupy the ribose pocket and interacts with Lys-394. In the model, the ring B isopropyl group occupies a hydrophobic back pocket. Ring A projects into the solvent-exposed space. The diethyl-amine and methoxy substituents are solvent exposed and may contribute to the orientation of the 7-azaindole in the flipped binding conformation.
Figure 3.

Putative binding mode of TCMDC-135051 1 in a PfCLK3 homology model.
We previously demonstrated that TCMDC-135051 showed selective inhibition of PfCLK3 when compared against the closely related human kinases (PRPF4B and CLK2), as well as the closest parasite kinase (PfCLK1), and other parasite kinases (PfPKG and PfCDPK1).13 To further assess kinase selectivity, and the potential for off-target toxicity, TCMDC-135051 was screened against 140 human kinases at 1 μM concentration. Only nine kinases were found to have less than 20% activity at this concentration (see the Supporting Information).
TCMDC-135051 Inhibits the Growth of Clinical Field Isolates
An important property for next-generation antimalarials is effectiveness against parasites that are resistant to currently used antimalarials. To determine whether the parent molecule TCMDC-135051 1 might offer such activity, we tested the efficacy of this compound against parasites that were collected from malaria patients in The Gambia. These parasite strains were sequenced and those that contained genetic markers of resistance were selected (Table 3). Parasites were then tested for resistance to pyrimethamine, a frontline antimalarial where resistance has been documented in parasites originating from The Gambia (Figure 4A).32
Table 3. Genotypes of Field Isolated Parasite Strains: Whole Genome Sanger Sequencing Identified Mutations in Three Parasite Genes (CRT, PfMDR1, and PfDHFR) Associated with Drug Resistancea.
| PfCRT | PfMDR1 | PfDHFR | ||||||
|---|---|---|---|---|---|---|---|---|
| sample ID | K76T | N86Y | Y184F | N1042D | N51I | C59R | S108N | I164L |
| GB0006 | X | X | X | N | X | X | N | L |
| GB0004 | K | N | Y | N | N | R | N | L |
| GB0026 | K | N | X | N | N | R | N | L |
| GB002 | T | N | X | N | I | C | N | L |
| GB0021 | K | N | F | N | X | X | N | L |
| GB0048 | X | N | F | N | I | C | N | L |
| GB0071 | X | N | Y | N | N | C | N | L |
| GB0087 | T | N | Y | N | I | C | N | L |
| GB0020 | T | Y | Y | N | I | C | N | L |
Shown are the amino acid changes associated with each of these three genes and whether the parasite strain contained the mutation.
Figure 4.

PRR of clinical isolates comparing pyrimethamine and TCMDC-135051 at EC50 and 10 times the EC50 of each drug. This represent the average of triplicate.
This analysis identified nine parasite isolates from patients with varying degrees of resistance to pyrimethamine (Figure 4A,B) and which contained mutations in one or more of the three genes; PfCRT (P. falciparum chloroquine-resistant transporter gene), PfMDR1 (P. falciparum multidrug-resistant gene 1), and PfDHFR (P. falciparum dihydrofolate reductase gene). The mutations identified in these genes (Table 3) have previously been associated with resistance to commonly used antimalarials, including chloroquine and pyrimethamine.33 An EC50 concentration of pyrimethamine was then determined using the laboratory strain parasite 3D7 (EC50 = 18 nM). Using this concentration in parasite reduction rate (PRR) assays, all isolates showed a reduction in parasitemia by at least 70% except for three isolates (GB0020, GB002, and GB0026), which showed resistance to pyrimethamine treatment. These three isolates uniquely contained triple mutations in the PfDHFR gene and a single mutation in PfCRT (Table 3). For these isolates, resistance was evident after 24 h of treatment with pyrimethamine where parasitemia was only reduced by 45, 64, and 36%, respectively (Figure 4A). Resistance to pyrimethamine of these three isolates was still evident after 72 h of drug treatment (Figure 4A) and in parasites exposed to 10× EC50 (180 nM) of pyrimethamine (Figure 4B).
In contrast, all isolates treated with the EC50 of TCMDC-135051, as determined in 3D7 parasites (EC50 = 180 nM), demonstrated total susceptibility, including isolates GB0020, GB002, and GB0026 (Figure 4C). This was true both at the EC50 concentration and 10× EC50 for TCMDC-135051 (Figure 4D). Hence in the field isolates tested, TCMDC-135051 showed equivalent activity against parasites that carried genetic markers of resistance and those that showed actual resistance to a frontline antimalarial. This indicates the possibility that the parent molecule, and potentially analogues of this molecule, would be active against naturally circulating malaria parasites harboring mutations that promoted resistance to current antimalarial drugs. The PRR assay was setup using clinical isolates previously cryopreserved in liquid nitrogen (LN2). These were thawed and placed in culture for at least one cycle (∼48 h ± 2). The life cycle stage of the parasites was monitored using a blood film, and greater than 95% of the parasites are expected to egress and form new invading rings. These new rings, mainly 0–3 h old postinvasion, were used as time point 0 hours (t = 0) for the assay. The parasites are then cultured with drugs for 24, 48, and 72 h. For 24 h treatment, the drug was removed by washing twice with wash media and for longer treatments (48 and 72 h time points), drug was replenished with fresh drug every 24 h. After the treatment period, parasites were grown in fresh media for an additional 48 h in freshly stained erythrocytes to allow invasion from any viable parasites.
Conclusions
In summary, we report the synthesis of hit PfCLK3 inhibitor TCMDC-135051 1 (PfCLK3 IC50 = 40 nM, 3D7 EC50 = 180 nM) and a series of related 7-azaindole-based analogues. Of the 14 analogues, 11 had low nanomolar activity and were further assessed in live parasite viability assays using the 3D7 (chloroquine-sensitive) strain of P. falciparum. Tetrazole analogue 30 was identified with improved in vitro kinase activity (PfCLK3, IC50 = 19 nM) and comparable activity in parasites 3D7 (EC50 = 271 nM). SAR was established for both ring A, highlighting the importance of H-bonding functionality in the 4-aryl position, and for the alkyl amino group on ring B. Together these data provide a good starting point for the hit to lead development of novel PfCLK3 inhibitors based on TCMDC-135051 (1).
Experimental Section
General Information
Chemicals and solvents were purchased from standard suppliers and used without additional purification. All glassware were dried with a flame under flushing argon gas or stored in the oven and allowed to cool under an inert atmosphere prior to use. Anhydrous solvents (THF, DCM, and Et2O) were obtained by passage through solvent filtration systems (Pure Solv), and solvents were transferred by the syringe. PET ether refers to petroleum (bp 40–60 °C, reagent grade, Fisher Scientific). All reactions were carried out under a blanket of nitrogen in an inert or dry atmosphere. Thin-layer chromatography (TLC) was performed using aluminium plates precoated with silica gel (0.25 mm, 60 A° pore-size) impregnated with a fluorescent indicator (254 nm). Visualization on TLC was achieved by the use of UV light (254 nm). Flash column chromatography was undertaken on silica gel (400–630 mesh). Proton nuclear magnetic resonance spectra (1H NMR) were recorded on an AVANCE III 400 Bruker (400 MHz). Proton chemical shifts are expressed in parts per million (ppm, δ scale) and are referenced to residual protium in the NMR solvent (CDCl3, δ 7.26; CD3OD, δ 3.31 and DMSO-d6, δ 2.50). The following abbreviations were used to describe peak patterns when appropriate: br = broad, s = singlet, d = doublet, t = triplet, q = quadruplet, sept = septet, and m = multiplet. Coupling constants, J, were reported in hertz unit (Hz). Carbon 13 nuclear magnetic resonance spectroscopy (13C NMR) was recorded on an AVANCE III 400 Bruker (101 MHz) and was fully decoupled by broad-band decoupling. Chemical shifts were reported in ppm referenced to the centerline of a triplet at 77.0, 49.0, and 39.5 ppm of CDCl3, CD3OD, and DMSO-d6. Low-resolution mass spectrometry was performed on a Thermo Scientific LCQ Fleet quadrupole mass spectrometer using electrospray ionization in the positive mode (ESI+), employing a 150 mm × 4 mm C18 column (Dr. Maisch Reprosil Gold). High-resolution mass spectrometry (HRMS) was performed on a Bruker microTOF-Q II (ESI+). Preparative high-performance liquid chromatography (HPLC) was carried out on a Dionex HPLC system equipped with Dionex P680 pumps and a Dionex UVD170U UV–vis detector (monitoring at 214 and 280 nm), using a Phenomenex, Gemini, C18, 5 μm, 250 × 21.2 mm column. Gradients were performed using solvents consisting of A (H2O + 0.1% TFA) and B (CH3CN + 0.1% TFA), and fractions were lyophilized on a Christ Alpha 2-4 LO plus freeze dryer. Final molecules were analyzed on a Shimadzu reverse-phase HPLC (RP-HPLC) system equipped with Shimadzu LC-20AT pumps, a SIL-20A autosampler, and a SPD-20A UV–vis detector (monitoring at 214 nm) using a Phenomenex, Aeris, 5 μm, peptide XB-C18, 150 × 4.6 mm column at a flow rate of 1 mL/min. RP-HPLC gradients were run using a solvent system consisting of solution A (5% CH3CN in H2O + 0.1% TFA) and B (5% H2O in CH3CN + 0.1% TFA). A gradient from 0 to 100% solution B over 20 min was run. Purity of all final compounds is >95%, as determined by RP-HPLC.
Experimental Procedures and Characterization Data
4-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (3)
To a solution of sodium hydride (1.83 g, 76.1 mmol, 3 equiv) and tetrabutylammonium bromide (0.25 g, 0.76 mmol, 0.03 equiv) in dichloromethane (80 mL) at 0 °C was added 4-bromo-1H-pyrrolo[2,3-b]pyridine, 2 (5 g, 25.4 mmol, 1 equiv). The mixture was then left to stir at 0 °C for 15 min. Toluene sulfonylchloride (5.81 g, 30.5 mmol, 1.2 equiv) in dichloromethane (20 mL) was slowly added over 5 min. The mixture was then left to warm up to room temperature and stirred for 1 h. The reaction was quenched by addition of water and extracted with dichloromethane. The organic layer was washed with brine and dried over magnesium sulfate. The residue was then purified by flash column chromatography (10% ethyl acetate–PET ether) to give 3 as a colorless solid (8.83 g, 99%); 1H NMR (400 MHz, CDCl3): δ 8.22 (d, J = 5.2 Hz, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 4.0 Hz, 1H), 7.35 (d, J = 5.3 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H), 6.64 (d, J = 4.0 Hz, 1H), 2.37 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 146.8, 145.5, 145.0, 135.1, 129.7, 128.2, 127.0, 125.7, 124.4, 122.1, 104.9, 21.7; HRMS m/z: calcd for C14H11BrN2NaO2S [M + Na]+, 372.9617; found, 372.9608 (Δ = 2.3 ppm).
4-Bromo-2-iodo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (4)
n-Butyllithium (2.5 M; 6.3 mL, 15.6 mmol, 1.1 equiv) was added dropwise to diisopropylamine (2.4 mL, 17.2 mmol, 1.2 equiv) in diethyl ether (30 mL) at −78 °C over a period of 5 min. The resulting solution was stirred at −78 °C for 60 min and then slowly added via cannula to a solution of 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine, 3 (5.0 g, 14.2 mmol, 1 equiv) and tetramethylethylenediamine (2.3 mL, 15.7 mmol, 1.1 equiv) in diethyl ether (170 mL) over a period of 10 min at −78 °C. The resulting solution was then stirred at −78 °C for 90 min. Iodine (5.4 g, 21.4 mmol, 1.5 equiv) was added in one portion, and the reaction mixture was stirred at −78 °C for 60 min. The reaction was quenched with saturated ammonium chloride solution, and the organic layer was washed with aqueous sodium thiosulfate and brine before drying over magnesium sulfate. The residue was then purified by column chromatography (20% ethyl acetate–PET ether) to give 4 as a colorless solid (5.59 g, 85%; 1H NMR (400 MHz, CDCl3): δ 8.11 (d, J = 5.2 Hz, 1H), 8.01 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 5.2 Hz, 1H), 7.22–7.19 (m, 2H), 6.96 (s, 1H), 2.30 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 149.1, 145.7, 144.7, 135.4, 129.8, 128.3, 125.3, 123.6, 122.4, 119.4, 21.7; HRMS m/z: calcd for C14H10BrIN2NaO2S [M + Na]+, 498.8583; found, 498.8602 (Δ = −3.8 ppm).
Method A: General Method of Suzuki Cross-Coupling
To a solution of desired aryl bromide (1.0 equiv) and tetrakis(triphenylphosphine)palladium(0) (0.05 equiv) in 1,4-dioxane was added boronic acid/ester (1.1 equiv) under a nitrogen atmosphere. Aqueous sodium carbonate (2 M, 7.0 equiv) was then added, and the reaction mixture was left to stir at 110 °C for 18 h. Solvent was removed under vacuum, and the crude was dissolved in ethyl acetate and poured into water and extracted with ethyl acetate. The organic layer was washed with brine before drying over magnesium sulfate and purified by flash column chromatography as indicated.
Method B: Reductive Amination of Aldehydes
To a solution of aryl aldehyde (1.0 equiv) in 1,4-dioxane was added amine (1.5 equiv), and the solution was allowed to stir for 2 min before the addition of sodium triacetoxyborohydride (2.5 equiv). The reaction mixture was stirred at room temperature for 18 h before quenching with ammonium hydroxide. The reaction mixture was extracted with ethyl acetate and washed with brine. The organic layer was dried over magnesium sulfate, and the residue was purified by flash column chromatography as indicated.
Method C: Deprotection of Azaindole
To a solution of protected 7-azaindole (1 equiv) in methanol was added potassium carbonate (3.5 equiv) and refluxed for 18 h. The reaction was poured into a mixture of EtOAc (10 mL) and H2O in a separatory funnel. Solvent was then removed under vacuum, and the residue was then purified by flash column chromatography as indicated.
Method D: Suzuki Cross-Coupling with Boronate Ester
To a 10 mL microwave vial containing the required bromo-7-azaindole (1 equiv) in 1,4-dioxane was added boronic acid/ester (1.1 equiv), Pd(dppf)Cl2·DCM complex (0.05 equiv) under a nitrogen atmosphere. The solution was purged with nitrogen for 5 min, and the reaction microwaved at 110 °C for 0.5 h. The reaction was allowed to cool to room temperature, and the mixture was filtered through Celite eluting with methanol. The filtrate was evaporated, and the resulting residue was purified by preparative HPLC: 10–95% acetonitrile in water + 0.1% TFA to give the desired products.
Method E: Synthesis of Boronate Ester
Boronate esters required for Suzuki coupling were prepared according to the procedure reported in the literature.34 To a solution of aryl bromide (1 equiv), bis(pinacolato)diboron (1.5 equiv) and potassium acetate (3 equiv) in 1,4-dioxane (20 mL), PdCI2(dppf)·CH2Cl2 complex (0.1 equiv) were added under nitrogen and stirred at 100 °C for 3 h. The reaction mixture was quenched with saturated NaHCO3 and extracted with ethyl acetate. The organic phase was dried (Na2SO4), filtered, and concentrated to dryness. The crude product was purified by chromatography (20% ethyl acetate–PET ether) to give the desired boronate esters.
3-[4-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-2-yl]-4-methoxybenzaldehyde (5)
Prepared according to method A. Purification by flash chromatography (50% ethyl acetate–PET ether) afforded 5 as yellow oil (1.72 g, 70%); 1H NMR (400 MHz, CDCl3): δ 9.90 (s, 1H), 8.15 (d, J = 5.2 Hz, 1H), 7.95 (dd, J = 8.5, 2.1 Hz, 1H), 7.84 (d, J = 2.1 Hz, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 5.3 Hz, 1H), 7.13 (d, J = 8.2 Hz, 2H), 7.04 (d, J = 8.5 Hz, 1H), 6.52 (s, 1H), 3.85 (s, 3H), 2.27 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 190.4, 163.5, 148.7, 145.1, 144.8, 137.6, 135.8, 134.4, 131.4, 129.4, 129.4, 128.1, 125.1, 123.3, 123.1, 122.3, 110.6, 107.9, 56.1, 21.6; HRMS m/z: calcd for C22H17BrN2NaO4S [M + Na]+, 506.9985; found, 506.9977 (Δ = 1.5 ppm).
N-(3-(4-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-2-yl)-4-methoxybenzyl)-N-Ethylethanamine (6a)
Prepared according to method B. Purification by flash chromatography (5% methanol–dichloromethane) afforded 6a as yellow oil (920 mg, 72%); 1H NMR (400 MHz, CD3OD): δ 8.15 (d, J = 5.3 Hz, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.65 (dd, J = 8.5, 2.4 Hz, 1H), 7.62 (d, J = 2.3 Hz, 1H), 7.47 (d, J = 5.3 Hz, 1H), 7.31 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.5 Hz, 1H), 6.65 (s, 1H), 4.45–4.35 (m, 2H), 3.81 (s, 3H), 3.31–3.23 (m, 4H), 2.36 (s, 3H), 1.38 (t, J = 7.3 Hz, 7H); 13C NMR (101 MHz, CD3OD): δ 159.3, 148.4, 145.7, 144.3, 138.2, 135.5, 133.7, 133.6, 129.1, 127.6, 124.7, 123.3, 122.3, 122.3, 120.8, 111.1, 107.6, 55.2, 54.9, 20.1; HRMS m/z: calcd for C26H28BrN3O3S [M + H]+, 541.1035; found, 542.1093 (Δ = 2.7 ppm).
1-({3-[4-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-2-yl]-4-methoxyphenyl}methyl)-N,N-dimethylamine (6b)
Prepared according to method B. Purification by flash chromatography (5% methanol–dichloromethane) afforded 6b as yellow oil (225 mg, 70%); 1H NMR (400 MHz, CD3OD): δ 8.17 (d, J = 5.3 Hz, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.65 (dd, J = 8.5, 2.3 Hz, 1H), 7.61 (d, J = 2.3 Hz, 1H), 7.50 (d, J = 5.3 Hz, 1H), 7.33 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.5 Hz, 1H), 6.66 (s, 1H), 4.27 (d, J = 14.4 Hz, 2H), 3.83 (s, 3H), 2.84 (s, 7H), 2.38 (s, 3H); 13C NMR (101 MHz, CD3OD): δ 156.2, 147.9, 142.1, 137.0, 131.0, 129.6, 129.5, 124.1, 122.6, 119.3, 118.8, 111.5, 98.8, 62.6, 55.0, 43.6; HRMS m/z: calcd for C24H25BrN3O3S [M + H]+, 514.0795; found, 514.0790 (Δ = 0.9 ppm).
1-({3-[4-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-2-yl]-4 methoxyphenyl}methyl)pyrrolidine (6c)
Prepared according to method B. Purification by flash chromatography (5% methanol–dichloromethane) afforded 6c as yellow oil (330 mg, 97%); 1H NMR (400 MHz, CD3OD): δ 8.13 (d, J = 5.2 Hz, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.65–7.67 (m, 2H), 7.42 (d, J = 5.3 Hz, 1H), 7.28 (d, J = 8.3 Hz, 2H), 7.15 (d, J = 9.1 Hz, 1H), 6.63 (s, 1H), 4.44–4.29 (m, 1H), 3.78 (s, 3H), 2.32 (s, 3H), 2.10–2.06 (p, 4H), 1.95 (s, 4H); 13C NMR (101 MHz, CD3OD): δ 176.8, 159.1, 148.4, 145.6, 144.3, 138.6, 135.6, 129.2, 127.6, 124.6, 123.3, 122.8, 122.3, 122.1, 110.9, 107.5, 56.9, 54.9, 52.8, 22.5, 21.5, 20.2; HRMS m/z: calcd for C26H27BrN3O3S [M + H]+, 540.0951; found, 540.0934 (Δ = 3.2 ppm).
4-({3-[4-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-2-yl]-4-methoxyphenyl}methyl)morpholine (6d)
Prepared according to method B. Purification by flash chromatography (5% methanol–dichloromethane) afforded 6d as yellow oil (270 mg, 80%); 1H NMR (400 MHz, CD3OD): δ 8.03 (m, 3H), 7.71 (d, J = 7.9 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.31 (d, J = 4.8 Hz, 1H), 7.23–7.18 (m, 3H), 6.96 (s, 1H), 4.36 (s, 2H), 4.04 (s, 3H), 3.34 (t, J = 6.2 Hz, 4H), 2.08 (t, J = 6.2 Hz, 4H), 1.99 (s, 3H); 13C NMR (101 MHz, CD3OD): δ 157.5, 147.9, 142.5, 140.4, 136.2, 131.8, 130.4, 128.5, 125.6, 124.3, 123.6, 122.4, 120.1, 118.9, 112.2, 99.4, 62.9, 57.2, 55.2, 53.2, 22.5; HRMS m/z: calcd for C26H27BrN3O4S [M + H]+, 556.0900; found, 556.0902 (Δ = −0.2 ppm).
N-(3-(4-Bromo-1H-pyrrolo[2,3-b]pyridin-2-yl)-4-methoxybenzyl)-N-ethylethanamine (7a)
Prepared according to method C. Purification by flash chromatography (10% methanol–dichloromethane) afforded 7a as colorless oil (560 mg, 79%); 1H NMR (400 MHz, CD3OD): δ 8.00 (d, J = 5.3 Hz, 1H), 7.83 (d, J = 2.3 Hz, 1H), 7.36 (dd, J = 8.5, 2.2 Hz, 1H), 7.29 (d, J = 5.3 Hz, 1H), 7.13 (d, J = 8.5 Hz, 1H), 6.91 (s, 1H), 4.00 (s, 3H), 3.75 (s, 2H), 3.31 (p, J = 1.6 Hz, 1H), 2.71 (q, J = 7.2 Hz, 4H), 1.15 (t, J = 7.2 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 156.3, 147.9, 142.1, 136.9, 131.2, 129.7, 124.11, 122.5, 119.4, 118.8, 111.6, 98.8, 56.1, 54.9, 9.4; HRMS m/z: calcd for C19H22BrN3O [M + H]+, 387.0946; found, 388.1008 (Δ = 2.7 ppm).
1-[(3-{4-Bromo-1H-pyrrolo[2,3-b]pyridin-2-yl}-4-methoxyphenyl)methyl]dimethylamine (7b)
Prepared according to method C. Purification by flash chromatography (10% methanol–dichloromethane) afforded 7b as colorless oil (280 mg, 92%); 1H NMR (400 MHz, CD3OD): δ 7.91 (d, J = 5.3 Hz, 1H), 7.70 (d, J = 2.2 Hz, 1H), 7.23 (dd, J = 8.4, 2.2 Hz, 1H), 7.20 (d, J = 5.3 Hz, 1H), 7.04 (d, J = 8.5 Hz, 1H), 6.82 (s, 1H), 3.91 (s, 3H), 3.44 (s, 2H), 2.21 (s, 6H); 13C NMR (101 MHz, CD3OD): δ 156.3, 147.9, 142.1, 137.0, 131.0, 129.7, 129.5, 124.1, 122.6, 119.3, 118.8, 111.5, 98.8, 62.6, 54.9, 43.6; HRMS m/z: calcd for C17H19BrN3OS [M + H]+, 360.0706; found, 360.0701 (Δ = 1.4 ppm).
1-[(3-{4-Bromo-1H-pyrrolo[2,3-b]pyridin-2-yl}-4-methoxyphenyl)methyl]pyrrolidine (7c)
Prepared according to method C. Purification by flash chromatography (10% methanol–dichloromethane) afforded 7c as yellow oil (165 mg, 77%); 1H NMR (400 MHz, CD3OD): δ 7.88 (d, J = 5.2 Hz, 1H), 7.84 (d, J = 2.3 Hz, 1H), 7.36 (dd, J = 8.5, 2.3 Hz, 1H), 7.16 (dd, J = 8.5, 3.4 Hz, 1H), 7.04 (d, J = 8.6 Hz, 1H), 6.81 (s, 1H), 4.06 (s, 2H), 3.89 (s, 3H), 3.04 (t, J = 6.8 Hz, 4H), 1.91 (p, J = 3.2 Hz, 4H); 13C NMR (101 MHz, CD3OD): δ 157.0, 148.0, 142.4, 136.5, 131.3, 130.0, 126.4, 124.2, 122.4, 119.9, 118.9, 112.0, 99.1, 58.1, 55.1, 53.4, 22.6; HRMS m/z: calcd for C19H21BrN3O [M + H]+, 386.0900; found, 386.0903 (Δ = 0.8 ppm).
4-[(3-{4-Bromo-1H-pyrrolo[2,3-b]pyridin-2-yl}-4-methoxyphenyl)methyl]morpholine (7d)
Prepared according to method C. Purification by flash chromatography (10% methanol–dichloromethane) afforded 7d as yellow oil (269 mg, 80%); 1H NMR (400 MHz, DMSO-d6): δ 12.18 (s, 1H), 8.08 (d, J = 5.1 Hz, 1H), 7.80 (d, J = 2.0 Hz, 1H), 7.65–7.60 (m, 2H), 7.58–7.55 (m, 1H), 7.33 (d, J = 5.1 Hz, 1H), 7.32 (d, J = 2.0 Hz, 1H), 7.15 (d, J = 8.5 Hz, 1H), 6.89 (d, J = 2.2 Hz, 1H), 3.94 (s, 3H), 3.58 (t, 4H), 3.47 (s, 2H), 2.38 (t, 4H); 13C NMR (101 MHz, DMSO-d6): δ 156.2, 149.1, 143.5, 136.8, 132.5, 130.9, 129.7, 129.2, 123.4, 122.1, 119.4, 119.1, 112.4, 100.2, 66.7, 62.3, 56.2, 53.6; HRMS m/z: calcd for C19H21BrN3O2 [M + H]+, 402.0812; found, 402.0810 (Δ = 0.5 ppm).
4-(2-(5-((Diethylamino)methyl)-2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-isopropylbenzoic Acid, 1
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 1 in a (85 mg, 68%); 1H NMR (400 MHz, CD3OD): δ 8.39 (d, J = 5.7 Hz, 1H), 8.01 (d, J = 2.2 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 1.8 Hz, 1H), 7.74 (dd, J = 8.0, 1.8 Hz, 1H), 7.58 (dd, J = 8.6, 2.3 Hz, 1H), 7.52 (d, J = 5.8 Hz, 1H), 7.32 (d, J = 8.6 Hz, 1H), 7.31 (s, 1H), 4.37 (s, 2H), 4.07 (s, 3H), 3.94 (p, J = 6.9 Hz, 1H), 3.25 (dq, J = 14.2, 7.1 Hz, 4H), 1.39–1.34 (m, 12H); 13C NMR (101 MHz, CD3OD): δ 169.8, 157.9, 150.1, 145.6, 143.9, 140.0, 137.8, 136.8, 132.9, 131.8, 131.0, 130.3, 126.4, 125.6, 122.1, 121.8, 119.6, 114.9, 112.5, 100.3, 55.3, 55.2, 29.4, 23.0, 7.6; HRMS m/z: calcd for C29H33N3O3 [M + H]+, 471.2522; found, 472.2576 (Δ = 2.1 ppm).
4-(2-{5-[(Dimethylamino)methyl]-2-methoxyphenyl}-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-(propan-2-yl)benzoic Acid (8a)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 8a in a (11 mg, 29%); 1H NMR (400 MHz, CD3OD): δ 8.27 (d, J = 5.4 Hz, 1H), 7.89 (d, J = 2.3 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 1.7 Hz, 1H), 7.63 (dd, J = 8.0, 1.8 Hz, 1H), 7.44 (dd, J = 8.6, 2.3 Hz, 2H), 7.32 (d, J = 5.5 Hz, 1H), 7.23 (d, J = 8.6 Hz, 1H), 7.13 (s, 1H), 4.23 (s, 2H), 3.97 (s, 3H), 3.89–3.81 (m, 1H), 2.79 (s, 6H), 1.27 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 169.9, 158.1, 150.2, 146.7, 142.6, 139.6, 138.4, 135.1, 133.2, 132.2, 131.1, 130.4, 126.5, 125.7, 122.3, 120.4, 119.2, 115.0, 112.5, 100.5, 60.1, 55.3, 41.4, 29.5, 23.0; HRMS m/z: calcd for C27H29N3O3 [M + H]+, 470.2438; found, 470.2433 (Δ = 1.1 ppm).
4-(2-{2-Methoxy-5-[(pyrrolidin-1-yl)methyl]phenyl}-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-(propan-2-yl)benzoic Acid (8b)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 8b in a (27 mg, 37%); 1H NMR (400 MHz, CD3OD): δ 8.27 (br s, 1H), 7.90 (s, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.79 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.46 (d, J = 8.6 Hz, 1H), 7.36 (s, 1H), 7.21–7.15 (m, 2H), 4.29 (s, 2H), 3.96 (s, 3H), 3.85–3.81 (m, 1H), 3.42 (br s, 2H), 3.14 (br s, 2H), 2.08 (br s, 2H), 1.93 (br s, 2H), 1.26 (d, J = 6.8 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 169.9, 157.9, 150.1, 144.5, 140.5, 137.4, 132.1, 131.5, 130.4, 130.3, 126.3, 125.5, 123.4, 119.9, 112.4, 100.0, 57.2, 55.2, 53.3, 29.4, 23.0, 22.4; HRMS m/z: calcd for C29H32N3O3 [M + H]+, 470.2438; found, 470.2433 (Δ = 1.1 ppm).
4-(2-{2-Methoxy-5-[(morpholin-4-yl)methyl]phenyl}-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-(propan-2-yl)benzoic Acid (8c)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 8c in a (34 mg, 60%); 1H NMR (400 MHz, DMSO-d6): δ 8.43 (d, J = 5.8 Hz, 1H), 8.04 (d, J = 2.3 Hz, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.93 (d, J = 1.8 Hz, 1H), 7.78 (dd, J = 8.0, 1.8 Hz, 1H), 7.61 (dd, J = 8.6, 2.2 Hz, 1H), 7.56 (d, J = 5.7 Hz, 1H), 7.36 (d, J = 8.5 Hz, 1H), 7.32 (s, 1H), 4.41 (s, 2H), 4.10 (s, 3H), 4.07 (s, 2H), 3.98–3.93 (m, 1H), 3.76 (s, 2H), 3.44 (s, 2H), 3.26 (s, 2H), 1.39 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz DMSO-d6): δ 170.3, 169.7, 149.6, 147.1, 136.0, 133.0, 133.0, 132.2, 132.1, 131.6, 131.3, 130.6, 128.3, 126.5, 125.9, 121.7, 120.6, 112.9, 100.5, 63.8, 59.4, 56.4, 51.2, 49.1, 29.4, 24.4, 24.3; HRMS m/z: calcd for C29H32N3O4 [M + H]+, 486.2387; found, 486.2384 (Δ = 0.8 ppm).
Ethyl 4-(2-(5-((Diethylamino)methyl)-2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-isopropylbenzoate (9)
Purification by preparative HPLC 10–95% MeCN/H2O to give 9 in a (36 mg, 98%); 1H NMR (400 MHz, CD3OD): δ 8.39 (d, J = 5.6 Hz, 1H), 8.01 (d, J = 2.2 Hz, 1H), 7.92 (d, J = 8.2 Hz, 2H), 7.77–7.72 (m, 1H), 7.57 (dd, J = 8.6, 2.2 Hz, 1H), 7.49 (d, J = 5.7 Hz, 1H), 7.33 (d, J = 8.6 Hz, 1H), 7.28 (s, 1H), 4.42 (q, J = 7.1 Hz, 2H), 4.37 (s, 2H), 4.07 (s, 3H), 3.82 (p, J = 6.9 Hz, 1H), 3.25 (p, J = 7.4 Hz, 4H), 1.50–1.29 (m, 16H); 13C NMR (101 MHz, CD3OD): δ 168.1, 157.9, 149.9, 140.7, 138.8, 137.2, 132.6, 131.1, 130.9, 130.1, 126.4, 125.6, 122.0, 120.1, 114.9, 112.5, 99.9, 60.9, 55.3, 29.6, 22.9, 13.2, 7.7; HRMS m/z: calcd for C31H37N3O3 [M + H]+, 499.2835; found, 500.2913 (Δ = 4.1 ppm).
3-[4-Bromo-1-(4-methylbenzenesulfonyl)-1H-pyrrolo[2,3-b]pyridin-2-yl]-4-methoxybenzonitrile (10)
Prepared according to method B. Purification by flash chromatography (50% ethyl acetate–PET ether) afforded 10 as yellow oil (586 mg, 87%; 1H NMR (400 MHz, DMSO-d6): δ 8.23 (d, J = 5.2 Hz, 1H), 8.03–7.99 (m, 2H), 7.80 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 5.2 Hz, 1H), 7.41–7.37 (m, 2H), 7.34–7.29 (m, 2H), 6.81 (s, 1H), 3.84 (s, 3H), 2.34 (s, 3H); 13C NMR (101 MHz, DMSO- d6): δ 161.8, 148.3, 145.9, 145.7, 143.5, 137.1, 136.4, 135.5, 134.8, 130.2, 128.2, 125.0, 123.0, 119.3, 112.4, 109.5, 108.3, 103.1, 56.6, 21.6; HRMS m/z: calcd for C22H16BrN3NaO3S [M + Na]+, 503.9988; found, 503.9973 (Δ = 3.0 ppm).
3-{4-Bromo-1H-pyrrolo[2,3-b]pyridin-2-yl}-4-methoxybenzonitrile (11)
Prepared according to method C. Purification by flash chromatography (10% methanol–dichloromethane) afforded 11 as yellow oil (327 mg, 59%); 1H NMR (400 MHz, DMSO-d6): δ 12.24 (s, 1H), 8.29 (d, J = 2.1 Hz, 1H), 8.06 (d, J = 5.1 Hz, 1H), 7.81 (dd, J = 8.7, 2.1 Hz, 1H), 7.33–7.29 (m, 2H), 6.96 (s, 1H), 3.98 (s, 3H); 13C NMR (101 MHz, DMSO-d6): δ 207.0, 160.3, 149.1, 144.4, 134.3, 132.5, 124.1, 121.1, 119.4, 119.3, 113.7, 103.8, 101.8, 56.9, 31.2; HRMS m/z: calcd for C15H11BrN3O [M + H]+, 328.0080; found, 328.0076 (Δ = 1.1 ppm).
4-{2-[5-(Aminomethyl)-2-methoxyphenyl]-1H-pyrrolo[2,3-b]pyridin-4-yl}-2-(propan-2-yl)benzoic Acid (12)
Prepared according to method D. The filtrate residue was dissolved in methanol (10 mL), CoCl2·6H2O (3 equiv) was then added before cooling to 0 °C. NaBH4 (10 equiv) was slowly added to the reaction, and the mixture was allowed to stir at room temperature for further 1 h before filtration through Celite and purification by preparative HPLC 10–95% MeCN/H2O to give 13 (26 mg, 60%); 1H NMR (400 MHz, DMSO-d6): δ 12.01 (s, 1H), 8.30 (d, J = 5.0 Hz, 1H), 8.17 (br s, 2H), 7.94 (d, J = 2.3 Hz, 1H), 7.82–7.80 (m, 2H), 7.62 (dd, J = 8.0, 1.8 Hz, 1H), 7.41 (dd, J = 8.6, 2.2 Hz, 1H), 7.24 (d, J = 5.0 Hz, 1H), 7.18 (d, J = 8.6 Hz, 1H), 7.09 (s, 1H), 3.98 (q, J = 5.6 Hz, 2H), 3.89 (s, 3H), 3.83–3.76 (m, 1H), 1.25 (d, J = 6.9 Hz, 6H).; 13C NMR (101 MHz, DMSO-d6): δ 169.7, 157.0, 149.6, 149.2, 142.9, 141.4, 140.4, 136.6, 131.4, 130.6, 130.6, 129.7, 126.7, 126.6, 125.9, 120.1, 119.0, 115.2, 112.7, 100.0, 56.3, 42.4, 29.4, 24.3; HRMS m/z: calcd for C25H26N3O3 [M + H]+, 416.1969; found, 416.1968 (Δ = 0.2 ppm).
4-Bromo-2-(2-methoxyphenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (13)
Prepared according to method A. Purification by flash chromatography (50% ethyl acetate–PET ether) afforded 14 as colorless oil (960 mg, 32%); 1H NMR (400 MHz, CDCl3): δ 8.14 (d, J = 5.3 Hz, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.41 (td, J = 8.2, 1.7 Hz, 1H), 7.29–7.24 (m, 2H), 7.12 (d, J = 8.1 Hz, 2H), 6.99 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 8.2 Hz, 1H), 6.45 (s, 1H), 3.73 (s, 3H), 2.27 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 158.3, 148.7, 144.8, 144.4, 139.4, 136.1, 131.1, 130.9, 129.2, 128.2, 124.8, 123.5, 122.1, 121.8, 120.0, 110.4, 107.3, 55.5, 21.6; HRMS m/z: calcd for C21H17BrN2NaO3S [M + Na]+, 479.0035; found, 479.0035 (Δ = 0.0 ppm).
4-Bromo-2-(2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridine (14)
Prepared according to method C. Purification by flash chromatography (10% methanol–dichloromethane) afforded 15 as colorless oil (300 mg 80%); 1H NMR (400 MHz, CDCl3): δ 10.38 (s, 1H), 8.09 (d, J = 5.2 Hz, 1H), 7.92 (dd, J = 7.8, 1.6 Hz, 1H), 7.41–7.34 (m, 1H), 7.30–7.25 (m, 2H), 7.14–7.07 (m, 2H), 6.89 (s, 1H), 4.04 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 156.1, 148.1, 143.0, 137.0, 129.9, 128.4, 124.3, 122.2, 121.6, 119.4, 119.3, 112.0, 97.8, 55.9; HRMS m/z: calcd for C14H12BrN2O [M + H]+, 303.0128; found, 303.0122 (Δ = 1.8 ppm).
4-[2-(2-Methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl]-2-(propan-2-yl)benzoic Acid (15)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 16 in a (30 mg, 59%); 1H NMR (400 MHz, DMSO-d6): δ 12.05 (s, 1H), 8.32 (d, J = 5.0 Hz, 1H), 7.92 (dd, J = 7.8, 1.7 Hz, 1H), 7.90 (d, J = 1.8 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.68 (dd, J = 8.1, 1.8 Hz, 1H), 7.38 (ddd, J = 8.7, 7.4, 1.7 Hz, 1H), 7.27 (d, J = 5.0 Hz, 1H), 7.20–7.18 (m, 2H), 7.08 (td, J = 7.6, 1.0 Hz, 1H), 3.92 (s, 3H), 3.90–3.83 (m, 1H), 1.32 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, DMSO-d6): δ 169.7, 157.1, 149.9, 149.6, 141.7, 136.5, 131.1, 130.7, 129.9, 126.5, 125.9, 121.2, 100.3, 45.4, 29.4; HRMS m/z: calcd for C24H23N2O3 [M + H]+, 387.17034; found, 387.1710 (Δ = −1.7 ppm).
3-(4-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-2-yl)-4-(methoxymethoxy)benzaldehyde (16)
Prepared according to method A. Purification by flash chromatography (50% ethyl acetate–PET ether) afforded 17 as colorless oil (850 mg, 77%); 1H NMR (400 MHz, CDCl3): δ 9.92 (s, 1H), 8.16 (d, J = 5.3 Hz, 1H), 7.92 (dd, J = 8.5, 2.1 Hz, 1H), 7.84 (d, J = 2.1 Hz, 1H), 7.69 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.5 Hz, 1H), 7.29 (d, J = 5.3 Hz, 1H), 7.12 (d, J = 8.1 Hz, 3H), 6.53 (s, 1H), 5.26–5.22 (m, 2H), 3.46 (s, 3H), 2.27 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 190.6, 161.7, 148.6, 145.2, 144.9, 137.7, 135.5, 134.4, 131.0, 130.0, 129.4, 128.0, 125.1, 123.6, 122.3, 113.9, 108.0, 95.1, 56.9, 21.6; HRMS m/z: calcd for C23H19BrN2NaO5S [M + Na]+, 537.0090; found, 537.0098 (Δ = −1.7 ppm).
({3-[4-Bromo-1-(4-methylbenzenesulfonyl)-1H-pyrrolo[2,3-b]pyridin-2-yl]-4-(methoxymethoxy)phenyl}methyl)diethylamine (17)
Prepared according to method B. Purification by flash chromatography (5% methanol–dichloromethane) afforded 18 as colorless oil (672 mg, 87%); 1H NMR (400 MHz, CD3OD): δ 8.16 (d, J = 5.3 Hz, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 7.9 Hz, 2H), 7.50 (d, J = 5.3 Hz, 1H), 7.36 (d, J = 8.5 Hz, 1H), 7.31 (d, J = 8.2 Hz, 2H), 6.68 (s, 1H), 5.18 (br s, 1H), 5.28 (br s, 1H), 4.20 (d, J = 5.4 Hz, 2H), 3.43 (s, 3H), 3.10 (q, J = 7.1 Hz, 4H), 2.37 (s, 3H), 1.33 (t, J = 7.2 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 156.7, 148.4, 145.7, 144.3, 138.7, 135.5, 133.1, 132.9, 129.2, 127.6, 124.7, 123.3, 122.8, 122.4, 114.1, 107.5, 94.7, 55.4, 55.4, 46.2, 20.1, 8.4; HRMS m/z: calcd for C27H31BrN3O4S [M + H]+, 572.1213; found, 572.1208 (Δ = 0.9 ppm).
[(3-{4-Bromo-1H-pyrrolo[2,3-b]pyridin-2-yl}-4-(methoxymethoxy)phenyl)methyl]diethylamine (18)
Prepared according to method C. Purification by flash chromatography (10% methanol–dichloromethane) afforded 19 as yellow oil (420 mg, 69%); 1H NMR (400 MHz, CD3OD): δ 8.03 (d, J = 5.2 Hz, 1H), 7.84 (d, J = 2.1 Hz, 1H), 7.34 (dd, J = 8.5, 2.1 Hz, 1H), 7.31–7.28 (m, 2H), 6.96 (s, 1H), 5.39 (s, 2H), 4.88 (s, 3H), 3.73 (s, 2H), 3.50 (s, 3H), 2.70 (q, J = 7.2 Hz, 4H), 1.15 (t, J = 7.2 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 153.9, 148.0, 142.3, 136.7, 130.9, 130.5, 129.7, 124.2, 122.6, 120.4, 118.9, 115.2, 99.3, 94.7, 56.1, 55.4, 46.1, 9.5; HRMS m/z: calcd for C21H25BrN2O2 [M + H]+, 418.1113; found, 418.1112 (Δ = −0.2 ppm).
4-[2-(2-Methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl]-2-(propan-2-yl)benzoic Acid (19)
Prepared according to method D. The filter residue was then stirred in a mixture of acetonitrile/HCl (3:1) for 30 min before filtering through Celite and purification by preparative HPLC 10–95% MeCN/H2O to give 21 in a (72 mg, 70%); 1H NMR (400 MHz, CD3OD): δ 8.30 (d, J = 5.3 Hz, 1H), 7.92 (d, J = 2.2 Hz, 1H), 7.89 (d, J = 8.1 Hz, 1H), 7.84 (d, J = 1.6 Hz, 1H), 7.68 (dd, J = 8.1, 1.7 Hz, 1H), 7.46 (d, J = 5.7 Hz, 1H), 7.3–7.31 (m, 2H), 7.04 (d, J = 8.4 Hz, 1H), 4.24 (s, 2H), 3.85 (sept, J = 6.9 Hz, 1H), 3.19–3.12 (m, 4H), 1.30–1.27 (m, 12H); 13C NMR (101 MHz, CD3OD): δ 169.8, 156.5, 150.2, 139.7, 138.9, 135.4, 132.7, 132.1, 130.8, 130.4, 126.6, 125.7, 121.0, 117.4, 117.1, 115.0, 99.7, 55.4, 46.4, 29.5, 23.0; HRMS m/z: calcd for C28H32N3O3 [M + H]+, 458.2438; found, 458.2430 (Δ = 1.8 ppm).
3-(4-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-2-yl)benzaldehyde (20)
Prepared according to method A. Purification by flash chromatography (50% ethyl acetate–PET ether) afforded 22 as colorless oil (500 mg, 76%); 1H NMR (400 MHz, CDCl3): δ 10.04 (s, 1H), 8.23 (d, J = 5.2 Hz, 1H), 7.96 (t, J = 1.8 Hz, 1H), 7.93 (d, J = 7.7 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.59 (t, J = 7.7 Hz, 1H), 7.33 (d, J = 5.3 Hz, 1H), 7.14 (d, J = 8.3 Hz, 2H), 6.56 (s, 1H), 2.29 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 191.8, 149.5, 145.5, 145.2, 141.0, 136.0, 135.9, 135.2, 133.3, 130.6, 130.3, 129.6, 128.5, 127.9, 125.3, 123.6, 122.9, 109.1, 21.7; HRMS m/z: calcd for C21H15BrN2NaO3S [M + Na]+, 476.9879; found, 476.9884 (Δ = −1.0 ppm).
({3-[4-Bromo-1-(4-methylbenzenesulfonyl)-1H-pyrrolo[2,3-b]pyridin-2-yl]-4-(methoxymethoxy)phenyl}methyl)diethylamine (21)
Prepared according to method B. Purification by flash chromatography (5% methanol–dichloromethane) afforded 23 as colorless oil (154 mg, 83%); 1H NMR (400 MHz, CD3OD): δ 8.22 (d, J = 5.3 Hz, 1H), 7.87 (d, J = 1.9 Hz, 1H), 7.75–7.70 (m, 2H), 7.68 (d, J = 7.4 Hz, 1H), 7.62 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 5.3 Hz, 1H), 7.29 (d, J = 8.4 Hz, 2H), 6.79 (s, 1H), 4.50 (s, 2H), 3.35–3.30 (m, 4H), 2.35 (s, 3H), 1.42 (t, J = 7.3 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 156.7, 148.4, 145.7, 144.3, 138.7, 135.5, 133.1, 132.9, 129.2, 127.6, 124.7, 123.3, 122.8, 122.4, 114.1, 107.5, 94.7, 55.4, 55.4, 46.2, 20.1, 8.4; HRMS m/z: calcd for C25H27BrN3O2S [M + H]+, 512.1002; found, 512.1013 (Δ = −2.3 ppm).
[(3-{4-Bromo-1H-pyrrolo[2,3-b]pyridin-2-yl}phenyl)methyl]diethylamine (22)
Prepared according to method C. Purification by flash chromatography (10% methanol–dichloromethane) afforded 24 as colorless oil (95 mg, 89%); 1H NMR (400 MHz, CD3OD): δ 7.92 (d, J = 5.3 Hz, 1H), 7.78 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 7.20 (d, J = 5.3 Hz, 1H), 6.76 (s, 1H), 3.70 (s, 2H), 2.62 (q, J = 7.2 Hz, 4H), 1.06 (t, J = 7.1 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 148.9, 142.4, 139.7, 131.5, 129.7, 128.9, 126.8, 124.7, 124.2, 123.2, 118.9, 96.9, 56.8, 46.3, 9.6; HRMS m/z: calcd for C18H21BrN3 [M + H]+, 358.0913; found, 358.0907 (Δ = 1.7 ppm).
4-(2-{3-[(Diethylamino)methyl]phenyl}-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-(propan-2-yl)benzoic Acid (23)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 25 in a (30 mg, 70%); 1H NMR (400 MHz, CD3OD): δ 8.25 (s, 1H), 7.94 (s, 1H), 7.91 (d, J = 6.9 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.73 (s, 1H), 7.62 (s, 2H), 7.53 (s, 2H), 7.45 (d, J = 7.7 Hz, 1H), 7.28 (d, J = 5.2 Hz, 1H), 7.04 (s, 1H), 4.32 (s, 2H), 3.83 (sept, J = 13.9, 6.9 Hz, 1H), 3.16 (br s, 4H), 1.27 (q, J = 7.2 Hz, 12H); 13C NMR (101 MHz, CD3OD): δ 170.0, 150.1, 132.3, 130.8, 130.6, 130.3, 129.9, 128.1, 127.0, 126.2, 125.5, 115.1, 111.9, 97.8, 55.7, 46.7, 29.4, 23.0, 7.6; HRMS m/z: calcd for C28H32N3O2 [M + H]+, 442.2489; found, 442.2476 (Δ = 3.0 ppm).
3-(4-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-2-yl)-2-methoxybenzaldehyde (24)
Prepared according to method A. Purification by flash chromatography (50% ethyl acetate–PET ether) afforded 26 as colorless oil (990 mg, 72%); 1H NMR (400 MHz, CDCl3): δ 10.49 (s, 1H), 8.28 (d, J = 5.3 Hz, 1H), 8.07–7.90 (m, 3H), 7.59 (dd, J = 7.4, 1.8 Hz, 1H), 7.41 (d, J = 5.3 Hz, 1H), 7.37–7.30 (m, 1H), 7.25 (d, J = 6.7 Hz, 2H), 6.66 (s, 1H), 3.75 (s, 3H), 2.38 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 189.6, 162.0, 148.7, 145.4, 145.0, 137.1, 137.1, 135.6, 130.2, 129.4, 129.0, 128.6, 127.3, 125.2, 123.7, 123.1, 122.5, 108.1, 63.6, 21.7; HRMS m/z: calcd for C22H17BrN2O4SNa [M + Na]+, 507.9990; found, 508.0068 (Δ = 0.2 ppm).
N-(3-(4-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-2-yl)-2-methoxybenzyl)-N-ethylethanamine (25)
Prepared according to method B. Purification by flash chromatography (50% ethyl acetate–PET ether) afforded 27 as yellow oil (765 mg, 81%; 1H NMR (400 MHz, CD3OD): δ 8.18 (d, J = 5.3 Hz, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.69 (dd, J = 7.7, 1.7 Hz, 1H), 7.63 (dd, J = 7.6, 1.7 Hz, 1H), 7.51 (d, J = 5.3 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H), 7.33–7.27 (m, 2H), 6.85 (s, 1H), 4.56 (d, J = 13.3 Hz, 1H), 4.28 (d, J = 13.3 Hz, 1H), 3.58 (s, 3H), 2.35 (s, 3H), 1.39 (t, J = 7.3 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 178.1, 158.6, 148.6, 145.9, 144.8, 138.4, 135.2, 133.7, 132.8, 129.2, 127.8, 126.4, 124.9, 124.2, 124.1, 123.2, 122.6, 108.0, 60.3, 50.2, 22.4, 20.2, 8.1; HRMS m/z: calcd for C26H28BrN3O3S [M + H]+, 541.1035; found, 542.1095 (Δ = 3.6 ppm).
N-(3-(4-Bromo-1H-pyrrolo[2,3-b]pyridin-2-yl)-2-methoxybenzyl)-N-Ethylethanamine (26)
Prepared according to method C. Purification by flash chromatography (5% methanol–dichloromethane) afforded 28 as yellow oil (630 mg, 85%); 1H NMR (400 MHz, CD3OD): δ 8.12 (d, J = 5.4 Hz, 1H), 7.92 (dd, J = 7.8, 1.7 Hz, 1H), 7.59 (dd, J = 7.6, 1.7 Hz, 1H), 7.46–7.37 (m, 2H), 7.03 (s, 1H), 4.43 (s, 2H), 3.69 (s, 3H), 3.32–3.28 (m, 4H), 1.43 (t, J = 7.3 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 157.0, 147.6, 142.1, 135.7, 132.8, 131.9, 125.6, 125.4, 125.3, 124.5, 123.2, 119.2, 100.5, 60.6, 50.9, 7.7; HRMS m/z: calcd for C19H22BrN3O [M + H]+, 387.0946; found, 388.1008 (Δ = 2.7 ppm).
4-(2-(3-((Diethylamino)methyl)-2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-isopropylbenzoic Acid (27)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 29 in a (30 mg, 74%); 1H NMR (400 MHz, CD3OD): δ 8.41 (s, 1H), 7.98–7.92 (m, 2H), 7.89 (d, J = 1.7 Hz, 1H), 7.73 (dd, J = 8.1, 1.7 Hz, 1H), 7.58 (dd, J = 7.6, 1.6 Hz, 1H), 7.47–7.43 (m, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.23 (s, 1H), 4.43 (s, 2H), 3.94 (p, J = 6.8 Hz, 1H), 3.72 (s, 3H), 3.31–3.25 (m, 5H), 1.42 (t, J = 7.3 Hz, 6H), 1.36 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 169.9, 157.1, 150.1, 140.6, 136.3, 132.8, 131.9, 131.4, 130.3, 126.3, 125.5, 125.4, 124.6, 100.5, 60.7, 50.8, 29.4, 23.0, 7.7; HRMS m/z: calcd for C29H33N3O3 [M + H]+, 471.2522; found, 472.2579 (Δ = 3.4 ppm).
4-(2-(5-((Diethylamino)methyl)-2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-methylbenzoic Acid (28)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 30 in a (50 mg, 75%); 1H NMR (101 MHz, CD3OD): δ 8.27 (d, J = 5.6 Hz, 1H), 8.04 (d, J = 8.7 Hz, 1H), 7.94 (d, J = 2.3 Hz, 1H), 7.70–7.61 (m, 2H), 7.46 (dd, J = 8.6, 2.3 Hz, 1H), 7.36 (d, J = 5.6 Hz, 1H), 7.23 (d, J = 8.6 Hz, 1H), 7.16 (s, 1H), 4.27 (s, 2H), 3.99 (s, 3H), 3.15 (p, J = 7.2 Hz, 4H), 2.63 (s, 3H), 1.27 (t, J = 7.3 Hz, 6H); 13C NMR (400 MHz, CD3OD): δ 168.9, 157.8, 140.9, 140.3, 136.5, 133.0, 131.5, 131.3, 131.2, 125.9, 122.2, 119.6, 115.1, 112.5, 99.7, 55.3, 20.6, 7.6; HRMS m/z: calcd for C27H29N3O3 [M + H]+, 443.2209; found, 444.2214 (Δ = 1.4 ppm).
4-(2-(5-((Diethylamino)methyl)-2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)benzoic Acid (29)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 31 in a (100 mg, 85%); 1H NMR (400 MHz, CD3OD): δ 8.28 (d, J = 5.5 Hz, 1H), 8.16 (d, J = 8.5 Hz, 2H), 7.95 (d, J = 2.3 Hz, 1H), 7.87 (d, J = 8.6 Hz, 2H), 7.46 (dd, J = 8.6, 2.3 Hz, 1H), 7.38 (d, J = 5.6 Hz, 1H), 7.23 (d, J = 8.6 Hz, 1H), 7.17 (s, 1H), 4.27 (s, 2H), 3.99 (s, 3H), 3.15 (p, J = 7.3 Hz, 4H), 1.27 (t, J = 7.3 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 168.0, 157.6, 148.6, 143.1, 142.4, 140.9, 136.3, 132.0, 130.9, 130.6, 129.9, 129.3, 128.3, 122.1, 120.8, 114.9, 112.4, 98.6, 55.5, 55.2, 7.6; HRMS m/z: calcd for C26H27N3O3 [M + H]+, 429.2052; found, 430.2056 (Δ = 1.1 ppm).
N-Ethyl-N-(3-(4-(3-isopropyl-4-(1H-tetrazol-5-yl)phenyl)-1H-pyrrolo[2,3-b]pyridin-2-yl)-4-methoxybenzyl)ethanamine (30)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 32 in a (27 mg, 62%); 1H NMR (400 MHz, CD3OD): δ 8.39 (d, J = 5.8 Hz, 1H), 8.01 (d, J = 2.3 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 1.8 Hz, 1H), 7.74 (dd, J = 8.0, 1.9 Hz, 1H), 7.58 (dd, J = 8.6, 2.3 Hz, 1H), 7.52 (d, J = 5.8 Hz, 1H), 7.32 (d, J = 8.7 Hz, 1H), 7.31 (s, 1H), 4.37 (s, 2H), 4.07 (s, 3H), 3.94 (p, J = 6.9 Hz, 1H), 3.25 (dd, J = 12.2, 7.0 Hz, 4H), 1.42–1.33 (m, 12H); 13C NMR (101 MHz, CD3OD): δ 169.8, 157.9, 150.1, 145.6, 143.9, 140.0, 137.8, 136.8, 132.9, 131.8, 131.0, 130.3, 126.4, 125.6, 122.1, 121.8, 119.6, 114.9, 112.5, 100.3, 55.3, 55.2, 29.4, 23.0, 7.6; HRMS m/z: calcd for C29H33N7O [M + H]+, 495.2747; found, 496.2829 (Δ = 0.6 ppm).
N-Ethyl-N-(3-(4-(3-isopropylphenyl)-1H-pyrrolo[2,3-b]pyridin-2-yl)-4-methoxybenzyl)ethanamine (31)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 33 in a (67 mg, 82%); 1H NMR (101 MHz, CD3OD): δ 8.37 (d, J = 5.9 Hz, 1H), 8.03 (d, J = 2.2 Hz, 1H), 7.75–7.68 (m, 2H), 7.61–7.54 (m, 2H), 7.50 (dd, J = 16.9, 6.8 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 4.37 (s, 2H), 4.08 (s, 3H), 3.25 (p, J = 7.5 Hz, 4H), 3.08 (p, J = 6.9 Hz, 1H), 1.43–1.31 (m, 12H); 13C NMR (400 MHz, CD3OD): δ 157.9, 149.9, 137.2, 132.8, 131.1, 128.9, 127.7, 126.5, 126.1, 122.1, 119.8, 114.9, 112.5, 100.3, 55.3, 34.1, 23.1, 7.6; HRMS m/z: calcd for C28H33N3O [M + H]+, 427.2624; found, 428.2624 (Δ = 3.8 ppm).
5-(2-(5-((Diethylamino)methyl)-2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-2-isopropylbenzoic Acid (32)
Prepared according to method D. Purification by preparative HPLC 10–95% MeCN/H2O to give 34 in a (55 mg, 78%); 1H NMR (400 MHz, CD3OD): δ 8.41 (d, J = 5.7 Hz, 1H), 8.26 (d, J = 2.0 Hz, 1H), 7.99 (ddd, J = 9.3, 8.0, 1.9 Hz, 2H), 7.73 (d, J = 8.2 Hz, 1H), 7.60 (dd, J = 7.6, 1.6 Hz, 1H), 7.53 (d, J = 5.7 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.36 (s, 1H), 4.43 (s, 2H), 3.92 (p, J = 6.9 Hz, 1H), 3.73 (s, 3H), 3.31–3.25 (m, 4H), 1.42 (t, J = 7.3 Hz, 6H), 1.34 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CD3OD): δ 169.8, 157.0, 151.0, 145.4, 144.8, 137.8, 136.9, 134.4, 133.1, 131.9, 131.6, 131.3, 129.8, 127.2, 125.4, 124.9, 124.7, 121.6, 114.9, 100.8, 60.7, 50.9, 29.4, 22.8, 7.7; HRMS m/z: calcd for C29H33N3O3 [M + H]+, 471.2522; found, 472.2579 (Δ = 3.4 ppm).
Assay
P. falciparum Culture and Synchronization
P. falciparum cultures were maintained in RPMI-1640 media (Invitrogen) supplemented with 0.2% sodium bicarbonate, 0.5% AlbuMAX II, 2.0 mM l-glutamine (Sigma), and 10 mg/L gentamycin. For continuous culture, the parasites were kept at 4% hematocrit in human erythrocytes from 0 + blood donors and between 0.5 and 3% parasitemia maintained in an incubator at 37 °C, 5% carbon dioxide (CO2), 5% oxygen (O2), and 90% nitrogen (N2). To obtain highly synchronous ring-stage parasites for the drug assays, cultures were double synchronized using Percoll and sorbitol synchronization, as previously described.35,36 First, highly segmented schizonts were enriched by centrifugation on a 70% Percoll (GE Healthcare) cushion gradient. The Schizont pellet was collected and washed before fresh erythrocytes were added to a final hematocrit of 4%. The schizonts were incubated for about 1–2 h shaking continuously to allow egress and reinvasion of new erythrocytes. Residual schizonts were then removed by treating the pellet with sorbitol to generate highly synchronous 1–2 h old ring-stage parasites.
Determining the EC50 of Compound Inhibitors and Drugs Ex Vivo
To determine the EC50 of the molecules in parasites (P. falciparum 3D7) ex vivo, the molecules were diluted 1 in 3 from a starting concentration of 100 μM for 12 dilution points. In total, 50 mL of freshly diluted drugs, at twice the required final concentrations, were aliquoted into black 96-well plates. To the drug plates, 50 μL of parasites prepared at 8% hematocrit at parasitemia (0.3–0.5%) were added and mixed by pipetting up and down several times giving a final culture volume of 100 μL at the required drug concentration (top concentration of 100 μM) and 4% hematocrit. To the “no drug” control, growth media was added, and uninfected erythrocytes were included on the plate as blank. The outer wells were filled with media to reduce evaporation from the experimental wells, and the plates were incubated for 50 h (±2 h) to allow the parasites sufficient time to reinvade before they are collected and frozen. To quantify growth inhibition, the plates were thawed at room temperature for at least 1 h, and 100 μL of lysis buffer (20 mM Tris-HCl; 5 mM EDTA; 0.004% saponin and triton X-100) in PBS containing Sybr Green I (1 μL in 5 mL) was added to each well and mixed by pipetting up and down several times and incubated for 1 h in the dark with shaking. Using a Fluoroskan/ClarioStar plate reader at excitation of 485 nm and emission of 538 nm, plate absorbances were acquired. The data were normalized against the controls, and graphs were generated using GraphPad prism 8 to determine the EC50 values using the nonlinear regression log (inhibitor) versus response (three parameter) curve.
TR-FRET to Determine the IC50 of the Inhibitors with Full-Length PfCLK3 Recombinant Protein
The TR-FRET assays, a high-throughput inhibition assay, as described previously,13 was used to determine the potency of the small molecules generated against full-length PfCLK3 recombinant protein in a kinase buffer (containing 50 mM HEPES, 10 mM MgCl2, 2 mM DTT, 0.01% Tween 20, and 1 mM EGTA), with the ULight-labeled peptide substrate MBP peptide (sequence: CFFKNIVTPRTPPPSQGK). First, in a 10 μL reaction volume, 5 μL of twice the required enzyme concentration (50 nM) and 2.5 μL of four times the required substrate concentration mix containing cold ATP, and the serially diluted drugs were mixed in a black 384-plate well plate and incubated at 37 °C for 1 h. The reaction was stopped after incubation by adding the stopping/detection solution (containing 30 mM EDTA in 1× Lance detection buffer and 3 nM Europium-labeled antiphospho-specific antibody) and incubated for another hour at RT before phosphorylation signals were measured using the ClarioStar.
For each test compound,
percent inhibition (response) which was calculated using the formula: 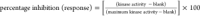 was plotted against
the log molar concentration
of compound to calculate the IC50 (potency) of each inhibitor
molecule and plotted using GraphPad prism software. All experiments
were carried out in triplicates, and the data presented were the standard
error of the mean (SEM) of three independent experiments run in triplicates.
was plotted against
the log molar concentration
of compound to calculate the IC50 (potency) of each inhibitor
molecule and plotted using GraphPad prism software. All experiments
were carried out in triplicates, and the data presented were the standard
error of the mean (SEM) of three independent experiments run in triplicates.
PfCLK3 phosphorylation of substrate results in the Europium-labeled antiphospho-specific antibody recognizing the phosphorylated site on the substrate. The Europium donor fluorophore is excited at 320 or 340 nm, and energy is transferred to the ULight acceptor dye on the substrate, which finally results in the emission of light at 665 nm. The level of ULight peptide phosphorylation correlates with the intensity of the emission. For normalization, a no kinase and a no inhibitor reaction wells were included, and all experiments were conducted in triplicates. Drug dilutions, protein concentrations, and incubation times were the same for easy comparison of results.
Microsomal Stability
Compounds were incubated at 37 °C at a concentration of 1 μM with CD1 mouse liver microsomes (GIBCO, Thermo Fisher Scientific) in a suspension of 50 mM potassium phosphate buffer (pH 7.4) with a final protein concentration of 0.5 mg/mL. The reaction was started by the addition of excess NADPH and then quenched at several time points starting from time 0, then at 3, 6, 9, 15, and 30 min addition of acetonitrile to an aliquot of the sample. An internal standard was added to each sample before centrifugation to remove any precipitates before monitoring loss of parent compound by HPLC analysis using Shimadzu LC-20A (Shimadzu, UK). Prism (Graphpad, USA) was used to fit an exponential decay for substrate depletion and subsequently rate constant (k) from the peak area of the parent compound to the internal standard at each time point. The rate of intrinsic clearance (CLint) was then calculated according to the methods of Obach using the equation37
where V is the incubation volume (volume/mg protein), and microsomal protein yield is assumed to be 52.5 mg protein/g liver with verapamil used as a positive control.
Distribution Coefficient (log D7.4)
Distribution coefficient (log D7.4) was estimated by correlation of the compounds’ chromatographic retention properties to those of 10 standard compounds with known distribution coefficients ranging from −0.5 to 5.5 at pH 7.4. A fast gradient HPLC methodology was used based on the method developed by Valkó et al.38
Kinase Screen Method
Each enzyme is assayed in its linear range with 0.3 μM substrate in 50 mM Tris pH 7.5, 0.1 mM EGTA, 0.01 mM DTT, and relevant Mg/ATP (5, 20, or 50 μM) for 30 min at room temp. Assays are stopped by the addition of 3% orthophosphoric acid and harvested onto a p81 filter paper using the PerkinElmer unifilter harvester. Once dried, they are read on a PerkinElmer Topcount NXT scintillation counter for 30 s/well.39
Acknowledgments
We thank the University of Glasgow (A.G.J. and A.B.T.), EPSRC (Research Project grant EP/N034260/2) (A.M.), and MRC Developmental Gap Fund (A.S.-A.) for financial support of this research. O.J. thanks the MRC Toxicology Unit and MRC Unit The Gambia for a PhD studentship. K.M. thanks the University of Glasgow for a Lord Kelvin Adam Smith Studentship. M.M.A. thanks the University of Glasgow for a Lord Kelvin Adam Smith Fellowship. The authors also thank Andrew Monaghan (HRMS, University of Glasgow) for providing technical assistance.
Glossary
Abbreviations
- PfCLK3
P. falciparum cyclin-dependent like kinase
- TCMDC
Tres cantos antimalarial set
- TR-FRET
time resolve fluorescence energy transfer
- SRPK
serine–arginine-rich protein kinase
- SNAr
nucleophilic aromatic substitution
- PfPKG
P. falciparum protein kinase G
- PfCDPK1
P. falciparum calcium-dependent protein kinase 1
- PfCRT
P. falciparum chloroquine-resistant transporter gene
- PfMDR1
P. falciparum multidrug-resistant gene 1
- PfDHFR
P. falciparum dihydrofolate reductase gene.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00451.
Experimental procedures and characterization data for all compounds; copies of 1H, 13C, NMR spectra for all compounds; log D7.4 (distribution co-efficient), metabolic stability, and analytical HPLC traces for final compounds; and P. falciparum culture, synchronization, and TR-FRET assay data (PDF)
Molecular formula strings (CSV)
Author Contributions
⊥ A.M. and O.J. have contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Organization, WHO . World Malaria Report, 2018.
- Straimer J.; Gnädig N. F.; Witkowski B.; Amaratunga C.; Duru V.; Ramadani A. P.; Dacheux M.; Khim N.; Zhang L.; Lam S.; Gregory P. D.; Urnov F. D.; Mercereau-Puijalon O.; Benoit-Vical F.; Fairhurst R. M.; Menard D.; Fidock D. A. K13-propeller mutations confer artemisinin resistance in plasmodium falciparum clinical isolates. Science 2015, 347, 428–431. 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariey F.; Witkowski B.; Amaratunga C.; Beghain J.; Langlois A.-C.; Khim N.; Kim S.; Duru V.; Bouchier C.; Ma L.; Lim P.; Leang R.; Duong S.; Sreng S.; Suon S.; Chuor C. M.; Bout D. M.; Ménard S.; Rogers W. O.; Genton B.; Fandeur T.; Miotto O.; Ringwald P.; Le Bras J.; Berry A.; Barale J.-C.; Fairhurst R. M.; Benoit-Vical F.; Mercereau-Puijalon O.; Ménard D. A molecular marker of artemisinin-resistant plasmodium falciparum malaria. Nature 2014, 505, 50–55. 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaratunga C.; Lim P.; Suon S.; Sreng S.; Mao S.; Sopha C.; Sam B.; Dek D.; Try V.; Amato R.; Blessborn D.; Song L.; Tullo G. S.; Fay M. P.; Anderson J. M.; Tarning J.; Fairhurst R. M. Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect. Dis. 2016, 16, 357–365. 10.1016/s1473-3099(15)00487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leang R.; Taylor W. R. J.; Bouth D. M.; Song L.; Tarning J.; Char M. C.; Kim S.; Witkowski B.; Duru V.; Domergue A.; Khim N.; Ringwald P.; Menard D. Evidence of plasmodium falciparum malaria multidrug resistance to artemisinin and piperaquine in western Cambodia: dihydroartemisinin-piperaquine open-label multicenter clinical assessment. Antimicrob. Agents Chemother. 2015, 59, 4719–4726. 10.1128/aac.00835-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerig C.; Abdi A.; Bland N.; Eschenlauer S.; Dorin-Semblat D.; Fennell C.; Halbert J.; Holland Z.; Nivez M.-P.; Semblat J.-P.; Sicard A.; Reininger L. Malaria: targeting parasite and host cell kinomes. Biochim. Biophys. Acta 2010, 1804, 604–612. 10.1016/j.bbapap.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Doerig C.; Rayner J. C.; Scherf A.; Tobin A. B. Post-translational protein modifications in malaria parasites. Nat. Rev. Microbiol. 2015, 13, 160–172. 10.1038/nrmicro3402. [DOI] [PubMed] [Google Scholar]
- Solyakov L.; Halbert J.; Alam M. M.; Semblat J. P.; Dorin-Semblat D.; Reininger L.; Bottrill A. R.; Mistry S.; Abdi A.; Fennell C.; Holland Z. Global kinomic and phospho-proteomic analyses of the human malaria parasite plasmodium falciparum. Nat. Commun. 2011, 2, 565. 10.1038/ncomms1558. [DOI] [PubMed] [Google Scholar]
- Talevich E.; Mirza A.; Kannan N. Structural and evolutionary divergence of eukaryotic protein kinases in Apicomplexa. BMC Evol. Biol. 2011, 11, 321. 10.1186/1471-2148-11-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Fu X.-D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. 10.1007/s00412-013-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern S.; Agarwal S.; Huber K.; Gehring A. P.; Strödke B.; Wirth C. C.; Brügl T.; Abodo L. O.; Dandekar T.; Doerig C.; Fischer R. Inhibition of the SR protein-phosphorylating CLK kinases of plasmodium falciparum impairs blood stage replication and malaria transmission. PloS One 2014, 9, e105732 10.1371/journal.pone.0105732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S.; Kern S.; Halbert J.; Przyborski J. M.; Baumeister S.; Dandekar T.; Doerig C.; Pradel G. Two nucleus-localized CDK-like kinases with crucial roles for malaria parasite erythrocytic replication are involved in phosphorylation of splicing factor. J. Cell. Biochem. 2011, 112, 1295–1310. 10.1002/jcb.23034. [DOI] [PubMed] [Google Scholar]
- Alam M. M.; Sanchez-Azqueta A.; Janha O.; Flannery E. L.; Mahindra A.; Mapesa K.; Char A. B.; Sriranganadane D.; Brancucci N. M. B.; Antonova-Koch Y.; Crouch K.; Simwela N. V.; Millar S. B.; Akinwale J.; Mitcheson D.; Solyakov L.; Dudek K.; Jones C.; Zapatero C.; Doerig C.; Nwakanma D. C.; Vázquez M. J.; Colmenarejo G.; Lafuente-Monasterio M. J.; Leon M. L.; Godoi P. H. C.; Elkins J. M.; Waters A. P.; Jamieson A. G.; Álvaro E. F.; Ranford-Cartwright L. C.; Marti M.; Winzeler E. A.; Gamo F. J.; Tobin A. B. Validation of the protein kinase PfCLK3 as a multistage cross-species malarial drug target. Science 2019, 365, eaau1682 10.1126/science.aau1682. [DOI] [PubMed] [Google Scholar]
- Irie T.; Sawa M. 7-Azaindole: A versatile scaffold for developing kinase inhibitors. Chem. Pharm. Bull. 2018, 66, 29–36. 10.1248/cpb.c17-00380. [DOI] [PubMed] [Google Scholar]
- Ghose A. K.; Herbertz T.; Pippin D. A.; Salvino J. M.; Mallamo J. P. Knowledge based prediction of ligand binding modes and rational inhibitor design for kinase drug discovery. J. Med. Chem. 2008, 51, 5149–5171. 10.1021/jm800475y. [DOI] [PubMed] [Google Scholar]
- Nakano H.; Hasegawa T.; Kojima H.; Okabe T.; Nagano T. Design and synthesis of potent and selective Pim kinase inhibitors by targeting unique structure of ATP-binding pocket. ACS Med. Chem. Lett. 2017, 8, 504–509. 10.1021/acsmedchemlett.6b00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mérour J.-Y.; Buron F.; Plé K.; Bonnet P.; Routier S. The azaindole framework in the design of kinase inhibitors. Molecules 2014, 19, 19935–19979. 10.3390/molecules191219935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey P. D.; Russo A. A.; Polyak K.; Gibbs E.; Hurwitz J.; Massagué J.; Pavletich N. P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995, 376, 313–320. 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- Cabrera D. G.; Horatscheck A.; Wilson C. R.; Basarab G.; Eyermann C. J.; Chibale K. Plasmodial kinase inhibitors: license to cure?. J. Med. Chem. 2018, 61, 8061–8077. 10.1021/acs.jmedchem.8b00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams N. D.; Adams J. L.; Burgess J. L.; Chaudhari A. M.; Copeland R. A.; Donatelli C. A.; Drewry D. H.; Fisher K. E.; Hamajima T.; Hardwicke M. A.; Huffman W. F.; Koretke-Brown K. K.; Lai Z. V.; McDonald O. B.; Nakamura H.; Newlander K. A.; Oleykowski C. A.; Parrish C. A.; Patrick D. R.; Plant R.; Sarpong M. A.; Sasaki K.; Schmidt S. J.; Silva D. J.; Sutton D.; Tang J.; Thompson C. S.; Tummino P. J.; Wang J. C.; Xiang H.; Yang J.; Dhanak D. Discovery of GSK1070916, a potent and selective inhibitor of Aurora B/C kinase. J. Med. Chem. 2010, 53, 3973–4001. 10.1021/jm901870q. [DOI] [PubMed] [Google Scholar]
- Verbiscar A. J. Synthesis of 1-p-chlorobenzyl-7-azaindole-3-.alpha.-piperidylmethanol as a potential antimalarial agent. J. Med. Chem. 1972, 15, 149–152. 10.1021/jm00272a008. [DOI] [PubMed] [Google Scholar]
- Wang T.; Bisson W. H.; Mäser P.; Scapozza L.; Picard D. Differences in conformational dynamics between plasmodium falciparum and human Hsp90 orthologues enable the structure-based discovery of pathogen-selective inhibitors. J. Med. Chem. 2014, 57, 2524–2535. 10.1021/jm401801t. [DOI] [PubMed] [Google Scholar]
- Barberis C.; Moorcroft N.; Arendt C.; Levit M.; Moreno-Mazza S.; Batchelor J.; Mechin I.; Majid T. Discovery of N -substituted 7-azaindoles as PIM1 kinase inhibitors - Part I. Bioorg. Med. Chem. Lett. 2017, 27, 4730–4734. 10.1016/j.bmcl.2017.08.069. [DOI] [PubMed] [Google Scholar]
- Wang T.; Yin Z.; Zhang Z.; Bender J. A.; Yang Z.; Johnson G.; Yang Z.; Zadjura L. M.; D’Arienzo C. J.; DiGiugno Parker D.; Gesenberg C.; Yamanaka G. A.; Gong Y.-F.; Ho H.-T.; Fang H.; Zhou N.; McAuliffe B. V.; Eggers B. J.; Fan L.; Nowicka-Sans B.; Dicker I. B.; Gao Q.; Colonno R. J.; Lin P.-F.; Meanwell N. A.; Kadow J. F. Inhibitors of Human Immunodeficiency Virus Type 1 (HIV-1) Attachment. 5. An Evolution from Indole to Azaindoles Leading to the Discovery of 1-(4-Benzoylpiperazin-1-yl)-2-(4,7-dimethoxy-1H-pyrrolo[2,3-c]pyridin-3-yl)ethane-1,2-dione (BMS-488043), a Drug Candidate That Demonstrates Antiviral Activity in HIV-1-Infected Subjects. J. Med. Chem. 2009, 52, 7778–7787. 10.1021/jm900843g. [DOI] [PubMed] [Google Scholar]
- Bollag G.; Tsai J.; Zhang J.; Zhang C.; Ibrahim P.; Nolop K.; Hirth P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discovery 2012, 11, 873–886. 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Ibrahim P. N.; Zhang J.; Burton E. A.; Habets G.; Zhang Y.; Powell B.; West B. L.; Matusow B.; Tsang G.; Shellooe R.; Carias H.; Nguyen H.; Marimuthu A.; Zhang K. Y. J.; Oh A.; Bremer R.; Hurt C. R.; Artis D. R.; Wu G.; Nespi M.; Spevak W.; Lin P.; Nolop K.; Hirth P.; Tesch G. H.; Bollag G. Design and pharmacology of a highly specific dual FMS and KIT kinase inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 5689–5694. 10.1073/pnas.1219457110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addie M.; Ballard P.; Buttar D.; Crafter C.; Currie G.; Davies B. R.; Debreczeni J.; Dry H.; Dudley P.; Greenwood R.; Johnson P. D.; Kettle J. G.; Lane C.; Lamont G.; Leach A.; Luke R. W. A.; Morris J.; Ogilvie D.; Page K.; Pass M.; Pearson S.; Ruston L. Discovery of 4-Amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (AZD5363), an Orally Bioavailable, Potent Inhibitor of Akt Kinases. J. Med. Chem. 2013, 56, 2059–2073. 10.1021/jm301762v. [DOI] [PubMed] [Google Scholar]
- Burrows J. N.; Hooft van Huijsduijnen R.; Möhrle J. J.; Oeuvray C.; Wells T. N. Designing the next generation of medicines for malaria control and eradication. Malar. J. 2013, 12, 187. 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows J. N.; Duparc S.; Gutteridge W. E.; van Huijsduijnen R. H.; Kaszubska W.; Macintyre F.; Mazzuri S.; Möhrle J. J.; Wells T. N. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017, 16, 26. 10.1186/s12936-017-1809-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoi P. H. C., Santiago A. S., Ramos P. Z., Fala A. M., Salmazo A. P. T., Counago R. M., Righetto G. L., Silva P. N. B., Gileadi O., Guimaraes C. R. W., Massirer K. B., Arruda P., Elkins J. M., Edwards A. M.. Crystal structure of the human PRPF4B in complex with Rebastinib; Structural Genomics Consortium (SGC)2018.
- Liddle J.; Bamborough P.; Barker M. D.; Campos S.; Chung C.-W.; Cousins R. P. C.; Faulder P.; Heathcote M. L.; Hobbs H.; Holmes D. S.; Ioannou C.; Ramirez-Molina C.; Morse M. A.; Osborn R.; Payne J. J.; Pritchard J. M.; Rumsey W. L.; Tape D. T.; Vicentini G.; Whitworth C.; Williamson R. A. 4-Phenyl-7-azaindoles as potent, selective and bioavailable IKK2 inhibitors demonstrating good in vivo efficacy. Bioorg. Med. Chem. Lett. 2012, 22, 5222. 10.1016/j.bmcl.2012.06.065. [DOI] [PubMed] [Google Scholar]
- Nwakanma D. C.; Duffy C. W.; Amambua-Ngwa A.; Oriero E. C.; Bojang K. A.; Pinder M.; Drakeley C. J.; Sutherland C. J.; Milligan P. J.; MacInnis B.; Kwiatkowski D. P.; Clark T. G.; Greenwood B. M.; Conway D. J. Changes in malaria parasite drug resistance in an endemic population over a 25-year period with resulting genomic evidence of selection. J. Infect. Dis. 2013, 209, 1126. 10.1093/infdis/jit618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amambua-Ngwa A.; Okebe J.; Mbye H.; Ceesay S.; El-Fatouri F.; Joof F.; Nyang H.; Janha R.; Affara M.; Ahmad A.; Kolly O. Sustained ex vivo susceptibility of plasmodium falciparum to artemisinin derivatives but increasing tolerance to artemisinin combination therapy partner quinolines in The Gambia. Antimicrob. Agents Chemother. 2017, 61, e00759 10.1128/aac.00759-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chausset-Boissarie L.; Àrvai R.; Cumming G. R.; Guénée L.; Kündig E. P. Asymmetric synthesis of (+)-vertine and (+)-lythrine. Org. Biomol. Chem. 2012, 10, 6473–6479. 10.1039/c2ob25880c. [DOI] [PubMed] [Google Scholar]
- Harris P. K.; Yeoh S.; Dluzewski A. R.; O’donnell R. A.; Withers-Martinez C.; Hackett F.; Bannister L. H.; Mitchell G. H.; Blackman M. J. Molecular identification of a malaria merozoite surface sheddase. PLoS Pathog. 2005, 1, e29 10.1371/journal.ppat.0010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncalés M.; Vidal J.; Torres P. A.; Herreros E. In Vitro culture of plasmodium falciparum obtention of synchronous asexual erythrocytic stages. Open J. Epidemiol. 2015, 05, 71–80. 10.4236/ojepi.2015.51010. [DOI] [Google Scholar]
- Obach R. S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [PubMed] [Google Scholar]
- Valkó K.; Bevan C.; Reynolds D. Chromatographic hydrophobicity index by fast-gradient RP-HPLC: a high-throughput alternative to log P/log D. Anal. Chem. 1997, 69, 2022–2029. 10.1021/ac961242d. [DOI] [PubMed] [Google Scholar]
- Bain J.; Plater L.; Elliott M.; Shpiro N.; Hastie C. J.; Mclauchlan H.; Klevernic I.; Arthur J. S. C.; Alessi D. R.; Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007, 408, 297–315. 10.1042/bj20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.