

Abstract
Our group has previously reported several indolecarboxamides exhibiting potent antitubercular activity. Herein, we rationally designed several arylcarboxamides based on our previously reported homology model and the recently published crystal structure of the mycobacterial membrane protein large 3 (MmpL3). Many analogues showed considerable anti-TB activity against drug-sensitive (DS) Mycobacterium tuberculosis (M. tb) strain. Naphthamide derivatives 13c and 13d were the most active compounds in our study (MIC: 6.55, 7.11 μM, respectively), showing comparable potency to the first line anti-tuberculosis (anti-TB) drug ethambutol (MIC: 4.89 μM). In addition to the naphthamide derivatives, we also identified the quinolone-2-carboxamides and 4-arylthiazole-2-carboxamides as potential MmpL3 inhibitors in which compounds 8i and 18b had MIC values of 9.97 and 9.82 μM, respectively. All four compounds retained their high activity against multidrug-resistant (MDR) and extensively drug-resistant (XDR) M. tb strains. It is worth noting that the two most active compounds 13c and 13d also exhibited the highest selective activity towards DS, MDR and XDR M. tb strains over mammalian cells [IC50 (Vero cells) ≥ 227 μM], indicating their potential lack of cytotoxicity. The four compounds were docked into the MmpL3 active site and were studied for their drug-likeness using Lipinski's rule of five.
Synthesis and pharmacological evaluation of arylcarboxamide derivatives based on an antimycobacterial indole-2-carboxamide scaffold. The most active compounds demonstrated activities against MDR and XDR M. tb strains.
1. Introduction
Tuberculosis (TB) is an infectious disease with a ubiquitous mortality worldwide caused by Mycobacterium tuberculosis (M. tb), which predominantly affects the lungs.1 The 2019 World Health Organisation (WHO) report revealed that approximately one-quarter of the global population are latently infected with M. tb. According to the report, 10.0 million people fell ill with TB in 2018, a number that has become relatively stable over the past years. Globally, 1.2 million HIV-negative patients died from TB in 2018 in addition to 0.25 million deaths among HIV-positive people. This inexorable global burden of TB makes it one of the top ten leading causes of death worldwide and number one infectious disease killer, surpassing HIV/AIDS.1 The onerous current treatment regimen requires 6 months minimum administration of a cocktail of the first-line anti-TB drugs: isoniazid (INH), rifampicin (RIF), pyrazinamide (PZA) and ethambutol (EMB) for the treatment of drug-sensitive (DS) TB.1,2 This drawn-out duration of therapy and high pill count in addition to their accompanied side effects, resulted in an incomplete eradication of TB due to the poor patient compliance and ultimately led to the emergence of drug-resistant TB.3 Notably, the treatment period of the second-line anti-TB drugs used for multi- and extensively-drug resistant TB strains (MDR- and XDR-TB, respectively) may be extended up to two years accompanied with their limited efficacy, drug toxicity and higher price compared to the first line regimen.4 Accordingly, anti-TB drugs with a novel mode of action to overcome the existing resistance to the current drugs, and shorter course of treatment, are urgently required.
The mycobacterial membrane protein large 3 (MmpL3) which is currently considered as one of the most druggable M. tb targets, is responsible for the translocation of mycolic acids (MAs) across the plasma membrane.5,6 The MAs biosynthesis and incorporation on the mycobacterial cell wall was previously illustrated in our review article.7 Summarised into five distinct steps, the first four steps of the process entail the biosynthesis of the MAs precursor, trehalose monomycolate (TMM), in the cytoplasm.7 The last step involves flipping and release of the formed TMM across the inner membrane into the periplasm by the membrane transporter MmpL3.6 Thereafter, these MAs accumulate in the M. tb cell envelope constituting the major lipid component of the outer coating of mycobacteria, rendering an extremely hydrophobic and impermeable bilayer.8,9 This formidable protective layer insulates the mycobacteria against exogenous compounds, including many antibiotics, and the host's immune system, underpinning its fundamental role in mycobacterial growth and survival in the host.8–10 Hence, lipophilicity of the anti-TB derivatives (Clog P) has been proposed to have positive correlation with their anti-TB activity.11–16 In other words, anti-TB derivatives with increased lipophilicity (high Clog P) are endowed with facilitated diffusion through the lipid-rich cell wall of M. tb and thereby enhanced efficacy.8 The role of MmpL3 in shuttling TMM across the cytoplasmic membrane was proven in M. smegmatis when the conditional depletion of MmpL3 led to an intracellular accumulation of TMM and loss of cell wall mycolation.17–19 In addition, downregulation of MmpL3 expression led to an abolition of cell division and consequently rapid cell death.20,21 Therefore, inactivation of MmpL3 prohibits this pivotal step in the MAs biosynthesis machinery, collapsing the permeability barrier and validating MmpL3 as a promising target for anti-TB drugs.22,23 Several MmpL3 inhibitors have been developed, including indolecarboxamides,14,24–27 benzimidazoles,28 adamantyl ureas,17 diamines,18,29 diphenyl pyrroles,30 benzothiazoles,31,32 spirocycles,33,34 and tetrahydropyrazolopyrimidines.33,35 Recent literature revealed that some of the well-known MmpL3 inhibitors indeed directly interact with MmpL3, whereas others impact the proton motive force in M. tb.36,37 Our group has previously identified several indolecarboxamides as potent antimycobacterial agents targeting MmpL3.14,25,27 The preliminary phenotypic screening identified the indole-2-carboxamide 1 (Fig. 1) as a highly potent anti-TB scaffold (MIC = 0.93 μM).27,38 Further modifications on the indolecarboxamide core were then initiated in which a conspicuous increase in the activity was obtained when the cyclohexyl group was replaced by a cyclooctyl 2, or an adamantyl ring.25 More recently, the utmost potency was achieved when the 4,6-dimethyl group on the indole core was replaced with 4,6-dihalo-substituents (compounds 3 and 4, Fig. 1). The methyl groups present a potential metabolic susceptibility (liable to metabolic oxidation), whereas the dihalo groups are more resistant while conferring similar lipophilicity.14
Fig. 1. Hit compound 1, lead compounds 2, 3 and 4.

A putative homology model for the MmpL3 transporter has also been generated to gain an approximate interpretation of the way the indole-2-carboxamides may interact with MmpL3.14 This homology model proposed the existence of three putative binding subpockets in which our lead compound 3 is stabilised. Despite the high activity of compounds 3 and 4in vitro, unfortunately they turned out to be inactive in the in vivo studies.14 Very recently, the crystal structure of MmpL3 in M. smegmatis was reported by two different groups which serves as an excellent paradigm for the M. tb counterpart.39,40 This crystal structure revealed that the MmpL3 inhibitor binding pocket is divided into five subsites (S1–S5) with the S4 subsite being the only hydrophilic one. The indole-2-carboxamides accommodated the S3–S5 subsites, resonating with our previously reported homology model.14,40
In this study, we describe the synthesis, in vitro biological evaluation and structure–activity relationship (SAR) of a number of arylamides rationally designed as potential antitubercular agents using the indole-2-carboxamides 3 and 4 as lead compounds. All the final compounds were screened in vitro against M. tb H37Rv strain. The top four potent compounds in our study were further evaluated for their antimycobacterial activity against Mycobacterium abscessus (M. abs), Mycobacterium avium (M. avium), MDR and XDR M. tb strains. In parallel, the cytotoxicity of these four compounds was also tested in Vero cells. Due to the analogy between the indole-2-carboxamides identified as MmpL3 inhibitors and our designed compounds, a docking analysis was performed on the most active compounds using the MmpL3 crystal structure40 in complex with ICA38 4. Their drug-likeness was also assessed to predict their oral bioavailability.
2. Results and discussion
2.1. Compound design
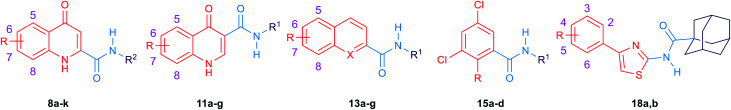
The general strategy for the chemical modifications of the lead compound 3 is illustrated in Fig. 2 and aimed at enhancing the drug-like properties, particularly the lipophilicity and solubility, while retaining the anti-TB activity. The first approach was to investigate the activity of several heterocyclic and hydrocarbon scaffolds as a replacement of the indole ring, while keeping the important amide linkage that is capable of hydrogen bonding with the side chain of Asp645.40 It is noteworthy that Asp645 is a crucial component of the two Asp–Tyr pairs which are implicated in the proton relay process.40 This is consistent with the abrogation of the growth of mycobacteria in culture upon mutation of these two pairs in MmpL3.41 Therefore, disrupting the two Asp–Tyr pairs blocks the proton motive force for substrate translocation. Towards this, bioisosteric replacements of the indole core by other aromatic moieties42 have been employed. Hence, several quinolines 8a–k, 11a–g and 13a,b as well as naphthalene derivatives 13c–g were chosen (Fig. 2). In addition, o-hydroxy- and o-aminobenzamides 15a–d were investigated due to their potential in mimicking the NH group of the indole-2-carboxamides. We also scrutinised the activity of the 4-arylthiazole scaffold 18a,b in which the substituted benzene ring is oriented in an extended way that could maximise its hydrophobic contacts with the S3 hydrophobic subsite. Throughout the indole replacement investigations, the N-linked hydrophobic moiety containing a lipophilic bulky adamantane or cyclooctane ring was maintained, as they are known to be essential for occupying the S5 hydrophobic subsite.40
Fig. 2. A diagram indicating the main subpockets of MmpL3 in which the indole-2-carboxamide scaffold is stabilised through hydrophobic interactions and hydrogen bonding, and the strategies adopted for the replacement of the indole ring in lead compound 3.

The second approach was to introduce an extra one, two or three atom spacers to the amide linker while retaining both the 4,6-dihalosubstituted indole ring and the bulky hydrophobic adamantane ring. The reason behind this strategy was to investigate whether these extra atom spacers will participate in forming H-bonds with the hydrophilic residues in the S4 hydrophilic subsite, leading to an improved binding. These extra spacer/s may also push the N-linked lipophilic moiety deeper to the S5 hydrophobic subsite maximising the hydrophobic interactions with the surrounding residues.
2.2. Chemistry
The synthetic routes to obtain the target arylamides 8a–k, 11a–g, 13a–g, 15a–d, 18a,b, 21, 23, 24 and 27 are depicted in Schemes 1–4. The methyl 4-oxo-1,4-dihydroquinoline-2-carboxylates 6a–h were obtained through a one-pot solvent-free hydroamination reaction43 between commercially available anilines 5a–g and dimethyl acetylenedicarboxylate followed by an intramolecular Friedel–Crafts reaction catalysed by polyphosphoric acid (PPA) (Scheme 1). Subsequent basic hydrolysis afforded the carboxylic acids 7a–h, which were then coupled with the corresponding amines in the presence of hydroxybenzotriazole hydrate (HOBt) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) as coupling agents and N,N-diisopropylethylamine (DIPEA) as a base to form final compounds 8a–k. Anilines 5a–g were also reacted with 2-(ethoxymethylene)malonic acid diethyl ester (EMME) and the intermediate malonates obtained were cyclised under relatively extreme conditions by heating at 250 °C in diphenyl ether in a one pot reaction according to Gould–Jacobs procedure (Scheme 2). Subsequent basic hydrolysis of the ethyl 4-oxo-1,4-dihydroquinoline-3-carboxylates 9a–g resulted in the carboxylic acids 10a–g. These were then coupled with 1-adamantyl amine in the presence of O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) as a coupling agent and DIPEA as a base to give the desired compounds 11a–g. It is noteworthy that as observed by 1H NMR and 13C NMR, the 4-hydroxy-quinoline tautomeric form was predominantly manifested in the quinoline-2-carboxamide series (8a–k) with only compounds 8a,h–j appeared in the 4-quinolone form. In the 1H NMR spectra of the quinoline-2-carboxamides, the signals of the proton at position 3 of the 4-quinolone tautomers appeared at an upper magnetic field than the 4-hydroxyquinoline counterparts. While in the 13C NMR spectra, the signals of the carbon at position 4 of the 4-quinolones were more deshielded than those of the corresponding 4-hydroxyquinolines. The preponderance of one of these tautomers was found to be dependent on the NMR solvent used, in which in DMSO, the 4-hydroxy form is predominantly observed for the 2-substituted-quinolines.44 On the contrary, also in line with another literature report,45 only the 4-quinolone tautomer was detected in the quinolone-3-carboxamide series 11a–g.
Scheme 1. General synthetic procedure for compounds 8a–k.

Scheme 2. General synthetic procedure for compounds 11a–g.

Scheme 3. General synthetic procedure for compounds 13a–g, 15a–d and 18a,b.

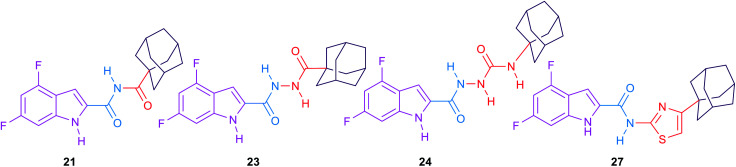
Scheme 4. General synthetic procedure for compounds 21, 23, 24 and 27.

For the preparation of analogues 13a–g and 15a–d commercially available carboxylic acids 12a–e and 14a,b, respectively, were reacted with their corresponding amines under standard amide coupling conditions (Scheme 3). On the other hand, the aminothiazoles 17a,b were prepared from the commercially available acetophenone derivatives 16a,b using iodine and thiourea. Compounds 18a,b were in turn obtained via amide coupling of the aminothiazoles 17a,b with 1-adamantanecarboxylic acid.
The synthetic route of the novel indole derivatives is outlined in Scheme 4. The synthesis started with 4,6-difluoroindole-2-carboxylic acid (19) which was reacted with 1,1′-carbonyldiimidazole (CDI) and either ammonia or hydrazine hydrate to yield the corresponding carboxamide 20 and acetohydrazide 22, respectively. The corresponding amide 20 was reacted in pyridine with 1-adamantanecarbonyl chloride, which was freshly prepared by treating 1-adamantanecarboxylic acid with oxalyl chloride, to yield the corresponding imide 21. On the other hand, the corresponding acetohydrazide 22 underwent two reactions. Firstly, it was reacted with 1-adamantanecarboxylic acid in a standard amide coupling condition forming compound 23. Secondly, the reaction of 22 with 1-adamantyl isocyanate afforded the corresponding compound 24. Finally, the 4-(1-adamantyl)-2-aminothiazole 26, which was prepared by heating 1-acetyladamantane 25 with iodine and thiourea, was subjected to amide coupling conditions with the carboxylic acid derivative 19 to yield compound 27, incorporating a thiazole ring next to the amide linker.
2.3. Biological evaluation and SAR analysis
Target compounds 8a–k, 11a–g, 13a–g, 15a–d, 18a,b, 21, 23, 24 and 27 were screened in vitro against M. tb H37Rv strain to obtain the minimum inhibitory concentration (MIC) values using the Microplate Alamar Blue Assay (MABA)46 as shown in Tables 1 and 2. The calculated Clog P values of the designed compounds were estimated using ChemDraw 16.0 to correlate their lipophilicity with the activity displayed. The first round of SAR investigation was focused on the role of the replacement of the indole core with other scaffolds. First, we probed the biological activity of 4-oxoquinoline heterocyclic scaffold as a replacement to the indole core (Table 1). Nevertheless, in the quinoline-2-carboxamide series 8a–k, the 4-hydroxy-quinoline form was the main tautomer in which compounds 8b–g,k showed a drastic drop in the activity (MIC ≥ 81.78 μM) compared to the lead compound 3 (MIC = 0.011 μM). It is noteworthy that the 8-hydroxy-quinoline derivative 8k was equally potent to the 8-chloro counterpart 8c although its Clog P is less than half that of 8c. A similar trend in activity was observed in the 4-quinolone tautomers 8a,h exhibiting a dramatic attenuation of activity (MIC > 89.67 and 40.89 μM, respectively). However, the bromo-substituted 4-quinolone tautomers 8i (MIC = 9.97 μM) and 8j (MIC = 19.93 μM) were the most active compounds in this series, with two-fold and four-fold less activity than the first line anti-TB drug ethambutol (MIC = 4.89 μM), respectively. These data suggest that the 4-quinolone tautomer is likely preferred over the 4-hydroxy-quinoline form for antimycobacterial activity. In addition, the most favourable sites of substitution on the quinolone ring are positions 5 and 7 which resembles the data previously obtained from the indole-2-carboxamides.14,25 In line with our pursuit of determining the activity of the quinolone scaffold, we scrutinised on shifting the carboxamide moiety to position 3 on the quinolone ring as shown in compounds 11a–g (Table 1). Despite the sole formation of the 4-quinolone tautomer in this series, all the tested compounds 11a–g did not show any improved anti-TB activity (MIC ≥ 79.74 μM). The attenuation in the activity of compounds 11f,g (MIC ≥ 81.78, ≥ 79.74 μM, respectively) compared to its 2-carboxamide analogues 8h,j (MIC = 40.89, 19.93 μM, respectively) is also consistent with our previous findings on the indole ring in which positioning the carboxamide group at position 2 was superior to position 3.14,25 It is noteworthy that all the substituted quinoline derivatives 8a–k and 11a–g have lower lipophilicity than the lead indoleamides (compound 3 and 4) which typically possess Clog P values of greater than 5. However, the wide gap in the activity between the quinolones and the previously reported indoles suggests that expanding the indole ring with an extra carbonyl group in case of the 4-quinolone ring is not favourable for their anti-TB activities.
In vitro anti-TB activity of compounds 8a–k, 11a–g, 13a–g, 15a–d, 18a,b.
| ||||||
|---|---|---|---|---|---|---|
| Analogue | R | R1 | X | MICa (μg mL−1) | MIC (μM) | Clog Pb |
| 8a | 6-Cl | 1-Adamantane | — | >32 | >89.67 | 2.94 |
| 8b | 8-Cl | 1-Adamantane | — | ≥32 | ≥89.67 | 2.94 |
| 8c | 8-Cl | Cyclooctane | — | >32 | >96.14 | 3.43 |
| 8d | 7,8-Dichloro | 1-Adamantane | — | ≥32 | ≥81.78 | 3.55 |
| 8e | 7,8-Dichloro | Cyclooctane | — | ≥32 | ≥87.13 | 4.04 |
| 8f | 6,8-Dichloro | Cyclooctane | — | ≥32 | ≥87.13 | 4.16 |
| 8g | 5,8-Dichloro | 1-Adamantane | — | ≥32 | ≥81.78 | 3.67 |
| 8h | 5,7-Dichloro | 1-Adamantane | — | 16 | 40.89 | 3.67 |
| 8i | 5-Br | 1-Adamantane | — | 4 | 9.97 | 3.09 |
| 8j | 7-Br | 1-Adamantane | — | 8 | 19.93 | 3.09 |
| 8k | 8-OH | 1-Adamantane | — | >32 | >94.56 | 1.60 |
| 11a | 6-Cl | 1-Adamantane | — | ≥32 | ≥89.67 | 3.20 |
| 11b | 8-Cl | 1-Adamantane | — | >32 | >89.67 | 3.20 |
| 11c | 7,8-Dichloro | 1-Adamantane | — | >32 | >81.78 | 3.81 |
| 11d | 6,8-Dichloro | 1-Adamantane | — | ≥32 | ≥81.78 | 3.93 |
| 11e | 5,8-Dichloro | 1-Adamantane | — | ≥32 | ≥81.78 | 3.93 |
| 11f | 5,7-Dichloro | 1-Adamantane | — | >32 | >81.78 | 3.93 |
| 11g | 7-Br | 1-Adamantane | — | ≥32 | ≥79.74 | 3.09 |
| 13a | H | Cyclooctane | N | 16 | 56.66 | 4.99 |
| 13b | H | 1-Adamantane | N | 16 | 52.22 | 4.50 |
| 13c | H | Cyclooctane | CH | 2 | 7.11 | 5.18 |
| 13d | H | 1-Adamantane | CH | 2 | 6.55 | 4.69 |
| 13e | 5-Br | 1-Adamantane | CH | ≥32 | ≥83.26 | 5.70 |
| 13f | 6-Br | 1-Adamantane | CH | ≥32 | ≥83.26 | 5.70 |
| 13g | 6-OCH3 | 1-Adamantane | CH | ≥32 | ≥95.40 | 4.82 |
| 13h | H | Cycloheptane | N | — | 111 (ref. 47) | 4.62 |
| 15a | NH2 | 1-Adamantane | — | >32 | >94.32 | 4.85 |
| 15b | NH2 | Cyclooctane | — | >32 | >101 | 5.34 |
| 15c | OH | 1-Adamantane | — | 32 | 94.05 | 5.46 |
| 15d | OH | Cyclooctane | — | 32 | 101 | 5.95 |
| 18a | 4-Cl | — | — | 32 | 85.81 | 5.87 |
| 18b | 2,4-Dichloro | — | — | 4 | 9.82 | 6.34 |
| Isoniazid | — | — | — | 0.04 | 0.29 | −0.66 |
| Ethambutol | — | — | — | 1 | 4.89 | 0.11 |
The lowest concentration of drug reducing at least 90% of bacterial growth by the Microplate Alamar Blue Assay (MABA). The reported MIC values are an average of three individual measurements.
Calculated using ChemDraw 16.0.
In vitro anti-TB activity of the linker analogues, compounds 2 and 3, isoniazid and ethambutol.
| |||
|---|---|---|---|
| Analogue | MICa (μg mL−1) | MIC (μM) | Clog Pb |
| 21 | 8 | 22.32 | 4.34 |
| 23 | >32 | >85.70 | 3.98 |
| 24 | 32 | 82.38 | 3.13 |
| 27 | >32 | >77.39 | 7.13 |
| 2 | 0.004 | 0.013 | 5.59 |
| 3 | 0.004 | 0.011 | 5.67 |
| Isoniazid | 0.04 | 0.29 | −0.66 |
| Ethambutol | 1 | 4.89 | 0.11 |
The lowest concentration of drug reducing at least 90% of bacterial growth by the microplate alamarBlue assay (MABA). The reported MIC values are an average of three individual measurements.
Calculated using ChemDraw 16.0.
Aiming to improve the water solubility by introducing an ionisable nitrogen atom and to extend our investigation on the effect of replacing the indole ring with a quinoline nucleus, we synthesised two quinoline-2-carboxamide derivatives 13a,b. The N-cyclooctylquinoline-2-carboxamide 13a was equipotent to the adamantane counterpart (MIC = 56.66 and 52.22 μM, respectively). Gonec et al. have previously investigated the anti-TB activity of similar quinoline derivatives.47 Their cycloheptyl derivative 13h, which showed the highest activity in their series of compounds, is two-fold less potent than our cyclooctyl counterpart 13a (MIC = 111 and 56.66 μM, respectively). The authors hypothesised that increasing the bulkiness of the substituent on the amide nitrogen is directly related to the increase of the anti-TB activity, consistent with our previous findings on the indole ring.25,47 Surprisingly, however, they encountered a solubility issue when increasing the bulkiness of the amide N-cycloaliphatic ring in which they reported the cyclooctyl derivative to be devoid of anti-TB activity. The authors linked its lack of activity to its precipitation in the test media.47 They also reported several unsubstituted-naphthalene-2-carboxamides in which the derivatives bearing a cycloheptyl or cyclooctyl ring were bereft of anti-TB activity. Contrary to their report, re-evaluation of the N-cyclooctyl-2-naphthamide 13c led us to the identification of the most active compounds in the current series, together with its adamantyl derivative 13d, with MIC values of 7.11 and 6.55 μM, respectively (Table 1), which are very similar to the activity of ethambutol. The higher anti-TB activity of the naphthalene-2-carboxamide derivatives 13c,d compared to the quinoline-2-carboxamide counterparts 13a,b contradicts the previous findings reported by Gonec et al. The activity of several substituted naphthamides was further investigated in which the increase in lipophilicity in the various tested naphthamides was not accompanied by a rise of activity (13a,bvs.13e–g).
Further attempts to replace the indole core included the evaluation of the 2-substituted-3,5-dichlorobenzene ring in which we preserved the dichloro substitution besides introducing either a hydroxyl group or an amino group oriented in a way similar to the NH group of the indole-2-carboxamide 3. Unfortunately, these modifications resulted in a drastic decrease in activity in compounds 15a–d, with MIC value of ≥94.05 μM, despite their high lipophilicity (Clog P = 4.85–5.95). These data further highlight the necessity of the 4,6-disubstituted-indole ring in its entirety to elicit a strong anti-TB activity. Moreover, we probed the activity of two 4-arylthiazole derivatives 18a,b in which the aryl group is oriented in an extended way that might maximise its hydrophobic interactions with the S3 hydrophobic subsite. The 4-(2,4-dichlorophenyl)thiazole 18b derivative was two-fold less active than ethambutol, while the 4-chloro counterpart 18a showed a dramatic loss of activity (MIC = 9.82, 85.81 μM, respectively). Of note is the fact that compound 18b is more lipophilic than 18a.
The second round of SAR investigation was focused on keeping the dihalosubstituted-indole nucleus intact as it was proven to be superior in terms of anti-TB activity. We examined the effect of varying the chain length connecting the indole core and the adamantane ring as shown in compounds 21, 23, 24 and 27. The biological activities of these analogues were found to be independent of their lipophilicity (Table 2). The imide derivative 21 showed the highest activity (MIC = 22.32 μM) in this series compared to the 4 and 5 atom linker derivatives 23 and 24 which exhibited MIC values of >85.70 and 82.38, respectively. Additionally, introducing a thiazole ring next to the amide linker in compound 27 resulted in an inferior activity (MIC > 77.39 μM) compared to the imide derivative 21, although its Clog P is higher than that of 21.
Overall, although lipophilicity (high Clog P) of some of the designed compounds was correlated with high anti-TB activity, other lipophilic analogues did not display such a positive trend. Accordingly, it seems that lipophilicity is not the sole parameter influencing the anti-TB activity in our series of compounds. The four most active derivatives 8i, 13c, 13d and 18b (MIC ≤ 4 μg mL−1) were chosen for further anti-mycobacterial studies, cytotoxicity evaluation and docking analysis.
2.4. In vitro activity of the most active compounds against M. abs, M. avium and clinical isolates of M. tb as well as their cytotoxicity evaluation
The most active compounds in our study, namely 8i, 13c, 13d and 18b were further evaluated for their in vitro activity against M. abs, M. avium and a panel of clinical isolates of M. tb (Table 3). All four compounds were inactive against M. abs and M. avium strains (MIC > 157 μM), suggesting their selective activity against M. tb. On the other hand, within the tested panel of M. tb strains, originally obtained from pulmonary TB patients, one was drug sensitive (DS) (V4207), two were MDR M. tb (V2475, KZN494), and two were XDR M. tb (R506, TF274). To our delight, all four compounds maintained their high activity against the susceptible M. tb strain H37Rv in all the tested drug resistant strains. Of note is the fact that all four compounds showed a two-fold increase in activity against the XDR M. tb strains (R506, TF274) (Table 3). These high activities against the MDR and XDR TB highlight the potential use of these compounds to treat drug-resistant M. tb and the likelihood that they will not exhibit cross resistance with currently used medications. In addition, Vero cells were used to assess the cytotoxicity of the most active compounds against mammalian cells, and a selectivity index (SI) was subsequently calculated (Table 3). Compounds 13c and 13d exhibited higher IC50 values (IC50 ≥ 227 μM, respectively) against Vero cells than compounds 8i and 18b (IC50 = 9.82–39.87 μM, respectively), resulting in high SI index values (Table 3). The aforementioned results accordingly indicate the potent antitubercular activity of compounds 13c and 13d towards M. tb DS, MDR and XDR strains and their potential lack of toxicity toward mammalian cells.
Activity [MIC (μM)] of the top four active compounds on selected clinical isolates of M. tb and toxicity on Vero cells [IC50 (μM)].
| M. abs (μM) | M. avium (μM) | M. tb MIC (μM) | IC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| V4207/DS | V2475/MDRa | KZN494/MDRa | R506/XDRb | TF274/XDRb | Vero cells | SIc | |||
| 8i | >159 | >159 | 9.97 | 9.97 | 9.97 | 4.98 | 4.98 | 39.87 | 4 |
| 13c | >227 | >227 | 7.11 | 7.11 | 7.11 | 3.55 | 3.55 | ≥227 | ≥32 |
| 13d | >209 | >209 | 6.55–13.10 | 6.55 | 6.55–13.10 | 3.27 | 3.27 | ≥419 | ≥64 |
| 18b | >157 | >157 | 9.82 | 19.64 | 9.82 | 4.91–9.82 | 4.91 | 9.82 | 1 |
Resistant to isoniazid and rifampin.
Resistant to isoniazid, rifampin, levofloxacin, ofloxacin, and kanamycin.
Selectivity index (SI) = IC50(Vero)/MIC.
2.5. Molecular docking of the most active compounds into MmpL3
In order to gain insight into whether the top-ranked active compounds (MIC ≤ 4 μg mL−1) have the potential to fit in the MmpL3 binding site and compare their binding mode to the indoleamides binding profile, a molecular modeling study was performed using the crystal structure of MmpL3 in complex with ICA38 4 (6AJJ).40 Docking of compounds 8i, 13c, 13d and 18b as well as lead compound 3 and ICA38 4 as reference ligands into the crystal structure of MmpL3 was carried out using MOE 2008.10 modeling software (Molecular Operating Environment).48 The evaluation of each docking simulation was based on the London dG scores of the generated poses after molecular mechanics refinement indicating the interaction energy between the ligand and the MmpL3 binding site. The docking scores for the top ranked poses of the aforementioned compounds are presented in Table 4. In order to validate the docking protocol, the co-crystallised ICA38 ligand 4 was redocked into MmpL3 active site and the docking pose was compared with the initial pose using root mean square deviation (RMSD). ICA38 docked almost at the same position (RMSD = 0.477 Å) with docking score of = −22.71 kcal mol−1 (Table 4). The 4,6-dichloroindole nucleus is positioned in the S3 hydrophobic subsite, forming hydrophobic interactions with the surrounding residues. The amide linker is inserted in the S4 hydrophilic subsite in which the amide nitrogen hydrogen bonds to the side chain of Asp645 (distance = 2.54 Å). The S5 hydrophobic subsite accommodates the spirocarbocyclic group, forming hydrophobic contacts with the surrounding residues. In addition, lead compound 3 adopted a similar binding mode to ICA38 in the MmpL3 active site (Fig. 3A). An additional H-bond between the indole NH of lead compound 3 and Asp645 is formed (distance = 2.75 Å), Fig. 3A. Consequently, both compound 3 and ICA38 inhibit the MmpL3 by occupying the proton translocation channel and blocking the proton motive force for substrate translocation by disrupting the two Asp–Tyr pairs for proton relay.40 Docking of the 4-quinolone derivative 8i pictured the MmpL3 active site overlaid with both 8i and ICA38 (Fig. 3B). In addition to the H-bond formed between the amide NH and Asp645, the quinolone NH in compound 8i H-bonds to Asp645 (distance = 2.48, 2.68 Å, respectively), in a similar manner to lead compound 3.
Docking score and H-bond interactions of lead compound 3, ICA38 and synthesized compounds 8i, 13c, 13d and 18b in the MmpL3 binding pocket.
| Compounds | Docking score (kcal mol−1) | No. of H-bonds | Distance | Amino acids | Ligand atom |
|---|---|---|---|---|---|
| 8i | −14.05 | 2 | 2.48 | Asp645 | NH of amide |
| 2.68 | Asp645 | NH of quinolone | |||
| 13c | −11.97 | 1 | 2.49 | Asp645 | NH of amide |
| 13d | −12.01 | 1 | 2.39 | Asp645 | NH of amide |
| 18b | −13.99 | 1 | 2.39 | Asp645 | NH of amide |
| 3 | −13.16 | 2 | 2.39 | Asp645 | NH of amide |
| 2.75 | Asp645 | NH of indole | |||
| ICA38 | −22.71 | 1 | 2.54 | Asp645 | NH of amide |
Fig. 3. Lead compound 3 and compound 8i adopt a similar binding mode, overlaid onto the active site of MmpL3 together with ICA38. (A) Superposition of lead compound 3 (dark cyan), and ICA38 (light brown) in the MmpL3 binding pocket. (B) Superposition of 8i (dark cyan) onto ICA38 (light brown) in the MmpL3 binding pocket.

Interestingly, the orientation of the most active compounds in our study 13c and 13d inside the active site resembled the ICA38 one. They also exhibited a docking score of −11.97 and −12.01 kcal mol−1, respectively. The naphthalene moiety occupies the S3 hydrophobic subsite, overlapping with the indole moiety of ICA38. Meanwhile, the amide NH H-bonds with Asp645 in the S4 hydrophilic subsite (distance = 2.49 and 2.39 Å, respectively), Fig. 4. Hence, similar to ICA38, these compounds likely disrupt the two Asp–Tyr pairs that play pivotal roles in proton transportation.
Fig. 4. Superposition of 13c (A), 13d (B) (dark cyan), and ICA38 (light brown) in the MmpL3 binding pocket, showing the inhibitors all have similar binding positions.

Surprisingly, the thiazole derivative 18b recognised and showed high binding affinity to the same binding pocket of ICA38 (docking score = −13.99 kcal mol−1). In addition, it is oriented in an extended conformation in the active site, adopting a nearly superimposed orientation with ICA38 (Fig. 5A). The 2,4-dichlorophenyl moiety of 18b is embedded in the S3 hydrophobic subsite occupied by the indole ring of ICA38 and the amide NH H-bonds to Asp645 (distance = 2.39 Å). Again, this might explain its in vitro anti-TB activity due to occupying the proton translocation channel similar to ICA38. It is also noteworthy that the eight-fold improvement of the activity of the dichlorophenyl derivative 18b compared with the monochlorophenyl counterpart 18a might be attributed to the increase in the electron-withdrawing substituents. Accordingly, the phenyl π-electron density in the former one is less than the latter and the electronic repulsion is reduced, possibly favoring the hydrophobic interactions with the surrounding residues.
Fig. 5. 18b binding mode and overlay of 8i, 13d and 18b in the MmpL3 binding pocket. (A) Superimposition of 18b (dark cyan), and ICA38 (light brown) in the MmpL3 active site, oriented in a similar way and having a similar binding mode as ICA38. (B) Superimposition of new classes of inhibitors in the MmpL3 active site with similar binding positions as ICA38. ICA38 (light brown), 8i (dark red), 13d (dark grey), and 18b (magenta).

Overall, the tested novel classes of compounds show a similar binding mode and orientation as lead compound 3 and ICA38 in the same binding pocket (Fig. 5B). In addition, the N-linked hydrophobic moiety in these four compounds, is lodged in the S5 hydrophobic subsite overlapping with the spirocarbocyclic group of ICA38 (Fig. 3–5).
2.6. ADME profiling
The bioavailability of the most active compounds 8i, 13c, 13d and 18b as well as lead compound 3 and ICA38 4 was predicted using ACD/Labs Percepta 2016 Build 2911 (13 Jul 2016) (Table 5). We particularly evaluated the drug-likeness of the compounds via assessing their compliance to Lipinski's “rule of five”.49,50 It is important to note, however, that although both lead compounds 3 and ICA38 4 fulfilled all these rules, these compounds were reported to have poor in vivo bioavailability and therefore were not pursued for further studies.14,51
Bioavailability of ICA38 and synthesized compounds 8i, 13c, 13d and 18b using ACD/Labs Percepta 2016 Build 2911 (13 Jul 2016). MW: molecular weight, HBD: H-bond donors, HBA: H-bond acceptors, log P: octanol–water partition coefficient, NRB: number of rotatable bonds, TPSA: topological polar surface area, PPB: plasma protein binding.
| Analogues | MW | HBD | HBA | Clog P (Chemdraw) | log P | NRB | TPSA | Solubility (mg mL−1) | Caco-2 (×10−6 cm s−1) | PPB (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 8i | 401.3 | 2 | 4 | 3.09 | 4.66 | 2 | 58.20 | 0.007 | 186 | 98 |
| 13c | 281.4 | 1 | 2 | 5.18 | 5.13 | 2 | 29.10 | 0.002 | 190 | 95 |
| 13d | 305.4 | 1 | 2 | 4.69 | 4.91 | 2 | 29.10 | 0.007 | 207 | 97 |
| 18b | 407.4 | 1 | 3 | 6.34 | 5.99 | 3 | 70.23 | 0.008 | 77 | 99 |
| 3 | 363.3 | 2 | 3 | 5.67 | 5.49 | 2 | 44.89 | 0.001 | 91 | 96 |
| ICA38 | 346.4 | 2 | 3 | 5.89 | 5.17 | 2 | 44.89 | 0.0007 | 132 | 96 |
The four compounds that showed the highest in vitro anti-TB activity in our study 8i, 13c, 13d and 18b are lipophilic, exhibiting high Clog P values (Clog P = 4.66–5.99 in ACD Percepta). In addition, their Caco-2 permeability values ranged from 77–207 cm s−1, so they all are expected to permeate the cell membrane. Hence, the in vitro anti-TB activity of these compounds might be related to their high lipophilicity, and therefore their ability to cross the notoriously known hydrophobic outer membrane. In addition, all the compounds are predicted to have higher solubility than the lead compounds 3 and ICA38 (Table 5). Further studies are needed to ensure the in vivo bioavailability and anti-TB activity of these compounds.
3. Conclusion
In the present work we rationally designed and evaluated the anti-TB activity of a set of arylcarboxamide derivatives 8a–k, 11a–g, 13a–g, 15a–d and 18a,b. Firstly, we replaced the indole ring in lead compound 3 with quinoline, naphthalene, 3,5-dichlorophenyl and 4-arylthiazole. The SAR of these analogues led us to infer that: (a) the 4,6-disubstituted-indole ring is the most beneficial moiety for potent anti-TB activity in which replacing it with 4-quinolone ring 8a–k and 11a–g, or o-hydroxy- and o-aminobenzamides 15a–d led to a remarkable decrease of activity; (b) the 4-quinolone tautomers exhibited higher anti-TB activities compared to their 4-hydroxy-quinoline counterparts, with compound 8i showing the highest activity in this series; (c) the carboxamide substitution at position 2 of the 4-quinolone ring is superior to position 3; (d) replacing the indole nucleus in lead compound 3 with naphthalene 13c,d or 4-arylthiazole 18b scaffolds was tolerated. Four compounds 8i, 13c, 13d and 18b exhibited decent in vitro anti-TB activities against DS M. tb strains, comparable to the activity of ethambutol. The antimycobacterial activity of these four compounds remained the same or two-fold increased when tested against MDR and XDR M. tb strains, respectively. However, none of these four compounds was active against M. abs or M. avium strains, corroborating their distinct activity against M. tb. In addition, compounds 13c and 13d showed higher IC50 values against Vero cells than 8i and 18b and thus exhibiting high selectivity indices (SI), suggesting their potential lack of cytotoxicity. Taking into account that MmpL3 is the most relevant potential enzymatic target of our compounds, molecular docking simulations were conducted. Compounds 8i, 13c, 13d and 18b accommodated the MmpL3 active site that was previously reported to harbour the indoleamides, displaying a binding mode similar to ICA38 and lead compound 3. In addition, we investigated the effect of extending the length of the linker connecting the 4,6-dihalosubstituted-indole and the adamantane ring. The three-atom linker derivative 21 was the most tolerated extension in this set of compounds (21, 23, 24 and 27).
Overall, the superimposed orientation of the quinolone-2-carboxamides, naphthamides, and 4-arylthiazole-2-carboxamides with ICA38 4 in the same MmpL3 binding pocket supported their good in vitro potency against tested M. tb strains. These findings establish the quinolone, naphthalene, and 4-arylthiazole derivatives as promising novel classes of anti-TB compounds that potentially target MmpL3 and can be further evaluated in vivo.
4. Experimental section
4.1. Chemistry
4.1.1. General information
The following carboxylic acids, xanthurenic acid (7i), quinoline-2-carboxylic acid (12a), and 2-naphthoic acid (12b) were purchased from Sigma-Aldrich, while 5-bromo-2-naphthoic acid (12c), 6-bromo-2-naphthoic acid (12d), 6-methoxy-2-naphthoic acid (12e), 2-amino-3,5-dichlorobenzoic acid (14a), 3,5-dichloro-2-hydroxybenzoic acid (14b), 4,6-difluoroindole-2-carboxylic acid (19), and 1-acetyladamantane (25) were purchased from FluoroChem. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance III spectrometer at 400 and 100 MHz, respectively, with TMS as an internal standard. Standard abbreviations indicating multiplicity were as follows: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quadruplet, m = multiplet and br = broad. HRMS experiments were performed on a Thermo Scientific Q-Exactive Orbitrap mass spectrometer. TLC was performed with Analtech silica gel F TLC plates (200 microns, 20 × 20 cm). Flash chromatography was performed using a Teledyne Isco CombiFlash Rf system with RediSep columns or manually using SiliCycle UltraPure Silica Gels [40–63 μm (230–400 mesh)]. Final compounds were purified by preparative HPLC unless otherwise stated. The preparative HPLC employed an Omega 5 μm Polar C18 (21.2 mm × 150 mm) column, with detection at 254 and 280 nm on a Shimadzu SPD-20A detector, flow rate = 25.0 mL min−1. Method 1: 40–100% acetonitrile/H2O in 10 min; 100% acetonitrile in 10 min; 100–40% acetonitrile/H2O in 10 min. Method 2: 50–100% acetonitrile/H2O in 10 min; 100% acetonitrile in 10 min; 100–50% acetonitrile/H2O in 10 min. Method 3: 60–100% acetonitrile/H2O in 10 min; 100% acetonitrile in 10 min; 100–60% acetonitrile/H2O in 10 min. Both solvents contained 0.05 vol% of trifluoroacetic acid (TFA). Purities of final compounds were established by analytical HPLC, which was carried out using the Waters 1525 HPLC system with a Phenomenex 5μ C18 (2) (150 × 4.6 mm), on a Waters 2487 dual wavelength detector. Analytical HPLC method: flow rate = 1 mL min; gradient elution over 30 min, from 100% H2O to 100% acetonitrile with 0.05% TFA. The purity of all tested compounds was >95% as determined by the method described above.
4.1.2. General procedure for the synthesis of 6a–h
A mixture of the appropriate aromatic amine 5a–g (1 mmol) and diethylacetylenedicarboxylate (1.5 mmol) was stirred at room temperature until completion of the reaction as monitored by TLC (usually 24–48 h). PPA (2 mL) was then added to the reaction mixture and heated at 90 °C for 12 h. The reaction mixture was cooled, and 10 mL of water was added to it with stirring and the resulting precipitate was collected by filtration, washed with water (3 × 20 mL) and dried. The crude product was purified by flash column chromatography on SiO2 with CH2Cl2/MeOH gradient as the eluent to obtain the quinoline-2-carboxylic acid methyl ester derivatives 6a–h in yields ranging from 30% to 92%. 1H NMR data of compounds 6a–c matched those reported previously in the literature.52,53 A mixture of the 5-bromo and 7-bromo isomers 6g,h were obtained in 1 : 1 ratio from 3-bromoaniline as a starting material and they were separated via recrystallization from dichloromethane (DCM).
Methyl 6-chloro-4-oxo-1,4-dihydroquinoline-2-carboxylate (6a)52
Buff solid, yield: 76%. 1H NMR (DMSO-d6) δ 3.97 (s, 3H), 6.65 (s, 1H), 7.76 (dd, J = 9.0, 2.4 Hz, 1H), 7.97–8.00 (m, 2H), 12.26 (s, 1H).
Methyl 8-chloro-4-hydroxyquinoline-2-carboxylate (6b)53
Buff solid, yield: 70%. 1H NMR (DMSO-d6) δ 3.95 (s, 3H), 7.19–7.78 (m, 2H), 7.98 (dd, J = 7.5, 1.3 Hz, 1H), 8.14 (d, J = 8.1 Hz, 1H), 12.25 (br s, 1H).
Methyl 7,8-dichloro-4-hydroxyquinoline-2-carboxylate (6c)53
Buff solid, yield: 50%. 1H NMR (DMSO-d6) δ 3.94 (s, 3H), 7.48 (s, 1H), 7.70 (d, J = 8.3 Hz, 1H), 8.07 (d, J = 8.9 Hz, 1H), 12.35 (br s, 1H).
Methyl 6,8-dichloro-4-hydroxyquinoline-2-carboxylate (6d)
Buff solid, yield: 73%. 1H NMR (DMSO-d6) δ 3.94 (s, 3H), 7.47 (br s, 1H), 8.12 (s, 2H), 12.36 (br s, 1H); 13C NMR (DMSO-d6) δ 53.4, 106.5, 121.2, 124.1, 131.1, 131.5, 134.9, 143.5, 149.9, 162.9, 165.3.
Methyl 5,8-dichloro-4-hydroxy quinoline-2-carboxylate (6e)
Buff solid, yield: 85%. 1H NMR (DMSO-d6) δ 3.95 (s, 3H), 7.54 (br s, 2H), 7.86 (d, J = 8.2 Hz, 1H), 12.32 (br s, 1H); 13C NMR (DMSO-d6) δ 53.4, 107.7, 120.7, 128.3, 129.6, 130.6, 133.1, 146.7, 149.5, 164.2, 165.4.
Methyl 5,7-dichloro-4-oxo-1,4-dihydroquinoline-2-carboxylate (6f)
Buff solid, yield: 92%. 1H NMR (DMSO-d6) δ 3.95 (s, 3H), 6.54 (s, 1H), 7.37 (s, 1H), 7.93 (d, J = 1.2 Hz, 1H), 12.00 (s, 1H); 13C NMR (DMSO-d6) δ 54.1, 113.4, 118.4, 121.0, 126.2, 134.0, 136.5, 137.2, 143.4, 162.6, 176.7.
Methyl 5-bromo-4-oxo-1,4-dihydroquinoline-2-carboxylate (6g)
The title compound was obtained by following the general procedure using 3-bromoaniline as a starting material. The crude product was purified by flash column chromatography with CH2Cl2/MeOH gradient as the eluent to obtain a mixture of compounds 6g and 6h, followed by crystallisation from DCM to give 6g as an off white solid; yield: 30%. 1H NMR (DMSO-d6) δ 3.96 (s, 3H), 6.59 (s, 1H), 7.41–7.61 (m, 2H), 7.95 (dd, J = 8.2, 1.3 Hz, 1H), 12.02 (s, 1H); 13C NMR (DMSO-d6) δ 54.0, 112.4, 119.7, 120.1, 122.9, 130.8, 133.0, 137.2, 143.1, 162.6, 177.1.
Methyl 7-bromo-4-oxo-1,4-dihydroquinoline-2-carboxylate (6h)
As previously mentioned, after obtaining a mixture of 6g and 6h, they were separated via crystallisation from DCM, affording 6h as a light brown solid; yield: 30%. It was difficult to characterise its chemical structure by 13C NMR due its very poor solubility even in DMSO-d6. 1H NMR (DMSO-d6) δ 3.97 (s, 3H), 6.64 (s, 1H), 7.52 (dd, J = 8.6, 1.6 Hz, 1H), 7.99 (d, J = 8.6 Hz, 1H), 8.17 (d, J = 1.4 Hz, 1H), 12.10 (s, 1H).
4.1.3. General procedure for the synthesis of 7a–h
The appropriate ester 6a–h (1 mmol) was refluxed for 4 h in a mixture of 10% sodium hydroxide (5 mL) and methanol (5 mL). After cooling, the mixture was acidified by adding 2 M HCl and the solid obtained was collected by filtration, excessively washed with water and dried to obtain the quinoline-2-carboxylic acid derivatives in yields ranging from 90% to 99%. The crude product was used for next reaction without further purification.
4.1.4. General procedure for the synthesis of 8a–k
To a solution of the appropriate carboxylic acid 7a–i (1 mmol) in anhydrous dichloromethane (DCM) or dimethylformamide (DMF, 10 mL), hydroxybenzotriazole hydrate (HOBt, 1.2 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl, 1.2 mmol) were added at room temperature (rt) under an argon atmosphere. After stirring for 10 min, the corresponding amine (1.2 mmol) and N,N-diisopropylethylamine (DIPEA, 1.5 equiv.) were added, and the reaction mixture was stirred at room temperature until disappearance of the starting material (usually 60–72 h). After this time water (10 mL) was added, and the mixture was extracted with EtOAc (3 × 25 mL). The organic layers were separated, washed with water (5 × 25 mL), brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography using CH2Cl2/MeOH gradient to obtain the quinoline-2-carboxamides 8a–k in yields ranging from 25% to 76%, which were further purified by preparative HPLC.
N-(1-Adamantanyl)-6-chloro-4-oxo-1,4-dihydroquinoline-2-carboxamide (8a)
White solid, yield: 56%. 1H NMR (DMSO-d6) δ 1.66 (s, 6H), 2.08 (s, 9H), 6.91 (s, 1H), 7.72 (dd, J = 8.9, 2.4 Hz, 1H), 7.95 (d, J = 9.0 Hz, 1H), 8.01 (d, J = 2.1 Hz, 1H), 8.18 (s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.4, 41.0, 52.7, 107.0, 123.5, 126.0, 129.3, 132.6, 140.1, 145.1, 158.4, 161.7, 174.7; HRMS (ESI) m/z calcd for C20H21ClN2O2 ([M + H]+) m/z 357.1364; found 357.1356.
N-(1-Adamantanyl)-8-chloro-4-hydroxyquinoline-2-carboxamide (8b)
White solid, yield: 70%. 1H NMR (DMSO-d6) δ 1.69 (s, 6H), 2.10 (s, 9H), 7.54 (s, 2H), 7.97 (d, J = 7.5 Hz, 1H), 8.14 (d, J = 8.3 Hz, 1H), 8.20 (s, 1H), 12.20 (s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.3, 41.3, 51.2, 102.3, 122.1, 123.3, 126.8, 131.1, 132.5, 143.8, 152.2, 162.5, 163.8; HRMS (ESI) m/z calcd for C20H21ClN2O2 ([M + H]+) m/z 357.1364; found 357.1357.
8-Chloro-N-cyclooctyl-4-hydroxyquinoline-2-carboxamide (8c)
White solid, yield: 66%. 1H NMR (DMSO-d6) δ 1.44–1.91 (m, 14H), 3.93–4.14 (m, 1H), 7.52 (br s, 2H), 7.95 (d, J = 7.3 Hz, 1H), 8.12 (d, J = 7.2 Hz, 1H), 8.38 (br s, 1H), 12.21 (br s, 1H); 13C NMR (DMSO-d6) δ 23.7, 25.5, 27.2, 32.0, 49.5, 102.8, 122.1, 123.4, 126.8, 131.2, 132.7, 144.1, 152.0, 162.7, 163.7; HRMS (ESI) m/z calcd for C18H21ClN2O2 ([M + H]+) m/z 333.1364; found 333.1364.
N-(1-Adamantanyl)-7,8-dichloro-4-hydroxyquinoline-2-carboxamide (8d)
White solid, yield: 74%. 1H NMR (DMSO-d6) δ 1.68 (s, 6H), 2.09 (s, 9H), 7.56 (br s, 1H), 7.70 (d, J = 8.9 Hz, 1H), 8.10 (d, J = 9.0 Hz, 1H), 8.15 (s, 1H), 12.40 (s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.3, 41.3, 51.3, 102.7, 121.7, 122.8, 127.6, 130.4, 134.5, 144.8, 153.2, 162.2, 164.1; HRMS (ESI) m/z calcd for C20H20Cl2N2O2 ([M + H]+) m/z 391.0975; found 391.0964.
7,8-Dichloro-N-cyclooctyl-4-hydroxyquinoline-2-carboxamide (8e)
White solid, yield: 76%. 1H NMR (DMSO-d6) δ 1.39–1.92 (m, 14H), 3.90–4.15 (m, 1H), 7.55 (br s, 1H), 7.70 (d, J = 7.8 Hz, 1H), 8.10 (d, J = 8.8 Hz, 1H), 8.36 (br s, 1H), 12.25 (br s, 1H); 13C NMR (DMSO-d6) δ 23.8, 25.5, 27.2, 32.1, 49.6, 103.2, 121.7, 122.9, 127.6, 130.4, 134.6, 145.0, 153.0, 162.5, 163.9; HRMS (ESI) m/z calcd for C18H20Cl2N2O2 ([M + H]+) m/z 367.0975; found 367.0967.
6,8-Dichloro-N-cyclooctyl-4-hydroxyquinoline-2-carboxamide (8f)
White solid, yield: 50%. 1H NMR (DMSO-d6) δ 1.36–1.91 (m, 14H), 3.94–4.12 (m, 1H), 7.55 (br s, 1H), 8.08 (s, 2H), 8.36 (br s, 1H), 12.44 (br s, 1H); 13C NMR (DMSO-d6) δ 23.7, 25.5, 27.2, 32.0, 49.5, 103.6, 121.1, 123.6, 130.5, 131.2, 134.3, 142.7, 152.3, 162.4, 163.0; HRMS (ESI) m/z calcd for C18H20Cl2N2O2 ([M + H]+) m/z 367.0975; found 367.0966.
N-(1-Adamantanyl)-5,8-dichloro-4-hydroxyquinoline-2-carboxamide (8g)
White solid, yield: 38%. 1H NMR (DMSO-d6) δ 1.69 (s, 6H), 2.09 (s, 9H), 7.47–7.74 (m, 2H), 7.91 (d, J = 7.8 Hz, 1H), 8.13 (s, 1H), 12.40 (s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.4, 41.4, 51.2, 104.5, 120.3, 128.6, 128.9, 130.2, 132.1, 145.5, 152.0, 162.1, 164.9; HRMS (ESI) m/z calcd for C20H20Cl2N2O2 ([M + H]+) m/z 391.0975; found 391.0971.
N-(1-Adamantanyl)-5,7-dichloro-4-oxo-1,4-dihydroquinoline-2-carboxamide (8h)
White solid, yield: 32%. 1H NMR (DMSO-d6) δ 1.65 (s, 6H), 2.07 (s, 9H), 6.77 (s, 1H), 7.37 (s, 1H), 7.96 (s, 1H), 8.18 (s, 1H), 11.73 (s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.4, 41.0, 53.0, 110.5, 118.4, 120.4, 125.9, 134.0, 136.1, 141.7, 143.5, 161.1, 177.1; HRMS (ESI) m/z calcd for C20H20Cl2N2O2 ([M + H]+) m/z 391.0975; found 391.0965.
N-(1-Adamantanyl)-5-bromo-4-oxo-1,4-dihydroquinoline-2-carboxamide (8i)
White solid, yield: 25%. 1H NMR (DMSO-d6) δ 1.67 (s, 6H), 2.09 (s, 9H), 6.74 (br s, 1H), 7.45–7.52 (m, 2H), 7.93 (dd, J = 8.4, 1.0 Hz, 1H), 8.18 (s, 1H), 11.69 (s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.4, 41.0, 52.8, 109.3, 119.4, 120.1, 122.2, 130.5, 132.5, 141.6, 143.0, 161.5, 177.3; HRMS (ESI) m/z calcd for C20H21BrN2O2 ([M + H]+) m/z 401.0859; found 401.0849.
N-(1-Adamantanyl)-7-bromo-4-oxo-1,4-dihydroquinoline-2-carboxamide (8j)
White solid, yield: 32%. 1H NMR (DMSO-d6) δ 1.67 (s, 6H), 2.09 (s, 9H), 6.81 (br s, 1H), 7.51 (s, 1H), 7.99 (d, J = 8.4 Hz, 1H), 8.11–8.25 (m, 2H), 11.80 (br s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.4, 41.0, 52.8, 108.3, 122.1, 124.6, 126.0, 127.2, 127.3, 141.4, 143.0, 161.6, 177.9; HRMS (ESI) m/z calcd for C20H21BrN2O2 ([M + H]+) m/z 401.0859; found 401.0848.
N-(1-Adamantanyl)-4,8-dihydroxyquinoline-2-carboxamide (8k)
White solid, yield: 42%. 1H NMR (DMSO-d6) δ 1.68 (s, 6H), 2.08 (s, 3H), 2.14 (s, 6H), 7.09 (dd, J = 7.6, 0.9 Hz, 1H), 7.37 (s, 1H), 7.45–7.68 (m, 2H), 8.68 (br s, 1H), 10.10 (br s, 1H); 13C NMR (DMSO-d6) δ 29.4, 36.5, 41.4, 51.9, 102.4, 112.3, 122.6, 127.5, 138.4, 138.8, 150.3, 153.9, 162.8, 163.4; HRMS (ESI) m/z calcd for C20H21BrN2O2 ([M + H]+) m/z 339.1703; found 339.1693.
4.1.5. General procedure for the synthesis of 9a–g
A mixture of the appropriate aromatic amine 5a–g (1 mmol) and 2-ethoxymethylene-malonic acid diethyl ester (2 mmol) was heated at 100 °C until completion of the reaction as monitored by TLC (usually 12–18 h). Diphenyl ether (3 mL) was then added to the reaction mixture and heated at 250 °C for 4 h. After the reaction mixture was cooled at room temperature, 10 mL of hexane was added to it with stirring and the formed precipitate was collected by filtration, washed with hexane (3 × 10 mL), recrystallized from DMF to provide the quinoline-3-carboxylic acid ethyl ester derivatives in yields ranging from 37% to 90%. 1H NMR data of compounds 9a–f matched those which were reported in the literature.54–56 The 7-bromo derivative 9g was the only isomer that could be isolated as a product from the starting material 3-bromoaniline and it was difficult to characterise its chemical structure by 1H NMR due its poor solubility even in DMSO-d6 as previously reported.57
6-Chloro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (9a)56
Buff solid, yield: 64%. 1H NMR (DMSO-d6) δ 1.28 (t, J = 7.1 Hz, 3H), 4.22 (q, J = 7.1 Hz, 2H), 7.66 (d, J = 8.8 Hz, 1H), 7.74 (dd, J = 8.8, 2.5 Hz, 1H), 8.08 (d, J = 2.2 Hz, 1H), 8.58 (s, 1H), 12.39 (br s, 1H).
8-Chloro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (9b)56
Buff solid, yield: 60%. 1H NMR (DMSO-d6) δ 1.28 (t, J = 7.1 Hz, 3H), 4.23 (q, J = 7.1 Hz, 2H), 7.42 (t, J = 7.9 Hz, 1H), 7.89 (dd, J = 7.7, 1.3 Hz, 1H), 8.13 (dd, J = 8.1, 1.3 Hz, 1H), 8.43 (s, 1H), 11.92 (br s, 1H).
7,8-Dichloro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (9c)55
Buff solid, yield: 86%. 1H NMR (DMSO-d6) δ 1.28 (t, J = 7.1 Hz, 3H), 4.23 (q, J = 7.1 Hz, 2H), 7.64 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 8.42 (s, 1H), 11.99 (br s, 1H).
6,8-Dichloro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (9d)55
Buff solid, yield: 90%. 1H NMR (DMSO-d6) δ 1.28 (t, J = 7.1 Hz, 3H), 4.23 (q, J = 7.1 Hz, 2H), 8.06 (d, J = 2.4 Hz, 1H), 8.08 (d, J = 2.4 Hz, 1H), 8.43 (s, 1H), 12.11 (s, 1H).
5,8-Dichloro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (9e)55
Buff solid, yield: 37%. 1H NMR (DMSO-d6) δ 1.27 (t, J = 7.0 Hz, 3H), 4.22 (q, J = 7.0 Hz, 2H), 7.38 (d, J = 8.4 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 8.32 (s, 1H), 11.73 (s, 1H).
5,7-Dichloro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (9f)54,55
Buff solid, yield: 37%. 1H NMR (DMSO-d6) δ 1.28 (t, J = 7.1 Hz, 3H), 4.21 (q, J = 7.1 Hz, 2H), 7.30 (d, J = 2.0 Hz, 1H), 7.53 (d, J = 2.0 Hz, 1H), 8.43 (s, 1H).
7-Bromo-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (9g)57
Buff solid, yield: 75%.
4.1.6. General procedure for the synthesis of 10a–g
The quinoline-3-carboxylic acid ethyl esters 9a–g (1 mmol) were refluxed for 4 h in a mixture of 10% sodium hydroxide (5 mL) and ethanol (5 mL). After cooling, the mixture was acidified by adding 2 M HCl and the solid obtained was collected by filtration, excessively washed with water and dried to obtain the quinoline-3-carboxylic acid derivatives 10a–g in yields ranging from 80% to 99%. The crude product was used for next reaction without further purification.
4.1.7. General procedure for the synthesis of 11a–g
To a solution of the appropriate carboxylic acid 10a–g (1 mmol) in anhydrous DMF (10 mL), O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU, 2 mmol), 1-aminoadamantane (1.2 mmol), and DIPEA (3 mmol) were added and the reaction mixture was stirred at rt for 72 h. Water (10 mL) was then added to the reaction mixture and extracted with EtOAc (3 × 25 mL). The organic layers were separated, washed with water (5 × 25 mL), brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by manual column chromatography using CH2Cl2/MeOH gradient to obtain the quinoline-3-carboxamides 11a–g in yields ranging from 51% to 89% and further purified by recrystallisation from DCM.
N-(1-Adamantanyl)-6-chloro-4-oxo-1,4-dihydroquinoline-3-carboxamide (11a)
White solid, yield: 84%. 1H NMR (DMSO-d6) δ 1.67 (s, 6H), 2.06 (s, 9H), 7.72 (d, J = 8.8 Hz, 1H), 7.79 (dd, J = 8.8, 2.4 Hz, 1H), 8.17 (d, J = 2.4 Hz, 1H), 8.71 (s, 1H), 9.85 (s, 1H), 12.76 (s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.5, 41.9, 51.0, 112.5, 121.9, 124.8, 127.7, 129.9, 133.1, 138.2, 144.1, 163.2, 175.4; HRMS (ESI) m/z calcd for C20H21ClN2O2 ([M + H]+) m/z 357.1364; found 357.1362.
N-(1-Adamantanyl)-8-chloro-4-oxo-1,4-dihydroquinoline-3-carboxamide (11b)
White solid, yield: 56%. 1H NMR (DMSO-d6) δ 1.67 (s, 6H), 2.06 (s, 9H), 7.47 (t, J = 7.9 Hz, 1H), 7.95 (dd, J = 7.7, 1.3 Hz, 1H), 8.22 (dd, J = 8.2, 1.3 Hz, 1H), 8.64 (s, 1H), 9.80 (s, 1H), 12.24 (s, 1H); 13C NMR (DMSO-d6) δ 29.4, 36.5, 41.9, 51.1, 112.9, 122.7, 125.3, 125.4, 128.2, 132.9, 136.1, 144.4, 162.9, 176.3; HRMS (ESI) m/z calcd for C20H21ClN2O2 ([M + H]+) m/z 357.1364; found 357.1359.
N-(1-Adamantanyl)-7,8-dichloro-4-oxo-1,4-dihydroquinoline-3-carboxamide (11c)
White solid, yield: 68%. 1H NMR (DMSO-d6) δ 1.69 (s, 6H), 2.08 (s, 9H), 7.62 (d, J = 8.7 Hz, 1H), 8.21 (d, J = 9.0 Hz, 1H), 8.66 (s, 1H), 9.72 (s, 1H), 12.29 (s, 1H); 13C NMR (DMSO-d6) δ 29.4, 36.5, 41.9, 51.1, 113.2, 121.4, 125.9, 126.0, 126.5, 136.3, 137.8, 145.1, 162.8, 175.9; HRMS (ESI) m/z calcd for C20H20Cl2N2O2 ([M + H]+) m/z 391.0975; found 391.0971.
N-(1-Adamantanyl)-6,8-dichloro-4-oxo-1,4-dihydroquinoline-3-carboxamide (11d)
White solid, yield: 89%. 1H NMR (DMSO-d6) δ 1.67 (s, 6H), 2.06 (s, 9H), 8.05–8.22 (m, 2H), 8.63 (s, 1H), 9.70 (s, 1H), 12.41 (s, 1H); 13C NMR (DMSO-d6) δ 29.4, 36.5, 41.8, 51.2, 113.2, 124.2, 124.5, 128.7, 129.6, 132.4, 135.1, 144.6, 162.6, 175.2; HRMS (ESI) m/z calcd for C20H20Cl2N2O2 ([M + H]+) m/z 391.0975; found 391.0973.
N-(1-Adamantanyl)-5,8-chloro-4-oxo-1,4-dihydroquinoline-3-carboxamide (11e)
White solid, yield: 51%. 1H NMR (DMSO-d6) δ 1.67 (s, 6H), 2.05 (s, 9H), 7.43 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 8.4 Hz, 1H), 8.58 (s, 1H), 9.67 (s, 1H), 12.07 (s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.5, 41.8, 51.1, 114.2, 121.9, 123.9, 128.0, 132.5, 132.7, 138.5, 144.1, 162.6, 176.3; HRMS (ESI) m/z calcd for C20H20Cl2N2O2 ([M + H]+) m/z 391.0975; found 391.0976.
N-(1-Adamantanyl)-5,7-dichloro-4-oxo-1,4-dihydroquinoline-3-carboxamide (11f)
White solid, yield: 64%. 1H NMR (DMSO-d6) δ 1.67 (s, 6H), 2.05 (s, 9H), 7.53 (d, J = 2.0 Hz, 1H), 7.65 (d, J = 2.1 Hz, 1H), 8.66 (s, 1H), 9.74 (s, 1H), 12.66 (s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.5, 41.8, 51.0, 114.1, 118.1, 121.5, 127.4, 134.9, 136.5, 142.6, 143.9, 162.9, 176.0; HRMS (ESI) m/z calcd for C20H20Cl2N2O2 ([M + H]+) m/z 391.0975; found 391.0974.
N-(1-Adamantanyl)-7-bromo-4-oxo-1,4-dihydroquinoline-3-carboxamide (11g)
White solid, yield: 75%. 1H NMR (DMSO-d6) δ 1.67 (s, 6H), 2.06 (s, 9H), 7.61 (dd, J = 8.7, 1.6 Hz, 1H), 7.88 (d, J = 1.8 Hz, 1H), 8.14 (d, J = 8.7 Hz, 1H), 8.70 (s, 1H), 9.86 (s, 1H), 12.59 (s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.5, 41.9, 51.0, 112.7, 121.6, 125.5, 126.4, 128.2, 128.3, 140.5, 144.3, 163.2, 176.2; HRMS (ESI) m/z calcd for C20H21BrN2O2 ([M + H]+) m/z 401.0859; found 401.0859.
4.1.8. General procedure for the synthesis of 13a–g and 15a–d
Title compounds were prepared following the general amide coupling protocol used for obtaining compounds 8a–k. The crude product was purified by flash column chromatography using CH2Cl2/MeOH gradient to obtain the title compounds in yields ranging from 31% to 92%. Compounds 13a–d were further purified by recrystallisation from acetone. Compounds 13e,f and 15a,c,d were already >95% pure after flash chromatography, while compounds 13g and 15b were further purified by preparative HPLC. 1H NMR data of compounds 13a–c matched those which were reported in the literature.47,58
N-Cyclooctylquinoline-2-carboxamide (13a)47
White solid, yield: 46%. 1H NMR (DMSO-d6) δ 1.41–1.98 (m, 14H), 3.96–4.21 (m, 1H), 7.72 (dd, J = 8.1, 7.0 Hz, 1H), 7.87 (dd, J = 8.3, 7.0 Hz, 1H), 8.08 (d, J = 8.1 Hz, 1H), 8.16 (dd, J = 8.4, 6.1 Hz, 2H), 8.56 (d, J = 8.5 Hz, 1H), 8.60 (d, J = 8.3 Hz, 1H).
N-(1-Adamantanyl)-2-quinoline-2-carboxamide (13b)58
White solid, yield: 65%. 1H NMR (DMSO-d6) δ 1.69 (s, 6H), 2.10 (s, 3H), 2.13 (s, 6H), 7.71 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.86 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 8.05–8.16 (m, 4H), 8.56 (d, J = 8.4 Hz, 1H).
N-Cyclooctyl-2-naphthamide (13c)47
White solid, yield: 56%. 1H NMR (DMSO-d6) δ 1.42–1.89 (m, 14H), 3.94–4.21 (m, 1H), 7.54–7.63 (m, 2H), 7.89–8.04 (m, 4H), 8.38 (d, J = 7.8 Hz, 1H), 8.42 (s, 1H).
N-(1-Adamantanyl)-2-naphthamide (13d)
White solid, yield: 60%. 1H NMR (DMSO-d6) δ 1.68 (s, 6H), 2.07 (s, 3H), 2.12 (s, 6H), 7.62–7.53 (m, 2H), 7.76 (s, 1H), 7.85 (dd, J = 8.6, 1.6 Hz, 1H), 7.93–8.01 (m, 3H), 8.36 (s, 1H); 13C NMR (DMSO-d6) δ 29.4, 36.6, 41.4, 52.1, 125.1, 127.0, 127.71, 127.74, 128.0 (2C), 129.2, 132.6, 133.8, 134.4, 166.6; HRMS (ESI) m/z calcd for C21H23NO ([M + H]+) m/z 306.1852; found 306.1844.
N-(1-Adamantanyl)-5-bromo-2-naphthamide (13e)
White solid, yield: 52%. 1H NMR (DMSO-d6) δ 1.69 (s, 6H), 2.09 (s, 3H), 2.14 (s, 6H), 7.50 (dd, J = 8.5, 7.5 Hz, 1H), 7.58 (dd, J = 7.0, 1.0 Hz, 1H), 7.69 (dd, J = 8.5, 7.1 Hz, 1H), 7.93 (dd, J = 7.4, 0.9 Hz, 1H), 8.05–8.14 (m, 2H), 8.21 (d, J = 8.5 Hz, 1H); 13C NMR (DMSO-d6) δ 29.4, 36.6, 41.4, 52.3, 122.5, 126.0, 126.1, 127.5, 127.7 (2C), 130.8, 131.5, 131.6, 137.6, 168.2; HRMS (ESI) m/z calcd for C21H22BrNO ([M + H]+) m/z 384.0958; found 384.0956.
N-(1-Adamantanyl)-6-bromo-2-naphthamide (13f)
White solid, yield: 31%. 1H NMR (DMSO-d6) δ 1.68 (s, 6H), 2.07 (s, 3H), 2.11 (s, 6H), 7.69 (dd, J = 8.7, 1.9 Hz, 1H), 7.80 (s, 1H), 7.88–7.99 (m, 3H), 8.25 (s, 1H), 8.37 (s, 1H); 13C NMR (DMSO-d6) δ 29.4, 36.6, 41.4, 52.1, 121.1, 126.2, 127.3, 127.7, 130.0, 130.1, 131.1, 131.4, 134.4, 135.5, 166.4; HRMS (ESI) m/z calcd for C21H22BrNO ([M + H]+) m/z 384.0958; found 384.0960.
N-(1-Adamantanyl)-6-methoxy-2-naphthamide (13g)
White solid, yield: 52%. 1H NMR (DMSO-d6) δ 1.67 (s, 6H), 2.07 (s, 3H), 2.11 (s, 6H), 3.89 (s, 3H), 7.21 (dd, J = 9.0, 2.5 Hz, 1H), 7.35 (d, J = 2.4 Hz, 1H), 7.66 (s, 1H), 7.83 (s, 2H), 7.90 (d, J = 9.0 Hz, 1H), 8.30 (s, 1H); 13C NMR (DMSO-d6) δ 29.4, 36.6, 41.4, 52.0, 55.7, 106.3, 119.6, 125.5, 126.8, 127.6, 127.9, 130.8, 131.5, 136.0, 158.8, 166.6; HRMS (ESI) m/z calcd for C22H25NO ([M + H]+) m/z 336.1958; found 336.1951.
N-(1-Adamantanyl)-2-amino-3,5-dichlorobenzamide (15a)
White solid, yield: 86%. 1H NMR (DMSO-d6) δ 1.65 (s, 6H), 2.05 (s, 9H), 6.30 (s, 2H), 7.46 (s, 2H), 7.78 (s, 1H); 13C NMR (DMSO-d6) δ 29.4, 36.5, 41.1, 52.4, 118.3, 119.7, 120.0, 127.8, 130.7, 144.1, 167.2; HRMS (ESI) m/z calcd for C17H20Cl2N2O ([M + H]+) m/z 339.1025; found 339.1022.
2-Amino-3,5-dichloro-N-cyclooctylbenzamide (15b)
White solid, yield: 92%. 1H NMR (DMSO-d6) δ 1.49–1.77 (m, 14H), 3.88–4.04 (m, 1H), 6.52 (s, 2H), 7.49 (d, J = 2.4 Hz, 1H), 7.55 (d, J = 2.4 Hz, 1H), 8.33 (d, J = 7.7 Hz, 1H); 13C NMR (DMSO-d6) δ 24.0, 25.6, 27.3, 31.9, 49.7, 118.3, 118.4, 119.9, 127.4, 131.0, 144.5, 166.2; HRMS (ESI) m/z calcd for C15H20Cl2N2O ([M + H]+) m/z 315.1025; found 315.1020.
N-(1-Adamantanyl)-3,5-dichloro-2-hydroxybenzamide (15c)
White solid, yield: 50%. 1H NMR (DMSO-d6) δ 1.66 (s, 6H), 2.08 (s, 9H), 7.67 (d, J = 2.3 Hz, 1H), 8.04 (d, J = 2.4 Hz, 1H), 8.51 (s, 2H), 13.59 (br s, 1H); 13C NMR (DMSO-d6) δ 29.3, 36.3, 40.9, 53.5, 118.0, 121.9, 122.8, 126.8, 133.1, 156.4, 168.2; HRMS (ESI) m/z calcd for C17H19Cl2NO2 ([M + H]+) m/z 340.0866; found 340.0864.
3,5-Dichloro-N-cyclooctyl-2-hydroxybenzamide (15d)
White solid, yield: 63%. 1H NMR (DMSO-d6) δ 1.42–1.79 (m, 14H), 4.02–4.08 (m, 1H), 7.68 (d, J = 1.2 Hz, 1H), 8.03 (d, J = 1.6 Hz, 1H), 9.19 (s, 1H), 13.87 (br s, 1H); 13C NMR (DMSO-d6) δ 23.9, 25.6, 27.1, 31.8, 50.2, 117.0, 122.1, 122.7, 126.2, 133.3, 156.5, 167.3; HRMS (ESI) m/z calcd for C15H19Cl2NO2 ([M + H]+) m/z 316.0866; found 316.0861.
4.1.9. General procedure for the synthesis of 17a,b
A mixture of the corresponding acetophenone 16a,b (1 mmol), thiourea (2 mmol) and iodine (1 mmol) was heated at 100 °C for 3–5 h. Then the reaction mixture was cooled, washed with diethyl ether to remove the unreacted acetophenone and iodine. The residue was dissolved in hot water, filtered to remove sulphone, and the filtrate was basified with aqueous NH4OH to yield the corresponding 2-amino-4-(substituted-phenyl)-1,3-thiazoles 17a,b. The crude product was purified by flash column chromatography on SiO2 with CH2Cl2/MeOH gradient as the eluent to obtain the title compounds 17a,b in yields 61% and 54%, respectively. 1H NMR data of compounds 17a,b matched the ones reported in the literature.59,60
2-Amino-4-(4-chlorophenyl)-1,3-thiazole (17a)
Yellowish white solid, yield: 61%. 1H NMR (DMSO-d6) δ 7.06 (s, 1H), 7.07 (s, 2H), 7.41 (d, J = 8.6 Hz, 2H), 7.80 (d, J = 8.6 Hz, 2H).59
2-Amino-4-(2,4-dichlorophenyl)-1,3-thiazole (17b)
Yellowish white solid, yield: 54%. 1H NMR (DMSO-d6) δ 7.09 (s, 2H), 7.10 (s, 1H), 7.45 (dd, J = 8.5, 2.2 Hz, 1H), 7.64 (d, J = 2.1 Hz, 1H), 7.87 (d, J = 8.5 Hz, 1H).60
4.1.10. General procedure for the synthesis of 18a,b
Title compounds were prepared following the general amide coupling procedure used for preparing compounds 8a–k. The crude product was purified by flash chromatography using CH2Cl2/MeOH gradient to obtain the thiazole-2-carboxamides 18a,b in yields 41% and 30%, respectively, and were further purified by preparative HPLC.
N-(4-(4-Chlorophenyl)thiazol-2-yl)adamantane-1-carboxamide (18a)
White solid, yield: 41%. 1H NMR (DMSO-d6) δ 1.69 (s, 6H), 1.95 (s, 6H), 2.01 (s, 3H), 7.48 (d, J = 8.6 Hz, 2H), 7.63 (s, 1H), 7.92 (d, J = 8.6 Hz, 2H), 11.83 (s, 1H); 13C NMR (DMSO-d6) δ 28.0, 36.3, 38.0, 41.1, 109.2, 127.9, 129.2, 132.6, 133.8, 148.0, 159.2, 176.8; HRMS (ESI) m/z calcd for C20H21ClN2OS ([M + H]+) m/z 373.1136; found 373.1127.
N-(4-(2,4-Dichlorophenyl)thiazol-2-yl)adamantane-1-carboxamide (18b)
White solid, yield: 30%. 1H NMR (DMSO-d6) δ 1.69 (s, 6H), 1.95 (s, 6H), 2.01 (s, 3H), 7.52 (dd, J = 8.5, 2.2 Hz, 1H), 7.62 (s, 1H), 7.70 (d, J = 2.2 Hz, 1H), 7.89 (d, J = 8.5 Hz, 1H), 11.90 (s, 1H); 13C NMR (DMSO-d6) δ 28.0, 36.3, 38.0, 41.1, 113.7, 128.0, 130.2, 132.3, 132.7, 132.8, 133.2, 144.7, 158.4, 176.8; HRMS (ESI) m/z calcd for C20H20Cl2N2OS ([M + H]+) m/z 407.0746; found 407.0737.
4.1.11. General procedure for the synthesis of 20 and 22
A mixture of 4,6-difluoro-1H-indole-2-carboxylic acid (19, 1 mmol) and 1,1′-carbonyldiimidazole (CDI) (1.5 mmol) in anhydrous DMF (10 mL) was stirred at rt for two hours, followed by the addition of aqueous NH4OH or hydrazine hydrate solution (5 mmol) and stirring was continued for 12–16 h at rt. Water (25 mL) was then added to the reaction mixture and extracted with EtOAc (3 × 25 mL). The organic layers were separated, washed with water (5 × 25 mL), brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography on SiO2 with CH2Cl2/MeOH gradient as the eluent to obtain the title compounds in yields 98% and 74%, respectively.
4,6-Difluoro-1H-indole-2-carboxamide (20)
Buff solid, yield: 98%. 1H NMR (400 MHz, DMSO) δ 6.86 (td, J = 10.5, 2.0 Hz, 1H), 7.01 (dd, J = 9.4, 1.9 Hz, 1H), 7.22 (s, 1H), 7.44 (s, 1H), 8.03 (s, 1H), 11.97 (s, 1H); 13C NMR δ 95.0 (dd, J = 25.8, 4.5 Hz), 95.6 (dd, J = 29.7, 23.3 Hz), 99.3, 113.6 (d, J = 21.8 Hz), 133.3 (d, J = 3.2 Hz), 138.1 (dd, J = 15.2, 13.2 Hz), 156.2 (dd, J = 248.6, 15.6 Hz), 159.6 (dd, J = 238.4, 12.1 Hz), 162.5.
4,6-Difluoro-1H-indole-2-carbohydrazide (22)
Buff solid, yield: 74%. 1H NMR (400 MHz, DMSO) δ 4.52 (s, 2H), 6.87 (td, J = 10.4, 1.9 Hz, 1H), 7.02 (d, J = 9.4 Hz, 1H), 7.16 (s, 1H), 9.86 (s, 1H),12.04 (s, 1H); 13C NMR δ 95.0 (dd, J = 25.9, 4.5 Hz), 95.6 (dd, J = 29.7, 23.3 Hz), 98.0, 113.6 (d, J = 22.5 Hz), 132.0 (d, J = 3.3 Hz), 138.0 (dd, J = 15.2, 13.2 Hz), 156.1 (dd, J = 248.5, 15.5 Hz), 159.5 (dd, J = 238.4, 12.3 Hz), 160.8.
N-Adamantane-1-carbonyl-4,6-difluoro-1H-indole-2-carboxamide (21)
To a solution of 1-adamantanecarboxylic acid (1.5 mmol) in DCM (10 mL), DMF (0.1 mL) and oxalyl chloride (3 mmol) were added. After stirring for 2 h at rt, the mixture was concentrated under vacuum. The residue was dissolved in pyridine (5 mL), followed by addition of compound 20 (1 mmol) and the reaction mixture was refluxed for 16 h. After cooling, 2 M HCl (25 mL) was added to the reaction mixture and extracted with EtOAc (3 × 25 mL). The organic layers were separated, washed with brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography on SiO2 using CH2Cl2/MeOH gradient to obtain the title compound and was further purified by preparative HPLC. White solid, yield: 42%. 1H NMR (400 MHz, DMSO) δ 1.71 (s, 6H), 1.95 (s, 6H), 2.02 (s, 3H), 6.95 (t, J = 10.3 Hz, 1H), 7.05 (d, J = 8.6 Hz, 1H), 7.52 (s, 1H), 10.01 (s, 1H), 12.26 (s, 1H); 13C NMR δ 28.0, 36.2, 37.7, 42.5, 95.2 (dd, J = 26.0, 4.5 Hz), 96.3 (dd, J = 30.0, 23.1 Hz), 103.2, 113.3 (d, J = 22.0 Hz), 131.7 (d, J = 3.2 Hz), 139.0 (dd, J = 15.3, 12.6 Hz), 156.7 (dd, J = 250.2, 15.7 Hz), 160.0, 160.5 (dd, J = 240.3, 12.1 Hz), 176.4; HRMS (ESI) m/z calcd for C20H20F2N2O2 ([M + H]+) m/z 359.1566; found 359.1559.
N′-Adamantane-1-carbonyl-4,6-difluoro-1H-indole-2-carbohydrazide (23)
Title compound was prepared following the standard amide coupling procedure used for preparing compounds 8a–k. The crude product was purified by flash chromatography on SiO2 using CH2Cl2/MeOH gradient and was further purified by preparative HPLC. White solid, yield: 48%. 1H NMR (400 MHz, DMSO) δ 1.69 (s, 6H), 1.88 (s, 6H), 1.99 (s, 3H), 6.89 (td, J = 10.4, 1.6 Hz, 1H), 7.06 (d, J = 9.2 Hz, 1H), 7.29 (s, 1H), 9.50 (s, 1H), 10.28 (s, 1H), 12.05 (s, 1H); 13C NMR δ 28.0, 36.5, 39.0, 40.1, 95.1 (dd, J = 25.9, 4.2 Hz), 95.9 (dd, J = 29.5, 23.5 Hz), 99.4, 113.6 (d, J = 21.9 Hz), 131.3 (d, J = 3.2 Hz), 138.2 (dd, J = 15.3, 13.1 Hz), 156.2 (dd, J = 249.0, 15.6 Hz), 159.8 (dd, J = 238.7, 12.1 Hz), 160.2, 176.9; HRMS (ESI) m/z calcd for C20H21F2N3O2 ([M + H]+) m/z 374.1675; found 374.1665.
N-(1-Adamantanyl)-2-(4,6-difluoro-1H-indole-2-carbonyl)hydrazine-1-carboxamide (24)
A mixture of compound 22 and 1-adamantylisocyanate in ethanol was refluxed for 16 h. The reaction mixture was then concentrated under vacuum, and water (50 mL) was added to the residue. The formed precipitate was filtered off, dried, purified by flash chromatography on SiO2 using CH2Cl2/MeOH gradient and was further purified by preparative HPLC. White solid, yield: 54%. 1H NMR (400 MHz, DMSO) δ 1 1H NMR (400 MHz, DMSO) δ 1.61 (s, 6H), 1.91 (s, 6H), 2.00 (s, 3H), 5.96 (s, 1H), 6.89 (td, J = 10.4, 1.7 Hz, 1H), 7.03 (dd, J = 9.3, 1.5 Hz, 1H), 7.27 (d, J = 1.5 Hz, 1H), 7.72 (s, 1H), 10.18 (s, 1H), 12.09 (s, 1H); 13C NMR δ 29.4, 36.5, 42.2, 50.4, 95.1 (dd, J = 25.9, 4.3 Hz), 95.8 (dd, J = 29.8, 23.2 Hz), 99.3, 113.6 (d, J = 21.8 Hz), 131.3 (d, J = 3.2 Hz), 138.2 (dd, J = 15.2, 13.2 Hz), 156.2 (dd, J = 248.8, 15.5 Hz), 157.1, 159.9 (dd, J = 238.7, 11.5 Hz), 160.9; HRMS (ESI) m/z calcd for C20H22F2N4O2 ([M + H]+) m/z 389.1784; found 389.1772.
4-(1-Adamantanyl)-2-amino-1,3-thiazole (26)
The title compound was prepared following the general procedure used for preparing compounds 17a,b. The crude product was purified by flash column chromatography on SiO2 with CH2Cl2/MeOH gradient as the eluent. 1H NMR data of compound 26 matched the reported one in the literature.61 White solid, yield: 63%. 1H NMR (DMSO-d6) δ 1.55–1.75 (m, 6H), 1.80 (s, 6H), 1.98 (s, 3H), 5.99 (s, 1H), 6.74 (s, 2H).
N-(4-(1-Adamantanyl)thiazol-2-yl)-4,6-difluoro-1H-indole-2-carboxamide (27)
A mixture of compound 26 and 4,6-difluoro-1H-indole-2-carboxylic acid (19) were then reacted following the general amide coupling protocol used in preparing compounds 8a–k. The crude product was purified by flash chromatography on SiO2 using CH2Cl2/MeOH gradient to obtain the title compound and was further purified by recrystallization from diethyl ether. Yellowish white solid, yield: 30%. 1H NMR (400 MHz, DMSO) δ1H NMR (400 MHz, DMSO) δ 1.82–1.62 (m, 6H), 1.93 (s, 6H), 2.05 (s, 3H), 6.73 (s, 1H), 6.93 (td, J = 10.3, 1.7 Hz, 1H), 7.07 (dd, J = 9.3, 1.2 Hz, 1H), 7.73 (s, 1H), 12.29 (br s, 1H); 13C NMR δ 28.4, 36.3, 36.8, 42.1, 95.2 (dd, J = 26.1, 4.3 Hz), 96.1 (dd, J = 29.8, 23.1 Hz), 101.9, 105.5, 113.8 (d, J = 21.8 Hz), 131.0, 138.8 (dd, J = 15.4, 12.7 Hz), 156.5 (dd, J = 249.8, 15.7 Hz), 157.9, 159.0, 160.3 (dd, J = 239.9, 12.2 Hz), 161.2; HRMS (ESI) m/z calcd for C22H21F2N3OS ([M + H]+) m/z 414.1446; found 414.1434.
4.2. Biology
MIC was determined using the MABA assay as previously reported in the literature.38,46 MABA format was also used in the cytotoxicity evaluation on Vero Cells.38
4.3. Molecular modeling studies
All the molecular modeling and docking simulations were done on Molecular Operating Environment MOE software version 2008.10.48 The structures of compounds 8i, 13c, 13d and 18b were drawn in ChemDraw Ultra 16.0 and saved as .mol file, then opened inside the MOE program. They were then 3D protonated and geometrically optimised using MMFF94 × forcefield with gradient 0.05. The X-ray crystal structure of MmpL3 bound to the ICA38 (PDB: 6AJJ) was obtained from the protein data bank. The target binding pocket was prepared for docking by: (1) removing co-crystallised ligand molecules and (2) 3D protonating the enzyme, in which hydrogens and partial charges were added to the system for optimisation. In order to validate the docking protocol, ICA38 was docked into the active site of 6AJJ. The generated conformers were docked into the same binding pocket of ICA38 with MOE-DOCK using Triangle Matcher placement method and scored using London dG scoring function. A molecular mechanics force field refinement was applied on the generated top 30 poses. For each ligand–enzyme complex, the top-ranked pose with the best docking score (i.e. the lowest binding free energy) was selected.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We gratefully acknowledge Curtin University for the funding of Curtin International Postgraduate Research Scholarship (CIPRS) for SSRA. We thank the support of NIH grants AI 27856 and HL 133190 for WRB. We also acknowledge ARC Discovery Early Career Researcher Award DE160100482 for HG.
References
- World Health Organization, Global Tuberculosis Report, Geneva, 2019 [Google Scholar]
- Tiberi S. Scardigli A. Centis R. D'Ambrosio L. Munoz-Torrico M. Salazar-Lezama M. A. Spanevello A. Visca D. Zumla A. Migliori G. B. Caminero Luna J. A. Int. J. Infect. Dis. 2017;56:181–184. doi: 10.1016/j.ijid.2016.10.026. [DOI] [PubMed] [Google Scholar]
- Al-Humadi H. W. Al-Saigh R. J. Al-Humadi A. W. Front. Pharmacol. 2017;8:689. doi: 10.3389/fphar.2017.00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgiu G. Centis R. D'Ambrosio L. Migliori G. B. Cold Spring Harbor Perspect. Med. 2015;5:a017822. doi: 10.1101/cshperspect.a017822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Upadhyay A. Fontes F. L. North E. J. Wang Y. Crans D. C. Grzegorzewicz A. E. Jones V. Franzblau S. G. Lee R. E. Crick D. C. Jackson M. Antimicrob. Agents Chemother. 2014;58:6413–6423. doi: 10.1128/AAC.03229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. Meshcheryakov V. A. Poce G. Chng S. S. Proc. Natl. Acad. Sci. U. S. A. 2017;114:7993–7998. doi: 10.1073/pnas.1700062114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsayed S. S. R. Beh C. C. Foster N. R. Payne A. D. Yu Y. Gunosewoyo H. Curr. Mol. Pharmacol. 2019;12:27–49. doi: 10.2174/1874467211666181025141114. [DOI] [PubMed] [Google Scholar]
- Brennan P. J. Nikaido H. Annu. Rev. Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- Nataraj V. Varela C. Javid A. Singh A. Besra G. S. Bhatt A. Mol. Microbiol. 2015;98:7–16. doi: 10.1111/mmi.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K. Wang C. Besra G. S. Clin. Microbiol. Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida M. V. Saraiva M. F. de Souza M. V. da Costa C. F. Vicente F. R. Lourenco M. C. Bioorg. Med. Chem. Lett. 2007;17:5661–5664. doi: 10.1016/j.bmcl.2007.07.073. [DOI] [PubMed] [Google Scholar]
- Hu Y. Q. Zhang S. Zhao F. Gao C. Feng L. S. Lv Z. S. Xu Z. Wu X. Eur. J. Med. Chem. 2017;133:255–267. doi: 10.1016/j.ejmech.2017.04.002. [DOI] [PubMed] [Google Scholar]
- Palmer B. D. Thompson A. M. Sutherland H. S. Blaser A. Kmentova I. Franzblau S. G. Wan B. Wang Y. Ma Z. Denny W. A. J. Med. Chem. 2010;53:282–294. doi: 10.1021/jm901207n. [DOI] [PubMed] [Google Scholar]
- Stec J. Onajole O. K. Lun S. Guo H. Merenbloom B. Vistoli G. Bishai W. R. Kozikowski A. P. J. Med. Chem. 2016;59:6232–6247. doi: 10.1021/acs.jmedchem.6b00415. [DOI] [PubMed] [Google Scholar]
- Tong A. S. T. Choi P. J. Blaser A. Sutherland H. S. Tsang S. K. Y. Guillemont J. Motte M. Cooper C. B. Andries K. Van den Broeck W. Franzblau S. G. Upton A. M. Denny W. A. Palmer B. D. Conole D. ACS Med. Chem. Lett. 2017;8:1019–1024. doi: 10.1021/acsmedchemlett.7b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uh E. Jackson E. R. San Jose G. Maddox M. Lee R. E. Lee R. E. Boshoff H. I. Dowd C. S. Bioorg. Med. Chem. Lett. 2011;21:6973–6976. doi: 10.1016/j.bmcl.2011.09.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzegorzewicz A. E. Pham H. Gundi V. A. Scherman M. S. North E. J. Hess T. Jones V. Gruppo V. Born S. E. Kordulakova J. Chavadi S. S. Morisseau C. Lenaerts A. J. Lee R. E. McNeil M. R. Jackson M. Nat. Chem. Biol. 2012;8:334–341. doi: 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahlan K. Wilson R. Kastrinsky D. B. Arora K. Nair V. Fischer E. Barnes S. W. Walker J. R. Alland D. Barry 3rd C. E. Boshoff H. I. Antimicrob. Agents Chemother. 2012;56:1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela C. Rittmann D. Singh A. Krumbach K. Bhatt K. Eggeling L. Besra G. S. Bhatt A. Chem. Biol. 2012;19:498–506. doi: 10.1016/j.chembiol.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degiacomi G. Benjak A. Madacki J. Boldrin F. Provvedi R. Palu G. Kordulakova J. Cole S. T. Manganelli R. Sci. Rep. 2017;7:43495. doi: 10.1038/srep43495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Obregon-Henao A. Wallach J. B. North E. J. Lee R. E. Gonzalez-Juarrero M. Schnappinger D. Jackson M. Antimicrob. Agents Chemother. 2016;60:5198–5207. doi: 10.1128/AAC.00826-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M. McNeil M. R. Brennan P. J. Future Microbiol. 2013;8:855–875. doi: 10.2217/fmb.13.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North E. J. Jackson M. Lee R. E. Curr. Pharm. Des. 2014;20:4357–4378. doi: 10.2174/1381612819666131118203641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondreddi R. R. Jiricek J. Rao S. P. Lakshminarayana S. B. Camacho L. R. Rao R. Herve M. Bifani P. Ma N. L. Kuhen K. Goh A. Chatterjee A. K. Dick T. Diagana T. T. Manjunatha U. H. Smith P. W. J. Med. Chem. 2013;56:8849–8859. doi: 10.1021/jm4012774. [DOI] [PubMed] [Google Scholar]
- Onajole O. K. Pieroni M. Tipparaju S. K. Lun S. Stec J. Chen G. Gunosewoyo H. Guo H. Ammerman N. C. Bishai W. R. Kozikowski A. P. J. Med. Chem. 2013;56:4093–4103. doi: 10.1021/jm4003878. [DOI] [PubMed] [Google Scholar]
- Rao S. P. Lakshminarayana S. B. Kondreddi R. R. Herve M. Camacho L. R. Bifani P. Kalapala S. K. Jiricek J. Ma N. L. Tan B. H. Ng S. H. Nanjundappa M. Ravindran S. Seah P. G. Thayalan P. Lim S. H. Lee B. H. Goh A. Barnes W. S. Chen Z. Gagaring K. Chatterjee A. K. Pethe K. Kuhen K. Walker J. Feng G. Babu S. Zhang L. Blasco F. Beer D. Weaver M. Dartois V. Glynne R. Dick T. Smith P. W. Diagana T. T. Manjunatha U. H. Sci. Transl. Med. 2013;5:214ra168. doi: 10.1126/scitranslmed.3007355. [DOI] [PubMed] [Google Scholar]
- Lun S. Guo H. Onajole O. K. Pieroni M. Gunosewoyo H. Chen G. Tipparaju S. K. Ammerman N. C. Kozikowski A. P. Bishai W. R. Nat. Commun. 2013;4:2907. doi: 10.1038/ncomms3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. A. Grant S. S. Kawate T. Iwase N. Shimizu M. Wivagg C. Silvis M. Kazyanskaya E. Aquadro J. Golas A. Fitzgerald M. Dai H. Zhang L. Hung D. T. ACS Chem. Biol. 2012;7:1377–1384. doi: 10.1021/cb300151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacksteder K. A. Protopopova M. Barry 3rd C. E. Andries K. Nacy C. A. Future Microbiol. 2012;7:823–837. doi: 10.2217/fmb.12.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa V. Poce G. Canseco J. O. Buroni S. Pasca M. R. Biava M. Raju R. M. Porretta G. C. Alfonso S. Battilocchio C. Javid B. Sorrentino F. Ioerger T. R. Sacchettini J. C. Manetti F. Botta M. De Logu A. Rubin E. J. De Rossi E. Antimicrob. Agents Chemother. 2012;56:324–331. doi: 10.1128/AAC.05270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groote M. A. Jackson M. Gonzalez-Juarrero M. Li W. Young C. L. Wong C. Graham J. Day J. Hoang T. Jarvis T. C. Ribble W. Sun X. Ochsner U. A. Front. Microbiol. 2018;9:2231. doi: 10.3389/fmicb.2018.02231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poce G. Cocozza M. Alfonso S. Consalvi S. Venditti G. Fernandez-Menendez R. Bates R. H. Barros Aguirre D. Ballell L. De Logu A. Vistoli G. Biava M. Eur. J. Med. Chem. 2018;145:539–550. doi: 10.1016/j.ejmech.2017.12.075. [DOI] [PubMed] [Google Scholar]
- Remuinan M. J. Perez-Herran E. Rullas J. Alemparte C. Martinez-Hoyos M. Dow D. J. Afari J. Mehta N. Esquivias J. Jimenez E. Ortega-Muro F. Fraile-Gabaldon M. T. Spivey V. L. Loman N. J. Pallen M. J. Constantinidou C. Minick D. J. Cacho M. Rebollo-Lopez M. J. Gonzalez C. Sousa V. Angulo-Barturen I. Mendoza-Losana A. Barros D. Besra G. S. Ballell L. Cammack N. PLoS One. 2013;8:e60933. doi: 10.1371/journal.pone.0060933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballell L. Bates R. H. Young R. J. Alvarez-Gomez D. Alvarez-Ruiz E. Barroso V. Blanco D. Crespo B. Escribano J. Gonzalez R. Lozano S. Huss S. Santos-Villarejo A. Martin-Plaza J. J. Mendoza A. Rebollo-Lopez M. J. Remuinan-Blanco M. Lavandera J. L. Perez-Herran E. Gamo-Benito F. J. Garcia-Bustos J. F. Barros D. Castro J. P. Cammack N. ChemMedChem. 2013;8:313–321. doi: 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioerger T. R. O'Malley T. Liao R. Guinn K. M. Hickey M. J. Mohaideen N. Murphy K. C. Boshoff H. I. Mizrahi V. Rubin E. J. Sassetti C. M. Barry 3rd C. E. Sherman D. R. Parish T. Sacchettini J. C. PLoS One. 2013;8:e75245. doi: 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli J. M. Yazidi A. Yang L. Fabre L. Li W. Jacques B. Angala S. K. Rouiller I. Zgurskaya H. I. Sygusch J. Jackson M. ACS Infect. Dis. 2016;2:702–713. doi: 10.1021/acsinfecdis.6b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Stevens C. M. Pandya A. N. Darzynkiewicz Z. Bhattarai P. Tong W. Gonzalez-Juarrero M. North E. J. Zgurskaya H. I. Jackson M. ACS Infect. Dis. 2019;5:1001–1012. doi: 10.1021/acsinfecdis.9b00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieroni M. Tipparaju S. K. Lun S. Song Y. Sturm A. W. Bishai W. R. Kozikowski A. P. ChemMedChem. 2011;6:334–342. doi: 10.1002/cmdc.201000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C. C. Klenotic P. A. Bolla J. R. Purdy G. E. Robinson C. V. Yu E. W. Proc. Natl. Acad. Sci. U. S. A. 2019;116:11241–11246. doi: 10.1073/pnas.1901346116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B. Li J. Yang X. Wu L. Zhang J. Yang Y. Zhao Y. Zhang L. Yang X. Yang X. Cheng X. Liu Z. Jiang B. Jiang H. Guddat L. W. Yang H. Rao Z. Cell. 2019;176:636–648. doi: 10.1016/j.cell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- Bernut A. Viljoen A. Dupont C. Sapriel G. Blaise M. Bouchier C. Brosch R. de Chastellier C. Herrmann J. L. Kremer L. Mol. Microbiol. 2016;99:866–883. doi: 10.1111/mmi.13283. [DOI] [PubMed] [Google Scholar]
- Ancizu S. Castrillo N. Pérez-Silanes S. Aldana I. Monge A. Delagrange P. Caignard D.-H. Galiano S. Molecules. 2012;17(7):7737–7757. doi: 10.3390/molecules17077737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. Guo J.-H. Fu H.-M. Yuan M.-L. Yang L.-J. Tetrahedron Lett. 2015;56:3777–3781. doi: 10.1016/j.tetlet.2015.04.060. [DOI] [Google Scholar]
- Kurasawa Y. Yoshida K. Yamazaki N. Iwamoto K. Hamamoto Y. Kaji E. Sasaki K. Zamami Y. J. Heterocycl. Chem. 2012;49:1323–1331. doi: 10.1002/jhet.922. [DOI] [Google Scholar]
- Stern E. Muccioli G. G. Millet R. Goossens J. F. Farce A. Chavatte P. Poupaert J. H. Lambert D. M. Depreux P. Henichart J. P. J. Med. Chem. 2006;49:70–79. doi: 10.1021/jm050467q. [DOI] [PubMed] [Google Scholar]
- Collins L. Franzblau S. G. Antimicrob. Agents Chemother. 1997;41:1004–1009. doi: 10.1128/AAC.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonec T. Bobal P. Sujan J. Pesko M. Guo J. Kralova K. Pavlacka L. Vesely L. Kreckova E. Kos J. Coffey A. Kollar P. Imramovsky A. Placek L. Jampilek J. Molecules. 2012;17:613–644. doi: 10.3390/molecules17010613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOE, Chemical Computing Group, lnc., Montreal, http://www.chemcomp.com [Google Scholar]
- Lipinski C. A. Lombardo F. Dominy B. W. Feeney P. J. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Drug Discovery Today: Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- McKerrow J. H. Lipinski C. A. Int. J. Parasitol.: Drugs Drug Resist. 2017;7:248–249. doi: 10.1016/j.ijpddr.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K. Yokomizo A. Naito H. Haginoya N. Kobayashi S. Yoshino T. Nagata T. Mochizuki A. Osanai K. Watanabe K. Kanno H. Ohta T. Bioorg. Med. Chem. 2009;17:8206–8220. doi: 10.1016/j.bmc.2009.10.023. [DOI] [PubMed] [Google Scholar]
- Zewge D. Chen C. Y. Deer C. Dormer P. G. Hughes D. L. J. Org. Chem. 2007;72:4276–4279. doi: 10.1021/jo070181o. [DOI] [PubMed] [Google Scholar]
- López Rivilli M. J. Moyano E. L. Yranzo G. I. Tetrahedron Lett. 2010;51:478–481. doi: 10.1016/j.tetlet.2009.10.123. [DOI] [Google Scholar]
- Podányi B. Keresztúri G. Vasvári-Debreczy L. Hermecz I. Tóth G. Magn. Reson. Chem. 1996;34:972–978. doi: 10.1002/(SICI)1097-458X(199611)34:11<972::AID-OMR994>3.0.CO;2-9. [DOI] [Google Scholar]
- Stern E. Muccioli G. G. Bosier B. Hamtiaux L. Millet R. Poupaert J. H. Henichart J. P. Depreux P. Goossens J. F. Lambert D. M. J. Med. Chem. 2007;50:5471–5484. doi: 10.1021/jm070387h. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Xiao X. Su T. Wu J. Ren J. Zhu J. Zhang X. Cao R. Du R. Eur. J. Med. Chem. 2017;140:239–251. doi: 10.1016/j.ejmech.2017.09.017. [DOI] [PubMed] [Google Scholar]
- Vanejevs M. Jatzke C. Renner S. Muller S. Hechenberger M. Bauer T. Klochkova A. Pyatkin I. Kazyulkin D. Aksenova E. Shulepin S. Timonina O. Haasis A. Gutcaits A. Parsons C. G. Kauss V. Weil T. J. Med. Chem. 2008;51:634–647. doi: 10.1021/jm0611298. [DOI] [PubMed] [Google Scholar]
- Vogt D. Weber J. Ihlefeld K. Bruggerhoff A. Proschak E. Stark H. Bioorg. Med. Chem. 2014;22:5354–5367. doi: 10.1016/j.bmc.2014.07.044. [DOI] [PubMed] [Google Scholar]
- Hanke T. Dehm F. Liening S. Popella S. D. Maczewsky J. Pillong M. Kunze J. Weinigel C. Barz D. Kaiser A. Wurglics M. Lammerhofer M. Schneider G. Sautebin L. Schubert-Zsilavecz M. Werz O. J. Med. Chem. 2013;56:9031–9044. doi: 10.1021/jm401557w. [DOI] [PubMed] [Google Scholar]
- Kouatly O. Geronikaki A. Kamoutsis C. Hadjipavlou-Litina D. Eleftheriou P. Eur. J. Med. Chem. 2009;44:1198–1204. doi: 10.1016/j.ejmech.2008.05.029. [DOI] [PubMed] [Google Scholar]