

SUMMARY
The developmental mechanisms that orchestrate differentiation of specific nephron segments are incompletely understood and the factors that maintain their terminal differentiation after nephrogenesis remain largely unknown. Here, the transcription factor AP-2β is shown to be required for the differentiation of distal tubule precursors into early-stage distal convoluted tubules (DCTs) during nephrogenesis. In contrast, its downstream target KCTD1 is essential for terminal differentiation of early-stage DCTs into mature DCTs, and impairment of their terminal differentiation due to lack of KCTD1 leads to a severe salt-losing tubulopathy. Moreover, sustained KCTD1 activity in the adult maintains mature DCTs in this terminally differentiated state and prevents renal fibrosis by repressing β-catenin activity, whereas KCTD1 deficiency leads to severe renal fibrosis. Thus, the AP-2β/KCTD1 axis links a developmental pathway in the nephron to the induction and maintenance of terminal differentiation of DCTs that actively prevents their dedifferentiation in the adult and protects against renal fibrosis.
Keywords: AP-2β, KCTD1, distal nephron, distal convoluted tubule, terminal differentiation, salt-losing tubulopathy, renal fibrosis, β-catenin
eTOC blurb
Marneros shows that the transcription factor AP-2β induces differentiation of distal tubule precursors into early-stage distal convoluted tubules (DCTs), whereas its downstream target KCTD1 is required for their terminal differentiation into mature DCTs. KCTD1 loss causes immature DCTs, leading to a salt-losing tubulopathy followed by renal fibrosis via β-catenin hyperactivation.
Graphical Abstract
INTRODUCTION
Genetic studies in mice and single-cell RNA-Seq data of embryonic kidneys have shown that during nephrogenesis nephron progenitor cells (NPCs) differentiate in a highly orchestrated manner into distinct nephrogenic lineages (podocyte and renal corpuscle precursors, proximal tubule precursors, Loop of Henle precursors and distal tubule precursors) that form subsequently specific segments of the nephron (Lindstrom et al., 2018a; Lindstrom et al., 2018b; McMahon, 2016). A time-dependent cell fate acquisition model of nephron patterning has been proposed, in which proximal-distal cell fate acquisition of NPCs occurs in a progressive manner depending on the time of their recruitment (Lindstrom et al., 2018a). Specification of proximal-distal fates during nephron patterning involves specific signaling pathways (Wnt, Bmp, Notch, Fgf signaling) and transcription factors (including Pou3f3, Lhx1, Irx2, Hnf1b, Foxc2, Mafb) (Cheng et al., 2007; Grieshammer et al., 2005; Heliot et al., 2013; Kobayashi et al., 2005; Lindstrom et al., 2015; Moriguchi et al., 2006; Nakai et al., 2003; Reggiani et al., 2007; Takemoto et al., 2006). Thus, specific nephron segments in the adult kidney can be traced to particular precursor populations in the embryonic kidney. However, it remains largely unknown which specific transcription factors or transcriptional regulators act as essential switches required for the formation of nephron segments from their specified precursor populations. Moreover, a critical unanswered question is whether these differentiation programs are inactivated once nephron segments are formed or whether their activity is sustained after nephrogenesis to maintain the terminal differentiation state. If the latter were the case, it would suggest that loss of such a differentiation program in the adult may lead to de-differentiation of specific nephron segments and impairment of their function. For example, the distal convoluted tubule (DCT), a distal nephron segment with important roles for salt reabsorption and urinary concentration, is derived from distal tubule precursors, but it is not known through which molecular mechanisms its differentiation is induced during nephrogenesis and whether these mechanisms actively maintain its terminal differentiation state in the adult.
Here, we show that the transcription factor AP-2β is essential for differentiation of distal tubule precursors into early-stage DCTs during nephrogenesis. Moreover, we show that AP-2β induces expression of the transcriptional regulator KCTD1, which we find to be required for the subsequent terminal differentiation of early-stage DCTs into mature DCTs. Impaired terminal differentiation of DCTs in KCTD1 mutants causes a severe salt-losing tubulopathy with a diminished ability to concentrate urine. Importantly, we demonstrate that the AP-2β/KCTD1 axis is not only essential for the induction of differentiation processes of DCTs, but that sustained AP-2β/KCTD1 activity is required to maintain terminal differentiation of mature DCTs. Inducible loss of AP-2β or KCTD1 in the adult leads to partial de-differentiation of DCTs and a salt-losing tubulopathy. The impaired terminal differentiation of DCTs due to lack of KCTD1 is associated with sustained hyperactivation of canonical β-catenin signaling that promotes progressive renal fibrosis and chronic kidney disease (CKD)-like pathologies, and attenuating this increase in β-catenin activity can inhibit renal fibrosis.
RESULTS
The transcription factor AP-2β is required for the formation of early-stage DCTs from distal tubule precursors
We hypothesized that the formation of specific distal nephron segments from distal tubule precursors during nephrogenesis is orchestrated by key transcription factors expressed mainly in distal tubule precursors but not in other cells of the developing nephron. We analyzed single-cell RNA-Seq data from embryonic and adult kidneys and found the transcription factor AP-2β to be predominantly expressed in distal tubule precursors and its derivatives (Lindstrom et al., 2018a; Park et al., 2018; Ransick et al., 2019), which we confirmed in immunolabeling experiments (Figs. 1A, 1B and 1G). Single-cell RNA-Seq data showed overlapping distal nephron expression of AP-2β and the transcriptional regulator KCTD1, and we observed co-localization of KCTD1 and AP-2β in distal nephron epithelial nuclei (Figs. 1B and 1C). Previous in vitro data showed that AP-2β interacts with KCTD1 (Ding et al., 2009). We generated KCTD1 reporter mice and confirmed that KCTD1 is expressed specifically in distal nephron segments (thick ascending limbs of Henle [TALs], DCTs, connecting tubules [CTs] and collecting ducts [CDs]) (Fig. 1C; Fig. S1A–C). Moreover, we found that AP-2β induces the expression of KCTD1 in kidney cells in vitro as well as in kidneys in vivo, where inducible inactivation of AP-2β resulted in reduced renal expression of KCTD1 (Fig. 1D).
Figure 1: AP-2β is required for the formation of early-stage DCTs from distal tubule precursors.

A. Immunolabeling for AP-2β in human kidneys shows that it is expressed in distal nephron epithelial cells, including THP+ TALs (yellow arrows) and AQP2+ CTs/CDs (white arrows). Scale bars, 100 μm.
B. KCTD1 (red) co-localizes with AP-2β (green) in nuclei of distal nephron epithelial cells (PNA+) in human kidneys. Scale bar, 50 μm.
C. Left: KCTD1 is expressed in NCC+ DCTs (white arrow, adult KCTD1−/+ mouse kidney), NKCC2+ TALs (white arrow, adult KCTD1−/+ mouse kidney) and in Aqp2+ principle cells of CTs/CDs (green arrows, adult KCTD1−/− mouse kidney), whereas little expression of KCTD1 is detected in Aqp2− intercalated cells of CTs/CDs (blue arrows). β-galactosidase immunolabeling identifies cells that would normally express KCTD1. Red: β-gal+ tubules; green: NCC, NKCC2 or Aqp2; phalloidin in white; DAPI in blue. Scale bars, 20 μm.
Right: KCTD1 (green, arrows) shows nuclear localization in NKCC2+ TALs and NCC+ DCTs in adult human kidneys. Scale bars, 20 μm.
D. AP-2β acts as an inducer of KCTD1 expression. Top: KCTD1 promoter-driven luciferase activity is increased when AP-2β is overexpressed (pAP2β compared to pCMV control vector group; n=6/group) but decreased with knockdown of AP-2β (siAP2β compared to scrambled siRNA control [siCTRL]; n=6/group) (HEK293 cells; luciferase normalized to secreted alkaline phosphatase [SEAP]). Bottom: SiRNA-mediated knockdown of AP-2β reduces KCTD1 expression (HEK293 cells; semiquantitative RT-PCR normalized to housekeeping gene 36b4; n=6/group). Induced inactivation of AP-2β in adult mice results in reduced renal KCTD1 expression (assessed at 4-months of age in β-actinCreERT2+TFAP2Bfl/fl mice treated with TAM at 6-weeks of age; n=5–6/group).
E. Left: Reduced kidney size with distal nephron dilatation is seen in P0 Six2Cre+TFAP2Bfl/fl mice compared to WT littermate controls. Scale bar, 500 μm. Middle: magnification from kidneys shown left: arrowheads point to dilated distal nephron epithelium in a kidney from a P0 Six2Cre+TFAP2Bfl/fl mouse. Scale bar, 200 μm. Right: Nephrogenic zone (red double arrows) without major abnormalities in kidney of a P0 Six2Cre+TFAP2Bfl/fl mouse. Scale bar, 200 μm. H&E staining.
F. Reduced kidney weight (KW) to body weight (BW) ratio in P0 Six2Cre+TFAP2Bfl/fl mice compared to littermate controls.
G. Characterization of kidneys of P0 Six2Cre+TFAP2Bfl/fl mice compared to littermate controls by immunolabelings for markers of specific nephron segments. Kidneys of P0 Six2Cre+TFAP2Bfl/fl mice show a complete absence of Pvalb protein (weak green signal is autofluorescence of a large vessel) and an almost complete absence of NCC (only rare dilated NCC+ tubules are seen [red arrow]), consistent with a lack of early-stage DCTs, whereas kidneys of P0 controls show regular NCC+Pvalb+ early-stage DCTs (yellow arrowheads). Pou3f3+ distal tubule precursors are observed in kidneys of P0 Six2Cre+TFAP2Bfl/fl mice as seen in controls (white arrowheads), in which a similar expression pattern is detected for AP-2β (red arrowhead). TALs (THP+NKCC2+) form but show irregular dilatation (green arrowheads). CDs (Aqp2+) (orange arrowheads) and PTs (LTL+Slc3a1+) (white arrows) show a normal morphology in kidneys of P0 Six2Cre+TFAP2Bfl/fl mice. No significant abnormalities are observed in the distribution of Six2+Cited1+ NPCs (yellow arrows) in the nephrogenic zone of kidneys of P0 Six2Cre+TFAP2Bfl/fl mice. Scale bars, 50 μm.
H. Western blot of whole kidney lysates of a P0 Six2Cre+TFAP2Bfl/fl mouse and a control littermate shows normal protein levels of the distal tubule precursor marker Pou3f3 but absence of the DCT marker Pvalb in the P0 Six2Cre+TFAP2Bfl/fl mouse kidney.
I. Mice with inactivation of AP-2β in DCTs (PvalbCre+TFA2Bfl/fl mice) form Pvalb+ DCTs (white arrowheads). Rhodamine-conjugated peanut agglutinin (PNA; red) labels distal nephron epithelial cells, whereas contiguous red labeling is seen in PTs (seen at this magnification here). Scale bars, 50 μm.
Graphs represent data as mean ± SEM. Semiquantitative RT-PCRs performed in triplicate. P-values are shown (two-tailed, unpaired t-test).
These findings suggest that AP-2β and KCTD1 are candidate genes to control the differentiation and function of specific distal nephron segments. Indeed, AP-2β null mice show abnormal distal nephron dilatation at birth, and Finlay-Marks syndrome patients with mutations in KCTD1 often have small kidneys and develop progressive kidney abnormalities (Marneros et al., 2013; Moser et al., 2003; Moser et al., 1997; Naik et al., 2012; Park et al., 2018; Plessis et al., 1997; Steinberg et al., 1990; Wang et al., 2018). However, a detailed characterization of the kidneys of AP-2β null mice has not been performed previously and the role of AP-2β during nephrogenesis remains unknown. Similarly, the absence of KCTD1 null mice has precluded a functional analysis of KCTD1 in the kidney.
To test the roles of AP-2β and KCTD1 for kidney development and renal function we inactivated their genes in NPCs by generating Six2Cre+TFAP2Bfl/fl mice and Six2Cre+KCTD1fl/fl mice (Six2Cre+ mice induce Cre-mediated excision of floxed alleles in NPCs, thereby targeting the entire nephron except the CDs (Kobayashi et al., 2008); we confirmed this Cre activity in Six2Cre+(ROSA)26Sortm3(CAG-EYFP) reporter mice), and also established KCTD1−/− mice (Figs. S1 and S2).
Six2Cre+TFAP2Bfl/fl mice had reduced kidney size and died shortly after birth, whereas targeting AP-2β in CTs/CDs in Aqp2Cre+TFAP2Bfl/fl mice resulted in viable mice with no major kidney abnormalities (Aqp2Cre+ mice target principle cells of CTs/CDs and both AP-2β and KCTD1 are detected in CTs/CDs mainly in principle cells (Ransick et al., 2019)) (Figs. 1C, 1E and 1F; Fig. S1F and S3A). Analysis of kidneys of P0 Six2Cre+TFAP2Bfl/fl mice showed absence of early-stage DCTs despite the formation of distal tubule precursors (Pou3f3+): immunolabeling showed no NCC+Pvalb+ early-stage DCTs in these kidneys (Fig. 1G). Inactivation of AP-2β did not impair early stages of nephrogenesis, as Six2+Cited1+ NPCs were seen, as were pretubular aggregates, renal vesicles, comma-shaped bodies and S-shaped bodies (Figs. 1E and 1G). Moreover, glomeruli, proximal tubules (PTs) (LTL+Slc3a1+) and CTs/CDs (Aqp2+) formed normally (Figs. 1E and 1G). TALs (NKCC2+THP+) formed but showed irregular dilatation (Fig. 1G). Western blotting confirmed absence of the DCT differentiation marker parvalbumin (Pvalb) in kidney lysates of P0 Six2Cre+TFAP2Bfl/fl mice, whereas the distal tubule precursor marker Pou3f3 was not diminished (Fig. 1H). These data show that in the absence of AP-2β distal tubule precursors can form but do not develop into early-stage DCTs.
This conclusion is supported by our finding that inactivation of AP-2β in early-stage DCTs (in PvalbCre+TFAP2Bfl/fl mice) does not prevent their proper formation (Fig. 1I), whereas inactivation of AP-2β in distal tubule precursors (in Six2Cre+TFAP2Bfl/fl mice) prevents formation of early-stage DCTs. We confirmed in PvalbCre+(ROSA)26Sortm3(CAG-EYFP) reporter mice that they efficiently target floxed alleles only in DCT1s (Grimm et al., 2017), consistent with Pvalb being a late-stage DCT marker that is expressed only in proximal DCTs [DCT1s] but not in distal tubule precursors (Fig. S1F). Collectively, our data demonstrate that AP-2β activity in distal tubule precursors is essential for the formation of early-stage DCTs during nephrogenesis.
KCTD1 is required for terminal differentiation of early-stage DCTs into mature DCTs
A marker of DCT terminal differentiation is the growth factor EGF, which is expressed specifically in TALs/DCTs after the onset of terminal differentiation after nephrogenesis (after ~P3) (Lindstrom et al., 2018a; Park et al., 2018; Ransick et al., 2019; Salido et al., 1990) (Figs. 2A and 2B). Thus, EGF expression distinguishes early-stage DCTs from DCTs undergoing terminal differentiation during the postnatal maturation phase.
Figure 2: KCTD1 induces terminal differentiation of early-stage DCTs into mature DCTs.

A. Semiquantitative RT-PCRs of whole WT kidneys show that EGF expression is detected at P8 (ongoing terminal differentiation phase) but is absent at P3 (late nephrogenesis phase) and has high expression levels in the adult. In contrast, no EGF expression is detected at P8 in kidneys of Six2Cre+KCTD1fl/fl mice and is hardly detectable in adult kidneys of KCTD1−/− or Six2Cre+KCTD1fl/fl mice. N.D. = not detected.
B. Kidneys of 3.5-week-old Six2Cre+KCTD1fl/fl mice show no EGF and reduced immunolabeling for NCC and NKCC2 compared to WT control littermates in TALs/DCTs (EGF: green; NCC, NKCC2: red). Green and red channels of the same image are shown of co-immunolabeling for EGF and NKCC2 or NCC demonstrating that EGF expression normally occurs in either NCC+ DCTs or NKCC2+ TALs in WT kidneys (arrows). EGF is not detected in other nephron segments. Scale bars, 40 μm.
C. Dilated Pvalb+ DCT1s in 3.5-week-old Six2Cre+KCTD1fl/fl mice (white arrows) but not in WT littermates (yellow arrows), whereas DCT1s are not yet dilated in P8 Six2Cre+KCTD1fl/fl mice. Immunolabeling for the endothelial cell marker CD31 shows no major difference in kidney vascularization between Six2Cre+KCTD1fl/fl mice and littermate controls. Scale bars, 50 μm.
D. Young adult EGF KO mice do not phenocopy the kidney abnormalities seen in mice lacking KCTD1. Western blot of whole kidney lysates of 3-months-old EGF KOs versus control littermates shows normal NCC, Pvalb or NKCC2 protein levels. Six-week-old EGF KO mice show no increase in BUN levels and normal kidney histology. Scale bars, 100 μm.
E. Left: No tubule dilatation or blebbing are detected in the kidneys of P7 Six2Cre+KCTD1fl/fl mice (Toluidine blue staining of 1μm plastic sections). Scale bars, 20 μm. Right: At P17 distal tubules of Six2Cre+KCTD1fl/fl mice show dilatation and epithelial blebbing (black arrow) from apical sites of epithelial cells (red arrow). PTs appear normal (yellow arrow). Scale bars, 10 μm.
F. Kidneys of P7 Six2Cre+KCTD1fl/fl mice and littermate controls (KCTD1fl/fl mice) show no apparent anatomical or histological differences with formation of proper distal nephron segments (TAL marker NKCC2 in green; PNA, red). Scale bars, 500 μm.
G. Serum creatinine levels (normalized to BW) are increased in P8 Six2Cre+KCTD1fl/fl mice compared to littermate controls. KCTD1-deficient kidneys show a disproportionate postnatal growth retardation with a decreasing KW/BW ratio with age progression. Adult Six2Cre+KCTD1fl/fl mice have smaller kidneys than their WT littermates (2-months-old kidneys).
H. Kidneys of 3.5-week-old Six2Cre+KCTD1fl/fl mice show basophilic primitive distal nephron tubules (black arrows) in the renal cortex and dilated DCTs with epithelial blebbing (red arrow), which are not seen in kidneys of control littermates (WT). Scale bars, 20 μm.
I. Left: In 4-months-old KCTD1−/− mice dilated TALs/DCTs with epithelial blebs are seen (white arrow), while primitive basophilic tubules persist (black arrow), some showing dilatation (red arrow). A tubulointerstitial inflammatory infiltrate is seen (green arrow). Right: Lack of KCTD1 leads to dilated distal nephron epithelial tubules (PNA+, red epithelial cells; yellow arrowheads) that would normally express KCTD1 (β-gal+ in KCTD1−/− mice, green). Epithelial blebs are derived from distal nephron epithelium that lacks KCTD1 (show green color of β-gal+-derived cells) (white arrow). PTs show normal morphology (yellow arrow). Scale bars, 50 μm.
J. Labeling of KCTD1−/− mouse kidneys. Mice contain a lacZ cassette in the endogenous KCTD1 locus, so β-galactosidase activity represents cells that would normally express KCTD1, while antibodies or lectins label specific nephron segments. KCTD1 expression was observed in the TALs, DCTs, AQP2+ cells of the CTs and CDs, but not in the PTs, glomeruli or non-epithelial cells of the kidney.
DBA (dolichos biflorus agglutinin) lectin marks CTs/CDs (red, arrow). PNA (peanut agglutinin) lectin marks distal nephron epithelium (TALs, DCTs and CTs/CDs [red, arrow]; contiguous red labeling seen in PTs). LTL (lotus tetragonolobus) lectin marks PTs (green). Anti-Aqp2 immunolabeling marks Aqp2+ cells of CTs and CDs (green). Anti-Tamm-Horsefall protein (THP; green, arrow) immunolabeling marks the TAL. 9-months-old KCTD1−/− mice. Scale bars, 40 μm.
Graphs represent data as mean ± SEM. Semiquantitative RT-PCRs performed in triplicate with n>6 samples/group. P-values are shown (two-tailed, unpaired t-test).
See also Figures S1–S3.
In contrast to the lack of early-stage DCTs in Six2Cre+TFAP2Bfl/fl mice, Six2Cre+KCTD1fl/fl mice or KCTD1−/− mice formed early-stage DCTs but demonstrated a block in their terminal differentiation into mature DCTs and no EGF-expressing DCTs were detected in their kidneys (Figs. 2A–2C). Notably, loss of EGF serves as a marker of impaired terminal differentiation of DCTs but its deficiency is not the cause of this differentiation defect or of the functional kidney abnormalities in mice lacking KCTD1, as adult EGF−/− mice showed normal terminal differentiation of DCTs and did not phenocopy the renal abnormalities seen in mice lacking KCTD1 (Figs. 2D and S3B). Moreover, administration of EGF to Six2Cre+KCTD1fl/fl mice during the terminal differentiation phase following nephrogenesis did not rescue the renal phenotype in these mice (Fig. S3C).
Consistent with the presence of early-stage DCTs in Six2Cre+KCTD1fl/fl mice or KCTD1−/− mice, lack of KCTD1 did not result in the early postnatal lethality seen in Six2Cre+TFAP2Bfl/fl mice. Instead, Six2Cre+KCTD1fl/fl mice or KCTD1−/− mice manifested postnatal abnormalities in the DCTs with progressive age-dependent dilatation and epithelial blebbing, which were seen after the onset of the phase when postnatal terminal differentiation normally occurs (Figs. 2C, 2E–2J). Persistence of immature distal nephron tubules was also observed (Figs. 2H and 2I). These progressive terminal differentiation defects of DCTs were associated with morphological TAL abnormalities, whereas glomeruli, PTs or CDs showed no major defects in adult Six2Cre+KCTD1fl/fl mice or KCTD1−/− mice (Figs. 2J and 3A). As with AP-2β, loss of KCTD1 in CTs/CDs (in Aqp2Cre+KCTD1fl/fl mice) did not result in kidney abnormalities, revealing a critical function of KCTD1 that is limited to TALs/DCTs (Fig. S3D; Tables S1 and S2). The observed TAL/DCT defects progressed with age and resulted in large cystic abnormal TALs/DCTs in aged mice lacking KCTD1 (Figs. 2J and 3A).
Figure 3: KCTD1 deficiency impairs terminal differentiation of DCTs and leads to an age-dependent increase in DCT abnormalities and a salt-losing tubulopathy.

A. Immunolabeling for DCT (Pvalb, NCC) and PT markers (LTL, Slc3a1) in kidneys of 1- and 7-month-old KCTD1−/− and WT littermate mice. DCTs show a progressive cystic dilatation with progressive age in KCTD1−/− mice (yellow arrows), whereas PTs show no morphological abnormalities (white arrows). PNA (red) labels distal nephron epithelial cells, whereas contiguous red labeling is seen in PTs (seen at this magnification here). Scale bars, 100 μm.
B. Renal transcript levels of NCC, NKCC2 and parvalbumin (Pvalb) increase with terminal differentiation and growth of distal nephrons in WT mice (P3 < P8 < 2-months-old kidneys). Their expression is reduced in P8 Six2Cre+KCTD1fl/fl kidneys and severely diminished in kidneys of 2-months-old Six2Cre+KCTD1fl/fl mice or KCTD1−/− mice compared to WT littermates. Kidneys of 8-months-old Six2Cre+KCTD1fl/fl mice have hardly detectable NCC, NKCC2 and parvalbumin transcripts.
C. Postnatal growth retardation in KCTD1−/− mice (KO) compared to WT littermates (male mice are shown). Representative images of gender- and age-matched littermates show that Six2Cre+KCTD1fl/fl mice and KCTD1−/− are smaller than their controls and that this size and weight difference is maintained with age. Left image shows an 8-week-old Six2Cre+KCTD1fl/fl mouse and a control littermate. Right image shows a 12-months-old KCTD1−/− mouse and a WT littermate.
D. Increases in BUN, water intake, and urine production are seen in 2-month-old Six2Cre+KCTD1fl/fl mice and in 4-month-old β-actinCreERT2+TFAP2Bfl/fl mice or β-actinCreERT2+KCTD1fl/fl mice treated with TAM at 6-weeks of age. Inactivation of KCTD1 at P9 results in a more severe kidney phenotype than when KCTD1 is inactivated in the adult (at 6-weeks of age).
E. Reduced urine osmolality in KCTD1−/− mice cannot be corrected by administration of the antidiuretic dDAVP, whereas it increases urine osmolality in WT littermates. The strong increase in urine osmolality that occurs in WT mice with 24-hour water deprivation is not seen in KCTD1−/− mice. This diminished ability of KCTD1−/− mice to concentrate urine results in a severe dehydration with loss of ~25% of BW after water deprivation for 24 hours. The increased loss of urine in KCTD1 mutants leads to an increase in serum renin levels and secondary hyperaldosteronism. KCTD1 deficiency in the kidney leads to a hypochloremic metabolic alkalosis (reduced serum HCO3− and Cl− and increased blood pH). Results from groups of 2months-old mice are shown.
Graphs represent data as mean ± SEM. Semiquantitative RT-PCRs performed in triplicate with n>6 samples/group. P-values are shown (two-tailed, unpaired t-test).
KCTD1 deficiency impairs terminal differentiation of DCTs and leads to an age-dependent increase in DCT abnormalities and a salt-losing tubulopathy
Consistent with the impaired terminal differentiation of DCTs lacking KCTD1, the morphological TAL/DCT defects in these mice were associated with a diminished postnatal increase in TAL/DCT-specific differentiation markers and electrolyte transporters (e.g. NCC [DCT], NKCC2 [TAL], Pvalb [DCT1]) (Fig. 3B). Their relative loss resulted in a salt-losing tubulopathy with impaired ability to concentrate urine and a postnatal growth retardation (Figs. 3C–3E; Tables S1 and S2). Six2Cre+KCTD1fl/fl mice and KCTD1−/− mice developed key clinical features observed in patients with severe salt-losing tubulopathies due to combined defects in TALs/DCTs, including a hypochloremic metabolic alkalosis with hyperaldosteronism, polydipsia and polyuria with hypoosmolar urine, which could not be corrected with administration of the antidiuretic dDAVP (Figs. 3C–3E; Tables S1 and S2). This excessive urinary water loss resulted in severe dehydration when these mice were deprived of water for 24 hours (Fig. 3E). The fractional excretion of electrolytes in the urine was increased, with high urinary sodium wasting in both Six2Cre+KCTD1fl/fl mice and KCTD1−/− mice (Table S2). These manifestations are at least in part explained by the diminished levels of the Na+-Cl− co-transporter NCC in DCTs and of the Na+-K+−2Cl− co-transporter NKCC2 in TALs of these mice, as impaired function of these transporters has been shown to result in salt-losing tubulopathies (Al Shibli and Narchi, 2015; Simon et al., 1996a; Simon et al., 1996b).
We hypothesized that the reduced ability of the kidney to reabsorb salt and concentrate urine due to the TAL/DCT defects would lead to secondary changes in the expression of genes involved in salt/water reabsorption in CTs/CDs in order to partially counter the severe water and salt loss. Indeed, expression of channels that increase sodium reabsorption in the CTs/CDs, such as SCNN1B, SCNN1G and EnAC, were increased in adult KCTD1−/− mice and Six2Cre+KCTD1fl/fl mice, but not in the early postnatal phase (P8) (Figs. S6A–C). The Cl−/HCO−3 exchanger pendrin was also increased in adult mice lacking KCTD1, which plays an important role for salt reabsorption in the setting of NCC loss (Figs. S6A–C) (Soleimani et al., 2012). Thus, NCC and NKCC2 expression was diminished already in the early postnatal period in mice lacking KCTD1, while the increased expression of sodium channels in CTs/CDs occurred subsequently, likely to partially counter the urinary concentration defect in the TALs/DCTs. However, this compensatory increase in sodium channel expression was not sufficient to prevent the severe salt-losing tubulopathy and urinary concentration defect that occur due to impaired TAL/DCT function as a consequence of KCTD1 loss.
AP-2β and KCTD1 are critical inducers of DCT differentiation markers
KCTD1’s role as an inducer of DCT terminal differentiation was confirmed by RNA-Seq of kidneys of P8 Six2Cre+KCTD1fl/fl mice, a time when nephrons are undergoing terminal differentiation: these data showed that DCT differentiation markers (e.g. NCC [Slc12a3], Pvalb and EGF) were significantly downregulated (Figs. 4A and 4B; Fig. S4). Similarly, RNA-Seq of kidneys from mice with inactivation of AP-2β in the adult showed reduced expression of KCTD1 and a downregulation of DCT signature genes (Figs. 4A and 4B; Fig. S5), which was confirmed by semiquantitative RT-PCR (Figs. 4E and S6A–C). Thus, AP-2β-mediated KCTD1 expression is critical not only for the induction of the terminal differentiation state of DCTs during the postnatal terminal differentiation phase but also for the maintenance of the terminal differentiation state in the adult (Figs. 4E and 4F).
Figure 4: AP-2β and KCTD1 are critical inducers of DCT differentiation markers.

A. Top: Volcano plot of differentially expressed genes (DEGs) in kidneys of P8 Six2Cre+KCTD1fl/fl mice versus control littermates shows that among the most significantly downregulated genes are markers of DCTs (Pvalb [DCT1], EGF [TAL/DCT] and NCC [Slc12a3, DCT]; arrows). Middle: Volcano plot of DEGs in kidneys of 4-months-old β-actinCreERT2+TFAP2Bfl/fl mice versus control littermates treated with TAM at 6 weeks of age. DCT markers are among the most significantly down-regulated DEGs (e.g. Pvalb [DCT1], NCC [DCT], EGF [TAL/DCT], SFRP1 [TAL/DCT]; arrows). Bottom: Scatter plot comparing shared DEGs of kidneys from P8 Six2Cre+KCTD1fl/fl mice (vs controls) and from 4-months-old β-actinCreERT2+TFAP2Bfl/fl mice (versus control littermates, both treated with TAM at 6 weeks of age). Both groups show a significant downregulation of DCT markers Pvalb, NCC (Slc12a3), and EGF (arrows, red dots).
B. Left: Heatmap shows shared DEGs between kidneys of mice with induced inactivation of AP-2β in the adult (4-months-old β-actinCreERT2+TFAP2Bfl/fl mice treated with TAM at 6-weeks of age) and kidneys of P8 Six2Cre+KCTD1fl/fl mice. Right: Their nephron segment- or cell type-specific expression pattern in the kidney is shown based on single-cell RNA-Seq data from adult mice (Park et al., 2018). Downregulated DEGs are mainly genes of DCTs, such as EGF, Slc12a3 (NCC) and Pvalb (black arrowheads). Top: Heatmaps also show that KCTD1 transcript levels are reduced in kidneys with induced inactivation of AP-2β (blue arrowhead), confirming our finding that AP-2β induces KCTD1 expression in the kidney (Fig. 1D), and that AP-2β and KCTD1 are predominantly expressed in the distal nephron. The increase in AP-2β transcripts in kidneys with inducible inactivation of AP-2β is likely a compensatory response, as the Cre-mediated excision of floxed alleles results in AP-2β transcripts that lack exon 6 (critical for DNA binding) and do not produce functional AP-2β (see Figure S2A). Thus, these mice lack functional AP-2β despite the increase in transcript levels of AP-2β. Values in heatmaps represent Z-scores. Endo: endothelial, vascular and descending loop of Henle; Podo: podocytes; PT: proximal tubules; LOH: loop of Henle (including TAL); DCT: distal convoluted tubule; CD_PC: collecting duct, principle cells; CD_IC: intercalated cells; CD_Trans: collecting duct, transient cells; Novel1: novel cell type 1; Fib: fibroblast; Macro: macrophage; Neutro: neutrophil; B_lymph: B lymphocyte; T_lymph: T lymphocyte; NK: natural killer cell; Novel 2: novel cell type 2.
C. Inactivating KCTD1 specifically in DCT1s (PvalbCre+KCTD1fl/fl mice) results in a decrease in renal NCC and parvalbumin transcripts and protein levels. Results in 2-months-old PvalbCre+KCTD1fl/fl mice and littermate control mice.
D. Dilated distal nephron tubules are seen in aged PvalbCre+KCTD1fl/fl mice (yellow arrows). Representative H&E images from kidneys of 14-months-old PvalbCre+KCTD1fl/fl mice and controls. Scale bars, 100 μm.
E. AP-2β and KCTD1 expression are detected only in TALs, DCTs, CTs and CDs. Targeting AP-2β proximal to the CDs in Six2Cre+TFAP2Bfl/fl mice results in lack of DCTs and abnormal TALs, whereas its inactivation in principle cells of CTs/CDs (Aqp2Cre+TFAP2Bfl/fl mice) has no significant phenotype. Inactivation of AP-2β postnatally (TAM-treated β-actinCreERT2+TFAP2Bfl/fl mice) leads also to TAL/DCT defects. The kidney abnormalities that are observed in KCTD1−/− mice are also observed in Six2Cre+KCTD1fl/fl mice (targeting KCTD1 in the nephron except the CDs), but no kidney abnormalities are observed in Aqp2Cre+KCTD1fl/fl mice (targeting KCTD1 in the CTs/CDs). Thus, lack of essential functions of AP-2β and KCTD1 in the TALs/DCTs is responsible for the observed kidney phenotypes in KCTD1−/−, Six2Cre+KCTD1fl/fl mice, Six2Cre+TFAP2Bfl/fl mice and mice with inactivation of AP-2β in the adult.
Top: Mice with inactivation of AP-2β in the adult show significant downregulation of DCT genes, whereas pendrin that is expressed in the CTs/CDs is upregulated.
Bottom: Six2Cre+KCTD1fl/fl mice show the earliest changes in expression in the DCTs (downregulation of NCC, parvalbumin, EGF and SFRP1). Subsequent upregulation of pendrin in CTs/CDs is seen.
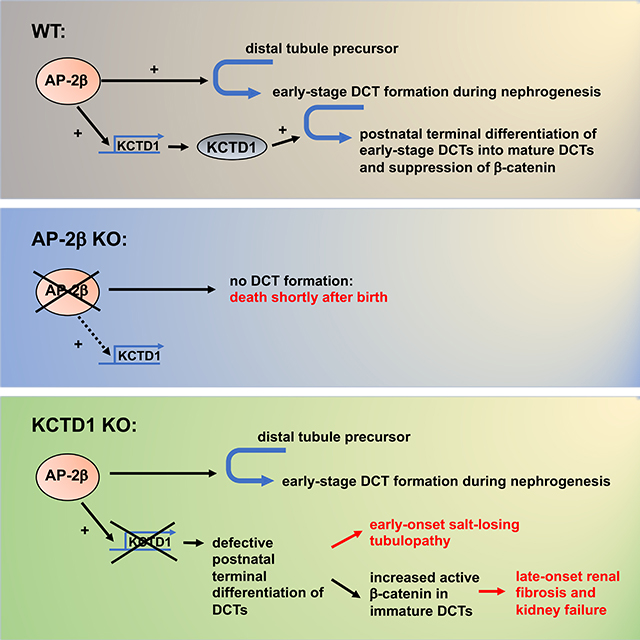
F. Proposed model of the roles of AP-2β and KCTD1 in kidney development. AP-2β induces differentiation of distal tubule precursors into early-stage DCTs during nephrogenesis, whereas KCTD1 induces terminal differentiation of these early-stage DCTs (NCC+Pvalb+EGF−) into mature DCTs (NCC+Pvalb+EGF+). PT: proximal tubule; NPC: nephron progenitor cells. NP: nephron precursor.
Graphs represent data as mean ± SEM. Semiquantitative RT-PCRs performed in triplicate. P-values are shown (two-tailed, unpaired t-test).
See also Figures S3–S6 and supplemental files.
Our model predicts that KCTD1 promotes the postnatal terminal differentiation of DCTs but is not required for transition of distal tubule precursors into early-stage DCTs during nephrogenesis. Thus, we expect that inactivation of KCTD1 specifically in early-stage DCTs after they have formed from distal tubule precursors would result in reduced terminal differentiation of DCTs but would not affect the formation of early-stage DCTs. To demonstrate this, we genetically inactivated KCTD1 in early-stage DCTs but not in distal tubule precursors (in PvalbCre+KCTD1fl/fl mice) and found a terminal differentiation defect with downregulation of DCT differentiation markers in adult mice similarly as observed in adult Six2Cre+KCTD1fl/fl mice (Figs. 4C and 4D). This observation is consistent with a key role of KCTD1 in inducing the terminal differentiation of early-stage DCTs into mature DCTs (Figs. 4E and 4F).
AP-2β and KCTD1 maintain the terminal differentiation state of DCTs in the adult
To provide further evidence that KCTD1 is critical for the induction of terminal differentiation of early-stage DCTs during the nephron maturation phase, we inactivated KCTD1 at various postnatal time-points. Loss of KCTD1 at P0 phenocopied the morphological kidney abnormalities observed in KCTD1−/− mice or Six2Cre+KCTD1fl/fl mice, confirming that the observed kidney abnormalities in mice lacking KCTD1 are mainly due to postnatal functions of KCTD1 in the distal nephron (Figs. 5A and S6E). When KCTD1 was inactivated at later stages of the nephron maturation phase (e.g. at P9 or P13) these morphological distal nephron defects where less severe, in support of the critical role of KCTD1 during early postnatal maturation of the distal nephron (Fig. 5A). However, even when KCTD1 was inactivated at P9, assessment of these mice at 6-weeks of age showed a salt-losing tubulopathy with increased BUN, polyuria, polydipsia, and hypochloremia, which was associated with attenuation of DCT differentiation markers (Figs. 3D and 5D–F). Thus, the first two weeks after birth are a critical temporal window for proper KCTD1-mediated terminal differentiation of DCTs.
Figure 5: AP-2β and KCTD1 maintain the terminal differentiation state of DCTs in the adult.

A. Induced inactivation of KCTD1 at P0 leads to severe TAL/DCT defects (green arrows) and resembles the morphological kidney abnormalities seen in Six2Cre+KCTD1fl/fl mice or KCTD1−/− mice. Inactivation at P9 leads to less severe TAL/DCT defects (yellow arrows), whereas inactivation of KCTD1 at P13 results in even less kidney abnormalities (red arrows). H&E of kidneys of 7-months-old mice are shown. Scale bars, 50 μm.
B. TAL/DCT dilatation (black arrows) and epithelial blebbing (green arrows) are observed in mice in which AP-2β or KCTD1 were inactivated in the adult (β-actinCreERT2+TFAP2Bfl/fl mice or β-actinCreERT2+KCTD1fl/fl mice treated with tamoxifen [TAM] at 6-weeks, 10-weeks or even at 5-months of age). Inducible inactivation of both AP-2β and KCTD1 in the same mouse also results in progressive TAL/DCT defects in the adult. The age of each mouse is indicated. Scale bars, 50 μm.
C. Inducible inactivation of AP-2β at 6-weeks and 10-weeks of age and assessment of kidney function at 5-months- and 10-months of age respectively: loss of AP-2β in the adult results in increased BUN levels, water intake and urine production.
D.-F. Renal KCTD1 expression is maintained in the adult (D, left). Expression analyses show a correlation of the increases in BUN and the polyuria and polydipsia seen in mice lacking KCTD1 or with inducible inactivation of AP-2β or KCTD1 in the adult (Fig. 3D) with diminished expression of DCT markers Pvalb, NCC, EGF, and SFRP1. This partial de-differentiation of DCTs due to inducible inactivation of AP-2β or KCTD1 in the adult leads also to hypochloremia.
Graphs represent data as mean ± SEM. Semiquantitative RT-PCRs performed in triplicate. P-values are shown (two-tailed, unpaired t-test).
Our RNA-Seq data, showing that DCT terminal differentiation markers were reduced in mice in which AP-2β was inactivated in the adult (Figs. 4A and 4B), strongly suggest that the AP-2β/KCTD1 axis plays an important role not only for the formation of terminally differentiated DCTs during the postnatal maturation phase, but that it is also needed to maintain DCTs in their fully terminally differentiated state in the adult. Indeed, inducible inactivation of AP-2β, KCTD1 or both together in the adult after the nephron maturation phase is concluded resulted in an attenuation of terminal differentiation markers of DCTs and in progressive DCT dilatation, consistent with a partial de-differentiation of DCTs in the adult (Figs. 5B–5F and S6E). This loss of maintenance of the terminal differentiation state resulted in a salt-losing tubulopathy with a reduced ability to concentrate urine, increased BUN, hypochloremia, increased water intake and urine production (Figs. 3D, 5C and 5F; Tables S1 and S2). Notably, the morphological and functional DCT abnormalities and the degree of the salt-losing tubulopathy were less severe in these mice than in KCTD1−/−, Six2Cre+KCTD1fl/fl mice or when KCTD1 was inactivated during the early postnatal maturation phase (Fig. 3D). Thus, AP-2β-mediated KCTD1 expression is not only essential for the induction of terminal differentiation of DCTs but is also required for maintaining this terminal differentiation state in the adult.
Progressive renal fibrosis due to lack of KCTD1
Cystic enlargement of TALs/DCTs progressed with age and resulted in multiple cortical cysts in mice lacking KCTD1 (Figs. 6A and 6B; Fig. S7A). Increased expression of genes associated with fibrosis (TGF-β1, PAI-1 and Col1a1) was already observed at 2-months of age in kidneys of mice lacking KCTD1 (Fig. 6C). At 4 months and progressing with age, kidneys of KCTD1−/− mice and Six2Cre+KCTD1fl/fl mice developed a severe tubulointerstitial fibrosis with strong accumulation of type I collagen and PDGFRβ+ myofibroblasts in the renal cortex (Figs. 6A–6D). Extensive fibrosis and inflammation occurred particularly adjacent to dilated TALs/DCTs and thin-walled cortical cysts (Figs. 6D and S7A). Progressive fibrosis resulted in shrunken cystic kidneys with diminished renal cortex, whereas the renal medulla was largely unaffected (Figs. 6A and 6B). The age-dependent worsening of renal fibrosis and cyst formation led to progressive deterioration of renal function with an age-dependent strong increase in BUN and serum creatinine (Fig. 6E; Table S1). Moreover, declining renal function resulted in end-stage renal disease with systemic manifestations that are also seen in patients with advanced chronic kidney disease (CKD) and renal fibrosis, such as chronic anemia, hypertension and cardiac hypertrophy, eventually causing renal failure and death at ~8–12 months of age (Figs. 6E, 6F; Figs. S7B–S7D; Tables S1–S2).
Figure 6: Progressive renal fibrosis due to lack of KCTD1.

A. With progressive age kidneys of mice lacking KCTD1 (both in Six2Cre+KCTD1fl/fl mice and in KCTD1−/− mice) show a loss of renal cortex with fibrosis and cyst formation. The renal medulla is affected only at late stages of this process. Blue double arrows indicate size of renal cortex, whereas black double arrows indicate size of renal medulla in 7-months-old mice. Representative H&E images. Scale bar, 1mm.
B. KCTD1 deficiency leads to shrunken fibrotic kidneys with progressive cystic degeneration of the renal cortex in aged mice. Bisected kidneys of a 12-months-old KCTD1−/− and a control littermate mouse are shown. Large cortical cysts can be seen in kidneys of 12-months-old KCTD1−/− mice (green arrow). The renal cortex is diminished in size (white arrow), whereas the medulla remains intact (black arrow).
C. Increased renal expression of the profibrotic growth factor TGF-β1 and of fibrosis markers PAI-1 and Col1a1 precede the manifestation of severe fibrosis in kidneys of 2-months-old KCTD1−/− mice.
D. Left image: Increased cortical fibrosis in mice lacking KCTD1 in the kidney, with increased accumulation of type I collagen (green, arrow) and F4/80+ macrophages (red). Scale bar, 50 μm. Right image: Cortical fibrosis shows strong accumulation of PDGFRβ+ myofibroblasts (red, arrow), surrounding ectatic β-gal+ distal nephron tubules (white). Scale bar, 100 μm. Images of 4-months-old kidneys of KCTD1−/− mice and of WT littermates.
E. Progressive age-dependent increases in BUN levels demonstrate deteriorating renal function in Six2Cre+KCTD1fl/fl mice as they get older (red dotted line), whereas this increase in BUN is not observed in control littermates (blue dotted line).
F. Renal fibrosis in aged 10-months-old KCTD1−/− mice is associated with systolic hypertension (increased AP max [aortic pressure in mmHg]) (left) and cardiac hypertrophy (increased heart weight [HW]/body weight [BW]) (right), which is not observed in 4-months-old KCTD1−/− mice that have not manifested severe renal fibrosis yet.
G. Clinical signs of CKD in affected family members of a family with autosomal dominant Finlay-Marks syndrome who carry a c.89C>A missense mutation that leads to a p.Ala30Glu (A30E) amino acid change in KCTD1. Rising serum creatinine levels of one affected individual (III-4) are shown over a 20-year period, occuring due to progressive deterioration of kidney function and worsening CKD with increasing age. Filled circles or boxes indicate individuals that have a clinical diagnosis of Finlay-Marks syndrome and were found to carry the KCTD1 c.89C>A missense mutation (+), whereas this mutation was not present in tested unaffected family members (−). Grey dots represent individuals who underwent exome sequencing in the original study (Marneros et al., 2013). ND: not determined (no clinical data were available).
Graphs represent data as mean ± SEM. Semiquantitative RT-PCRs performed in triplicate with n>6 samples/group. P-values are shown (two-tailed, unpaired t-test).
See also Figure S7.
Progressive CKD in Finlay-Marks syndrome patients with a missense mutation in KCTD1
Our findings in KCTD1−/− and Six2Cre+KCTD1fl/fl mice suggest that proper KCTD1 function is required to prevent renal fibrosis and CKD-like pathologies in the kidney. As KCTD1 shows the same distal nephron expression pattern in both mouse and human kidneys (Fig. 1C) (Lindstrom et al., 2018a; Park et al., 2018; Ransick et al., 2019), we hypothesized that KCTD1 also plays a role in preventing renal fibrosis in human kidneys and that individuals with missense mutations in KCTD1 would likely develop renal fibrosis and CKD with age progression. Notably, several Finlay-Marks syndrome patients that carry KCTD1 missense mutations have been reported to have renal abnormalities, but their renal function has previously not been carefully evaluated (Marneros et al., 2013; Naik et al., 2012; Plessis et al., 1997; Steinberg et al., 1990). Affected family members of the largest kindred with Finlay-Marks syndrome identified to date carry a c.89C>A missense mutation in KCTD1 (Marneros et al., 2013). Clinical analysis of their renal function shows that they develop CKD with renal fibrosis and hypertension in their ~20s or ~30s, which deteriorates with progressive age and leads to renal failure in some affected family members (Fig. 6G). Family members without this mutation did not develop kidney abnormalities. The age-dependent progression of CKD and renal fibrosis in patients with the c.89C>A KCTD1 missense mutation is reflected in deteriorating kidney function with increasing age that leads to a steady increase in serum creatinine levels, which resembles the progressive renal impairment associated with worsening renal fibrosis seen in mice lacking KCTD1 (Figs. 6E and 6G). These clinical data provide a human disease correlate to the renal fibrosis seen in mice lacking KCTD1, strengthening the clinical relevance of the findings in KCTD1-deficient mice and suggesting that KCTD1 has similar functions in mouse and human kidneys.
KCTD1 deficiency leads to renal β-catenin hyperactivation
Next, we tested how lack of KCTD1 leads to progressive renal fibrosis. Recent in vitro studies suggested that KCTD1 can act as an endogenous β-catenin inhibitor by enhancing its proteasomal degradation (Li et al., 2014). Furthermore, this study showed that some of the KCTD1 missense mutations causing Finlay-Marks syndrome, including the aforementioned c.89C>A missense mutation, attenuate the ability of KCTD1 to inhibit β-catenin signaling activity in vitro (Li et al., 2014; Marneros et al., 2013; Naik et al., 2012; Plessis et al., 1997; Steinberg et al., 1990). Notably, previous studies showed that a sustained increase in Wnt/β-catenin activity promotes renal fibrosis and cyst formation (Brack et al., 2007; Humphreys, 2018; Liu et al., 2007; Saadi-Kheddouci et al., 2001; Satoh et al., 2012; Zhou et al., 2016; Zhou et al., 2013). This suggests that renal fibrosis in this Finlay-Marks syndrome family and in KCTD1-deficient mice may occur at least in part due to a chronic increase in renal β-catenin activity, which we tested next.
Indeed, adult KCTD1−/− mice, Six2Cre+KCTD1fl/fl mice or mice with inducible inactivation of KCTD1 in the adult all showed strongly increased renal β-catenin activity with increased phosphorylation of GSK-3β [Ser9] and increased levels of active β-catenin (Figs. 7A–7C). Notably, kidneys of mice lacking KCTD1 had an increase in both active and total β-catenin levels (Fig. 7B), consistent with activated Wnt signaling preventing proteasomal degradation of cytosolic β-catenin. These findings provide in vivo confirmation for the previous in vitro observation that KCTD1 can function as an endogenous inhibitor of β-catenin activity (Li et al., 2014).
Figure 7: Lack of KCTD1 causes β-catenin hyperactivation that promotes renal fibrosis.

A. The increase in active β-catenin levels in kidneys of 2-month-old Six2Cre+KCTD1fl/fl mice or KCTD1−/− mice is associated with increased GSK-3β phosphorylation at Ser9 in the same kidneys, consistent with increased canonical Wnt/β-catenin signaling activity in kidneys due to KCTD1 deficiency.
B. Induced inactivation of KCTD1 (TAM-treated β-actinCreERT2+KCTD1fl/fl mice) at P7 or P9 (during the nephron terminal differentiation phase) leads to diminished protein levels of the DCT marker NCC, the TAL marker NKCC2 and of EGF (expressed in TALs and DCTs). Their decrease is associated with increased canonical Wnt/β-catenin signaling (increased GSK3β phosphorylation and increased total and active β-catenin), as well as increased p70 S6 kinase (Thr389) phosphorylation and 4E-BP1 (Thr37/46) phosphorylation when assessed at 2-months of age. These findings in kidneys of mice with induced inactivation of KCTD1 at P7 or P9 are similar to those of age-matched Six2Cre+KCTD1fl/fl mice. Kidney lysates of 2-months-old mice.
C. Induced inactivation of KCTD1 in 6-week-old mice and evaluation of their kidneys at 7-months of age shows that de-differentiation of DCTs in adult kidneys with loss of NCC and parvalbumin is associated with an increase in renal active β-catenin levels.
D. Kidneys of P8 Six2Cre+KCTD1fl/fl mice show loss of the DCT differentiation marker NCC whereas active β-catenin levels are not increased at that time.
E. Heterozygous KCTD1 mice show no high β-catenin signaling activity in their NCC+ DCTs (no TCF/Lef:H2B-GFP reporter activity in a 5-months-old KCTD1−/WTTCF/Lef:H2B-GFP mouse kidney; green arrow). In contrast, littermate KCTD1−/−TCF/Lef:H2B-GFP mice that lack KCTD1 in the distal nephron epithelium show GFP+ green nuclei (representing increased β-catenin signaling activity) in dilated DCTs (white arrows), often at sites with diminished NCC protein. Scale bars, 100 μm.
F. Strong β-catenin signaling activity (GFP+ green nuclei; arrows) is observed in β-gal+ (red) distal nephron epithelial cells that lack KCTD1 expression in adult KCTD1−/−TCF/Lef:H2B-GFP mice (β-gal+ cells are cells that would normally express KCTD1). Adjacent β-gal− PTs that do not normally express KCTD1 show no green nuclei (red arrow). 5-months-old KCTD1−/TCF/Lef:H2B-GFP reporter mouse is shown. Scale bar, 50 μm.
G. Increased staining for phospho-histone γH2A.X (Ser139) in NCC+ DCTs in 4-months-old KCTD1−/− mice (white arrow), whereas this staining is not observed in adjacent PTs that normally do not express KCTD1 (blue arrow). Staining for phospho-histone γH2A.X (Ser139) is also not observed in kidneys of WT littermates. Kidneys of a 4-months-old KCTD1−/− mouse and of a WT littermate are shown. Scale bar, 20 μm.
H. Kidneys of 8-months-old Six2Cre+KCTD1fl/flβ-cateninWT/fl mice have a lesser increase in active β-catenin levels (serving as a partial rescue of the increased β-catenin signaling activity due to KCTD1 deficiency) and less phospho-4E-BP1 (Thr37/46) and phospho-p70 S6 kinase (Thr389), as well as less type I collagen (a fibrosis marker), when compared to age-matched Six2Cre+KCTD1fl/fl mice.
I. Six2Cre+KCTD1fl/flβ-cateninWT/fl mice show an improved kidney function (reduced increases in BUN and creatinine serum levels) when compared to Six2Cre+KCTD1fl/fl mice. 8–9-months-old mice are shown. Renal fibrosis is strongly inhibited in kidneys of 8-months-old Six2Cre+KCTD1fl/flβ-cateninWT/fl mice compared to age-matched Six2Cre+KCTD1fl/fl mice. Renal transcript levels of PAI-1 and of TGF-β1, both associated with fibrosis, are strongly increased in Six2Cre+KCTD1fl/fl mice, whereas Six2Cre+KCTD1fl/flβ-cateninWT/fl mice have reduced renal PAI-1 and TGF-β1 levels when compared to those in Six2Cre+KCTD1fl/fl mice. Quantification of Trichrome-stained kidneys (fibrotic areas stain blue) of 8-months-old Six2Cre+KCTD1fl/flβ-cateninWT/fl mice and Six2Cre+KCTD1fl/fl mice shows that cortical fibrosis is strongly inhibited in Six2Cre+KCTD1fl/flβ-cateninWT/fl mice. Representative images of Trichrome-stained kidneys of 8-months-old Six2Cre+KCTD1fl/flβ-cateninWT/fl mice and Six2Cre+KCTD1fl/fl mice show that inhibition of β-catenin hyperactivation strongly reduces kidney fibrosis and cyst formation. Lower images are magnifications of the boxed cortical areas of the upper images. Scale bars: upper images 2 mm, lower images 100 μm.
Graphs represent data as mean ± SEM. Semiquantitative RT-PCRs performed in triplicate with n>6 samples/group. P-values are shown (two-tailed, unpaired t-test).
Notably, single-cell RNA-Seq data showed that terminally differentiated TALs/DCTs are the main site of expression of the Wnt antagonist SFRP1 in the adult nephron and that SFRP1 is the only Wnt antagonist to be expressed at high levels in TALs/DCTs (Lindstrom et al., 2018a; Park et al., 2018; Ransick et al., 2019). The TAL/DCT terminal differentiation defect due to loss of KCTD1 leads to severely diminished renal expression of SFRP1 in adult kidneys (Fig. 5F). However, this loss of SFRP1 is not a major cause of the active β-catenin increase in kidneys of mice lacking KCTD1. SFRP1−/− mice did not show an increase in renal active β-catenin levels and thus the increase in canonical β-catenin activity in kidneys of KCTD1−/− mice is not a consequence of SFRP1 deficiency (Fig. S7E). Moreover, kidneys of aged SFRP1−/− mice had normal TAL/DCT differentiation marker protein levels and normal kidney histology and developed no renal fibrosis. These results are consistent with previous studies in SFRP1−/− mice (Matsuyama et al., 2014; Trevant et al., 2008). Thus, loss of SFRP1 serves as a marker of impaired terminal differentiation of TALs/DCTs but does not significantly affect renal canonical β-catenin signaling activity.
The increase in renal canonical Wnt/β-catenin signaling activity was also seen in kidneys of adult mice in which KCTD1 was inactivated during the postnatal terminal differentiation phase at P7 or at P9 (Figs. 5D–F and 7B). Moreover, induced inactivation of KCTD1 in 6-week-old mice, when TALs/DCTs are fully differentiated, resulted in their partial de-differentiation with loss of TAL/DCT differentiation markers and an increase in renal active β-catenin levels (Figs. 5F and 7C). Thus, the increase in renal canonical Wnt/β-catenin signaling activity is a consequence of KCTD1 deficiency during the postnatal terminal differentiation phase or in the fully differentiated state in the adult and is not due to lack of KCTD1 function during nephrogenesis.
Notably, whereas the DCT terminal differentiation defect in KCTD1−/− mice or Six2Cre+KCTD1fl/fl mice occurred during the postnatal nephron maturation phase, renal β-catenin hyperactivation occurred subsequently (Fig. 7D). This observation is consistent with the increase in β-catenin signaling activity occurring in adult kidneys that lack KCTD1 as a secondary downstream consequence of the distal nephron terminal differentiation defect.
In order to confirm that DCT epithelial cells that lack KCTD1 show increased β-catenin activity, KCTD1−/− mice were crossed with TCF/Lef:H2B-GFP mice that serve as reporter mice for cellular β-catenin activation (Figs. 7E and 7F). Consistent with the findings in whole kidneys, increased β-catenin signaling activity (GFP+ cells) was also observed in dilated β-gal+ distal nephron tubules of adult mice lacking KCTD1 (KCTD1−/− mice express lacZ from the endogenous KCTD1 locus and, thus, β-gal+ cells represent cells that would normally express KCTD1 but lack functional KCTD1), but not in β-gal− PTs (that do not express KCTD1) (Fig. 7F). Increased numbers of GFP+ cells were detected in dilated DCTs of KCTD1−/−TCF:LEF-GFP reporter mice that showed a terminal differentiation defect: GFP+ cells represent cells with high β-catenin signaling activity and were particularly prevalent in dilated DCTs at sites with diminished expression of the differentiation marker NCC (Fig. 7E). In contrast, mice heterozygous for KCTD1 that had normal DCTs (KCTD1WT/−TCF:LEF-GFP reporter mice) showed no GFP+ cells in their DCTs (Fig. 7E). Our findings suggest that chronic β-catenin hyperactivation in the context of immature or partially de-differentiated DCT epithelium due to KCTD1 deficiency results in progressive renal fibrosis and cyst formation, leading to clinical features of CKD.
Sustained increase in β-catenin activity in the distal nephron results in increased TGF-β1/mTOR activity and progressive renal fibrosis that can be inhibited by targeting β-catenin
A chronic increase in canonical Wnt/β-catenin activity has been reported to lead to premature cell senescence that promotes organ fibrosis through secretion of profibrotic growth factors such as TGF-β1, and continuously increased Wnt/β-catenin activity has been linked to renal fibrosis and cyst formation (Brack et al., 2007; Humphreys, 2018; Saadi-Kheddouci et al., 2001; Satoh et al., 2012; Zhou et al., 2016; Zhou et al., 2013). Consistent with these findings, kidneys of Six2Cre+KCTD1fl/fl mice and KCTD1−/− mice with a sustained increase in Wnt/β-catenin activity showed progressive signs of premature senescence of TAL/DCT epithelial cells and increased expression of TGF-β1 and of fibrosis markers (e.g. PAI-1, Col1a1) that preceded the progressive cystic enlargement and severe renal fibrosis (Fig. 6C). Immunolabeling for phospho-histone γH2A.X (Ser139), which is associated with DNA damage and premature senescence, was strongly increased in TALs/DCTs of 4-months-old KCTD1−/− mice compared to littermate controls, but not in other nephron segments (Fig. 7G). The same 4-month-old KCTD1−/− kidneys with increased phospho-histone γH2A.X (Ser139) labeling also showed onset of renal fibrosis (Fig. 6D). Similarly, an increase in p21 (CDKN1A) has been associated with premature senescence, and renal expression of CDKN1A was significantly increased in 2-month-old Six2Cre+KCTD1fl/fl mice, KCTD1−/− mice or mice in which KCTD1 was inactivated at P9 (Fig. S6D), preceding the manifestation of renal fibrosis. In contrast, kidneys of P8 Six2Cre+KCTD1fl/fl mice did not show increased CDKN1A expression (Fig. S6B). Similarly, inducible inactivation of AP-2β in the adult also resulted in increased CDKN1A expression (Fig. S6C).
Next, we tested whether Six2Cre+KCTD1fl/flβ-cateninWT/fl mice, which are heterozygous for β-catenin and also lack KCTD1 have an attenuated increase in renal β-catenin signaling activity and whether this results in an inhibition of renal fibrosis (Fig. 7H). Indeed, Six2Cre+KCTD1fl/flβ-cateninWT/fl mice had a reduced increase in renal β-catenin signaling activity, resulting in reduced renal fibrosis and cyst formation, no chronic anemia, and improved kidney function when compared to Six2Cre+KCTD1fl/fl littermates (Figs. 7H and 7I, S7B and S7C; Table S1). Whereas kidneys of 8-month-old Six2Cre+KCTD1fl/fl mice had small shrunken and fibrotic kidneys with strongly Trichrome-positive (blue) fibrotic areas in the renal cortex that was strongly reduced in thickness, 8-month-old littermate Six2Cre+KCTD1fl/flβ-cateninWT/fl mice had only mild cortical fibrosis and preserved much of their renal cortex (Fig. 7I). BUN and serum creatinine levels in 8-month-old Six2Cre+KCTD1fl/flβ-cateninWT/fl mice showed a significantly smaller increase over healthy control mice when compared to the increases seen in 8-month-old Six2Cre+KCTD1fl/fl mice (Fig. 7I; Table S1). As KCTD1 is only expressed in the distal nephron and not in glomeruli or PTs, and as inactivation of KCTD1 in CTs/CDs (in Aqp2Cre+KCTD1fl/fl mice) did not result in renal fibrosis even at an advanced age (Fig. S3D), the severe CKD-like pathologies and renal fibrosis in Six2Cre+KCTD1fl/fl mice can be attributed to aberrant hyperactivation of Wnt/β-catenin activity due to lack of KCTD1 in TALs/DCTs.
TGF-β1 is a major inducer of kidney fibrosis and promotes expression of profibrotic genes, such as PAI-1 and collagen I (Col1a1), and TGF-β1 expression and signaling can be increased by canonical Wnt/β-catenin signaling (Baarsma et al., 2011; Carthy et al., 2011). Consistent with these reports, the increase in renal canonical Wnt/β-catenin signaling due to lack of KCTD1 resulted in increased TGF-β1 and PAI-1 expression in kidneys of both KCTD1−/− mice and Six2Cre+KCTD1fl/fl mice, whereas heterozygosity for β-catenin attenuated this increase in TGF-β1 and PAI-1 expression, in part explaining the inhibition of renal fibrosis in Six2Cre+KCTD1fl/flβ-cateninWT/fl mice (Fig. 7I).
TGF-β1 can promote fibrosis through activation of mTOR-signaling, which results in activation of p70 S6 kinase via phosphorylation of Thr389 and in phosphorylation of 4E-BP1 at Thr37/46 that leads to stimulation of protein synthesis and cell proliferation. Notably, increased mTOR activation has been implicated in renal fibrosis and cystogenesis (Grahammer et al., 2014). Indeed, the increase in TGF-β1 expression in kidneys lacking KCTD1 was associated with increased protein levels of phosphorylated p70 S6 kinase (Thr389) and 4E-BP1 (Thr37/46) in their kidneys, preceding the manifestation of kidney fibrosis (Figs. 7B and 7H). Consistent with a role of increased Wnt/β-catenin signaling in promoting renal fibrosis and cystogenesis in part through increased mTOR activation, kidneys of Six2Cre+KCTD1fl/flβ-cateninWT/fl mice with reduced β-catenin activity and diminished fibrosis also had less mTOR activity (Fig. 7H).
DISCUSSION
The distal nephron plays essential roles in salt reabsorption and urinary concentration, in part due to the functions of the salt transporters NKCC2 in the TALs and NCC in the DCTs (Al Shibli and Narchi, 2015; Simon et al., 1996a; Simon et al., 1996b). Which molecular mechanisms induce the formation of these distal nephron segments has been unknown.
Based on (1) the overlapping distal nephron expression pattern of AP-2β and KCTD1 and our finding that AP-2β induces expression of KCTD1, (2) reports of renal abnormalities in patients with missense mutations in KCTD1, and (3) the observation of distal nephron defects in AP-2β null mice, we considered AP-2β and KCTD1 to be strong candidates for inducing differentiation programs of specific distal nephron segments during nephrogenesis. Notably, AP-2β null mice have previously been reported to have cystic dilatation of distal nephron epithelia and died shortly after birth (Moser et al., 2003; Moser et al., 1997; Wang et al., 2018). However, detailed characterization of their phenotype has not been undertaken in order to understand which specific nephron segments are affected and to determine the functional roles of AP-2β for the formation of specific nephron segments. Moreover, AP-2β has previously not been targeted in a cell type-specific manner in the kidney and the downstream molecular mechanisms through which AP-2β may regulate differentiation of distal nephron epithelia has not been investigated. Similarly, the in vivo role of KCTD1 in the kidney has remained unknown, in part due to the lack of KCTD1 null mice. Of note, a recent report described that a KCTD1 (I27N) mutation induced by ENU-mutagenesis resulted in perinatal lethality and heterozygosity for this mutation resulted in increased BUN (Kumar et al., 2017). However, no further characterization of kidney function or histology were shown in this report, which also did not assess the effects of this mutation on KCTD1 activity. Furthermore, we observed even in 12-months old heterozygous KCTD1−/WT mice no increase in BUN and these mice had normal kidneys (Table S1), raising the possibility that the reported ENU-mutagenesis mutant strain may harbor additional mutations that impair kidney function.
Thus, in order to assess the in vivo roles of AP-2β and KCTD1 for kidney development and function, we generated several genetic mouse models that allow for cell type-specific or inducible inactivation of AP-2β and KCTD1 in the kidney. Analyses of these mice showed that AP-2β is essential for the formation of early-stage DCTs from distal tubule precursors during nephrogenesis, whereas KCTD1 promotes terminal differentiation of early-stage DCTs into mature DCTs. Moreover, sustained KCTD1 activity in the adult is required to maintain DCTs in a terminally differentiated state.
After nephrogenesis is concluded, nephrons undergo terminal differentiation and growth, during which nephron segments achieve their full functional capacities (Kopan et al., 2014; Rumballe et al., 2011). Through which molecular mechanisms postnatal terminal differentiation and growth of distal nephron segments are controlled has remained largely unexplored. The duration of this time period is not fully clear, but previous studies suggested that terminal differentiation of nephron segments occurs mainly during a maturation phase in the first two weeks after birth (Rumballe et al., 2011). We show that the transcriptional regulator KCTD1, whose expression we found to be induced by AP-2β, is required for the differentiation of early-stage DCTs into terminally differentiated mature DCTs. By generating mice in which we inactivated KCTD1 at various time-points postnatally we demonstrated that the terminal differentiation of DCTs occurs mainly during the first two weeks postnatally, as DCT differentiation defects showed a progressive attenuation of severity the later KCTD1 was inactivated during that two-week postnatal maturation phase. However, once DCTs are fully differentiated, AP-2β-mediated KCTD1 expression is not simply switched off and both AP-2β and KCTD1 continue to be expressed in the distal nephron epithelium. This suggests that the AP-2β/KCTD1 axis also functions in terminally differentiated DCTs in the adult and that its role is not limited to inducing DCT differentiation. Indeed, we inactivated both AP-2β and KCTD1 in the adult at a time when nephrogenesis and terminal differentiation are concluded and found that sustained AP-2β/KCTD1 activity is required to maintain DCTs in a terminally differentiated state. Loss of AP-2β or KCTD1 in the adult phenocopies morphological and functional DCT defects that we observed in KCTD1−/− mice or Six2Cre+KCTD1fl/fl mice, albeit with a less severe manifestation. This suggests that lack of KCTD1 during the early postnatal maturation phase impairs terminal differentiation of DCTs that causes a severe salt-losing tubulopathy, whereas lack of KCTD1 in the adult leads to a partial de-differentiation of previously terminally differentiated DCT epithelium, explaining the less severe phenotype.
Previous data have attributed critical roles of both epithelial de-differentiation as well as β-catenin hyperactivation to the pathogenesis of renal fibrosis (Brack et al., 2007; Humphreys, 2018; Liu et al., 2007; Saadi-Kheddouci et al., 2001; Satoh et al., 2012; Zhou et al., 2016; Zhou et al., 2013). As inactivation of KCTD1 results in a DCT terminal differentiation defect that is accompanied by increased β-catenin activity in DCTs, we hypothesized that the combination of an epithelial terminal differentiation defect and aberrant β-catenin hyperactivation drive renal fibrosis with progressive age. Indeed, the critical role of the sustained high β-catenin activity for the progression of renal fibrosis is demonstrated in a partial genetic rescue experiment where reducing this β-catenin activity in the same cells that lack KCTD1 (Six2Cre+KCTD1fl/flβ-cateninfl/WT mice) could inhibit renal fibrosis. This suggests that approaches to promote or maintain terminal differentiation of nephron epithelium and to inhibit β-catenin hyperactivation are likely to have therapeutic benefit in the prevention or treatment of renal fibrosis.
Collectively, our data identify AP-2β/KCTD1 to be critical for both the induction as well as the maintenance of DCT differentiation programs in the kidney, and also link this developmental pathway to the pathogenesis of renal fibrosis in the adult. These findings further demonstrate that understanding epithelial differentiation mechanisms in the kidney may provide targets for therapeutic interventions to prevent adult-onset renal diseases, as is shown here that β-catenin inhibition can attenuate the manifestation of renal fibrosis.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alexander G. Marneros (amarneros@mgh.harvard.edu).
Materials Availability
Mouse lines generated in this study are available upon request.
Data and Code Availability
RNA-Seq data has been deposited to the GEO database: GSE126326 and GSE130864.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
In order to assess the physiological roles of KCTD1 in vivo, KCTD1-reporter mice, KCTD1−/− mice, and conditional KCTD1 KO mice were generated (Figs. S1A–1D). Two mouse ES cell clones (ES cell clones EPD0820–5-E03 and EPD0820–5-C01) were obtained from EUCOMM (Kctd1tm1a(EUCOMM)Wtsi) and correct targeting of the KCTD1 locus was confirmed (including by 5’ and 3’ long-range PCR and qPCR for neo). The tm1a allele is a “KO first allele: lacZ reporter-tagged insertion with conditional potential” (Skarnes et al., 2011). It inserts between exon 2 and 3 of the KCTD1 gene, disrupting the critical BTB domain and after removal of the floxed region (loxP sites flank exon 3 of KCTD1) after crossing these mice with β-actin-Cre+ mice (tm1b allele). Subsequently the Cre allele was crossed out through intercrossing these mice. Lack of functional KCTD1 expression (exon 3-containing KCTD1 transcripts) in the kidneys of KCTD1−/− mice was confirmed by RT-PCR and semiquantitative RT-PCR (one primer within exon 3) (Figure S2C). Notably, both mouse lines (derived from either the ES cell clone EPD0820–5-E03 or EPD0820–5-C01) had identical kidney phenotypes, providing independent confirmation in two mouse strains that the observed kidney abnormalities are a direct consequence of KCTD1 deficiency.
The Kctd1tm1a allele after β-actinCre+ excision of the floxed region maintains a lacZ cassette, allowing to monitor β-galactosidase expression from the endogenous KCTD1 locus as a way to identify cells that normally express KCTD1 (Fig. 1C). Heterozygous KCTD1WT/lacZ mice were utilized as reporter mice, as they express β-galactosidase in cells that normally express KCTD1. The expression pattern of β-galactosidase (using anti-β-galactosidase antibodies or X-Gal staining) was also confirmed by immunolabeling with anti-KCTD1 antibodies in kidney sections. In addition, the mice generated from the Kctd1tm1a(EUCOMM)Wtsi ES cell clones were crossed with a FLP-deleter mouse strain, which results in the removal of the lacZ and neoR cassette that are flanked by FRT sites, and thereby allowing for normal KCTD1 expression, but maintaining loxP sites flanking the critical exon 3 of KCTD1 (tm1c). These mice represent KCTD1fl/fl mice. These mice were crossed with several Cre lines (Six2Cre+ mice [IMSR Cat# JAX:009606, RRID:IMSR_JAX:009606 (Kobayashi et al., 2008)], PvalbCre+ mice (IMSR Cat# JAX:017320, RRID:IMSR_JAX:017320) (Grimm et al., 2017), and Aqp2Cre+ mice [IMSR Cat# JAX:006881, RRID:IMSR_JAX:006881; using only female Aqp2Cre+ mice for matings]) in order to obtain mice that are homozygous for the KCTD1fl/fl allele and heterozygous for the corresponding Cre to establish cell type-specific conditional KCTD1 KO mice. Cre activity and specificity were confirmed by crossing these strains with B6.Cg-Gt(ROSA)26Sortm3(CAG-EYFP)Hze/J (Ai3) reporter mice (IMSR Cat# JAX:007903, RRID:IMSR_JAX:007903) (Figs. S1E and S1F). Efficient Cre-mediated recombination and removal of exon 3 of KCTD1 was confirmed by RNA-Seq and semiquantitative RT-PCR of kidney lysates from Six2Cre+KCTD1fl/fl mice (Figs. S2B and S2C). KCTD1fl/fl mice were also crossed with β-actinCre+ mice to generate KCTD1 KO mice without the lacZ cassette (tm1d). Notably, β-actinCre+KCTD1fl/fl mice (tm1d), as well as KCTD1−/− mice (tm1b) that contained the lacZ cassette derived from either ES cell clone (EPD0820–5-E03 and EPD0820–5-C01) showed the same kidney phenotypes. All reported kidney phenotypes occurred in all of these KCTD1 mouse strains in both male and female mice. Induced inactivation of KCTD1 in the adult was achieved in β-actinCreERT2+KCTD1fl/fl mice (generated by crossing CAGGCreERT2+ mice [IMSR Cat# JAX:004453, RRID:IMSR_JAX:004453] (Hayashi and McMahon, 2002) with KCTD1fl/fl mice) with injection of tamoxifen (TAM; T5648, Sigma Aldrich) (9mg/40gm BW daily for 5 consecutive days IP), a tamoxifen administration regimen that has been shown to efficiently and uniformly activate Cre recombinase in the kidney (Hayashi and McMahon, 2002). P0, P7, P9 or P13 β-actinCreERT2+KCTD1fl/fl mice were injected with 1mg tamoxifen/40gm BW once IP, which also efficiently and uniformly activates Cre recombinase in the kidney of mice at that age (confirmed by qPCR) (Hayashi and McMahon, 2002). Efficient Cre-mediated removal of exon 3 of KCTD1 with this treatment regimen was confirmed by semiquantitative RT-PCR (one primer within exon 3) (Fig. S2C). SFRP1−/− and EGF−/− mice have previously been reported (Luetteke et al., 1999; Satoh et al., 2006). TCF/Lef:H2B-GFP reporter mice (IMSR Cat# JAX:013752, RRID:IMSR_JAX:013752) were used to assess β-catenin signaling activity in vivo (Ferrer-Vaquer et al., 2010). Floxed β-catenin mice (IMSR Cat# JAX:022775, RRID:IMSR_JAX:022775) were crossed with Six2Cre+KCTD1fl/fl mice to generate either Six2Cre+KCTD1fl/flβ-cateninfl/WT mice or Six2Cre+KCTD1fl/WTβ-cateninfl/WT mice. TFAP2Bfl/fl mice were kindly provided by Dr. Trevor Williams. The generation of these mice has previously been described: Cre-mediated recommendation results in removal of exon 6 that is critical for DNA binding, resulting in TFAP2B without transcription factor activity (Van Otterloo et al., 2018). RNA-Seq of kidney lysates from TAM-treated β-actinCreERT2+TFAP2Bfl/fl mice confirmed efficient Cre-mediated removal of exon 6 of TFAP2B (Fig. S2A). For all animal studies, institutional approval was granted and international guidelines for the care and use of laboratory research animals were followed. ARRIVE guidelines for reporting of animal studies were followed. Summary of all mouse strains used in this study is provided in Table S3.
Characterization of renal abnormalities in a family with Finlay-Marks syndrome
A large family with autosomal-dominant inheritance of Finlay-Marks syndrome (also known as Scalp-Ear-Nipple syndrome [OMIM# 181270]) with a c.89C>A missense mutation that leads to a p.Ala30Glu (A30E) amino acid change has previously been described (Marneros et al., 2013). After obtaining institutional IRB approval, clinical information regarding the manifestation of kidney abnormalities was gathered from affected individuals of this family as well as from their physicians.
METHOD DETAILS
Metabolic cages, ELISAs, serum and urine chemistries
Single mice were kept for 24-hours in metabolic cages (3600M021, Techniplast) and urine and stool production, as well as water and food intake were measured. Treatments were performed with dDAVP (0.4μg/kg intraperitoneally) and effects on these parameters were assessed. Mouse serum chemistries were determined with a Dri-Chem7000 chemistry analyzer (Heska). Urine chemistries were determined with a Roche ModP analyzer and urine osmolality with an Advanced Instruments Micro Osmometer Model 3300. CBC was obtained with the HemaTrue analyzer (Heska). Blood gas analysis was performed with an IRMA TRUpoint capillary blood analysis system (Lifehealth). Mouse serum renin was measured with a mouse renin 1 ELISA kit (EMREN1, Thermo Fisher), according to the manufacturer’s instructions. Aldosterone assays were performed with a commercially available radioimmunoassay kit (ACTIVE® Aldosterone RIA, Beckman Coulter, IMMUNOTECH, Prague, Czech Republic). Anti-aldosterone antibody-coated tubes, 125I-labeled aldosterone radioligand, 7 calibrators ranging from 0 to 4448 pmol/L, and 2 controls were provided by the manufacturer. The assays were performed according to the kit protocol with the modification of an overnight incubation step at room temperature.
Immunolabeling of kidney sections and morphological kidney analyses
For morphological analysis of mouse kidneys they were fixed in 4% paraformaldehyde. Kidneys were bisected and then processed and embedded in paraffin for histological analysis. H&E, PAS and Trichrome stainings were performed according to standard protocols. The other kidney half was embedded in 30% sucrose and subsequently in OCT for immunolabeling experiments. For immunolabeling, 7μm kidney sections were permeabilized in 0.5% Triton X-100 and subsequently blocked with serum in which the secondary antibodies were raised. The following primary antibodies were used: rabbit anti-β-galactosidase (Molecular Probes Cat# A-11132, RRID:AB_221539), rabbit anti-AP-2β (Cell Signaling Technology Cat# 2509, RRID:AB_2058198), rabbit anti-Pou3f3 (Thermo Fisher Scientific Cat# PA5–64311, RRID:AB_2645790), mouse anti-Six2 (Abnova Corporation Cat# H00010736-M01, RRID:AB_436993), rabbit anti-Cited1 (MyBiosource, MBS2025863), chicken anti-β-galactosidase (Abcam Cat# ab9361, RRID:AB_307210), rat anti-CD31 (BD Biosciences Cat# 550274, RRID:AB_393571), rat anti-F4/80 (conjugated with Alexa647, BioLegend Cat# 123121, RRID:AB_893492), goat anti-KCTD1 (Santa Cruz Biotechnology Cat# sc-84861, RRID:AB_2234293), rabbit anti-Slc3a1 (Proteintech Cat# 16343–1-AP, RRID:AB_2239419), goat anti-Aqp2 (Santa Cruz Biotechnology Cat# sc-9882, RRID:AB_2289903), rabbit anti-NCC (Millipore Cat# AB3553, RRID:AB_571116), goat anti-THP (Santa Cruz Biotechnology Cat# sc-19554, RRID:AB_793728), rabbit anti-GFP (Thermo Fisher Scientific Cat# A-11122, RRID:AB_221569), rabbit anti-NKCC2 (Cell Signaling Technologies Cat# 38436, RRID:AB_2799134), rabbit anti-parvalbumin (Abcam Cat# ab11427, RRID:AB_298032), rabbit anti-collagen I (Acris Antibodies GmbH Cat# R1038, RRID:AB_978381), rat anti-PDGFRβ (Thermo Fisher Scientific Cat# 14–1402-82, RRID:AB_467493), rabbit anti-phospho-histone H2A.X (Ser139) (Cell Signaling Technology Cat# 9718, RRID:AB_2118009), and goat anti-EGF (R&D Systems Cat# AF2028, RRID:AB_355111). Phalloidin conjugated with Alexa488 (Molecular Probes Cat# A-12379, RRID:AB_2315147) or Alexa647 (Thermo Fisher Scientific Cat# A22287, RRID:AB_2620155) were used for cytoskeletal staining at a dilution of 1:100. DAPI was used to stain nuclei (Thermo Fisher Scientific Cat# D3571, RRID:AB_2307445). Secondary Alexa-488/555/647 antibodies were used at a dilution of 1:200 (Thermo Fisher). Controls included stainings with no primary antibody or with IgG control primary antibodies. Lectins at a dilution of 1:100 were from Vector Laboratories: rhodamine-conjugated peanut agglutinin (Vector Laboratories Cat# RL-1072, RRID:AB_2336642) [epithelial staining of distal nephron epithelial cells; contiguous staining of PTs], rhodamine-conjugated Dolichos Biflorus Agglutinin (Vector Laboratories Cat# RL-1032, RRID:AB_2336396) [labels CTs/CDs], fluorescein-conjugated Lotus Tetragonolobus Lectin (Vector Laboratories Cat# FL-1321, RRID:AB_2336559) [labels PTs]. Antibodies against NCC, NKCC2, THP, KCTD1, AQP2 and AP-2β were also used for immunolabeling of frozen sections of human kidneys with the addition of a rabbit anti-AP2β antibody from Atlas antibodies (Atlas Antibodies Cat# HPA034683, RRID:AB_10670966). For semithin and ultrathin sections, bisected kidneys were fixed in 1.25% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4). After postfixation in 4% osmium tetroxide, and dehydration steps, tissues were embedded in TAAB epon (Marivac Ltd.) and 1μm thin sections were used for toluidine blue staining and light microscopy.
Cell culture and Western blotting
Kidneys were lysed in NP40 lysis buffer (Life Technologies) with 1mM PMSF and protease inhibitor cocktail (Complete, Roche) using the Qiagen TissueLyser-II. After centrifugation, the protein concentrations in the supernatant were determined with a Bradford assay. Equal amounts of protein were loaded onto NuPage 4–12% Bis-Tris gels (Life Technologies) and blotted to nitrocellulose membranes. Equal protein loading was assessed using a rabbit polyclonal anti-β-actin antibody (Cell Signaling Technology Cat# 4970, RRID:AB_2223172). The following primary antibodies were used: antibodies against NKCC2 (38436, Cell Signaling Technology), NCC (Millipore Cat# AB3553, RRID:AB_571116), parvalbumin (Abcam Cat# ab11427, RRID:AB_298032), Pou3f3 (Proteintech Cat# 18999–1-AP, RRID:AB_2284130), active β-catenin (non-phospho Ser33/37/Thr41) (Cell Signaling Technology Cat# 8814S, RRID:AB_11127203), total β-catenin (Cell Signaling Technology Cat# 8480, RRID:AB_11127855), phospho GSK-3β (Ser9) (Cell Signaling Technology Cat# 9322S, RRID:AB_2115196), total GSK-3β (Cell Signaling Technology Cat# 9315, RRID:AB_490890), total 4E-BP1 (Cell Signaling Technology Cat# 9644, RRID:AB_2097841), phospho 4E-BP1 (Thr37/46) (Cell Signaling Technology Cat# 2855, RRID:AB_560835), p70 S6 kinase (Thr389) (Cell Signaling Technology Cat# 9234, RRID:AB_2269803), and collagen I (Acris Antibodies GmbH Cat# R1038, RRID:AB_978381). HRP-conjugated secondary antibodies were used and chemiluminescence signal was determined with the SuperSignal WestPico chemiluminescent substrate (Pierce).
HEK293 cells were used for transfection experiments with Lipofectamine 2000 (Life Technologies). HEK293 cells were transfected with expression plasmids driving luciferase expression by the human KCTD1 promoter (HRPM39938-PG04, GeneCopoeia). These expression plasmids also express secreted alkaline phosphatase, which allows for normalization for transfection efficiency (luciferase activity was normalized for secreted alkaline phosphatase activity using the Secrete-Pair Dual Luminescence Assay kit (LF031, GeneCopoeia). Overexpression plasmids for human AP-2β (RC204504) or control plasmid (PS100001) were from Origene. One day after transfection the medium was changed, and luciferase and secreted alkaline phosphatase activity were assessed two days later. SiRNA-mediated knockdown of HEK293 cells was performed using lipofectamine 2000 and ON-TARGETplus human TFAP2B SMARTpool siRNAs (L-017730–02-0005 ON-TARGETplus Human TFAP2B (7021) siRNA–SMARTpool; GE Healthcare Dharmacon Inc.) or control siRNA and RNA was isolated 48 hours later with Trizol reagent.
RNA-Seq and semiquantitative RT-PCR
RNA was isolated from kidneys or cells with Trizol reagent (Life technologies). For RNA-Seq experiments RNA was further purified with the Directzol RNA MiniPrep kit from Zymo (R2050). Kidneys of the following groups were used for RNA-Seq: (1) P8 Six2Cre+KCTD1fl/fl mice and littermate KCTD1fl/fl control mice (n=4 per group); (2) 4-months-old β-actinCreERT2+TFAP2Bfl/fl mice versus control littermates treated with TAM at 6 weeks of age (n=4 per group). For library construction, 200ng of total RNA using the NEBNext Ultra Directional RNA Library Prep Kit for Ilumina (E7420L) was used. 15 cycles of PCR were done. Quality control of libraries was done via Bioanalyzer High Sensitivity DNA and Tape Station High Sensitivity D5000 Screen Tape. Quantification of the samples was done via qPCR using the BioRad CFX96. The reagent kit for qPCR was the Kapa Library Quantification Kit for Illumina (KK4824). Sequencing was performed on Illumina HiSeq 2500 instrument, resulting in approximately 30 million of 50 bp reads per sample.
For gene expression studies, cDNA was obtained using the Transcriptor First strand synthesis kit utilizing hexamer primers (Roche). Semiquantitative RT-PCR was performed using a LightCycler 480 system with the LightCycler 480 SYBR Green I master mix according to standard procedures (45 amplification cycles) (Roche Applied Science). Primers for 36b4 were used as a normalization control. Concentrations were determined using a standard dilution curve. Experiments for all samples were performed in triplicate with n>7 mice/experimental group. Primer sequences for semiquantitative RT-PCR are listed in Table S4.
Blood pressure measurements and hemodynamic pressure/volume (PV)-loop experiments
Aortic blood pressure measurements and hemodynamic PV-loop experiments were performed, as previously reported (Marneros, 2018). For hemodynamic measurements (PV-loop experiments), age-matched male mice were anesthetized with isoflurane inhalation (initially 4–5% in an induction chamber then 1.5% via a nose cone). Body temperature was maintained at 36–37°C using a temperature controller. A 1.2-Fr high-fidelity pressure catheter (Scisence catheters) was inserted into the carotid artery. Pressure signals were recorded at 10kHz and analyzed using PowerLab software (Chart 5, ADInstruments). After aortic blood pressure measurements, baseline PV-loop measurements were obtained (Marneros, 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-Seq data analysis
Sequencing reads were mapped in a splice-aware fashion to the Ensembl annotation of the mouse GRCm37/mm9 transcriptome using STAR (Dobin et al., 2013). Read counts over transcripts were calculated using HTSeq (Anders et al., 2015), followed by the differential expression analysis using EdgeR (Robinson et al., 2010). Genes were claddified as differentially expressed (DEGs) based on the cutoffs of 1.5-fold change in expression value and false discovery rates (FDR) below 0.05. Heatmaps of DEGs represent Z-scores of log2RPKM values. Nephron segment- or cell type-specific expression pattern of DEGs is shown in heatmaps based on single-cell RNA-Seq data from mouse kidneys (Z-scores) (Park et al., 2018). RNA-Seq data has been deposited to the GEO database: GSE126326 and GSE130864.
Statistics
An unpaired two-tailed Student’s t-test was used for statistical analyses. P-values <0.05 were considered to be statistically significant. P-values are indicated. All graphs show mean ± SEM.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-β-galactosidase | Thermo Fisher Scientific | Cat# A-11132, RRID:AB_221539 |
| rabbit anti-AP-2β | Cell Signaling Technology | Cat# 2509, RRID:AB_2058198 |
| rabbit anti-Pou3f3 | Thermo Fisher Scientific | Cat# PA5-64311, RRID:AB_2645790 |
| mouse anti-Six2 | Abnova Corporation | Cat# H00010736-M01, RRID:AB_436993 |
| rabbit anti-Cited1 | MyBiosource | MBS2025863 |
| chicken anti-β-galactosidase | Abcam | Cat# ab9361, RRID:AB_307210 |
| rat anti-CD31 | BD Biosciences | Cat# 550274, RRID:AB_393571 |
| rat anti-F4/80 conjugated with Alexa647 | BioLegend | Cat# 123121, RRID:AB_893492 |
| goat anti-KCTD1 | Santa Cruz Biotechnology | Cat# sc-84861, RRID:AB_2234293 |
| goat anti-Aqp2 | Santa Cruz Biotechnology | Cat# sc-9882, RRID:AB_2289903 |
| rabbit anti-NCC | Millipore | Cat# AB3553, RRID:AB_571116 |
| goat anti-THP | Santa Cruz Biotechnology | Cat# sc-19554, RRID:AB_793728 |
| rabbit anti-GFP | Thermo Fisher Scientific | Cat# A-11122, RRID:AB_221569 |
| rabbit anti-NKCC2 | Cell Signaling Technologies | Cat# 38436, RRID:AB_2799134 |
| rabbit anti-parvalbumin | Abcam | Cat# ab11427, RRID:AB_298032 |
| rabbit anti-collagen I | Acris Antibodies | Cat# R1038, RRID:AB_978381 |
| rat anti-PDGFRβ | Thermo Fisher Scientific | Cat# 14-1402-82, RRID:AB_467493 |
| rabbit anti-phospho-histone H2A.X (Ser139) | Cell Signaling Technology | Cat# 9718, RRID:AB_2118009 |
| goat anti-EGF | R&D Systems | Cat# AF2028, RRID:AB_355111 |
| rabbit anti-β-actin antibody | Cell Signaling Technology | Cat# 4970, RRID:AB_2223172 |
| rabbit anti-Pou3f3 | Proteintech | Cat# 18999-1-AP, RRID:AB_2284130 |
| rabbit anti-Slc3a1 | Proteintech | Cat# 16343-1-AP, RRID:AB_2239419 |
| rabbit anti-active β-catenin (non-phospho Ser33/37/Thr41) | Cell Signaling Technology | Cat# 8814S, RRID:AB_11127203 |
| rabbit anti-total β-catenin | Cell Signaling Technology | Cat# 8480, RRID:AB_11127855 |
| rabbit anti-phospho GSK-3β (Ser9) | Cell Signaling Technology | Cat# 9322S, RRID:AB_2115196 |
| rabbit anti-total GSK-3β | Cell Signaling Technology | Cat# 9315, RRID:AB_490890 |
| rabbit anti-total 4E-BP1 | Cell Signaling Technology | Cat# 9644, RRID:AB_2097841 |
| rabbit anti-phospho 4E-BP1 (Thr37/46) | Cell Signaling Technology | Cat# 2855, RRID:AB_560835 |
| rabbit anti-p70 S6 kinase (Thr389) | Cell Signaling Technology | Cat# 9234, RRID:AB_2269803 |
| rabbit anti-AP2β | Atlas Antibodies | Cat# HPA034683, RRID:AB_10670966 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| human kidney frozen sections | Zyagen | NC1132314 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| tamoxifen | Sigma Aldrich | T5648 |
| recombinant mouse EGF | Peprotech | 315-09 |
| Critical Commercial Assays | ||
| Secrete-Pair Dual Luminescence Assay kit | GeneCopoeia | LF031 |
| NEBNext Ultra Directional RNA Library Prep Kit for Ilumina | NEB | E7420L |
| mouse renin ELISA kit | Thermo Fisher Scientific | EMREN1 |
| aldosterone RIA kit | IMMUNOTECH | ACTIVE® Aldosterone RIA |
| Deposited Data | ||
| RNA-Seq data | this paper | GEO: GSE126326 and GSE130864 |
| Experimental Models: Cell Lines | ||
| KCTD1 ES cell clones EPD0820-5-E03 and EPD0820-5-C01 | EUCOMM | Kctd1tm1a(EUCOMM)Wtsi |
| Experimental Models: Organisms/Strains | ||
| KCTD1−/− mice and KCTD1fl/fl mice | Marneros lab, this paper | |
| Six2Cre+ mice | Kobayashi et al., 2008 | JAX:009606 |
| PvalbCre+ mice | Grimm et al., 2017 | JAX:017320 |
| Aqp2Cre+ mice | Jackson Labs | JAX:006881 |
| B6.Cg-Gt(ROSA)26Sortm3(CAG-EYFP)Hze/J (Ai3) | Jackson Labs | JAX:007903 |
| CAGGCreERT2+ mice | Hayashi and McMahon, 2002 | JAX:004453 |
| SFRP1−/− mice | Satoh et al., 2006 | |
| EGF−/− mice | Luetteke et al., 1999 | |
| TFAP2Bfl/fl mice | Van Otterloo et al., 2018 | |
| TCF/Lef:H2B-GFP reporter mice | Ferrer-Vaquer et al., 2010 | JAX:013752 |
| β-catenin floxed mice | Jackson Labs | JAX:022775 |
| Oligonucleotides | ||
| ON-TARGETplus human TFAP2B SMARTpool siRNAs | GE Healthcare Dharmacon Inc. | L-017730-02-0005 |
| semiquantitative RT-PCR primers | Life Technologies | See Table S3 |
| Recombinant DNA | ||
| overexpression plasmids for human AP-2β | Origene | RC204504 |
| control plasmid | Origene | PS100001 |
| plasmids driving luciferase expression by the human KCTD1 promoter | GeneCopoeia | HRPM39938-PG04 |
| Software and Algorithms | ||
| STAR | Dobin et al., 2013 | |
| HTSeq | Anders et al., 2015 | |
| EdgeR | Robinson et al., 2010 | |
| Other | ||
| rhodamine-conjugated peanut agglutinin | Vector Laboratories | Cat# RL-1072, RRID:AB_2336642 |
| rhodamine-conjugated Dolichos Biflorus Agglutinin | Vector Laboratories | Cat# RL-1032, RRID:AB_2336396 |
| fluorescein-conjugated Lotus Tetragonolobus Lectin | Vector Laboratories | Cat# FL-1321, RRID:AB_2336559 |
| metabolic cages | Techniplast | 3600M021 |
| phalloidin conjugated with Alexa488 | Thermo Fisher Scientific | Cat# A-12379, RRID:AB_2315147 |
| phalloidin conjugated with Alexa647 | Thermo Fisher Scientific | Cat# A22287, RRID:AB_2620155 |
HIGHLIGHTS.
AP-2β is required for distal tubule precursor differentiation into early-stage DCTs
KCTD1 leads to terminal differentiation of early-stage DCTs into mature DCTs
Lack of KCTD1 results in immature DCTs in the adult and a salt-losing tubulopathy
Loss of KCTD1 leads to renal fibrosis through β-catenin hyperactivation
ACKNOWLEDGMENTS
The author would like to thank Karin Strittmatter for technical assistance, Dr. Trevor Williams for providing TFAP2Bfl/fl mice and Dr. Sallie Schneider for providing SFRP1−/− kidneys. The author is grateful for the participation of all members and care givers of the family with Scalp-Ear-Nipple syndrome. Services of the following core facilities is acknowledged: Next-Generation Sequencing core at Massachusetts General Hospital (Drs. Ruslan Sadreyev and Murat Cetinbas) and cardiac physiology core at Beth Israel Deaconess Hospital (Drs. Peter Kang and Qingen Ke). This study was supported by institutional funds from Massachusetts General Hospital and by funding from the NIH (R01DK118134, R01DK121178, R21AG063377) to AGM.
Footnotes
DECLARATION OF INTERESTS: The author declares no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Al Shibli A, and Narchi H (2015). Bartter and Gitelman syndromes: Spectrum of clinical manifestations caused by different mutations. World J Methodol 5, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baarsma HA, Menzen MH, Halayko AJ, Meurs H, Kerstjens HA, and Gosens R (2011). beta-Catenin signaling is required for TGF-beta1-induced extracellular matrix production by airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 301, L956–965. [DOI] [PubMed] [Google Scholar]
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, and Rando TA (2007). Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317, 807–810. [DOI] [PubMed] [Google Scholar]
- Carthy JM, Garmaroudi FS, Luo Z, and McManus BM (2011). Wnt3a induces myofibroblast differentiation by upregulating TGF-beta signaling through SMAD2 in a beta-catenin-dependent manner. PLoS One 6, e19809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HT, Kim M, Valerius MT, Surendran K, Schuster-Gossler K, Gossler A, McMahon AP, and Kopan R (2007). Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 134, 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Luo C, Zhou J, Zhong Y, Hu X, Zhou F, Ren K, Gan L, He A, Zhu J, et al. (2009). The interaction of KCTD1 with transcription factor AP-2alpha inhibits its transactivation. Journal of cellular biochemistry 106, 285–295. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Vaquer A, Piliszek A, Tian G, Aho RJ, Dufort D, and Hadjantonakis AK (2010). A sensitive and bright single-cell resolution live imaging reporter of Wnt/ss-catenin signaling in the mouse. BMC Dev Biol 10, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahammer F, Wanner N, and Huber TB (2014). mTOR controls kidney epithelia in health and disease. Nephrol Dial Transplant 29 Suppl 1, i9–i18. [DOI] [PubMed] [Google Scholar]
- Grieshammer U, Cebrian C, Ilagan R, Meyers E, Herzlinger D, and Martin GR (2005). FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 132, 3847–3857. [DOI] [PubMed] [Google Scholar]
- Grimm PR, Coleman R, Delpire E, and Welling PA (2017). Constitutively Active SPAK Causes Hyperkalemia by Activating NCC and Remodeling Distal Tubules. J Am Soc Nephrol 28, 2597–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, and McMahon AP (2002). Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244, 305–318. [DOI] [PubMed] [Google Scholar]
- Heliot C, Desgrange A, Buisson I, Prunskaite-Hyyrylainen R, Shan J, Vainio S, Umbhauer M, and Cereghini S (2013). HNF1B controls proximal-intermediate nephron segment identity in vertebrates by regulating Notch signalling components and Irx1/2. Development 140, 873–885. [DOI] [PubMed] [Google Scholar]
- Humphreys BD (2018). Mechanisms of Renal Fibrosis. Annu Rev Physiol 80, 309–326. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kwan KM, Carroll TJ, McMahon AP, Mendelsohn CL, and Behringer RR (2005). Distinct and sequential tissue-specific activities of the LIM-class homeobox gene Lim1 for tubular morphogenesis during kidney development. Development 132, 2809–2823. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, and McMahon AP (2008). Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Chen S, and Little M (2014). Nephron progenitor cells: shifting the balance of self-renewal and differentiation. Curr Top Dev Biol 107, 293–331. [DOI] [PubMed] [Google Scholar]
- Kumar S, Rathkolb B, Sabrautzki S, Krebs S, Kemter E, Becker L, Beckers J, Bekeredjian R, Brommage R, Calzada-Wack J, et al. (2017). Standardized, systemic phenotypic analysis reveals kidney dysfunction as main alteration of Kctd1 (I27N) mutant mice. J Biomed Sci 24, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chen C, Wang F, Huang W, Liang Z, Xiao Y, Wei K, Wan Z, Hu X, Xiang S, et al. (2014). KCTD1 suppresses canonical Wnt signaling pathway by enhancing beta-catenin degradation. PLoS One 9, e94343. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lindstrom NO, De Sena Brandine G, Tran T, Ransick A, Suh G, Guo J, Kim AD, Parvez RK, Ruffins SW, Rutledge EA, et al. (2018a). Progressive Recruitment of Mesenchymal Progenitors Reveals a Time-Dependent Process of Cell Fate Acquisition in Mouse and Human Nephrogenesis. Dev Cell 45, 651–660 e654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom NO, Guo J, Kim AD, Tran T, Guo Q, De Sena Brandine G, Ransick A, Parvez RK, Thornton ME, Basking L, et al. (2018b). Conserved and Divergent Features of Mesenchymal Progenitor Cell Types within the Cortical Nephrogenic Niche of the Human and Mouse Kidney. J Am Soc Nephrol 29, 806–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom NO, Lawrence ML, Burn SF, Johansson JA, Bakker ER, Ridgway RA, Chang CH, Karolak MJ, Oxburgh L, Headon DJ, et al. (2015). Integrated beta-catenin, BMP, PTEN, and Notch signalling patterns the nephron. Elife 3, e04000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, et al. (2007). Augmented Wnt signaling in a mammalian model of accelerated aging. Science 317, 803–806. [DOI] [PubMed] [Google Scholar]
- Luetteke NC, Qiu TH, Fenton SE, Troyer KL, Riedel RF, Chang A, and Lee DC (1999). Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 126, 2739–2750. [DOI] [PubMed] [Google Scholar]
- Marneros AG (2018). Effects of chronically increased VEGF-A on the aging heart. FASEB J 32, 1550–1565. [DOI] [PubMed] [Google Scholar]
- Marneros AG, Beck AE, Turner EH, McMillin MJ, Edwards MJ, Field M, de Macena Sobreira NL, Perez AB, Fortes JA, Lampe AK, et al. (2013). Mutations in KCTD1 cause scalp-ear-nipple syndrome. American journal of human genetics 92, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama M, Nomori A, Nakakuni K, Shimono A, and Fukushima M (2014). Secreted Frizzled-related protein 1 (Sfrp1) regulates the progression of renal fibrosis in a mouse model of obstructive nephropathy. J Biol Chem 289, 31526–31533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP (2016). Development of the Mammalian Kidney. Curr Top Dev Biol 117, 31–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi T, Hamada M, Morito N, Terunuma T, Hasegawa K, Zhang C, Yokomizo T, Esaki R, Kuroda E, Yoh K, et al. (2006). MafB is essential for renal development and F4/80 expression in macrophages. Mol Cell Biol 26, 5715–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser M, Dahmen S, Kluge R, Grone H, Dahmen J, Kunz D, Schorle H, and Buettner R (2003). Terminal renal failure in mice lacking transcription factor AP-2 beta. Lab Invest 83, 571–578. [DOI] [PubMed] [Google Scholar]
- Moser M, Pscherer A, Roth C, Becker J, Mucher G, Zerres K, Dixkens C, Weis J, Guay-Woodford L, Buettner R, et al. (1997). Enhanced apoptotic cell death of renal epithelial cells in mice lacking transcription factor AP-2beta. Genes Dev 11, 1938–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik P, Kini P, Chopra D, and Gupta Y (2012). Finlay-Marks syndrome: report of two siblings and review of literature. Am J Med Genet A 158A, 1696–1701. [DOI] [PubMed] [Google Scholar]
- Nakai S, Sugitani Y, Sato H, Ito S, Miura Y, Ogawa M, Nishi M, Jishage K, Minowa O, and Noda T (2003). Crucial roles of Brn1 in distal tubule formation and function in mouse kidney. Development 130, 4751–4759. [DOI] [PubMed] [Google Scholar]
- Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, and Susztak K (2018). Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessis G, Le Treust M, and Le Merrer M (1997). Scalp defect, absence of nipples, ear anomalies, renal hypoplasia: another case of Finlay-Marks syndrome. Clin Genet 52, 231–234. [DOI] [PubMed] [Google Scholar]
- Ransick A, Lindstrom NO, Liu J, Zhu Q, Guo JJ, Alvarado GF, Kim AD, Black HG, Kim J, and McMahon AP (2019). Single-Cell Profiling Reveals Sex, Lineage, and Regional Diversity in the Mouse Kidney. Dev Cell 51, 399–413 e397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiani L, Raciti D, Airik R, Kispert A, and Brandli AW (2007). The prepattern transcription factor Irx3 directs nephron segment identity. Genes Dev 21, 2358–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumballe BA, Georgas KM, Combes AN, Ju AL, Gilbert T, and Little MH (2011). Nephron formation adopts a novel spatial topology at cessation of nephrogenesis. Dev Biol 360, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadi-Kheddouci S, Berrebi D, Romagnolo B, Cluzeaud F, Peuchmaur M, Kahn A, Vandewalle A, and Perret C (2001). Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene 20, 5972–5981. [DOI] [PubMed] [Google Scholar]
- Salido EC, Lakshmanan J, Shapiro LJ, Fisher DA, and Barajas L (1990). Expression of epidermal growth factor in the kidney and submandibular gland during mouse postnatal development. An immunocytochemical and in situ hybridization study. Differentiation 45, 38–43. [DOI] [PubMed] [Google Scholar]
- Satoh M, Nagasu H, Morita Y, Yamaguchi TP, Kanwar YS, and Kashihara N (2012). Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am J Physiol Renal Physiol 303, F1641–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh W, Gotoh T, Tsunematsu Y, Aizawa S, and Shimono A (2006). Sfrp1 and Sfrp2 regulate anteroposterior axis elongation and somite segmentation during mouse embryogenesis. Development 133, 989–999. [DOI] [PubMed] [Google Scholar]
- Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, and Lifton RP (1996a). Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13, 183–188. [DOI] [PubMed] [Google Scholar]
- Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, et al. (1996b). Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12, 24–30. [DOI] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimani M, Barone S, Xu J, Shull GE, Siddiqui F, Zahedi K, and Amlal H (2012). Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci U S A 109, 13368–13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg RD, Ethington J, and Esterly NB (1990). Lumpy scalp syndrome. Int J Dermatol 29, 657–658. [DOI] [PubMed] [Google Scholar]
- Takemoto M, He L, Norlin J, Patrakka J, Xiao Z, Petrova T, Bondjers C, Asp J, Wallgard E, Sun Y, et al. (2006). Large-scale identification of genes implicated in kidney glomerulus development and function. EMBO J 25, 1160–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevant B, Gaur T, Hussain S, Symons J, Komm BS, Bodine PV, Stein GS, and Lian JB (2008). Expression of secreted frizzled related protein 1, a Wnt antagonist, in brain, kidney, and skeleton is dispensable for normal embryonic development. J Cell Physiol 217, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Otterloo E, Li H, Jones KL, and Williams T (2018). AP-2alpha and AP-2beta cooperatively orchestrate homeobox gene expression during branchial arch patterning. Development 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ji W, Zhu D, Wang W, Chen Y, Zhang Z, and Li F (2018). Tfap2b mutation in mice results in patent ductus arteriosus and renal malformation. J Surg Res 227, 178–185. [DOI] [PubMed] [Google Scholar]
- Zhou D, Tan RJ, Fu H, and Liu Y (2016). Wnt/beta-catenin signaling in kidney injury and repair: a double-edged sword. Lab Invest 96, 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Li Y, Zhou D, Tan RJ, and Liu Y (2013). Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. J Am Soc Nephrol 24, 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data has been deposited to the GEO database: GSE126326 and GSE130864.