

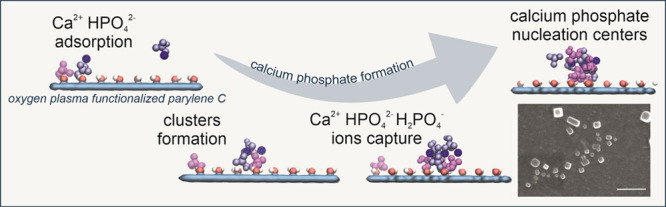
Abstract
Interactions at the solid–body fluid interfaces play a vital role in bone tissue formation at the implant surface. In this study, fully atomistic molecular dynamics (MD) simulations were performed to investigate interactions between the physiological components of body fluids (Ca2+, HPO42–, H2PO4–, Na+, Cl–, and H2O) and functionalized parylene C surface. In comparison to the native parylene C (−Cl surface groups), the introduction of −OH, −CHO, and −COOH surface groups significantly enhances the interactions between body fluid ions and the polymeric surface. The experimentally observed formation of calcium phosphate nanocrystals is discussed in terms of MD simulations of the calcium phosphate clustering. Surface functional groups promote the clustering of calcium and phosphate ions in the following order: −OH > −CHO > −Cl (parent parylene C) ≈ −COO–. This promoting role of surface functional groups is explained as stimulating the number of Ca2+ and HPO42– surface contacts as well as ion chemisorption. The molecular mechanism of calcium phosphate cluster formation at the functionalized parylene C surface is proposed.
Keywords: molecular dynamics, polymer surface, parylene C, functional groups, calcium phosphate, nucleation mechanism
Introduction
Polymers have been widely used for the last 3 decades in medical applications, such as coated transducers, neural prosthesis, catheters, and parts of orthopedic implants.1−5 A polymer vividly explored in this context and meeting sophisticated criteria for biomaterials is parylene C (poly(chloro-para-xylene)). Applied as a metal implant coating, parylene C provides a variety of benefits, such as corrosion protection and stability in body fluid environment.6,7 It was shown that parylene C can be applied in biomedical devices as a versatile coating serving as a multifunctional anticorrosive,8 biocompatible,9 and anti-infection/therapeutic layer.10−12 Notably, these functions are achievable on all classes of biomaterials, that is, ceramics,13 metallic,6 and polymeric.14
When a biomaterial is placed in the human body, it first interacts with a biological fluid consisting of water enriched with ions, sugars, and proteins. Upon such exposure, the oriented adsorption of molecules creates a conditioned surface that is responsible for the subsequent cell–surface interactions.15 One of the strategies to improve surface biocompatibility, in this context, is an incorporation of the functional groups of various chemical nature at the biomaterial surface, such as −OH, −COOH, −NH2, and −F.10,16,17 The functional groups can support adhesion of proteins responsible for cell attachment and formation of focal adhesion sites (oxygen-containing groups),10,18 prevent coagulation (nitrogen-containing groups covalently immobilize heparin),16 or add a surface antibacterial activity (fluorine-containing groups).19 Because of a plethora of existing possibilities, there are three questions that need to be addressed in order to obtain the biomaterial tailored for the desired site in the body: (a) what is the required function? (b) Should functional groups be introduced? If yes, which one? (c) What will be the most effective surface coverage?
For the biocompatibility of the adherent cells, such as osteoblasts, the parylene C surface can be successfully transformed from hydrophobic to hydrophilic using oxygen plasma treatment.20 This modification results in the introduction of oxygen-containing functional groups (−OH, −CHO, −COOH) and change in surface nanotopography.10,20,21 Such surface functional groups, in particular, −OH and −COOH, have been described to be crucial for initial steps of osteogenesis, which induces wound healing and consequently osseointegration.22−24
The clinical success of orthopedic implant coating relies on the quick and efficient formation of the bone tissue at an implanted surface. Calcium phosphate minerals are the main components of bone, constituting ∼50% of the mass of a normal adult bone. Initially amorphous, calcium phosphate matures through several consecutive intermediate stages to form crystalline hydroxyapatite.25 Therefore, the calcium phosphate formation is considered as a descriptor of the successful implant osseointegration and an established experimental tool used for functional correlation for in vitro studies.26 Based on experimental results, biomineralization is described by two sequential processes, including nucleation and crystal growth, during which the initial nucleation site must reach a critical size, enabling crystal growth and mineralization. Thus, it is extremely important to ensure specific surface properties required for the nucleation of calcium phosphate formation, which is considered as an initial step in the bone tissue formation.27 Providing an osteo-friendly environment for mesenchymal stem cells28 is essential for, in particular, cell adhesion, differentiation, and the eventual tissue formation at the interface.15 Therefore, solid–water interfaces play a vital role in biomedicine, providing a natural playground for most biochemical reactions and physiological processes.15 This is the reason why the functionalization of surfaces, with the aim of optimizing the implant–tissue interface, is vital. It should be mentioned that in the literature the classic nucleation model has been extended using the results of cryo-transmission electron microscopy, several in situ analysis techniques, and ab initio calculations pointing out the importance of the prenucleation complexes ([Ca(HPO4)3]4–) and postnucleation clusters of [Ca2(HPO4)3]2–, which precipitate as amorphous calcium phosphate.29,30 It seems that molecular dynamics (MD) simulations involving a large number of molecules can give a more comprehensive realistic picture of the biomineralization. Indeed, several approaches have been proposed,31−35 but the obtained results do not provide the univocal description of calcium phosphate formation at the biomaterial surfaces.
Calcium phosphate growth on polymeric surfaces is a challenge, mostly because of the hydrophobicity of such surfaces. This results in low calcium phosphate nucleation rates and, if eventually formed, poorly adhered crystals.36 The importance of the surface functionalization of orthopedic implants with calcium phosphate, prior to implantation, is based on partial solubilization of calcium phosphate in body fluids. This phenomenon increases locally at the bone–implant interface, where high ion concentrations induce protein adsorption and consecutive cell adhesion and proliferation.37
Although numerous experimental studies were performed, in order to evaluate calcium phosphate nucleation on biomaterial surfaces made of ceramics38 and polymers,39 only a few of them combined the experiments with theoretical modeling such as MD simulations.40,41 Recent advances in both surface engineering of biomaterials and MD simulations have generated a wealth of knowledge about the factors influencing the biocompatibility.31,42−46 The constant development of high-performance computing and the increasing power of supercomputers made MD the predestined method for the evaluation of the initial stages of novel biomaterials design. Furthermore, MD is often the only method, which gives an atomistic insight into the mechanisms of material biocompatibility.
In our previous study of the water–parylene C interface,47 MD simulations gave insights into the impact of the oxygen plasma treatment on the parylene C biocompatibility. It was estimated that substitution of 50–60% native −Cl groups with −OH groups, with plasma treatment, leads to increased wettability and biocompatibility of the parylene C surface. Although there are studies on parylene osseointegration potential,10,48 neither the native material nor oxygen-plasma-modified parylene has ever been investigated toward osseointegration potential at the molecular level description.
Assessing the biocompatibility of designed materials has usually focused on applying in vitro methodologies to establish the risk for adverse impact on the host. Surely, such methods present a largely simplified view of the complex in vivo conditions; nevertheless, they provide insight into potential tissue and cellular responses. The typical in vitro biocompatibility tests include the evaluation of material cytotoxicity, cell attachment, and cell response to substrate chemistry and topography. However, none of the applied methods gives feedback on molecular interactions at the interface. For this reason, MD simulations can be applied as a useful tool in understanding the processes at the biointerface.
The aim of this study is to gain an in-depth atomistic insight into the experimentally observed formation of calcium phosphate at the functionalized parylene C–body fluid interface. We employ the MD simulations to reveal the role of surface functional groups (−OH, −CHO, −COO–) formed during the oxygen plasma treatment in the nucleation process of calcium phosphate formation. Such an approach, to our best knowledge, is used for the first time and provides general guidelines for the optimal functionalization of implant coating surfaces, which can be easily extended for other polymeric materials.
Materials and Methods
Atomistic MD Simulations
Atomistic MD simulations were performed to investigate early stages of hydroxyapatite formation on the modified and unmodified surfaces of parylene C. Four different functional groups, corresponding to different ways of surface modification, were considered here, namely, chloride (−Cl), hydroxyl (−OH), aldehyde (−CHO), and carboxyl (−COO–) groups. The simulation box, of size 11.622 nm (the x-direction) × 12.720 nm (the y-direction) × 5 nm (the z-direction), contained two model surfaces interacting with a water slab composed of 2000 water molecules (corresponding to the water density ≈ 1 g/cm3). Each system was first energy-minimized using the steepest descent algorithm and then equilibrated in the NVT ensemble until both the potential energy of the system and the pressure stabilized. Finally, the production run of 200 ns was performed. All the analyses were performed on the last 100 ns of the production run. Further details of the model are published elsewhere.47 Artificial body fluid ions (H2PO4–, HPO42–, Na+, Ca2+, Cl–) were added to the water slab in the following quantities: 16 Ca2+, 16 H2PO4–, 8 HPO42–, 56 Na+, and 56 Cl– corresponding to the solution with 20 mmol/L of CaHPO4, 20 mmol/L of Ca(H2PO4)2, and 150 mmol/L of NaCl. Ion concentrations are the same as in the Dulbecco’s phosphate-buffered saline (DPBS; Lonza) used in the experiments. Force-field parameters for the modified and unmodified parylene C surface were taken from ref (47). The TIP3P model was used for water.49 The electronic continuum correction (ECC) has been applied to all ions as described in refs (50) and (51). In such ECC force fields, the electronic polarization is taken into account in a mean-field way through scaling the total charge of the ions by a factor of 0.75. This correction has been shown to improve ion pairing properties of salt solutions and the interaction of ions with biomolecules.52 The scaled force-field parameters for sodium, calcium, and chloride were previously published and tested.53−55 In the case of phosphate ions used here, we introduced ECC by scaling the atomic charges obtained from the general Amber force field.56 The calculated free energies of calcium–phosphate interactions (see Figure S5, Supporting Information) are in agreement with both experimental and recently reported force-field results.42,57 No ECC was applied for −COO– surface groups as it was shown to only partially reproduce experimental data.58 The ion force-field parameters are given in the Supporting Information. The force field of the parylene surface model is given in ref (47).
The atomistic MD simulations were carried out in an NVT ensemble using GROMACS 5.1.x software.59 The temperature was kept at 310 K using a v-rescale thermostat60 with a temperature coupling constant of 0.5 ps. Functional groups, carbon surface atoms, and water with ions were coupled to three separate thermostats. Long-range electrostatic interactions beyond a nonbonded interaction cutoff of 1.0 nm were treated by the particle mesh Ewald scheme61 with a Fourier spacing of 0.12 nm. A long-range dispersion correction to the energy and pressure was added. The LINCS algorithm62 was used to constrain all covalent bonds, allowing a time step of 2 fs. For water, the SETTLE method63 was applied. Periodic boundary conditions were applied in all dimensions.
Sample Preparation
Oxygen Plasma Functionalization
To modify the parylene C surface, oxygen plasma treatment was carried out using a Diener electronic Femto plasma system (Diener Electronic GmbH, Nagold, Germany) at 50 W and an oxygen partial pressure of 0.2 mbar. The time of exposure to oxygen plasma was 8 min, which is the optimal modification in terms of biocompatibility, according to our previous studies.9,10
Hydroxyapatite Deposition
Unmodified parylene C and oxygen-plasma-modified parylene C were incubated for 24, 48, and 72 h in PBS (Lonza) at 37 °C with gentle shaking. After each time interval, the buffer was removed, and samples were air-dried and used for characterization. All of the samples were prepared in triplicates.
Sample Characterization
Scanning Electron Microscopy
Images of parylene C surfaces and formed hydroxyapatite crystallites were taken using a Hitachi S-4700 scanning electron microscope. All of the samples were coated with Au prior to the observations.
X-ray Photoelectron Spectroscopy
The surface composition of oxygen-plasma-modified parylene C was checked using X-ray photoelectron spectroscopy (XPS) in an ultrahigh vacuum system, equipped with an SES R4000 (Gammadata Scienta) analyzer. The monochromatic Al Kα source (1486.6 eV) operated at 350 W was applied. The spectra were obtained at a takeoff angle of 90°. The vacuum in the spectrometer chambers was better than 5 × 10–9 mbar. The acquired XPS spectra were recorded using Casa-XPS 2.3.15 software. All spectra were calibrated using the adventitious C 1s peak with a fixed value of 285 eV. Surface concentrations of parylene C components were determined by the integration of narrow scan C 1s and O 1s maxima.
Laser Desorption/Ionization Mass Spectrometry
The surface analysis of unmodified and oxygen-plasma-treated parylene C was performed on a Bruker Ultraflex time-of-flight (TOF) mass spectrometer with a SCOUT-MTP ion source (Bruker Daltonics, Bremen, Germany) in reflector mode and equipped with a 337 nm nitrogen laser. The acceleration voltage was 25 kV, and the reflector voltage was 26.3 kV. Each surface was placed on a modified stainless steel LDI plate. The spectrum obtained for each sample is an accumulation of 1000 shots. The mass-to-charge (m/z) ratio range was set to scan from 60 to 2000 m/z.
X-ray Diffraction
The X-ray diffraction (XRD) patterns of parylene C foils were recorded using a Rigaku MiniFlex diffractometer with Cu Kα radiation at 10 mA and 10 kV, a 2θ step scan of 0.02, and a counting time of 1 s per step. The diffraction measurements were performed in the characteristic region of 2θ = 3–40°, and the crystallite size was estimated from Sherrer’s formula.
X-ray Fluorescence Spectrometry
For elemental X-ray fluorescence (XRF) analysis, an ARL Quant’X spectrometer, calibrated with the dedicated series of metal standards, was used. The X-rays of 4–50 kV (1 kV step), with the beam size of 8 mm generated with the Rh anode, were used. The detector used was a 3.5 mm Si(Li) drifted crystal with a Peltier cooling (−88 °C).
Results and Discussion
The biocompatible oxygen-plasma-modified parylene C, one of the most recently explored polymeric coatings, was used in the experimental studies. This material is well characterized in terms of physicochemical surface properties9,20 and interaction with biological moieties.10,21,47 The samples, used in this study, were obtained using oxygen plasma parameters optimized in our previous studies, that is, t = 8 min, p = 0.2 mbar, and P = 50 W.9,10 The basic characterization of the treated samples is summarized in Figure 1. After the plasma treatment, the parylene C surface becomes rough in the nanoscale, with nanocorrugations in the range of 60–200 nm (Figure 1A), and hydrophilic (Figure 1B), with a water contact angle θW of 0.1° and the corresponding surface free energy (SFE) of 72.9 mJ/m2 with 48.6 and 24.3 mJ/m2 polar (γsp) and dispersive (γs) components, respectively.47 The chemical nature of the introduced functional groups was identified with the surface-sensitive techniques, XPS and LDI-TOF-MS, as summarized in Figure 1C,D.20,64,65 In general, the oxygen plasma treatment results in multitype groups formed at the polymeric surface, as can be inferred from the XPS C 1s spectra (Figure 1C) with marked characteristic binding energies. The C 1s peak shape indicates the successful insertion of oxygen because, except 284.9 eV maximum, which corresponds to C–C/C=C bonds, two additional maxima are present at 286.1 and 289.4 eV, which correspond to C–O and C=O groups, respectively.20
Figure 1.
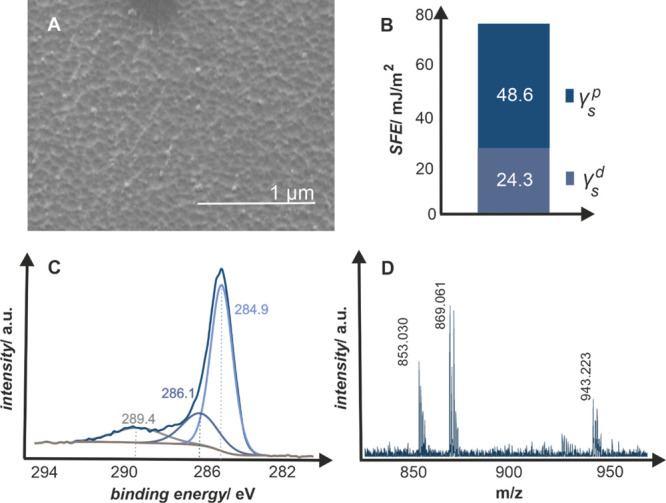
Oxygen-plasma-modified parylene C surface: (A) SEM morphology, (B) polar and dispersive components of the SFE determined from the contact angle measurements, (C) C 1s XPS profile deconvoluted in characteristic binding energies for C–O and C=O groups at 286.1 and 289.4 eV, respectively, and (D) LDI-TOF mass spectrum with marked peaks for six parylene C repeating units alone (853.030 and 869.061 m/z) and with the attached −OH and −COOH chain ends (943.223 m/z).
The results were complemented with the LDI-TOF-MS technique (Figure 1D). Each line in the 800–1000 m/z spectra represents a polymeric chain fragment, which was ionized and desorbed from the oxygen-plasma-modified parylene C surface. Signals at 853.030 and 869.061 m/z indicate six monomer units of parylene C (chemical formula C48H44Cl6) with Na+ and K+ adducts, respectively, while signal at 943.223 m/z corresponds to six monomer units with chain ends of −OH and −COOH groups (chemical formula C50H46Cl6O3) and K+ adduct.
The introduction of oxygen to the surface of polymeric materials has tremendous practical implications for the biological performance.15 The oxygen-plasma-modified parylene C proved to be biocompatible and effectively limits the microbial biofilm formation.10 The main application of parylene C is an anticorrosive conformal coating of the orthopedic metal implants. Hence, the investigation of the body fluid–implant interface, implicating the bone–implant integration, is an inseparable aspect of the coating biocompatibility. One of the straightforward descriptors of in vitro osseointegration potential is based on the calcium phosphate (CaP) formation on the implant surface. Here, the calcium phosphate formation was evaluated using the DPBS buffer commonly employed in biological experiments for its osmolarity and ion concentration matching those of the human body fluids.
Upon exposure to DPBS, the crystallites of calcium phosphate are formed on the surfaces of parylene C after 24 h of incubation. The representative results are summarized in Figure 2, where scanning electron microscopy (SEM) images (Figure 2A) of the calcium phosphate crystallites on unmodified and oxygen-plasma-modified parylene C are presented. The formed calcium phosphate moieties significantly differ in morphology. On unmodified parylene C, there are few, large crystallites 1–3 μm in diameter, in contrast with the oxygen-plasma-modified samples, where a bigger number of crystallites, with significantly smaller sizes 200–500 nm, can be observed. The results of SEM observations are visualized in the size distribution histograms (Figure 2B) that clearly illustrate the effect of parylene C modification on the formed crystals. While on unmodified parylene C, the crystallites are polydisperse and in the wide range of sizes (0–2.4 μm); crystallites formed on the oxygen-plasma-modified parylene C are in a significantly narrower range of sizes (0–0.7 μm), with the majority being between 200 and 400 nm. As can be inferred from XRD patterns (Figure 2C), both types of samples support the growth of calcium phosphate crystallites. On both diffractograms, two types of maxima are present: one at 2θ = 13.4° characteristic for the crystalline part of parylene C and the other at 2θ = 31.4° diagnostic for calcium phosphate (P63/m space groups), which correspond to the hexagonal hydroxyapatite structure (Ca10(PO4)6(OH)2 with the Ca/P ratio of 1.67).66 The XRD results are in line with SEM observations, as the diffraction maximum (211)67 is less pronounced and slightly broader for the calcium phosphate crystallites formed at oxygen-plasma-modified parylene C.
Figure 2.
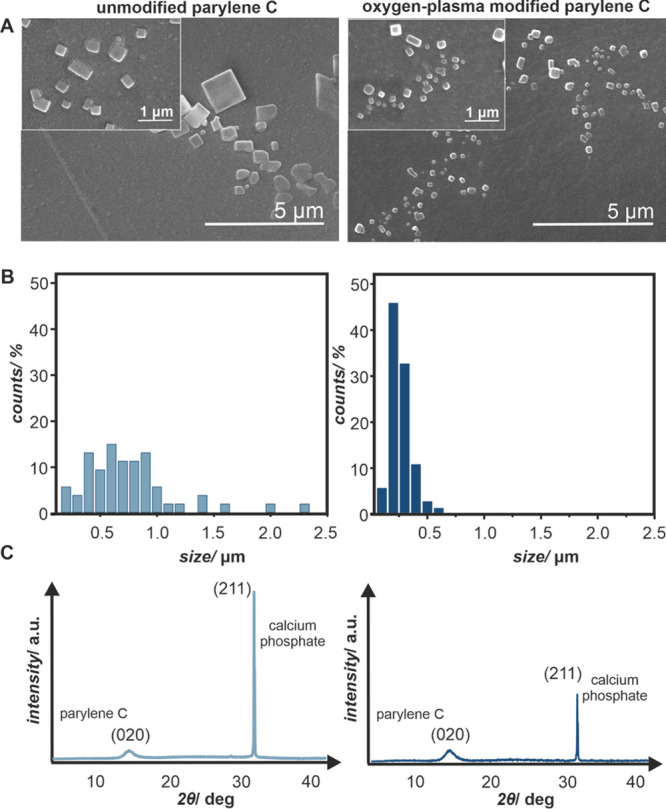
(A) SEM images of calcium phosphate crystallites formed after 72 h of incubation with DPBS on the unmodified and oxygen-plasma-modified parylene C surfaces, (B) histograms quantifying the differences in size distribution of the crystallites, and (C) XRD patterns with characteristic diffraction peaks for the crystalline part of parylene C 2θ = 13.4° and calcium phosphate 2θ = 31.4°.
In order to estimate the crystallite sizes and compare them to those observed in SEM (Figure 2), the XRD data were used (Figure 3A), while the chemical compositions of the formed crystallites were confirmed with XRF measurements (Figure 3B). The changes were recorded for 7 days in order to investigate the growth kinetics of the calcium phosphate on the parylene C surfaces. As summarized in Figure 3, the oxygen-plasma modification of parylene C has a strong effect on calcium phosphate formation. Native parylene C samples support the growth of calcium phosphate and allow the rapid formation of large crystallites, while oxygen plasma modification significantly diminishes the process; nevertheless, it ensures constant crystallite growth over the course of incubation time. Although the general trend is the same, there is a large difference in the crystallite sizes observed in SEM and estimated from XRD. The likely explanation is the crystalline–amorphous nature of the formed calcium phosphate and invisibility of the amorphous component to the X-rays. The amorphousness of the formed calcium phosphate is important from the biological point of view because it is known that at the beginning of the bone mineralization, the initially amorphous, mineral particles are transformed into a crystalline mineral, which eventually turns into hydroxyapatite.68,69
Figure 3.

(A) Changes in calcium phosphate crystallites size and (B) calcium content on the surfaces of unmodified and oxygen-plasma-modified parylene C as a function of incubation time in the artificial body fluids (DPBS).
The investigated surfaces show different kinetics of calcium phosphate crystal growth, which has important implications, when biocompatibility is concerned. Large crystals of calcium phosphate are less stable at the native surface, and after 14 days of mineralization studies (data not shown), the overgrown calcium phosphate crystallites fall off the parylene C surface. In contrast, the oxygen-plasma-modified parylene surface, exhibiting the nanotopography, promotes the anchorage of the smaller crystallites within the nanocorrugations. Moreover, the systematic growth of calcium phosphate crystals over the course of time (Figure 3) is desired in the context of the bone-implant integration process, which usually takes place over weeks.
As described in the Introduction section, the chemical nature and surface coverage of the oxygen-containing groups may lead to specific biological responses, particularly in the context of osseointegration. Therefore, understanding the interactions between ions present in the body fluid (Ca2+, HxPO4Δ−), leading to the formation of calcium phosphate precipitates, is of key importance for designing novel biomaterial surfaces. Specifically, the polar surface functional groups may play the role of sites, where the crystallization process initiates.70 To optimize the implant–tissue interface, the understanding of the calcium phosphate nucleation process at the molecular level is necessary and can be provided by MD simulations.
Models of unmodified and oxygen-plasma-modified parylene C surfaces were constructed as in our previous study.47 These models were successfully used for assessing water–surface interactions, and the obtained results were in good agreement with experiments. As visualized in Figure 4A, the unmodified parylene C surface contains native −Cl groups, whereas the oxygen plasma modification is mimicked by exchanging them for oxygen-containing groups, such as −OH, −CHO, and −COO–. The rationale for these substitutions is based on the surface characterization via XPS and LDI-TOF-MS (Figure 1). In this study, the model is extended by the introduction of the ions present in DPBS (H2PO4–, HPO42–, Na+, Ca2+, Cl–) to the aqueous phase in the simulation box. This extension allows us to model the interactions between parylene surfaces and the artificial body fluid. The overview of the simulated system is shown in Figure 4B, where a representative snapshot of the OH-functionalized parylene C surface in contact with water molecules and artificial body fluid ions is presented.
Figure 4.
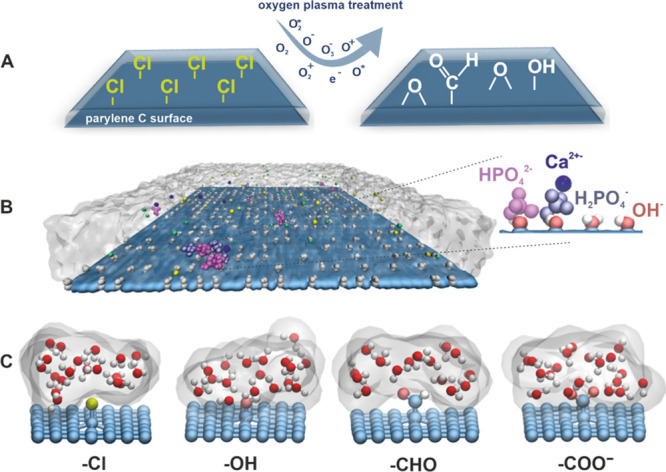
(A) Scheme of modification of the parylene C surface via oxygen plasma treatment, (B) model of the parylene C surface decorated with −OH groups used in MD simulations (the whole simulation box is shown in the Supporting Information, Figure S1), and (C) representative fragments of decorated parylene C surfaces showing interaction of various surface groups with water molecules. Gray shadowing based on isovalues of water density illustrates the hydrophilic properties of the parylene C surfaces.
In Figure 4C, individual fragments of decorated parylene surfaces, showing the interaction of various surface groups with water molecules, are presented in atomistic resolution. Qualitative differences in wetting properties emerging from the chemical character of the surface moieties are visualized in terms of isovalues of water density, represented as gray shadowing. A more in-depth insight into the water– and ion–surface interactions may be obtained from the mass density profile analysis (Figure 5). The profiles quantify the concentration of several important moieties as a function of the distance from the surface averaged over simulated surface area and time. In particular, HPO42– and H2PO4– (red and gray curves) adsorb relatively well on oxygen-plasma-modified parylene C, in contrast to the native parylene C. The reason for that is an overall hydrophobic character of its native surface, as indicated by the water density profiles (Owater, light blue curve), with the first water peak being the smallest among all considered surfaces. However, the hydrophilicity alone is not a good indicator of surface–phosphate affinity, as shown by the −COO–-decorated surface for which a weak binding of HPO42– and H2PO4– occurs; this is caused by the competitive binding of Na+. Importantly, Ca2+ (pink curve) and HPO42– (red curve) are almost completely depleted near the unmodified parylene C surface. In contrast, the Ca2+ and HPO42– binding takes place in all of the hydrophilized versions of parylene C, which is further corroborated by the analysis of the contacts per surface (see the Supporting Information, Figure S2).
Figure 5.

Number density profile comparison between unmodified parylene C (with parent −Cl groups) and oxygen-plasma-modified parylene C (−OH, −CHO, −COO–). For all of the oxygen-containing groups, the presented surface coverage is 50%. Profiles of water oxygen and surface groups were scaled down for presentation purposes.
The number density profiles clearly indicate that there is colocalization of phosphate and calcium ions, as their corresponding density profiles significantly overlap. To quantify the effect of colocalization, we calculated the number of ion–ion contacts for all considered surfaces; the data are presented in Figure 6. The number of contacts has been calculated using the standard GROMACS tool (gmx mindist). A contact was recorded when the distance between the center of mass of any two ions was equal to or within 1.0 nm. Ca2+–HPO42– pairs dominate in most of the systems, with the exception of the surface covered by −COO–, where a competitive binding of Ca2+ with surface carboxyl groups occurs (data not shown). Notably, both Ca2+–H2PO4– and H2PO4––HPO42– contacts, which are important for calcium phosphate formation, are enhanced at the surface covered by −OH groups.
Figure 6.
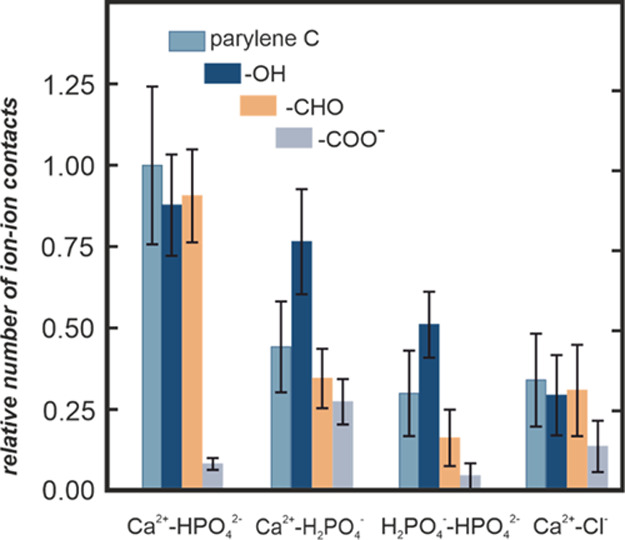
Relative number of ion–ion contacts for the considered unmodified and modified parylene C surfaces calculated from MD simulations for the equilibrated system. Error bars correspond to standard deviation.
The number density profiles and the number of surface contacts presented in Figures 5 and 6, respectively, provide the information averaged in the surface plane. In order to get laterally resolved information and to identify different binding motifs and nucleation of calcium phosphate, the simulation snapshots were visually analyzed; representative snapshots are presented in Figure 7. Further, to pinpoint individual steps leading to nucleation and to elucidate the nucleation mechanism, ion–surface and ion–ion contacts were analyzed in time along the simulated trajectories as a function of simulation time (the data are presented in Figures S3 and S4).
Figure 7.
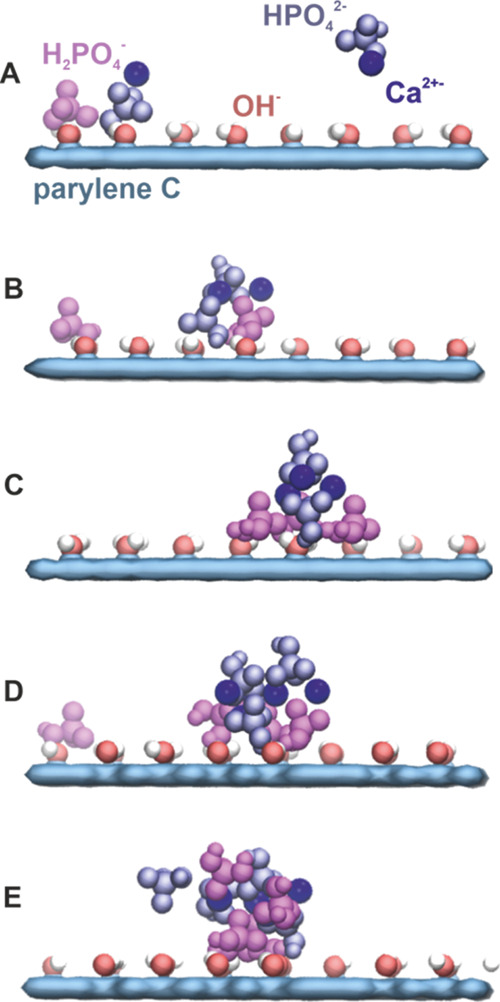
Nucleation model of the calcium phosphate crystal at the oxygen-plasma −OH-modified parylene C surface. Characteristic snapshots taken along a single atomistic MD simulation represent the subsequent stages of the nucleation process (A–E) (detailed description in the text).
As shown in Figure 7, for the oxygen-plasma −OH-modified parylene C surface, most of H2PO4– ions initially adsorb at the surface accompanied by Ca2+ and some HPO42– ions. The adsorbed ions are mobile and laterally diffuse at the parylene surface. Majority of HPO42– ions initially predominantly stay above the surface in the water phase (Figure 7A). Subsequently, individual ions adsorb at the surface and form Ca2+-mediated clusters with H2PO42– (Figure 7B). The adsorbed surface clusters grow, mostly, by capturing the surface diffusing HPO42–, H2PO4–, and Ca2+ (Figure 7C). The clusters further grow by capturing ions from both the surface and aqueous phase (Figure 7D). At this stage, the small surface clusters are to some extent dynamic, undergoing structural rearrangements; they consist of coclustered HPO42–, H2PO4–, and Ca2+ ions (Figure 7E). We hypothesize that these coclustered structures are early-stage calcium phosphate nucleation centers. Regarding lateral mobility of adsorbed species, a rigorous analysis of the diffusion coefficients was not possible because of a low concentration of ions and hence a limited diffusion statistic. It can be roughly estimated that the adsorbed species diffuse on the length scale of about 10 nm over the 200 ns long trajectories.
In the case of the −CHO-modified parylene C surface, the processes occurring at the surface are the same but somewhat faster. In particular, the steady increase of Ca2+–HPO42– contacts (Supporting Information, Figure S4C) is due to the formation of these ion pairs in water, which only subsequently slowly adsorb at the surface. In the case of the surface covered by −COO–, the surface is practically polluted by Ca2+ ions. HPO42– adsorbs first but without forming Ca2+–HPO42– clusters. The subsequently adsorbed H2PO4– is mostly laterally immobilized, often clustered with Ca2+, but not with HPO42–. Hence, calcium phosphate nucleation centers do not form. Of note, the surface covered by −COO– groups should be treated only as a qualitative model of a strongly polar and hydrophilic surface.
HPO42– does not adsorb at the unmodified parylene C surface. The Ca2+–HPO42– and H2PO4––HPO42– contacts, visible in Figure S4D, are due to ion pairs in water. This shows that the unmodified parylene C surface does not promote the calcium phosphate formation on the timescale considered in MD.
The obtained results clearly explain the necessity of parylene C functionalization in order to obtain the biocompatible surface for bone–tissue contact. The preferred type of oxygen-containing groups is −OH, responsible for the calcium phosphate nucleation, as confirmed by MD simulations (Ca2+–H2PO4– cluster formation). The superiority of surface hydroxyl groups, as adsorption centers for calcium phosphate, might be related to their high dipole moments and small size, which results in strong surface–ion interactions without sterical hindrance.
It should be noted that the exact mechanism and initial stages of hydroxyapatite nucleation in bulk water solutions are still debated and were recently intensively studied by detailed molecular simulations.31−35 In particular, the formation of initial Ca2+–(HPO42–)n clusters, probably kinetically trapped, was reported, and their role is being studied. It was also observed that the same charge phosphate ions could form clusters mediated by cations. Our work focuses on the very onset of hydroxyapatite clustering at the decorated surfaces. Only small clusters are studied here because of a limited concentration of the species mimicking the human body fluids. Still, in relation to the studies in bulk solutions, our simulations confirm that Ca2+–HPO42– interactions are crucial for the formation of clusters at the surface. Overall, it can be concluded that by changing the delicate balance between interactions involving all present ions, the surface can alter the clustering mechanisms involving calcium and phosphate ions.
In this study, we took a synergetic approach by complementing experimental results with MD simulations. This allowed us to investigate the parylene C-simulated body fluid interactions at both molecular and microscopic scales. Such methodology is indispensable for a detailed description of the calcium phosphate formation onset and the effect of oxygen-containing surface functional groups. As a result, we were able to identify the contribution of the formed groups to the overall biocompatibility increase of the oxygen-plasma-modified parylene C, as well as premises for surface functionalization. As reported before, the presence of the −OH groups at the surface is crucial for wettability47 and biocompatibility10 of parylene C. Here, for the first time, the role of surface functional groups for the nucleation process of calcium phosphate on parylene C is reported and described at the atomistic level with the use of MD simulations. Such a combined experimental and theoretical approach, presented in this work, paves the way for rational design and functionalization of polymeric biomaterials toward effective osseointegration.
Conclusions
In this paper, we address the role of functional groups on the surface of parylene C in calcium phosphate formation as the descriptor of the polymeric coating–bone tissue compatibility. The functional groups were introduced via oxygen plasma treatment, and their chemical nature was characterized by XPS and LDI-TOF. The experiments revealed the dramatic difference in calcium phosphate crystallite formation upon contact with the simulated body fluid (DPBS). At the unmodified parylene C surface, after 7 days of incubation, the crystallites were larger and unstable, whereas at the plasma-modified surface, the desired, more uniform coverage of smaller crystallites <0.7 μm was observed. To understand the effect of functionalization and elucidate the role of functional groups in the calcium phosphate nucleation mechanism, we applied atomistic MD simulations. The simulation results revealed differences in the osseointegration potential of the surface functional groups: −OH > −CHO > −Cl (parent parylene C) ≈ −COO–. The superiority of the −OH groups was identified as the most effective sites for calcium phosphate nucleation. The advantage of the combined experimental and theoretical approach is pointed out as effective for biointerface design and fabrication.
Acknowledgments
We acknowledge the financial support of the Academy of Finland Center of Excellence program (grant no. 307415 (W.K.)). CSC-IT Centre for Science (Espoo, Finland; project numbers tty3995 and tty8088) and the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533) are acknowledged for excellent computational resources. L.C. acknowledges the support from NF Neuron and the Czech Science Foundation (grant 17-06792S)
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.9b20877.
Whole simulation box, comparison of number of contacts per surface unit for Ca2+ and HPO42– ions, ion–surface contacts per surface unit as a function of simulation time, ion–ion contacts per surface unit as a function of simulation time, free energy profiles showing the potential of mean force between phosphate and calcium ions, and force-field parameters for ions used in this work (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Keum H.; Kim J. Y.; Yu B.; Yu S. J.; Kim J.; Jeon H.; Lee D. Y.; Im S. G.; Jon S. Prevention of Bacterial Colonization on Catheters by a One-Step Coating Process Involving an Antibiofouling Polymer in Water. ACS Appl. Mater. Interfaces 2017, 9, 19736–19745. 10.1021/acsami.7b06899. [DOI] [PubMed] [Google Scholar]
- Kim Y. H.; Park J.; Koo H.; Kim M. S.; Jung S.-D. Fluoropolymer-Based Flexible Neural Prosthetic Electrodes for Reliable Neural Interfacing. ACS Appl. Mater. Interfaces 2017, 9, 43420–43428. 10.1021/acsami.7b12364. [DOI] [PubMed] [Google Scholar]
- Green R.; Abidian M. R. Conducting Polymers for Neural Prosthetic and Neural Interface Applications. Adv. Mater. 2015, 27, 7620–7637. 10.1002/adma.201501810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose C. G.; Hartline B. E.; Clanton T. O.; Lowe W. R.; McGarvey W. C.. Polymers in Orthopaedic Surgery. Advanced Polymers in Medicine; Springer International Publishing: Cham, 2015; pp 129–145. [Google Scholar]
- Kuppusami S.; Oskouei R. H. Parylene Coatings in Medical Devices and Implants: A Review. Univers. J. Biomed. Eng. 2015, 3, 9–14. 10.13189/ujbe.2015.030201. [DOI] [Google Scholar]
- Cieślik M.; Zimowski S.; Gołda M.; Engvall K.; Pan J.; Rakowski W.; Kotarba A. Engineering of Bone Fixation Metal Implants Biointerface - Application of Parylene C as Versatile Protective Coating. Mater. Sci. Eng., C 2012, 32, 2431–2435. 10.1016/j.msec.2012.07.018. [DOI] [Google Scholar]
- Hassler C.; von Metzen R. P.; Ruther P.; Stieglitz T. Characterization of Parylene C as an Encapsulation Material for Implanted Neural Prostheses. J. Biomed. Mater. Res., Part B 2010, 9999B, 266. 10.1002/jbm.b.31584. [DOI] [PubMed] [Google Scholar]
- Cieślik M.; Kot M.; Reczyński W.; Engvall K.; Rakowski W.; Kotarba A. Parylene Coatings on Stainless Steel 316L Surface for Medical Applications - Mechanical and Protective Properties. Mater. Sci. Eng., C 2012, 32, 31–35. 10.1016/j.msec.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Golda-Cepa M.; Engvall K.; Kotarba A. Development of Crystalline–Amorphous Parylene C Structure in Micro- and Nano-Range towards Enhanced Biocompatibility: The Importance of Oxygen Plasma Treatment Time. RSC Adv. 2015, 5, 48816–48821. 10.1039/c5ra06366c. [DOI] [Google Scholar]
- Golda-Cepa M.; Chorylek A.; Chytrosz P.; Brzychczy-Wloch M.; Jaworska J.; Kasperczyk J.; Hakkarainen M.; Engvall K.; Kotarba A. Multifunctional PLGA/Parylene C Coating for Implant Materials: An Integral Approach for Biointerface Optimization. ACS Appl. Mater. Interfaces 2016, 8, 22093–22105. 10.1021/acsami.6b08025. [DOI] [PubMed] [Google Scholar]
- Staufert S.; Gutzwiller P.; Mushtaq F.; Hierold C. Surface Nanostructuring of Ti6Al4V Surfaces for Parylene-C Coatings with Ultradurable Adhesion. ACS Appl. Nano Mater. 2018, 1, 1586–1594. 10.1021/acsanm.8b00081. [DOI] [Google Scholar]
- Golda-Cepa M.; Chytrosz P.; Chorylek A.; Kotarba A. One-Step Sonochemical Fabrication and Embedding of Gentamicin Nanoparticles into Parylene C Implant Coating: Towards Controlled Drug Delivery. Nanomedicine 2018, 14, 941–950. 10.1016/j.nano.2018.01.012. [DOI] [PubMed] [Google Scholar]
- Xie X.; Rieth L.; Williams L.; Negi S.; Bhandari R.; Caldwell R.; Sharma R.; Tathireddy P.; Solzbacher F. Long-Term Reliability of Al2O3 and Parylene C Bilayer Encapsulated Utah Electrode Array Based Neural Interfaces for Chronic Implantation. J. Neural. Eng. 2014, 11, 026016. 10.1088/1741-2560/11/2/026016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B. J.; Meng E. Micromachining of Parylene C for BioMEMS. Polym. Adv. Technol. 2016, 27, 564–576. 10.1002/pat.3729. [DOI] [Google Scholar]
- Gentleman M. M.; Gentleman E. The Role of Surface Free Energy in Osteoblast–Biomaterial Interactions. Int. Mater. Rev. 2014, 59, 417–429. 10.1179/1743280414Y.0000000038. [DOI] [Google Scholar]
- Yang Y.; Qi P.; Wen F.; Li X.; Xia Q.; Maitz M. F.; Yang Z.; Shen R.; Tu Q.; Huang N. Mussel-Inspired One-Step Adherent Coating Rich in Amine Groups for Covalent Immobilization of Heparin: Hemocompatibility, Growth Behaviors of Vascular Cells, and Tissue Response. ACS Appl. Mater. Interfaces 2014, 6, 14608–14620. 10.1021/am503925r. [DOI] [PubMed] [Google Scholar]
- Brancato L.; Decrop D.; Lammertyn J.; Puers R.; Brancato L.; Decrop D.; Lammertyn J.; Puers R. Surface Nanostructuring of Parylene-C Coatings for Blood Contacting Implants. Materials 2018, 11, 1109. 10.3390/ma11071109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trantidou T.; Rao C.; Barrett H.; Camelliti P.; Pinto K.; Yacoub M. H.; Athanasiou T.; Toumazou C.; Terracciano C. M.; Prodromakis T. Selective Hydrophilic Modification of Parylene C Films: A New Approach to Cell Micro-Patterning for Synthetic Biology Applications. Biofabrication 2014, 6, 025004. 10.1088/1758-5082/6/2/025004. [DOI] [PubMed] [Google Scholar]
- Lin J.; Chen X.; Chen C.; Hu J.; Zhou C.; Cai X.; Wang W.; Zheng C.; Zhang P.; Cheng J.; Guo Z.; Liu H. Durably Antibacterial and Bacterially Antiadhesive Cotton Fabrics Coated by Cationic Fluorinated Polymers. ACS Appl. Mater. Interfaces 2018, 10, 6124–6136. 10.1021/acsami.7b16235. [DOI] [PubMed] [Google Scholar]
- Gołda M.; Brzychczy-Włoch M.; Faryna M.; Engvall K.; Kotarba A. Oxygen Plasma Functionalization of Parylene C Coating for Implants Surface: Nanotopography and Active Sites for Drug Anchoring. Mater. Sci. Eng., C 2013, 33, 4221–4227. 10.1016/j.msec.2013.06.014. [DOI] [PubMed] [Google Scholar]
- Golda-Cepa M.; Brzychczy-Wloch M.; Engvall K.; Aminlashgari N.; Hakkarainen M.; Kotarba A. Microbiological Investigations of Oxygen Plasma Treated Parylene C Surfaces for Metal Implant Coating. Mater. Sci. Eng., C 2015, 52, 273–281. 10.1016/j.msec.2015.03.060. [DOI] [PubMed] [Google Scholar]
- Cheruthazhekatt S.; Černák M.; Slavíček P.; Havel J. Gas Plasmas and Plasma Modified Materials in Medicine. J. Appl. Biomed. 2010, 8, 55–66. 10.2478/V10136-009-0013-9. [DOI] [Google Scholar]
- Nandakumar A.; Tahmasebi Birgani Z.; Santos D.; Mentink A.; Auffermann N.; van der Werf K.; Bennink M.; Moroni L.; van Blitterswijk C.; Habibovic P. Surface Modification of Electrospun Fibre Meshes by Oxygen Plasma for Bone Regeneration. Biofabrication 2012, 5, 015006. 10.1088/1758-5082/5/1/015006. [DOI] [PubMed] [Google Scholar]
- Jacobs T.; Morent R.; De Geyter N.; Dubruel P.; Leys C. Plasma Surface Modification of Biomedical Polymers: Influence on Cell-Material Interaction. Plasma Chem. Plasma Process. 2012, 32, 1039–1073. 10.1007/s11090-012-9394-8. [DOI] [Google Scholar]
- Kini U.; Nandeesh B. N.. Physiology of Bone Formation, Remodeling, and Metabolism. Radionuclide and Hybrid Bone Imaging; Springer Berlin Heidelberg: Berlin, Heidelberg, 2012; pp 29–57. [Google Scholar]
- Huang L.; Zhou B.; Wu H.; Zheng L.; Zhao J. Effect of Apatite Formation of Biphasic Calcium Phosphate Ceramic (BCP) on Osteoblastogenesis Using Simulated Body Fluid (SBF) with or without Bovine Serum Albumin (BSA). Mater. Sci. Eng., C 2017, 70, 955–961. 10.1016/j.msec.2016.05.115. [DOI] [PubMed] [Google Scholar]
- Kim H. D.; Amirthalingam S.; Kim S. L.; Lee S. S.; Rangasamy J.; Hwang N. S. Biomimetic Materials and Fabrication Approaches for Bone Tissue Engineering. Adv. Healthcare Mater. 2017, 6, 1700612. 10.1002/adhm.201700612. [DOI] [PubMed] [Google Scholar]
- Shih Y.-R. V.; Hwang Y.; Phadke A.; Kang H.; Hwang N. S.; Caro E. J.; Nguyen S.; Siu M.; Theodorakis E. A.; Gianneschi N. C.; Vecchio K. S.; Chien S.; Lee O. K.; Varghese S. Calcium Phosphate-Bearing Matrices Induce Osteogenic Differentiation of Stem Cells through Adenosine Signaling. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 990–995. 10.1073/PNAS.1321717111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habraken W. J. E. M.; Tao J.; Brylka L. J.; Friedrich H.; Bertinetti L.; Schenk A. S.; Verch A.; Dmitrovic V.; Bomans P. H. H.; Frederik P. M.; Laven J.; van der Schoot P.; Aichmayer B.; de With G.; DeYoreo J. J.; Sommerdijk N. A. J. M.. Ion-Association Complexes Unite Classical and Non-Classical Theories for the Biomimetic Nucleation of Calcium Phosphate. Nat. Commun. 2013, 4. 10.1038/ncomms2490. [DOI] [PubMed] [Google Scholar]
- Xie B.; Halter T. J.; Borah B. M.; Nancollas G. H. Tracking Amorphous Precursor Formation and Transformation during Induction Stages of Nucleation. Cryst. Growth Des. 2014, 14, 1659–1665. 10.1021/cg401777x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancardi G.; Terranova U.; de Leeuw N. H. Calcium Phosphate Prenucleation Complexes in Water by Means of Ab Initio Molecular Dynamics Simulations. Cryst. Growth Des. 2016, 16, 3353–3358. 10.1021/acs.cgd.6b00327. [DOI] [Google Scholar]
- Garcia N. A.; Malini R. I.; Freeman C. L.; Demichelis R.; Raiteri P.; Sommerdijk N. A. J. M.; Harding J. H.; Gale J. D. Simulation of Calcium Phosphate Prenucleation Clusters in Aqueous Solution: Association beyond Ion Pairing. Cryst. Growth Des. 2019, 19, 6422–6430. 10.1021/acs.cgd.9b00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti Malini R.; Freeman C. L.; Harding J. H. Interaction of Stable Aggregates Drives the Precipitation of Calcium Phosphate in Supersaturated Solutions. CrystEngComm 2019, 21, 6354–6364. 10.1039/c9ce00658c. [DOI] [Google Scholar]
- Mancardi G.; Hernandez Tamargo C. E.; Di Tommaso D.; De Leeuw N. H. Detection of Posner’s Clusters during Calcium Phosphate Nucleation: A Molecular Dynamics Study. J. Mater. Chem. B 2017, 5, 7274–7284. 10.1039/c7tb01199g. [DOI] [PubMed] [Google Scholar]
- Yang X.; Wang M.; Yang Y.; Cui B.; Xu Z.; Yang X. Physical Origin Underlying the Prenucleation-Cluster-Mediated Nonclassical Nucleation Pathways for Calcium Phosphate. Phys. Chem. Chem. Phys. 2019, 21, 14530–14540. 10.1039/c9cp00919a. [DOI] [PubMed] [Google Scholar]
- da Silva L. C. E.; Más B. A. M.; Duek E. A. R.; Landers R.; Bertran C. A.; Gonçalves M. C. Amphiphilic Nucleating Agents to Enhance Calcium Phosphate Growth on Polymeric Surfaces. Langmuir 2017, 33, 3855. 10.1021/acs.langmuir.6b04562. [DOI] [PubMed] [Google Scholar]
- Surmenev R. A.; Surmeneva M. A.; Ivanova A. A. Significance of Calcium Phosphate Coatings for the Enhancement of New Bone Osteogenesis – A Review. Acta Biomater. 2014, 10, 557–579. 10.1016/J.ACTBIO.2013.10.036. [DOI] [PubMed] [Google Scholar]
- Wei Q.; Wang Y.; Chai W.; Zhang Y.; Chen X. Molecular Dynamics Simulation and Experimental Study of the Bonding Properties of Polymer Binders in 3D Powder Printed Hydroxyapatite Bioceramic Bone Scaffolds. Ceram. Int. 2017, 43, 13702–13709. 10.1016/j.ceramint.2017.07.082. [DOI] [Google Scholar]
- Karaman O.; Kumar A.; Moeinzadeh S.; He X.; Cui T.; Jabbari E. Effect of Surface Modification of Nanofibres with Glutamic Acid Peptide on Calcium Phosphate Nucleation and Osteogenic Differentiation of Marrow Stromal Cells. J. Tissue Eng. Regener. Med. 2016, 10, E132–E146. 10.1002/term.1775. [DOI] [PubMed] [Google Scholar]
- Wu L. N. Y.; Genge B. R.; Wuthier R. E. Analysis and Molecular Modeling of the Formation, Structure, and Activity of the Phosphatidylserine-Calcium-Phosphate Complex Associated with Biomineralization. J. Biol. Chem. 2007, 283, 3827. 10.1074/jbc.M707653200. [DOI] [PubMed] [Google Scholar]
- Tang R.; Darragh M.; Orme C. A.; Guan X.; Hoyer J. R.; Nancollas G. H. Control of Biomineralization Dynamics by Interfacial Energies. Angew. Chem., Int. Ed. 2005, 44, 3698–3702. 10.1002/anie.200500153. [DOI] [PubMed] [Google Scholar]
- Demichelis R.; Garcia N. A.; Raiteri P.; Innocenti Malini R.; Freeman C. L.; Harding J. H.; Gale J. D. Simulation of Calcium Phosphate Species in Aqueous Solution: Force Field Derivation. J. Phys. Chem. B 2018, 122, 1471–1483. 10.1021/acs.jpcb.7b10697. [DOI] [PubMed] [Google Scholar]
- Tian T.; Liao J.; Zhou T.; Lin S.; Zhang T.; Shi S.-R.; Cai X.; Lin Y. Fabrication of Calcium Phosphate Microflowers and Their Extended Application in Bone Regeneration. ACS Appl. Mater. Interfaces 2017, 9, 30437–30447. 10.1021/acsami.7b09176. [DOI] [PubMed] [Google Scholar]
- Duman E.; Şahin Kehribar E.; Ahan R. E.; Yuca E.; Şeker U. Ö. Ş. Biomineralization of Calcium Phosphate Crystals Controlled by Protein–Protein Interactions. ACS Biomater. Sci. Eng. 2019, 5, 4750. 10.1021/acsbiomaterials.9b00649. [DOI] [PubMed] [Google Scholar]
- Li M.; Wang L.; Putnis C. V. Energetic Basis for Inhibition of Calcium Phosphate Biomineralization by Osteopontin. J. Phys. Chem. B 2017, 121, 5968–5976. 10.1021/acs.jpcb.7b04163. [DOI] [PubMed] [Google Scholar]
- Xue Z.; Yang M.; Xu D. Nucleation of Biomimetic Hydroxyapatite Nanoparticles on the Surface of Type I Collagen: Molecular Dynamics Investigations. J. Phys. Chem. C 2019, 123, 2533. 10.1021/acs.jpcc.8b10342. [DOI] [Google Scholar]
- Golda-Cepa M.; Kulig W.; Cwiklik L.; Kotarba A. Molecular Dynamics Insights into Water-Parylene C Interface: Relevance of Oxygen Plasma Treatment for Biocompatibility. ACS Appl. Mater. Interfaces 2017, 9, 16685–16693. 10.1021/acsami.7b03265. [DOI] [PubMed] [Google Scholar]
- Tsai Y.-T.; Huang C.-W.; Liu H.-Y.; Huang M.-C.; Sun T.-P.; Chen W.-C.; Wu C.-Y.; Ding S.-T.; Chen H.-Y. Enhanced Bone Morphogenic Property of Parylene-C. Biomater. Sci. 2016, 4, 1754–1760. 10.1039/C6BM00664G. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Kohagen M.; Mason P. E.; Jungwirth P. Accounting for Electronic Polarization Effects in Aqueous Sodium Chloride via Molecular Dynamics Aided by Neutron Scattering. J. Phys. Chem. B 2016, 120, 1454–1460. 10.1021/acs.jpcb.5b05221. [DOI] [PubMed] [Google Scholar]
- Martinek T.; Duboué-Dijon E.; Timr Š.; Mason P. E.; Baxová K.; Fischer H. E.; Schmidt B.; Pluhařová E.; Jungwirth P. Calcium Ions in Aqueous Solutions: Accurate Force Field Description Aided by Ab Initio Molecular Dynamics and Neutron Scattering. J. Chem. Phys. 2018, 148, 222813. 10.1063/1.5006779. [DOI] [PubMed] [Google Scholar]
- Kirby B. J.; Jungwirth P. Charge Scaling Manifesto: A Way of Reconciling the Inherently Macroscopic and Microscopic Natures of Molecular Simulations. J. Phys. Chem. Lett. 2019, 10, 7531–7536. 10.1021/acs.jpclett.9b02652. [DOI] [PubMed] [Google Scholar]
- Kohagen M.; Mason P. E.; Jungwirth P. Accurate Description of Calcium Solvation in Concentrated Aqueous Solutions. J. Phys. Chem. B 2014, 118, 7902–7909. 10.1021/jp5005693. [DOI] [PubMed] [Google Scholar]
- Pluhařová E.; Mason P. E.; Jungwirth P. Ion Pairing in Aqueous Lithium Salt Solutions with Monovalent and Divalent Counter-Anions. J. Phys. Chem. A 2013, 117, 11766–11773. 10.1021/jp402532e. [DOI] [PubMed] [Google Scholar]
- Melcr J.; Martinez-Seara H.; Nencini R.; Kolafa J.; Jungwirth P.; Ollila O. H. S. Accurate Binding of Sodium and Calcium to a POPC Bilayer by Effective Inclusion of Electronic Polarization. J. Phys. Chem. B 2018, 122, 4546–4557. 10.1021/acs.jpcb.7b12510. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Busenberg E.; Niel Plummer L. Thermodynamics of Magnesian Calcite Solid-Solutions at 25°C and 1 Atm Total Pressure. Geochim. Cosmochim. Acta 1989, 53, 1189–1208. 10.1016/0016-7037(89)90056-2. [DOI] [Google Scholar]
- Antila H.; Buslaev P.; Favela-Rosales F.; Ferreira T. M.; Gushchin I.; Javanainen M.; Kav B.; Madsen J. J.; Melcr J.; Miettinen M. S.; Määttä J.; Nencini R.; Ollila O. H. S.; Piggot T. J. Headgroup Structure and Cation Binding in Phosphatidylserine Lipid Bilayers. J. Phys. Chem. B 2019, 123, 9066–9079. 10.1021/acs.jpcb.9b06091. [DOI] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- Miyamoto S.; Kollman P. A. SETTLE: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. 10.1002/jcc.540130805. [DOI] [Google Scholar]
- Golda M.; Brzychczy-Włoch M.; Faryna M.; Engvall K.; Kotarba A. Oxygen Plasma Functionalization of Parylene C Coating for Implants Surface: Nanotopography and Active Sites for Drug Anchoring. Mater. Sci. Eng., C 2013, 33, 4221–4227. 10.1016/j.msec.2013.06.014. [DOI] [PubMed] [Google Scholar]
- Golda-Cepa M.; Aminlashgari N.; Hakkarainen M.; Engvall K.; Kotarba A. LDI-MS Examination of Oxygen Plasma Modified Polymer for Designing Tailored Implant Biointerfaces. RSC Adv. 2014, 4, 26240–26243. 10.1039/c4ra02656j. [DOI] [Google Scholar]
- Rincón-López J.; Hermann-Muñoz J.; Giraldo-Betancur A.; De Vizcaya-Ruiz A.; Alvarado-Orozco J.; Muñoz-Saldaña J. Synthesis, Characterization and In Vitro Study of Synthetic and Bovine-Derived Hydroxyapatite Ceramics: A Comparison. Materials 2018, 11, 333. 10.3390/ma11030333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers R.; Nielsen-Preiss S.; Ferguson V.; Gotolli G.; Moore J. J.; Kleebe H.-J. Osteoblast-like Cell Mineralization Induced by Multiphasic Calcium Phosphate Ceramic. Mater. Sci. Eng., C 2006, 26, 1333–1337. 10.1016/j.msec.2005.08.028. [DOI] [Google Scholar]
- Mahamid J.; Sharir A.; Addadi L.; Weiner S. Amorphous Calcium Phosphate Is a Major Component of the Forming Fin Bones of Zebrafish: Indications for an Amorphous Precursor Phase. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 12748–12753. 10.1073/pnas.0803354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Nancollas G. H. Pathways to Biomineralization and Biodemineralization of Calcium Phosphates: The Thermodynamic and Kinetic Controls. Dalton Trans. 2009, 15, 2665–2672. 10.1039/b815887h. [DOI] [PubMed] [Google Scholar]
- Cui W.; Li X.; Xie C.; Zhuang H.; Zhou S.; Weng J. Hydroxyapatite Nucleation and Growth Mechanism on Electrospun Fibers Functionalized with Different Chemical Groups and Their Combinations. Biomaterials 2010, 31, 4620–4629. 10.1016/j.biomaterials.2010.02.050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.