

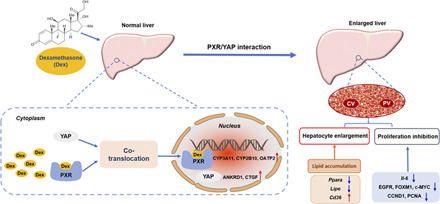
Abstract
Dexamethasone (Dex), a widely prescribed anti-inflammatory drug, was reported to induce liver enlargement (hepatomegaly) in clinical practice and in animal models. However, the underlying mechanisms are not elucidated. Dex is a known activator of pregnane X receptor (PXR). Yes-associated protein (YAP) has been implicated in chemically induced liver enlargement. Here, the roles of PXR and YAP pathways were investigated in Dex-induced hepatomegaly. Upregulation of PXR downstream proteins, including cytochrome P450 (CYP) 3A11, 2B10, and organic anion transporter polypeptide 2 (OATP2) (also known as SLCO1A4 and SLC21A5 in mouse), indicated PXR signaling was activated after high dose of Dex (50 mg/kg, i.p.), and Dex at 100 μM activated PXR in the dual-luciferase reporter gene assay. Dex also increased the expression of total YAP, nuclear YAP, and YAP downstream proteins, including connective tissue growth factor and cysteine-rich angiogenic inducer 61, indicating activation of the YAP pathway. Furthermore, nuclear translocation of YAP was promoted by activation of PXR. However, hepatocyte proliferation was inhibited with significant decrease in the expression of proliferation-related proteins cyclin D1 and proliferating cell nuclear antigen as well as other regulatory factors, such as forkhead box protein M1, c-MYC, and epidermal growth factor receptor. The inhibitory effect of Dex on hepatocyte proliferation was likely due to its anti-inflammation effect of suppression of inflammation factors. β-catenin staining revealed enlarged hepatocytes, which were mostly attributable to the accumulation of lipids, such as triglycerides. In summary, high-dose Dex increased liver size accompanied by enlarged hepatocytes, and this was due to the activation of PXR/YAP and their effects on lipid accumulation but not hepatocyte proliferation. These findings provide new insights for understanding the mechanism of Dex-induced hepatomegaly.
SIGNIFICANCE STATEMENT
This study identified the roles of pregnane X receptor (PXR) and yes-associated protein (YAP) pathways in dexamethasone (Dex)-induced hepatomegaly. Dex induced PXR/YAP activation, enlarged hepatocytes, and promoted liver enlargement with lipid accumulation, such as triglycerides. However, hepatocyte proliferation was inhibited by the anti-inflammatory effect of Dex. These findings provide new insights for understanding the mechanism of Dex-induced hepatomegaly.
Introduction
Dexamethasone (Dex), 16α-methyl 9α-fluoroprednisolone, is a synthetic glucocorticoid. Owing to its potency for anti-inflammation, Dex has been widely applied in clinic for decades as a treatment of diverse inflammatory and autoimmune diseases (e.g., rheumatic arthritis, asthma, allergy, and transplantation rejection) (Rhen and Cidlowski, 2005; Ericson-Neilsen and Kaye, 2014). Dex also has an influence on the physiologic process of metabolism of endogenous compounds, including lipids (Harasim-Symbor et al., 2016), glucose (Cui et al., 2019), and bile acids (Glantz et al., 2005). Most of the physiologic effects of Dex are mediated through the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily of transcription factors (Petta et al., 2016). Apart from GR, Dex is also an agonist of the nuclear receptor pregnane X receptor (PXR) and can induce specific drug-metabolizing enzymes, such as cytochrome P450 (CYP) 3A4 (Cheng et al., 2005; Buckley and Klaassen, 2009). Different dose and frequency of Dex treatment can lead to various side effects, which may be due to activation of GR or PXR. It was reported that high-dose Dex treatment induced hepatomegaly accompanied by increased glycogen and lipids contents in clinical cases (Verrips et al., 1998). Consistently, a similar phenomenon was observed in animal models (Thatcher and Caldwell, 1994; Micuda et al., 2007). However, the related mechanisms of the Dex-induced hepatomegaly remain unknown and need to be clarified.
In mammals, the liver plays a crucial role in xenobiotic metabolism including drugs and chemicals, which puts an emphasis on maintaining a constant liver mass and size. As a rapid response system that is responsible for the stability of normal homeostasis and tight control of biologic parameters, the liver regulates its size not only by genetic factors but by a range of stimuli, such as hormonal fluctuations during pregnancy and lactation, acute-phase proteins stimulated by infections by viruses and bacteria, and enzymes induced by xenobiotics (Botts et al., 2010). The response of the liver to these different stimuli may include an increase in the size and capacity of function of the liver due to an increase in the size and number of hepatocytes, which is also defined as hepatocyte hypertrophy and hepatocellular hyperplasia (Maronpot et al., 2010).
Because of the flexible pocket domain, PXR can bind to a variety of structurally diverse ligands, including steroids, bile acids, and antibiotics, such as rifampicin (RIF) (Kliewer et al., 2002). PXR is expressed in the liver and plays a vital role in controlling the transport and metabolism of xenobiotics as well as maintaining the homeostasis of certain endobiotics (Kliewer et al., 1998; Xie et al., 2000). Pregnenolone-16α-carbonitrile, a PXR agonist, induces remarkable liver enlargement. Most recently, PXR was found to regulate liver size by interaction with yes-associated protein (YAP) (Jiang et al., 2019), indicating a critical role for YAP in the liver enlargement induced by PXR activation. YAP, the core component of Hippo-YAP pathway, plays an important role in controlling organ growth, including the size of liver (Dong et al., 2007). Hyperactivation of YAP can cause hepatomegaly with increased numbers of hepatocytes (Patel et al., 2017). A previous study also showed that high expression of YAP could result in potent cell enlargement (Tumaneng et al., 2012).
Based on the information mentioned above, we hypothesized that Dex-induced liver enlargement may be related to the activation of PXR and its interaction with YAP. Therefore, this study aimed to investigate whether PXR and YAP are involved in this process and explore the changes of lipids in Dex-induced hepatomegaly.
Materials and Methods
Chemicals and Reagents.
Dex with 98% purity (Cat. D137736) and corn oil (Cat. C116025) were purchased from Aladdin Biotechnology (Aladdin Industrial Corporation, Shanghai, China). RIF with 97% purity (Cat. R3501) was acquired from Sigma-Aldrich (Sigma, St. Louis, MO). Rabbit monoclonal anti–cyclin (CCN) D1 (Cat. 2978T), anti-CCNE1 (Cat. 20808S), anti–proliferating cell nuclear antigen (PCNA) (Cat. 13110S), anti-c-MYC (Cat. 13987S), anti-β-catenin (CTNNB1) (Cat. 8480S), anti-YAP (Cat. 14074S), and anti-β-actin (Cat. 4970S) antibodies were all obtained from Cell Signaling Technology (MA). Mouse monoclonal anti-CYP3A4 (Cat. sc-53850) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal anti-CYP2B6 (Cat. A1463) antibody was purchased from ABclonal Technology (Wuhan, China). Rabbit polyclonal anti-organic anion transporter polypeptide 2 (OATP2) (Cat. DF4534) antibody was obtained from Affinity Biosciences (Changzhou, China). Rabbit polyclonal anti-CCNA1 (Cat. D220507), anti–phosphorylated YAP (Cat. D151452), anti–ankyrin repeat domain 1 (Cat. D121628), anti-cysteine-rich angiogenic inducer 61 (Cat. D122190), anti–connective tissue growth factor (Cat. D160212), anti–epidermal growth factor receptor (EGFR) (Cat. D260292), anti–mesenchymal-epithelial transition factor (MET) (Cat. D160981), and anti–lamin B1 (Cat. D220926) antibodies were acquired from Sangon Biotechnology (Sangon Tech, Shanghai, China). Rabbit polyclonal anti–forkhead box protein (FOX) M1 antibody (Cat. 13147-1-AP) was obtained from Proteintech Group, Inc (Wuhan, China). Peroxidase-conjugated anti-rabbit IgG antibody (Cat. 7074S) and anti-mouse (Cat. 7076S) were purchased from Cell Signaling Technology. For the use of immunohistochemical staining, rabbit monoclonal Ki67 antibody (Cat. ab16667) was purchased from Abcam, and purified mouse monoclonal CTNNB1 antibody (Cat. 610153) was from BD Biosciences (San Jose, CA). For the staining of co-location, rabbit polyclonal anti-PXR (Cat. sc-25381) and mouse monoclonal anti-YAP (Cat. MAB8094) were obtained from Santa Cruz and R&D Systems (MN), respectively. 4′,6-Diamidino-2-phenylindole (Cat. C1002) was purchased from Beyotime Technology (Shanghai, China). Secondary antibodies, including anti-rabbit IgG Alexa Fluor 647 (Cat. 4414S) and anti-mouse IgG Alexa Fluor 488 (Cat. 4408S), were from Cell Signaling Technology. The pSG5-hPXR expression vector was generously provided by Dr. Steven Kliewer (University of Texas Southwestern Medical Center, Dallas, TX). The pGL3-CYP3A4-XREM luciferase reporter construct was generously provided by Dr. Jeff Staudinger (University of Kansas, Lawrence, KS).
Experimental Animals and Treatments.
Adult male C57BL/6J mice (6–8 weeks old, 18–22 g) were purchased from Guangdong Medical Laboratory Animal Center. The animal room was specific-pathogen-free. Mice were housed under a standard 12-hour light/12-hour dark cycle and 55%–60% humidity at 22–24°C with free access to water and a standard rodent chow. All animal experiments were operated in compliance with the guidelines of the Institutional Animal Care and Use Committee of Sun Yat-sen University (Guangzhou, China).
For Dex treatment, all the mice were randomized to two groups. Dex at the dose of 50 mg/kg or corn oil (vehicle) at the dose of 0.1 ml/10 g was administered intraperitoneally once daily for 5 days. Livers were harvested at 24 hours after the last injection. The body weight and liver weight of mice were both measured for calculation of liver-to-body-weight ratios. A portion of liver was rapidly fixed in 10% buffered formalin for histologic examination. The rest of the tissues were immediately frozen by lipid nitrogen and stored at −80°C for further study.
Histologic Analysis.
Liver tissues were fixed in 10% buffered formalin solution. After being embedded in paraffin, specimens were cut into sections about 4 μm–thick each. Sections were deparaffinized by different concentrations of xylene and ethyl alcohol and then stained with H&E solutions (Servicebio, Wuhan, China). For immunohistochemical staining, paraffin-embedded sections were stained with primary antibodies against Ki67, PCNA, and CTNNB1 after heat-induced epitope retrieval in citrate buffer. H&E staining and immunohistochemical staining were all visualized by an Olympus inverted microscope (Olympus IX73; Olympus Corporation, Japan).
Oil Red O Staining.
The fixed liver tissue was removed from 10% buffered formalin solution to a 15% sucrose solution at 4°C for dewatering and sinking, and this was followed by a 30% sucrose solution. After being coated with optimal cutting temperature compound embedding agent, the section was cut into 6-8 μm–thick sections after optimal cutting temperature compound turned white and hardened. The frozen sections were washed with PBS buffer and stained with oil red O working solution. Hematoxylin solution was used to dye the nuclei.
Measurement of Triglyceride Contents.
Triglyceride (TG) contents of liver tissue were measured by TG detection assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Generously, liver tissues were homogenized (weight of liver tissues: volume of PBS = 1:9) and centrifuged at a speed of 2500 rpm for 10 minutes at 4°C. The procedure of TG contents analysis strictly followed the protocol that the manufacture provided. In brief, 2.5 μl distilled water, calibration solution (2.26 mM TG solution), and sample tested were added to 250 μl working solution in 96-well plates, respectively. After being mixed and incubated at 37°C for 10 minutes, the absorbance of the mixture was measured at a wavelength of 500 nm. The calculation formula provided in the guidelines was applied for the analysis of hepatic total TG levels.
CCK8 Test.
Human embryonic kidney (HEK) 293T cells (from American Type Culture Collection) were cultured in Dulbecco’s modified Eagle’s medium containing 10% FBS (Gibco) and 100 U penicillin/streptomycin. Cells were seeded into 96-well plates at 1 × 104 cells per well. After being incubated overnight, the cells were treated with DMSO or Dex (6.25, 25, 100, and 400 μM) for 24 hours. Then 10 μl CCK8 (Dojindo laboratories, Shanghai, China) (Cat. CK04) was added to each well, which was followed by 2–4 hours incubation. Optical density value was measured at a wavelength of 450 nm. This experiment was repeated three times independently.
Dual-Luciferase Reporter Gene Assays.
Dual-luciferase reporter gene assays were performed according to our previously reported protocols (Zeng et al., 2017). In general, HEK293T cells were seeded into 96-well plates at 1.2 × 104 cells per well and incubated overnight, and this was followed by the transfection of 100 ng pGL3-CYP3A4-XREM-Luc, 3 ng pGL4.54-TK, and 50 ng pSG5-hPXR. The transfection processed according to MegaTran 1.0 instructions (OriGene). The transfected mixture was replaced by serum-free medium Opti-MEM (Gibco) 6 hours later. Transfected cells were then treated with Dex (6.25, 25, 100, and 400 μM) and human PXR (hPXR) positive agonist RIF (10 μM) for 24 hours. Luciferase activity was examined by a tube luminometer applied with the Dual Reporter Assay System (Berthold Technologies, Germany) following manufacturer’s protocols. With Renilla activity as control, firefly luciferase activity was normalized to Renilla activity for each well.
HepG2 Culture and Immunofluorescence Double Staining.
HepG2 culture and immunofluorescence double staining were performed in accordance with previous procedure with slight modifications (Jiang et al., 2019). Briefly, HepG2 cells (from American Type Culture Collection) were maintained in Dulbecco’s modified Eagle’s medium containing 10% FBS and 100 U penicillin/streptomycin. Cells (1 × 104) were seeded into glass-bottom cell culture dish, which was followed by DMSO or 100 μM Dex treatment for 6 hours. Then cells were fixed in 4% paraformaldehyde for 30 minutes and 0.5% Triton X-100 for 10 minutes. After being blocked by 10% goat serum for 1 hour and incubated with rabbit polyclonal anti-PXR and mouse monoclonal anti-YAP antibodies overnight at 4°C, the cells were stained with fluorescent secondary antibodies, including anti-rabbit IgG Alexa Fluor 647 and anti-mouse IgG Alexa Fluor 488 for 1 hour away from light. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Images were captured by a confocal microscope (Olympus FV3000; Olympus, Japan), and the immunofluorescence double staining of PXR and YAP was quantified by Image J software.
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis.
RNA isolation and qRT-PCR analysis of hepatic mRNA expression were performed as described in our previous study (Guan et al., 2019). Mouse glyceraldehyde-3-phosphate dehydrogenase mRNA was used to normalize total RNA levels. The specific primer sequences were designed by Primer Bank and listed in Supplemental Table 1.
Protein Extraction and Western Blot Analysis.
Liver total and nuclear/cytosol protein extracts were prepared as described previously as well as the conduction of Western blot (Jiang et al., 2019). Briefly, 40 μg of protein lysate from liver samples of different groups was used. After being separated by 10% SDS-PAGE gel, the protein was transferred onto polyvinylidene difluoride membranes (0.45-μm; GE Healthcare Life Sciences, Piscataway, NJ). Blots were blocked in 5% skim milk or 5% BSA, which was followed by incubation with appropriate primary antibodies overnight at 4°C. The detection of specific protein bands and the analysis of the intensity was conducted by an electro-chemiluminescence detection kit (GE Healthcare Life Sciences) and Quantity One software, respectively.
Liquid Chromatography/Mass Spectrometry and Lipidomic Analysis.
Liver lipids were extracted according to a previous method (Li et al., 2018). In short, 20 mg of liver samples was homogenized with PBS, and lipids were extracted by 1.2 ml of cold methanol/methyl-tert-butyl ether/H2O (4:5:5, v/v/v). After being vortexed thoroughly and centrifuged, the upper supernatant of the mixture was transferred into a clean tube and dried in a vacuum oven. The extracts were resuspended in 1 ml mixture of methanol/isopropanol (1:1, v/v) and centrifuged, and this was followed by 100 μl upper supernatant added into the injection bottle for further ultra-high-performance liquid chromatography coupled with electrospray ionization high-resolution mass spectrometry (UHPLC-ESI-HRMS) analysis. The chromatographic separation system and mass spectrometry (MS) analysis were similar with the conditions we reported before (Li et al., 2018). Briefly, the sample was analyzed by Ultimate 3000 UHPLC system (Dionex Corporation, Sunnyvale, CA). Chromatographic separation was performed on an Ascentis Express C18 2.7 µm column (100 mm × 2.1 mm; Sigma-Aldrich) with gradient elution of 50% acetonitrile (v/v in water with 5 mM ammonium formate and 0.1% formic acid) and 95% isopropanol (v/v in acetonitrile with 5 mM ammonium formate and 0.1% formic acid) at a flow rate of 0.3 ml/min. Mass spectrometry was performed under electrospray positive (ESI+) and negative (ESI−) ionization modes. The main parameters for MS/MS were the same to our previous report (Fu et al., 2019). Lipidomic data were processed using Lipid Search software (Thermo Scientific, San Jose, CA). The structure of lipids was analyzed by comparing accurate mass number and characteristic fragment ion information with the database of the mass spectrum of lipids. The preprocessed data were exported into SIMCA 13.0 software (Umetrics, Kinnelon, NJ) followed by data analysis using principal component analysis (PCA) and orthogonal partial least squares discriminant analysis (OPLS-DA).
Statistical Analysis.
Each group consisted at least five animals. All experimental data are presented as the means ±S.D. SPSS19.0 was used for statistical analysis and GraphPad Prism 6.0 was used for graph preparation. Two-tailed Student’s test or nonparametric Mann-Whitney U test was used to assess the differences between groups. Statistical significance is represented as: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
Dex Induces Liver Enlargement.
A recent study revealed that Dex significantly induced liver enlargement, but the underlying details remain unknown (Jiang et al., 2019). After intraperitoneal injection of 0.1 ml/10 g of corn oil or 50 mg/kg of Dex for 5 days (Fig. 1A), the liver-to-body-weight ratio was increased (4.80% ± 0.63% in the vehicle group and 6.14% ± 0.53% in the Dex group), which was a significant rise of 27.92% in the Dex-treated group compared with that of the vehicle-treated group (Fig. 1, B and C). The size of hepatocytes was measured by CTNNB1 staining. A remarkable increase was seen in cell size around the central vein (CV) area in the Dex group compared with that of the vehicle group. (Fig. 1D). Quantitative analysis suggested that the size of hepatocytes was 894.05 ± 97.66 μm in the Dex group, which was significantly larger than that in the vehicle group (721.74 ± 12.75 μm)(Fig. 1E). There was no significant difference in hepatocyte size around the portal vein (PV) area (Supplemental Fig. 1, A and B). These results indicated that Dex (50 mg/kg) promoted hepatomegaly accompanied with hepatocyte enlargement.
Fig. 1.

Dex significantly induces liver enlargement in mice. (A) Mice were intraperitoneally treated with vehicle (corn oil, 0.1 ml/10 g) or Dex (50 mg/kg per day) for 5 days. (B) Representative morphologic pictures of mice livers of the vehicle or Dex group. (C) Liver-to-body-weight ratios. Data are expressed as mean ± S.D. (n = 5). (D) CTNNB1 staining of representative liver samples measuring the size of hepatocytes around the CV area. Scale bar, 50 μm. (E) Quantification of the size of hepatocytes around the CV area. Data are expressed as mean ± S.D. (n = 3). *P < 0.05 compared with the vehicle group.
Dex Is a PXR Agonist.
Given that PXR activation was reported to induce liver enlargement, and high dose of Dex can activate PXR (Staudinger et al., 2001), whether Dex-induced hepatomegaly is related to PXR activation needs to be explored. We first measured the effect of Dex on PXR downstream genes. The protein levels of CYP3A11, CYP2B10, and OATP2 (also known as SLCO1A4 and SLC21A5 in mouse) were significantly increased after treatment with Dex (Fig. 2, A and B), suggesting activation of PXR by Dex. Furthermore, the dual-luciferase reporter gene assays were applied to investigate whether hPXR could be transactivated by Dex. In response to positive control RIF, a classic hPXR agonist, luciferase activity of the hPXR-PXRE-luciferase reporter gene assay was markedly increased to 2.74-fold compared with that of the DMSO-treated group, whereas Dex enhanced hPXR reporter gene luciferase activity to 1.45-fold at micromolar concentrations (up to 100 μM) without cellular toxicity (Fig. 2C; Supplemental Fig. 2). Taken together, these results indicated that Dex was able to transactivate hPXR and modulate downstream gene of mouse PXR (mPXR), suggesting that Dex is a PXR agonist.
Fig. 2.

Dex activates mPXR and hPXR. (A and B) Western blot and quantification of mPXR downstream proteins from vehicle- or Dex-treated mice livers. Data are expressed as mean ± S.D. (n = 3). *P < 0.05; **P < 0.01 compared with the vehicle group. (C) Dual-luciferase reporter gene assay was used to determine the effect of RIF or Dex on hPXR activation in HEK293T cells. Data are expressed as mean ± S.D. (n = 5). **P < 0.01; ***P < 0.001; ****P < 0.0001 compared with the control group.
Dex Upregulates YAP Expression via PXR Activation.
The YAPsignaling pathway plays a vital role in controlling organ size (Tordjmann, 2011). Recently, we revealed that YAP is involved in the PXR-induced liver enlargement and there is a potential interaction between PXR and YAP (Jiang et al., 2019). In the current study we determined whether the hepatic YAP pathway contributed to Dex-induced hepatomegaly. Western blot results showed that the protein level of phosphorylated YAP was decreased to 71.18% of the vehicle group, whereas the protein levels of total YAP and nuclear YAP were increased to 1.44 and 1.40 times compared with those of the vehicle group, respectively. This indicated that the increased level of YAP was not only due to nuclear translocation but also due to the upregulation of total YAP (Fig. 3, A and B). To further confirm the effect of Dex on the activation of YAP pathway, protein expression of YAP-targeted genes was measured. Protein levels of ankyrin repeat domain 1 and connective tissue growth factor were significantly increased, whereas cysteine-rich angiogenic inducer 61 expression was unchanged compared with that of the vehicle-treated group (Fig. 3, C and D). Next, to identify whether PXR and YAP translocated to nucleus together after Dex treatment, immunofluorescence double staining was performed in HepG2 cells. After Dex treatment, the contents of PXR and YAP were significantly increased in the nucleus of HepG2 cells, indicating that Dex induced PXR and YAP nuclear translocation (Fig. 3, E and F). These data support the hypothesis that Dex, as a PXR activator, could promote YAP nuclear translocation, activate the YAP pathway, and then upregulate its downstream target genes.
Fig. 3.

Effect of Dex on YAPsignaling pathway. (A and B) Western blot analysis and quantification of total YAP, nuclear YAP and cytoplasmic phosphorylated YAP protein of liver samples after a 5-day treatment with the vehicle or Dex. (C and D) Western blot analysis and quantification of YAP downstream proteins of liver samples after the vehicle or Dex treatment. Data are expressed as mean ± S.D. (n = 3). *P < 0.05; **P < 0.01 compared with the vehicle group. (E) Confocal microscopy displaying PXR and YAP distribution in HepG2 cells treated with DMSO or 100 μM of Dex for 6 hours. Scale bar, 40 μm. (F) Quantification of immunofluorescence double staining of YAP and PXR. Data are expressed as mean ± S.D. (n = 3). *P < 0.05 compared with the control group. ANKRD1, ankyrin repeat domain 1; CTGF, connective tissue growth factor; CYR61, cysteine-rich angiogenic inducer 61; DAPI, 4′,6-diamidino-2-phenylindole; p-YAP, phosphorylated YAP.
Dex Inhibits Hepatocyte Proliferation Partially Because of Its Anti-Inflammation Effect.
Hyperactivation of YAP can provoke the proliferation of hepatocytes by upregulating proliferation-related protein (Patel et al., 2017). Moreover, liver enlargement is partially due to the proliferation of hepatocytes (Maronpot et al., 2010). To determine whether hepatocyte proliferation was involved in Dex-induced liver enlargement, the marker of cell proliferation Ki67 was analyzed in liver samples by immunohistochemistry. There were no visible Ki67 positive cells in either the vehicle-treated group or the Dex-treated group around the CV and PV areas (Fig. 4A), whereas a reduced number of PCNA (another cellular proliferation marker) positive cells were observed in livers of mice treated with Dex (Fig. 4B), indicating that Dex could not promote hepatocyte proliferation and hepatocyte proliferation may be not involved in Dex-induced liver enlargement. To examine whether Dex attenuated the proliferative response, protein levels of proliferation-related proteins were measured. The expression of CCND1 and PCNA protein was decreased markedly; however, CCNA1 and CCNE1 were unchanged compared with those of the vehicle-treated group (Fig. 4, C and D), suggesting that the cell cycle was impaired. Gene and protein expression of other regulators involved in cell proliferation, such as MET, EGFR, CTNNB1, c-MYC, and FOXM1 protein were also measured to determine potential signals involved in Dex-induced hepatomegaly. As expected, EGFR, c-MYC, and FOXM1 were significantly lower than that of the vehicle-treated group at both the mRNA and protein levels, whereas the expression of CTNNB1 and MET remained unchanged (Fig. 4, E and F; Supplemental Fig. 3).
Fig. 4.

Effect of Dex on hepatocyte proliferation. (A and B) Immunohistochemistry staining of Ki67 and PCNA in mice treated with the vehicle or Dex. Scale bar, 100 μm. (C–F) Western blot analysis and quantification of proliferation-related protein in mice after a 5-day treatment with the vehicle or Dex. Data are expressed as mean ± S.D. (n = 3). *P < 0.05; **P < 0.01; ****P < 0.0001 compared with the vehicle group. (G) qRT-PCR analysis of inflammatory factors in mice in response to the vehicle or Dex treatment. Data are expressed as mean ± S.D. (n = 5). *P < 0.05; ***P < 0.001 compared with the vehicle group.Ifng, interferon γ.
Next, the possible mechanism of the Dex-induced inhibition of hepatocyte proliferation was investigated. Previous studies showed the importance of inflammation factors, such as tumor necrosis factor (TNF) α and interleukin 6 (IL6), on the regulation of liver growth both in the acute phase and normal status, suggesting that inflammatory factors and cytokines play important roles in hepatocyte proliferation (Nagy et al., 1998; Zimmers et al., 2003; Han et al., 2018). Considering the therapeutic effect of Dex as an anti-inflammation drug, we further measured the expression of inflammation factors. Dex treatment suppressed the mRNA expression level of inflammation factors, including Il6, Tnfa, and interferon γ, indicating that Dex abrogated hepatocyte proliferation in part by suppressing the production of inflammatory factors (Fig. 4G).
Overall, hepatocyte proliferation did not significantly contribute to the Dex-induced hepatomegaly. Dex may counteract the PXR/YAP-induced hepatocyte proliferation partially by the suppression of inflammatory factors.
Dex-Induced Liver Enlargement Is Involved with Lipid Accumulation.
Dex was previously shown to influence the metabolism of hepatic lipids (Harasim-Symbor et al., 2016). On the other hand, the enlargement of liver and hepatocytes need energy supplied by lipid metabolism. Therefore, we further evaluated the effect of Dex on the liver lipid homeostasis. Prominent microvesicular and macrovesicular fatty changes were observed clearly in the H&E-stained livers in Dex-treated mice (Fig. 5A). Oil red O staining further confirmed that the abundance of neutral lipid droplets, stained in red and orange, accumulated in the cytoplasm of hepatocytes after Dex treatment (Fig. 5B). Dex significantly increased the levels of TG contents approximately 9-fold higher than that of the vehicle-treated group from 0.04 to 0.34 mmol/g protein (Supplemental Fig. 4), suggesting that accumulation of lipids may contribute to the increase in liver weight.
Fig. 5.

Effect of Dex on the lipid profiles and lipid homeostasis. (A) H&E staining of representative liver samples from the vehicle- or Dex-treated mice. Scale bar, 100 μm. (B) Oil red O staining of representative liver samples of mice treated with the vehicle or Dex. Scale bar, 100 μm. (C and D) PCA scatter plots, (E and F) OPLS-DA score plots, and (G and H) S-plots under positive and negative ion modes of lipid profiles from livers of mice treated with the vehicle or Dex. For PCA and OPLS-DA plots (C–F), blue diamonds represent the vehicle-treated mice, red diamonds represent the Dex-treated mice, and pink diamonds represent the quality controls. For S-plots (G and H), the specific calculated lipid components with P value <0.05, variable importance in projection (VIP) >1 were highlighted by red diamonds. (I–M) Altered lipid contents and species in liver samples of mice treated with the vehicle or Dex. (I and J) TG; (K) phosphatidyl ethanolamine (PE); (L) phosphatidylcholine (PC); and (M) phosphatidylinositol (PI). (N) qRT-PCR analysis of TG metabolic genes in mice treated with the vehicle or Dex. Data are expressed as mean ± S.D. (n = 4 or 5). *P < 0.05; **P < 0.01; ***P < 0.001 compared with the vehicle group.
To evaluate the effect of Dex on lipid profiles in liver, lipidomic analysis was performed on liver samples. A noticeable separation was found in the PCA scatter plots acquired from both positive (Fig. 5C) and negative (Fig. 5D) ion modes, implicating distinct discrimination in lipid profiles between the vehicle-treated and Dex-treated groups. LipidResearch software and OPLS-DA score plot (Fig. 5, E and F) were performed to identify altered lipids in two groups from the database of the mass spectrum of lipids. Lipids with variable importance in projection values >1 were highlighted with red diamonds in the S-plots (Fig. 5, G and H). Upon the false discovery rate test, four lipid species with P < 0.05 were selected (Fig. 5, I–M). Among these classes of lipids, the robustly rising trends of TGs were similar in this specific species no matter whether they were composed of unsaturated/saturated fatty acids or different lengths of fatty acids (Fig. 5, I and J), which was consistent with the biochemical results mentioned above. However, the trend changes of the other three lipids in liver samples varied even in the same species. Six lipids were reduced, and two were upregulated in phosphatidyl ethanolamine after treatment with Dex (Fig. 5K). Eleven phosphatidylcholine species were decreased, and one was increased (Fig. 5L). Among the species of phosphatidylinositol, four lipids were altered markedly, two of which were increased, and two were downregulated (Fig. 5M). Additionally, the hepatic mRNA levels of TG metabolism–related genes were measured (Fig. 5N). Expression of diacylglycerol acyltransferase (Dgat) 1 and Dgat2 mRNAs encoding regulators of the final step of TG synthesis was unchanged, whereas a notable reduction was observed on the mRNAs encoding hormone-sensitive lipase (also known as Hsl or Lipe). There was a downward trend in adipose triglyceride lipase (also known as Atgl or Pnpla2) mRNA but with no significance. Besides, expression of the fatty acid transporter CD36 encoding Cd36 mRNA was increased markedly, suggesting a larger flux of exogenous fatty acids. At the same time, a reduction was observed on the mRNA level of peroxisome proliferator-activated receptor α (Ppara), which is responsible for the regulation of peroxisomal and mitochondrial fatty acid β-oxidation. Taken together, lipolysis and fatty acid β-oxidation were suppressed, whereas the uptake of fatty acids was enhanced, which ultimately contributed to the accumulation of TG. In short, lipidomics and gene analysis suggested that Dex had significant influence on the hepatic lipid metabolism in mice, with much more TGs accumulating in the liver, and may eventually lead to liver enlargement.
These data together indicated that Dex significantly enlarged hepatocytes by TG accumulation and then resulted in hepatomegaly.
Discussion
Liver enlargement (hepatomegaly) is characterized by an increased weight or/and size of the liver. It was reported that high-dose Dex treatment of three children induced significant hepatomegaly (Verrips et al., 1998). A similar phenomenon was observed in other clinical steroid treatment cases as well as in rodents (Iancu et al., 1986; Thatcher and Caldwell, 1994; Micuda et al., 2007). Consistently in the current study, the liver was significantly enlarged after intraperitoneal dosing of Dex at 50 mg/kg. The increased liver weight was manifested with enlarged hepatocytes around the CV area and obvious fat vacuoles in the liver tissue. Dex induced hepatocyte enlargement around CV area, but there was no significant difference in the size of hepatocytes around PV area. Under physiologic conditions, a higher rate of fatty acid oxidation was found in the PV area, and more lipid synthesis was found in the CV area (Katz et al., 1983; Hijmans et al., 2014), suggesting that Dex-induced hepatomegaly may be related to the lipid accumulation.
Hepatomegaly and hepatocyte hypertrophy are often attributed to the change of cellular contents, including glycogen excess, water retention, and lipids accumulation (Maronpot et al., 2010). The current study revealed the increase in intracellular mass, especially with lipids, plays an important role in Dex-induced hepatomegaly. Lipidomics further revealed alterations in specific altered lipids. Increased TGs were found to account for the majority of lipid changes, which were confirmed by biochemical analysis and histochemical staining. TG metabolism in the liver is regulated by multiple metabolic enzymes, transporters, and transcriptional factors. DGAT1 and DGAT2, catalytic enzymes for the acylation of diacylglycerol (DG) that are the final step of TG synthesis (Coleman and Lee, 2004), were found unchanged at the mRNA level, indicating the synthesis of TGs was not affected by Dex. Adipose triglyceride lipase regulates TG lipolysis in the adipose tissue, and the resulting DG molecules are then hydrolyzed by LIPE to release monoglycerides (Alves-Bezerra and Cohen, 2017). As expected, gene expression of Lipe was reduced, indicating that the lipolysis was inhibited. Combined with the decreased Ppara and increased Cd36 mRNA that regulate fatty acids β-oxidation (Aoyama et al., 1998) and the fatty acids uptake, respectively (Buqué et al., 2010), these results indicated that lipolysis and β-oxidation were suppressed, whereas the uptake of fatty acids was enhanced, which ultimately contributed to the accumulation of TGs. Similar results were found in a previous report (Lemke et al., 2008), which uncovered the role of GR-hairy enhancer of split 1 axis in the regulation of Dex-induced hepatic lipid accumulation. Several other signaling pathways were revealed recently. Notably, mitogen-activated protein kinase phosphatase 3 was confirmed to be the downstream gene of FOXO1 and partially mediated the lipid accumulation by Dex (Feng et al., 2014). microRNA-17-5p, as a regulator of PPARα, was found to attenuate Dex-induced excessive lipids (Du et al., 2015).
Although Dex is a classic ligand of GR and a vital regulator of the homeostasis of glucose and lipids, the effect of Dex on other nuclear receptors should not be ignored. For example, Dex activates PXR under certain conditions. PXR activation by pegnenolone-16α-carbonitrile could lead to significant liver enlargement. A recent report noted that Dex-induced liver enlargement was related to activation of PXR signaling (Wang et al., 2018). As the crucial regulator for xenobiotic metabolism, PXR modulates the expression of a series of drug-metabolizing enzymes and transporters, such as CYP3A11, CYP2B10, and OATP2 (also known as SLCO1B1, LST1, OATP-C and SLC21A6 in human, and known as SLCO1A4 and SLC21A5 in mouse) (Kliewer, 2003; Wang et al., 2003; Hartley et al., 2004). In human hepatocytes, nanomolar of Dex was found to enhance the expression of CYP3A4, a human homolog of the mouse Cyp3a11 gene, by activation of PXR, whereas Dex could activate PXR under micromolar concentrations (Pascussi et al., 2000). In vivo studies also showed that Dex could activate PXR at a higher dose >10 mg/kg (Hunter et al., 2017). In the current study, PXR activation by Dex was identified by the increased protein expression of PXR-targeted genes, such as Cyp3a11, Cyp2b10, and Oatp2 (also known as Slco1a4, Oatp1a4 and Slc21a5 in mouse) in vivo. Dex was able to activate hPXR in vitro as well. These data together demonstrated that Dex indeed is a PXR agonist in both mice and humans. Furthermore, PXR was activated under the indicated dosage in the animal experiments, which was consistent with previous reports (Schuetz et al., 2000; Scheer et al., 2010). PXR also plays a crucial role in sustaining the homeostasis of endobiotics, such as lipids, and recent studies have investigated the effects of PXR on the regulation of nonalcoholic fatty liver disease (Cave et al., 2007). PXR can directly bind to FOXA2 to prevent its binding with the promoters of carnitine palmitoyltransferase 1A and mitochondrial 3-hydroxy-3-methylglutarate-CoA synthase 2, thereby inhibiting fatty acid β oxidation and ketogenesis (Nakamura et al., 2007). In addition, CD36 was confirmed to be induced by PXR and positively related to increased fatty acids uptake and TG accumulation in the liver (Zhou et al., 2006), implicating a possible role of PXR in the Dex-induced TG accumulation during liver enlargement. Furthermore, YAP has been recently reported to accelerate lipid accumulation (Jeong et al., 2018; Liu et al., 2019) and promote the development and progression of nonalcoholic fatty liver disease with interaction of transforming growth factor-βsignaling pathways (Chen et al., 2018) in vivo, suggesting the close relationship between lipid accumulation and the PXR/YAP activation.
YAP is the critical regulator of liver size. With overexpression of hepatic YAP, the size of liver can be promoted to approaching 4–5-fold its normal size (Dong et al., 2007). Recently, the contribution of YAP signaling in PXR-induced liver enlargement was uncovered (Jiang et al., 2019). Since PXR was activated by Dex in the current study, YAP signaling may be involved in Dex-induced hepatomegaly. The upregulation of total YAP, nuclear YAP, and YAP downstream proteins together confirmed that YAP signaling was activated in response to Dex treatment and Dex-induced PXR activation. YAP activation was also proven to suppress the expression of gluconeogenic genes promoted by administration of glucocorticoids, such as Dex (Hu et al., 2017). In the present study, excessive glucose may increase the activation of YAP signaling, which ultimately attenuated the Dex-induced flux of glucose. PXR and YAP were found to translocate to the HepG2 cell nucleus 6 hours after the Dex treatment, which was consistent with our previous finding and suggested a potential interaction between PXR and YAP (Jiang et al., 2019). Interestingly, the best time point to observe this co-localization was 6 hours after Dex treatment instead of 48 hours.
YAP activation could evoke cell proliferation, which is also thought to be involved in the PXR-induced liver enlargement (Huang et al., 2005). Intriguingly, the present results showed a different trend after Dex treatment. The staining of proliferation markers Ki67 and PCNA revealed no induction of proliferation around the CV and PV areas by Dex. The downregulation of CCND1, a member of cyclins, controls the entry of cell cycle, suggested that DNA synthesis and mitosis of hepatic cells were inhibited. The markable reduction of CCND1 and PCNA have revealed that proliferation response was arrested. Several proliferation-related factors were further assessed. Wnt/β-catenin signaling is another vital pathway that regulates organ size, and it is considered to be evidently activated by YAP activation (Camargo et al., 2007). However, no difference was noted between vehicle- and Dex-treated mice in the expression of CTNNB1, which may involve the interplay of Hippo/Wnt signaling. Receptor tyrosine kinases, including MET and EGFR as the upstream regulators of cyclins including CCND1, were found to partially mediate hepatocyte proliferation (Bhushan et al., 2019). c-MYC was also reported to be the regulator of hepatocyte proliferation (Baena et al., 2005). Consistently, a reduction was both observed in the protein and mRNA levels of c-MYC in Dex-treated mice. Additionally, FOXM1, as the downstream targeted gene of MET, EGFR, and c-MYC, is one of the major transcription factors that controls the cell cycle, including progression to S-phase and mitosis (Blanco-Bose et al., 2008; Francica et al., 2016; Stoll et al., 2016). In the present study, decreased EGFR, c-MYC, and FOXM1 indicated their possible roles in the impaired cell cycle and proliferation inhibition by Dex. Inflammatory factors, such as IL6, were capable of rescuing the inhibition of hepatocyte proliferation by Dex in the acute phase (Han et al., 2018) and inducing hepatomegaly by continuous administration in physical stasis (Zimmers et al., 2003). Considering the potency of the anti-inflammatory effect of Dex, we hypothesized that YAP signaling–promoted proliferation was partially counteracted by Dex-induced anti-inflammatory effect. Here, Dex treatment suppressed mRNA expression level of inflammation factors, including Il6, Tnfa, and interferon γ, indicating that Dex abrogated hepatocyte proliferation in part by suppressing the production of inflammatory factors. These data together demonstrated that Dex-induced liver enlargement is mainly caused by liver cell enlargement instead of hepatocyte proliferation, and Dex may counteract the YAP-induced hepatocyte proliferation partially by the suppression of inflammation factors.
In summary, the current study demonstrated that Dex, at a high dose used to treat inflammatory diseases, can induce liver cell enlargement and hepatomegaly possibly by activating PXR and then inducing nuclear translocation of YAP and activating YAP signaling. Although it has been proven that PXR-induced liver enlargement is YAP-dependent (Jiang et al., 2019), whether Dex-induced hepatomegaly is PXR-dependent still needs to be confirmed using PXR loss-of-function models, such as Pxr knockout mice. Furthermore, Dex-induced hepatomegaly was not due to hepatocyte proliferation likely because of its anti-inflammation effect of suppression of inflammation factors. The Dex-induced hepatocyte enlargement was mostly attributable to the accumulation of lipids, such as triglycerides. These findings provide new insights for understanding the mechanism of Dex-induced hepatomegaly and provide more information on its clinical use in the future.
Abbreviations
- CCN
cyclin
- CTNNB1
β-catenin
- CV
central vein
- Dex
dexamethasone
- DG
diacylglycerol
- DGAT
DG acyltransferase
- EGFR
epidermal growth factor receptor
- FOX
forkhead box protein
- GR
glucocorticoid receptor
- HEK
human embryonic kidney
- hPXR
human PXR
- IL6
interleukin 6
- LIPE
hormone-sensitive lipase
- LST
liver-specific organic anion transporter
- MET
mesenchymal-epithelial transition factor
- mPXR
mouse PXR
- MS
mass spectrometry
- OATP2
organic anion transporter polypeptide 2
- OPLS-DA
orthogonal partial least squares discriminant analysis
- PCA
principal component analysis
- PCNA
proliferating cell nuclear antigen
- PPARα
peroxisome proliferator-activated receptor α
- PV
portal vein
- PXR
pregnane X receptor
- qRT-PCR
quantitative real-time polymerase chain reaction
- RIF
rifampicin
- SLC
solute carrier family
- SLCO
solute carrier organic anion transporter family
- TG
triglyceride
- TNF
tumor necrosis factor
- UHPLC-ESI-HRMS
ultra-high-performance liquid chromatography coupled with electrospray ionization high-resolution mass spectrometry analysis
- YAP
yes-associated protein
Authorship Contributions
Participated in research design: Bi, Huang, Jiao.
Conducted experiments: Jiao, Yao, Zhao, Zhou, Gao, Fan, Chen, Li, Jiang, Yang.
Performed data analysis: Jiao, Yao.
Wrote or contributed to the writing of the manuscript: Jiao, Bi, Gonzalez.
Note Added in Proof—Alternative names of OATP2 included and funding footnote updated to include support provided by the Intramural Research Program of the National Institutes of Health National Cancer Institute not included in the Fast Forward version published June 19, 2020. The article text and funding footnote have now been corrected.
Footnotes
The work was supported by the Natural Science Foundation of China [Grant 81973392], the National Key Research and Development Program [Grant 2017YFE0109900], the Natural Science Foundation of Guangdong [Grant 2017A030311018], the 111 project [Grant B16047], the Key Laboratory Foundation of Guangdong Province [Grant 2017B030314030], the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program [Grant 2017BT01Y093], and the National Engineering and Technology Research Center for New drug Druggability Evaluation [Seed Program of Guangdong Province, Grant 2017B090903004] and the Intramural Research Program of the National Institutes of Health National Cancer Institute.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Alves-Bezerra M, Cohen DE. (2017) Triglyceride metabolism in the liver. Compr Physiol 8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. (1998) Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem 273:5678–5684. [DOI] [PubMed] [Google Scholar]
- Baena E, Gandarillas A, Vallespinós M, Zanet J, Bachs O, Redondo C, Fabregat I, Martinez-A C, de Alborán IM. (2005) c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc Natl Acad Sci USA 102:7286–7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan B, Stoops JW, Mars WM, Orr A, Bowen WC, Paranjpe S, Michalopoulos GK. (2019) TCPOBOP-induced hepatomegaly and hepatocyte proliferation are attenuated by combined disruption of MET and EGFR signaling. Hepatology 69:1702–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Bose WE, Murphy MJ, Ehninger A, Offner S, Dubey C, Huang W, Moore DD, Trumpp A. (2008) C-Myc and its target FoxM1 are critical downstream effectors of constitutive androstane receptor (CAR) mediated direct liver hyperplasia. Hepatology 48:1302–1311. [DOI] [PubMed] [Google Scholar]
- Botts S, Ennulat D, Francke-Carroll S, Graham M, Maronpot RR, Mohutsky M. (2010) Introduction to hepatic drug metabolizing enzyme induction in drug safety evaluation studies. Toxicol Pathol 38:796–798. [DOI] [PubMed] [Google Scholar]
- Buckley DB, Klaassen CD. (2009) Induction of mouse UDP-glucuronosyltransferase mRNA expression in liver and intestine by activators of aryl-hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor, peroxisome proliferator-activated receptor alpha, and nuclear factor erythroid 2-related factor 2. Drug Metab Dispos 37:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buqué X, Martínez MJ, Cano A, Miquilena-Colina ME, García-Monzón C, Aspichueta P, Ochoa B. (2010) A subset of dysregulated metabolic and survival genes is associated with severity of hepatic steatosis in obese Zucker rats. J Lipid Res 51:500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. (2007) YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol 17:2054–2060. [DOI] [PubMed] [Google Scholar]
- Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, McClain C. (2007) Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem 18:184–195. [DOI] [PubMed] [Google Scholar]
- Chen P, Luo Q, Huang C, Gao Q, Li L, Chen J, Chen B, Liu W, Zeng W, Chen Z. (2018) Pathogenesis of non-alcoholic fatty liver disease mediated by YAP. Hepatol Int 12:26–36. [DOI] [PubMed] [Google Scholar]
- Cheng X, Maher J, Dieter MZ, Klaassen CD. (2005) Regulation of mouse organic anion-transporting polypeptides (Oatps) in liver by prototypical microsomal enzyme inducers that activate distinct transcription factor pathways. Drug Metab Dispos 33:1276–1282. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Lee DP. (2004) Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res 43:134–176. [DOI] [PubMed] [Google Scholar]
- Cui A, Fan H, Zhang Y, Zhang Y, Niu D, Liu S, Liu Q, Ma W, Shen Z, Shen L, et al. (2019) Dexamethasone-induced Krüppel-like factor 9 expression promotes hepatic gluconeogenesis and hyperglycemia. J Clin Invest 129:2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130:1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du WW, Liu F, Shan SW, Ma XC, Gupta S, Jin T, Spaner D, Krylov SN, Zhang Y, Ling W, et al. (2015) Inhibition of dexamethasone-induced fatty liver development by reducing miR-17-5p levels. Mol Ther 23:1222–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson-Neilsen W, Kaye AD. (2014) Steroids: pharmacology, complications, and practice delivery issues. Ochsner J 14:203–207. [PMC free article] [PubMed] [Google Scholar]
- Feng B, He Q, Xu H. (2014) FOXO1-dependent up-regulation of MAP kinase phosphatase 3 (MKP-3) mediates glucocorticoid-induced hepatic lipid accumulation in mice. Mol Cell Endocrinol 393:46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francica P, Nisa L, Aebersold DM, Langer R, Bladt F, Blaukat A, Stroka D, Martínez MR, Zimmer Y, Medová M. (2016) Depletion of FOXM1 via MET targeting underlies establishment of a DNA damage-induced senescence program in gastric cancer. Clin Cancer Res 22:5322–5336. [DOI] [PubMed] [Google Scholar]
- Fu K, Wang C, Gao Y, Fan S, Zhang H, Sun J, Jiang Y, Liu C, Guan L, Liu J, et al. (2019) Metabolomics and lipidomics reveal the effect of hepatic Vps33b deficiency on bile acids and lipids metabolism. Front Pharmacol 10:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz A, Marschall HU, Lammert F, Mattsson LA. (2005) Intrahepatic cholestasis of pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid. Hepatology 42:1399–1405. [DOI] [PubMed] [Google Scholar]
- Guan L, Chen Y, Wang Y, Zhang H, Fan S, Gao Y, Jiao T, Fu K, Sun J, Yu A, et al. (2019) Effects of carnitine palmitoyltransferases on cancer cellular senescence. J Cell Physiol 234:1707–1719. [DOI] [PubMed] [Google Scholar]
- Han R, Zhang F, Wan C, Liu L, Zhong Q, Ding W. (2018) Effect of perfluorooctane sulphonate-induced Kupffer cell activation on hepatocyte proliferation through the NF-κB/TNF-α/IL-6-dependent pathway. Chemosphere 200:283–294. [DOI] [PubMed] [Google Scholar]
- Harasim-Symbor E, Konstantynowicz-Nowicka K, Chabowski A. (2016) Additive effects of dexamethasone and palmitate on hepatic lipid accumulation and secretion. J Mol Endocrinol 57:261–273. [DOI] [PubMed] [Google Scholar]
- Hartley DP, Dai X, He YD, Carlini EJ, Wang B, Huskey SE, Ulrich RG, Rushmore TH, Evers R, Evans DC. (2004) Activators of the rat pregnane X receptor differentially modulate hepatic and intestinal gene expression. Mol Pharmacol 65:1159–1171. [DOI] [PubMed] [Google Scholar]
- Hijmans BS, Grefhorst A, Oosterveer MH, Groen AK. (2014) Zonation of glucose and fatty acid metabolism in the liver: mechanism and metabolic consequences. Biochimie 96:121–129. [DOI] [PubMed] [Google Scholar]
- Hu Y, Shin DJ, Pan H, Lin Z, Dreyfuss JM, Camargo FD, Miao J, Biddinger SB. (2017) YAP suppresses gluconeogenic gene expression through PGC1α. Hepatology 66:2029–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, Pan D. (2005) The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122:421–434. [DOI] [PubMed] [Google Scholar]
- Hunter SR, Vonk A, Mullen Grey AK, Riddick DS. (2017) Role of glucocorticoid receptor and pregnane X receptor in dexamethasone induction of rat hepatic aryl hydrocarbon receptor nuclear translocator and NADPH-cytochrome P450 oxidoreductase. Drug Metab Dispos 45:118–129. [DOI] [PubMed] [Google Scholar]
- Iancu TC, Shiloh H, Dembo L. (1986) Hepatomegaly following short-term high-dose steroid therapy. J Pediatr Gastroenterol Nutr 5:41–46. [DOI] [PubMed] [Google Scholar]
- Jeong S-H, Kim H-B, Kim M-C, Lee J-M, Lee JH, Kim J-H, Kim J-W, Park W-Y, Kim S-Y, Kim JB, et al. (2018) Hippo-mediated suppression of IRS2/AKT signaling prevents hepatic steatosis and liver cancer. J Clin Invest 128:1010–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Feng D, Ma X, Fan S, Gao Y, Fu K, Wang Y, Sun J, Yao X, Liu C, et al. (2019) Pregnane X receptor regulates liver size and liver cell fate by yes-associated protein activation in mice. Hepatology 69:343–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz NR, Fischer W, Giffhorn S. (1983) Distribution of enzymes of fatty acid and ketone body metabolism in periportal and perivenous rat-liver tissue. Eur J Biochem 135:103–107. [DOI] [PubMed] [Google Scholar]
- Kliewer SA. (2003) The nuclear pregnane X receptor regulates xenobiotic detoxification. J Nutr 133 (7 Suppl):2444S–2447S. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Goodwin B, Willson TM. (2002) The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev 23:687–702. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterström RH, et al. (1998) An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92:73–82. [DOI] [PubMed] [Google Scholar]
- Lemke U, Krones-Herzig A, Berriel Diaz M, Narvekar P, Ziegler A, Vegiopoulos A, Cato AC, Bohl S, Klingmüller U, Screaton RA, et al. (2008) The glucocorticoid receptor controls hepatic dyslipidemia through Hes1. Cell Metab 8:212–223. [DOI] [PubMed] [Google Scholar]
- Li J, Gao Y, Guan L, Zhang H, Sun J, Gong X, Li D, Chen P, Ma Z, Liang X, et al. (2018) Discovery of phosphatidic acid, phosphatidylcholine, and phosphatidylserine as biomarkers for early diagnosis of endometriosis. Front Physiol 9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ren H, Zhou Y, Shang L, Zhang Y, Yang F, Shi X. (2019) The hypoxia conditioned mesenchymal stem cells promote hepatocellular carcinoma progression through YAP mediated lipogenesis reprogramming. J Exp Clin Cancer Res 38:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maronpot RR, Yoshizawa K, Nyska A, Harada T, Flake G, Mueller G, Singh B, Ward JM. (2010) Hepatic enzyme induction: histopathology. Toxicol Pathol 38:776–795. [DOI] [PubMed] [Google Scholar]
- Micuda S, Fuksa L, Mundlova L, Osterreicher J, Mokry J, Cermanova J, Brcakova E, Staud F, Pokorna P, Martinkova J. (2007) Morphological and functional changes in p-glycoprotein during dexamethasone-induced hepatomegaly. Clin Exp Pharmacol Physiol 34:296–303. [DOI] [PubMed] [Google Scholar]
- Nagy P, Kiss A, Schnur J, Thorgeirsson SS. (1998) Dexamethasone inhibits the proliferation of hepatocytes and oval cells but not bile duct cells in rat liver. Hepatology 28:423–429. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Moore R, Negishi M, Sueyoshi T. (2007) Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J Biol Chem 282:9768–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascussi JM, Drocourt L, Fabre JM, Maurel P, Vilarem MJ. (2000) Dexamethasone induces pregnane X receptor and retinoid X receptor-alpha expression in human hepatocytes: synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol Pharmacol 58:361–372. [DOI] [PubMed] [Google Scholar]
- Patel SH, Camargo FD, Yimlamai D. (2017) Hippo signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology 152:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petta I, Dejager L, Ballegeer M, Lievens S, Tavernier J, De Bosscher K, Libert C. (2016) The interactome of the glucocorticoid receptor and its influence on the actions of glucocorticoids in combatting inflammatory and infectious diseases. Microbiol Mol Biol Rev 80:495–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhen T, Cidlowski JA. (2005) Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N Engl J Med 353:1711–1723. [DOI] [PubMed] [Google Scholar]
- Scheer N, Ross J, Kapelyukh Y, Rode A, Wolf CR. (2010) In vivo responses of the human and murine pregnane X receptor to dexamethasone in mice. Drug Metab Dispos 38:1046–1053. [DOI] [PubMed] [Google Scholar]
- Schuetz EG, Schmid W, Schutz G, Brimer C, Yasuda K, Kamataki T, Bornheim L, Myles K, Cole TJ. (2000) The glucocorticoid receptor is essential for induction of cytochrome P-4502B by steroids but not for drug or steroid induction of CYP3A or P-450 reductase in mouse liver. Drug Metab Dispos 28:268–278. [PubMed] [Google Scholar]
- Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, et al. (2001) The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA 98:3369–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll SW, Stuart PE, Swindell WR, Tsoi LC, Li B, Gandarillas A, Lambert S, Johnston A, Nair RP, Elder JT. (2016) The EGF receptor ligand amphiregulin controls cell division via FoxM1. Oncogene 35:2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher NJ, Caldwell J. (1994) Origins of hepatomegaly produced by dexamethasone (DEX), pregnenolone 16 alpha-carbonitrile (PCN) and phenobarbitone (PB) in female Sprague-Dawley rats. Biochem Soc Trans 22:132S. [DOI] [PubMed] [Google Scholar]
- Tordjmann T. (2011) Hippo signalling: liver size regulation and beyond. Clin Res Hepatol Gastroenterol 35:344–346. [DOI] [PubMed] [Google Scholar]
- Tumaneng K, Schlegelmilch K, Russell RC, Yimlamai D, Basnet H, Mahadevan N, Fitamant J, Bardeesy N, Camargo FD, Guan KL. (2012) YAP mediates crosstalk between the Hippo and PI(3)K–TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol 14:1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrips A, Rotteveel JJ, Lippens R. (1998) Dexamethasone-induced hepatomegaly in three children. Pediatr Neurol 19:388–391. [DOI] [PubMed] [Google Scholar]
- Wang H, Faucette SR, Gilbert D, Jolley SL, Sueyoshi T, Negishi M, LeCluyse EL. (2003) Glucocorticoid receptor enhancement of pregnane X receptor-mediated CYP2B6 regulation in primary human hepatocytes. Drug Metab Dispos 31:620–630. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang F, Lu Z, Jin X, Zhang Y. (2018) Semi-quantitative profiling of bile acids in serum and liver reveals the dosage-related effects of dexamethasone on bile acid metabolism in mice. J Chromatogr B Analyt Technol Biomed Life Sci 1095:65–74. [DOI] [PubMed] [Google Scholar]
- Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, Nelson MC, Neuschwander-Tetri BA, Brunt EM, Guzelian PS, Evans RM. (2000) Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature 406:435–439. [DOI] [PubMed] [Google Scholar]
- Zeng H, Jiang Y, Chen P, Fan X, Li D, Liu A, Ma X, Xie W, Liu P, Gonzalez FJ, et al. (2017) Schisandrol B protects against cholestatic liver injury through pregnane X receptors. Br J Pharmacol 174:672–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, Ren S, Evans RM, Xie W. (2006) A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem 281:15013–15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmers TA, McKillop IH, Pierce RH, Yoo JY, Koniaris LG. (2003) Massive liver growth in mice induced by systemic interleukin 6 administration. Hepatology 38:326–334. [DOI] [PubMed] [Google Scholar]