

Abstract

Coibamide A (CbA) is a marine natural product with potent antiproliferative activity against human cancer cells and a unique selectivity profile. Despite promising antitumor activity, the mechanism of cytotoxicity and specific cellular target of CbA remain unknown. Here, we develop an optimized synthetic CbA photoaffinity probe (photo-CbA) and use it to demonstrate that CbA directly targets the Sec61α subunit of the Sec61 protein translocon. CbA binding to Sec61 results in broad substrate-nonselective inhibition of ER protein import and potent cytotoxicity against specific cancer cell lines. CbA targets a lumenal cavity of Sec61 that is partially shared with known Sec61 inhibitors, yet profiling against resistance conferring Sec61α mutations identified from human HCT116 cells suggests a distinct binding mode for CbA. Specifically, despite conferring strong resistance to all previously known Sec61 inhibitors, the Sec61α mutant R66I remains sensitive to CbA. A further unbiased screen for Sec61α resistance mutations identified the CbA-resistant mutation S71P, which confirms nonidentical binding sites for CbA and apratoxin A and supports the susceptibility of the Sec61 plug region for channel inhibition. Remarkably, CbA, apratoxin A, and ipomoeassin F do not display comparable patterns of potency and selectivity in the NCI60 panel of human cancer cell lines. Our work connecting CbA activity with selective prevention of secretory and membrane protein biogenesis by inhibition of Sec61 opens up possibilities for developing new Sec61 inhibitors with improved drug-like properties that are based on the coibamide pharmacophore.
Introduction
Natural products are a rich source of bioactive and specific chemical probes and serve as starting points for development of new therapeutics once their mechanism of action and cellular targets have been identified.1,2 Coibamide A (CbA)3 is an N-methyl-stabilized lariat depsipeptide (Figure 1) isolated from a Caldora species4 of marine cyanobacterium collected in Panama. CbA potently inhibits cell proliferation, migration, and invasive capacity, and in early assessments of the in vivo activity of the natural product, or simplified analogue, inhibited tumor growth in subcutaneous xenograft models of human glioblastoma and breast cancer.5,6 Further, CbA rapidly induces a macroautophagy stress response in mammalian cells, and a phase-specific G1 cell-cycle block prior to cell death.5,7 The observed biological profile and distinct pattern of selectivity against cell lines of the National Cancer Institute (NCI) 60 human tumor cell line panel has generated considerable interest in CbA, resulting in development of total synthesis methods and revision of the absolute configuration of the natural product.8−10
Figure 1.
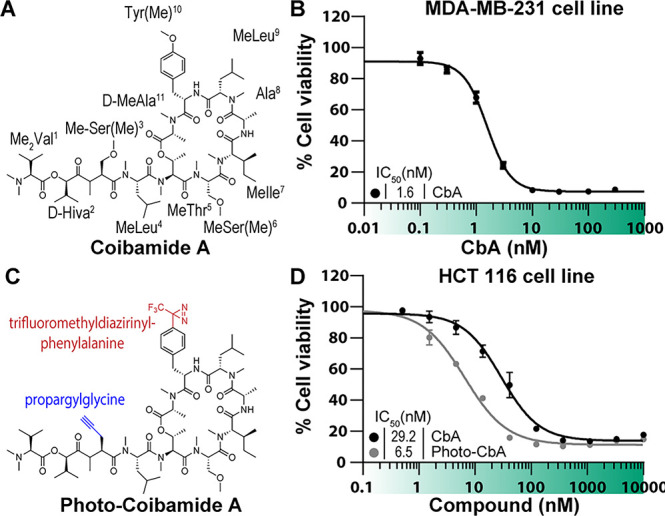
Cytotoxicity of synthetic and Pra-containing coibamides. (A) Structure of CbA. (B) Human MDA-MB-231 breast cancer cells were treated with increasing concentrations of synthetic CbA or vehicle (0.1% DMSO) and cell viability assessed at 72 h by an MTS end-point assay. (C) Structure of Photo-CbA. (D) Human HCT116 cells were treated with increasing concentrations of synthetic or photo-CbA in 0.1% DMSO, and cell viability assessed at 72 h by Alamar Blue assay.
CbA inhibits expression of the integral membrane receptor, vascular endothelial growth factor receptor 2 (VEGFR-2), and its secreted ligand vascular endothelial growth factor A (VEGF-A). It induces mTOR-independent autophagy in a manner similar to apratoxin A (AprA), a previously characterized inhibitor of protein import into the early secretory pathway,5 despite yielding different cytotoxic profiles against cell lines of the NCI-60 tumor cell line panel.3,11 Protein secretion is a complicated multistep process12 that begins when nascent secretory proteins are synthesized in the cytosol. Small molecule probes with a defined mechanism have allowed dissection of the basic function of the secretory pathway13 and provided new insights into the mechanism of protein transport into the endoplasmic reticulum.14−17 Such probes can also serve as therapeutic lead scaffolds for targeting diseases where the secretory pathway plays a central role.18 The first step in protein secretion is entry into the endoplasmic reticulum (ER), after which newly synthesized secretory polypeptides undergo distinct maturation steps that enable correctly folded proteins to exit the ER and be targeted to their correct final destinations. Previously reported natural products that prevent protein entry into the secretory pathway include, in addition to AprA,14 HUN-7293 (pestahivin)19,20 and related synthetic cotransins,19−21 mycolactone A/B,15,22,23 decatransin,16 ipomoeassin F (IpoF),24 and eeyarestatin I compounds.25 However, the critical step inhibited by CbA during biogenesis of VEGFR-2 and VEGF-A and the direct cellular target of CbA remain unknown.
In the current study, we explore the structure–activity relationship (SAR) of CbA to develop an optimized CbA photoaffinity probe (photo-CbA), which allowed us to identify the Sec61α subunit of the Sec61 protein translocation channel as the direct cellular binding target of CbA. Sec61 binding prevents cellular production of a broad range of secreted and integral membrane proteins that depend on Sec61 for their cotranslational biogenesis. The CbA binding site on Sec61α near the lumenal plug domain seems to be only partially overlapping to that of previously described substrate-nonselective Sec61 inhibitors AprA and mycolactone, suggesting that CbA interacts with Sec61 through unique interactions. CbA also has differential growth inhibitory potential against a panel of cancer cells relative to AprA and IpoF.
Results and Discussion
Synthesis of CbA and Its Photoaffinity Derivatization
Obtaining sufficient quantities of CbA for detailed mechanism of action studies from field-collected material is highly challenging,3 and thus we first set out to establish a total synthesis for this N-methylated peptidic macrocycle (Figure 1A) using a modification of a previously reported method8 (see the Supporting Information). Briefly, we initially constructed the middle part of CbA (MeThr5–MeIle7: fragment 2) on (2-Cl)Trt resin by standard Fmoc-solid phase peptide synthesis, and then conjugated the N-terminal four residues (Me2Val1–MeLeu4: fragment 1). After coupling of d-MeAla11 onto the Thr5 hydroxy group, the remaining sequence (Ala8–Tyr(Me)10: fragment 3) was appended. EDCI/HOAt-mediated cyclization of the open-chain precursor, which was obtained by cleavage from the resin by treatment with HFIP, afforded the expected CbA.8 Having a robust source of synthetic CbA, we proceeded with biological characterization of the synthetic product in human MDA-MB-231 triple negative breast cancer cells. These cells were previously identified as highly sensitive (IC50 = 2.8 nM) to natural CbA,3 and we observed consistent cytotoxic potency (IC50 = 1.6 nM) for the synthetic CbA (Figure 1B).
Similar to the previously reported photocotransin,26 we aimed to install a diazirine group in CbA for photoactivated cross-linking to the binding target and an alkyne handle for in situ click chemistry coupling to fluorescent or affinity tags. All-l-CbA was reported to have moderate micromolar cytotoxicity against three cancer cell lines,27 while [d-MeAla11]-all-l-CbA displayed high nanomolar activity against four cancer cell lines.9 Given the adverse effect of the l-MeAla11 configuration for CbA activity, we anticipated that the neighboring Tyr(Me)10 could be involved in target interactions and could be substituted with the nearly isosteric 4-[3-(trifluoromethyl)-3H-diazirin-3-yl]phenylalanine (Tdf) side chain. This substitution is further supported by the loss of activity for an AprA analogue in which the MeTyr is epimerized.28
Positioning of the clickable alkyne amino acid was informed by a structure–activity relationship study to identify the optimal position for insertion of a propargylglycine (Pra) residue. In total, we synthesized six Pra/MePra-containing CbA analogues using the identical protocol with on-resin fragment condensation. Comparative antiproliferative testing in A549 lung cancer cells led to selection of residue position 3 for MePra in the targeted photoprobe (Figure S1). Dual modifications with MePra3 and Tdf10 afforded a potent photoaffinity probe, photocoibamide (Photo-CbA) with an alkyne handle (IC50 = 6.5 nM against HCT116 cells; Figure 1D).
Photo-CbA Directly Targets the Sec61α Subunit of the Protein Translocon
To identify the direct photo-CbA photo-cross-linking partner in an unbiased manner, we incubated live human A431 carcinoma cells with photo-CbA either in the presence or in the absence of parent CbA, followed by photolysis in intact cells. Detection of photo-cross-linked adducts was then carried out following installation of a TAMRA fluorophore by Cu(I) catalyzed Click chemistry under denaturing conditions and in-gel fluorescence scanning. This revealed a single cross-linked band of approximately 37 kDa in molecular weight, which was efficiently competed by addition of excess unmodified CbA (Figure 2A, first 2 lanes). A prior observation that CbA prevents biogenesis and ER import of VEGFR-2 during or following protein translation5 suggests that CbA may target a component of the ER protein biogenesis machinery. The observed 37 kDa molecular weight corresponds to that of the Sec61α subunit, which is the essential subunit of the protein translocon that forms the conduit through which newly synthesized proteins enter the secretory pathway.12 As Sec61α is the direct target of established highly selective natural products that inhibit ER import such as cotransins, AprA, decatransin, and mycolactone,29 we speculated that CbA could prevent VEGFR-2 expression by directly inhibiting Sec61. Repeating the photo-cross-linking assay against cells in which the endogenous locus of Sec61α has been edited to introduce additional sequence bearing a 3xFLAG epitope that increases the molecular weight by approximately 10 kDa revealed that the photo-cross-linked product shifted size accordingly (Figure 2A, last 2 lanes). As further validation that photo-CbA is cross-linking to Sec61α, a nonglycosylated protein, we used endoglycosidase H treatment to strip glycans from proteins in our sample. While the abundant and glycosylated Sec61 translocon component, Translocation Associated Membrane Protein 1 (TRAM), shifted to a smaller molecular weight species, the photo-cross-linked product remained at approximately 37 kDa, indicating that the target of photo-CbA is a non glycosylated protein (Figure S26).
Figure 2.

Photo-cross-linking of photo-CbA to Sec61α and CbA stabilization of Sec61 in cells. Photocotransin (CT7) or photocoibamide (pCbA) cross-linking to cells or sheep rough microsomes (SRM). Samples were photolyzed and the covalent adduct detected by click-chemistry to TAMRA-azide reporter and in-gel fluorescence scanning and Western blotting. (A) A431 cell lines were first incubated with CbA or carrier, followed by incubation with photo-CbA, photolysis, and click chemistry. Following SDS PAGE, lysates were queried for in gel fluorescence and subsequently transferred for anti-Sec61α and anti-FLAG Western Blot. Arrows indicate Sec61 (with or without a 10 kDa insert), star indicates nonspecific WB signal, triangle indicates free TAMRA dye within the gel. (B) As in A but in SRMs. Sample without UV irradiation shows the nonspecific background labeling of photo-CbA. (C) SRMs were first incubated with indicated concentrations of AprA, or Myco, followed by incubation with photo-CbA, photolysis, and click chemistry. (D) CT7 cross-linking to microsomes in the presence or absence of 10 μM CbA. (E) Stabilization of intracellular Sec61α by CbA. Isothermal concentration–response analysis of Sec61α in the presence or absence of CbA (0.01 nM to 3 μM), OSU-03012 (0.03 nM to 10 μM), or 0.1% DMSO. Intact U87-MG glioblastoma cells were treated as indicated and subjected to heating at 51 °C for 3 min. Heat-treated cell suspensions were snap frozen, lysates cleared by centrifugation, and the soluble fraction analyzed by SDS-PAGE and Western blotting. Note that Sec61α migrated above the 40 kDa molecular weight marker in several human cell lines using a standard Western blot protocol (Abcam ab183046; Figure S26C). (F) Quantification of immunoblot data shown in E. Sec61α band intensity was normalized to tubulin, and data points were fitted using nonlinear regression analysis.
To compare the binding of photo-CbA to known inhibitors of Sec61α, we then investigated cross-linking of photo-CbA and CT7, a potent and specific photoaffinity inhibitor of Sec61α,26 in isolated ER microsomes. Incubation with photo-CbA yielded a single band of approximately 37 kDa apparent molecular weight as per the in vivo result, which could be competed out in a concentration dependent manner by the addition of excess unmodified CbA (Figure 2B). We then tested whether photo-CbA cross-linking can be competed with known Sec61 ligands, AprA or mycolactone.14,15,28 Both compounds prevented photo-CbA cross-linking in a concentration-dependent manner; similarly, addition of excess CbA competed for cross-linking by cotransin CT7, both consistent with the notion that the photo-CbA cross-linked adduct is with Sec61α (Figure 2C and D)
We next used cellular thermal shift assays as an independent test of the ability of CbA to engage with Sec61 in cells. The feasibility of this approach was first interrogated by establishing a melting curve for Sec61α by analysis of soluble protein fractions by Western blot following a heat challenge (Figure S26). For these studies, intact human U87-MG glioblastoma cells were subjected to temperatures ranging from 45 to 72 °C in the presence and absence of CbA, AprA, or an unrelated PDK-1 and putative immunoglobulin binding protein (BiP) inhibitor, OSU-03012. Both CbA and AprA stabilized Sec61α, resulting in the continued detection of the presumed ligand-bound protein at a higher temperature range (60–66 °C) than for the relatively weak immunoreactivity observed for either vehicle- or OSU-03012-treated samples (Figure S26). On the basis of these results, a fixed temperature (51 °C) was then selected for isothermal dose–response fingerprinting of Sec61α in the presence, or absence, of increasing concentrations of CbA (0.01 nM to 3 μM) or OSU-03012 (0.03 nM to 10 μM). Under these conditions, CbA stabilized Sec61α in a concentration-dependent manner with half-maximal stabilization observed at ∼0.2 nM concentration, whereas no apparent stabilization of Sec61α was observed with OSU-03012 (Figure 2E and F).
Collectively, these data provide robust evidence indicating that CbA directly and specifically interacts with the Sec61α subunit of the ER protein translocation channel. All currently known natural product inhibitors of Sec61 bind at the same lumenal Sec61 cavity,29 and our photo-cross-linking data (Figure 2) suggests that also CbA binds Sec61α at this or a partially overlapping region.
Coibamide Reversibly Inhibits Biogenesis of Secreted and Membrane Proteins
The Sec61 translocon facilitates a key step in protein maturation by mediating the insertion of substrate proteins into the ER membrane or across it into the lumenal space.12 To investigate the global impact of CbA on cellular protein biogenesis, we performed metabolic labeling with 35S-methionine/cysteine in HCT116 colon carcinoma cells and investigated the processing of newly synthesized proteins in the presence or absence of CbA. In these experiments, even micromolar CbA concentrations did not result in reduction of the production levels of total cellular proteins (Figure 3A). However, production of glycosylated and secreted proteins was severely inhibited by CbA in a concentration-dependent manner, and full inhibition was observed with 100 nM CbA (Figure 3B and C). Collectively, CbA treatment does not impact cellular protein synthesis but instead prevents cotranslational protein glycosylation and net nascent protein secretion, which both require function of the Sec61 translocon. In this experiment, the effect of CbA on nascent protein synthesis was similar to that of AprA, a previously described substrate-nonselective inhibitor of ER protein translocation.14
Figure 3.

CbA specifically inhibits biogenesis of secretory and membrane proteins. (A to C) HCT-116 cells were labeled with 35S-Met and 35S-Cys in the presence of increasing concentrations of CbA. CHX/CA denotes control samples treated with cycloheximide and chloramphenicol to inhibit protein synthesis by cytosolic and mitochondrial ribosomes. Molecular weights are shown in kDa. (A) The collected cells were homogenized and analyzed by SDS-PAGE and autoradiography. (B) As in A but the samples are glycosylated protein fractions isolated with ConA-lectin affinity. (C) As in A but the samples are TCA-precipitated culture medium from the same experiment. (D) HEK-293T cells transiently expressing human VCAM1 were treated with 3 nM CbA, AprA, or vehicle (0.1% DMSO) at 5 h post-transfection and protein expression analyzed by Western Blotting at 24 h. (E) As in D, cells were treated with CbA (3 nM) or vehicle (0.1% DMSO) at 5 h post-transfection, incubated for a further 24 h, after which CbA was diluted 6-fold by the addition of fresh medium before the cells were harvested at the indicated time points. In D and E, the arrow indicates mature VCAM1 in several glycosylation states; the star denotes PARP-1.
We next set out to investigate the effects of CbA on the biogenesis of type I integral membrane proteins using human vascular cell adhesion molecule 1 (VCAM1) as a model protein. Biogenesis of this protein is potently inhibited by cotransin, a highly substrate-selective inhibitor of VCAM1 membrane insertion.19,20 We transiently expressed human VCAM1 in HEK293T cells and treated the cells with either CbA (3 nM), AprA (3 nM), or vehicle (0.1% DMSO) for 24 h. Immunoblot analysis of whole-cell lysates harvested after compound treatment revealed a strong reduction in VCAM1 expression in cells treated with either CbA or AprA (Figure 3D). Importantly, we did not observe evidence of proteolytic processing of poly[ADP-ribose] polymerase 1 (PARP-1), suggesting that caspase-dependent cell death is not involved in inhibition of VCAM1 expression. Further, dilution of CbA to subnanomolar concentrations, by the addition of fresh culture medium, resulted in a time-dependent reversal of VCAM1 expression inhibition, demonstrating that CbA inhibits Sec61-mediated protein biogenesis in a reversible manner (Figure 3E).
Coibamide Inhibits Sec61-Mediated Translocation in a Substrate-Nonselective Manner
Earlier studies have revealed that cotransin downregulates VCAM1 by preventing its Sec61-mediated ER insertion and causing the cytosolic displacement of the nascent VCAM1 polypeptide and subsequent cytosolic degradation by the ubiquitin proteasome system.20 Thus, we set out to test whether CbA inhibits VCAM1 expression via a similar mechanism. Here, treating cells with CbA resulted in downregulation of VCAM1 expression, which was rescued in a time-dependent manner by treatment with bortezomib (BtZ), a specific inhibitor of the proteasome (Figure 4A). Therefore, CbA appears to inhibit VCAM1 expression during or after nascent VCAM1 synthesis, which could involve stages of ER targeting, membrane insertion, maturation, or protein trafficking. The observed accumulation of immature unglycosylated VCAM1 forms following CbA and BtZ treatment indicate that CbA interferes with proper VCAM1 maturation (Figure 4A).
Figure 4.

CbA inhibits translocation of a range of Sec61 substrates. (A and B) The star indicates the unprocessed form of the protein, and the arrow points to the processed, mature form. Molecular weights of the standards are shown in kDa. (A) Cells transiently expressing VCAM1 were treated 5 h post-transfection with either 1 or 3 nM CbA in the presence or absence of 20 nM proteasomal inhibitor bortezomib (BtZ). Whole cell lysates were analyzed by Western Blotting after 24 h. (B and C) For the in vitro translocation assay (IVT), the indicated proteins were translated in the presence of microsomes, 35S-Met, and the indicated inhibitors. (B) Translocation of glycosylated proteins was assessed by change in migration in SDS-PAGE. Endoglycosidase treatment (EndoH) was used to demonstrate that the altered migration was due to glycosylation. Yeast α-factor, mouse elongation of very long chain fatty acids protein 3 (CIG30), beta-lactamase (β-Lac). (C) Translocation of nonglycosylated proteins was demonstrated by treatment with proteinase K (PK). The Hamster binding immunoglobulin protein (BiP) and bovine prolactin (Prl). The star indicates protein degradation products following PK digestion, and the arrow points to the intact protein.
To dissect the biochemical mechanism by which CbA prevents maturation of nascent secretory proteins, we used a reconstituted mammalian translation system supplemented with isolated sheep rough microsomes (SRM).30 In these experiments, CbA did not influence protein translation of any of the diverse Sec61 substrate proteins tested, which included secreted proteins (yeast α-factor, β-lactamase, BiP, preprolactin) and a polytopic membrane protein (CIG30). We assayed the inhibitory effect of CbA on protein processing in vitro and compared the effects with those of AprA and cotransin analogue CT8 (Figure 4 B). CbA prevents both glycosylation (yeast α-factor) and signal peptide cleavage (β-lactamase). Processing of secreted proteins was inhibited, while CIG30, a multipass membrane protein, was resistant against CbA and all other translocation inhibitors tested (CT8, AprA) and is also known to be resistant toward mycolactone,15 consistent with the critical CIG30 dependence for the ER membrane protein complex (EMC) instead of Sec61 for its biogenesis31
To further investigate the CbA-sensitive stage of protein translocation, we again used the reconstituted in vitro translation and translocation system and queried the accessibility of Sec61 substrate proteins to exogenous proteinase K (PK) in the presence or absence of SRM. In these reactions, nascent polypeptides of ER translocated BiP and preprolactin (Prl) are shielded from protease digestion in the presence of SRM, yet are cleaved in the absence of SRM or when microsomes are solubilized with detergent indicative of membrane translocation (Figure 4C). However, the addition of 1 μM CbA, CT8, or AprA into SRM-containing reactions renders the nascent polypeptides sensitive to PK (Figure 4C), indicating that these compounds prevent ER entry of the newly synthesized proteins. Collectively, our data (Figures 3D, 4) indicate that CbA potently prevents ER insertion and subsequent processing of a wide range of secreted and integral membrane proteins with the notable exception of membrane proteins with N-terminal transmembrane segments of the type III topology.
Coibamide Binding to Sec61 Is Distinct from Other Translocation Inhibitors
In an effort to map the location of the CbA binding site on Sec61α, we used a genetic selection approach in mammalian cells and attempted to identify specific resistance-conferring point mutations. Such chemogenetic screens are a powerful means to discover novel mutations and mechanisms of action for cell-active small molecules and have been used to identify point mutations in SEC61A1 that confer specific resistance to cytotoxicity of different Sec61 inhibitors.14,16,17,32 We exposed EMS mutagenized HCT116 colon carcinoma cells to 50–100 nM CbA (IC50 ∼ 29 nM), which resulted in a majority of the cells dying, yet during selection, six colonies grew and were isolated. Monoclonal cell lines derived from the colonies exhibited strong resistance to CbA (up to 100-fold desensitization) and to an even greater degree to AprA (Figure 5A). Sequencing the coding regions of the SEC61A1 gene revealed a single nucleotide transition encoding for the heterozygous Sec61α mutation S71P. In contrast to CbA selection, previous unbiased resistance mapping screens in HCT116 cells with cotransin, AprA, and dectransin all revealed a range of Sec61α mutations conferring specific resistance to the tested Sec61 inhibitors. To assay the effect of previously identified Sec61α mutations, we tested a panel of mutations in naive HEK293 FRT cells where the mutant Sec61α proteins are stably expressed from an exogenous locus at a similar level as endogenous Sec61α.17 As we reported earlier, all of the tested mutations conferred strong resistance to AprA,14 but only moderate resistance was observed for CbA (Figure 5B). This finding is surprising especially for the R66I mutation that confers essentially complete resistance for all known Sec61 inhibitors,14−16,24 yet only causes moderate (∼7-fold) desensitization to CbA (Figure 5B). The Sec61α mutations S82P and T86M that confer resistance to other Sec61 inhibitors, but not CbA, are clustered on the lumenal end of the Sec61 lateral gate, whereas mutations R66I and S71P are located in different parts of the Sec61 plug domain (Figure 6). Taken together, these mutational results suggest that CbA may interact with Sec61 in a unique manner or possibly bind a different conformation of the channel.
Figure 5.

CbA mode of inhibition is specific compared to known Sec61 inhibitors. (A) Cell viability was measured by Alamar Blue assay (mean ± SD, n = 4, all cell lines assayed simultaneously) for HCT-116 cell lines isolated on the basis of CbA resistance in the presence of concentration series of indicated compounds. (B) Cell viability was measured by Alamar Blue assay (mean ± SD, n = 4, all cell lines assayed simultaneously) for HEK293FRT cells stably expressing Sec61α mutants in the presence of concentration series of indicated compounds. (C) Comparison of published NCI60 data for CbA,3 AprA,11 and IpoF.33 The heat map was derived by plotting each GI50 value divided by the median GI50 for that compound. X indicates a cell line that was not tested. Closed arrowhead indicates cell lines with most notable differences across the three compounds.
Figure 6.

Structure of Sec61 complex and location of resistance point mutations. Lateral and ER lumenal views in closed and open conformations of the Sec61 complex are shown. Lateral gate helices 2 + 3 (light blue) and 7 + 8 (light coral) as well as the plug region (light green) are indicated on the Sec61α core subunit (light gray). Mutations in Sec61α at residues 82 and 86 in helix 2 are shown in purple, lumenal plug residues 66 and 71 are shown in teal. The open but stalled Sec61 complex is shown with bound preprolactin signal sequence (brown). Structural coordinates were obtained from the Protein Data Bank, IDs 3J7Q and 3JC2.
Summary
Here, we report the comprehensive identification of target interactions of coibamide A (CbA), a cytotoxic marine natural product, with the ability to inhibit biogenesis of secreted and integral membrane proteins. By developing an isosteric photoaffinity probe of CbA, we demonstrate that the main binding target of CbA in mammalian cells is the Sec61α subunit of the Sec61 protein translocon complex. Metabolic labeling experiments in cells and biochemical ER translocation experiments indicate that CbA inhibits cotranslational Sec61-facilitated ER translocation in a substrate-nonselective manner. Finally, through an unbiased mutational mapping approach, we demonstrate that the cytotoxic potential of CbA for mammalian cells results from Sec61 inhibition, likely through binding of a Sec61 site partially overlapping with other substrate-selective and nonselective Sec61 inhibitors.
The Sec61 translocon forms a membrane channel which facilitates the essential ER membrane translocation or membrane integration step during biogenesis of secretory or integral membrane proteins, respectively. As rapidly proliferating cancer cells display heightened dependence on protein synthesis, pharmacological targeting of cellular proteostatic pathways, including the ER protein biogenesis machinery, has potential for the development of new therapeutic strategies,34 prompting an interest in finding new privileged scaffolds to target critical proteostasis factors. Recent work has identified many structurally distinct natural product small molecules that appear to have evolved independently in distinct micro-organisms to target Sec61 as a way to modulate or prevent biogenesis of secreted or integral membrane proteins. Intriguingly, all of these inhibitors appear to target Sec61 at its lumenal cavity near the Sec61 lateral gate and plug domains (Figure 6) whether they inhibit production of Sec61 substrate proteins in a substrate-selective (cotransins)19,20 or substrate-nonselective (AprA, mycolactone, IpoF, decatransin)14−16,22,24 manner.
The lumenal Sec61 cavity where all reported inhibitors bind has been outlined by mutations identified in several independent studies in both mammalian and yeast chemogenomic screens (reviewed in ref (29)). Together the mutations outline a general binding pocket at the lumenal end of the Sec61α subunit, which appears to be at least partially shared by all five published inhibitors. Yet, a distinct pattern of resistance has been observed for some of these inhibitors,14,24 suggesting that despite occupying the same general binding cavity, they utilize different specific interactions with Sec61 or bind different conformations of the inherently dynamic channel. Specifically, Sec61α mutations R66I and S82P confer potent resistance to all other known Sec61 inhibitors, while only conferring mild resistance to CbA. This suggests that CbA may bind Sec61 in a unique way, possibly by targeting a conformation in which the plug has moved to a different position. This notion is also supported by failure of our unbiased screen to identify common Sec61α mutations, which have been identified for several inhibitors earlier in multiple resistance mutation screens.14,16,17,24 Future structural studies will be required to definitively characterize the binding modes of Coibamide A and other Sec61 inhibitors and can provide a basis for understanding means for inhibiting Sec61 in a substrate-selective manner.
This study and earlier work reveal an expansion of the binding site for Sec61 inhibition by diverse natural and synthetic inhibitors and prompt the question whether the observed binding differences translate to different cellular phenotypes. So far only two inhibitor classes, cotransins and CADA, have been reported to inhibit biogenesis of Sec61 client proteins with substrate selectivity. All the other reported natural product inhibitors prevent biogenesis of nearly all Sec61 dependent secreted and membrane proteins. Preventing synthesis of key proteins required for cancer cell survival and proliferation forms the basis for targeting cancer cells with substrate-selective Sec61 inhibitors.29,36
Also, the substrate-nonselective inhibitors AprA and coibamide A have demonstrated a therapeutic possibility to target cancer cells in in vivo models of human cancer,5,28,33,37 albeit with a limited therapeutic window. It remains unclear how structurally different Sec61 inhibitors with a seemingly identical biochemical inhibition mechanism could be leveraged for the design of cell type selective therapeutic lead scaffolds. To investigate this, we examined the published comparative growth inhibitory phenotypes across the cell lines assayed in the USA National Cancer Institute panel of 60 cancer cell lines (NCI-60)38 against CbA,3 AprA,11 and IpoF.39 This correlative analysis revealed that each of the three compounds was designated as “COMPARE-negative,” suggestive of having distinct cytotoxic mechanisms. While not all data from the two testing events for each compound were available, there were activity data from one test event for at least 55 cell lines in each case, out of a total of 61 different cell lines tested across the three compounds. Because all three compounds appear to cause cytotoxicity by targeting the same site on Sec61 in a substrate-nonselective manner, it is remarkable that the NCI-60 panel fails to recognize them as a mechanistic set. Comparison of the relative sensitivities of the tested cell lines reveals notable examples of differential cell targeting despite a lack of common trends at the level of histological cell types (Figure 5C). For example, the SF268 CNS cancer cell line ranks in the top 10 most sensitive to CbA (GI50 1.5 nM), yet it is in the 10 least sensitive cell lines to AprA (GI5051 nM) and is also less sensitive to IpoF (GI50120 nM). Further, HCC-2998 colon cancer cells are highly sensitive to CbA (GI50 < 1 nM) and AprA (GI50 3.7 nM), yet they are one of the most resistant of the cell lines exposed to IpoF (GI50 > 1000 nM). Finally, the renal cell line TK-10 appears to be much more sensitive to AprA and IpoF over CbA. Collectively, the NCI-60 profiling data reveal surprising cell type selectivity for these three compounds that induce cytotoxicity through binding and global inhibition of ER protein import. Possible explanations for the observed differences in cell type specificities include differences in compound bioavailability, differential cell export by diverse multidrug efflux pumps, and differences in the cellular Sec61 inhibition mechanism by the ability to, for example, target specific Sec61 cofactor complexes. Together, these findings support the notion that structurally distinct Sec61 binding inhibitors could be developed to target specific diseased cells and tissues. In support of this notion, changes in the structure of AprA and CbA have yielded compound variants with reduced general cytotoxicity, while retaining efficacy in human tumor xenograft models.7,33,40 Further, modifications to the structure of mycolactone demonstrated that its cytotoxic and anti-inflammatory effects can at least partially be dissociated from each other.41 Finally, modifications to the side-chains of the substrate-selective Sec61 inhibitor cotransin altered the range of inhibited Sec61 substrates.21
Taken together, our work adds the structurally distinct cyclic peptide CbA to the class of potent Sec61 inhibitors whose on-target interactions at the Sec61α lateral plug region prevent biogenesis of secreted protein factors and integral membrane proteins. Our work expands the class of structurally unique chemotypes that inhibit Sec61 and permit targeting of distinct cell types, which could be particularly relevant in diseases such as cancer, inflammation, and certain viral diseases, where Sec61-facilitated protein biogenesis contributes to disease progression. Finally, convergent evolution that resulted in the appearance of multiple classes of inhibitors presumably targeting diverse eukaryotic Sec61 channel orthologs highlights the important role that modulating protein biogenesis of extracellular proteins has for diverse microbial cells in distinct ecological niches.
Methods
General Method for Synthetic Coibamide Compounds
1H NMR spectra were recorded using a JEOL ECA-500 spectrometer. Chemical shifts are reported in δ (ppm) relative to Me4Si (in CDCl3) as an internal standard. 13C NMR spectra were recorded using a JEOL ECA-500 spectrometer and referenced to the residual solvent signal (δ 77.00 ppm). High resolution mass spectra (HRMS) were recorded on a Shimadzu LC-ESI-IT-TOF-MS instrument. Optical rotations were measured using a JASCO P-1020 polarimeter. For flash chromatography, Wakogel C-200E (Wako) was employed. For analytical HPLC, a Cosmosil 5C18-ARII column (4.6 × 250 mm, Nacalai Tesque, Inc.) was employed with a linear gradient of CH3CN (with 0.1% (v/v) TFA) in H2O at a flow rate of 1 mL/min, and eluting products were detected by UV at 220 nm. Preparative HPLC was performed using a Cosmosil 5C18-ARII preparative column (20 × 250 mm, Nacalai Tesque, Inc.) at a flow rate of 8 mL/min.
Reagents for Chemical Biology
The production and purification of translocation inhibitors have been described previously: CT7 and CT8,26 AprA,42 and mycolactone.15 OSU-03012 (AR12) was purchased from Millipore-Sigma. Sheep rough microsomes were isolated as previously described.43
DNA Constructs and Transfections
DNA constructs encoding in vitro transcription templates for the cell-free translocation assays were PCR amplified using 5′-primers containing either T7 or SP6 promoter, a Kozak sequence, and a region complementary to the 5′-end of the gene. The 3′-primers contained a stop-codon and a region complementary to the 3′-end of the gene. For analysis of VCAM1 expression, full length VCAM1 in pCDNA3.120 was transiently expressed in HEK293T cells using jetPRIME Transfection Reagent (Polyplus transfection) 5 h before treatment.
SDS-PAGE, Autoradiography, and Western Blot
SDS-PAGE was performed either with Tris/Tricine or with TGX stain-free polyacrylamide gels (Bio-Rad). For autoradiography, the dried gels were exposed to a storage phosphor screen (GE Healthcare) and imaged on a Typhoon phosphorimager (GE Healthcare). For Western blotting, proteins were transferred to nitrocellulose membranes (Bio-Rad). Following blocking of the membranes with Odyssey Blocking Buffer (PBS; LI-COR Biosciences) or 5% (w/v) nonfat dry milk in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl (TBS) plus 0.1% Tween-20 (TBS-Tween), the membranes were first incubated with the appropriate primary antibodies and then with the appropriate secondary antibodies and finally imaged on an Odyssey infrared fluorescent scanner (LI-COR Biosciences) or a MyECL image analysis system (Thermo Fisher Scientific). All antibodies were from commercial sources and used according to the recommendations of the manufacturer.
Cell-Free Translation/Translocation Assays
In vitro transcription, translation, and translocation assays were done as previously described.14,30 The indicated genes were transcribed with T7 or SP6 polymerase, translated at 32 °C in the presence of microsomes, 35S-Met, and indicated inhibitors. The translocation was confirmed either with endoglycosidase (EndoH) treatment or protease protection assay with proteinase K (PK). All the samples were TCA-precipitated before gel analysis. The synthesized proteins were detected by SDS-PAGE and autoradiography.
Photoaffinity Labeling
The photoaffinity labeling and click chemistry with microsomes were done as previously described.17 Sheep rough microsomes (SRMs) containing 100 nM Sec61 were first incubated for 30 min with indicated inhibitors, then with 100 nM photocoibamide or photocotransin CT7 for 10 min and cross-linking performed by UV-irradiation for 10 min. After denaturation with SDS, copper catalyzed Click chemistry was used to label the cross-linked adducts with the fluorescent group. The labeled proteins were analyzed by SDS-PAGE and in-gel fluorescence. For in cell photo-cross-linking, 1.5 × 105 A431 cells were plated into each well on a 12-well plate and grown for 24 h. After washing the cells twice with Dulbecco’s PBS, media containing either the competing, unlabeled CbA, or vehicle (0.1% DMSO) were added. The cells were incubated in the cell incubator for 1 h, photo-CbA added, and the incubation continued for 30 min. The wells were washed twice with Dulbecco’s PBS and photolyzed with UV irradiation (50 W, 365 nm) for 10 min. Cells were collected by scraping, pelleted, and resuspended into 50 mM HEPES, pH 7.5, 100 mM KOAc, 250 mM sucrose, 2 mM Mg(OAc)2, 1% Triton X-100, and protease inhibitor (Pierce Protease Inhibitor, EDTA-free). The lysed cells were centrifuged at 21 100g for 10 min, the supernatant collected, and the proteins denatured by adding 1.1% SDS. The click chemistry and gel analysis were done as with the reactions containing SRMs.
Cell Culture and Cell Viability Assays, Pulse-Labeling of Cells
A431, HEK293T, and HEK293FRT cells were cultured in DMEM supplemented with 10% FBS. HCT-116 cells were cultured in McCoy’s 5A media supplemented with 10% FBS. U87-MG cells were cultured in Minimum Essential Media (MEM) with 10% FBS, l-glutamine (2 mM), and 1% penicillin and streptomycin. MDA-MB-231 cells were cultured in MEM with Earl’s salts plus 1% penicillin/streptomycin and 10% FBS. All cell lines were incubated at 37 °C under 5% CO2. The activity of synthetic coibamide and photoaffinity analogues was tested in MDA-MB-231 and A549 cells after 72 h by MTS assay. All other cell viability assays were performed by seeding PerkinElmer Viewplate-96 plates at a density of 2.5 × 103 cells per well. Cells were allowed 24 h to adhere and then treated with indicated concentrations of inhibitors or vehicle (0.1% DMSO) for a further 72 h. Viability was estimated by the addition of Alamar Blue (Life Technologies) and measuring fluorescence after a further 4 h. Pulse-labeling experiments were performed by seeding six-well plates with 5 × 105 cells per well and allowing 24 h for cells to adhere. Cells were washed twice with PBS, then exchanged into Met-Cys free media with indicated concentrations of inhibitors for 30 min. 100 μCi of PerkinElmer EXPRE35S35S Protein Labeling Mix was then added per well. Media wer collected, and the cells were harvested by scraping them into ice cold PBS after 30 min. Total protein was acquired by RIPA extraction from the cell pellet. Glycosylated protein was acquired by Concanavalin A purification, and secreted protein was acquired by TCA precipitation of the media.
Cellular Thermal Shift Assay
Assays were carried out according to a method previously described by Jafari and co-workers.44 Briefly, a melting curve for Sec61α was established in U87-MG glioblastoma cells. Whole cells were treated as indicated for 1 h, harvested using trypsin, and collected by gentle centrifugation at 300g for 3 min. Cell pellets were resuspended in PBS supplemented with PMSF and benzamidine to a final cell density of ∼2 × 106 cells/mL. The resulting cell suspension was equally distributed into PCR tubes and subjected to a range of temperatures (45 to 72 °C) using a Veriti 96-well thermal cycler. Cells were heated to the designated temperature for 3 min and immediately snap-frozen in liquid nitrogen. All samples were subjected to two freeze–thaw cycles. Lysates were cleared by centrifugation at 18,000g for 20 min, at 4 °C, and the supernatants were carefully transferred to new tubes for immunoblot analysis. For isothermal concentration–response fingerprinting of Sec61α, U87-MG cells were harvested, as above, and resuspended in fresh medium to a cell density of ∼4 × 107 cells/mL. CbA, OSU-03012, or DMSO (final 0.1%) in 50 μL of cell medium was added to 15 μL of the cell suspension and incubated at 37 °C for 30 min. Cells were then subjected to a 51 °C heat treatment in a Veriti 96-well thermal cycler for 3 min and immediately snap-frozen in liquid nitrogen. The samples went through two freeze–thaw cycles, centrifugation, and preparation for immunoblot analysis as described above.
Resistant Cell Line Isolation
For obtaining resistant cell lines, HCT-116 cells were mutagenized by incubation with ethylmethanesulfonate (EMS) at a concentration of 2000 μg/mL for 60 min. Cells were allowed 48 h to recover, then cultured in the presence of 50–100 nM coibamide A for 14 days, after which cell colonies were isolated by disc cloning and cultured further in drug-free media. Total RNA was isolated using Trizol reagent according to the manufacturer’s instructions. Total cDNA was synthesized using anchored oligo(dT) primers and SuperScript IV reverse transcriptase (Invitrogen). Different cDNAs were amplified with Phusion polymerase (Thermo Fisher Scientific) and sequenced bidirectionally by Sanger sequencing.
Acknowledgments
This work was supported by the Academy of Finland (grants 289737 and 314672 to V.O.P., 299762 to J.K.) and Sigrid Juselius Foundation (V.O.P.), and in part by a Discovery Grant from the American Brain Tumor Association (J.E.I.); NIH Fogarty International Center ICBG grant TW006634-06 (K.L.M.); NIGMS R01GM132649 (J.E.I., K.L.M., S.O., V.O.P.); NCCIH T32AT010131 (D.R.M.); JSPS KAKENHI (24659004); AMED (JP17am0101092j0001; JP18lm0203006j0002); Takeda Science Foundation. The slaughterhouse Vainion Teurastamo is acknowledged for providing the sheep pancreas for isolating the microsomes. We are grateful to T. Matsuda and T. Kitamura (Kyoto University) for their technical assistance.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00325.
Supporting methods, compound characterization data (Figures S1–S25), and supplemental biological experiment data (Figure S26 and Tables S1 and S2) (PDF)
Author Contributions
A.O.P., D.T., J.E.I., U.R., and V.O.P designed the chemical biological and pharmacological experiments. S.K. and S.O. designed and synthesized the CbA probes. A.O.P., D.T., J.K., S.Kaw., S.Kaz., T.M., M.N., S.K., S.O., J.D.S., D.R.M., X.W., and V.O.P. conducted the experiments. A.O.P., D.T., J.E.I., K.L.M., S.O., V.O.P., C.C.T., and W.K.V. analyzed and interpreted the data. A.O.P., D.T., J.E.I., K.L.M., S.O., and V.O.P. drafted or revised the article.
Author Contributions
# These authors contributed equally
The authors declare no competing financial interest.
Supplementary Material
References
- Newman D. J.; Cragg G. M. (2016) Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661. 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Carlson E. E. (2010) Natural products as chemical probes. ACS Chem. Biol. 5, 639–653. 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R. A.; Goeger D. E.; Hills P.; Mooberry S. L.; Huang N.; Romero L. I.; Ortega-Barría E.; Gerwick W. H.; McPhail K. L. (2008) Coibamide A, a potent antiproliferative cyclic depsipeptide from the Panamanian marine cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 130, 6324–6325. 10.1021/ja801383f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engene N.; Paul V. J.; Byrum T.; Gerwick W. H.; Thor A.; Ellisman M. H. (2013) Five chemically rich species of tropical marine cyanobacteria of the genus Okeania gen. nov. (Oscillatoriales, Cyanoprokaryota). J. Phycol. 49, 1095–1106. 10.1111/jpy.12115. [DOI] [PubMed] [Google Scholar]
- Serrill J. D.; Wan X.; Hau A. M.; Jang H. S.; Coleman D. J.; Indra A. K.; Alani A. W. G.; McPhail K. L.; Ishmael J. E. (2016) Coibamide A, a natural lariat depsipeptide, inhibits VEGFA/VEGFR2 expression and suppresses tumor growth in glioblastoma xenografts. Invest. New Drugs 34, 24–40. 10.1007/s10637-015-0303-x. [DOI] [PubMed] [Google Scholar]
- Hau A. M.; Greenwood J. A.; Löhr C. V.; Serrill J. D.; Proteau P. J.; Ganley I. G.; McPhail K. L.; Ishmael J. E. (2013) Coibamide A induces mTOR-independent autophagy and cell death in human glioblastoma cells. PLoS One 8, e65250 10.1371/journal.pone.0065250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G.; Wang W.; Ao L.; Cheng Z.; Wu C.; Pan Z.; Liu K.; Li H.; Su W.; Fang L. (2018) Improved Total Synthesis and Biological Evaluation of Coibamide A Analogues. J. Med. Chem. 61, 8908–8916. 10.1021/acs.jmedchem.8b01141. [DOI] [PubMed] [Google Scholar]
- Yao G.; Pan Z.; Wu C.; Wang W.; Fang L.; Su W. (2015) Efficient Synthesis and Stereochemical Revision of Coibamide A. J. Am. Chem. Soc. 137, 13488–13491. 10.1021/jacs.5b09286. [DOI] [PubMed] [Google Scholar]
- Nabika R.; Suyama T. L.; Hau A. M.; Misu R.; Ohno H.; Ishmael J. E.; McPhail K. L.; Oishi S.; Fujii N. (2015) Synthesis and biological evaluation of the [d-MeAla(11)]-epimer of coibamide A. Bioorg. Med. Chem. Lett. 25, 302–306. 10.1016/j.bmcl.2014.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sable G. A.; Park J.; Kim H.; Lim S.-J.; Jang S.; Lim D. (2015) Solid-Phase Total Synthesis of the Proposed Structure of Coibamide A and Its Derivative: Highly Methylated Cyclic Depsipeptides. Eur. J. Org. Chem. 2015, 7043–7052. 10.1002/ejoc.201500697. [DOI] [Google Scholar]
- Luesch H.; Chanda S. K.; Raya R. M.; DeJesus P. D.; Orth A. P.; Walker J. R.; Izpisúa Belmonte J. C.; Schultz P. G. (2006) A functional genomics approach to the mode of action of apratoxin A. Nat. Chem. Biol. 2, 158–167. 10.1038/nchembio769. [DOI] [PubMed] [Google Scholar]
- Rapoport T. A.; Li L.; Park E. (2017) Structural and Mechanistic Insights into Protein Translocation. Annu. Rev. Cell Dev. Biol. 33, 369–390. 10.1146/annurev-cellbio-100616-060439. [DOI] [PubMed] [Google Scholar]
- Klausner R. D.; Donaldson J. G.; Lippincott-Schwartz J. (1992) Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116, 1071–1080. 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paatero A. O.; Kellosalo J.; Dunyak B. M.; Almaliti J.; Gestwicki J. E.; Gerwick W. H.; Taunton J.; Paavilainen V. O. (2016) Apratoxin Kills Cells by Direct Blockade of the Sec61 Protein Translocation Channel. Cell Chem. Biol. 23, 561–566. 10.1016/j.chembiol.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Baron L.; Paatero A. O.; Morel J.-D.; Impens F.; Guenin-Macé L.; Saint-Auret S.; Blanchard N.; Dillmann R.; Niang F.; Pellegrini S.; Taunton J.; Paavilainen V. O.; Demangel C. (2016) Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J. Exp. Med. 213, 2885–2896. 10.1084/jem.20160662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junne T.; Wong J.; Studer C.; Aust T.; Bauer B. W.; Beibel M.; Bhullar B.; Bruccoleri R.; Eichenberger J.; Estoppey D.; Hartmann N.; Knapp B.; Krastel P.; Melin N.; Oakeley E. J.; Oberer L.; Riedl R.; Roma G.; Schuierer S.; Petersen F.; Tallarico J. A.; Rapoport T. A.; Spiess M.; Hoepfner D. (2015) Decatransin, a new natural product inhibiting protein translocation at the Sec61/SecYEG translocon. J. Cell Sci. 128, 1217–1229. 10.1242/jcs.165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon A. L.; Paavilainen V. O.; Sharma A.; Hegde R. S.; Taunton J. (2014) An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. eLife 3, e01483 10.7554/eLife.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejeans N.; Manié S.; Hetz C.; Bard F.; Hupp T.; Agostinis P.; Samali A.; Chevet E. (2014) Addicted to secrete - novel concepts and targets in cancer therapy. Trends Mol. Med. 20, 242–250. 10.1016/j.molmed.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Besemer J.; Harant H.; Wang S.; Oberhauser B.; Marquardt K.; Foster C. A.; Schreiner E. P.; de Vries J. E.; Dascher-Nadel C.; Lindley I. J. D. (2005) Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature 436, 290–293. 10.1038/nature03670. [DOI] [PubMed] [Google Scholar]
- Garrison J. L.; Kunkel E. J.; Hegde R. S.; Taunton J. (2005) A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature 436, 285–289. 10.1038/nature03821. [DOI] [PubMed] [Google Scholar]
- Maifeld S. V.; MacKinnon A. L.; Garrison J. L.; Sharma A.; Kunkel E. J.; Hegde R. S.; Taunton J. (2011) Secretory protein profiling reveals TNF-α inactivation by selective and promiscuous Sec61 modulators. Chem. Biol. 18, 1082–1088. 10.1016/j.chembiol.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B. S.; Hill K.; McKenna M.; Ogbechi J.; High S.; Willis A. E.; Simmonds R. E. (2014) The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog. 10, e1004061 10.1371/journal.ppat.1004061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George K. M.; Chatterjee D.; Gunawardana G.; Welty D.; Hayman J.; Lee R.; Small P. L. (1999) Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science 283, 854–857. 10.1126/science.283.5403.854. [DOI] [PubMed] [Google Scholar]
- Zong G.; Hu Z.; O’Keefe S.; Tranter D.; Iannotti M. J.; Baron L.; Hall B.; Corfield K.; Paatero A. O.; Henderson M. J.; Roboti P.; Zhou J.; Sun X.; Govindarajan M.; Rohde J. M.; Blanchard N.; Simmonds R.; Inglese J.; Du Y.; Demangel C.; High S.; Paavilainen V. O.; Shi W. Q. (2019) Ipomoeassin F Binds Sec61α to Inhibit Protein Translocation. J. Am. Chem. Soc. 141, 8450–8461. 10.1021/jacs.8b13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamayun I.; O’Keefe S.; Pick T.; Klein M.-C.; Nguyen D.; McKibbin C.; Piacenti M.; Williams H. M.; Flitsch S. L.; Whitehead R. C.; Swanton E.; Helms V.; High S.; Zimmermann R.; Cavalié A. (2019) Eeyarestatin Compounds Selectively Enhance Sec61-Mediated Ca2+ Leakage from the Endoplasmic Reticulum. Cell Chem. Biol. 26, 571–583. e6. 10.1016/j.chembiol.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon A. L.; Garrison J. L.; Hegde R. S.; Taunton J. (2007) Photo-leucine incorporation reveals the target of a cyclodepsipeptide inhibitor of cotranslational translocation. J. Am. Chem. Soc. 129, 14560–14561. 10.1021/ja076250y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W.; Qiu H.-B.; Chen Y.-J.; Xi J.; Yao Z.-J. (2014) Total synthesis of proposed structure of coibamide A, a highly N- and O-methylated cytotoxic marine cyclodepsipeptide. Tetrahedron Lett. 55, 6109–6112. 10.1016/j.tetlet.2014.09.047. [DOI] [Google Scholar]
- Huang K.-C.; Chen Z.; Jiang Y.; Akare S.; Kolber-Simonds D.; Condon K.; Agoulnik S.; Tendyke K.; Shen Y.; Wu K.-M.; Mathieu S.; Choi H.-W.; Zhu X.; Shimizu H.; Kotake Y.; Gerwick W. H.; Uenaka T.; Woodall-Jappe M.; Nomoto K. (2016) Apratoxin A Shows Novel Pancreas-Targeting Activity through the Binding of Sec 61. Mol. Cancer Ther. 15, 1208–1216. 10.1158/1535-7163.MCT-15-0648. [DOI] [PubMed] [Google Scholar]
- Van Puyenbroeck V.; Vermeire K. (2018) Inhibitors of protein translocation across membranes of the secretory pathway: novel antimicrobial and anticancer agents. Cell. Mol. Life Sci. 75, 1541–1558. 10.1007/s00018-017-2743-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A., Mariappan M., Appathurai S., and Hegde R. S. (2010) In Vitro Dissection of Protein Translocation into the Mammalian Endoplasmic Reticulum, in Protein Secretion: Methods and Protocols (Economou A., Ed.), pp 339–363, Humana Press, Totowa, NJ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitwood P. J.; Juszkiewicz S.; Guna A.; Shao S.; Hegde R. S. (2018) EMC Is Required to Initiate Accurate Membrane Protein Topogenesis. Cell 175, 1507–1519. e16. 10.1016/j.cell.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor T. M.; Miller R. M. (2017) Leveraging Chemotype-Specific Resistance for Drug Target Identification and Chemical Biology. Trends Pharmacol. Sci. 38, 1100–1109. 10.1016/j.tips.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.-Y.; Liu Y.; Cai W.; Luesch H. (2014) Improved total synthesis and biological evaluation of potent apratoxin S4 based anticancer agents with differential stability and further enhanced activity. J. Med. Chem. 57, 3011–3029. 10.1021/jm4019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Teuber V.; Albert-Gasco H.; Auyeung V. C.; Papa F. R.; Mallucci G. R.; Hetz C. (2019) Small Molecules to Improve ER Proteostasis in Disease. Trends Pharmacol. Sci. 40, 684–695. 10.1016/j.tips.2019.07.003. [DOI] [PubMed] [Google Scholar]
- Vermeire K.; Bell T. W.; Van Puyenbroeck V.; Giraut A.; Noppen S.; Liekens S.; Schols D.; Hartmann E.; Kalies K.-U.; Marsh M. (2014) Signal peptide-binding drug as a selective inhibitor of co-translational protein translocation. PLoS Biol. 12, e1002011 10.1371/journal.pbio.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Saenz A.; Sandhu M.; Carrasco Y.; Maglathlin R. L.; Taunton J.; Moasser M. M. (2015) Targeting HER3 by interfering with its Sec61-mediated cotranslational insertion into the endoplasmic reticulum. Oncogene 34, 5288–5294. 10.1038/onc.2014.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W.; Ratnayake R.; Gerber M. H.; Chen Q.-Y.; Yu Y.; Derendorf H.; Trevino J. G.; Luesch H. (2019) Development of apratoxin S10 (Apra S10) as an anti-pancreatic cancer agent and its preliminary evaluation in an orthotopic patient-derived xenograft (PDX) model. Invest. New Drugs 37, 364–374. 10.1007/s10637-018-0647-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker R. H. (2006) The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 6, 813–823. 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Zong G.; Whisenhunt L.; Hu Z.; Shi W. Q. (2017) Synergistic Contribution of Tiglate and Cinnamate to Cytotoxicity of Ipomoeassin F. J. Org. Chem. 82, 4977–4985. 10.1021/acs.joc.7b00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.-Y.; Liu Y.; Luesch H. (2011) Systematic Chemical Mutagenesis Identifies a Potent Novel Apratoxin A/E Hybrid with Improved in Vivo Antitumor Activity. ACS Med. Chem. Lett. 2, 861–865. 10.1021/ml200176m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenin-Macé L.; Baron L.; Chany A.-C.; Tresse C.; Saint-Auret S.; Jönsson F.; Le Chevalier F.; Bruhns P.; Bismuth G.; Hidalgo-Lucas S.; Bisson J.-F.; Blanchard N.; Demangel C. (2015) Shaping mycolactone for therapeutic use against inflammatory disorders. Sci. Transl. Med. 7, 289ra85. 10.1126/scitranslmed.aab0458. [DOI] [PubMed] [Google Scholar]
- Gutiérrez M.; Suyama T. L.; Engene N.; Wingerd J. S.; Matainaho T.; Gerwick W. H. (2008) Apratoxin D, a potent cytotoxic cyclodepsipeptide from papua new guinea collections of the marine cyanobacteria Lyngbya majuscula and Lyngbya sordida. J. Nat. Prod. 71, 1099–1103. 10.1021/np800121a. [DOI] [PubMed] [Google Scholar]
- Vermeire K.; Allan S.; Provinciael B.; Hartmann E.; Kalies K.-U. (2015) Ribonuclease-neutralized pancreatic microsomal membranes from livestock for in vitro co-translational protein translocation. Anal. Biochem. 484, 102–104. 10.1016/j.ab.2015.05.019. [DOI] [PubMed] [Google Scholar]
- Jafari R.; Almqvist H.; Axelsson H.; Ignatushchenko M.; Lundbäck T.; Nordlund P.; Martinez Molina D. (2014) The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.