

Abstract

In this work, the first application of 3,5-dimethyl-4-nitroisoxazole as a vinylogous pronucleophile in the allylic–allylic alkylation of Morita–Baylis–Hillman (MBH) carbonates is described. The reaction has been realized under nucleophilic catalysis conditions with dimeric cinchona alkaloids, providing excellent enantiocontrol of the process. The usefulness of the products thus obtained has been confirmed in selected chemoselective reactions. The most important one involves the transformation of the isoxazole moiety into a carboxylic acid group, thus opening access to dicarboxylic acid derivatives.
Introduction
The asymmetric synthesis of simple, chiral building blocks possessing important functional groups (that can be subsequently utilized for their further chemical and stereochemical diversifications) constitutes an important task in the contemporary organic synthesis.1 Carboxylic acids and their α,β-unsaturated derivatives belong to such a category of compounds with widespread applications in organic and polymer sciences (Scheme 1).2 Dicarboxylic acids bearing two carboxylic moieties in their structure are an interesting group of carboxylic acid derivatives with molecules containing such a structural motif being exploited in target-oriented synthesis.3
Scheme 1. Importance of Carboxylic Acids and Their Derivatives.

Allylic alkylation with the participation of Morita–Baylis–Hillman (MBH) carbonates realized under nucleophilic catalysis constitutes a facile route for the synthesis of acrylic acid derivatives bearing a chiral substituent in the α-position.4 The synthetic potential of allylic alkylations realized with MBH-carbonates has been recently significantly expanded by employing various vinylogous pronucleophiles in this reaction.5 In such a manner, allylic–allylic alkylations have been introduced, leading to various synthetically relevant products bearing two allylic moieties connected through the C–C bond in a stereocontrolled manner.
In continuation of our studies related to the development of novel vinylogous reactivities,6,7 we became particularly interested in 3,5-dimethyl-4-nitroisoxazole (1, Scheme 2). This compound and its alkylidene derivatives have recently emerged as a useful class of compounds capable of participating in various C–C bond forming reactions.8 One relevant feature of this group of reactants is the possibility to transform the isoxazole ring system to the carboxylic acid group. As a consequence, they are considered as carboxylic acid surrogates. To our surprise, such pronucleophiles have never before been explored in the allylic–allylic alkylation with MBH-carbonates 2 realized under nucleophilic catalysis conditions (Scheme 2, top). It was anticipated that the initially obtained product 3 could be utilized as a direct precursor of dicarboxylate 4. Notably, an alternative strategy for the preparation of 4 can be based on the allylic alkylation of MBH-carbonates with malonates or related systems, followed by the hydrolysis and monodecarboxylation. While the first step of the sequence is known in the literature, the development of the enantioselective protocol remains a challenge or requires a specific substitution pattern.9 Therefore, the development of an alternative strategy is highly appealing.
Scheme 2. Synthetic Objectives of Our Study and the Anticipated Mechanistic Scenario.

Herein, we present our studies on the development of the first allylic–allylic alkylation using 3,5-dimethyl-4-nitroisoxazole (1) as a vinylogous pronucleophile and the application of the products 3 obtained for the synthesis of enantiomerically enriched dicarboxylates 4. The anticipated mechanistic scenario for the key step of the devised strategy should be initiated through two independent activation processes (Scheme 2, bottom). The first one involving the corresponding MBH-carbonate 2 relies on the SN2′ reaction of 2 with a nucleophilic catalyst to give intermediate A. The reaction proceeds with the concomitant release of carbon dioxide and tert-butoxide anion. In a parallel transformation 3,5-dimethyl-4-nitroisoxazole (1) is deprotonated to give anion B that is stabilized through mesomeric effect. A subsequent second SN2′ reaction between A and B yields target product 3 and releases the nucleophilic catalyst that can re-enter the catalytic cycle.
Results and Discussion
The optimization studies were performed using isoxazole 1 and MBH-carbonate 2a as model reactants (Table 1 and Supporting Information). Initially, quinine 5a was used as a catalyst, and to our delight, the formation of the desired product 3a was observed (Table 1, entry 1). However, both the yield and enantioselectivity of the reaction were low. Therefore, a catalyst screening was performed employing various cinchona-alkaloids-derived systems 5b–g. While cupreine derivative 5b catalyzed the reaction with similar efficiency (Table 1, entry 2), the thiourea derivative 5c failed to induce any reactivity (Table 1, entry 3). Therefore, we turned our attention to dimeric catalysts 5d–g (Table 1, entries 4–7) as their ability to act as nucleophilic catalysts in allylic alkylation reactions is well recognized. We were pleased to observe that the use of 5f led to an increase in enantioselectivity of the developed reaction (Table 1, entry 6). In order to further improve the results, the solvent screening was initiated (Table 1, entries 6, 8–12). Among solvents tested, chloroform proved the best reaction medium that enabled to increase the yield of the reaction significantly (Table 1, entry 8). In the course of further studies, various experiments aiming at fine-tuning of the reaction conditions were performed (Table 1, entries 13–19). It was found that neither dilution of the reaction mixture (Table 1, entry 13) nor an increase in concentration (Table 1, entry 14) had a beneficial effect on its outcome. Subsequently, the temperature effect was checked (Table 1, entries 8, 15–17). Interestingly, while the decrease in temperature resulted in sluggish conversion (Table 1, entry 15), its increase led to an improvement of reactivity (Table 1, entries 16 and 17) with 40 °C, providing also a better outcome in terms of enantioselectivity of the process (Table 1, entry 16). Finally, the influence of the relative reactant ratios was checked; however, it resulted in deteriorated results (Table 1, entries 18 and 19).
Table 1. Enantioselective Allylic–allylic Alkylation Using 3,5-Dimethyl-4-nitroisoxazole 1 as a Vinylogous Pronucleophile: Optimization Studiesa.

| entry | solvent (catalyst) | T [°C] | yield [%]b | erc |
|---|---|---|---|---|
| 1 | CH2Cl2 (5a) | rt | 26 | 60:40 |
| 2 | CH2Cl2(5b) | rt | 22 | 81:19 |
| 3 | CH2Cl2 (5c) | rt | <5 | nd |
| 4 | CH2Cl2 (5d) | rt | 22 | 79:21 |
| 5 | CH2Cl2 (5e) | rt | 41 | 88:12 |
| 6 | CH2Cl2 (5f) | rt | 43 | 90:10 |
| 7 | CH2Cl2 (5g) | rt | 38 | 76:24 |
| 8 | CHCl3 (5f) | rt | 69 | 91:9 |
| 9 | ClCH2CH2Cl (5f) | rt | 40 | 86:14 |
| 10 | toluene (5f) | rt | 19 | 89:11 |
| 11 | THF (5f) | rt | 36 | 87:13 |
| 12 | CH3CN (5f) | rt | 80 | 89:11 |
| 13d | CHCl3 (5f) | rt | 41 | 89:11 |
| 14e | CHCl3 (5f) | rt | 66 | 90:10 |
| 15 | CHCl3 (5f) | 10 | 20 | 92:8 |
| 16 | CHCl3 (5f) | 40 | 84 | 96:4 |
| 17 | CHCl3 (5f) | 60 | 78 | 90:10 |
| 18f | CHCl3 (5f) | 40 | 81 | 91:9 |
| 19g | CHCl3 (5f) | 40 | 79 | 93:7 |
Reactions performed on a 0.1 mmol scale using 1 (1 equiv) and 2a (1 equiv) in 0.4 mL of the solvent.
Isolated yields are given.
Determined by a chiral stationary phase HPLC.
Reaction was performed in 0.8 mL of CH2Cl2.
Reaction was performed in 0.2 mL of CH2Cl2.
Reaction was performed using 1 (1.5 equiv).
Reaction was performed using 2a (1.5 equiv).
With the optimization studies accomplished (Table 1, entry 16), the scope of the developed methodology was studied (Table 2). It was found that the presence of electron-withdrawing groups on the aromatic ring in 2 did not significantly influence the reaction outcome (Table 2, entries 2–6). In all of the cases, high yields and enantioselectivities were obtained. Similarly, the reaction proved unbiased toward the position of substituent with para-, meta- and ortho-substitution being well-tolerated (Table 2, entries 4–6). Furthermore, it was found that the introduction of electron-donating groups had no deteriorative effect on the reaction (Table 2, entries 7 and 8). The disubstituted aromatic ring, heteroaromatic group, and alkenyl moiety were also able to be present in 2 as demonstrated in the synthesis of 3i–k (Table 2, entries 9–11). Finally, the possibility to modify the ester group in 2 was checked (Table 2, entries 12 and 13) with excellent enantiocontrol achieved when tert-butyl ester was employed (Table 2, entry 13). The reaction also proved possible to scale up (5 mmol, Table 1, entry 14) with 3a obtained in lower yield (54%) (for information on additional scope experiments, see Supporting Information).
Table 2. Enantioselective Allylic–Allylic Alkylation Using 3,5-Dimethyl-4-nitroisoxazole 1 as a Vinylogous Pronucleophile: Scope Studiesa.

| entry | R1 | R2 | yield [%]b | erc |
|---|---|---|---|---|
| 1 | Ph (3a) | Me | 84 | 96:4 |
| 2 | 4-CF3C6H4 (3b) | Me | 82 | 90:10 |
| 3 | 4-BrC6H4 (3c) | Me | 86 | 97:3 |
| 4 | 4-ClC6H4 (3d) | Me | 90 | 98:2 |
| 5 | 3-ClC6H4 (3e) | Me | 89 | 96:4 |
| 6 | 2-ClC6H4 (3f) | Me | 83 | 94:6 |
| 7 | 4-CH3C6H4 (3g) | Me | 92 | 97:3 |
| 8 | 4-CH3OC6H4 (3h) | Me | 90 | 96:4 |
| 9 | 3,4-(OCH2O)C6H3 (3i) | Me | 89 | 96:4 |
| 10 | 2-furyl (3j) | Me | 84 | 98:2 |
| 11 | E-Prop-1-enyl (3k) | Me | 84 | 95:5 |
| 12 | Ph (3l) | Et | 85 | 98:2 |
| 13 | Ph (3m) | tBu | 90 | 99:1 |
| 14d | Ph (3a) | Me | 58 | 96:4 |
Reactions performed on a 0.1 mmol scale using 1 (1 equiv) and 2a (1 equiv) in 0.4 mL of the solvent.
Isolated yields are given.
Determined by a chiral stationary phase HPLC.
Reaction was performed on a 5 mmol scale.
The absolute configuration of the representative products 3a,g was established by comparison of their experimental electronic circular dichroism (ECD) spectrum with the calculated ones (for details, see Supporting Information).10 A perfect consistency between experimental and theoretical data (see Figure 1) unambiguously confirmed the R absolute configuration of the model compounds tested. The absolute stereochemistry of the products 3b–f,h–m was assigned through analogy.
Figure 1.
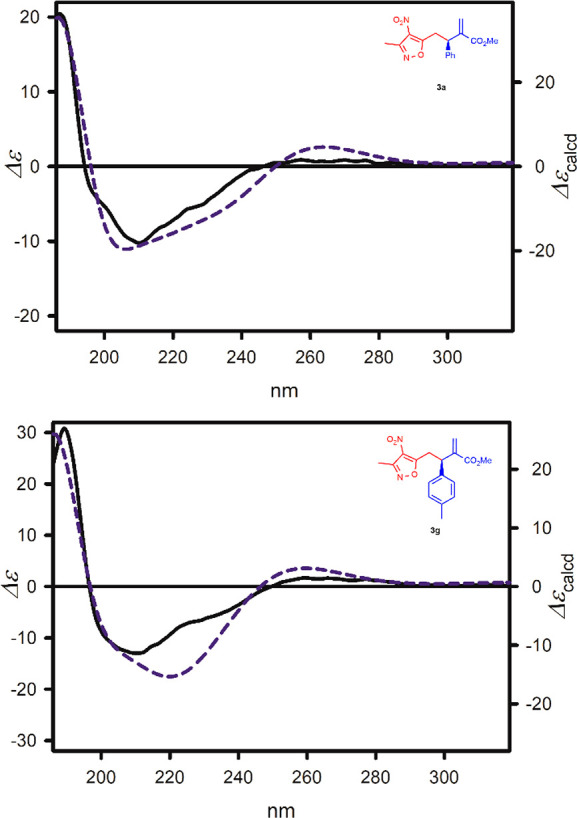
ECD spectra of 3a and 3g, measured in cyclohexane (solid black lines) and calculated at the TD-CAM-B3LYP/6-311++G(2d,2p) level and ΔΔG-based Boltzmann average (dashed blue lines). Wavelengths were corrected to match the experimental UV maximum.
In order to demonstrate the synthetic usefulness of the allylic–allylic alkylation products 3, acrylate 3a was subjected to selected transformations (Scheme 3). Initially, the possibility to transform the 4-nitroisoxazole moiety in 3a into the carboxylic group was attempted (Scheme 3, top). It proved possible employing sodium hydroxide aqueous solution. The dicarboxylic acid initially obtained was directly subjected to esterification to afford dimethyl ester 4a in an 83% yield. Importantly, the optical purity introduced in the organocatalytic step was almost entirely maintained despite basic reaction conditions. In the course of further studies, it was found that the reduction of the nitro group in 3a was possible to be conducted chemoselectively when Zn in acetic acid was used as a reducing agent (Scheme 3, bottom). The reaction again occurred with the preservation of the optical purity of 3a.
Scheme 3. Enantioselective Allylic–Allylic Alkylation Using 3,5-Dimethyl-4-nitroisoxazole 1 as a Vinylogous Pronucleophile: Synthetic Applications.

Conclusions
In conclusion, we have demonstrated that 3,5-dimethyl-4-nitroisoxazole 1 serves as a new vinylogous pronucleophile in the allylic–allylic alkylation reaction. The reaction proceeded efficiently in the presence of dimeric cinchona alkaloid derivative 5f as the nucleophilic catalyst initiating the reaction sequence. Target products bearing an interesting functionalization potential were obtained with high yields and with excellent enantiocontrol. The possibility to transform the 4-nitroisoxazole moiety into the carboxylic group was demonstrated.
Experimental Section
General Information
Unless otherwise noted, analytical grade solvents and commercially available reagents were used without further purification. NMR spectra were acquired on a Bruker Ultra Shield 700 instrument, running at 700 MHz for 1H and 176 MHz for 13C, respectively. Chemical shifts (δ) are reported in ppm relative to residual solvent signals (CDCl3: 7.26 ppm for 1H NMR, 77.16 ppm for 13C NMR). Mass spectra were recorded on a Bruker Maxis Impact spectrometer using electrospray (ES+) ionization (referenced to the mass of the charged species). Analytical thin-layer chromatography (TLC) was performed using precoated aluminum-backed plates (Merck Kieselgel 60 F254) and visualized by ultraviolet irradiation. Silica gel (Silica gel 60, 230–400 mesh, Fluka) was used for flash chromatography (FC). MBH carbonates 2 were prepared according to literature procedures.11 Cinchona-alkaloid-derived catalyst 5c was synthesized following the literature procedure.12
General Procedure for the Synthesis of Compounds 3a–m
An ordinary screw-cap vial was charged with a magnetic stir bar, the corresponding 3,5-dimethyl-4-nitroisoxazole 1 (0.1 mmol, 1 equiv), the corresponding MBH carbonate 2 (0.1 mmol, 1 equiv), catalyst 5f (0.01 mmol, 0.1 equiv), and CHCl3 (0.4 mL). The reaction mixture was stirred at 40 °C in an aluminum heating block and monitored by 1H NMR spectroscopy. After 24 h, the mixture was directly purified by FC on silica gel to afford a target product 3.
(R)-Methyl 4-(3-Methyl-4-nitroisoxazol-5-yl)-2-methylene-3-phenylbutanoate (3a)
Following the general procedure, 3a was isolated by FC on silica (hexane/ethyl acetate 96:4) in an 84% yield (28.5 mg) as a pale-yellow oil (910.4 mg, 58% yield of 3a was obtained when the reaction was performed on a 5 mmol scale). 1H NMR (700 MHz, CDCl3): δ 7.32–7.28 (m, 2H), 7.27–7.22 (m, 3H), 6.41 (s, 1H), 5.79 (d, J = 1.4 Hz, 1H), 4.60 (t, J = 8.1 Hz, 1H), 3.80–3.77 (m, 2H), 3.71 (s, 3H), 2.52 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 172.8, 166.5, 155.6, 141.5, 139.9, 129.6, 128.9 (2C), 127.8 (2C), 127.6, 125.8, 52.2, 44.1, 32.4, 11.7. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H16N2O5Na, 339.0952; found, 339.0951. The er was determined by HPLC using a Chiralpak IG column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 11.6 min, τminor = 10.5 min (96:4 er). [α]D20 −173.1 (c = 0.8, CHCl3).
(R)-Methyl 4-(3-Methyl-4-nitroisoxazol-5-yl)-2-methylene-3-(4-(trifluoromethyl)phenyl)-butanoate (3b)
Following the general procedure, 3b was isolated by FC on silica (hexane/ethyl acetate 96:4) in an 82% yield (33.4 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.41 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 6.40 (s, 1H), 5.77 (d, J = 1.2 Hz, 1H), 4.54 (td, J = 8.2, 1.2 Hz, 1H), 3.74 (d, J = 8.1 Hz, 2H), 3.69 (s, 3H), 2.51 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 172.2, 166.1, 155.8, 144.1, 140.1, 130.2 (q, J = 30.7 Hz), 128.2 (2C), 128.0, 126.4, 125.9 (q, J = 3.7 Hz, 2C), 125.5, 124.1 (q, J = 272.1 Hz), 52.4, 43.9, 32.1, 11.7. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H15F3N2O5Na, 407.0826; found, 407.0828. The er was determined by HPLC using a Chiralpak IG column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 8.8 min, τminor = 11.5 min (90:10 er). [α]D20 −146.8 (c = 0.7, CHCl3).
(R)-Methyl 3-(4-Bromophenyl)-4-(3-methyl-4-nitroisoxazol-5-yl)-2-methylenebutanoate (3c)
Following the general procedure, 3c was isolated by FC on silica (hexane/ethyl acetate 96:4) in an 86% yield (35.9 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.41 (d, J = 8.5 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 6.40 (s, 1H), 5.77 (d, J = 1.3 Hz, 1H), 4.55–4.53 (m, 1H), 3.74 (d, J = 8.1 Hz, 2H), 3.69 (s, 3H), 2.51 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 172.4, 166.3, 155.7, 141.1, 139.0, 132.0 (2C), 129.6 (2C), 126.0 (2C), 121.6, 52.3, 43.6, 32.2, 11.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H15BrN2O5Na, 417.0057/419.0037; found, 417.0069/419.0044. The er was determined by HPLC using a Chiralpak IF column [hexane/iPrOH (80:20)]; flow rate 1.0 mL/min; τmajor = 18.0 min, τminor = 15.9 min (97:3 er). [α]D20 −151.2 (c = 0.9, CHCl3).
(R)-Methyl 3-(4-Chlorophenyl)-4-(3-methyl-4-nitroisoxazol-5-yl)-2-methylenebutanoate (3d)
Following the general procedure, 3d was isolated by FC on silica (hexane/ethyl acetate 96:4) in a 90% yield (33.6 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.26–7.25 (m, 2H), 7.18 (d, J = 8.5 Hz, 2H), 6.40 (s, 1H), 5.77 (d, J = 1.2 Hz, 1H), 4.61–4.47 (m, 1H), 3.74 (d, J = 8.1 Hz, 2H), 3.69 (s, 3H), 2.51 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 172.4, 166.3, 155.7, 141.2, 138.5, 133.5, 129.2 (2C), 129.1 (2C), 126.0 (2C), 52.3, 43.6, 32.3, 11.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H15ClN2O5Na, 373.0562; found, 373.0570. The er was determined by HPLC using a Chiralpak IG column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 14.6 min, τminor = 11.8 min (98:2 er). [α]D20 −132.1 (c = 0.8, CHCl3).
(R)-Methyl 3-(3-Chlorophenyl)-4-(3-methyl-4-nitroisoxazol-5-yl)-2-methylenebutanoate (3e)
Following the general procedure, 3e was isolated by FC on silica (hexane/ethyl acetate 96:4) in an 89% yield (33.2 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.24–7.20 (m, 3H), 7.14 (dt, J = 7.3, 1.8 Hz, 1H), 6.42 (s, 1H), 5.79 (d, J = 1.2 Hz, 1H), 4.55 (td, J = 8.2, 1.1 Hz, 1H), 3.77 (dd, J = 15.1, 8.1 Hz, 1H), 3.73 (dd, J = 15.1, 8.2 Hz, 1H), 3.70 (s, 3H), 2.52 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 172.3, 166.2, 155.7, 142.1, 140.8, 134.7, 130.2, 129.6, 128.0, 127.9, 126.3, 126.1, 52.4, 43.8, 32.3, 11.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H15ClN2O5Na, 373.0562; found, 373.0566. The er was determined by HPLC using a Chiralpak IF column [hexane/iPrOH (80:20)]; flow rate 1.0 mL/min; τmajor = 7.6 min, τminor = 6.8 min (96:4 er). [α]D20 −142.3 (c = 0.7, CHCl3).
(S)-Methyl 3-(2-Chlorophenyl)-4-(3-methyl-4-nitroisoxazol-5-yl)-2-methylenebutanoate (3f)
Following the general procedure, 3f was isolated by FC on silica (hexane/ethyl acetate 96:4) in an 83% yield (30.1 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.34 (dd, J = 7.9, 1.4 Hz, 1H), 7.30 (dd, J = 7.8, 1.7 Hz, 1H), 7.24 (td, J = 7.6, 1.4 Hz, 1H), 7.18 (td, J = 7.6, 1.7 Hz, 1H), 6.45 (s, 1H), 5.71 (d, J = 1.4 Hz, 1H), 5.17–5.03 (m, 1H), 3.82 (dd, J = 15.1, 7.7 Hz, 1H), 3.69 (s, 3H), 3.67 (dd, J = 15.1, 8.3 Hz, 1H), 2.52 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 172.3, 166.3, 155.7, 140.6, 137.5, 134.3, 130.2, 129.6, 128.8, 128.4, 127.3, 126.8, 52.3, 40.4, 31.7, 11.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H15ClN2O5Na, 373.0562; found, 373.0569. The er was determined by HPLC using a Chiralpak IC column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 11.0 min, τminor = 10.3 min (94:6 er). [α]D20 −129.5 (c = 0.8, CHCl3).
(R)-Methyl 4-(3-Methyl-4-nitroisoxazol-5-yl)-2-methylene-3-(p-tolyl)butanoate (3g)
Following the general procedure, 3g was isolated by FC on silica (hexane/ethyl acetate 96:4) in a 92% yield (32.5 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.12 (d, J = 8.1 Hz, 2H), 7.08 (d, J = 8.0 Hz, 2H), 6.36 (s, 1H), 5.75 (d, J = 1.2 Hz, 1H), 4.54 (t, J = 8.1 Hz, 1H), 3.74 (dd, J = 8.1, 1.5 Hz, 2H), 3.69 (s, 3H), 2.50 (s, 3H), 2.29 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 173.0, 166.6, 155.6, 141.7, 137.2, 136.9, 129.6, 129.5, 127.7, 127.6, 125.8, 125.5, 52.3, 43.7, 32.5, 21.3, 11.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H18N2O5Na, 353.1108; found, 353.1112. The er was determined by HPLC using a Chiralpak IG column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 12.8 min, τminor = 11.2 min (97:3 er). [α]D20 −186.8 (c = 0.8, CHCl3).
(R)-Methyl 3-(4-Methoxyphenyl)-4-(3-methyl-4-nitroisoxazol-5-yl)-2-methylenebutanoate (3h)
Following the general procedure, 3h was isolated by FC on silica (hexane/ethyl acetate 96:4) in a 90% yield (33.2 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.14 (d, J = 8.7 Hz, 2H), 6.81 (d, J = 8.7 Hz, 2H), 6.35 (s, 1H), 5.74 (d, J = 1.2 Hz, 1H), 4.53 (td, J = 8.2, 1.2 Hz, 1H), 3.77 (s, 3H), 3.73 (dd, J = 8.1, 2.5 Hz, 2H), 3.69 (s, 3H), 2.50 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 173.1, 166.7, 159.0, 155.7, 141.9, 132.0, 129.0 (2C), 125.5 (2C), 114.3 (2C), 55.5, 52.3, 43.5, 32.7, 11.9. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H18N2O6Na, 369.1057; found, 369.1054. The er was determined by HPLC using a Chiralpak IG column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 20.3 min, τminor = 18.9 min (96:4 er). [α]D20 −172.6 (c = 0.7, CHCl3).
(R)-Methyl 3-(Benzo[d][1,3]dioxol-5-yl)-4-(3-methyl-4-nitroisoxazol-5-yl)-2-methylenebutanoate (3i)
Following the general procedure, 3i was isolated by FC on silica (hexane/ethyl acetate 96:4) in an 89% yield (34.1 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 6.72–6.69 (m, 2H), 6.68 (dd, J = 8.0, 1.7 Hz, 1H), 6.37 (s, 1H), 5.92 (s, 2H), 5.75 (d, J = 1.3 Hz, 1H), 4.50 (td, J = 8.2, 1.4 Hz, 1H), 3.71 (d, J = 1.2 Hz, 1H), 3.70 (s, 3H), 3.70 (d, J = 1.6 Hz, 1H), 2.51 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 172.8, 166.5, 155.7, 148.0, 147.0, 141.6, 133.7, 129.6, 125.4, 121.1, 108.5, 108.2, 101.3, 52.3, 43.7, 32.6, 11.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H16N2O7Na, 383.0850; found, 383.0855. The er was determined by HPLC using a Chiralpak IG column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 14.6 min, τminor = 17.0 min (96:4 er). [α]D20 −146.3 (c = 0.7, CHCl3).
(S)-Methyl 3-(Furan-2-yl)-4-(3-methyl-4-nitroisoxazol-5-yl)-2-methylenebutanoate (3j)
Following the general procedure, 3j was isolated by FC on silica (hexane/ethyl acetate 96:4) in an 84% yield (27.6 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.32 (dd, J = 1.8, 0.8 Hz, 1H), 6.39 (s, 1H), 6.28 (dd, J = 3.2, 1.8 Hz, 1H), 6.13 (dt, J = 3.1, 0.8 Hz, 1H), 5.74 (d, J = 1.0 Hz, 1H), 4.72–4.61 (m, 1H), 3.83 (dd, J = 15.0, 8.5 Hz, 1H), 3.76 (s, 3H), 3.73 (dd, J = 15.0, 7.3 Hz, 1H), 2.53 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 172.3, 166.3, 155.7, 153.0, 142.4, 139.3, 127.7, 110.6 (2C), 107.1, 52.4, 38.2, 31.1, 11.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C14H14N2O6Na, 329.0745; found, 329.0748. The er was determined by HPLC using a Chiralpak IC column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 19:5 min, τminor = 18.1 min (98:2 er). [α]D20 −151.2 (c = 0.9, CHCl3).
(R,E)-Methyl 3-((3-Methyl-4-nitroisoxazol-5-yl)methyl)-2-methylenehex-4-enoate (3k)
Following the general procedure, 3k was isolated by FC on silica (hexane/ethyl acetate 96:4) in an 84% yield (25.5 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 6.23 (d, J = 0.7 Hz, 1H), 5.64 (t, J = 0.8 Hz, 1H), 5.57–5.46 (m, 2H), 3.80–3.77 (m, 1H), 3.77 (s, 3H), 3.49 (dd, J = 7.6, 2.6 Hz, 2H), 2.54 (s, 3H), 1.63 (d, 3H, J = 4.5 Hz,). 13C{1H} NMR (176 MHz, CDCl3): δ 172.4, 166.4, 155.8, 153.1, 142.5, 139.4, 127.8, 110.7, 107.2, 52.5, 38.3, 31.2, 11.9. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H16N2O5Na, 303.0952; found, 303.0947. The er was determined by HPLC using a Chiralpak IC column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 6.7 min, τminor = 7.6 min (95:5 er). [α]D20 −152.6 (c = 0.8, CHCl3).
(R)-Ethyl 4-(3-Methyl-4-nitroisoxazol-5-yl)-2-methylene-3-phenylbutanoate (3l)
Following the general procedure, 3l was isolated by FC on silica (hexane/ethyl acetate 96:4) in an 85% yield (30.0 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.29–7.26 (m, 2H), 7.23–7.21 (m, 3H), 6.39 (s, 1H), 5.74 (d, J = 1.2 Hz, 1H), 4.59–4.56 (m, 1H), 4.16–4.10 (m, 2H), 3.77 (d, J = 8.1 Hz, 2H), 2.50 (s, 3H), 1.21 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 172.9, 166.1, 155.6, 141.8, 140.1, 129.6, 128.8 (2C), 127.8 (2C), 127.5, 125.5, 61.2, 44.1, 32.4, 32.1, 14.2. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H18N2O5Na, 353.1108; found, 353.1112. The er was determined by HPLC using a Chiralpak IG column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 6.7 min, τminor = 7.5 min (98:2 er). [α]D20 −186.1 (c = 0.8, CHCl3).
(R)-tert-Butyl 4-(3-Methyl-4-nitroisoxazol-5-yl)-2-methylene-3-phenylbutanoate (3m)
Following the general procedure, 3m was isolated by FC on silica (hexane/ethyl acetate 96:4) in a 90% yield (34.3 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.29–7.26 (m, 2H), 7.22–7.19 (m, 3H), 6.30 (t, J = 0.7 Hz, 1H), 5.64 (dd, J = 1.4, 0.6 Hz, 1H), 4.51 (t, J = 8.1 Hz, 1H), 3.74 (dd, J = 8.1, 0.6 Hz, 2H), 2.50 (s, 3H), 1.35 (s, 9H). 13C{1H} NMR (176 MHz, CDCl3): δ 173.0, 165.3, 155.6, 143.2, 140.4, 128.7 (3C), 127.8 (2C), 127.4, 124.7, 81.6, 44.2, 32.5, 28.0 (3C), 11.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C19H22N2O5Na, 381.1421; found, 381.1427. The er was determined by HPLC using a Chiralpak IG column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 6.0 min, τminor = 7.6 min (99:1 er). [α]D20 −197.1 (c = 0.9, CHCl3).
Procedure for the Synthesis of Compound 4a
An ordinary screw-cap vial was charged with a magnetic stir bar and compound 3a (0.1 mmol) and treated with a freshly made 5 M aqueous solution of NaOH (0.1 mL, 5 equiv, 0.5 mmol). The resulting solution was heated at 60 °C in an oil bath for 2 h and then allowed to reach room temperature. Isolation of free carboxylic acid involved the following: cooling the aqueous solution of compound sodium salt, adjustment of pH to 3, and extraction with EtOAc (2 × 5 mL). Combined organic layers were dried over anhydrous MgSO4. The solvent was removed under reduced pressure. The crude mixture was dissolved in MeOH (3 mL) and cooled to 0 °C. Thionyl chloride (0.1 mL) was added dropwise, and the resulting solution was stirred overnight, allowing to warm gradually to room temperature. The solvent was removed under reduced pressure, and the crude mixture was diluted with EtOAc (10 mL) and washed with a saturated aqueous solution of NaHCO3 (2 × 5 mL) and brine (2 × 5 mL). The organic layer was dried over anhydrous MgSO4 and the solvent removed under reduced pressure.
(R)-Dimethyl 2-Methylene-3-phenylpentanedioate (4a)
Following the general procedure, pure product 4a was isolated by FC on silica gel (CH2Cl2/MeOH 98:2) in an 83% yield (22.5 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.28 (t, J = 7.6 Hz, 2H), 7.24–7.22 (m, 2H), 7.22–7.18 (m, 1H), 6.33 (s, 1H), 5.66 (d, J = 1.2 Hz, 1H), 4.45 (t, J = 7.8 Hz, 1H), 3.68 (s, 3H), 3.61 (s, 3H), 2.93 (dd, J = 15.6, 7.9 Hz, 1H), 2.81 (dd, J = 15.6, 7.9 Hz, 1H). 13C{1H} NMR (176 MHz, CDCl3): δ 172.0, 166.9, 142.5, 141.3, 128.6 (2C), 128.0 (2C), 127.1, 124.8, 52.1, 51.9, 42.9, 39.5. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C14H16O4Na, 271.0941; found, 271.0949. The er was determined by HPLC using a Chiralpak IC column [hexane/iPrOH (90:10)]; flow rate 1.0 mL/min; τmajor = 12.8 min, τminor = 11.6 min (94:6 er). [α]D20 −149.4 (c = 0.8, CHCl3).
Procedure for the Synthesis of Compound 6a
To the solution of 3a (87.6 mg, 0.1 mmol, 1.0 equiv) in acetic acid (1.6 mL) was added zinc dust (101.4 mg, 1.6 mmol, 16 equiv) slowly. After 2 h at room temperature, the mixture was filtered over Celite, diluted with ethyl acetate (5 mL), and then washed with sodium bicarbonate solution (2 × 5 mL). The combined organic phase was dried over MgSO4 and concentrated in vacuo.
(R)-Methyl 4-(4-Amino-3-methylisoxazol-5-yl)-2-methylene-3-phenylbutanoate (6a)
Following the general procedure, pure product 6a was isolated by FC on silica gel (hexane/ethyl acetate gradient from 3:1 to 1:1) in an 81% yield (23.3 mg) as a pale-yellow oil. 1H NMR (700 MHz, CDCl3): δ 7.47–7.02 (m, 5H), 6.34 (s, 1H), 5.75 (s, 1H), 4.29 (t, J = 8.0 Hz, 1H), 3.66 (s, 3H), 3.28 (dd, J = 14.7, 6.7 Hz, 1H), 3.08 (dd, J = 14.7, 9.1 Hz, 1H), 2.12 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ 167.1, 155.2, 155.0, 142.3, 141.3, 128.7 (2C), 128.0 (2C), 127.2, 125.3, 121.9, 52.1, 45.3, 30.6, 9.4. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H19N2O3, 287.1391; found, 287.1392. The er was determined by HPLC using a Chiralpak IA column [hexane/iPrOH (80:20)]; flow rate 1.0 mL/min; τmajor = 5.2 min, τminor = 4.7 min (94:6 er). [α]D20 −127.2 (c = 0.9, CHCl3).
Acknowledgments
This project was realized within the Sonata Bis program (2015/18/E/ST5/00309) from the National Science Centre, Poland. All calculations were performed in Poznań Supercomputing and Networking Center.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.9b02530.
A full account of screening results, additional scope studies, details of absolute configuration assignments (computational details), copies of 1H and 13C NMR spectra, and HPLC chromatograms (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Dedication
Dedicated to Professor Grzegorz Mlostoń (University of Łódź) on the occasion of his 70th birthday.
Supplementary Material
References
- a Jacobsen E. N.; Pfaltz A.; Yamamoto H.. Comprehensive Asymmetric Catalysis; Springer: Berlin, 1999. [Google Scholar]; b Mikami K.; Lautens M.. New Frontiers in Asymmetric Catalysis; Wiley-Interscience: NJ, 2007. [Google Scholar]
- Badea G.-I.; Radu G. L.. Carboxylic Acid - Key Role in Life Sciences; Intech Open, 2018. [Google Scholar]
- For examples, see:; a Chinchilla R.; Galindo N.; Nájera C. Dimethyl 2-(tosylmethyl) fumarate: An allyl sulfone as electrophilic reagent for the synthesis of itaconate ester derivatives. Tetrahedron 1996, 52, 1035. 10.1016/0040-4020(95)00937-X. [DOI] [Google Scholar]; b Pierik A. J.; Ciceri D.; Bröker G.; Edwards C. H.; McFarlane W.; Winter J.; Buckel W.; Golding B. T. Rotation of the exo-methylene group of (R)-3-methylitaconate catalyzed by coenzyme B12-dependent 2-methyleneglutarate mutase from Eubacteriumbarkeri. J. Am. Chem. Soc. 2002, 124, 14039. 10.1021/ja020340f. [DOI] [PubMed] [Google Scholar]; c Nishimura H.; Murayama K.; Watanabe T.; Honda Y.; Watanabe T. Absolute configuration of ceriporic acids, the iron redox-silencing metabolites produced by a selective lignin-degrading fungus. Chem. Phys. Lipids 2009, 159, 77. 10.1016/j.chemphyslip.2009.03.006. [DOI] [PubMed] [Google Scholar]; d Nakahashi A.; Miura N.; Monde K.; Tsukamoto S. Stereochemical studies of hexylitaconic acid, an inhibitor of p53–HDM2 interaction. Bioorg. Med. Chem. Lett. 2009, 19, 3027. 10.1016/j.bmcl.2009.04.057. [DOI] [PubMed] [Google Scholar]; e Steward K. M.; Corbett M. T.; Goodman C. G.; Johnson J. S. Asymmetric synthesis of diverse glycolic acid scaffolds via dynamic kinetic resolution of α-ketoesters. J. Am. Chem. Soc. 2012, 134, 20197. 10.1021/ja3102709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review, see:; Liu T.-Y.; Xie M.; Chen Y.-C. Organocatalytic asymmetric transformations of modified Morita–Baylis–Hillman adducts. Chem. Soc. Rev. 2012, 41, 4101. 10.1039/c2cs35017c. [DOI] [PubMed] [Google Scholar]
- For selected applications of vinylogous reactants in allylic alkylation with MBH carbonates, see:; a Cui H.-L.; Peng J.; Feng X.; Du W.; Jiang K.; Chen Y.-C. Dual organocatalysis: asymmetric allylic–allylic alkylation of α,α-dicyanoalkenes and Morita–Baylis–Hillman carbonates. Chem. - Eur. J. 2009, 15, 1574. 10.1002/chem.200802534. [DOI] [PubMed] [Google Scholar]; b Feng J.; Li X.; Cheng J.-P. An asymmetric allylic alkylation reaction of 3-alkylidene oxindoles. Chem. Commun. 2015, 51, 14342. 10.1039/C5CC06182B. [DOI] [PubMed] [Google Scholar]; c Zhao S.; Zhao Y.-Y.; Lin J.-B.; Xie T.; Liang Y.-M.; Xu P.-F. Organocatalyzed asymmetric vinylogous allylic–allylic alkylation of Morita–Baylis–Hillman carbonates with olefinicazlactones: facile access to chiral multifunctional α-amino acid derivatives. Org. Lett. 2015, 17, 3206. 10.1021/acs.orglett.5b01066. [DOI] [PubMed] [Google Scholar]; d Kayal S.; Mukherjee S. Catalytic enantioselective vinylogous allylic alkylation of coumarins. Org. Lett. 2017, 19, 4944. 10.1021/acs.orglett.7b02421. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Fuson R. C. The Principleof Vinylogy. Chem. Rev. 1935, 16, 1. 10.1021/cr60053a001. [DOI] [Google Scholar]; b Pansare S. V.; Paul E. K. The organocatalytic vinylogous aldol reaction: recent advances. Chem. - Eur. J. 2011, 17, 8770. 10.1002/chem.201101269. [DOI] [PubMed] [Google Scholar]; c Schneider C.; Abels F. Catalytic, enantioselective vinylogous Michael reactions. Org. Biomol. Chem. 2014, 12, 3531. 10.1039/c4ob00332b. [DOI] [PubMed] [Google Scholar]; d Hepburn H. B.; Dell’Amico L.; Melchiorre P. Enantioselective vinylogous organocascade reactions. Chem. Rec. 2016, 16, 1787. 10.1002/tcr.201600030. [DOI] [PubMed] [Google Scholar]; e Battistini L.; Curti C.; Rassu G.; Sartori A.; Zanardi F. Enolizable alkylidene heterocyclic and carbocyclic carbonyl systems: valuable vinylogous donor substrates in synthesis. Synthesis 2017, 49, 2297. 10.1055/s-0036-1589487. [DOI] [Google Scholar]; f Chauhan P.; Kaya U.; Enders D. Advances in organocatalytic 1,6-addition reactions: enantioselective construction of remote stereogenic centers. Adv. Synth. Catal. 2017, 359, 888. 10.1002/adsc.201601342. [DOI] [Google Scholar]; g Li H.; Yin L. Recent progress on direct catalytic asymmetric vinylogous reactions. Tetrahedron Lett. 2018, 59, 4121. 10.1016/j.tetlet.2018.10.012. [DOI] [Google Scholar]
- For selected, recent examples, see:; a Romaniszyn M.; Gronowska K.; Albrecht Ł. 2-Substituted 1,4-naphthoquinones in [6 + 4]-cycloaddition with 8,8-dicyanoheptafulvene. J. Org. Chem. 2019, 84, 9929. 10.1021/acs.joc.9b01091. [DOI] [PubMed] [Google Scholar]; b Skrzyńska A.; Drelich P.; Frankowski S.; Albrecht Ł. Synthesis of γ,γ-disubstituted butenolides through a doubly vinylogous organocatalytic cycloaddition. Chem. - Eur. J. 2018, 24, 16543. 10.1002/chem.201804650. [DOI] [PubMed] [Google Scholar]; c Kowalczyk D.; Albrecht Ł. Vinylogous nucleophiles bearing the endocyclic double bond in the allylic alkylation with Morita-Baylis-Hillman carbonates. Adv. Synth. Catal. 2018, 360, 406. 10.1002/adsc.201701185. [DOI] [Google Scholar]; d Skrzyńska A.; Romaniszyn M.; Pomikło D.; Albrecht Ł. The application of 2-benzyl-1,4-naphthoquinones as pronucleophiles in aminocatalytic synthesis of tricyclic derivatives. J. Org. Chem. 2018, 83, 5019. 10.1021/acs.joc.8b00170. [DOI] [PubMed] [Google Scholar]; e Przydacz A.; Kowalczyk R.; Albrecht Ł. Brønsted-base-catalyzed remote cascade reactivity of 2,4-dienones–asymmetric synthesis of tetrahydrothiophenes. Org. Biomol. Chem. 2017, 15, 9566. 10.1039/C7OB02397A. [DOI] [PubMed] [Google Scholar]; f Hejmanowska J.; Jasinski M.; Wojciechowski J.; Mloston G.; Albrecht Łu. The first organocatalytic, ortho-regioselective inverse-electron-demand hetero-Diels–Alder reaction. Chem. Commun. 2017, 53, 11472. 10.1039/C7CC06518C. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Adamo M. F. A.; Nagabelli M. Synthesis of homochiral dihydroxy-4-nitroisoxazolines via one-pot asymmetric dihydroxylation-reduction. Org. Lett. 2008, 10, 1807. 10.1021/ol800397z. [DOI] [PubMed] [Google Scholar]; b Baschieri A.; Bernardi L.; Ricci A.; Suresh S.; Adamo M. F. A. Catalytic asymmetric conjugate addition of nitroalkanes to 4-nitro-5-styrylisoxazoles. Angew. Chem., Int. Ed. 2009, 48, 9342. 10.1002/anie.200905018. [DOI] [PubMed] [Google Scholar]; c Zhang J.; Liu X.; Ma X.; Wang R. Organocatalyzed asymmetric vinylogous Michael addition of α,β-unsaturated γ-butyrolactam. Chem. Commun. 2013, 49, 9329. 10.1039/c3cc44059a. [DOI] [PubMed] [Google Scholar]; d Li Y.; Lopez-Delgado F. J.; Jørgensen D. K. B.; Nielsen R. P.; Jiang H.; Jørgensen K. A. Trienamine-mediated asymmetric [4 + 2]-cycloaddition of α,β-unsaturated ester surrogates applying 4-nitro-5-styrylisoxazoles. Chem. Commun. 2014, 50, 15689. 10.1039/C4CC08171D. [DOI] [PubMed] [Google Scholar]; e Disetti P.; Moccia M.; Illera D. S.; Suresh S.; Adamo M. F. A. Catalytic enantioselective addition of isocyanoacetate to 3-methyl-4-nitro-5-styrylisoxazoles under phase transfer catalysis conditions. Org. Biomol. Chem. 2015, 13, 10609. 10.1039/C5OB01880C. [DOI] [PubMed] [Google Scholar]; f Zhang Y.; Wei B.; Lin H.; Cui W.; Zeng X.; Fan X. Organocatalyzed asymmetric vinylogous Michael reactions of 3,5-dialkyl-substituted 4-nitroisoxazoles: adirect method for the synthesis of chiral isoxazolederivatives. Adv. Synth. Catal. 2015, 357, 1299. 10.1002/adsc.201400833. [DOI] [Google Scholar]; g Zhu B.; Lee R.; Yin Y.; Li F.; Coote M. L.; Jiang Z. Enantioselective vinylogous amination of 5-alkyl-4-nitroisoxazoles with a dipeptide-based guanidiniumphase-transfer catalyst. Org. Lett. 2018, 20, 429. 10.1021/acs.orglett.7b03759. [DOI] [PubMed] [Google Scholar]; h Li S.-W.; Du Y.; Kang Q. A chiral-at-metal asymmetric catalyzed vinylogous Michael addition of ortho-methyl aromatic nitro compounds for isoxazole derivative synthesis. Org. Chem. Front. 2019, 6, 2775. 10.1039/C9QO00676A. [DOI] [Google Scholar]
- a Baidya M.; Remennikov G. Y.; Mayer P.; Mayr H. SN2′ versus SN2 reactivity: control of regioselectivity in conversions of Baylis–Hillman adducts. Chem. - Eur. J. 2010, 16, 1365. 10.1002/chem.200902487. [DOI] [PubMed] [Google Scholar]; b Basavaiah D.; Reddy B. S.; Lingam H. Baylis–Hillman acetates in carbocyclic synthesis: a convenient protocol for synthesis of densely substituted indenes. Tetrahedron 2013, 69, 1994. 10.1016/j.tet.2012.12.069. [DOI] [Google Scholar]; c Zheng Y.; Wang J.; Xia P.-J.; Zhao Q.-l.; Xiao J.-A.; Xiang H.-Y.; Chen X.-Q.; Yang H. Organocatalytic asymmetric allylic alkylation of Morita–Baylis–Hillman carbonates with diethyl 2-aminomalonate assisted by in situ protection. J. Org. Chem. 2017, 82, 12202. 10.1021/acs.joc.7b02064. [DOI] [PubMed] [Google Scholar]; For a related study, see:; d Trost B. M.; Brennan M. K. Palladium-catalyzed regio-and enantioselective allylic alkylation of bisallylic carbonates derived from Morita-Baylis-Hillman adducts. Org. Lett. 2007, 9, 3961. 10.1021/ol701585b. [DOI] [PubMed] [Google Scholar]
- Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Baldwin J. E.; Hoefle G. A.; Lever O. W. Jr. α-Methoxyvinyllithium and related metalated enol ethers. Practical reagents for nucleophilic acylation. J. Am. Chem. Soc. 1974, 96, 7125. 10.1021/ja00829a064. [DOI] [Google Scholar]
- a Yang W.; Du D.-M. Highly enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramides as hydrogen bonding organocatalysts. Org. Lett. 2010, 12, 5450. 10.1021/ol102294g. [DOI] [PubMed] [Google Scholar]; b Vakulya B.; Varga S.; Csámpai A.; Soós T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts. Org. Lett. 2005, 7, 1967. 10.1021/ol050431s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.