

Abstract
Semiconducting polymers are versatile materials for solar energy conversion and have gained popularity as photocatalysts for sunlight-driven hydrogen production. Organic polymers often contain residual metal impurities such as palladium (Pd) clusters that are formed during the polymerization reaction, and there is increasing evidence for a catalytic role of such metal clusters in polymer photocatalysts. Using transient and operando optical spectroscopy on nanoparticles of F8BT, P3HT, and the dibenzo[b,d]thiophene sulfone homopolymer P10, we demonstrate how differences in the time scale of electron transfer to Pd clusters translate into hydrogen evolution activity optima at different residual Pd concentrations. For F8BT nanoparticles with common Pd concentrations of >1000 ppm (>0.1 wt %), we find that residual Pd clusters quench photogenerated excitons via energy and electron transfer on the femto-nanosecond time scale, thus outcompeting reductive quenching. We spectroscopically identify reduced Pd clusters in our F8BT nanoparticles from the microsecond time scale onward and show that the predominant location of long-lived electrons gradually shifts to the F8BT polymer when the Pd content is lowered. While a low yield of long-lived electrons limits the hydrogen evolution activity of F8BT, P10 exhibits a substantially higher hydrogen evolution activity, which we demonstrate results from higher yields of long-lived electrons due to more efficient reductive quenching. Surprisingly, and despite the higher performance of P10, long-lived electrons reside on the P10 polymer rather than on the Pd clusters in P10 particles, even at very high Pd concentrations of 27000 ppm (2.7 wt %). In contrast, long-lived electrons in F8BT already reside on Pd clusters before the typical time scale of hydrogen evolution. This comparison shows that P10 exhibits efficient reductive quenching but slow electron transfer to residual Pd clusters, whereas the opposite is the case for F8BT. These findings suggest that the development of even more efficient polymer photocatalysts must target materials that combine both rapid reductive quenching and rapid charge transfer to a metal-based cocatalyst.
Introduction
Solar hydrogen production via photocatalysis provides a pathway to generate hydrogen as a carbon-free energy carrier in a clean and renewable way. The key requirement is a highly active and stable photocatalyst that can act as a light absorber and catalyze the desired chemical reactions—for example, direct hydrogen production from water. Polymeric photocatalysts have gained recently in popularity as an alternative to traditional inorganic photocatalysts, particularly because they provide a high degree of synthetic flexibility, thus allowing for tailoring of their frontier orbital energies. Rather than performing overall water splitting, most such organic photocatalysts are currently employed to couple the hydrogen evolution reaction to a sacrificial organic oxidation.1,2 To this end, the photocatalyst is suspended in a mixture of water and an organic electron donor, which serve as the proton and electron sources, respectively. However, the detailed catalytic mechanisms of such organic photocatalysts are still mostly unexplored, and a growing body of evidence suggests that residual metal impurities can act as cocatalysts for the hydrogen evolution reaction in polymeric materials.3−5
The field of polymer photocatalysts for hydrogen evolution was effectively started by carbon nitrides in 2009,6 which have since seen significant improvements7,8 with external quantum efficiencies (EQEs) reaching values of up to 60%.9 In the last 5 years, conjugated polymers have been investigated extensively as photocatalysts for solar hydrogen evolution. Current optimized conjugated polymer systems can reach EQEs exceeding 20%10,11 and offer benefits over carbon nitrides such as highly tunable frontier orbital energy levels. This tunability allows for tailoring of their optical absorption and their charge transfer driving force to specific target reactions, and the resulting ability to harvest more visible photons is considered as vital to achieve the hydrogen evolution activities required for practical applications.12,13 Other advantages include the possibility to create push–pull structures by incorporating electron-donating and -withdrawing units, thus facilitating intramolecular charge transfer and charge separation, or bulk heterojunctions where charge separation takes place at the interface between two materials with different ionization potentials/electron affinities.14,15 The versatility of polymeric photocatalysts has given rise to a variety of platforms such as conjugated microporous polymers, covalent triazine-based frameworks, and linear conjugated polymers.16−23 Several recent reviews provide an overview of this rapidly evolving field of research.2,24−28
One of the less well understood aspects of polymer photocatalysts is the role of the residual metal left over from the polymerization reaction. It is not clear how much residual metal clusters contribute to the ability of these organic materials to catalyze multielectron reactions or how the interaction of the metal with the polymer varies from system to system. In other photocatalytic systems that perform multielectron reactions, such as proton or CO2 reduction, inorganic materials or metal–organic molecules are often employed as cocatalysts. These cocatalysts incorporate transition metals, which can access a range of different oxidation states and are thus able to accumulate charge carriers to promote multielectron transfer reactions. In addition, cocatalysts can act as charge sinks to suppress recombination, as well as provide sites with favorable binding energies for reaction intermediates.29,30 There is a debate, therefore, about whether all-organic materials can function as efficient photocatalysts. Carbon nitrides, for example, do not typically contain residual metal from their syntheses, but they require the addition of cocatalysts for appreciable photocatalytic activities; very low performances are observed if no such cocatalyst is added.31,32 In contrast, metal-catalyzed synthesis routes such as Suzuki–Miyaura coupling, as widely used in the synthesis of conjugated polymers, produce polymers that contain inherent residual metal. Such residual metal clusters can act as cocatalysts and thus affect the photocatalytic performance of such polymers, although their precise role is not yet clear.
Polymers derived from metal-catalyzed synthesis routes can be difficult to purify because they are often not soluble in organic solvents, and even soluble products can be hard to purify using conventional purification techniques. For instance, conjugated polymers with dibenzo[b,d]thiophene sulfone groups incorporated into their backbones have shown some of the highest photocatalytic quantum yields in this class of materials10,11,16,20 but residual Pd is difficult to remove from these polymers due to their insolubility. Dibenzo[b,d]thiophene sulfone homopolymer P10 retains about 60% of its photocatalytic activity when it is synthesized using Ni(0)-mediated oxidative Yamamoto coupling instead of Pd-catalyzed Suzuki–Miyaura coupling.20 However, the catalytic role of residual nickel cannot be ruled out, especially given that efficient Ni-based molecular electrocatalysts exist.33 In a series of covalent triazine-based frameworks, polymers prepared via a metal-free acid-catalyzed route were largely inactive in comparison to their analogues made via Pd-catalyzed Suzuki-Miyaura coupling, suggesting that Pd acted as a cocatalyst in this study.5 Most recently, we have reported F8BT nanoparticles with well-defined variations in Pd concentration, which demonstrated a rapid decrease in hydrogen evolution activity when their residual Pd content was lowered to less than 100 ppm.3
Here, we use a combination of optical spectroscopic techniques to draw direct comparisons among F8BT, P3HT, and the dibenzo[b,d]thiophene sulfone homopolymer P10. We investigate the mechanism behind the dependence of photocatalytic activity on Pd content for these different polymers and place particular emphasis on the effect of Pd on the excited state of the polymer and on the accumulation of long-lived charges during catalysis. We selected F8BT because its Pd content can be tuned readily due to its solution processability, P10 because it exhibits a much higher hydrogen evolution activity than F8BT at comparable Pd concentrations, and P3HT because it contains residual Ni rather than Pd. For Pd concentrations typical of polymer photocatalysts (>1000 ppm), we find that electron transfer to Pd clusters strongly depends on the polymer: long-lived electrons rapidly localize on Pd clusters in F8BT nanoparticles but remain on the polymer for P10 even during charge accumulation under quasi-continuous illumination. We explore the effect of different Pd concentrations on photocatalytic hydrogen evolution for these materials and discuss potential causes and implications for the pronounced differences among them. Overall, this study demonstrates for the first time that the influence of Pd can be dramatically different for different conjugated polymer catalysts, opening the door for better catalyst design in the future.
Experimental Section
Sample Preparation
F8BT (Mn = 40 kg mol–1, Mw = 84 kg mol–1, Đ = 2.13) was synthesized, purified, and processed into nanoparticles as described previously.3 P3HT (Mn = 98 kg mol–1, Mw = 136 kg mol–1, Đ = 1.39) was synthesized via a Grignard metathesis (GRIM) polymerization following a previously reported procedure.34
Nanoparticles of P3HT and F8BT were formed using a reprecipitation process. In a typical preparation, a solution of F8BT or P3HT (0.50 mg mL–1) and poly(styrene-co-maleic anhydride) (0.10 mg mL–1) in tetrahydrofuran (5 mL) was filtered (0.2 μm PTFE syringe filter) and then rapidly injected into Milli-Q water (40 mL) under sonication in an ultrasonic bath. The mixture was sonicated for a further 2 min and then heated in an oil bath at 80 °C under constant nitrogen bubbling to remove the THF and concentrate the solution to 10 mL. Finally, the concentrated solution was filtered (0.45 μm glass fiber syringe filter) to remove large agglomerates, where less than 1% of the polymer mass was removed by the filter in each nanoparticle batch. Gradually reducing the volume of water used for reprecipitation from 40 to 1 mL increased the Z average nanoparticle diameter from 40 ± 10 to 189 ± 10 nm.
P10 was synthesized following a previously reported synthetic procedure:16,20 3,7-dibromodibenzo[b,d]thiophene sulfone (187 mg, 0.5 mmol), 3,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)dibenzo[b,d]thiophene sulfone (234 mg, 0.5 mmol), N,N-dimethylformamide (25 mL), and K2CO3 (aqueous, 2 M, 5 mL) were placed in a flask and degassed by N2 bubbling for 30 min. Tetrakis(triphenylphosphine)palladium(0) (6, 23, or 69 mg, corresponding to 0.5, 2, or 6 mol %, respectively) was added, and the mixtures were degassed for a further 10 min. The reaction mixtures were refluxed at 140 °C for 48 h under a nitrogen atmosphere. After they were cooled to room temperature, the reaction mixtures were poured into water (400 mL) and stirred for 30 min. The yellow-green solids were collected by filtration and washed with water (100 mL) and methanol (100 mL). The polymers were purified by Soxhlet extraction in methanol and chloroform before drying the solids under vacuum. The polymers were ground before use to give free-flowing yellow-green powders (207, 214, or 211 mg, yields 97–100%). P10 was also prepared via Yamamoto coupling following a previously reported protocol:20 a dried Schlenk flask was charged with 3,7-dibromodibenzo[b,d]thiophene sulfone (374 mg, 1.00 mmol) and 2,2′-bipyridine (344 mg, 2.20 mmol) and transferred into a nitrogen glovebox. Inside the glovebox the flask was charged with bis(cyclooctadiene)nickel(0) (660 mg, 2.40 mmol). Outside the glovebox 1,5-cyclooctadiene (338 mg, 2.20 mmol) and N,N-dimethylformamide (anhydrous, 20 mL) were added and the resulting suspension was heated to 80 °C under nitrogen for 2 days. After the mixture was cooled to room temperature, hydrochloric acid was added (concentrated, 20 mL) and the polymer was filtered off. The polymer was washed with water until neutral and then washed with methanol and tetrahydrofuran. Further purification of the polymer was carried out by Soxhlet extraction in chloroform before drying the solids under vacuum. The polymer was ground before use to give a free-flowing yellow powder (190 mg, 84%). The typical size of the resulting P10 particles in pure water is around 650 nm,20 and in line with previous studies, the dispersions were not filtered.
Pd nanoparticle films were prepared in a single aerosol-assisted chemical vapor deposition (AACVD) step. FTO substrates were heated inside the reactor from the carbon block below, to a deposition temperature of 350 °C. The heating, deposition, and cooling steps were conducted under a steady flow of N2 (3 L min–1). A precursor solution was prepared from [(NH4)2PdCl4] (0.003 mg) in methanol (50 mL) that was sonicated for 3 min. Aerosols were generated from portions of the precursor solution using an ultrasonic humidifier (Johnson Matthey Liquifog 2 MHz). Following deposition, the substrates were cooled to room temperature.
Hydrogen Evolution Experiments
The activity of P10 samples with different metal contents was measured as described previously.20 In brief, a quartz flask was charged with the polymer (25 mg) and an equal parts mixture of water, methanol, and triethylamine (25 mL), sealed with a septum, and sonicated for 10 min before degassing by nitrogen bubbling for 30 min. The sample was irradiated by a Newport 300 W Xe light source fitted with a λ > 420 nm filter. Gas samples were taken at intervals using a gastight syringe, and hydrogen was measured using a Bruker 450-GC gas chromatograph.
Metal Content
Metal contents were determined by inductively coupled plasma (ICP) spectroscopy on acid-digested polymer samples. F8BT and P3HT samples were digested in a microwave digester (UltraWAVE) in a mixture of aqua regia containing a small amount of hydrogen peroxide and sulfuric acid. The metal content of the digested F8BT and P3HT samples was determined using ICP-OES (Agilent 7200-ES) relative to standards of known concentration. P10 samples (5 mg) were digested in nitric acid (70 wt %, 10 mL) using a PerkinElmer Microwave Titan instrument prior to analysis and diluted to a minimum volume of 50 mL. Pd contents were determined by ICP-MS (PerkinElmer ICP MS NexION 2000), while the Ni content was determined by ICP-OES by the University of Liverpool Analytical Services.
Transient Absorption Spectroscopy (TAS): Femtosecond–Nanosecond Time Scale
The employed ultrafast transient absorption setup uses a regeneratively amplified Ti:sapphire laser (Solstice, Spectra-Physics), which produces 800 nm laser pulses with a width of 92 fs at a 1 kHz repetition rate. The transient absorption setup described in the following is commercially available (Helios, Ultrafast Systems). After the Solstice amplifier, each pulse is divided into what will become pump and probe pulses using a semitransparent mirror. The pump pulse is directed through an optical parametric amplifier (TOPAS Prime, Light Conversion) and a frequency mixer (NirUVis, Light Conversion), which allows for tuning of the excitation wavelength from the UV region up to the NIR region. The probe pulse is directed through a delay stage to delay it by an adjustable time period with respect to its corresponding pump pulse, thus defining the time at which the sample is probed. The maximum delay is ultimately defined by the total length of the delay stage and in this case corresponds to ∼6 ns. After the delay stage, the probe pulse is focused into a sapphire crystal, which transforms the monochromatic beam into a white light continuum. Depending on the inserted sapphire crystal, a visible (450–800 nm) or NIR probe continuum (850–1400 nm) is generated. The generated continuum pulses are then again divided before the sample using a semitransparent mirror, where one of the split off pulses probes through the sample and the other one serves as a reference to account for fluctuations and improve the signal to noise ratio. Each of the two continuum pulses is eventually focused into a separate multichannel spectrometer (InGaAs sensor) via optical fibers. The continuum pulse that probes through the sample is spatially overlapped with the pump pulse on the sample. These continuum pulses alternately record the transmission of the sample in the excited state and in the ground state by blocking every other pump pulse with an optical chopper rotating at 500 Hz. Pulse energies were measured using an energy meter (VEGA P/N 7Z01560, OPHIR Photonics) equipped with a 500 μm diameter aperture, roughly corresponding to the diameter of the probe beam. The pump beam was slightly larger than 500 μm at the sample position.
Transient Absorption Spectroscopy (TAS): Microsecond–Second Time Scale
The transient absorption setup employed for longer time scales uses a Nd:YAG laser (OPOTEK Opolette 355 II, 4–7 ns pulse width), which generates UV pulses (355 nm) or visible/NIR pulses (410–2200 nm, tunable via an optical parametric oscillator). The probe beam originates from a 100 W quartz halogen lamp driven by a stabilized power supply (Bantham IL1) which is sequentially directed through a first monochromator, the sample, and then a second monochromator before it impinges onto a Si photodiode detector (Hamamatsu S3071). Pump pulses are directed from the laser output to the sample via a liquid light guide and are overlapped with the probe beam at the position of the sample. Data acquisitions are triggered by scattered laser light using a photodiode (Thorlabs DET210) positioned close to the laser output. Appropriate long pass filters are positioned between the sample and detector to attenuate scattered laser light. A home-built LabVIEW-based software package acquires data on two different time scales simultaneously: the microsecond–millisecond signal is sampled using an oscilloscope (Tektronix DPO 2012B) after amplification (Costronics 1999 amplifier), whereas the millisecond–second signal is sampled without amplification using a DAQ card (National Instruments USB-6211 or USB-6361). Excitation fluences were measured using a pyroelectric energy sensor (Ophir Photonics PE9).
Time-Correlated Single-Photon Counting (TCSPC)
TCSPC experiments were carried out using a commercial TCSPC setup (Horiba DeltaFlex) equipped with a pulsed LED excitation source (Horiba NanoLED series) and a fast rise-time photomultiplier detector (Horiba PPD-650). The instrument response function (IRF) was measured at the wavelength of the excitation source. During all other measurements, a suitable long-pass filter was inserted between the sample and detector to block off scattered excitation light.
Photoinduced Absorption Spectroscopy (PIAS)
PIAS experiments were performed on the microsecond–second TAS setup with the following modifications: a high-power LED (LedEngin LZ1-10U600 365 nm; Cree XLamp XP-E 465 nm), which is driven by a high-precision DC power supply (TTi QL564P), is used as an excitation source. Again, a liquid light guide is used to direct the LED output to the sample. Light pulses are generated via a MOSFET transistor (STMicroelectronics STF8NM50N), where the gate is modulated by the DAQ card (National Instruments USB-6361). The DAQ card initiates a light pulse by switching the gate using a voltage signal, which simultaneously triggers the data acquisition. All data were sampled without prior amplification using the DAQ card. Excitation fluences were measured with a silicon photodiode power sensor (Thorlabs S120UV) in conjunction with a digital power meter (Thorlabs PM100).
Spectroelectrochemistry (SEC)
Spectroelectrochemical measurements were conducted using a home-built spectroelectrochemical cell inside an Agilent Cary 60 UV–vis spectrometer. This spectroelectrochemical cell contained a Pd nanoparticle film as a working electrode, a platinum-mesh counter electrode, a saturated Ag/AgCl reference electrode, and a pH 9 phosphate buffer as the electrolyte. UV–vis spectra were recorded at a series of applied potentials, and absorbance difference spectra were calculated by subtracting the open-circuit spectrum from each of the recorded spectra.
X-ray Absorption Spectroscopy (XAS)
The XAS spectra of the P10 powders were collected at the Taiwan Photon Source (TPS 44A) beamline end-station (energy, 3.0 GeV; ring current, 500 mA) of the National Synchrotron Radiation Research Center (NSRRC) in Hsinchu, Taiwan. The X-ray absorption near-edge structure (XANES) spectra of these samples were collected with a Lytle detector at 25 °C in the energy ranges of Pd (24350 eV) or Ni (8333 eV) K edges using correspondingly Pd and Ni foils as references. Palladium foil (Pd) and several nickel references (Ni, NiO, Ni(OH)2, and LiNiO4) with confirmed metallic valences were adopted as the standards in the linear regression calculation for the Pd or Ni valence identification. These XAS spectra were collected in three steps: pre-edge (energy range, −200 to −20 eV; interval, 5 eV/step), edge-jump/white line (energy range, −20–50 eV; interval, 0.35 eV/step) and postedge (energy range, 50–600 eV, interval, 0.06 eV/step), following the Pd and Ni K-edge energies as the data points.
Results
The absorbance and photoluminescence properties of our F8BT, P3HT, and P10 dispersions in H2O are characterized in Figure 1a–c. Approximate absorption onsets are 520, 645, and 475 nm for F8BT, P3HT, and P10, respectively, suggesting that F8BT and P10 absorb similar amounts of visible light. P10 suspensions exhibit a more pronounced scattering tail in comparison to suspensions of the other two polymers, which complicates an accurate quantification of their absorbance. Absorbance and photoluminescence spectra are in good agreement with literature reports for all three polymers, suggesting that the F8BT and P3HT nanoparticles used here are comparable to thin films of these polymers used for other applications such as organic photovoltaics or organic light-emitting diodes.35−37 P10 is primarily a photocatalyst, and its optical properties are in good agreement with those reported in our earlier study.20
Figure 1.

Optical properties and photoinduced absorption spectra of F8BT (1170 ppm of Pd), P3HT (<1 ppm Pd, 17 ppm of Ni), and P10 (2000 ppm Pd) dispersions. Absorbance and photoluminescence emission spectra of (a) F8BT, (b) P3HT, and (c) P10 dispersions in H2O. Photoluminescence spectra were obtained using excitation wavelengths of 465 nm for P3HT and 365 nm for P3HT and P10. Photoinduced absorption spectra upon constant illumination of nanoparticle suspensions of (d) F8BT in a 30 vol % DEA in water mixture, (e) P3HT in a 1/1/1 H2O/MeOH/TEA mixture, and (f) P10 in a 1/1/1 mixture of H2O/MeOH/TEA. The insets show the structures of the respective polymers. PIAS conditions: F8BT, 465 nm excitation, 25.6 mW cm–2 fluence; P3HT, 365 nm excitation, 7.1 mW cm–2 fluence; P10, 365 nm excitation, 12.5 mW cm–2 fluence.
We first compared dispersions of our F8BT, P3HT, and P10 particles in the presence of an electron-donating substrate. We used diethylamine (DEA) for F8BT and triethylamine (TEA) for P10 and P3HT, corresponding to their optimized photocatalytic reaction conditions and thus their highest yield of long-lived electron polarons. The use of a different scavenger for a given polymer would alter this polaron yield but be less likely to affect the hydrogen evolution reaction beyond the number of available reactants, as hydrogen evolution is driven by these polarons on a considerably longer time scale in comparison to the scavenger oxidation. Both F8BT and P10 were synthesized via Suzuki–Miyaura coupling as reported previously,3,20 whereas P3HT (95% regioregular) was synthesized via Grignard metathesis polymerization and therefore contains low amounts of residual Ni (17 ppm) but no detectable residual Pd. P10 is one of the most active polymer photocatalysts reported thus far, and we have recently identified the optical signature of its long-lived electron polarons (i.e., polymer-centered electrons) using transient absorption spectroscopy, which is characterized by a narrow absorption peak around 630 nm.20 Residual Pd in P10 (2000 ppm) is very difficult to remove, since this polymer is not solution-processable, and P10 is therefore first compared here to F8BT with a high Pd content (1170 ppm). Overall, P10 exhibits a hydrogen evolution rate of 2825 μmol h–1 g–1 at this Pd content and thus produces at least 20 times more hydrogen than F8BT under the reaction conditions.3 In contrast, P3HT is about 20 times less active for hydrogen evolution in comparison to F8BT, even in the presence of added platinum (Figure S1).
To investigate charge accumulation in these materials under operando catalytic conditions, we use light pulses with a duration of several seconds to bring the samples to a steady state. When this steady state is reached, an equilibrium between the generation, recombination, and reaction of long-lived charges is established. By recording the absorption spectra of the resulting equilibrium population of long-lived charges for each of the three polymers (photoinduced absorption spectroscopy, PIAS38), we obtain insights into which species accumulate before the rate-determining step of the reaction. In this way, kinetic limitations of the hydrogen evolution reaction can be identified for these materials.
As shown in Figure 1d, the PIAS spectrum for F8BT nanoparticles is characterized by an absorption that increases from longer to shorter probe wavelengths. Such an absorption feature is not common for charges in organic polymers and, as will be demonstrated below, is assigned to reduced Pd herein. This spectrum therefore suggests that long-lived electrons accumulate on the Pd clusters in these F8BT nanoparticles under constant illumination. In contrast to F8BT, the PIAS spectrum of P3HT exhibits a narrow peak at around 650 nm and an additional absorption feature toward the near-infrared region (Figure 1e), consistent with typical absorption features of electron polarons: polymer polarons can be expected to give rise to two absorption peaks, one of which is often found in the visible range while the second usually appears in the near-infrared range.39−41 In line with the absence of residual Pd in P3HT and its low residual Ni content, its polaron-like PIAS spectrum indicates that electrons generated by photoinduced hole transfer to the electron donor (reductive quenching) accumulate on the P3HT polymer.
The PIAS spectrum for P10 (Figure 1f) exhibits a narrow absorption peak at around 630 nm and thus is virtually identical to its electron polaron transient spectrum reported previously.20 Interestingly, this polaron-like absorption feature suggests that photogenerated electrons in the P10 system accumulate on the polymer (as for P3HT) rather than on the Pd clusters within the P10 particles, despite P10 containing even more residual Pd than the F8BT sample investigated here.
Due to this strong contrast in charge accumulation between F8BT and P10 at high Pd content, we next focused on a comparison of these two polymers. We previously demonstrated that residual Pd can be efficiently removed from F8BT using a combination of gel permeation chromatography (GPC) and washing with sodium N,N-diethyldithiocarbamate (DTC),3 which yielded a series of F8BT polymer batches with Pd concentrations of 1170, 195, 36, and <1 ppm (below the detection limit). The obtained F8BT polymer samples were then processed into nanoparticles with different Pd concentrations.
In our previous study, we found that these F8BT nanoparticles exhibited a sharply decreasing hydrogen evolution rate at Pd concentrations below 100 ppm when they were tested in the presence of DEA as an electron donor, and no hydrogen was detected at <1 ppm Pd.3 However, the hydrogen evolution activity of the <1 ppm Pd sample was recovered when defined amounts of Pd precursor were added back into the polymer before processing it into nanoparticles, thus demonstrating that the presence of a cocatalyst is essential for photocatalytic hydrogen evolution using F8BT.3 Here, we now elucidate the reasons behind this striking dependence of the hydrogen evolution activity of F8BT on its residual Pd content using transient optical spectroscopy before comparing our findings to those for P10.
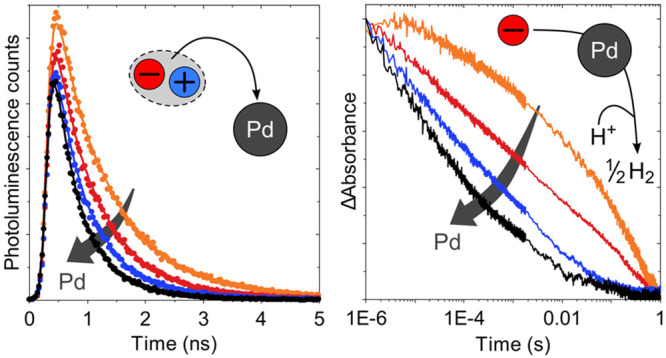
We first used time-correlated single photon counting (TCSPC) to investigate the effect of Pd on the population and lifetime of photogenerated excitons in F8BT nanoparticles at defined times after photoexcitation. To this end, we studied F8BT nanoparticles with a large variation in Pd content (<1–1170 ppm as described above) and a narrow size range (60–80 nm in diameter). We probed the photoluminescence of our F8BT nanoparticles in suspension at 545 nm following 465 nm excitation, close to their respective emission and visible absorption maxima (Figure 1a). Nanoparticle suspensions with the same absorbance at the excitation wavelength were used to ensure that different samples absorbed the same number of photons, and we counted emitted photons over a period of 10 s for each sample. This approach allowed us to quantitatively compare signal amplitudes between samples in addition to the kinetic profiles of the decay.
The photoluminescence decay kinetics shown in Figure 2a are characterized by a gradual decrease in lifetime with increasing Pd concentration, suggesting that Pd quenches the polymer excitons which give rise to the photoluminescence signal. As discussed in more detail below, this quenching process can be attributed to a combination of energy transfer and charge transfer from photogenerated excitons to the Pd clusters in these F8BT nanoparticles. By integrating the photoluminescence decays in Figure 2a up to 10 ns, we estimate that at least 54% of photogenerated excitons are quenched by Pd in unpurified F8BT nanoparticles (Table 1), implying that at least 54% of the generated excitons eventually reach a Pd cluster in this sample. An exciton diffusion length of ∼9.2 nm was determined for these F8BT nanoparticles (at <1 ppm Pd) from ultrafast transient absorption exciton–exciton annihilation experiments (Figure S2 and Table S1), which is in good agreement with literature values for exciton diffusion lengths in F8BT films reported in the range of 8–12 nm.42−44 Given the 60–80 nm diameter of these nanoparticles, we can conclude that several Pd clusters are present per particle, significantly more than those observable in our previously reported HRTEM data,3 which most likely are only able to resolve the largest clusters. On the basis of a comparison of the initial signal amplitudes, we determine that 23% of this quenching occurs at times faster than 200 ps (the time resolution of the TCPSC setup). Direct measurements of the exciton population using femtosecond–nanosecond transient absorption spectroscopy (TAS) suggest that 10% quenching takes place even before 100 fs in unpurified F8BT (Figure S3).
Figure 2.

Photoluminescence decays of F8BT nanoparticle suspensions probed at 545 nm upon 467 nm excitation. (a) Nanoparticles with different Pd concentrations in H2O and (b) nanoparticles with <1 ppm Pd in H2O and in a 30 vol % DEA mixture. All suspensions were prepared with an absorbance of 0.10 at the excitation wavelength to ensure that equal numbers of absorbed photons and emitted photons were counted over a period of 10 s for each sample, thus allowing a quantitative comparison of signal amplitudes. The full lines represent stretched exponential fits to the data with the same stretching exponent for all traces.
Table 1. Exciton Quenching in F8BT Nanoparticles via Pd Clusters or DEAa.
| Pd content (ppm) | aqueous medium | k (ns–0.87) | QYexc via IQ/I0 | QYexc via (k – k0)/k |
|---|---|---|---|---|
| 1170 | H2O | 2.3 | 0.54 | 0.48 |
| 195 | H2O | 1.8 | 0.44 | 0.33 |
| 36 | H2O | 1.5 | 0.28 | 0.20 |
| <1 | H2O | 1.2 | 0 | 0 |
| <1 | 30 vol % DEA in H2O | 1.7 | 0.24 | 0.29 |
Exciton decay rate constants k were obtained from stretched exponential fits to the data as shown in Figure 2, using y ∝ exp(−ktb), where the stretching exponent b = 0.87 was fixed for all decays to enable a comparison of k. Exciton quenching yields (QYexc) were estimated in two complementary ways: (i) via IQ/I0 by comparing the integrated signal intensities in the presence of a quencher IQ (with quenchers being Pd or DEA) to the integrated signal intensity in the absence of a quencher I0 or (ii) via (k – k0)/k by comparing changes in rate constants, where k0 represents the exciton decay in the absence of a quencher. In both cases, quenchers were taken as absent in the <1 ppm Pd sample in H2O.
While quenching via Pd takes place on the femtosecond–nanosecond time scale, exciton quenching via DEA, a reductive quenching process, occurs on the late-picosecond to early-nanosecond time scale for our F8BT nanoparticles (Figure 2b). For F8BT nanoparticles with <1 ppm Pd, we observe that 24% of photogenerated excitons are quenched in a 30 vol % DEA suspension relative to pure H2O. At higher Pd concentrations, exciton quenching via DEA is observed to be less efficient (Figure S4). For instance, for unpurified F8BT, little additional exciton quenching is observed when DEA is present, in good agreement with quenching via Pd being dominant at high Pd concentrations. This means that quenching via Pd constitutes a competitive pathway which quickly starts to dominate over reductive quenching via DEA when the Pd concentration increases.
As represented by the full lines in Figure 2, the photoluminescence decays fit well to the stretched exponential function I(t) ∝ exp(−ktb), from which nominal rate constants k can be obtained. Such stretched exponential decays are commonly observed in disordered materials such as polymers, where the degree of disorder gives rise to a distribution of lifetimes (and rate constants),45,46 in contrast to an ideal exponential decay, which characterizes a single lifetime. The stretching exponent was fixed to the unconstrained average of b = 0.87 for all decays, thus enabling comparisons of k between samples (Table 1). The decays were reconvoluted with the measured instrument response, and the extracted k values are thus independent of the chosen time zero. Since this is not the case for half-lifetimes t50%, which are often used to characterize decays in disordered materials, we thus use k to compare decay rates herein. Our analysis yields approximate rate constants of 2.3 ns–0.87 for the exciton decay in the presence of 1170 ppm of Pd as opposed to 1.7 ns–0.87 in the presence of 30 vol % of DEA (giving a rate constant of 0.5 ns–0.87 for reductive quenching) and suggests that exciton quenching via Pd starts to dominate over reductive quenching via DEA between 36 and 195 ppm of Pd for the F8BT nanoparticles investigated herein. These rate constants also provide an alternative way to calculate quenching yields, which have already been estimated via integration above, as (k – k0)/k. Quenching yields obtained in this way are in overall good agreement with those obtained via integration (Table 1).
We next identified photogenerated reaction intermediates on the time scale of proton reduction, typically reported at milliseconds to early seconds after photoexcitation,47−49 and investigated how the nature of these intermediates depends on the presence of Pd. Transient absorption spectra probed 100 μs to 100 ms after laser excitation are shown in Figure 3a–c for F8BT nanoparticles with different Pd concentrations, suspended in a 30 vol % DEA mixture. The transient absorption spectra underwent a pronounced change in shape as Pd was removed from the polymer: while the spectrum of unpurified F8BT is characterized by an absorption which increases from longer toward shorter probe wavelengths, a gradual transformation to a peak centered around 700–750 nm is observed as the Pd content decreases. This change in spectral shape suggests the presence of different species in the extreme cases of high and no Pd content, with a mixture of the two being observed at intermediate Pd concentrations. These different species were not separately observed in our previous study,3 and we thus characterize them in detail herein.
Figure 3.
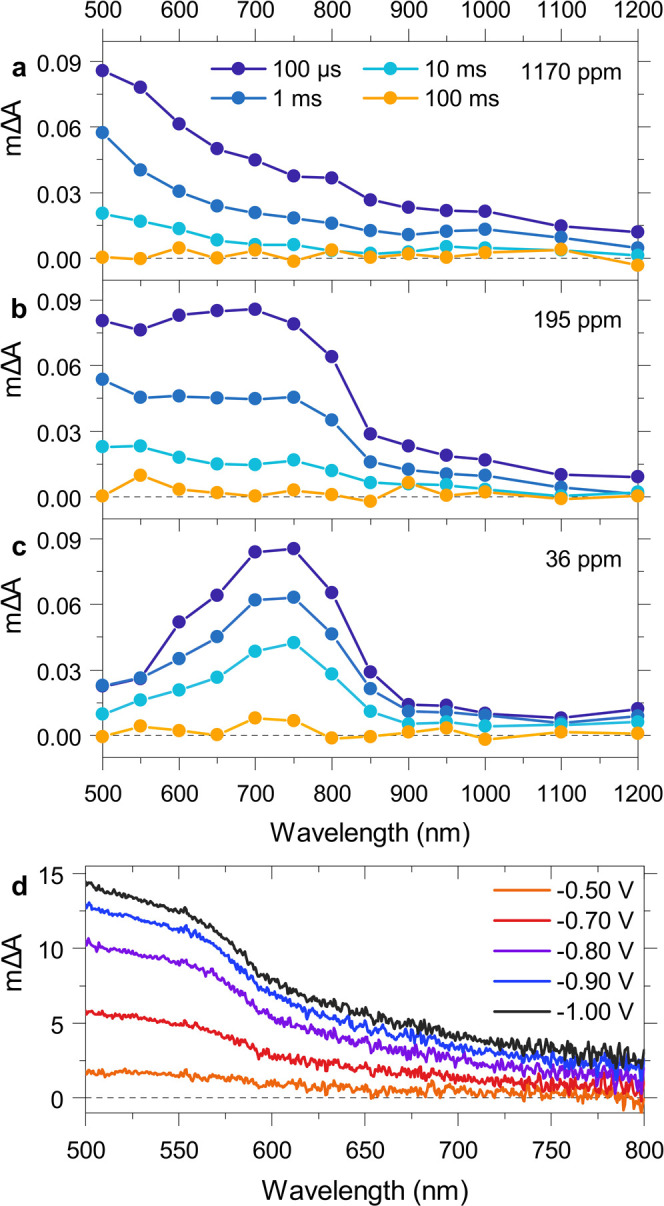
(a–c) Transient absorption spectra of F8BT nanoparticles with different Pd concentrations, suspended in an aqueous 30 vol % DEA mixture. Spectra were obtained using a 460 nm excitation fluence of 0.54 mJ cm–2 and were probed 100 μs and 1, 10, and 100 ms after excitation. All suspensions were prepared with an absorbance of 0.41 at the excitation wavelength to ensure an equal number of absorbed photons for different samples. (d) Absorbance difference spectra of Pd nanoparticles deposited on an FTO-coated glass substrate calculated between open circuit (−0.10 V vs Ag/AgCl) and a series of applied potentials between −0.50 and −1.0 V vs Ag/AgCl.
We assign the peak at 700–750 nm to polymer-centered electron polarons, generated via reductive quenching in the presence of DEA. This assignment is in reasonable agreement with a reported electron polaron absorption peak at 674 nm in F8BT films.50 The peaklike shape is generally consistent with absorption features expected for charged polymers39−41 as discussed above and is in line with the spectral signature of electron polarons in P10.20 In contrast, the increasing absorption toward shorter probe wavelengths observed at high Pd concentrations is not usually found for conjugated polymers, which indicates that it might be related to charges on the metallic Pd. To test the optical response of Pd upon electron injection, we deposited Pd nanoparticles on a conductive glass (FTO) substrate using chemical vapor deposition. As shown in Figure 3d, spectroelectrochemistry reveals that the application of increasingly negative potentials induces an absorption which increases toward shorter probe wavelengths, strongly resembling the transient spectra at high Pd concentration in Figure 3a. This strong resemblance suggests that the reduction of Pd gives rise to this spectral signature, and we therefore attribute its appearance in our transient measurements to photogenerated electrons on the Pd clusters in our F8BT nanoparticles. As shown above using the very similar PIAS spectrum in Figure 1d, this transient localization of electrons on Pd clusters then leads to electron accumulation on these Pd clusters under quasi-continuous illumination conditions.
To focus on the effect of Pd in the data above, we used F8BT nanoparticles within a narrow size range of 60–80 nm in diameter to minimize effects due to differences in surface area. We now turn to nanoparticles with much greater differences in diameter, all of which were prepared using F8BT with high Pd content (1170 ppm Pd), again adjusting nanoparticle concentrations to ensure matched sample absorption. Figure 4a shows a steadily increasing hydrogen evolution activity with decreasing size for particles smaller than ∼100 nm in diameter, while larger particles exhibit comparably small differences in activity. The photoluminescence lifetime and amplitude of smaller nanoparticles are reduced in comparison to larger ones even in pure H2O (Figure S5), pointing to enhanced exciton quenching via Pd for smaller particles. This enhanced exciton quenching indicates that Pd clusters might be located predominantly in the near-surface region of these F8BT nanoparticles, in line with Pd likely being more hydrophilic than the F8BT polymer itself.51Figure 4b shows higher transient absorption amplitudes for smaller particles, suggesting a larger yield of photogenerated electrons on Pd clusters for smaller particles.
Figure 4.

(a) Photocatalytic hydrogen evolution of unpurified F8BT nanoparticles with varying size, suspended in a 30 vol % DEA mixture. (b) Transient absorption decay kinetics of unpurified F8BT nanoparticles with variable size, suspended in a 30 vol % DEA mixture. The kinetics were probed at 750 nm (to avoid early time photoluminescence artifacts at shorter wavelengths) following 460 nm excitation using a fluence of 0.54 mJ cm–2. All suspensions were prepared with an absorbance of 0.41 at the excitation wavelength to ensure equal numbers of absorbed photons.
Motivated by the rapid localization of long-lived electrons on the Pd clusters within our F8BT nanoparticles, we investigated the transient dynamics of long-lived electrons in P10 at different Pd concentrations in a H2O/MeOH/TEA mixture. While Pd cannot be completely removed from P10 due to its insolubility, its Pd content can be tuned by using different amounts of Pd catalyst during the polymerization. In this way, three batches of P10 were synthesized using different amounts of [Pd(PPh3)4] catalyst in the Suzuki–Miyaura polycondensation reaction, which yielded P10 samples with residual Pd levels of 2000, 7000, and 27000 ppm Pd as determined by ICP-MS. These samples are compared here to P10Y, which is synthesized via an oxidative Yamamoto coupling using bis(cyclooctadiene)nickel(0), meaning that P10Y contains residual Ni (4000 ppm) rather than Pd. TEM images confirm the presence of visibly more metal clusters in P10 particles with higher metal content (Figure S7).
To confirm the states of Pd/Ni atoms in these samples, their fine structures (i.e., valence, coordination numbers, bond distances, and possible coordination configurations) were characterized using X-ray absorption spectroscopy (XAS). The relevant details are described in the Supporting Information. Using Pd/Ni metal foils and compounds with different Ni valences as references, these XAS measurements demonstrate that most Pd clusters in P10 are present as Pd(0) (Figure S9), whereas Ni species in P10Y primarily exist as Ni(II) (Figure S11). Our XAS experiments further reveal that Pd clusters in as-synthesized P10 have a coordination number of 3.3. This number is substantially lower than the coordination number in P10 following Pd photodeposition (7.7) or in Pd foil (8.7) (Table S2). In addition, the Pd–Pd bond distance in as-synthesized P10 (2.55 Å) is longer than that in P10 following Pd photodeposition (2.52 Å) or in Pd foil (2.45 Å), implying less Pd aggregation in as-synthesized P10. These coordination data suggest the presence of relatively small Pd clusters dispersed throughout the polymer particles in the P10 samples studied herein. Similarly to F8BT, we thus expect that only the largest Pd clusters are resolved in our TEM images.
In TAS, an electron polaron peak around 630 nm is observed for all P10 samples (Figure S14d), confirming that electrons remain on the P10 polymer even for the highest Pd concentration. As shown in Figure 5, the decay of these electron polarons is slowest in P10Y and continuously accelerates with increasing Pd concentration, which is reflected in a continuously decreasing half-lifetime of the polaron signal (Table 2). We attribute this accelerating polaron decay to faster catalysis in the presence of larger numbers of available Pd clusters. We also note that our previously reported P10 polaron decay in the presence of 4000 ppm of Pd20 falls nicely into this series.
Figure 5.

Transient absorption decay kinetics of long-lived electron polarons in P10 and F8BT. (a) P10 synthesized via a Ni-mediated Yamamoto coupling (P10Y) in comparison to P10 prepared with different amounts of Pd catalyst used in a Suzuki–Miyaura polycondensation reaction. (b) Electron polaron decay kinetics in P10 (2000 ppm of Pd) and F8BT (36 ppm of Pd) providing an estimate of the relative electron polaron yields in both materials, where the F8BT trace was recorded with 1.7 times more absorbed photons in comparison to those of P10. P10: 630 nm probe, 355 nm excitation using 0.32 mJ cm–2, 0.24 g L–1 in H2O/MeOH/TEA. F8BT: 700 nm probe, 460 nm excitation using 0.54 mJ cm–2, A = 0.42 at 460 nm in 30 vol % DEA.
Table 2. Overview of the P10 Samples Investigated Herein: Metal Content as Determined by ICP-OES; Hydrogen Evolution Rate (HER) in a Solvent Mixture Consisting of Equal Volumes of H2O, MeOH, and TEA; Half-Lifetime of the Electron Polaron Decay as Observed via Transient Absorption Spectroscopy, Defined with Respect to the Signal Amplitude at 1 μs.
| residual metal | metal content (ppm) | HER (μmol h–1 g–1) | t50% (s) |
|---|---|---|---|
| Ni | 4000 | 1897 | 2.3 × 10–2 |
| Pd | 2000 | 2825 | 1.3 × 10–3 |
| Pd | 7000 | 5332 | 1.5 × 10–4 |
| Pd | 27000 | 4906 | 5.0 × 10–5 |
In good agreement with this enhanced electron transfer to metal clusters inferred from our transient absorption data, the hydrogen evolution activity of P10 increases from 2825 μmol h–1 g–1 for 2000 ppm of Pd to 5332 μmol h–1 g–1 for 7000 ppm of Pd and then stays at almost the same level, within error, for 27000 ppm of Pd (see Table 2). In contrast, P10Y gives a lower hydrogen evolution reaction (HER) rate of 1897 μmol h–1 g–1,20 which, together with the slowest polaron decay in Figure 5, suggests that Ni is not as good a hydrogen evolution catalyst as Pd. This interpretation is in agreement with a higher electrocatalytic hydrogen evolution activity of Pd in comparison to Ni.52 To rule out that different catalyst loadings affect the photocatalytic activity via other pathways such as polymer chain length, a sample of P10 with 2000 ppm of Pd was tested after additional Pd was added by in situ photodeposition of [(NH4)2PdCl4]. This gave a HER rate of 5718 μmol h–1 g–1 for a Pd level of 17000 ppm, which fits the observed trend and therefore shows that different chain lengths are not the activity-determining factor in these different P10 batches.
While an activity plateau is reached at ∼100 ppm for F8BT,3 these data demonstrate that at least 2 orders of magnitude higher Pd concentrations are required for the observation of an activity plateau in P10. An activity plateau at such a high Pd concentration suggests that electron polarons in P10 generally struggle to transfer to the Pd clusters that catalyze hydrogen evolution in this material. This interpretation is further corroborated by a faster decay of long-lived electrons in our highest-performing F8BT sample (40 nm and 1170 ppm of Pd, Figure 4b) in comparison to those in P10, even at the highest Pd content of 27000 ppm (Figure 5).
Despite the faster charge transfer to Pd in F8BT, the overall hydrogen evolution activity of P10 is substantially higher. The higher activity of P10 is evident from the hydrogen evolution data shown in Figure 4a/Figure S15 and is also reflected in our previously reported EQEs of 11.6% for P10 (4000 ppm of Pd, measured at 420 nm20) vs <0.1% for F8BT (1170 ppm of Pd, measured at 340 nm3). As shown in Figure 5b, this activity gap can be linked to pronounced differences in the yields of long-lived electron polarons in these polymers: when the electron polarons are probed at their respective absorption maxima (630 nm for P10, 700 nm in F8BT), the optical signal from electron polarons generated in P10 is more than 1 order of magnitude higher than in F8BT, suggesting a substantially higher electron polaron yield in comparison to F8BT. We note that the number of absorbed photons for the F8BT sample used in this comparison was ∼1.7 times higher than that of P10, which makes the higher electron polaron yield in P10 even more striking. This comparison thus reveals that P10 generates long-lived electrons efficiently but their transfer to catalytic Pd sites is slow, whereas the opposite is the case for F8BT. This contrast will be discussed in further detail below.
Discussion
Many of the polymer photocatalysts reported in the literature have been synthesized using Pd-based catalysts, yet a detailed understanding of the role of resulting metal impurities in the process of hydrogen evolution has so far remained elusive. More detailed studies of this aspect have been impeded by the fact that most reported polymer photocatalysts, especially the more active ones, are difficult to purify because they are typically not solution-processable. Here we provide further evidence that residual Pd clusters serve as catalytically active sites in polymer photocatalysts and show that differences in the time scale of electron transfer to these residual Pd clusters translate into quantitatively different dependences of hydrogen evolution activity on residual Pd content. As a result, activity plateaus are observed at vastly different Pd concentrations for different polymers, which can be understood by measuring and comparing the charge carrier dynamics in these materials.
Our first photocatalytic system uses solution-processable F8BT to separate polymer purification from nanoparticle fabrication. This approach allows us to change the Pd content without affecting other properties, such as the molecular weight of the polymer or the morphology of the nanoparticles. Using this F8BT system, we found that the hydrogen evolution activity strongly decreases at Pd concentrations below ∼100 ppm of Pd, whereas an activity plateau was observed at Pd concentrations above this threshold,3 in qualitative agreement with another study on bipyridyl-based polymers.4 We investigated this striking dependence of activity on Pd content using transient absorption spectroscopy and found that the presence of Pd can lead to pronounced exciton quenching in F8BT. We consider two possible mechanisms for this exciton quenching process: energy transfer to Pd, which induces a relaxation to the ground state, or charge transfer to Pd, which corresponds to a charge separation process.
In the case of inorganic nanostructured materials with added Pt clusters, energy transfer has been found to dominate for photoexcited CdSe nanosheets,53,54 whereas CdS nanorods exhibit selective electron transfer to these Pt clusters.55,56 The preference for charge separation in the latter case has been ascribed to ultrafast hole trapping, thus enabling selective electron transfer to Pt.55,56 Although the photoluminescence spectrum of F8BT (Figure 1a) does not show evidence of deep hole trapping, it seems likely that Pd separates charges to some extent because charge yields generally appear to be higher when more excitons are quenched via Pd. For example, this is the case for the smaller particles in Figure 4b, and transient amplitudes in Figure 3a–c are comparable among F8BT nanoparticles with different Pd contents despite a change in the predominant location of long-lived electrons as a function of Pd content. However, the absorption coefficient of Pd-centered electrons is expected to be lower than that polymer-centered ones (30000 ± 3000 M–1 cm–1 at the absorption peak of electron polarons in F8BT50), suggesting that Pd-centered electrons must be present in higher yields to give rise to a transient absorption signal of magnitude similar to that of polymer-centered electrons.
While the branching ratio between energy transfer and charge transfer cannot be determined with certainty, it is striking that both pathways combined quench at least half of the photogenerated excitons in F8BT with high Pd content (>1000 ppm). We consider this estimation as a lower limit, because an even higher quenching yield would be obtained if the photoluminescence lifetime of our most purified sample were still affected by trace impurities of Pd below our ICP-OES resolution limit (1 ppm). Visible Pd clusters as observed via TEM (Figure S6) are present in much less than half of the nanoparticles, and it therefore seems likely that much smaller clusters are distributed throughout these nanoparticles—possibly preferentially near the nanoparticle surface, as suggested by the enhanced exciton quenching for smaller F8BT nanoparticles on dispersion in pure H2O (Figure S5).
Charge separation in organic photocatalyst systems is normally thought to be driven by reductive quenching via an electron donor in the solution phase. However, reductive quenching via the electron donor DEA only occurs from the late picosecond time scale onward, in contrast to the fast quenching via Pd on the femtosecond–nanosecond time scale (Figure 2b). As a result, exciton quenching via Pd starts to dominate over exciton quenching via DEA at Pd concentrations between 36 and 195 ppm of Pd, which is in line with the observed activity plateau at ∼100 ppm of Pd. The dominance of exciton quenching via Pd explains why the exciton lifetime is less and less affected by the presence of DEA when the Pd concentration is increased (Figure S4). Despite some degree of charge separation via Pd, appreciable yields of long-lived (up to the millisecond time scale) electrons are only observed in the presence of DEA, which highlights the importance of DEA in eliminating the hole as a recombination partner. Depending on the Pd concentration, these long-lived electrons are either situated on Pd clusters within a polymer nanoparticle or on the F8BT polymer itself. At high Pd contents, we observe transient spectral signatures very similar to those of reduced Pd nanoparticles on a conductive substrate (compare Figure 3a and Figure 3d), suggesting an assignment to electrons localized on the Pd clusters within our F8BT nanoparticles. This spectral signature gradually changes to a polaron-like absorption feature with increasing purification, indicating that electrons are situated more and more on the polymer itself when fewer Pd clusters are available.
By optically probing accumulated charges under quasi-continuous illumination conditions in F8BT (1170 ppm of Pd), P3HT (<1 ppm of Pd), and the highly active P10 (2000 ppm of Pd), we obtain further insights into the differences among these photocatalytic systems. The absorption spectrum of accumulated charges obtained upon illumination of unpurified F8BT with light pulses of several seconds duration (PIAS, Figure 1a) strongly resembles that of reduced Pd (Figure 3d), suggesting the accumulation of electrons on Pd clusters. This is in good agreement with our TAS data, where electrons in unpurified F8BT are observed on Pd even transiently (Figure 3a). P3HT, which is about 20 times less active for hydrogen evolution than F8BT even in the presence of added Pt (Figure S1), does not contain residual Pd and exhibits a spectral signature that suggests electron accumulation on the polymer itself. Finally, P10 is at least 20 times more active than F8BT20 but contains hard-to-remove residual Pd, since P10 is not solution-processable. Despite an even higher Pd concentration in comparison to unpurified F8BT, we find that photogenerated electrons in P10 accumulate on the polymer itself, as in the Pd-free P3HT.
This charge accumulation on the P10 polymer suggests that the transfer of photogenerated electron polarons to the Pd clusters that catalyze hydrogen evolution is relatively slow in this material—much slower than in F8BT, where electrons reside on Pd clusters on the pre-microsecond time scale at comparable Pd concentrations. This interpretation is corroborated by a decrease in electron polaron lifetime by orders of magnitude when the Pd concentration of P10 is increased (Table 2), which demonstrates that the addition of more Pd causes these electron polarons to react more quickly. In line with this faster reaction, a continuous increase in hydrogen evolution rate is observed at least up to 17000 ppm of Pd—in strong contrast to F8BT, where such an increase in activity is only found up to ∼100 ppm of Pd. These data reveal a striking difference between P10 and F8BT: electron polarons in P10 take a long time to transfer to catalytic Pd sites, and their transfer to Pd clusters can therefore be considered the kinetic bottleneck of hydrogen evolution on P10. For F8BT, in contrast, electron polarons encounter Pd clusters much more quickly, which implies that electrons already reside on Pd sites before the typical time scale of hydrogen evolution at milliseconds to seconds after photoexcitation.
To discuss the origin of these differences in charge transfer, we first consider the energetic alignment between polymers and metals. The HOMO|LUMO levels for P10,20 F8BT,57,58 and P3HT59 have been reported as −5.9|−2.9, −5.9|−3.3, and −5.2|−3.5 eV, respectively. The electron work function of Pd is 5.22–5.60 eV, while that of Ni is 5.04–5.35 eV,60 where the ranges represent work function measurements on different surface facets. Given these values, we observe that the work functions of Pd and Ni are energetically similar to the HOMO levels of the studied polymers. With electron polaron energy levels lying near the LUMOs, there exists a large driving force for the transfer of electron polarons from all three of these polymers to Pd and Ni, and we therefore rule out that the faster electron transfer to Pd in F8BT in comparison to P10 is due to differences in the energetic alignment between polymer and metal.
This leaves a number of possible reasons for the observed differences in charge transfer to Pd clusters in P10 and F8BT. (i) Pd distribution: due to the different synthetic protocols with different polymerization temperatures for F8BT and P10, it is possible that the resulting spatial distributions of Pd clusters might differ. The up to 54% exciton quenching observed in TCSPC suggests the presence of more and smaller Pd clusters than are evident from TEM in our F8BT nanoparticles (Figure S6), and the low coordination numbers in our XAS data (Table S2) similarly indicate the presence of small Pd clusters in P10 particles. While these results suggest that most Pd clusters are small and are finely dispersed in both F8BT and P10, a comprehensive treatment of this aspect will require mapping techniques with higher resolution. (ii) Carrier mobilities: whether electrons accumulate on polymer chains or on Pd clusters might also be related to the electron mobility of the respective polymer. Investigations of films of small-molecule semiconductors61,62 and conjugated polymers63 have demonstrated that the use of monomers with large dipole moments leads to a pronounced reduction in charge carrier mobility. On the basis of these results, a low electron mobility for P10 can be expected due to the large 5.7 D dipole moment20 of its dibenzo[b,d]thiophene sulfone unit, which is not shielded by any side chains. F8BT, in contrast, has been reported to exhibit reasonable electron mobilities on the order of 10–3 cm2 V–1 s–1.64,65 However, as charge carrier mobilities for polyfluorenes can vary strongly depending on the different preparation conditions of the polymer,64 an accurate comparison of mobilities between these two materials remains challenging. (iii) Saturation of Pd clusters: since P10 generates much higher numbers of long-lived electrons (see discussion below), it is possible that its Pd clusters become saturated with electrons and need to be discharged via proton reduction before accepting more electrons from the polymer. The number of Pd atoms (6 × 1021 cm–3 at 1000 ppm) is still considerably higher than the charge densities generated in our transient absorption experiments (∼1019 cm–3 of absorbed photons for a penetration depth of 100 nm), suggesting that this saturation is less likely, although clustering of Pd atoms and surface effects might ultimately render these numbers more comparable.
Despite slower charge transfer to Pd clusters in comparison to F8BT, P10 exhibits a substantially higher hydrogen evolution activity which can be assigned to 1 order of magnitude higher yields of long-lived electrons in comparison to F8BT (Figure 5b). While reductive quenching of F8BT excitons via DEA occurs on the late-picosecond–nanosecond time scale (Figure 2b), P10 exhibits a pronounced polaron-like transient signal with a half rise time of only 1–2 ps in the presence of TEA (Figure S14c).20 This fast appearance is likely linked to the polar sulfone groups in the backbone of P10, which make the polymer hydrophilic,20 allow water and scavenger molecules to penetrate into the several hundred nanometer large polymer particles, and therefore lead to faster reductive quenching than in the case of the largely hydrophobic F8BT. In contrast, reductive quenching of F8BT excitons occurs on the early-nanosecond time scale and competes less efficiently with F8BT exciton decay to ground and/or quenching at Pd metal clusters. The polar sulfone groups in P10 thus allow for faster and more efficient reductive quenching, although they most likely also decrease the mobilities of photogenerated electrons as discussed above.
We believe that the pronounced interaction between excitons and residual Pd clusters observed for F8BT herein also has implications for other fields that rely on conjugated polymers. For instance, organic light-emitting diodes (OLEDs) emit light by combining injected charges to excitons, which we anticipate will be affected by the presence of Pd clusters. In organic photovoltaics (OPVs), excitons normally separate at the interface between a donor and an acceptor material, in a way similar to the interface between an electron donor (e.g. DEA) and a polymer photocatalyst. Although charge separation in OPVs typically occurs significantly more quickly than is observed here for F8BT and DEA, we anticipate that the presence of Pd might affect the charge separation efficiency in these devices.
Conclusions
We have demonstrated that differences in the dependence of hydrogen evolution activity on residual Pd content in F8BT and the dibenzo[b,d]thiophene sulfone homopolymer P10 can be understood by tracking photogenerated reaction intermediates in these materials under transient and steady-state conditions. In Pd concentrations that are typical for polymers prepared via Pd-catalyzed cross coupling polymerizations (>1000 ppm; >0.1 wt %), residual Pd in F8BT quenches excitons efficiently. As a result, exciton quenching via Pd starts to dominate over reductive quenching via the electron donor DEA in the solution phase at Pd concentrations between 36 and 195 ppm. Exciton quenching via Pd appears to partially aid charge separation but also constitutes a competitive pathway for relaxation to the ground state. When exciton quenching via Pd dominates, long-lived electrons rapidly localize on Pd clusters within these polymer nanoparticles, whereas their predominant location gradually shifts to the F8BT polymer when the Pd concentration is lowered via additional purification steps. In contrast, long-lived electrons in P10 are located on the polymer up to Pd concentrations of at least 27000 ppm, similarly to P3HT, which contains no residual Pd. This difference in the predominant location of long-lived electrons between F8BT and P10 or P3HT is maintained in operando charge accumulation experiments, which reflects the constant-illumination conditions of photocatalytic activity measurements.
These data support the emerging notion that residual metal impurities are essential for substantial hydrogen evolution from polymer photocatalysts and suggest that the limited accessibility of such metal clusters for electron polarons can introduce a bottleneck for hydrogen evolution in conjugated polymers: for P10, the kinetic bottleneck appears to be the transfer of long-lived electrons to Pd sites, whereas for F8BT electrons are already localized on Pd sites before the typical time scale of hydrogen evolution. This difference is reflected in an activity plateau at around 100 ppm Pd in F8BT, whereas in P10 no such plateau is observed at least until 17000 ppm of Pd. Yet, P10 exhibits much higher activity than F8BT because of its more efficient reductive quenching pathway, which is a result of its hydrophilic sulfone-containing polymer backbone. The challenge is therefore to combine the advantages of both materials and to create polymers that exhibit both rapid reductive quenching as in P10 and rapid transfer to catalytic sites as in F8BT. Our results also demonstrate that residual Pd in polymer photocatalysts can affect the performance of these materials in ways beyond acting as a catalyst for proton reduction, which implies that residual Pd may also be important in other fields, such as organic light-emitting diodes and organic photovoltaics.
Acknowledgments
M.S. is grateful to Imperial College for a President’s Ph.D. Scholarship and to the EPSRC for a Doctoral Prize Fellowship. J.R.D. and I.M. acknowledge support from KAUST (project numbers OSR-2015-CRG4-2572 and OSR-2018-CRG7-3749.2). C.M.A., A.I.C., and R.S.S. acknowledge the Engineering and Physical Sciences Research Council (EPSRC, EP/N004884/1). L.F. thanks the EU for a Marie Curie fellowship (658270). S.C. thanks Imperial College London for a Schrödinger Scholarship. R.G. is grateful to the FRQNT for a postdoctoral award and NSERC Discovery Grant funding. C.-L.C. appreciates his colleague, Dr. Bing-Jian Su, for his kind support and assistance. All plotted data have been deposited on the open-access repository Zenodo and can be accessed via dx.doi.org/10.5281/zenodo.3932340.
Glossary
Abbreviations
- DEA
diethylamine
- DTC
N,N-diethyldithiocarbamate
- F8BT
poly(9,9-dioctylfluorene-alt-benzothiadiazole)
- PIAS
photoinduced absorption spectroscopy
- TAS
transient absorption spectroscopy
- TCSPC
time-correlated single-photon counting. XAS, X-ray absorption spectroscopy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c06104.
F8BT femtosecond–nanosecond TAS data, F8BT TCSPC data in the presence of DEA, F8BT exciton–exciton annihilation data for exciton diffusion length calculation and methodology description, F8BT TCSPC data for different particle sizes, P3HT hydrogen evolution data, P10 absorbance and photoluminescence data, P10 TCSPC data, P10 femtosecond–nanosecond TAS data, P10 microsecond–second TAS data, P10 hydrogen evolution data, F8BT and P10 TEM images, and XAS data (PDF)
Author Present Address
¶ R.S.S.: Department of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow G1 1XL, UK.
Author Present Address
△ I.M.: Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Oxford, OX1 3TA, UK.
Author Present Address
○ L.F.: Departament de Química, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, 08193 Barcelona, Spain.
Author Present Address
⊥ R.G.: Department of Chemistry, The University of British Columbia, Okanagan Campus, 3247 University Way, Kelowna, BC, Canada.
The authors declare no competing financial interest.
Supplementary Material
References
- Hutton G. A. M.; Martindale B. C. M.; Reisner E. Carbon Dots as Photosensitisers for Solar-Driven Catalysis. Chem. Soc. Rev. 2017, 46 (20), 6111–6123. 10.1039/C7CS00235A. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Vogel A.; Sachs M.; Sprick R. S.; Wilbraham L.; Moniz S. J. A.; Godin R.; Zwijnenburg M. A.; Durrant J. R.; Cooper A. I.; Tang J. Current Understanding and Challenges of Solar-Driven Hydrogen Generation Using Polymeric Photocatalysts. Nat. Energy 2019, 4 (9), 746–760. 10.1038/s41560-019-0456-5. [DOI] [Google Scholar]
- Kosco J.; Sachs M.; Godin R.; Kirkus M.; Francas L.; Bidwell M.; Qureshi M.; Anjum D.; Durrant J. R.; McCulloch I. The Effect of Residual Palladium Catalyst Contamination on the Photocatalytic Hydrogen Evolution Activity of Conjugated Polymers. Adv. Energy Mater. 2018, 8 (34), 1802181. 10.1002/aenm.201802181. [DOI] [Google Scholar]
- Li L.; Cai Z.; Wu Q.; Lo W.-Y.; Zhang N.; Chen L. X.; Yu L. Rational Design of Porous Conjugated Polymers and Roles of Residual Palladium for Photocatalytic Hydrogen Production. J. Am. Chem. Soc. 2016, 138 (24), 7681–7686. 10.1021/jacs.6b03472. [DOI] [PubMed] [Google Scholar]
- Meier C. B.; Sprick R. S.; Monti A.; Guiglion P.; Lee J.-S. M.; Zwijnenburg M. A.; Cooper A. I. Structure-Property Relationships for Covalent Triazine-Based Frameworks: The Effect of Spacer Length on Photocatalytic Hydrogen Evolution from Water. Polymer 2017, 126, 283–290. 10.1016/j.polymer.2017.04.017. [DOI] [Google Scholar]
- Wang X.; Maeda K.; Thomas A.; Takanabe K.; Xin G.; Carlsson J. M.; Domen K.; Antonietti M. A Metal-Free Polymeric Photocatalyst for Hydrogen Production from Water under Visible Light. Nat. Mater. 2009, 8 (1), 76–80. 10.1038/nmat2317. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Li G.; Lan Z.-A.; Lin L.; Savateev A.; Heil T.; Zafeiratos S.; Wang X.; Antonietti M. Optimizing Optical Absorption, Exciton Dissociation, and Charge Transfer of a Polymeric Carbon Nitride with Ultrahigh Solar Hydrogen Production Activity. Angew. Chem., Int. Ed. 2017, 56 (43), 13445–13449. 10.1002/anie.201706870. [DOI] [PubMed] [Google Scholar]
- Lin L.; Ou H.; Zhang Y.; Wang X. Tri-s-Triazine-Based Crystalline Graphitic Carbon Nitrides for Highly Efficient Hydrogen Evolution Photocatalysis. ACS Catal. 2016, 6 (6), 3921–3931. 10.1021/acscatal.6b00922. [DOI] [Google Scholar]
- Zhang G.; Lin L.; Li G.; Zhang Y.; Savateev A.; Zafeiratos S.; Wang X.; Antonietti M. Ionothermal Synthesis of Triazine-Heptazine-Based Copolymers with Apparent Quantum Yields of 60% at 420 Nm for Solar Hydrogen Production from “Sea Water. Angew. Chem., Int. Ed. 2018, 57 (30), 9372–9376. 10.1002/anie.201804702. [DOI] [PubMed] [Google Scholar]
- Aitchison C. M.; Sprick R. S.; Cooper A. I. Emulsion Polymerization Derived Organic Photocatalysts for Improved Light-Driven Hydrogen Evolution. J. Mater. Chem. A 2019, 7, 2490–2496. 10.1039/C8TA11383A. [DOI] [Google Scholar]
- Bai Y.; Wilbraham L.; Slater B. J.; Zwijnenburg M. A.; Sprick R. S.; Cooper A. I. Accelerated Discovery of Organic Polymer Photocatalysts for Hydrogen Evolution from Water through the Integration of Experiment and Theory. J. Am. Chem. Soc. 2019, 141 (22), 9063–9071. 10.1021/jacs.9b03591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata T.; Domen K. Particulate Photocatalysts for Water Splitting: Recent Advances and Future Prospects. ACS Energy Lett. 2019, 4 (2), 542–549. 10.1021/acsenergylett.8b02209. [DOI] [Google Scholar]
- Wang Z.; Li C.; Domen K. Recent Developments in Heterogeneous Photocatalysts for Solar-Driven Overall Water Splitting. Chem. Soc. Rev. 2019, 48 (7), 2109–2125. 10.1039/C8CS00542G. [DOI] [PubMed] [Google Scholar]
- Kosco J.; Bidwell M.; Cha H.; Martin T.; Howells C. T.; Sachs M.; Anjum D. H.; Gonzalez Lopez S.; Zou L.; Wadsworth A.; Zhang W.; Zhang L.; Tellam J.; Sougrat R.; Laquai F.; DeLongchamp D. M.; Durrant J. R.; McCulloch I. Enhanced Photocatalytic Hydrogen Evolution from Organic Semiconductor Heterojunction Nanoparticles. Nat. Mater. 2020, 19 (5), 559–565. 10.1038/s41563-019-0591-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Li X.; Sprick R. S.; Cooper A. I. Conjugated Polymer Donor-Molecular Acceptor Nanohybrids for Photocatalytic Hydrogen Evolution. Chem. Commun. 2020, 56 (50), 6790–6793. 10.1039/D0CC00740D. [DOI] [PubMed] [Google Scholar]
- Sprick R. S.; Bonillo B.; Clowes R.; Guiglion P.; Brownbill N. J.; Slater B. J.; Blanc F.; Zwijnenburg M. A.; Adams D. J.; Cooper A. I. Visible-Light-Driven Hydrogen Evolution Using Planarized Conjugated Polymer Photocatalysts. Angew. Chem., Int. Ed. 2016, 55 (5), 1792–1796. 10.1002/anie.201510542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Fernández-Terán R.; Zhang L.; Fernandes D. L. A.; Tian L.; Chen H.; Tian H. Organic Polymer Dots as Photocatalysts for Visible Light-Driven Hydrogen Generation. Angew. Chem., Int. Ed. 2016, 55 (40), 12306–12310. 10.1002/anie.201607018. [DOI] [PubMed] [Google Scholar]
- Pati P. B.; Damas G.; Tian L.; Fernandes D. L. A.; Zhang L.; Pehlivan I. B.; Edvinsson T.; Araujo C. M.; Tian H. An Experimental and Theoretical Study of an Efficient Polymer Nano-Photocatalyst for Hydrogen Evolution. Energy Environ. Sci. 2017, 10 (6), 1372–1376. 10.1039/C7EE00751E. [DOI] [Google Scholar]
- Tseng P.-J.; Chang C.-L.; Chan Y.-H.; Ting L.-Y.; Chen P.-Y.; Liao C.-H.; Tsai M.-L.; Chou H.-H. Design and Synthesis of Cycloplatinated Polymer Dots as Photocatalysts for Visible-Light-Driven Hydrogen Evolution. ACS Catal. 2018, 8 (9), 7766–7772. 10.1021/acscatal.8b01678. [DOI] [Google Scholar]
- Sachs M.; Sprick R. S.; Pearce D.; Hillman S. A. J.; Monti A.; Guilbert A. A. Y.; Brownbill N. J.; Dimitrov S.; Shi X.; Blanc F.; Zwijnenburg M. A.; Nelson J.; Durrant J. R.; Cooper A. I. Understanding Structure-Activity Relationships in Linear Polymer Photocatalysts for Hydrogen Evolution. Nat. Commun. 2018, 9 (1), 4968. 10.1038/s41467-018-07420-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L.-Y.; Jayakumar J.; Chang C.-L.; Lin W.-C.; Elsayed M. H.; Chou H.-H. Effect of Controlling the Number of Fused Rings on Polymer Photocatalysts for Visible-Light-Driven Hydrogen Evolution. J. Mater. Chem. A 2019, 7 (40), 22924–22929. 10.1039/C9TA06425G. [DOI] [Google Scholar]
- Hu Z.; Wang Z.; Zhang X.; Tang H.; Liu X.; Huang F.; Cao Y. Conjugated Polymers with Oligoethylene Glycol Side Chains for Improved Photocatalytic Hydrogen Evolution. iScience 2019, 13, 33–42. 10.1016/j.isci.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-L.; Lin W.-C.; Jia C.-Y.; Ting L.-Y.; Jayakumar J.; Elsayed M. H.; Yang Y.-Q.; Chan Y.-H.; Wang W.-S.; Lu C.-Y.; Chen P.-Y.; Chou H.-H. Low-Toxic Cycloplatinated Polymer Dots with Rational Design of Acceptor Co-Monomers for Enhanced Photocatalytic Efficiency and Stability. Appl. Catal. B Environ. 2020, 268, 118436. 10.1016/j.apcatb.2019.118436. [DOI] [Google Scholar]
- Dai C.; Liu B. Conjugated Polymers for Visible-Light-Driven Photocatalysis. Energy Environ. Sci. 2020, 13 (1), 24–52. 10.1039/C9EE01935A. [DOI] [Google Scholar]
- Vyas V. S.; Lau V. W.; Lotsch B. V. Soft Photocatalysis: Organic Polymers for Solar Fuel Production. Chem. Mater. 2016, 28 (15), 5191–5204. 10.1021/acs.chemmater.6b01894. [DOI] [Google Scholar]
- Zhang G.; Lan Z.-A.; Wang X. Conjugated Polymers: Catalysts for Photocatalytic Hydrogen Evolution. Angew. Chem., Int. Ed. 2016, 55 (51), 15712–15727. 10.1002/anie.201607375. [DOI] [PubMed] [Google Scholar]
- Rahman M. Z.; Kibria M. G.; Mullins C. B. Metal-Free Photocatalysts for Hydrogen Evolution. Chem. Soc. Rev. 2020, 49 (6), 1887–1931. 10.1039/C9CS00313D. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Chen Z.; Shi R.; Yang X.; Zhang T. Recent Advances in Conjugated Polymers for Visible-Light-Driven Water Splitting. Adv. Mater. 2020, 32, 1907296. 10.1002/adma.201907296. [DOI] [PubMed] [Google Scholar]
- Yang J.; Wang D.; Han H.; Li C. Roles of Cocatalysts in Photocatalysis and Photoelectrocatalysis. Acc. Chem. Res. 2013, 46 (8), 1900–1909. 10.1021/ar300227e. [DOI] [PubMed] [Google Scholar]
- Savateev A.; Antonietti M. Heterogeneous Organocatalysis for Photoredox Chemistry. ACS Catal. 2018, 8 (10), 9790–9808. 10.1021/acscatal.8b02595. [DOI] [Google Scholar]
- Ye S.; Wang R.; Wu M.-Z.; Yuan Y.-P. A Review on G-C3N4 for Photocatalytic Water Splitting and CO2 Reduction. Appl. Surf. Sci. 2015, 358, 15–27. 10.1016/j.apsusc.2015.08.173. [DOI] [Google Scholar]
- Pati P. B.; Damas G.; Tian L.; Fernandes D. L. A.; Zhang L.; Pehlivan I. B.; Edvinsson T.; Araujo C. M.; Tian H. An Experimental and Theoretical Study of an Efficient Polymer Nano-Photocatalyst for Hydrogen Evolution. Energy Environ. Sci. 2017, 10 (6), 1372–1376. 10.1039/C7EE00751E. [DOI] [Google Scholar]
- Helm M. L.; Stewart M. P.; Bullock R. M.; DuBois M. R.; DuBois D. L. A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 s-1 for H2 Production. Science (Washington, DC, U. S.) 2011, 333 (6044), 863–866. 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- Loewe R. S.; Ewbank P. C.; Liu J.; Zhai L.; McCullough R. D. Regioregular, Head-to-Tail Coupled Poly(3-Alkylthiophenes) Made Easy by the GRIM Method: Investigation of the Reaction and the Origin of Regioselectivity. Macromolecules 2001, 34 (13), 4324–4333. 10.1021/ma001677+. [DOI] [Google Scholar]
- Ohkita H. Charge Generation Dynamics in Polymer/Polymer Solar Cells Studied by Transient Absorption Spectroscopy. J. Photonics Energy 2011, 1 (1), 011118. 10.1117/1.3569105. [DOI] [Google Scholar]
- Donley C. L.; Zaumseil J.; Andreasen J. W.; Nielsen M. M.; Sirringhaus H.; Friend R. H.; Kim J.-S. Effects of Packing Structure on the Optoelectronic and Charge Transport Properties in Poly(9,9-Di- n -Octylfluorene- Alt -Benzothiadiazole). J. Am. Chem. Soc. 2005, 127 (37), 12890–12899. 10.1021/ja051891j. [DOI] [PubMed] [Google Scholar]
- Dimitriev O. P.; Blank D. A.; Ganser C.; Teichert C. Effect of the Polymer Chain Arrangement on Exciton and Polaron Dynamics in P3HT and P3HT:PCBM Films. J. Phys. Chem. C 2018, 122 (30), 17096–17109. 10.1021/acs.jpcc.8b05155. [DOI] [Google Scholar]
- Le Formal F.; Pastor E.; Tilley S. D.; Mesa C. A.; Pendlebury S. R.; Grätzel M.; Durrant J. R. Rate Law Analysis of Water Oxidation on a Hematite Surface. J. Am. Chem. Soc. 2015, 137 (20), 6629–6637. 10.1021/jacs.5b02576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Österbacka R.; An C. P.; Jiang X. M.; Vardeny Z. V. Two-Dimensional Electronic Excitations in Self-Assembled Conjugated Polymer Nanocrystals. Science (Washington, DC, U. S.) 2000, 287 (5454), 839–842. 10.1126/science.287.5454.839. [DOI] [PubMed] [Google Scholar]
- Baniya S.; Vardeny S. R.; Lafalce E.; Peygambarian N.; Vardeny Z. V. Amplitude-Mode Spectroscopy of Charge Excitations in PTB7 π-Conjugated Donor-Acceptor Copolymer for Photovoltaic Applications. Phys. Rev. Appl. 2017, 7 (6), 064031. 10.1103/PhysRevApplied.7.064031. [DOI] [Google Scholar]
- Fesser K.; Bishop A. R.; Campbell D. K. Optical Absorption from Polarons in a Model of Polyacetylene. Phys. Rev. B: Condens. Matter Mater. Phys. 1983, 27 (8), 4804–4825. 10.1103/PhysRevB.27.4804. [DOI] [Google Scholar]
- Leow C.; Ohnishi T.; Matsumura M. Diffusion Lengths of Excitons in Polymers in Relation to External Quantum Efficiency of the Photocurrent of Solar Cells. J. Phys. Chem. C 2014, 118 (1), 71–76. 10.1021/jp4079376. [DOI] [Google Scholar]
- Mikhnenko O. V.; Kuik M.; Lin J.; van der Kaap N.; Nguyen T.-Q.; Blom P. W. M. Trap-Limited Exciton Diffusion in Organic Semiconductors. Adv. Mater. 2014, 26 (12), 1912–1917. 10.1002/adma.201304162. [DOI] [PubMed] [Google Scholar]
- Groff L. C.; Wang X.; McNeill J. D. Measurement of Exciton Transport in Conjugated Polymer Nanoparticles. J. Phys. Chem. C 2013, 117 (48), 25748–25755. 10.1021/jp407065h. [DOI] [Google Scholar]
- Regan K. P.; Koenigsmann C.; Sheehan S. W.; Konezny S. J.; Schmuttenmaer C. A. Size-Dependent Ultrafast Charge Carrier Dynamics of WO 3 for Photoelectrochemical Cells. J. Phys. Chem. C 2016, 120 (27), 14926–14933. 10.1021/acs.jpcc.6b04390. [DOI] [Google Scholar]
- Soloviev V. N.; Eichhöfer A.; Fenske D.; Banin U. Size-Dependent Optical Spectroscopy of a Homologous Series of CdSe Cluster Molecules. J. Am. Chem. Soc. 2001, 123 (10), 2354–2364. 10.1021/ja003598j. [DOI] [PubMed] [Google Scholar]
- Reynal A.; Lakadamyali F.; Gross M. A.; Reisner E.; Durrant J. R. Parameters Affecting Electron Transfer Dynamics from Semiconductors to Molecular Catalysts for the Photochemical Reduction of Protons. Energy Environ. Sci. 2013, 6 (11), 3291. 10.1039/c3ee40961a. [DOI] [Google Scholar]
- Rodenberg A.; Orazietti M.; Probst B.; Bachmann C.; Alberto R.; Baldridge K. K.; Hamm P. Mechanism of Photocatalytic Hydrogen Generation by a Polypyridyl-Based Cobalt Catalyst in Aqueous Solution. Inorg. Chem. 2015, 54 (2), 646–657. 10.1021/ic502591a. [DOI] [PubMed] [Google Scholar]
- Pastor E.; Le Formal F.; Mayer M. T.; Tilley S. D.; Francàs L.; Mesa C. A.; Grätzel M.; Durrant J. R. Spectroelectrochemical Analysis of the Mechanism of (Photo)Electrochemical Hydrogen Evolution at a Catalytic Interface. Nat. Commun. 2017, 8, 14280. 10.1038/ncomms14280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird M. J.; Bakalis J.; Asaoka S.; Sirringhaus H.; Miller J. R. Fast Holes, Slow Electrons, and Medium Control of Polaron Size and Mobility in the DA Polymer F8BT. J. Phys. Chem. C 2017, 121 (29), 15597–15609. 10.1021/acs.jpcc.7b04602. [DOI] [Google Scholar]
- Gim S.; Cho K. J.; Lim H.-K.; Kim H. Structure, Dynamics, and Wettability of Water at Metal Interfaces. Sci. Rep. 2019, 9 (1), 14805. 10.1038/s41598-019-51323-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørskov J. K.; Bligaard T.; Logadottir A.; Kitchin J. R.; Chen J. G.; Pandelov S.; Stimming U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152 (3), J23. 10.1149/1.1856988. [DOI] [Google Scholar]
- Wu K.; Li Q.; Du Y.; Chen Z.; Lian T. Ultrafast Exciton Quenching by Energy and Electron Transfer in Colloidal CdSe Nanosheet-Pt Heterostructures. Chem. Sci. 2015, 6 (2), 1049–1054. 10.1039/C4SC02994A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Lian T. Exciton Dissociation Dynamics and Light-Driven H2 Generation in Colloidal 2D Cadmium Chalcogenide Nanoplatelet Heterostructures. Nano Res. 2018, 11 (6), 3031–3049. 10.1007/s12274-018-2024-x. [DOI] [Google Scholar]
- Wu K.; Zhu H.; Liu Z.; Rodríguez-Córdoba W.; Lian T. Ultrafast Charge Separation and Long-Lived Charge Separated State in Photocatalytic CdS-Pt Nanorod Heterostructures. J. Am. Chem. Soc. 2012, 134 (25), 10337–10340. 10.1021/ja303306u. [DOI] [PubMed] [Google Scholar]
- Wu K.; Zhu H.; Lian T. Ultrafast Exciton Dynamics and Light-Driven H 2 Evolution in Colloidal Semiconductor Nanorods and Pt-Tipped Nanorods. Acc. Chem. Res. 2015, 48 (3), 851–859. 10.1021/ar500398g. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Blom P. W. M. Electron and Hole Transport in Poly(Fluorene-Benzothiadiazole). Appl. Phys. Lett. 2011, 98 (14), 143504. 10.1063/1.3574907. [DOI] [Google Scholar]
- Zhang X.; Shen F.; Hu Z.; Wu Y.; Tang H.; Jia J.; Wang X.; Huang F.; Cao Y. Biomass Nanomicelles Assist Conjugated Polymers/Pt Cocatalysts To Achieve High Photocatalytic Hydrogen Evolution. ACS Sustainable Chem. Eng. 2019, 7, 4128. 10.1021/acssuschemeng.8b05637. [DOI] [Google Scholar]
- Al-Ibrahim M.; Roth H.-K.; Schroedner M.; Konkin A.; Zhokhavets U.; Gobsch G.; Scharff P.; Sensfuss S. The Influence of the Optoelectronic Properties of Poly(3-Alkylthiophenes) on the Device Parameters in Flexible Polymer Solar Cells. Org. Electron. 2005, 6 (2), 65–77. 10.1016/j.orgel.2005.02.004. [DOI] [Google Scholar]
- Haynes W. M.CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, 2016. [Google Scholar]
- Friederich P.; Meded V.; Poschlad A.; Neumann T.; Rodin V.; Stehr V.; Symalla F.; Danilov D.; Lüdemann G.; Fink R. F.; Kondov I.; von Wrochem F.; Wenzel W. Molecular Origin of the Charge Carrier Mobility in Small Molecule Organic Semiconductors. Adv. Funct. Mater. 2016, 26 (31), 5757–5763. 10.1002/adfm.201601807. [DOI] [Google Scholar]
- Friederich P.; Gómez V.; Sprau C.; Meded V.; Strunk T.; Jenne M.; Magri A.; Symalla F.; Colsmann A.; Ruben M.; Wenzel W. Rational In Silico Design of an Organic Semiconductor with Improved Electron Mobility. Adv. Mater. 2017, 29 (43), 1703505. 10.1002/adma.201703505. [DOI] [PubMed] [Google Scholar]
- Borsenberger P. M.; Bässler H. Concerning the Role of Dipolar Disorder on Charge Transport in Molecularly Doped Polymers. J. Chem. Phys. 1991, 95 (7), 5327–5331. 10.1063/1.461646. [DOI] [Google Scholar]
- Kreouzis T.; Bradley D. D. C.; Campbell A. J.. Hole and Electron Transport in Poly(9,9-Dioctylfluorene) and Poly (9,9-Dioctylfluorene- Co -Benzothiadiazole). In Organic Light-Emitting Materials and Devices VII; Kafafi Z. H., Lane P. A., Eds.; SPIE: 2004; Vol. 5214, p 141. 10.1117/12.506016. [DOI] [Google Scholar]
- Campbell A. J.; Bradley D. D. C.; Antoniadis H. Dispersive Electron Transport in an Electroluminescent Polyfluorene Copolymer Measured by the Current Integration Time-of-Flight Method. Appl. Phys. Lett. 2001, 79 (14), 2133–2135. 10.1063/1.1406143. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.