

Abstract
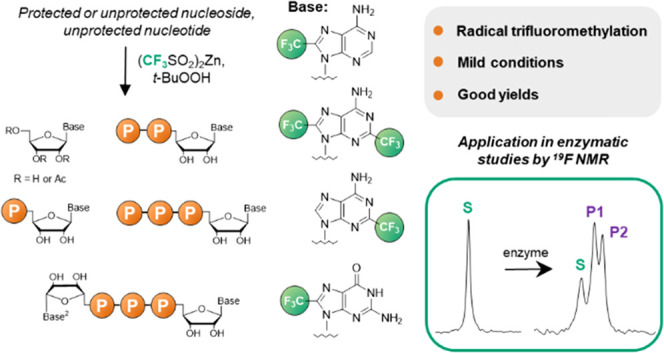
Protected guanosine and adenosine ribonucleosides and guanine nucleotides are readily functionalized with CF3 substituents within the nucleobase. Protected guanosine is trifluoromethylated at the C8 position under radical-generating conditions in up to 95% yield and guanosine 5′-oligophosphates in up to 35% yield. In the case of adenosine, the selectivity of trifluoromethylation depends heavily on the functional group protection strategy and leads to a set of CF3-modified nucleosides with different substitution patterns (C8, C2, or both) in up to 37% yield. Further transformations based on phosphorimidazolide chemistry afford various CF3-substituted mono- and dinucleoside oligophosphates in good yields. The utility of the trifluoromethylated nucleotides as probes for 19F NMR-based real-time enzymatic reaction monitoring is demonstrated with three different human nucleotide hydrolases (Fhit, DcpS, and cNIIIB). Substrate and product(s) resonances were sufficiently separated to enable effective tracking of each enzymatic activity of interest.
Introduction
Inserting a trifluoromethyl group (CF3) into an organic molecule is a common and effective approach for fine-tuning the properties of drug candidates1−3 and designing molecular probes for 19F nuclear magnetic resonance (NMR) experiments.4,5 The placement of the CF3 substituent in an organic compound can affect the neighboring functional groups and alter its overall polarity, acid–base properties, reactivity, and many other properties. Consequently, CF3 functionalization has been explored in medicinal chemistry as a strategy for modulating the biological activities of drug candidates by influencing metabolic stability, conformational equilibrium, lipophilicity, pharmacodynamics, and pharmacokinetics.1−3
The 19F nucleus has several properties that are beneficial for NMR spectroscopy, including spin of 1/2, one of the highest magnetic sensitivities (83% that of 1H), and high abundance (100%). Moreover, the wide chemical shift range (up to 400 ppm) and the absence of fluorine in natural compounds (low physiological content) simplify 19F NMR spectral analysis even for complex biomolecular mixtures. An additional advantage of using the CF3 group as a biomolecular NMR tag (instead of a single fluorine atom, for example) is the presence of 3 equivalent F atoms, which increases sensitivity. As such, fluorinated organic molecules and biopolymers have found application in NMR-based ligand screening assays such as fluorine chemical shift anisotropy and exchange for screening (FAXS)6,7 and fluorine atoms for biochemical screening (FABS),7,8 enzymatic assays, protein and nucleic acid structure studies, and others.9 Consequently, there is a high demand for synthetic transformations that assure the efficient and robust preparation of CF3-containing building blocks, preferably through late-stage, site-selective direct trifluoromethylation. Recent years have seen extensive developments in direct C–H trifluoromethylation methodologies, including electrophilic/nucleophilic reactions,10−14 photoredox-based reactions,15 metal-mediated reactions,16 and radical reactions with various “CF3” sources.11,17 Fluorinated nucleosides, nucleotides, and oligonucleotides18 are of particular interest as anticancer and antiviral compounds or probes for studying nucleic acid structures, interactions, and biological transformations.19−31 However, the synthesis of fluorinated nucleic acid components often poses complex challenges, especially in the case of purine derivatives.
Consequently, few methods that access trifluoromethylated purine nucleosides have been reported; hence, the properties of these compounds are underexplored. One of the early strategies involves the use of CF3-containing building blocks for the de novo synthesis of the six-membered purine ring.32 This approach has been applied by Langer33 and Pankiewicz34 for the preparation of CF3-substituted purine nucleosides as enzymatic inhibitors. The trifuoromethylation of halogenated purine ribosides was introduced by Kobayashi (Scheme 1)35 and requires the preparation of a CF3-copper complex as a CF3 source followed by reaction with an O-protected nucleoside bearing a halogen substituent at the C8-position. Although this synthesis is a multistep one, operationally demanding, and afforded moderate yields, its modified variants have become methods of choice for preparing purine ribosides trifluoromethylated at positions C8,36,37 C2,38−40 and C6.41
Scheme 1. Synthetic Approaches toward Trifluoromethylated Purines.

Montesarchio reported the direct trifluoromethylation of canonical nucleoside derivatives under mild conditions (Scheme 1)42 in moderate yields by taking advantage of CF3SO2Na as a CF3-radical precursor.43 The substrate scope included deoxyguanosine, deoxyadenosine, and inosine.
Baran reported (CF3SO2)2Zn as a versatile reagent for the C–H trifluoromethylation of heterocycles,44 including drugs bearing the purine motif.45 Inspired by this work, herein we aimed to develop (CF3SO2)2Zn-based protocols for the synthesis of protected and unprotected purine ribonucleosides and ribonucleotides. As a result, we report the synthesis of trifluoromethylated adenine and guanine nucleosides, nucleotides, and dinucleotides and evaluate their potential as 19F NMR probes for enzymatic reactions.
Results and Discussion
In a pilot experiment, we subjected guanosine (1, Table 1, entry 1) to the conditions described by Baran.44 The reaction was selective, although the isolated yield of 8-trifluoromethylguanosine (2) was low (5%), even at elevated temperatures (Table 1, entry 2). The effect of solvent mixtures (including the two-phase system) on the reaction was next evaluated. Application of the two-phase dichloromethane (DCM)/water system resulted in no product formation (Table 1, entry 3), while dimethyl sulfoxide (DMSO)/water and DMSO/10% AcOH (which was recently reported by Parish and Krska to be the optimal solvent for trifluoromethylation of complex compounds, such as peptides46) mixtures raised the yield to 20 and 25%, respectively (Table 1, entries 4 and 5). The highest conversion was achieved in 10% AcOH/DMSO with t-BuOOH added at 0 °C (Table 1, entry 6); 2 was isolated in 40% yield. While the reaction was selective for the expected product, the starting material was not fully consumed.
Table 1. Optimization of Trifluoromethylation of Guanosinea.

| entry | solvent | time [h] | temperature [°C] | yieldd [%] |
|---|---|---|---|---|
| 1 | DMSO | 72 | rt | 5 |
| 2 | DMSO | 24 | 60b | 7 |
| 3 | DCM/water, 1/1 | 72 | rt | 0 |
| 4 | DMSO/water, 1/1 | 72 | rt | 20 |
| 5 | DMSO/10% AcOH, 1/1 | 72 | rt | 25 |
| 6 | DMSO/10% AcOH, 1/1 | 72 | 0 to rtc | 40 |
General conditions: guanosine (0.2 mmol), solvent (2 mL), (CF3SO2)2Zn dihydrate (0.6 mmol), t-BuOOH (70% solution in water, 1 mmol).
The heating was started after addition of the whole t-BuOOH solution.
The solution of guanosine and (CF3SO2)2Zn was cooled to 0 °C prior to addition of the t-BuOOH solution. This temperature was maintained until whole t-BuOOH was added.
Isolated yield.
We next examined the application of similar conditions to the trifluoromethylation of guanosine 5′-oligophosphates (3–5, Table 2). As observed for guanosine, higher yields were obtained in AcOH/DMSO at low temperatures (Table 2, entries 1–4), and increasing the AcOH concentration resulted in a significant drop of yields (Table 2, entries 4–6). The optimized conditions afforded 8-trifluoromethyl-GMP (6) in 35% isolated yield (Table 2, entry 4), while GDP 4 and GTP 5 afforded the corresponding products 7 and 8 in 25 and 15% isolated yields, respectively (Table 2, entries 8 and 9). Prolonged reaction times led to the decomposition of products 7 and 8 through pyrophosphate bond hydrolysis. It is worth mentioning that the reaction outcome was insensitive toward the counterion of the nucleotide (Table 2, entry 4 vs entry 7). Surprisingly, adenosine, adenosine monophosphate, and diadenosine 5′,5′-triphosphate gave only traces of the trifluoromethylated compounds under the conditions examined.
Table 2. Optimization of Trifluoromethylation of Guanosine 5′-Oligophosphatesa.

| entry | substrate | solvent | temperature [°C] | product | yieldd [%] |
|---|---|---|---|---|---|
| 1 | 3 | DMSO | rt | 6 | traces |
| 2 | 3 | DMSO/water, 1/1 | rt | 6 | <5 |
| 3 | 3 | DMSO/10% AcOH, 1/1 | rt | 6 | 15 |
| 4 | 3 | DMSO/10% AcOH, 1/1 | 0 to rt | 6 | 35 |
| 5 | 3 | DMSO/20% AcOH, 1/1 | 0 to rt | 6 | 20 |
| 6 | 3 | DMSO/30% AcOH, 1/1 | 0 to rt | 6 | 10 |
| 7b | 3 | DMSO/10% AcOH, 1/1 | 0 to rt | 6 | 35 |
| 8c | 4 | DMSO/10% AcOH, 1/1 | 0 to rt | 7 | 25 |
| 9c | 5 | DMSO/10% AcOH, 1/1 | 0 to rt | 8 | 15 |
General conditions: triethylammonium salt of the respective nucleotide (0.1 mmol), solvent (1 mL), (CF3SO2)2Zn dihydrate (0.3 mmol), and t-BuOOH (70% solution in water, 0.5 mmol).
Sodium salt of 3 was used.
Reaction was stopped after 24 h due to the detection of decomposition products in the reaction mixture (high-performance liquid chromatography (HPLC) analysis).
Isolated yield.
Although the conditions developed for guanosine and its derivatives appear acceptable, incomplete conversion led to tedious product isolation. Moreover, the adenosine problem remained unsolved. To improve conversions and to simplify isolation procedures, we examined the reactions of protected nucleosides, since this strategy has been reported to be effective for purine 2′-deoxynucleosides.42
2′,3′,5′-Tri-O-acetylguanosine (9, Scheme 2) turned out to be an excellent starting material for this transformation, giving 10 in 95% isolated yield. The product was easily isolated by extraction followed by simple silica gel chromatography to remove trace impurities. Treatment of 10 with a MeNH2/EtOH solution afforded pure 2 in nearly quantitative yield by solvent evaporation; 2 was then transformed into monophosphate 6 under Yoshikawa conditions47 in the presence of lutidine (to avoid depurination). This reaction provided 6 in 90% yield after ion-exchange chromatography. Since all reactions in this sequence were selective and high yielding, we decided to repeat it without isolating the intermediates by chromatography, which resulted in 85% overall yield of 6 from 9 over three steps (Scheme 2), which is practical from a preparative perspective.
Scheme 2. Synthesis of 8-Trifluoromethylguanosine and Subsequent Transformations.

Monophosphate 6 was converted into its P-imidazolide derivative 6-Im by reacting it with imidazole in the presence of 2,2′-dithiodipyridine/triphenylphosphine (Scheme 2);486-Im decomposed upon storage, but when used directly after preparation, it afforded good yields in further transformations. For example, when 6-Im was reacted with triethylammonium phosphate or pyrophosphate in the presence of ZnCl2, diphosphate 7 or triphosphate 8 was produced in satisfactory yields, respectively (Scheme 2).
Protecting the OH groups of guanosine significantly improved the yield and simplified product isolation; consequently, we applied the same approach for adenosine. Indeed, when 2′,3′,5′-tri-O-acetyladenosine (11, Scheme 3) was subjected to similar conditions, the 8-trifluoromethylated product 12 was obtained in 37% yield. Further manipulation of the reaction conditions did not improve this result.
Scheme 3. Synthesis of Trifluoromethyladenosine 5′-Monophosphates.

A complex mixture of products was obtained when N6,2′,3′,5′-tri-O-tetraacetyladenosine 14 was subjected to similar conditions (Scheme 3). Chromatographic separation and analysis revealed the presence of three trifluoromethylated products: the C8- and C2-trifluoromethyladenosine derivatives 15 (9%) and 16 (10%) and the C2,C8-disubstituted adenosine derivative 17 (7%). To the best of our knowledge, 17 is the first reported example of an adenosine derivative bearing two trifluoromethyl substituents on the purine ring. Yields were not improved nor was the product distribution significantly altered by changing the reaction conditions (temperature, solvent). Treating compounds 12 and 15–17 with 33% MeNH2/EtOH afforded nucleosides 13, 18, and 19, which were phosphorylated to their respective monophosphates 20–22 in good yields (Scheme 3).
Monophosphate 20 was converted into its P-imidazolide derivative 20-Im (Scheme 4), which enabled efficient elongation of the phosphate chain to give 8-CF3 adenosine diphosphate 23 and triphosphate 24 in good yields.
Scheme 4. Synthesis of 8-Trifloromethyladenosine Oligophosphates.

Finally, we prepared a series of trifluoromethylated dinucleoside 5′,5′-triphosphates using some of the synthesized mononucleotides (Scheme 5). This was achieved by coupling together two nucleotide building blocks, one of which is activated as a phosphorimidazolide, in the presence of excess ZnCl2 to form a new pyrophosphate bond. The reaction of imidazolide 20-Im with diphosphate 23 led to a complex reaction mixture containing the desired dinucleotide 25, the monophosphate 20 (from the hydrolysis of 20-Im), and the coupling product of 20-Im and 20. Nevertheless, we isolated the desired dinucleotide 25 in 20% yield. The reaction of 20-Im with a subequimolar amount of triethylammonium phosphate proved to be more efficient; the in situ-formed diphosphate 23 readily reacted with excess 20-Im present in the reaction mixture to yield 25 in 63%. The reaction of imidazole-activated adenosine monophosphate ADP-Im with monophosphate 6 led to dinucleotide 26 in 40% yield, which could not be improved by reacting 6-Im with adenosine diphosphate. On the other hand, 6 reacted smoothly with GDP-Im to give dinucleotide 27 composed of two guanosines, one of which bears an 8-CF3 group. The presence of the electron-withdrawing 8-CF3 group in the guanosine structure dramatically decreased the nucleophilicity of the neighboring (N7) nitrogen. The drop in reactivity was evident when 27 was treated with iodomethane, affording exclusively the mRNA 5′ cap analogue 28, which was site-selectively methylated at the guanosine rather than 8-CF3-guanosine.
Scheme 5. Synthesis of Trifluoromethylated Dinucleotides.

To evaluate the usefulness of trifluoromethylated purine nucleotides as probes for enzymatic activity monitoring by 19F NMR, we subjected select compounds to three nucleotide-specific phosphohydrolases: human fragile histidine triad (hFhit), human decapping scavenger (hDcpS), and human cytosolic nucleotidase IIIB (hcNIIIB). These enzymes are of interest due to their function in regulation of the endogenous nucleotide metabolism and links to disease development, and as such, assays have been developed that allow monitoring their activity and the discovery of inhibitors. These include assays involving radioactivity,49,50 fluorogenic probes,31 malachite green (MG) assay,51 (2) and fluorescent resonance energy transfer (FRET) probes.52,53 (3) However, these assays have some limitations such as discontinuity, susceptibility to interference from UV–vis absorbing and emitting inhibitors, or high structural complexity of the probes. Thus, methods that enable robust and straightforward real-time monitoring of activity of these enzymes are still desired.
We first used compounds 25–27 to monitor the activity of the human fragile histidine triad (hFhit) pyrophosphatase, which unsymmetrically cleaves diadenosine 5′,5′-triphosphate (Ap3A) and other purine dinucleotides.54 hFhit is considered to be a tumor suppressor, and its function has been linked to substrate binding. Hence, efforts have been made to identify hFhit inhibitors52,53 but not by 19F NMR spectroscopy. We verified that hFhit accepts the bis(trifluoromethylated) Ap3A analog 25 as a substrate. Compound 25 exhibits a single narrow signal at −61.38 ppm in its 19F NMR spectrum, which corresponds to 6 equivalent fluorine atoms (Figure 1A). A decrease in intensity of this resonance was observed after adding 20 nM hFhit to a 100 μM buffered solution of 25, and two new slightly upfield-shifted signals appeared, consistent with substrate hydrolysis to give 20 (8-CF3-AMP, δF −61.39) and 23 (8-CF3-ADP, δF −61.40; Figure S1A). The signals of 20 and 23 were assigned on the basis of the 19F NMR spectra of synthetic references (Figure S1B-D). Despite the small differences in chemical shifts from the substrate, it was convenient to monitor the reaction progress by 19F NMR and observe the inhibitory effect of a previously identified compound53 (Figure 1A). Similar experiments were performed for unsymmetrical 8-trifluoromethylguanosine-containing dinucleotides 26 and 27 (100 μM each in the presence of 20 or 25 nM hFhit, respectively; Figure S2). 19F NMR spectroscopy revealed the formation of two trifluoromethylated products from each compound, namely, 8-CF3-GDP (7; δF −61.28) and 8-CF3-GMP (6; δF −61.27), albeit in different proportions (Figures S3). Under these comparable conditions, 25 and 26 are preferred substrates of hFhit, rather than 27, consistent with the known preference of the enzyme for adenine-containing dinucleotides.
Figure 1.

Monitoring enzymatic activity by 19F NMR spectroscopy. (A) 25 with hFhit, (B) 28 with hDcpS, and (C) 6 with hcNIIIB. For experimental details, see the Supporting Information.
We next investigated the decapping scavenger (hDcpS) and cytosolic nucleotidase IIIB (hcNIIIB), two enzymes involved in the cellular metabolism of N7-methylguanine nucleotides. hDcpS degrades cap moieties (m7GpppNn) released during 3′-to-5′ mRNA degradation55 and has been identified as a therapeutic target for spinal muscular atrophy and acute myeloid leukemia, which created demand for inhibitors.49,50,56,57 To verify that trifluoromethylated nucleotides can be used to study hDcpS activity, mRNA cap analog 28 (100 μM) was incubated with 80 nM enzyme in the absence and presence of RG3039, a potent inhibitor. 19F NMR analysis revealed that the substrate (δF −61.22) is site-selectively cleaved to release 7 (8-CF3-GDP; δF −61.25), consistent with the high specificity of DcpS for 7-methylguanosine, which controls the regioselectivity; inhibition by RG3039 was also clearly visible (Figure 1B).
hcNIIIB dephosphorylates m7GMP to 7-methylguanosine,58 and its inhibitors are potential modulators of mononucleotide metabolism and downstream RNA degradation pathways.51 hcNIIIB also hydrolyses electron-poor pyrimidine nucleotides, whereas GMP and AMP are very poor substrates.59 Since trifluoromethylation decreases the electron density in the purine, we tested 8-CF3-GMP (6) as an artificial substrate for hcNIIIB. Indeed, the substrate peak (δF −61.27) was observed to disappear when 6 at 100 μM was incubated with 120 nM hcNIIIB, and a product signal (δF −61.17) emerged, which was independently confirmed to be 8-trifluoromethylguanosine (2). The reaction was almost completely stopped by the hcNIIIB-specific inhibitor (Figure 1C).
Conclusions
In summary, we optimized the conditions for the synthesis of trifluoromethylated purine nucleotides and nucleosides using (CF3SO2)2Zn as a source of CF3 radicals. The synthesized compounds include trifluoromethylguanosine and trifluormethyladenosine, their 5′-mono, di-, and triphosphates, as well as several dinucleoside 5′,5′-triphosphates. The synthetized trifluoromethylated (di)nucleotides were successfully used as molecular probes to monitor the activities of three enzymes (hFhit and hDcpS pyrophosphatases and hcNIIIB phosphatase) by 19F NMR spectroscopy. The introduction of CF3 moieties into the purines in dinucleotide analogs does not prevent specific recognition by either hFhit or hDcpS. Interestingly, 8-CF3-GMP (6) acted as an m7GMP mimic, as manifested by its efficient dephosphorylation by cNIIIB. Substrate and product(s) resonances were sufficiently separated to enable effective monitoring of the enzymatic activity of interest, which opens possibilities for the development of 19F NMR-based inhibitor-discovery and evaluation assays. We envisage that the higher synthetic availability of trifluoromethylated guanine- and adenine-derived building blocks offered by our work will also pave the way for their use in more-complex biomolecular systems, such as oligonucleotides and nucleic acids, thereby facilitating studies on nucleic acid structure and function.
Experimental Section
General Information
All commercial reagents and solvents were used as received without additional purification. Guanosine and adenosine were purchased from Carbosynth. (CF3SO2)2Zn dihydrate and t-BuOOH (70% in water) were purchased from TCI. Anhydrous solvents were purchased from Sigma-Aldrich. Thin-layer chromatography (TLC) analysis was carried out on precoated Silica Gel 60 Å on aluminum foil with a fluorescence indicator (Sigma-Aldrich) and visualized under a UV lamp (254 nm).
Preparative Chromatography
Preparative chromatography (SiO2 and RP C18) was performed using a Reveleris X2 flash chromatography system (BUCHI) with FlashPure cartridges (4, 12, 24, 40 g). Conditioning methods, loading, and flow rates were set according to manufacturers’ guidelines. UV detection was performed at three wavelengths (254, 265, and 280 nm) simultaneously.
Ion-Exchange Chromatography
The synthesized nucleotides were purified by ion-exchange chromatography on a DEAE Sephadex A-25 (HCO3– form) column. After loading the column with the reaction mixture and washing it with water, the products were eluted using different linear gradients of triethylammonium bicarbonate (TEAB) in deionized water: 0–0.7 M for nucleoside monophosphates, 0–1.0 M for nucleoside diphosphates and dinucleotides, or 0–1.2 M for nucleoside triphosphates. Fractions containing the desired product were collected together after reversed-phase (RP) HPLC and spectrophotometric (at 260 nm) analysis. Evaporation under reduced pressure with repeated additions of 96% and then 99.8% ethanol resulted in isolation of nucleotide analogues as triethylammonium salts.
Analytical and Preparative HPLC
Analytical HPLC was performed on Agilent Tech. Series 1200 using a Supelcosil LC-18-T HPLC column (4.6 × 250 mm, flow rate 1.3 mL/min) with linear gradients of methanol in 0.05 M ammonium acetate buffer and UV detection at 254 nm. Analytical HPLC programs are included in the Supporting Information. Semipreparative HPLC was performed on the same apparatus equipped with a Discovery RP Amide C-16 HPLC column (25 cm × 21.2 mm, 5 μm, flow rate 5.0 mL/min) with linear gradients of MeCN in 0.05 M ammonium acetate buffer (pH 5.9) and UV detection at 260 nm.
Spectroscopic Analysis of the Synthesized Compounds
The structure and purity of the products were confirmed by high-resolution mass spectrometry using electrospray ionization (HRMS ESI) and NMR spectroscopy. Purity of water-soluble compounds was additionally confirmed by RP HPLC. Mass spectra were recorded on Thermo Scientific LTQ OrbitrapVelos (high-resolution spectra) and AB Sciex API 3200 (low-resolution spectra) spectrometers. NMR spectra were recorded on a Varian INOVA 400 or 500 MHz spectrometer equipped with a high-stability temperature unit using a 5 mm 4NUC probe at 25 °C. The chemical shifts were reported in ppm with the residual solvent peak as the internal standard. The 31P NMR chemical shifts were reported in ppm and referenced to 20% phosphoric acid in D2O as an external standard. The 19F chemical shifts were reported in ppm and referenced to CFCl3 (for spectra recorded in CDCl3 and DMSO-d6, 0.65 and −0.24 ppm, respectively) or NaF (for spectra recorded in D2O, −121.50 ppm) as an external standard. Signal assignments of compounds 2, 13, 16, and 17 were based on correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC), and heteronuclear multiple bond correlation (HMBC) spectra analysis.
Optimization of Trifluoromethylation of Guanosine: General Procedure
Guanosine (56 mg, 0.2 mmol) was dissolved/suspended in the solvent/solvent mixture (2 mL) followed by addition of (CF3SO2)2Zn dihydrate (219 mg, 0.6 mmol). To this mixture, t-BuOOH (70% solution in water, 130 μL, 1 mmol) in 10 aliquots (13 μL each) in 20 min intervals was added upon vigorous stirring. The reaction mixture was stirred at room temperature. The progress of the reaction was monitored by TLC analysis (10% MeOH in DCM) until no further progress could be detected. After the indicated time, the reaction mixture was diluted with water (approx. 20 mL) and extracted with DCM (3 × 10 mL). The combined organic fractions were washed with water (approx. 20 mL) and brine (approx. 20 mL) and dried over Na2SO4. The drying agent was filtered off and washed with DCM, and the filtrate was concentrated in vacuo. To the residue, 10 mL of MeOH and 0.5 g of silica were added. The slurry was concentrated in vacuo, and the dry residue was loaded on the preconditioned silica gel column (4 g). The product was eluted with the mixture of MeOH in DCM (0–10% linear gradient). The fractions containing the desired product were combined, concentrated in vacuo, coevaporated with diethyl ether (approx. 5 mL), and dried overnight under high vacuum, giving 2 as an off-white solid. Isolated yields and additional information are summarized in Table 1. 1H NMR (400 MHz, DMSO-d6) δ = 11.00 (bs, 1H, NH), 6.71 (bs, 2H, −NH2), 5.63 (d, J = 6.2 Hz, 1H, C1′), 5.49 (d, J = 6.22 Hz, 1H, C2′–OH), 5.11 (d, J = 4.9 Hz, 1H, C3′–OH), 5.05 (d, J = 5.9 Hz, 11.7 Hz, 1H, C2′), 4.91 (t, J = 6.0 Hz, 1H, C5′–OH), 4.16 (m, 1H, C3′), 3.89 (m, 1H, C4′), 3.71–3.63 (m, 1H, C5′), 3.57–3.49 (m, 1H, C5′); 13C {H} NMR (100 MHz, DMSO-d6) δ = 156.5 (C6), 154.4 (C2), 152.5 (C4), 133.3 (q, J = 39.0 Hz, C8), 118.6 (q, J = 269.9 Hz, CF3), 116.5 (C5), 89.4 (C1′), 86.4 (C4′), 70.7 (C2′), 70.6 (C3′), 61.9 (C5′); 19F NMR (376 MHz, DMSO-d6) δ =–59.87; HRMS (+) ESI m/z: [M + H]+ calcd for C11H13F3N5O5+ 352.0863; found 352.0861.
Optimization of Trifluoromethylation of Guanosine Phosphates: General Procedure
A triethylammonium salt of respective nucleotide (0.1 mmol) was dissolved in the indicated solvent (1 mL) followed by addition of (CF3SO2)2Zn dihydrate (110 mg, 0.3 mmol). To this mixture, t-BuOOH (70% solution in water, 65 μL, 0.5 mmol) in 10 aliquots (6.5 μL each) in 20 min intervals was added under vigorous stirring. The progress of the reaction was monitored by RP HPLC analysis until no further progress could be detected (usually 72 h) or decomposition of the starting material/products was detected. The reaction mixture was diluted with ethylenediamine tetraacetic acid (EDTA) solution (100 mg in 10 mL of water) and neutralized with 10% NaHCO3. The resulting mixture was loaded on a DEAE Sephadex A-25 column (HCO3– form, 10 g); the column was washed with water (50 mL) and then eluted using TEAB in deionized water (400 mL, linear gradient). The fractions containing the mixture of the desired product and starting material (UV and RP HPLC analyses) were combined, concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL), and the residue was dried overnight under high vacuum. The product was separated from the starting material and possible byproducts using preparative RP HPLC chromatography with a linear gradient of MeCN in 0.05 M ammonium acetate buffer (pH = 5.9). The fractions containing the pure product were concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL). The residue was dissolved in MQ water and freeze-dried. After repeated freeze-drying, the product was isolated as ammonium salt. Isolated yields and additional information are summarized in Table 2.
8-Trifluoromethylguanosine 5′-Monophosphate (6)
White foam. 1H NMR (400 MHz, D2O) δ = 5.89 (d, J = 6.1 Hz, 1H), 5.33 (t, J = 5.8 Hz, 1H), 4.59 (dd, J = 3.6 Hz, 5.7 Hz, 1H), 4.31–4.23 (m, 1H), 4.22–4.09 (m, 1H); 13C {H} NMR (125 MHz, D2O) δ = 161.0, 156.7, 155.5, 138 (q, J = 40.1 Hz), 120.6 (q, J = 270.3 Hz), 118.1, 91.9, 86.7, 73.7, 72.8, 67.20; 19F NMR (376 MHz, D2O) δ = −61.23; 31P NMR (162 MHz, D2O) δ = 1.75; HRMS (−) ESI m/z: [M – H]− calcd for C11H12F3N5O8P- 430.0381; found 430.0380.
8-Trifluoromethylguanosine 5′-Diphosphate (7)
White foam. 1H NMR (400 MHz, D2O) δ = 5.90 (d, J = 6.2 Hz, 1H), 5.38 (t, J = 5.9 Hz, 1H), 4.67 (dd, J = 3.1 Hz, 5.5 Hz, 1H), 4.35–4.27 (m, 2H), 4.26–4.17 (m, 1H); 19F NMR (376 MHz, D2O) δ −61.21; 31P NMR (162 MHz, D2O) δ = −9.85 (d, J = 20.7 Hz, 1P), −10.29 (dt, J = 5.9 Hz, 20.7 Hz, 1P); HRMS (−) ESI m/z: [M – H]− calcd for C11H13F3N5O11P2– 510.0044; found 510.0041.
8-Trifluoromethylguanosine 5′-Triphosphate (8)
White foam. 1H NMR (400 MHz, D2O) δ = 5.90 (d, J = 6.2 Hz, 1H), 5.39 (t, J = 5.9 Hz, 1H), 4.69 (dd, J = 2.3 Hz, 5.3 Hz, 1H), 4.38–4.22 (m, 3H); 19F NMR (376 MHz, D2O) δ = −61.20; 31P NMR (162 MHz, D2O) δ = −9.82 (d, J = 19.3 Hz, 1P), −10.35 (d, J = 19.7 Hz, 1P), −22.10 (t, J = 19.5 Hz, 1P); HRMS (−) ESI m/z: [M – H]− calcd for C11H14F3N5O14P3– 589.9708; found 589.9708.
2′,3′,5′-Tri-O-acetyl-8-trifluoromethylguanosine (10)
2′,3′,5′-Tri-O-acetylguanosine (9, 818 mg, 2 mmol) was dissolved in DMSO (12 mL) followed by addition of (CF3SO2)2Zn dihydrate (2200 mg, 6 mmol) upon vigorous stirring at room temperature. When the clear solution was formed (15–20 min), t-BuOOH (70% solution in water, 1.3 mL, 10 mmol) was added in 10 aliquots (130 μL each) in 20 min intervals. During addition of t-BuOOH, the reaction mixture started to become yellow. The reaction mixture was stirred for 24 h, after which TLC analysis (3% MeOH in DCM) indicated full consumption of the starting material. The reaction mixture was poured into 200 mL of water and extracted with DCM (3 × 50 mL). The combined organic layers were washed with water (3 x approx. 100 mL) and brine (approx. 100 mL) and dried over Na2SO4. The drying agent was filtered off and washed with DCM, and the filtrate was concentrated in vacuo. The oily residue was dissolved in a small amount of DCM and loaded on the preconditioned silica gel column (12 g). The product was eluted with the mixture of MeOH in DCM (5%, v/v). The fractions containing the product were combined, concentrated in vacuo, coevaporated with diethyl ether (approx. 10 mL), and dried overnight under high vacuum, giving 10 as an off-white solid (906 mg, 95%). 1H NMR (400 MHz, CDCl3) δ = 6.36 (bs, 1H), 5.95 (d, J = 4.6 Hz, 1H), 5.94–5.89 (m, 1H), 4.56–4.50 (m, 1H), 4.47–4.36 (m, 2H), 2.15 (s, 3H), 2.11 (s, 3H), 2.04 (s, 3H); 19F NMR (376 MHz, DMSO-d6) δ = −61.19; HRMS (+) ESI m/z: [M + H]+ calcd for C17H19F3N5O8+ 478.1180; found 478.1187.
Deprotection of 10
10 (906 mg, 1.9 mmol) was placed in a 50 mL round-bottom flask equipped with a rubber septum and flushed with a stream of argon. Then, MeNH2 (33% in EtOH, 10 mL) was added under a gentle flow of argon, and the resulting mixture was stirred for 4 h at room temperature after which TLC analysis (5% MeOH in DCM) indicated full consumption of the starting material. The reaction mixture was concentrated in vacuo. To the residue, approx. 20 mL of MeOH and 2 g of silica were added. The slurry was concentrated in vacuo, and the dry residue was loaded on the preconditioned silica gel column (10 g). The product was eluted with the mixture of MeOH in DCM (10%, v/v). The fractions containing the desired product were combined, concentrated in vacuo, coevaporated with diethyl ether (approx. 10 mL), and dried overnight under high vacuum, giving 2 as a yellowish solid (634 mg, 95%). The analytical data matched those obtained for 2 synthesized by direct trifluoromethylation of guanosine.
8-Trifluoromethylguanosine 5′-Monophosphate (6)
8-Trifluoromethylguanosine (2, 351 mg, 1 mmol) was dissolved in anhydrous (MeO)3PO (10 mL) under a gentle flow of argon. The resulting solution was cooled below 0 °C (ice/brine bath), followed by addition of 2,6-lutidine (350 μL, 3 mmol) and dropwise addition of freshly distilled POCl3 (280 μL, 3 mmol). During the reaction, a white precipitate was formed. The reaction mixture was stirred below 0 °C for 4 h, after which RP HPLC analysis indicated full consumption of the starting material. The reaction mixture was poured into cold, deionized water (approx. 100 mL) and neutralized with 10% NaHCO3. The resulting mixture was loaded on a DEAE Sephadex A-25 column (HCO3– form, 100 g); the column was washed thoroughly with water and then eluted using TEAB in deionized water (3600 mL, 0–0.7 M linear gradient). The fractions containing the pure product (UV and RP HPLC analyses) were combined, concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL). The residue was dissolved in MQ water, loaded on an RP C18 column (20 g), and eluted with 20% MeCN in 0.05 M ammonium acetate buffer (pH 5.9). The fractions containing the desired product were combined, concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN. The residue was dissolved in MQ water and freeze-dried. After repeated freeze-drying, the product was isolated as ammonium salt (419 mg, 90%). The analytical data matched those obtained for 6 synthesized by direct trifluoromethylation of guanosine monophosphate.
2′,3′,5′-Tri-O-acetyl-8-trifluoromethyl-adenosine (12)
2′,3′,5′-Tri-O-acetyladenosine (11, 393 mg, 1 mmol) was dissolved in DMSO (6 mL) followed by addition of (CF3SO2)2Zn dihydrate (1100 mg, 3 mmol) upon vigorous stirring at room temperature. When the clear solution was formed (15–20 min), t-BuOOH (70% solution in water, 0.65 mL, 5 mmol) was added in 10 aliquots (65 μL each) in 20 min intervals. During addition of t-BuOOH, the reaction mixture started to become yellow. The reaction mixture was stirred for 24 h, after which TLC analysis (3% MeOH in DCM) indicated no further progress of the reaction. The reaction mixture was poured into 100 mL of water and extracted with DCM (3 × 30 mL). The combined organic layers were washed with water (3 x approx. 100 mL) and brine (approx. 100 mL) and dried over Na2SO4. The drying agent was filtered off and washed with DCM, and the filtrate was concentrated in vacuo. To the oily residue, approx. 20 mL of DCM and 2 g of silica were added. The slurry was concentrated in vacuo, and the dry residue was loaded on the preconditioned silica gel column (24 g). The product was eluted with the mixture of MeOH in DCM (0–5% linear gradient). The fractions containing the product were combined, concentrated in vacuo, coevaporated with diethyl ether (approx. 10 mL), and dried overnight under high vacuum, giving 12 as an off-white solid (171 mg, 37%). 1H NMR (400 MHz, CDCl3) δ = 8.42 (s, 1H), 6.41 (dd, J = 4.7 Hz, 6.1 Hz, 1H), 6.09 (d, J = 4.7 Hz, 1H), 5.90 (m, 3H), 4.55 (m, 1H), 4.44–4.36 (m, 2H), 2.16 (s, 3H), 2.09 (s, 3H), 2.06 (s, 3H); 19F NMR (376 MHz, CDCl3) δ = −61.39; HRMS (+) ESI m/z: [M + H]+ calcd for C17H19F3N5O7+ 462.1231; found 462.1225.
Trifluoromethylation of N6,2′,3′,5′-Tri-O-tetraacetyladenosine
N6,2′,3′,5′-Tri-O-tetraacetyladenosine (14, 870 mg, 2 mmol) was dissolved in DMSO (12 mL) followed by addition of (CF3SO2)2Zn dihydrate (2200 mg, 6 mmol) upon vigorous stirring at room temperature. When the clear solution was formed (15–20 min), t-BuOOH (70% solution in water, 1.3 mL, 10 mmol) was added in 10 aliquots (135 μL each) in 20 min intervals. During addition of t-BuOOH, the reaction mixture started to become yellow. The reaction mixture was stirred for 24 h, after which TLC analysis (3% MeOH in DCM) indicated no further progress of the reaction. The reaction mixture was poured into 100 mL of water and extracted with DCM (3 × 30 mL). The combined organic layers were washed with water (3 x approx. 100 mL) and brine (approx. 100 mL) and dried over Na2SO4. The drying agent was filtered off and washed with DCM, and the filtrate was concentrated in vacuo. To the oily residue, approx. 20 mL of DCM and 2 g of silica were added. The slurry was concentrated in vacuo, and the dry residue was loaded on the preconditioned silica gel column (40 g). The column was eluted with the mixture of MeOH in DCM by stepwise changing of the MeOH concentration (0–3%). The products and the remaining starting material were washed from the column in the following order: 17, 15, 16, 14 (see Figure S4). The fractions containing each product were combined, concentrated in vacuo, coevaporated with diethyl ether (approx. 10 mL), and dried overnight under high vacuum.
N6,2′,3′,5′-Tri-O-tetraacetyl-8-trifluoromethyl-adenosine (15)
White solid (91 mg, 9%). 1H NMR (400 MHz, CDCl3) δ = 8.78 (s, 1H), 8.73 (bs, 1H), 6.40 (dd, J = 4.9 Hz, 6.1 Hz, 1H), 6.11 (d, J = 4.9 Hz, 1H), 5.86 (dd, J = 5.2 Hz, 6.1 Hz, 1H), 4.45 (dd, J = 3.5 Hz, 11.4 Hz, 1H), 4.47–4.36 (m 2H), 2.65 (s, 3H), 2.18 (s, 3H), 2.08 (s, 3H), 2.06 (s, 3H); 13C {H} NMR (100 MHz, CDCl3) δ = 170.6, 170.4, 169.6, 169.4, 154.6, 151.7, 151.0, 140.7 (q, J = 40.7 Hz) 120.2, 118.2 (q, J = 272.4 Hz), 88.1 (q, J = 2.4 Hz), 80.9, 72.0, 70.5, 62.86, 26.0, 20.8, 20.6, 20.4; 19F NMR (376 MHz, CDCl3) δ = −61.63; HRMS (+) ESI m/z: [M + H]+ calcd for C19H21F3N5O8+ 504.1337; found 504.1336.
N6,2′,3′,5′-Tri-O-tetraacetyl-2-trifluoromethyl-adenosine (16)
White solid (101 mg, 10%). 1H NMR (400 MHz, CDCl3) δ = 9.50 (bs, 1H), 8.48 (s, 1H), 6.26 (d, J = 5.3 Hz, 1H), 5.87 (dd, J = 5.5 Hz, 5.9 Hz, 1H), 5.68 (dd, J = 4.5 Hz, 5.6 Hz, 1H), 4.53–4.47 (m, 1H), 4.46–4.38 (m, 2H), 2.76 (s, 3H), 2.19 (s, 3H), 2.11 (s, 3H), 2.09 (s, 3H); 13C {H} NMR (100 MHz, CDCl3) δ = 171.9, 170.5, 169.7, 169.6, 150.9, 150.01 (q, J = 37.4 Hz), 149.9, 144.0, 122.7, 119.7 (q, J = 275.1 Hz), 87.1, 80.9, 73.6, 70.8, 63.2, 26.4, 20.8, 20.7, 20.4; 19F NMR (376 MHz, CDCl3) δ = −69.37; HRMS (+) ESI m/z: [M + H]+ calcd for C19H21F3N5O8+ 504.1337; found 504.1333.
N6,2′,3′,5′-Tri-O-tetraacetyl-2,8-ditrifluoromethyl-adenosine (17)
Yellowish solid (80 mg, 7%). 1H NMR (400 MHz, CDCl3) δ = 8.55 (bs, 1H), 6.21 (dd, J = 4.6 Hz, 6.2 Hz, 1H), 6.11 (d, J = 4.6 Hz, 1H), 5.83 (dd, J = 5.8 Hz, 6.2 Hz, 1H), 4.55 (dd, J = 4.0 Hz, 11.8 Hz, 1H), 4.48–4.42 (m, 1H), 4.41–4.35 (m, 1H), 2.77 (s, 3H), 2.18 (s, 3H), 2.08 (s, 3H), 2.04 (s, 3H); 19F NMR (376 MHz, CDCl3) δ = −61.79, −69. 71; HRMS (+) ESI m/z: [M + H]+ calcd for C20H20F6N5O8+ 572.1211; found 572.1200.
14 was recovered in 33% yield.
General Procedure for Deprotection of 12, 15, 16, and 17 and the Synthesis of 13, 18, and 19
Protected nucleoside was placed in a round-bottom flask equipped with a rubber septum and flushed with a stream of argon. Then, MeNH2 (33% in EtOH, 100 μL per each 10 mg of the starting material) was added under a gentle flow of argon, and the resulting mixture was stirred for 4 h at room temperature, after which TLC analysis (5% MeOH in DCM) indicated full consumption of the starting material. The reaction mixture was concentrated in vacuo. To the residue, a small amount of MeOH and silica were added. The slurry was concentrated in vacuo, and the dry residue was loaded on the preconditioned silica gel column. The product was eluted with the mixture of MeOH in DCM (5%, v/v). The fractions containing the desired product were combined, concentrated in vacuo, coevaporated with diethyl ether (approx. 10 mL), and dried overnight under high vacuum.
8-Trifluoromethyl-adenosine (13)
White solid (64 mg, starting from 92 mg (0.20 mmol) of 12, 95%). 1H NMR (400 MHz, DMSO-d6) δ = 8.26 (s, 1H, C2), 8.12–7.79 (m, 2H, −NH2), 5.79 (d, J = 6.8 Hz, 1H, C1′), 5.54 (dd, J = 3.8 Hz, 8.8 Hz, 1H, C5′–OH), 5.50 (d, J = 6.2 Hz, 1H, C2′–OH), 5.27 (d, J = 4.4 Hz, 1H, C3′–OH), 5.11 (m, 1H, C2′), 4.22 (m, 2H, C3′), 4.02 (m, 1H, C4′), 3.75–3.67 (m, 1H, C5′), 3.60–3.51 (m, 1H, C5′); 13C {H} NMR (100 MHz, DMSO-d6) δ = 157.33 (C6), 154.6 (C2), 150.0 (C4), 136.8 (q, J = 39.1 Hz, C8), 118.7 (q, J = 271.4 Hz, CF3), 118.0 (C5), 89.7 (C1′), 87.2 (C4′), 71.5 (C2′), 70.9 (C3′), 62.0 (C5′); 19F NMR (376 MHz, DMSO-d6) δ = −59.95; HRMS (+) ESI m/z: [M + H]+ calcd for C11H13F3N5O4+ 336.0914; found 336.0911.
2-Trifluoromethyl-adenosine (18)
White solid (32 mg, starting from 50 mg (0.10 mmol) of 16, 95%). 1H NMR (400 MHz, DMSO-d6) δ = 8.57 (s, 1H, C8), 7.97 (bs, 1H, −NH2), 5.91 (d, J = 6.1 Hz, 1H, C1′), 5.49 (d, J = 6.2 Hz, 1H, C2′–OH), 4.92 (d, J = 4.9 Hz, 1H, C3′–OH), 4.99 (dd, J = 5.5 Hz, 5.7 Hz, 1H, C5′–OH), 4.60 (m, 1H, C2′), 4.15 (m, 1H, C3′), 3.95 (m, 1H, C4′), 3.71–3.62 (m, 1H, C5′), 3.59–3.51 (m, 1H, C5′); 13C {H} NMR (100 MHz, DMSO-d6) δ = 156.3 (C6), 149.2 (q, J = 34.7 Hz, C2), 148.9 (C4), 141.7 (C8), 120.0 (q, J = 275.0 Hz, CF3), 119.85 (C5), 87.3 (C1′), 85.9 (C4′), 73.6 (C2′), 70.5 (C3′), 61.4 (C5′); 19F NMR (376 MHz, DMSO-d6) δ = −68.03 ppm. HRMS (+) ESI m/z: [M + H]+ calcd for C11H13F3N5O4+ 336.0914; found 336.0910.
2,8-Ditrifluoromethyl-adenosine (19)
Off-white solid (38 mg, starting from 57 mg (0.10 mmol) of 17, 95%). 1H NMR (400 MHz, DMSO-d6) δ = 8.64–8.38 (m, 2H, −NH2), 5.82 (d, J = 6.2 Hz, 1H, C1′), 5.53 (d, J = 5.9 Hz, 1H, C2′–OH), 5.32 (d, J = 4.8 Hz, 1H, C3′–OH), 5.09 (m, 1H, C2′), 4.77 (dd, J = 5.6 Hz, 6.6 Hz, 1H, 5′–OH), 4.25 (m, 1H, C3′), 3.96 (m, 1H, C4′), 3.77–3.67 (m, 1H, C5′), 3.59–3.49 (m, 1H, C5′); 13C {H} NMR (100 MHz, DMSO-d6) δ = 157.5 (C6), 151.1 (q, J = 35.7 Hz, C2), 149.8 (C4), 138.3 (q, J = 39.3 Hz, C8), 119.7 (q, J = 275.7 Hz, C2-CF3), 118.7 (C5), 118.3 (q, J = 271.0 Hz, C8-CF3), 89.9 (C1′), 86.7 (C4′), 71.4 (C2′), 70.6 (C3′), 61.6 (C5′); 19F NMR (376 MHz, DMSO-d6) δ = −60.23, −68.66; HRMS (+) ESI m/z: [M + H]+ calcd for C12H12F6N5O4+ 404.0788; found 404.0784.
General Procedure for Phosphorylation of 13, 18, and 19 and the Synthesis of 20, 21, and 22
Nucleoside (1 equiv) was placed in a round-bottom flask equipped with a rubber septum and flushed with a stream of argon. (MeO)3PO (0.1 M) was added under a gentle flow of argon, and the resulting solution was cooled below 0 °C (ice/brine bath) followed by addition of 2,6 lutidine (3 equiv) and dropwise addition of freshly distilled POCl3 (3 equiv). The reaction mixture was stirred below 0 °C until RP HPLC analysis indicated full consumption of the starting material (usually 3–4 h). The reaction mixture was poured into cold, deionized water (10 times the volume of the solvent used) and neutralized with 10% NaHCO3. The resulting mixture was loaded on a DEAE Sephadex A-25 column (HCO3– form, 10 g); the column was washed thoroughly with water and then eluted using TEAB in deionized water (400 mL, 0–0.7 M linear gradient). The fractions containing the pure product (UV and RP HPLC analyses) were combined, concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL). The residue was dissolved in MQ water and freeze-dried. The products were purified using preparative RP HPLC chromatography with a linear gradient of MeCN in 0.05 M ammonium acetate buffer (pH = 5.9). The fractions containing the pure product were concentrated in vacuo, coevaporated with 96% EtOH, and coevaporated with MeCN (approx. 50 mL). The residue was dissolved in MQ water and freeze-dried. After repeated freeze-drying, the products were obtained as ammonium salt.
8-Trifluoromethyl-adenosine 5′-Monophosphate (20)
White foam (58.6 mg, starting from 50.3 mg (0.15 mmol) of 13, 87%). 1H NMR (400 MHz, D2O) δ = 8.25 (s, 1H), 6.02 (d, J = 5.8 Hz, 1H), 5.28 (dd, J = 5.9 Hz, 6.1 Hz, 1H), 4.59 (dd, J = 4.7 Hz, 6.1 Hz, 1H), 4.26 (m, 1H), 4.16–4.01 (m, 2H); 13C {H} NMR (125 MHz, D2O) δ = 158.6, 157.2, 152.6, 140.8 (q, J = 40.3 Hz), 120.3 (q, J = 271.1 Hz), 120.0, 91.5, 86.4, 73.5, 72.1, 66.4; 19F NMR (376 MHz, D2O) δ = −61.32; 31P NMR (162 MHz, D2O) δ = 0.95; HRMS (−) ESI m/z: [M – H]− calcd for C11H12F3N5O7P- 414.0432; found 414.0432.
2-Trifluoromethyl-adenosine 5′-Monophosphate (21)
White foam (23.6 mg, starting from 23.5 mg (0.07 mmol) of 18, 75%). 1H NMR (400 MHz, D2O) δ = 8.54 (s, 1H), 6.16 (d, J = 5.3 Hz, 1H), 4,73 (m, 1H), 4.51 (m, 1H), 4.38 (m, 1H), 4.14 (m, 2H); 13C {H} NMR (125 MHz, D2O) δ = 158.4, 152.9 (q, J = 36.0 Hz), 151.5, 144.1, 122.2 (q, J = 274.6 Hz), 122.0, 90.1, 86.5, 77.2, 72.9, 67.0; 19F NMR (376 MHz, D2O) δ = −68.98; 31P NMR (162 MHz, D2O) δ = 1.15; HRMS (−) ESI m/z: [M – H]− calcd for C11H12F3N5O7P- 414.0432; found 414.0432.
2,8-Ditrifluoromethyl-adenosine 5′-Monophosphate (22)
White foam (7.2 mg, starting from 8.1 mg (0.02 mmol) of 19, 70%). 1H NMR (400 MHz, D2O) δ = 6.12 (d, J = 4.9 Hz, 1H), 5.27 (dd, J = 5.3 Hz, 5.9 Hz, 1H), 4.73 (dd, J = 5.1 Hz, 6.0 Hz, 1H), 4.33 (m, 1H), 4.27–4.09 (m, 2H); 13C {H} NMR (125 MHz, D2O) δ = 159.7, 154.5 (q, J = 36.4 Hz), 152.4, 142.7 (q, J = 40.8 Hz), 123.1 (q, J = 274.8 Hz), 121.3, 120.5 (q, J = 271.2 Hz),92.5, 86.9, 74.8, 72.9, 67.5; 19F NMR (376 MHz, D2O) δ = −61.53, −69.61; 31P NMR (162 MHz, D2O) δ = 1.44; HRMS (−) ESI m/z: [M – H]− calcd for C12H11F6N5O7P- 482.0306; found 482.0305.
General Procedure for the Synthesis of 6-Im and 20-Im
Monophosphate 6 or 20 (1 equiv) was dissolved in anhydrous dimethylformamide (DMF, 0.05 M) followed by addition of imidazole (20 equiv), 2,2′-dithiodipyridine (5 equiv), and trimethylamine (5 equiv). The resulting solution was stirred at room temperature for 15 min, and then triphenylphosphine (5 equiv) was added in one portion. The solution turned deep yellow immediately. The reaction mixture was stirred overnight at room temperature. Addition of a solution of anhydrous LiClO4 (5 equiv) in anhydrous acetone (10× volumes of DMF used) resulted in precipitation of the product as lithium salt. The suspension was cooled at 4 °C for approx. 2 h, and the precipitate was centrifuged and washed repeatedly with cold acetone until the supernatant was colorless. The resulting solid was additionally washed with cold diethyl ether and dried overnight under high vacuum. Yields + 95%. Caution: A substantial level of decomposition of 6-Im and 20-Im was observed while storage, even at −18 °C. To assure good yields of further reactions, they should be used immediately after preparation.
General Procedure for the Synthesis of 7, 8, 23, and 24
Freshly prepared P-imidazolide of the respective nucleotide (1 equiv) was dissolved in anhydrous DMF (0.05 M). Triethylammonium phosphate or triethylammonium pyrophosphate (8 equiv) was added followed by addition of anhydrous ZnCl2 (8 equiv). The reaction mixture was stirred at room temperature until HPLC analysis showed full consumption of the starting material (usually 24–30 h). The reaction mixture was diluted with EDTA solution (8.1 equiv in water, 10× volume of DMF used) and neutralized with 10% NaHCO3. The resulting mixture was loaded on a DEAE Sephadex A-25 column (HCO3– form, 10 g); the column was washed with water (approx. 50 mL) and then eluted using TEAB in deionized water (400 mL, linear gradient). The fractions containing the mixture of the desired product and the starting material (UV and RP HPLC analyses) were combined, concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL), and the residue was dried overnight under high vacuum. The product was purified using RP HPLC chromatography with a linear gradient of MeCN in 0.05 M ammonium acetate buffer (pH = 5.9). The fractions containing the pure product were concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL). The residue was dissolved in MQ water and freeze-dried. After repeated freeze-drying, the product was obtained as ammonium salt.
7 was isolated in 80% yield (9.4 mg starting from 10.0 mg (0.021 mmol) of 6-Im). 8 was isolated in 40% yield (5.5 mg starting from 10.0 mg (0.021 mmol) of 6-Im). For 7 and 8, the analytical data matched those obtained from the 7 and 8 synthesized by direct trifluoromethylation of guanosine diphosphate and triphosphate, respectively.
8-Trifluoromethyl-adenosine 5′-Diphosphate (23)
White foam (9.2 mg starting from 10.0 mg (0.021 mmol) of 20-Im, 80%). 1H NMR (400 MHz, D2O) δ = 8.36 (s, 1H), 6.05 (d, J = 5.6 Hz, 1H), 5.28 (dd, J = 5.7 Hz, 6.2 Hz, 1H), 4.65 (dd, J = 4.3 Hz, 5.8 Hz, 1H), 4.35–4.18 (m, 3H);19F NMR (376 MHz, D2O) δ = −61.43; 31P NMR (162 MHz, D2O) δ = −9.91 (d, J = 20.1 Hz, 1p), −10.34 (d, J = 20.3 Hz, 1P); HRMS (−) ESI m/z: [M – H]− calcd for C11H13F3N5O10P2– 494.0095; found 494.0095.
8-Trifluoromethyl-adenosine 5′-Triphosphate (24)
White foam (7.7 mg starting from 10.0 mg (0.021 mmol) of 20-Im, 60%). 1H NMR (400 MHz, D2O) δ = 8.37 (s, 1H), 6.08 (d, J = 6.0 Hz, 1H), 5.39 (dd, J = 6.0 Hz, 6.7 Hz, 1H), 4.68 (m, 1H), 4.40–4.19 (m, 3H); 19F NMR (376 MHz, D2O) δ = −61.31; 31P NMR (162 MHz, D2O) δ = −9.37 to −9.99 (m, 1P), −10.36 (d, J = 19.8 Hz, 1P), −22.2 (t, J = 19.8 Hz, 1P); HRMS (−) ESI m/z: [M – H]− calcd for C11H14F3N5O13P3– 573.9759; found: 573.9761.
Synthesis of Dinucleotide 25
Freshly prepared 20-Im (10.0 mg, 0.021 mmol) was dissolved in anhydrous DMSO (0.5 mL) followed by addition of triethylammonium phosphate (6.0 mg, 0.015 mmol). To this solution, anhydrous ZnCl2 was added (32.0 mg, 0.235 mmol). After approx. 2 h, HPLC analysis indicated formation of substantial amounts of 23 and the presence of 20 (formed by the hydrolysis of the starting material). An additional portion of 20-Im (10.0 mg, 0.021 mmol) was added, and the mixture was stirred overnight at room temperature. The reaction was stopped by addition of EDTA solution (80 mg, 0.238 mmol in 5 mL of water) and neutralized with 10% NaHCO3. The resulting mixture was loaded on a DEAE Sephadex A-25 column (HCO3– form, 10 g); the column was washed with water (approx. 50 mL) and then eluted using TEAB in deionized water (400 mL, 0–1 M linear gradient). The fractions containing the mixture of the desired product and traces of byproducts (UV and RP HPLC analyses) were combined, concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL), and the residue was dried overnight under high vacuum. The product was purified using RP HPLC chromatography with a linear gradient of MeCN in 0.05 M ammonium acetate buffer (pH = 5.9). The fractions containing the pure product were concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL). The residue was dissolved in MQ water and freeze-dried. After repeated freeze-drying, the product was obtained as ammonium salt (white foam, 8.9 mg, 63%). 1H NMR (400 MHz, D2O) δ = 8.05 (s, 2H), 5.77 (d, J = 6.0 Hz, 2H), 5.06 (dd, J = 5.8 Hz, 6.1 Hz, 2H), 4.47 (dd, J = 3.6 Hz, 5.7 Hz, 2H), 4.25–4.15 (m, 2H), 4.14–4.01 (m, 4H);19F NMR (376 MHz, D2O) δ = −61.28; 31P NMR (162 MHz, D2O) δ = −10.46 (d, J = 17.7 Hz, 2P), −22.01 (t, J = 17.8 Hz, 1P); HRMS (−) ESI m/z: [M – H]− calcd for C22H24F6N10O16P3– 891.0494; found 891.0502.
Synthesis of Dinucleotide 26
ADP-Im (sodium salt, 27.0 mg, 0.051 mmol) and 6 (ammonium salt, 17.0 mg, 0.036 mmol) were dissolved in anhydrous DMSO (2 mL) followed by addition of anhydrous ZnCl2 (86.0 mg, 0.632 mmol). The reaction mixture was stirred at room temperature for 72 h, at which time HPLC analysis indicated no further progress of the reaction. The reaction mixture was diluted with EDTA solution (235 mg, 0.642 mmol in 20 mL of water) and neutralized with 10% NaHCO3. The resulting mixture was loaded on a DEAE Sephadex A-25 column (HCO3– form, 10 g); the column was washed with water (approx. 50 mL) and then eluted using TEAB in deionized water (400 mL, 0–1 M linear gradient). The fractions containing the mixture of the desired product and traces of the starting material (UV and RP HPLC analyses) were combined, concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL), and the residue was dried overnight under high vacuum. The product was purified using RP HPLC chromatography with a linear gradient of MeCN in 0.05 M ammonium acetate buffer (pH = 5.9). The fractions containing the pure product were concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL). The residue was dissolved in MQ water and freeze-dried. After repeated freeze-drying, the product was obtained as ammonium salt (white foam, 12.8 mg, 40%). 1H NMR (400 MHz, D2O) δ = 8.46 (s, 1H), 8.27 (s, 1H), 6.00 (d, J = 5.5 Hz, 1H), 5.77 (d, J = 6.4 Hz, 1H), 5.30 (dd, J = 5.8 Hz, 6.2 Hz, 1H), 4.63 (dd, J = 5.4 H, 5.7 Hz, 1H), 4.59 (dd, J = 2.6 Hz, 5.4Hz, 1H), 4.50 (m, 1H), 4.42–4.16 (m, 6H); 19F NMR (376 MHz, D2O) δ = −61.14; 31P NMR (162 MHz, D2O) δ = −10.46 (t, J = 17.0 Hz, 2P), −22.16 (t, J = 18.9 Hz, 1P); HRMS (−) ESI m/z: [M – H]− calcd for C21H25F3N10O17P3– 839.0570; found 839.0571.
Synthesis of Dinucleotide 27
GDP-Im (sodium salt, 34.0 mg, 0.051 mmol) and 6 (ammonium salt, 17.0 mg, 0.036 mmol) were dissolved in anhydrous DMSO (2 mL) followed by addition of anhydrous ZnCl2 (86.0 mg, 0.632 mmol). The reaction mixture was stirred at room temperature for 48 h, at which time HPLC analysis indicated no further progress of the reaction. The reaction mixture was diluted with EDTA solution (235 mg, 0.642 mmol in 20 mL of water) and neutralized with 10% NaHCO3. The resulting mixture was loaded on a DEAE Sephadex A-25 column (HCO3– form, 10 g); the column was washed with water (approx. 50 mL) and then eluted using TEAB in deionized water (400 mL, 0–1 M linear gradient). The fractions containing the mixture of the desired product and traces of the starting material (UV and RP HPLC analyses) were combined, concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL), and the residue was dried overnight under high vacuum. The product was purified using RP HPLC chromatography with a linear gradient of MeCN in 0.05 M ammonium acetate buffer (pH = 5.9). The fractions containing the pure product were concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL). The residue was dissolved in MQ water and freeze-dried. After repeated freeze-drying, the product was obtained as ammonium salt (white foam, 27.1 mg, 83%). 1H NMR (400 MHz, D2O) δ = 8.11 (s, 1H), 5.82 (d, J = 6.5 Hz, 1H), 5.79 (d, J = 5.8 Hz, 1H), 5.36 (dd, J = 5.8 Hz, 6.6 Hz, 1H), 4.66 (dd, J = 5.5 Hz, 6.0 Hz, 1H), 4.59 (m, 1H), 4.49 (m, 1H), 4.39–4.17 (m, 6H); 19F NMR (376 MHz, D2O) δ = −61.06; 31P NMR (162 MHz, D2O) δ = −10.41 (t, J = 6.8 Hz, 1P), −10.50 (t, J = 6.5 Hz, 1P), −23.14 (t, J = 19.0 Hz, 1P); HRMS (−) ESI m/z: [M – H]− calcd for C21H25F3N10O18P3 855.0519; found 855.0524.
Synthesis of Dinucleotide 28
27 (9.0 mg, 0.01 mmol) was dissolved in anhydrous DMSO (0.5 mL) followed by addition of MeI (40 μL, excess). The reaction was stirred at room temperature until HPLC analysis indicated full consumption of the starting material (approx. 6 h). The reaction mixture was diluted with water (5 mL) and washed with diethyl ether (3 × 5 mL). The aqueous phase was freeze-dried, and the residue was purified using RP HPLC chromatography with a linear gradient of MeCN in 0.05 M ammonium acetate buffer (pH = 5.9). The fractions containing the pure product were concentrated in vacuo, coevaporated with 96% EtOH (approx. 50 mL), and coevaporated with MeCN (approx. 50 mL). The residue was dissolved in MQ water and freeze-dried. After repeated freeze-drying, the product was obtained as ammonium salt (white foam, 7.2 mg, 80%). 1H NMR (400 MHz, D2O) δ = 5.79–5.71 (m, 2H), 5.16 (m, 1H), 4.55 (m, 1H), 4.47 (m, 1H), 4.44–4.32 (m, 4H), 4.31–4.16 (m, 3H), 4.06 (bs, 3H); 19F NMR (376 MHz, D2O) δ = −61.17; 31P NMR (162 MHz, D2O) δ = −10.36 to −10.69 (m, 2P), −22.15 (t, J = 19.4 Hz, 1P); HRMS (−) ESI m/z: [M – H]− calcd for C22H27F3N10O18P3– 869.0675; found: 869.0681.
Stock Solutions of Studied (Di)Nucleotides
Compounds 6, 20, 23, 25, 26, 27, and 28 were dissolved in pure water (200–300 μL) and 13 was dissolved in DMSO (200 μL), and concentrations were estimated spectrophotometrically by measurement of absorbance at 260 nm in 0.1 M phosphate buffer pH 6.0 (6, 28) or 7.0 (23, 25, 26, 27). To calculate the exact concentrations, the following molar extinction coefficients [M–1 cm–1] were employed: ε = 11400 (6, 13, 20), ε = 15020 (23), ε = 27036 (25, 26), ε = 22600 (27), and ε = 21132 (28).
Sample Preparation for Enzymatic Studies
For studies with human Fhit compound 26 or 27 was diluted in a buffer containing 50 mM MES KOH pH 6.50, 1 mM MgCl2, and 10% D2O to a final concentration of 100 μM. For studies with the human DcpS compound 28 was diluted in a buffer containing 50 mM Tris-HCl pH 7.60, 0.2 M KCl, 0.5 mM EDTA, and 10% D2O to a final concentration of 100 μM. For studies with the human cNIIIB compound 6 was diluted in a buffer containing 20 mM HEPES KOH pH 7.50, 50 mM KCl, 5 mM MgCl2, and 10% D2O to a final concentration of 100 μM. RG3039 was purchased from KareBay Biochem. Human Fhit inhibitor (IN-A, 7,8-dihydro-7,7-dimethyl-10-(4-chlorophenyl)-5H-indeno[1,2-b]quinoline-9,11(6H,10H)-dione) was synthesized as previously described.60 All inhibitors were dissolved in DMSO, and the concentrations were established by mass (RG3039, IN-A) or spectrophotometrically by absorbance measurement at 260 nm in 0.1 M phosphate buffer pH 6.0 and by using a molar extinction coefficient equal to 11400 M–1 cm–1 (cNIIIB-specific inhibitor). To samples without an inhibitor, 0.3–0.5% DMSO was added (v/v) to match the solvent composition of all samples. All enzymatic reactions were performed at 30 °C.
19F NMR Spectroscopy
19F NMR spectra were recorded on a Bruker Avance III HD 500 MHz spectrometer equipped with a 5 mm PABBO BB/19F-1H/D Z-GRD probe at a frequency of 470.67 MHz in 5 mm NMR samples. Typical experimental parameters were chosen as follows: 19F excitation pulse, 15.1 μs; acquisition time, 1.2 s; relaxation delay, 1.0 s; number of scans, 32; spectral width, 32.8 ppm; spectral resolution, 0.83 Hz. The 19F NMR chemical shifts were reported to 0.1 M NaF in D2O (δF = −121.5 ppm) as an external standard. Before each enzymatic experiment, the sample without the enzyme was incubated inside a magnet at 30 °C for 5 min, then locked, tuned, shimmed, and initial 32 scans were recorded (see Figures 1 and S2, no enzyme spectra). To perform the kinetic experiment, the multi_zgvd command was applied with fixed delays (120 s), and the number of experiments was set to 14. The data were analyzed by MestReNova 12.0 and GraphPad Prism 8.0.
Protein Expression and Purification
The plasmids for expression of human Fhit, pSGA02_hFhit, and Arabidopsis Thaliana Fhit, pSGA02_AtFhit, were kindly provided by Dr. Pawel Bieganowski (Mossakowski Medical Research Centre, Polish Academy of Sciences).
Human Fhit
Full-length human Fhit was produced in the Escherichia coli BL21(DE3) RIL strain in an LB medium with ampicillin (100 μg/mL). The bacterial culture was grown to OD600 0.4, and the protein expression was induced by 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 30 °C during 6 h. For the protein purification, cells were lysed with 0.1 mg/mL lysozyme during 20 min on ice followed by sonication in a buffer A containing 20 mM Tris/HCl pH 7.5, 100 mM NaCl, and 2 mM dithiothreitol (DTT) with addition of protease inhibitors (10 μM leupeptin, 0.3 μM aprotinin, 1 μM pepstatin A, and 1 mM phenylmethylsulfonyl fluoride (PMSF)). The lysate was clarified by centrifugation at 35 000g for 40 min at 4 °C. Then, nucleic acids were removed from the supernatant by precipitation using 0.1% polyethyleneimine (PEI), and protein was clarified by multistep precipitation in ammonium sulfate. Initial precipitation in 20% ammonium sulfate removed nonsoluble proteins, and double precipitation in 70% ammonium sulfate isolated the main protein fraction. Finally, the pelleted protein was resuspended in buffer A, desalted on a HiPrep 26/10 column, and polished by gel filtration on a HiLoad 26/600 Superdex 75 pg column filled with buffer B containing 20 mM HEPES/NaOH pH 7.0, 150 mM NaCl, and 2 mM DTT. This multistep procedure allows one to gain more than 90% pure protein assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). The absorbance ratio A260/A280 for the final protein sample was 0.73. Human Fhit samples were concentrated on Amicon Ultra-15 10K filters up to 8.4 mg/mL or 16.8 g/mL, flash-frozen in liquid nitrogen, and stored at −80 °C in 50 or 100 μL aliquots in the presence of 10% glycerol. Molecular mass of the human Fhit monomer is 16 800 Da, and the protein concentration was determined spectrophotometrically using the extinction coefficient calculated from the amino acid composition, ε280 = 8480 M–1 cm–1 (Expasy Server).
Human DcpS Preparation
Human DcpS (hDcpS) was expressed as previously described but with minor modifications.61 The concentration of the protein was determined spectrophotometrically by assuming ε280 = 30 400 M–1 cm–1. The enzyme was stored at −80 °C in a storage buffer (50 mM Tris-HCl, pH 7.6, 200 mM NaCl, 1 mM DTT, 10% glycerol).
Human cNIIIB Preparation
Human cNIIIB (HscNIIIB) was expressed as previously described.57
Acknowledgments
We thank Dorota Kubacka and Mateusz Kozarski (University of Warsaw) for providing the reagents for the experiments with hcNIIIB. This work was supported by the Foundation for Polish Science (TEAM/2016–2/13) and by the Ministry of Science and Higher Education (0149/DIA/2014/43).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.9b03198.
1H NMR, 13C NMR, 19F NMR, 31P NMR, and HRMS spectra; HPLC profiles of new compounds; additional experimental details (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Mei H.; Han J.; Fustero S.; Medio-Simon M.; Sedgwick D. M.; Santi C.; Ruzziconi R.; Soloshonok V. A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. – Eur. J. 2019, 25, 11797–11819. 10.1002/chem.201901840. [DOI] [PubMed] [Google Scholar]
- Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Wang J.; Gu Z.; Wang S.; Zhu W.; Luis Acena J.; Soloshonok V. A.; Izawa K.; Liu H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently In Phase II-III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]
- Chen H.; Viel S.; Ziarelli F.; Peng L. F-19 NMR: a Valuable Tool for Studying Biological Events. Chem. Soc. Rev. 2013, 42, 7971–7982. 10.1039/c3cs60129c. [DOI] [PubMed] [Google Scholar]
- Cobb S. L.; Murphy C. D. F-19 NMR Applications in Chemical Biology. J. Fluorine Chem. 2009, 130, 132–143. 10.1016/j.jfluchem.2008.11.003. [DOI] [Google Scholar]
- Dalvit C.; Fagerness P. E.; Hadden D. T. A.; Sarver R. W.; Stockman B. J. Fluorine-NMR Experiments for High-Throughput Screening: Theoretical Aspects, Practical Considerations, and Range of Applicability. J. Am. Chem. Soc. 2003, 125, 7696–7703. 10.1021/ja034646d. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Mongelli N.; Papeo G.; Giordano P.; Veronesi M.; Moskau D.; Kummerle R. Sensitivity Improvement in F-19 NMR-Based Screening Experiments: Theoretical Considerations and Experimental Applications. J. Am. Chem. Soc. 2005, 127, 13380–13385. 10.1021/ja0542385. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Ardini E.; Flocco M.; Fogliatto G. P.; Mongelli N.; Veronesi M. A General NMR Method for Rapid, Efficient, and Reliable Biochemical Screening. J. Am. Chem. Soc. 2003, 125, 14620–14625. 10.1021/ja038128e. [DOI] [PubMed] [Google Scholar]
- Vulpetti A.; Dalvit C. Fluorine Local Environment: From Screening to Drug Design. Drug Discovery Today 2012, 17, 890–897. 10.1016/j.drudis.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Charpentier J.; Frueh N.; Togni A. Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents. Chem. Rev. 2015, 115, 650–682. 10.1021/cr500223h. [DOI] [PubMed] [Google Scholar]
- Ma J.-A.; Cahard D. Strategies for Nucleophilic, Electrophilic, and Radical Trifluoromethylations. J. Fluorine Chem. 2007, 128, 975–996. 10.1016/j.jfluchem.2007.04.026. [DOI] [Google Scholar]
- Barata-Vallejo S.; Lantano B.; Postigo A. Recent Advances in Trifluoromethylation Reactions with Electrophilic Trifluoromethylating Reagents. Chem. – Eur. J. 2014, 20, 16806–16829. 10.1002/chem.201404005. [DOI] [PubMed] [Google Scholar]
- Clarke S. L.; McGlacken G. P. Methyl Fluorosulfonyldifluoroacetate (MFSDA): an Underutilised Reagent for Trifluoromethylation. Chem. – Eur. J. 2017, 23, 1219–1230. 10.1002/chem.201602511. [DOI] [PubMed] [Google Scholar]
- Ni C.; Hu M.; Hu J. Good Partnership between Sulfur and Fluorine: Sulfur-Based Fluorination and Fluoroalkylation Reagents for Organic Synthesis. Chem. Rev. 2015, 115, 765–825. 10.1021/cr5002386. [DOI] [PubMed] [Google Scholar]
- Pan X.; Xia H.; Wu J. Recent Advances in Photoinduced Trifluoromethylation and Difluoroalkylation. Org. Chem. Front. 2016, 3, 1163–1185. 10.1039/C6QO00153J. [DOI] [Google Scholar]
- Furuya T.; Kamlet A. S.; Ritter T. Catalysis for Fluorination and Trifluoromethylation. Nature 2011, 473, 470–477. 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer A. A ″Renaissance″ in Radical Trifluoromethylation. Angew. Chem., Int. Ed. 2012, 51, 8950–8958. 10.1002/anie.201202624. [DOI] [PubMed] [Google Scholar]
- Guo F.; Li Q.; Zhou C. Synthesis and Biological Applications of Fluoro-Modified Nucleic Acids. Org. Biomol. Chem. 2017, 15, 9552–9565. 10.1039/C7OB02094E. [DOI] [PubMed] [Google Scholar]
- Fauster K.; Kreutz C.; Micura R. 2′-SCF3 Uridine - a Powerful Label for Probing Structure and Function of RNA by F-19 NMR Spectroscopy. Angew. Chem., Int. Ed. 2012, 51, 13080–13084. 10.1002/anie.201207128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granqvist L.; Virta P. 4′-C- (4-Trifluoromethyl-1H-1,2,3-Triazol-1-yl)methyl Thymidine as a Sensitive F-19 NMR Sensor for the Detection of Oligonucleotide Secondary Structures. J. Org. Chem. 2014, 79, 3529–3536. 10.1021/jo500326j. [DOI] [PubMed] [Google Scholar]
- Ishizuka T.; Zhao P.-Y.; Bao H.-L.; Xu Y. A. Multi-Functional Guanine Derivative for Studying the DNA G-Quadruplex Structure. Analyst 2017, 142, 4083–4088. 10.1039/C7AN00941K. [DOI] [PubMed] [Google Scholar]
- Kiviniemi A.; Virta P. Characterization of RNA Invasion by F-19 NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 8560–8562. 10.1021/ja1014629. [DOI] [PubMed] [Google Scholar]
- Ishizuka T.; Yamashita A.; Asada Y.; Xu Y. Studying DNA G-Quadruplex Aptamer by F-19 NMR. ACS Omega 2017, 2, 8843–8848. 10.1021/acsomega.7b01405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Košutić M.; Jud L.; Da Veiga C.; Frener M.; Fauster K.; Kreutz C.; Ennifar E.; Micura R. Surprising Base Pairing and Structural Properties of 2′-Trifluoromethylthio-Modified Ribonucleic Acids. J. Am. Chem. Soc. 2014, 136, 6656–6663. 10.1021/ja5005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhate N. B.; Barhate R. N.; Cekan P.; Drobny G.; Sigurdsson S. T. A Nonafluoro Nucleoside as a Sensitive F-19 NMR Probe of Nucleic Acid Conformation. Org. Lett. 2008, 10, 2745–2747. 10.1021/ol800872a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K.; Tsuda T.; Ito T.; Nishimoto S.-i. Probing DNA Mismatched and Bulged Structures by Using F-19 NMR Spectroscopy and Oligodeoxynucleotides With an F-19-Labeled Nucleobase. Chem. – Eur. J. 2013, 19, 15133–15140. 10.1002/chem.201302770. [DOI] [PubMed] [Google Scholar]
- Granqvist L.; Virta P. Characterization of G-Quadruplex/Hairpin Transitions of RNAs by F-19 NMR Spectroscopy. Chem. – Eur. J. 2016, 22, 15360–15372. 10.1002/chem.201602898. [DOI] [PubMed] [Google Scholar]
- Bhuma N.; Tahtinen V.; Virta P. Synthesis and Applicability of Base-Discriminating DNA-Triplex-Forming F-19 NMR Probes. Eur. J. Org. Chem. 2018, 605–613. 10.1002/ejoc.201701110. [DOI] [Google Scholar]
- Nakamura S.; Fujimoto K. Photo-Cross-Linking Using Trifluorothymidine and 3-Cyanovinylcarbazole Induced a Large Shifted F-19 MR Signal. Chem. Commun. 2015, 51, 11765–11768. 10.1039/C5CC02972D. [DOI] [PubMed] [Google Scholar]
- Olszewska A.; Pohl R.; Hocek M. Trifluoroacetophenone-Linked Nucleotides and DNA for Studying of DNA-Protein Interactions by F-19 NMR Spectroscopy. J. Org. Chem. 2017, 82, 11431–11439. 10.1021/acs.joc.7b01920. [DOI] [PubMed] [Google Scholar]
- a Ginersorolla A.; Bendich A. Fluorine-containing Pyrimidines and Purines – Synthesis and Properties of Trifluoromethyl Pyrimidines and Purines. J. Am. Chem. Soc. 1958, 80, 5744–5752. 10.1021/ja01554a041. [DOI] [Google Scholar]; b Baranowski M. R.; Nowicka A.; Rydzik A. M.; Warminski M.; Kasprzyk R.; Wojtczak B. A.; Wojcik J.; Claridge T. D. W.; Kowalska J.; Jemielity J. Synthesis of Fluorophosphate Nucleotide Analogues and Their Characterization as Tools for 19F NMR Studies. J. Org. Chem. 2015, 80, 3982–3997. 10.1021/acs.joc.5b00337. [DOI] [PubMed] [Google Scholar]
- Ginersorolla A.; Bendich A. Fluorine-containing pyrimidines and purines - synthesis and properties of trifluoromethyl pyrimidines and purines. J. Am. Chem. Soc. 1958, 80, 5744–5752. 10.1021/ja01554a041. [DOI] [Google Scholar]
- Iaroshenko V. O.; Ostrovskyi D.; Petrosyan A.; Mkrtchyan S.; Villinger A.; Langer P. Synthesis of Fluorinated Purine and 1-Deazapurine Glycosides as Potential Inhibitors of Adenosine Deaminase. J. Org. Chem. 2011, 76, 2899–2903. 10.1021/jo102579g. [DOI] [PubMed] [Google Scholar]
- Felczak K.; Vince R.; Pankiewicz K. W. NAD-Based Inhibitors with Anticancer Potential. Bioorg. Med. Chem. Lett. 2014, 24, 332–336. 10.1016/j.bmcl.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y.; Yamamoto K.; Asai T.; Nakano M.; Kumadaki I. Studies on Organic Fluorine-Compounds. 35. Trifluoromethylation of Pyrimidine-Nucleosides and Purine-Nucleosides with Trifluoromethyl-Copper Complex. J. Chem. Soc., Perkin Trans. 1 1980, 2755–2761. 10.1039/P19800002755. [DOI] [Google Scholar]
- Dong M.; Kirchberger T.; Huang X.; Yang Z. J.; Zhang L. R.; Guse A. H.; Zhang L. H. Trifluoromethylated Cyclic-ADP-Ribose Mimic: Synthesis of 8-Trifluoromethyl-N-1- (5″-O-phosphorylethoxy)methyl -5′-O-Phosphorylinosine-5′,5″-Cyclic Pyrophosphate (8-CF3-Cidpre) and its Calcium Release Activity in T Cells. Org. Biomol. Chem. 2010, 8, 4705–4715. 10.1039/C0OB00090F. [DOI] [PubMed] [Google Scholar]
- Huang X.; Dong M.; Liu J.; Zhang K.; Yang Z.; Zhang L.; Zhang L. Concise Syntheses of Trifluoromethylated Cyclic and Acyclic Analogues of cADPR. Molecules 2010, 15, 8689–8701. 10.3390/molecules15128689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M.; Gao Z. G.; Van Rompaey P.; Tchilibon S.; Kim S. K.; Harris B. A.; Gross A. S.; Duong H. T.; Van Calenbergh S.; Jacobson K. A. Modulation of Adenosine Receptor Affinity And Intrinsic Efficacy in Adenine Nucleosides Substituted at the 2-Position. Bioorg. Med. Chem. 2004, 12, 2995–3007. 10.1016/j.bmc.2004.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair V.; Buenger G. S. Novel, Stable Congeners of the Antiretroviral Compound 2′,3′-Dideoxyadenosine. J. Am. Chem. Soc. 1989, 111, 8502–8504. 10.1021/ja00204a026. [DOI] [Google Scholar]
- Nair V.; Purdy D. F.; Sells T. B. Synthesis of Congeners of Adenosine Resistant to Deamination by Adenosine-Deaminase. J. Chem. Soc., Chem. Commun. 1989, 878–879. 10.1039/c39890000878. [DOI] [Google Scholar]
- Véliz E. A.; Stephens O. M.; Beal P. A. Synthesis and Analysis of RNA Containing 6-Trifluoromethylpurine Ribonucleoside. Org. Lett. 2001, 3, 2969–2972. 10.1021/ol016295i. [DOI] [PubMed] [Google Scholar]
- Musumeci D.; Irace C.; Santamaria R.; Montesarchio D. Trifluoromethyl Derivatives of Canonical Nucleosides: Synthesis and Bioactivity Studies. MedChemComm 2013, 4, 1405–1410. 10.1039/c3md00159h. [DOI] [Google Scholar]
- Guyon H.; Chachignon H.; Cahard D. CF3SO2X (X = Na, Cl) as Reagents for Trifluoromethylation, Trifluoromethylsulfenyl-, -Sulfinyl and Sulfonylation. Part 1: Use Of CF3SO2Na. Beilstein J. Org. Chem. 2017, 13, 2764–2799. 10.3762/bjoc.13.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y.; Dixon J. A.; O’Hara F.; Funder E. D.; Dixon D. D.; Rodriguez R. A.; Baxter R. D.; Herle B.; Sach N.; Collins M. R.; Ishihara Y.; Baran P. S. Practical and Innate Carbon-Hydrogen Functionalization of Heterocycles. Nature 2012, 492, 95–99. 10.1038/nature11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q.; Gui J.; Pan C.-M.; Albone E.; Cheng X.; Suh E. M.; Grasso L.; Ishihara Y.; Baran P. S. Bioconjugation by Native Chemical Tagging of C–H Bonds. J. Am. Chem. Soc. 2013, 135, 12994–12997. 10.1021/ja407739y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiishi N.; Caldwell J. P.; Lin M.; Zhong W.; Zhu X. H.; Streckfuss E.; Kim H. Y.; Parish C. A.; Krska S. W. Protecting Group Free Radical C–H Trifluoromethylation Of Peptides. Chem. Sci. 2018, 9, 4168–4175. 10.1039/C8SC00368H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa M.; Kato T.; Takenishi T. A Novel Method for Phosphorylation of Nucleosides to 5′-Nucleotides. Tetrahedron Lett. 1967, 5065–5068. 10.1016/S0040-4039(01)89915-9. [DOI] [PubMed] [Google Scholar]
- Mukaiyama T.; Hashimoto M. Synthesis of Oligothymidylates and Nucleoside Cyclic Phosphates by Oxidation-Reduction Condensation. J. Am. Chem. Soc. 1972, 94, 8528–8532. 10.1021/ja00779a039. [DOI] [PubMed] [Google Scholar]
- Gogliotti R. G.; Cardona H.; Singh J.; Bail S.; Emery C.; Kuntz N.; Jorgensen M.; Durens M.; Xia B.; Barlow C.; Heier C. R.; Plasterer H. L.; Jacques V.; Kiledjian M.; Jarecki J.; Rusche J.; DiDonato C. J. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum. Mol. Genet. 2013, 22, 4084–4101. 10.1093/hmg/ddt258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.; Salcius M.; Liu S.-W.; Staker B. L.; Mishra R.; Thurmond J.; Michaud G.; Mattoon D. R.; Printen J.; Christensen J.; Bjornsson J. M.; Pollok B. A.; Kiledjian M.; Stewart L.; Jarecki J.; Gurney M. E. DcpS as a Therapeutic Target for Spinal Muscular Atrophy. ACS Chem. Biol. 2008, 3, 711–722. 10.1021/cb800120t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozarski M.; Kubacka D.; Wojtczak B. A.; Kasprzyk R.; Baranowski M. R.; Kowalska J. 7-Methylguanosine monophosphate analogues with 5′-(1,2,3-triazoyl) moiety: Synthesis and evaluation as the inhibitors of cNIIIB nucleotidase. Bioorg. Med. Chem. 2018, 26, 191–199. 10.1016/j.bmc.2017.11.032. [DOI] [PubMed] [Google Scholar]
- Hacker S. M.; Mortensen F.; Scheffner M.; Marx A. Selective Monitoring of the Enzymatic Activity of the Tumor Suppressor Fhit. Angew. Chem., Int. Ed. 2014, 53, 10247–10250. 10.1002/anie.201405259. [DOI] [PubMed] [Google Scholar]
- Lange S.; Hacker S. M.; Schmid P.; Scheffner M.; Marx A. Small-Molecule Inhibitors of the Tumor Suppressor Fhit. Chembiochem 2017, 18, 1707–1711. 10.1002/cbic.201700226. [DOI] [PubMed] [Google Scholar]
- Guranowski A.; Wojdyla A. M.; Pietrowska-Borek M.; Bieganowski P.; Khurs E. N.; Cliff M. J.; Blackburn G. M.; Blaziak D.; Stec W. J. Fhit proteins can also recognize substrates other than dinucleoside polyphosphates. FEBS Lett. 2008, 582, 3152–3158. 10.1016/j.febslet.2008.07.060. [DOI] [PubMed] [Google Scholar]
- Liu S. W.; Jiao X. F.; Liu H. D.; Gu M. G.; Lima C. D.; Kiledjian M. Functional analysis of mRNA scavenger decapping enzymes. RNA 2004, 10, 1412–1422. 10.1261/rna.7660804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meerbeke J. P.; Gibbs R. M.; Plasterer H. L.; Miao W.; Feng Z.; Lin M.-Y.; Rucki A. A.; Wee C. D.; Xia B.; Sharma S.; Jacques V.; Li D. K.; Pellizzoni L.; Rusche J. R.; Ko C.-P.; Sumner C. J. The DcpS inhibitor RG3039 improves motor function in SMA mice. Hum. Mol. Genet. 2013, 22, 4074–4083. 10.1093/hmg/ddt257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T.; Masuda T.; Canver M. C.; Seiler M.; Semba Y.; Shboul M.; Al-Raqad M.; Maeda M.; Schoonenberg V. A. C.; Cole M. A.; Macias-Trevino C.; Ishikawa Y.; Yao Q. M.; Nakano M.; Arai F.; Orkin S. H.; Reversade B.; Buonamici S.; Pinello L.; Akashi K.; Bauer D. E.; Maeda T. Genome-wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-mRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 2018, 33, 386–400. 10.1016/j.ccell.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monecke T.; Buschmann J.; Neumann P.; Wahle E.; Ficner R. Crystal Structures of the Novel Cytosolic 5′-Nucleotidase IIIB Explain Its Preference for m(7)GMP. PLoS One 2014, 9, e90915 10.1371/journal.pone.0090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschmann J.; Moritz B.; Jeske M.; Lilie H.; Schierhorn A.; Wahle E. Identification of Drosophila and Human 7-Methyl GMP-specific Nucleotidases. J. Biol. Chem. 2013, 288, 2441–2451. 10.1074/jbc.M112.426700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirini F.; Beigbaghlou S. S.; Atghia S. V.; Mousazadeh S. A. R. Multi-component one-pot synthesis of unsymmetrical dihydro-5H-indeno 1,2-b quinolines as new pH indicators. Dyes Pigm. 2013, 97, 19–25. 10.1016/j.dyepig.2012.11.002. [DOI] [Google Scholar]
- Rydzik A. M.; Lukaszewicz M.; Zuberek J.; Kowalska J.; Darzynkiewicz Z. M.; Darzynkiewicz E.; Jemielity J. Synthetic dinucleotide mRNA cap analogs with tetraphosphate 5′,5′ bridge containing methylenebis(phosphonate) modification. Org. Biomol. Chem. 2009, 7, 4763–4776. 10.1039/b911347a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.