

Abstract
Benzene exhibits a rich photochemistry which can provide access to complex molecular scaffolds that are difficult to access with reactions in the electronic ground state. While benzene is aromatic in its ground state, it is antiaromatic in its lowest ππ* excited states. Herein, we clarify to what extent relief of excited-state antiaromaticity (ESAA) triggers a fundamental benzene photoreaction: the photoinitiated nucleophilic addition of solvent to benzene in acidic media leading to substituted bicyclo[3.1.0]hex-2-enes. The reaction scope was probed experimentally, and it was found that silyl-substituted benzenes provide the most rapid access to bicyclo[3.1.0]hexene derivatives, formed as single isomers with three stereogenic centers in yields up to 75% in one step. Two major mechanism hypotheses, both involving ESAA relief, were explored through quantum chemical calculations and experiments. The first mechanism involves protonation of excited-state benzene and subsequent rearrangement to bicyclo[3.1.0]hexenium cation, trapped by a nucleophile, while the second involves photorearrangement of benzene to benzvalene followed by protonation and nucleophilic addition. Our studies reveal that the second mechanism is operative. We also clarify that similar ESAA relief leads to puckering of S1-state silabenzene and pyridinium ion, where the photorearrangement of the latter is of established synthetic utility. Finally, we identified causes for the limitations of the reaction, information that should be valuable in explorations of similar photoreactions. Taken together, we reveal how the ESAA in benzene and 6π-electron heterocycles trigger photochemical distortions that provide access to complex three-dimensional molecular scaffolds from simple reactants.
Introduction
Benzene is the archetype aromatic molecule.1−3 Yet, while its reactivity in the electronic ground state (S0) is well-established, there are many aspects that remain to be explored and exploited related to its photophysics and photochemistry. The inherent stability of benzene in the S0 state is attributed to its aromaticity, as given by Hückel’s 4n+2 rule. In contrast, it displays a marked reactivity in the lowest excited states,4−10 and this reactivity change can be attributed to a switch between aromaticity and antiaromaticity whereby benzene becomes antiaromatic in these states.11 The reversal of Hückel’s rule was derived qualitatively for the lowest ππ* triplet state (T1) by Baird in 1972,12,13 and later extended through quantum chemical computations to the lowest ππ* singlet excited state (S1) of benzene.14,15 The impact of excited-state aromaticity and antiaromaticity for a series of different photophysical and photochemical phenomena has been unraveled in recent years.16,17 We now explore to what extent these concepts can be used to rationalize a benzene photoreaction that provides access to complex three-dimensional molecular structures. We also show how the hypothesis on excited-state antiaromaticity (ESAA) relief applies to the photochemical rearrangements of two 6π-electron heterocycles (silabenzene and pyridinium ion) with S1 states of ππ* character.
A connection between the photoreactivity and relief of T1-state antiaromaticity through structural distortions has been revealed as the origin for triplet-state photoreactions of a few different benzene derivatives,18−21 and also shown to impact on the bimolecular photoreactivity of benzene and small polycyclic aromatic hydrocarbons (PAHs) in their T1 states.22 In contrast to the (sensitized) T1-state reactivity of benzene derivatives, herein we probe if structural rearrangements of benzene itself, observed upon direct irradiation to its S1 state, are also triggered by ESAA alleviation. This aspect of benzene photochemistry has never been analyzed earlier, yet in the analysis one should also consider what other factors impact on the shape of the S1-state potential energy surface (PES) of benzene.
When irradiated with 254 nm light, benzene rearranges in the S1 state to benzvalene and fulvene, and when irradiated by light in the range of 165–200 nm, it also forms Dewar benzene.23 Irradiation of benzene by 254 nm light in nucleophilic solvents leads to derivatives of bicyclo[3.1.0]hexene 1 (Scheme 1),24 a reaction that was established for 1,3,5-tri-tert-butylbenzene,24 benzene,25 and tert-butylbenzene several decades ago.25 However, the low reaction yields, the long reaction times, and the observed complex mixtures of products are severe drawbacks. Hence, the products of the photorearrangement of simple substituted benzenes have never been utilized synthetically in an applied context. A renewed analysis of the reaction, including considerations of excited-state antiaromaticity relief and the electronic structure of the S1 state, can provide insights that potentially are useful for synthetic method development. Here, it can be noted that the bicyclo[3.1.0]hexane scaffold is a useful building block for the construction of molecules with rigid three-dimensional shape and unique biological activity, such as glutamic acid analogues,26 β-arabidofuranosyl derivatives,27 nucleoside-based pharmaceuticals,28 and nucleic acid analogues.29 Thus, the photoreaction investigated herein turns a simple flat molecule into a complex three-dimensional scaffold with potential utility in one step.
Scheme 1. Photochemical Rearrangement and Addition of Nucleophilic Solvent to Benzene under Acidic Conditions Leading to Bicyclo[3.1.0]hexenyl Derivatives.

Formally, the photorearrangement of benzene to 4-methoxybicyclo[3.1.0]hex-2-ene (1, Nu = OMe) in methanol represents an addition of a proton and a methoxide ion onto benzene. The reaction is accelerated in acidic media,30 as compared to the neutral solution, and one can thus argue that it is catalyzed by acid and that protonation could play an important role in the reaction mechanism. Yet, if this is the case, how does protonation play a role?
The photochemical reaction of benzene in nucleophilic media leading to substituted bicyclo[3.1.0]hexenes previously gained considerable attention, and several different mechanisms were proposed.4,25,31−33 Our hypothesis is that the ESAA character of benzene impacts its photoreactivity in one of two ways: either through increased basicity (proton affinity) facilitating protonation of benzene in its S1 and T1 states followed by structural rearrangements (mechanism A, Scheme 2), or through the unimolecular rearrangement of S1-state antiaromatic benzene to the prefulvene conical intersection which leads to benzvalene (mechanism B). A third possible mechanism in which benzene is protonated in the S0 state, followed by excitation of the formed benzenium ion, occurs only in an extremely strong superacid, FSO3H-SbF5-SO2CIF.34 In fact, benzene in the S0 state remains unprotonated even in very strong acids such as triflic acid.35 Two other mechanistic proposals earlier turned out to be invalid—more specifically, mechanisms that involved either prefulvene as a reactive intermediate4,31 or a cis,cis,trans-cyclohexa-1,3,5-triene with Möbius topology.36
Scheme 2. The Two Mechanisms Explored, A and B, for the Photorearrangement of Benzene in Nucleophilic Media (here MeOH) Leading to 4-Methoxy-bicyclo[3.1.0]hex-2-ene (1).

Note that benzene in the T1 state can act as a triplet sensitizer of benzvalene 4, which ring-opens to the S0 state of benzene.
With this foundation we used a combined computational and experimental approach to reinvestigate the photochemical rearrangement of benzenes to bicyclo[3.1.0]hexenes in nucleophilic media under acidic conditions. A complex product with several stereogenic centers and further possibility of functionalization is formed as a result of a single photochemical step from simple and inexpensive starting materials. In this context, we demonstrate the potential of the reaction through preparation of an optically pure single isomer of a bicyclo[3.1.0]hexene derivative in two steps starting from benzene. Although the reaction still has apparent limitations for synthesis, the deepened mechanistic insights could allow for the future identification of conditions and reagents that pave the way to similar photoreactions with widened scopes for synthetic applications. Focus in our investigation is given to the impact of ESAA on the photoreactivity of simple substituted benzenes, and it is revealed that relief of S1-state antiaromaticity is a strongly contributing factor to the observed photochemical deformation of the benzene ring, which ultimately leads to the substituted bicyclo[3.1.0]hexenes.
Results and Discussion
We begin with an exploration of the reaction scope and the description of the isolation of an enantiomerically pure bicyclo[3.1.0]hexene derivative. The two mechanistic alternatives of Scheme 2 are subsequently investigated. We also consider two 6π-electron heterocycles, silabenzene and the pyridinium ion, and place the findings on these in relation to what we observe for substituted benzenes. A lower symmetry of the S1-state electronic structure than that of benzene is key to achieving a high selectivity in the photorearrangements, and such a symmetry lowering is provided by heteroatoms and certain substituents.
Reaction Scope
We first re-examined the synthetic potential of the reaction. The earliest studies reported low reaction yields (few percent) and long irradiation times (weeks).24,25 Since then, several substrates (benzene,25,33 toluene, tBu-benzene,25,37o-xylene, tri-tBu-benzene,24 benzonitriles,38−40 and trifluoromethylbenzene and toluenes39) have been reported to undergo the photoinduced rearrangement and addition of methanol,24,33,41 acetic acid,25,32 water,25,32 and 2,2,2-trifluoroethanol24,25,32,38−40 under acidic conditions (Figure 1). The quantum yields for several of the substrates were low (∼0.05).24,30 Also, the published reactions have never been used before for synthetic applications.
Figure 1.

Previously reported (i) substrates, (ii) nucleophiles for photoinduced rearrangement and addition to simple arenes, and (iii) observed products.
Irradiation of simple arenes leads not only to a mixture of regio- and stereoisomers of substituted bicyclo[3.1.0]hexenes but also to fulvene and its polymers4,31 as well as to phototransposed products.39,42 Furthermore, the bicyclic photoproducts 1 are unstable and decompose to fulvenes and polymeric material in acidic as well as in basic media.25 Moreover, different isomers of 1 are known to interconvert between each other, either by acid-catalyzed epimerization32 and racemization,32 by the triplet-benzene-sensitized vinyl cyclopropane rearrangement,33 or by the bicyclo[2.1.1]hexene rearrangement37 (Scheme 3, pathways a–d). Consequently, the photoproduct composition changes over the course of the reaction as secondary photochemical and thermal processes scramble the isomers, leading to complex mixtures. Still, some isomers are more stable than others, and they accumulate in the system after prolonged irradiation.
Scheme 3. Rearrangement Mechanisms of Bicyclo[3.1.0]hexenes: (a) Acid-Catalyzed Epimerization,32 (b) Acid-Catalyzed Racemization,32 (c) Sensitized Vinylcyclopropane Rearrangement,33 and (d) Sensitized Rearrangement through [2.1.1]Hexenyl Derivative 6(37).

We now optimized the reaction conditions for the rearrangement of benzene in acidic water and acetic acid, as well as that of tBu-benzene in acidic methanol (Scheme 4). The reaction yields were optimized by choosing an optimal (i) concentration of the substrate, (ii) amount and strength of the acid used, and (iii) irradiation time (for details see Supporting Information, section 2). We also ran the reaction in a FEP (fluorinated ethylene propylene) tubing flow reactor which minimized the irradiation of the reaction mixture and limited the secondary photoprocesses. Finally, we checked the substrate scope of the reaction by testing a series of simple substituted aromatic compounds (Scheme S1 and Table S3). However, for most of the tested substituents the reaction either did not proceed (polar groups, O- and S-containing arenes) or led to very complicated mixtures and/or to other photochemical transformations (anilines and halobenzenes).
Scheme 4. Main Photoaddition Products of the Tested Model Substrates.

Reaction conditions: 70 mM substrate, Rayonet reactor, FEP tube reactor, irradiation time 30 min, (a) 1 mM AcOH (aq.), (b) AcOH (conc.), (c) 1.25 mM dichloroacetic acid in MeOH. Isolated reaction yields of the major isomerization product are shown.
It should be noted that the limits in the substrate selectivity have previously been studied computationally for benzonitriles and anisoles.43 For methylbenzonitrile, prefulvenic biradical structures were found to be stabilized by the cyano group through π-conjugation, allowing for interconversion between the different isomers. In methylanisoles, on the other hand, the photoisomerization is not possible because biradical prefulvenic structures cannot be formed due to the lack of π-conjugation between the C4 atom and the methoxy group, which results in higher barriers in the S1 state. Yet, we found that one substituent, the silyl group, yields arenes with suitable properties for this reaction. Silyl-substituted benzenes react efficiently, and their reactivity increases with higher alkyl substitution at the silicon atom (for the reactivity of a series of methylated phenylsilanes PhSiHnMe3-n (n = 0, 1, 2, or 3), see Table S4 and Figure S1). Indeed, a test for a linear free energy relationship between the log(k/k0) and the Hammett σ constants of the silyl substituents of the four silyl-substituted benzenes reveals an increase in reaction rate with increased electron-donating ability of the substituent, yet the relationship is nonlinear, reflecting an impact of steric bulk on the reaction rate (see Figure S2).
Trimethylsilylbenzene (TMS-benzene) had the optimal reactivity, while dichloroacetic acid (DCA) was found to have the optimal acidity (more acidic media cause decomposition of the formed products; less acidic media slow down the photoreaction). Interestingly, the main silylated photoproduct 1d had a different constitution from the tBu-analogue 1c, which possibly can be rationalized by either (i) elimination of the TMS group from the other positions occurring in the photoequilibration or by different substituent effects, or (ii) stabilization of a transition-state structure or intermediate by the silyl group (vide infra).
While the substrate scope of the photorearrangement is limited and the reaction conditions need to be carefully optimized, the reaction generates highly complex three-dimensional structures from simple and easily accessible substrates in one step. Consequently, we attempted to prepare a single enantiomer of the bicyclo[3.1.0]hexenyl photoproduct. Whereas direct methods of enantioselective synthesis turned out to be unsuccessful due to racemization of the photoproduct in the acidic reaction media, we also attempted the chiral resolution of the product (±)-1a. Enzymatic kinetic resolution by esterification with Candida antarctica lipase B (CALB) was attempted, yet it could not be optimized to give high enantiomeric excess (ee) (Supporting Information, section 4.4). Instead we managed to prepare diastereomeric esters 7 with Boc-l-Phe (Scheme 5) which could be successfully separated by HPLC. Vibrational circular dichroism spectra44 were measured on the separated diastereoisomers in CDCl3 solution. Comparison of experimental and calculated spectra as well as an X-ray crystal structure determination enabled assignment of the absolute stereochemistry. Further details can be found in the Supporting Information, section 3.
Scheme 5. Synthesis of Diastereomeric Esters 7.

Mechanism Investigations
The two mechanistic pathways shown in Scheme 2 (pathways A and B) both involve excitation of benzene to S1 as the first step. One may consider intersystem crossing, but it has been shown experimentally through quenching with various triplet-state quenchers that the photorearrangement does not proceed in the T1 state,4 and we can confirm this computationally (vide infra). Moreover, when benzene is excited to the S2 state, Dewar benzene is formed in addition to benzvalene and fulvene.23 Hence, one can conclude that an S1-state process is likely. In this state, benzene either becomes protonated to form a benzenium cation that rearranges to the bicyclo[3.1.0]hexenium cation 3 (pathway A, Scheme 2), or rearranges to benzvalene 4(45) via the prefulvenic transition state 5 and an S1/S0 conical intersection (pathway B).46
Both mechanistic pathways involve relief of the S1-state antiaromatic character of benzene, but in different ways. Many pieces of evidence have been acquired for pathway A31,32 and pathway B,24,33 but no previous study has unambiguously confirmed one mechanism over the other. With regard to ESAA relief, any pathway that allows for disruption of the 6π-electron cycle which is antiaromatic in S1 will provide for such relief. Which pathway is followed depends on (i) the excited-state lifetime of S1-state benzene, as this determines the effective concentration of the reactive S1-state benzene, (ii) the concentration and strength of the acid, and (iii) the activation barriers for the different reaction steps. Thus, ESAA relief combined with other factors (concentrations and excited-state lifetimes) impacts which mechanism is followed. Further factors that can have an impact are the energy penalty for distortion of the σ-framework from the hexagon, which is ideal for sp2-hybridized C atoms, and the ability of substituents to reduce the antiaromaticity of a benzene ring in its S1 state, similar to what was recently found for the T1-state antiaromaticity.47
Mechanism A
In pathway A, the ESAA is alleviated through protonation, as this breaks the cyclic π-conjugation and transforms the S1-state benzene into an initially non-aromatic benzenium ion with a linear pentadienyl cationic segment with four π-electrons (Scheme 6). On the basis of such ESAA relief, we postulate that benzene is more basic and has a higher proton affinity in its antiaromatic S1 state than in its S0 state. Indeed, benzene has a reported proton affinity which is higher in the S1 state than in the S0 state by 26 kcal/mol.48 This is in line with an earlier observation on magnitude reversals in proton and hydride affinities of 4nπ- and (4n+2)π-electron (benz)annulenyl anions and cations, respectively, caused by switches in aromaticity/antiaromaticity when going from their lowest singlet to their lowest triplet states.49
Scheme 6. Formation of the S1-State Homoaromatic Structure through Relaxation of the Excited Benzenium Cation.

With regard to the benzenium cation in the S1 state, previous computations revealed that this species adopts a markedly puckered structure, placed close to an S1/S0 conical intersection.50 This puckered benzenium cation could decay to the S0 state and collapse to bicyclo[3.1.0]hexenium cation 3 (Scheme 6). Subsequent nucleophilic attack on this cation would occur from the less sterically hindered exo side. Now, why does the benzenium ion pucker in the S1 state? Recently, we explored the existence of homoaromaticity in the excited states, primarily in the T1 state, but a few molecules with S1-state homoaromaticity were also identified.51 Thus, a driving force for the puckering of the benzenium ion should be the attainment of excited-state 4π-electron homoaromaticity, which is also in line with the disrotatory path for a photochemically allowed electrocyclic ring-closure of a linear polyenyl chain with four π-electrons.52−54
One may ask if a T1-state homoaromatic benzenium ion could be involved, but as noted above, earlier experiments showed that the reaction proceeds in the S1 state.4 Further support for this conclusion is found through computations (Figure 2). Although the benzenium ion has a clear T1 homoaromatic structure,51 decay from this Cs-symmetric structure to the S0 state leads back to the C2v-symmetric benzenium ion 2 and not to the bicyclo[3.1.0]hexenium cation 3. Also, there is no stationary point in the T1 state that corresponds to a bicyclo[3.1.0]hexenium ion (the vertically excited T1-state structure of 3 is a non-stationary point 58.9 kcal/mol above the S0 state). Thus, if mechanism A is operative, it must involve formation of a benzenium ion in the S1 state. Now, to what extent does protonation of S1-state benzene lead to ESAA relief?
Figure 2.
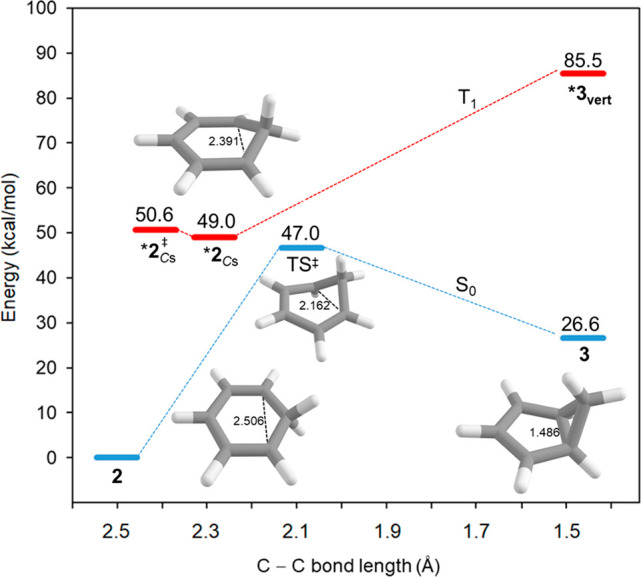
Reaction coordinate for benzenium cation (2) leading to bicyclo[3.1.0]hexenium cation (3) in the S0 and T1 states calculated at the (U)B3LYP/6-311+G(d,p) level.
S1-State Protonation and ESAA Relief
Clearly, excited-state protonation of benzene disrupts the antiaromaticity, as revealed by changes in the NICS(1)zz values [ΔNICS(1)zz = NICS(1; benzene)zz – NICS(1; benzenium cation)zz] between benzene and the benzenium ion in their S1 and T1 states when optimized with C2v symmetry. This reveals large reliefs in ESAA as the benzenium ion in C2v symmetry is non-aromatic (ΔNICS(1)zz = −55.6 and −28.3 ppm in S1 and T1, respectively, see Table 1; for DFT results, see Table S34). In contrast, protonation of benzene in the S0 state leads to a loss of aromaticity (ΔNICS(1)zz = 16.0 ppm). This antiaromaticity relief occurs upon protonation of benzene in the S1 and T1 states, while aromaticity loss upon protonation in the S0 state is reflected in the higher calculated proton affinities in the excited states (Table 1), in line with the earlier experimental finding.48 Furthermore, through comparison of the calculated proton affinities of benzene with those of cyclooctatetraene (COT, Table 1), it is revealed that the proton affinities of benzene in the two excited states are similar to that of COT in the S0 state. Interestingly, while COT upon protonation in S0 turns into the aromatic homotropylium ion,55 the benzenium ion puckers to Cs symmetry and turns homoaromatic in T1 and possibly also in S1. The same applies for the proton affinities of the planar D8h-symmetric COT in the S1 and T1 states when compared to those of benzene in S0 (Table 1). Opposite effects of aromaticity and antiaromaticity, when compared to benzene, are observed in the S0 and T1 states of COT, while its S1 state displays a different pattern according to NICS, likely due to methodological issues, as it follows the expected trend when other indices are used (see the Supporting Information, section 7.10).
Table 1. Calculated Gas-Phase Proton Affinities (PA, kcal/mol) and Changes in NICS (ΔNICS(1)zz, ppm) upon Protonation of Benzene and Cyclooctatetraene (COT) in the S0, S1, and T1 Statesa.
| S0 |
S1 |
T1 |
||||
|---|---|---|---|---|---|---|
| compound | PA | ΔNICS(1)zz | PA | ΔNICS(1)zz | PA | ΔNICS(1)zz |
| benzene | 183.1 | 16.0 | 214.7 (C2v, i)b | –55.6 | 217.0 (C2v, i)b | –28.3 (C2v, i)b |
| (181.3)d | 232.1 (Cs, HA)b | 218.4 (Cs, HA)b | –48.2 (Cs)b | |||
| (207.3)d | ||||||
| COT | 220.8 (Cs, HA)c | –6.7c | 182.8 | –18.7 | 195.0 | 15.3 |
Proton affinities at (U)- and (TD-)B3LYP levels and ΔNICS(1)zz values at CASSCF//B3LYP. NICS(1)zz at CASSCF//B3LYP, B3LYP//B3LYP, and CASSCF//CASSCF levels are given in Table S34. For further computational details and additional discussion, including electronic aromaticity index (MCI and FLU), see Table S36 and section 7.9 in the Supporting Information. ΔNICS(1)zz = NICS(1; non-protonated)zz – NICS(1; protonated)zz.
Benzenium cation in the T1 and S1 states has homoaromatic (HA) minimum (Cs), but the values for PA and ΔNICS related to the proton addition to the planar benzene are also given and indicated as “(C2v, i)” (i = imaginary frequency). The NICS(1)zz value of the Cs-symmetric S1 structure cannot be determined as it is too close to the conical intersection.
The COT and COTH+ minima are strongly puckered, and COTH+ has homoaromatic (HA) character. NICS(1)zz values for both puckered and planar structures are given in Table S34.
Experimental values from ref (48).
Fluorescence Quenching Attempts
Protonation of benzene in S1 seems plausible, but in order to examine mechanism A experimentally, we performed a fluorescence quenching study of benzene and TMS-benzene in anhydrous methanol. The fluorescence of benzene is known to be quenched by dissolved oxygen,56 a phenomenon which has not yet been fully explained.57,58 The quenching was therefore performed with degassed solutions. However, no fluorescence quenching was observed in a concentration range of the acid up to 1.25 M (anhydrous HCl in MeOH) for both TMS-benzene and benzene (Figures 3 and S32, respectively). This finding reveals that there is no interaction of the acid with benzene in its S1 state. As the photorearrangement occurs in the S1 state (vide supra),4 it can be concluded that excited-state protonation is not operational despite the enhanced proton affinity of S1-state benzene. On the other hand, in experiments with benzene dissolved in THF/9-BBN mixtures, where 9-BBN is used as a Lewis acidic co-solvent with a potential ability to coordinate to S1-state benzene, an increased fluorescence quenching is observed with higher concentration of 9-BBN (see Figure S33).
Figure 3.
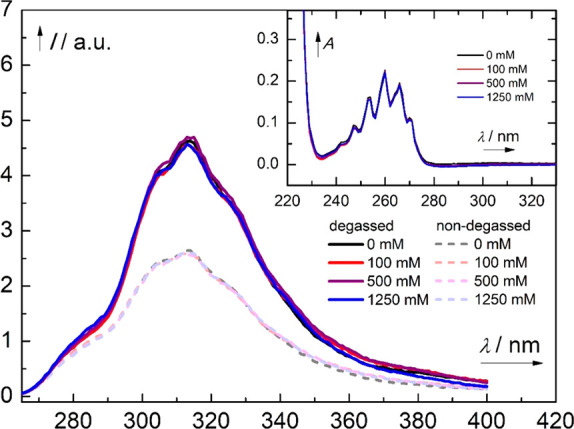
Fluorescence spectra of TMS-benzene (c = 1.0 mM, λexc = 254 nm) in anhydrous MeOH, degassed (solid line, purged with argon) and non-degassed (dashed line) in the absence and presence of anhydrous HCl (c = 0–1250 mM). The absorption spectra are shown in the inset.
So why is there no fluorescence quenching upon increased concentration of anhydrous HCl? The excited-state lifetime of benzene in the S1 state is 28 ns,59 and with the conditions used in our experiments, the effective concentration of S1-state benzene (based on its lifetime) can be estimated to merely ∼10–13 M (see Supporting Information, section 5.4). Thus, there is an extremely low concentration of benzene molecules in their S1 states available for protonation (note here also that protonation of benzene in the S0 state occurs only in exceptionally strong superacids).34,35 Thus, if mechanism A is followed, it should require an essentially barrierless S1-state protonation.
Indeed, protonation of benzene in its S1 state is barrierless according to our CASPT2//CASSCF calculations (Figure S37). Furthermore, the proton migration within MeOH should be rapid, as the free energy of activation for proton transfer in a (MeOH)8H+ cluster, used as protonated methanol clusters (MeOH)nH+ saturate at n = 8 or 9,60 is calculated to be merely 1.3 kcal/mol with an implicit solvation model. This resembles the experimentally determined activation barriers (2–3 kcal/mol) for proton migration in water and methanol.61,62 Additionally, concerted proton transfer in both solvents can involve tunneling.63,64 Thus, the computational data speak for mechanism A but the fluorescence quenching does the opposite. Clearly, there are features that make mechanism B more beneficial, the most apparent being the concentration of benzvalene in the S0 state (included in mechanism B), which is ∼10–3 M,45 i.e., 10 orders of magnitude higher than that of S1-state benzene (∼10–13 M). Protonation of benzvalene in the S0 state, provided it proceeds over a low activation barrier, can therefore be much more efficient than protonation of S1-state benzene. Indeed, it has been observed earlier that benzvalene is rapidly attacked by nucleophilic methanol to yield the methoxy adducts 1 in the S0 state.45
Mechanism B
This pathway begins with a unimolecular rearrangement to benzvalene 4 via the prefulvenic transition state, and the puckering of the benzene ring leads to attenuation of the cyclic π-conjugation and relief of ESAA. NICS(1)zz values computed at each point along the intrinsic reaction coordinate (IRC) in the S1 state show that antiaromaticity is alleviated along this pathway (Figure 4). Clearly, benzene in the S1 state is antiaromatic, as evidenced by a highly positive NICS(1)zz value of 99.5 ppm at the vertically excited structure and 80.9 ppm at the S1-state minimum. The prefulvenic transition state, on the other hand, is already moderately influenced by aromaticity, as the structure of highest energy along the S1-state CASPT2//CASSCF surface has a NICS(1)zz value of −10.1 ppm, while at the CASSCF transition-state NICS(1)zz = −21.2 ppm (for S0-state benzene NICS(1)zz = −27.4 ppm). Thus, puckering of benzene in S1 alleviates ESAA, yet is ESAA the factor that triggers the structural deformation?
Figure 4.
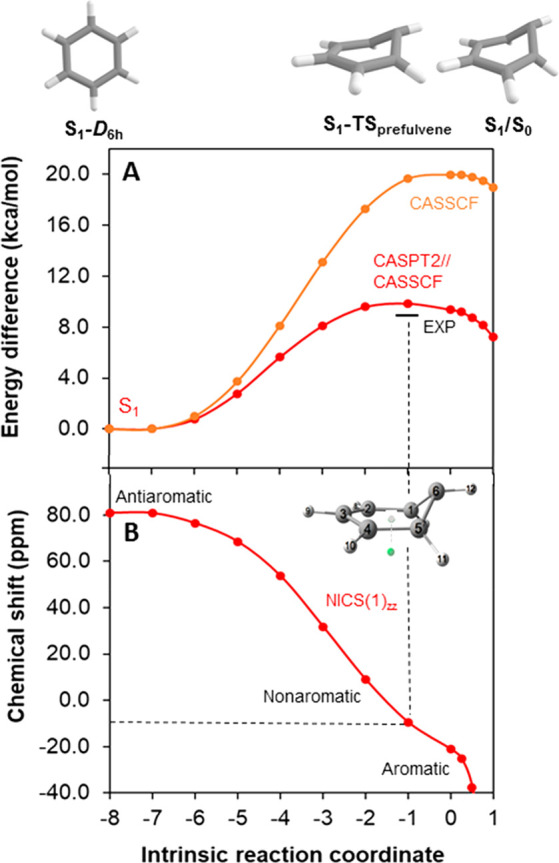
(A) Potential energy surface of benzene in the S1 state from S1 planar minimum (D6h symmetry) until the S1/S0 conical intersection. The experimentally determined activation energy (∼8.6 kcal/mol) given in ref (69) is indicated by a line. (B) The NICS(1)zz at the various structures along the IRC. Geometries were calculated at the SA4-CASSCF(6,6)/ANO-RCC-VTZP level, energies were obtained at the MS4-CASPT2(6,6)/ANO-RCC-VTZP level, and NICS(1)zz values were determined at the CASSCF(6,6)/6-31++G(d,p) level. The plane defined by the C1, C2, C4, and C5 atoms was used to define the central position of the ghost atom located 1 Å below the plane (displayed as a green dot in the inset) for NICS. For computational details and NICS values in the opposite direction, see Figure S47.
Origin of the Activation Barrier and ESAA Relief
The S1-state barrier observed computationally for the photochemical transformation of benzene to the prefulvenic S1/S0 conical intersection, leading to benzvalene, is well-established from experimental and theoretical studies.65−67 More specifically, the barrier lies ∼3000 cm–1 (8.6 kcal/mol) above the S1 planar minimum, and as soon as the excess vibrational energy is enough to overcome it, the non-radiative process known as “channel 3” is opened.68−75 Access to “channel 3” leads to the loss of fluorescence and opening of the pathway that leads to the prefulvenic conical intersection and to benzvalene. Now, in order to obtain a reasonable agreement between experiment and computations with regard to the activation barrier, both static and dynamic electron correlations have to be included in the computations—while a CASPT2//CASSCF calculation gives a barrier of 9.4 kcal/mol, the barrier is exaggerated at the CASSCF level (18–20 kcal/mol).65,67 Yet, why is there an activation barrier if ESAA is relieved?
Benzene in the S1 state (1B2u) is D6h symmetric and described to equal extents by two electron configurations coupled in-phase, while the out-of-phase combination constitutes the higher-energy 1E1u state. It has been concluded that the S1 state is strongly stabilized by the in-phase coupling.76,77 Upon distortion away from the D6h symmetry toward the prefulvenic structure, the S1 state (former 1B2u) increases in energy while the higher-energy (former 1E1u) state decreases in energy, leading to an avoided crossing and an activation barrier on the S1 PES along the puckering coordinate. Interestingly, it has been revealed that the activation barrier vanishes when going to the less symmetric pyrazine,77 and a lack of activation barrier has also been observed for silabenzene.78 We now confirm this feature for silabenzene at the CASPT2//CASSCF level (Figure 5), and also for the pyridinium cation, which has a photochemistry which is of higher synthetic value than that of pyrazine.79−84 Yet, at the lower CASSCF level, which contains only static electron correlation, there are still activation barriers, although they are smaller than for S1-state benzene. Thus, the activation barrier observed in the S1 state of benzene is traceable to its high symmetry (for comparisons between the S1 states of benzene, silabenzene, and pyridinium ion, see Table S19). Here, it should be pointed out that the structural distortion going from the S1-state minimum to the transition-state structure is much less unfavorable in S1 than in S0 because the energy difference between these two structures in S0 is 84.3 kcal/mol (see Figure S48), compared to 9.4 kcal/mol in the S1 state. This large difference in energy loss (74.9 kcal/mol) upon an identical distortion in S0 vs in S1 indicates that the puckering goes with aromaticity loss in S0 and antiaromaticity relief in S1. Yet, a precise assessment of the various contributions is not possible, as this energy penalty also includes energy contributions to distort the σ-orbital skeleton of the sp2-hybridized C atoms from its ideal hexagon (see Figure S48 and the related discussion in the Supporting Information for an estimate).
Figure 5.

Potential energy surfaces at the CASPT2//CASSCF and CASSCF levels from the S1 minimum to the S1/S0 conical intersection for (A) silabenzene and (B) the pyridinium cation. Respective NICS(1)zz for (C) silabenzene and (D) the pyridinium cation. For further computational details, see Supporting Information, section 7.2.
Similarly to benzene, drastic decreases in S1 antiaromaticity, determined by NICS(1)zz, are observed for the S1-state silabenzene and pyridinium ion when going from the CASSCF S1-state minima toward the S1/S0 conical intersections (Figure 5C,D). Notably, S1-state silabenzene at the CASSCF level has an optimized nonplanar structure with a considerably reduced antiaromaticity (33.3 ppm) compared to that of S1-state benzene at its D6h minimum (80.9 ppm). Yet, when regarding the NICS(1)zz of vertically excited silabenzene and the pyridinium ion (99.3 and 91.2 ppm, respectively), it is apparent that the ESAA relief prior to the CASSCF S1-state minimum of silabenzene is extensive. Clearly, ESAA alleviation seems to be a general process for 6π-electron cycles with S1 states of ππ* character. The photochemistry of both silabenzene and pyridinium salts is established (for a summary, see Supporting Information, section 6),85,86 and the mechanism and synthetic applications of pyridinium ion photochemistry (Scheme 7) have been reviewed extensively.79−84 Hence, the ESAA relief of 6π-electron cycles is a feature that triggers these molecules to undergo photorearrangements that can be of general synthetic utility. For example, 6-azabicyclo[3.1.0]hexene, formed photochemically from pyridinium salts, can efficiently be transformed to polysubstituted cyclopentenes by acid-promoted ring opening (Scheme 7).87,88
Scheme 7. Photochemistry of Pyridinium Salts and Acid-Promoted Ring Opening of the Bicyclic Product.

Substituent Effects
Now returning to benzene, with mechanism B and the fact that lowered symmetry lowers the activation barrier for puckering, we can explain why the TMS- and tBu-substituted benzenes have higher reaction rates than the parent benzene. First, the increased reaction rate of TMS-benzene is not an effect of an increase in S1-state antiaromaticity when the electron-donating ability increases because the calculated S1-state antiaromaticity of TMS-benzene is similar to that of benzene (NICS(1)zz = 78.8 and 80.9 ppm, respectively). Instead, when benzene, TMS-benzene, and tBu-benzene are calculated at the same computational level, the activation barrier for puckering of benzene in the S1 state is 9.9 kcal/mol, while the barriers for TMS-benzene and tBu-benzene are 7.6–9.7 and 7.8–9.9 kcal/mol, respectively, depending on the position of the substituent group (see Table S26). Thus, the lowest activation barrier for puckering of the S1-state TMS-benzene, leading toward the prefulvenic conical intersection, is ∼2 kcal/mol lower than for S1-state benzene. This difference becomes slightly larger when considering ΔG⧧, as the puckering toward the prefulvenic conical intersection with the TMS group at the 6-position is 3–4 kcal/mol lower than the free energies of activation along the other paths and ∼7 kcal/mol lower than that of benzene (Table S26). Additionally, the CAr–Si(Me3) bond at this transition-state structure of TMS-benzene is distinctly shorter (1.865 Å, Figure S43) than at the other transition-state structures (1.888–1.895 Å), indicating a hyperconjugative interaction.
As seen for silabenzene and the pyridinium ion, heteroatoms distort the description of the S1 state so that one of the two electron configurations that are degenerate in the S1 state of benzene becomes more dominant. This can also be achieved to a smaller extent by certain substituents at the benzene ring, such as the TMS group (Table S24). As a consequence, these substituted benzenes become guided by their electronic structures toward one of the transition-state structures, and at the transition state, as just noted, substituents that are able to interact hyperconjugatively provide for stabilization. Thus, there are benzene-to-prefulvenic S1-state pathways for the two substituted benzenes that involve slightly lower activation barriers than what is the case for the parent benzene. In summary, substituents that (i) lead to a difference in the weights of the two electron configurations that describe S1-state benzene and (ii) are able to interact through (hyper)conjugation with the C6 atom are useful as they will provide access to primarily one of the prefulvenic S1/S0 conical intersections. The TMS group is one such group; others will be reported in forthcoming studies.
Stereochemistry of Mechanism B
Based on mechanism B, how can the observed stereochemistry be rationalized? The preferential formation of exo adducts has previously been used as the main argument for the intermediacy of bicyclo[3.1.0]hexenium ion 3 (mechanism A in Scheme 2),31 as the nucleophile should attack from the sterically less hindered exo face. This was explored by Berson et al., who found the main observed photoproduct of irradiation of benzene in acidic D2O to be exo-8, which was found to be formed by epimerization of the primary photoproduct endo-8 (Scheme 8a).32 He also observed selective endo incorporation of deuterium in position 6 and no addition of deuterium from the exo site leading to 9. We now confirm this through a study of the photoreaction of benzene-d6 in aqueous solvent under acidic conditions (Scheme 8b). The only observed product was 10, which has the H atom in the 6-endo position and the hydroxyl group in the 4-exo position. This excludes any intermediacy of the benzenium cation (mechanism A), as its puckering would scramble the position of the deuterium/protium label in the 6 position (Scheme 8c). Instead, it is consistent with the concerted attack on the benzvalene tricyclic structure and the subsequent epimerization (Scheme 8d). This can be either concerted or stepwise involving a contact ion-pair. The epimerization goes via protonation of the MeO group, and the exo-8 isomer is lower in energy by 1.4 kcal/mol than the endo-8 when using methanol as implicit solvent (Figure S40). The epimerization of endo-8 to exo-8 is therefore a thermodynamically controlled process and does not scramble the isotope label at the 6-position.
Scheme 8. Isotopic Labeling Experiments of Photoaddition of Water to Benzene.

Indeed, the proton affinity of benzvalene in its S0 state is high: 240 kcal/mol in the gas phase at the B3LYP/6-311+G(d,p) level, i.e., higher than that of S1-state benzene and even higher than that of S1-state tBu- and TMS-benzene (see Table S38). This supports protonation of benzvalene in the S0 state, as its concentration in our experiments is 10 orders of magnitude higher than that of S1-state benzene (vide supra). Moreover, the addition reaction is stepwise according to our computations, progressing with an activation free energy of 7.4 kcal/mol in the first step. The MeOH2+ approaches from the endo face, leading to incorporation of the proton in the 6-endo position, in accord with the reported photolysis experiments using deuteriophosphoric acid.32 The activation free energies for the subsequent methanol addition are 10.9 kcal/mol (exo) and 15.5 kcal/mol (endo), respectively.
Cause of Low Quantum Yield
Finally, one can ask why is the quantum yield for the photoreaction not higher than 0.045?30 Experiments reveal that a photostationary state evolves in a solution with both benzene and benzvalene present. Furthermore, earlier experiments give E(T1; benzene) = 84.1 kcal/mol89 and E(T1; benzvalene) ≈ 65 kcal/mol,90 while our (U)B3LYP/6-311+G(d,p) computations give E(T1) for the two compounds at 83.5 and 60.3 kcal/mol, respectively. Also, the calculated energy needed to reach the vertically excited T1 state of benzvalene (79.3 kcal/mol) is slightly lower than the triplet energy of benzene (83.5 kcal/mol). As benzene has a non-negligible quantum yield for intersystem crossing (ΦISC = 0.25),91 it will exist in both its S1 and T1 states when excited. Thus, due to its high E(T1) and the energy match, benzene can transfer its T1 energy to benzvalene. Now, when benzvalene becomes excited to the T1 state, it will rearrange to T1-state benzene through a series of transformations, progressing over low activation barriers according to our calculations (Figure 6). Furthermore, this rearrangement of T1-state benzvalene to T1-state benzene, via a T1-state prefulvene intermediate instead of a transition state as on the T1 PES (vide supra), is highly exothermic (∼60 kcal/mol). Combined, this means that benzene has the ability to catalytically sensitize the back-reaction from benzvalene to benzene, i.e., act as a catalytic sensitizer. Thus, based on the computed S0 and T1 PESs, it becomes clear that the low quantum yield (Φ = 0.045)30 could be overcome if T1-state benzene (or a derivative) were unable to transfer its triplet energy to benzvalene.
Figure 6.

Back-reaction from benzvalene to benzene initiated by formation of T1-state benzvalene through triplet energy transfer from T1-state benzene (inset), and the T1 and S0 potential energy curves for its rearrangement to T1 benzene, calculated at the (U)B3LYP/6-311+G(d,p) level (electronic energies include ZPE corrections; vertically excited energies given in parentheses are purely electronic energies). Energies given in red are relative to S0-state benzene, and energies given in blue are relative to S0-state benzvalene.
To probe the ability of benzene to act as a catalytic sensitizer, we investigated the effect of the acid and benzene concentrations on the reaction rate and yield. Benzvalene has earlier been shown to give methanol adduct 1 in acidic methanol (Scheme 2),45 yet as just noted, benzvalene rearranges to benzene upon T1 benzene-induced sensitization.92 Indeed, high concentrations of TMS-benzene resulted in low yields of the bicyclic photoproducts (for details see Table S1), and the reaction also occurred inefficiently in pure methanol without addition of acid. Thus, benzvalene undergoes a slow reaction in neutral methanol, and this process is enhanced by acid catalysis. A way to increase both the reaction yield and the quantum yield for formation of the bicyclo[3.1.0]hexene would therefore be to identify (hetero)benzenes which in their T1 states are unable to transfer their triplet energies to the corresponding benzvalene analogues.
Taken together, the experimental and computational evidence reveals unequivocally that mechanism B is operative, knowledge that could be useful for the rationalization and development of similar photoreactions.
Conclusions and Outlook
It is known that benzene becomes antiaromatic in its S1 and T1 states,11,14−16 and that relief of this destabilizing antiaromatic character is a driving force for photochemical reactions of various benzene derivatives in their T1 states.20,22 Herein we show that S1-state antiaromaticity relief of the benzene molecule itself is strongly involved in the photochemical process that leads up to formation of substituted bicyclo[3.1.0]hex-2-enes in nucleophilic media under acidic conditions. Through combined quantum chemical and experimental studies, we deduce that the first step in the mechanism involves the puckering of S1-state benzene, which after a conical intersection leads to benzvalene. Importantly, the puckering of S1-state benzene to the prefulvenic S1/S0 conical intersection is strongly connected to relief of S1-state antiaromatic character, despite the fact that it passes over an activation barrier of 9.4 kcal/mol. Similar S1-state antiaromaticity relief occurs in silabenzene and the pyridinium ion—and in these species without activation barriers, a feature that results from a lowered symmetry of the electronic structure. Certain substituents at the benzene ring (e.g., a trimethylsilyl group) can, similarly to a heteroatom, lower the activation barrier so that S1-state TMS-benzene preferentially follows the path to the prefulvenic conical intersection with the TMS group in the 6-position.
When in the S0 state, nucleophilic attack by MeOH at the benzvalene yields the substituted bicyclo[3.1.0]hex-2-enes, with the highest yield (75%) for TMS-benzene. The synthetic accessibility to the complex bicyclo[3.1.0]hexenyl scaffold offers an interesting possibility for its further utilization in synthetic applications. Generation of three chiral centers in one simple step is often difficult in thermal chemistry. We show this to be a result of a process triggered by ESAA relief of benzene photoexcited to its S1 state.
We further reveal limiting factors which, if they can be bypassed or removed, indicate how the benzene photochemistry can be further enhanced, as there are obvious drawbacks with the reaction. First, rearrangements of the initially formed products lead to scrambling of substituents. Moreover, the quantum yield for product formation is low (Φ = 0.045),30 as benzene in the T1 state (ΦISC = 0.25) transfers its T1 energy to benzvalene, which subsequently rearranges in the T1 state and thus regenerates the sensitizer in a unique process of catalytic sensitization. One should identify a 6π-electron (hetero)cycle with lower T1 energy than the corresponding benzvalene analogue, hindering the re-aromatization.
In conclusion, our combined experimental and computational investigation puts an end to a long debate over the mechanistic photochemistry of benzene, reveals the impact of excited-state antiaromaticity relief in the S1 state of benzene, and opens up possibilities for new studies within the field of photochemistry of 6π-electron (hetero)cycles.
Acknowledgments
The Olle Engkvist Byggmästare Foundation is greatly acknowledged for postdoctoral fellowships to T.S., J.T., and W.R. (184-390 and 194-677). T.S. appreciates support from the Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences and the Czech Grant Academy (19-20467Y). H.O. is grateful to the Swedish Research Council (VR) for financial support (2015-04538 and 2019-05618) and to the Vinnova agency for an academia-industry exchange grant (2016-04572). R.A. is grateful to the Liljewalch Foundation for a travel grant allowing a research visit at FSU. I.A. is grateful for the support by the National Science Foundation (CHE-1465142). R.L. and I.F.G. are grateful to the Swedish Research Council (VR) for financial support (grant 2016-03398). M.N. is grateful for an Erasmus+ scholarship that allowed for a summer internship at UU. The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Center (NSC) in Linköping and at UPPMAX in Uppsala.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b13769.
Materials and methods, optimization and control experiments, synthetic procedures, reactivity studies, mechanistic experiments (fluorescence quenching, Stern–Volmer analysis, isotope labeling experiments, concentration calculations), computational details, Cartesian coordinates, structures, energies, electronic configurations and the respective weights (for CASPT2//CASSCF calculations), aromaticity indices (NICS(1)zz, MCI, and FLU values), and additional plots for PES and NICS(1)zz for benzene (list of compounds included: benzene and photochemical derivatives, protonated benzene, TMS-benzene, tBu-benzene, silabenzene, pyridinium cation, cyclooctatetraene, and protonated cyclooctatetraene), including Figures S1–S70 and Tables S1–S38 (PDF)
Author Contributions
∇ R.A. and J.T. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Gleiter R.; Haberhauer G.. Aromaticity and Other Conjugation Effects; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Schleyer P. v. R. Aromaticity. Chem. Rev. 2001, 101, 1115–1566. (special issue) 10.1021/cr0103221. [DOI] [PubMed] [Google Scholar]
- Martín N.; Scott L. T. Challenges in Aromaticity: 150 Years after Kekulé’s Benzene. Chem. Soc. Rev. 2015, 44, 6397–6400. 10.1039/C5CS90085A. [DOI] [PubMed] [Google Scholar]
- Bryce-Smith D.; Gilbert A. The Organic Photochemistry of Benzene—I. Tetrahedron 1976, 32, 1309–1326. 10.1016/0040-4020(76)85002-8. [DOI] [Google Scholar]
- Bryce-Smith D.; Gilbert A. The Organic Photochemistry of Benzene—II. Tetrahedron 1977, 33, 2459–2490. 10.1016/0040-4020(77)80069-0. [DOI] [Google Scholar]
- Mueller F.; Mattay J. Photocycloadditions: Control of Energy and Electron Transfer. Chem. Rev. 1993, 93, 99–117. 10.1021/cr00017a006. [DOI] [Google Scholar]
- Cornelisse J. The Meta Photocycloaddition of Arenes to Alkenes. Chem. Rev. 1993, 93, 615–669. 10.1021/cr00018a002. [DOI] [Google Scholar]
- Streit U.; Bochet C. G. The Arene-Alkene Photocycloaddition. Beilstein J. Org. Chem. 2011, 7, 525–542. 10.3762/bjoc.7.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann N. Photochemical Reactions of Aromatic Compounds and the Concept of the Photon as a Traceless Reagent. Photochem. Photobiol. Sci. 2012, 11, 1613–1641. 10.1039/c2pp25074h. [DOI] [PubMed] [Google Scholar]
- Remy R.; Bochet C. R. Arene-Alkene Cycloaddition. Chem. Rev. 2016, 116, 9816–9849. 10.1021/acs.chemrev.6b00005. [DOI] [PubMed] [Google Scholar]
- Papadakis R.; Ottosson H. The Excited State Antiaromatic Benzene Ring: A Molecular Mr. Hyde?. Chem. Soc. Rev. 2015, 44, 6472–6493. 10.1039/C5CS00057B. [DOI] [PubMed] [Google Scholar]
- Baird N. C. Quantum Organic Photochemistry. II. Resonance and Aromaticity in the Lowest 3ππ* State of Cyclic Hydrocarbons. J. Am. Chem. Soc. 1972, 94, 4941–4948. 10.1021/ja00769a025. [DOI] [Google Scholar]
- Ottosson H. Exciting Excited-State Aromaticity. Nat. Chem. 2012, 4, 969–971. 10.1038/nchem.1518. [DOI] [PubMed] [Google Scholar]
- a Karadakov P. B. Ground- and Excited-State Aromaticity and Antiaromaticity in Benzene and Cyclobutadiene. J. Phys. Chem. A 2008, 112, 7303–7309. 10.1021/jp8037335. [DOI] [PubMed] [Google Scholar]; b Karadakov P. B. Aromaticity and Antiaromaticity in the Low-Lying Electronic States of Cyclooctatetraene. J. Phys. Chem. A 2008, 112, 12707–12713. 10.1021/jp8067365. [DOI] [PubMed] [Google Scholar]; c Karadakov P. B.; Hearnshaw P.; Horner K. E. Magnetic Shielding, Aromaticity, Antiaromaticity, and Bonding in the Low-Lying Electronic States of Benzene and Cyclobutadiene. J. Org. Chem. 2016, 81, 11346–11352. 10.1021/acs.joc.6b02460. [DOI] [PubMed] [Google Scholar]; d Karadakov P. B.; Al-Yassiri M. A.H.; Cooper D. L. Magnetic Shielding, Aromaticity, Antiaromaticity and Bonding in the Low-Lying Electronic States of S2N2. Chem. - Eur. J. 2018, 24, 16791–16803. 10.1002/chem.201804292. [DOI] [PubMed] [Google Scholar]
- Feixas F.; Vandenbussche J.; Bultinck P.; Matito E.; Solà M. Electron Delocalization and Aromaticity in Low-lying Excited States of Archetypal Organic Compounds. Phys. Chem. Chem. Phys. 2011, 13, 20690–20703. 10.1039/c1cp22239b. [DOI] [PubMed] [Google Scholar]
- Rosenberg M.; Dahlstrand C.; Kilså K.; Ottosson H. Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations. Chem. Rev. 2014, 114, 5379–5425. 10.1021/cr300471v. [DOI] [PubMed] [Google Scholar]
- Oh J.; Sung Y. M.; Hong Y.; Kim D. Spectroscopic Diagnosis of Excited-State Aromaticity: Capturing Electronic Structures and Conformations upon Aromaticity Reversal. Acc. Chem. Res. 2018, 51, 1349–1358. 10.1021/acs.accounts.7b00629. [DOI] [PubMed] [Google Scholar]
- Kato H.; Brink M.; Möllerstedt H.; Piqueras M. C.; Crespo R.; Ottosson H. Z/E-Photoisomerizations of Olefins with 4nπ- or (4n + 2)π-Electron Substituents: Zigzag Variations in Olefin Properties along the T1 State Energy Surfaces. J. Org. Chem. 2005, 70, 9495–9504. 10.1021/jo051558y. [DOI] [PubMed] [Google Scholar]
- Villaume S.; Ottosson H. Aromaticity Changes along the Lowest-Triplet-State Path for C=C Bond Rotation of Annulenyl-Substituted Olefins Probed by the Electron Localization Function. J. Phys. Chem. A 2009, 113, 12304–12310. 10.1021/jp904335j. [DOI] [PubMed] [Google Scholar]
- Mohamed R. K.; Mondal S.; Jorner K.; Delgado T. F.; Lobodin V. V.; Ottosson H.; Alabugin I. V. The Missing C1–C5 Cycloaromatization Reaction: Triplet State Antiaromaticity Relief and Self-Terminating Photorelease of Formaldehyde for Synthesis of Fulvenes from Enynes. J. Am. Chem. Soc. 2015, 137, 15441–15450. 10.1021/jacs.5b07448. [DOI] [PubMed] [Google Scholar]
- Ayub R.; Papadakis R.; Jorner K.; Zietz B.; Ottosson H. Cyclopropyl Group: An Excited-State Aromaticity Indicator?. Chem. - Eur. J. 2017, 23, 13684–13695. 10.1002/chem.201701404. [DOI] [PubMed] [Google Scholar]
- Papadakis R.; Li H.; Bergman J.; Lundstedt A.; Jorner K.; Ayub R.; Haldar S.; Jahn B. O.; Denisova A.; Zietz B.; Lindh R.; Sanyal B.; Grennberg H.; Leifer K.; Ottosson H. Metal-Free Photochemical Silylations and Transfer Hydrogenations of Benzenoid Hydrocarbons and Graphene. Nat. Commun. 2016, 7, 12962. 10.1038/ncomms12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman P. J.; Kent J. E.; O’Dwyer M. F.; Griffith D. W. T. Photochemistry of Benzene Isomers. 2. Benzvalene and Dewar Benzene. J. Phys. Chem. 1981, 85, 2731–2733. 10.1021/j150619a008. [DOI] [Google Scholar]
- Kaplan L.; Ritscher J. S.; Wilzbach K. E. The Photochemical 1,3-Addition of Alcohols to Benzenes. J. Am. Chem. Soc. 1966, 88, 2881–2882. 10.1021/ja00964a067. [DOI] [Google Scholar]
- Farenhorst E.; Bickel A. F. Photochemical Reactions of Benzene with Acids. Tetrahedron Lett. 1966, 7, 5911–5913. 10.1016/S0040-4039(00)76107-7. [DOI] [Google Scholar]
- Stadler H. Synthesis of Novel, Chiral Bicyclo[3.1.0]hex-2-ene Amino Acid Derivatives as Useful Synthons in Medicinal Chemistry. Helv. Chim. Acta 2015, 98, 1189–1201. 10.1002/hlca.201500163. [DOI] [Google Scholar]
- Li J.; Lowary T. L. Synthesis of Bicyclo[3.1.0]hexane Derivatives as Conformationally Restricted Analogues of β-Arabinofuranosyl and α-Galactofuranosyl Rings. Org. Lett. 2008, 10, 881–884. 10.1021/ol703041y. [DOI] [PubMed] [Google Scholar]
- Bernstein D. I.; Bravo F. J.; Clark J. R.; Earwood J. D.; Rahman A.; Glazer R.; Cardin R. D. N-Methanocarbathymidine Is More Effective than Acyclovir for Treating Neonatal Herpes Simplex Virus Infection in Guinea Pigs. Antiviral Res. 2011, 92, 386–388. 10.1016/j.antiviral.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier M. A.; Choi Y.; Gaus H.; Barchi J. J.; Marquez V. E.; Manoharan M. Synthesis and Characterization of Oligonucleotides Containing Conformationally Constrained Bicyclo[3.1.0]hexane Pseudosugar Analogs. Nucleic Acids Res. 2004, 32, 3642–3650. 10.1093/nar/gkh667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilzbach K. E.; Harkness A. L.; Kaplan L. Photochemical Rearrangement of Benzene-1,3,5-d3. J. Am. Chem. Soc. 1968, 90, 1116–1118. 10.1021/ja01007a004. [DOI] [Google Scholar]
- Bryce-Smith D.; Gilbert A.; Longuet-Higgins H. C. Cationic Intermediates in the Photochemistry of Benzene. Chem. Commun. (London) 1967, 0 (5), 240b–241. 10.1039/c1967000240b. [DOI] [Google Scholar]
- Berson J. A.; Hasty N. M. Solvolytic Behavior of Bicyclo[3.1.0]hex-3-en-2-yl Derivatives. Mechanism of Photolysis of Benzene in Hydroxylic Media. J. Am. Chem. Soc. 1971, 93, 1549–1551. 10.1021/ja00735a066. [DOI] [Google Scholar]
- Kaplan L.; Rausch D. J.; Wilzbach K. E. Photosolvation of Benzene. Mechanism of Formation of Bicyclo[3.1.0]hex-3-en-2-yl and of Bicyclo[3.1.0]hex-2-en-6-yl Derivatives. J. Am. Chem. Soc. 1972, 94, 8638–8640. 10.1021/ja00779a089. [DOI] [Google Scholar]
- Olah G. A.; Liang G.; Jindal S. P. Stable Carbocations. CLXXXVIII. Bicyclo[3.1.0]hexenyl Cations. J. Org. Chem. 1975, 40, 3259–3263. 10.1021/jo00910a021. [DOI] [Google Scholar]
- Fãrcaşiu D.; Miller G. Lack of Protonation of Benzene and Toluene in Trifluoromethanesulfonic Acid and Its Significance for Evaluating Superacid Strengths. J. Phys. Org. Chem. 1989, 2, 425–427. 10.1002/poc.610020508. [DOI] [Google Scholar]
- Farenhorst E. A Possible Common Intermediate in the Photochemical Reactions of Benzene. Tetrahedron Lett. 1966, 7, 6465–6469. 10.1016/S0040-4039(00)76127-2. [DOI] [Google Scholar]
- Izawa Y.; Tomioka H.; Kagami T.; Sato T. Photochemical 1,3-Addition of Methanol to tButylbenzene. J. Chem. Soc., Chem. Commun. 1977, 0 (21), 780–781. 10.1039/c39770000780. [DOI] [Google Scholar]
- Foster J.; Pincock A. L.; Pincock J. A.; Thompson K. A. Photochemical Addition of 2,2,2-Trifluoroethanol to Benzonitrile and p-, m-, and o-Methylbenzonitrile. J. Am. Chem. Soc. 1998, 120, 13354–13361. 10.1021/ja982712j. [DOI] [Google Scholar]
- Foster J.; Pincock A. L.; Pincock J. A.; Rifai S.; Thompson K. A. The Photoequilibration of the Ortho-, Meta-, and Para-Isomers of Substituted Benzenes in Acetonitrile and the Photoaddition of 2,2,2-Trifluoroethanol (TFE) to the Same Isomers: A Survey. Can. J. Chem. 2000, 78, 1019–1029. 10.1139/v00-094. [DOI] [Google Scholar]
- Howell N.; Pincock J. A.; Stefanova R. The Phototransposition in Acetonitrile and the Photoaddition of 2,2,2-Trifluoroethanol to the Six Isomers of Dimethylbenzonitrile. J. Org. Chem. 2000, 65, 6173–6178. 10.1021/jo000701e. [DOI] [PubMed] [Google Scholar]
- Izawa Y.; Tomioka H.; Kagami T.; Sato T. Photochemical 1,3-Addition of Methanol to tButylbenzene. J. Chem. Soc., Chem. Commun. 1977, 0 (21), 780–781. 10.1039/c39770000780. [DOI] [Google Scholar]
- Gonzalez C. M.; Pincock J. A. Steric Effects in the Phototransposition Reactions of Dialkylbenzenes. Can. J. Chem. 2006, 84, 10–20. 10.1139/v05-258. [DOI] [Google Scholar]
- Xu X.; Cao Z.; Zhang Q. What Definitively Controls the Photochemical Activity of Methylbenzonitriles and Methylanisoles? Insights from Theory. J. Phys. Chem. A 2007, 111, 5775–5783. 10.1021/jp071975+. [DOI] [PubMed] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Cheesman J. R.. VCD Spectroscopy for Organic Chemists; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Wilzbach K. E.; Ritscher J. S.; Kaplan L. Benzvalene, the Tricyclic Valence Isomer of Benzene. J. Am. Chem. Soc. 1967, 89, 1031–1032. 10.1021/ja00980a053. [DOI] [Google Scholar]
- Palmer I. J.; Ragazos I. N.; Bernardi F.; Olivucci M.; Robb M. A. An MC-SCF Study of the S1 and S2 Photochemical Reactions of Benzene. J. Am. Chem. Soc. 1993, 115, 673–682. 10.1021/ja00055a042. [DOI] [Google Scholar]
- Baranac-Stojanović M. Substituent Effect on Triplet State Aromaticity of Benzene. J. Org. Chem. 2020, 85, 4289–4297. 10.1021/acs.joc.9b03472. [DOI] [PubMed] [Google Scholar]
- Freiser B. S.; Beauchamp J. L. Acid-base Properties of Molecules in Excited Electronic States Utilizing Ion Cyclotron Resonance Spectroscopy. J. Am. Chem. Soc. 1977, 99, 3214–3225. 10.1021/ja00452a003. [DOI] [Google Scholar]
- Rosenberg M.; Ottosson H.; Kilså K. Proton and Hydride Affinities in Excited States: Magnitude Reversals in Proton and Hydride Affinities between the Lowest Singlet and Triplet States of Annulenyl and Benzannulenyl Anions and Cations. J. Org. Chem. 2010, 75, 2189–2196. 10.1021/jo9025868. [DOI] [PubMed] [Google Scholar]
- Rode M. F.; Sobolewski A. L.; Dedonder C.; Jouvet C.; Dopfer O. Computational Study on the Photophysics of Protonated Benzene. J. Phys. Chem. A 2009, 113, 5865–5873. 10.1021/jp902729m. [DOI] [PubMed] [Google Scholar]
- Jorner K.; Jahn B. O.; Bultinck P.; Ottosson H. Triplet State Homoaromaticity: Concept, Computational Validation and Experimental Relevance. Chem. Sci. 2018, 9, 3165–3176. 10.1039/C7SC05009G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward R. B.; Hoffmann R. Stereochemistry of Electrocyclic Reactions. J. Am. Chem. Soc. 1965, 87, 395–397. 10.1021/ja01080a054. [DOI] [Google Scholar]
- Dewar M. J. S. A Molecular Orbital Theory of Organic Chemistry – VIII: Aromaticity and Electrocyclic Reactions. Tetrahedron 1966, 22, 75–92. 10.1016/S0040-4020(01)82171-2. [DOI] [Google Scholar]
- Zimmerman H. E. On Molecular Orbital Correlation Diagrams, the Occurrence of Mobius Systems in Cyclization Reactions, and Factors Controlling Ground- and Excited-State Reactions. I. J. Am. Chem. Soc. 1966, 88, 1564. 10.1021/ja00959a052. [DOI] [Google Scholar]
- von Rosenberg J. L.; Mahler J. E.; Pettit R. The Bicyclo[5.1.0]Octadienyl Cation, A New Stable Carbonium Ion. J. Am. Chem. Soc. 1962, 84, 2842–2843. 10.1021/ja00873a051. [DOI] [Google Scholar]
- Brewer T. L. Oxygen Quenching of the Aromatic Excited Singlet State. J. Am. Chem. Soc. 1971, 93, 775–776. 10.1021/ja00732a037. [DOI] [Google Scholar]
- Morikawa A.; Cvetanović R. J. Quenching of Benzene Fluorescence by Oxygen. J. Chem. Phys. 1970, 52, 3237–3239. 10.1063/1.1673464. [DOI] [Google Scholar]
- Schmidt R. Photosensitized Generation of Singlet Oxygen. Photochem. Photobiol. 2006, 82, 1161–1177. 10.1562/2006-03-03-IR-833. [DOI] [PubMed] [Google Scholar]
- Montalti M.; Credi A.; Prodi L.; Gandolfi M. T.. Handbook of Photochemistry, 3rd ed.; CRC Press: Boca Raton, FL, 2006. [Google Scholar]
- Fifen J. J.; Nsangou M.; Dhaouadi Z.; Motapon O.; Jaidane N.-J. Solvation Energies of the Proton in Methanol. J. Chem. Theory Comput. 2013, 9, 1173–1181. 10.1021/ct300669v. [DOI] [PubMed] [Google Scholar]
- Agmon N. Proton Solvation and Proton Mobility. Isr. J. Chem. 1999, 39, 493–502. 10.1002/ijch.199900054. [DOI] [Google Scholar]
- Agmon N. The Grotthuss Mechanism. Chem. Phys. Lett. 1995, 244, 456–462. 10.1016/0009-2614(95)00905-J. [DOI] [Google Scholar]
- Keutsch F. N.; Saykally R. J. Water Clusters: Untangling the Mysteries of the Liquid, One Molecule at a Time. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 10533–10540. 10.1073/pnas.191266498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhn M. Hydrogen Bond Dynamics in Alcohol Clusters. Adv. Chem. Phys. 2008, 142, 1–57. 10.1002/9780470475935.ch1. [DOI] [Google Scholar]
- Palmer I. J.; Ragazos I. N.; Bernardi F.; Olivucci M.; Robb M. A. An MC-SCF Study of the S1 and S2 Photochemical Reactions of Benzene. J. Am. Chem. Soc. 1993, 115, 673–682. 10.1021/ja00055a042. [DOI] [Google Scholar]
- Thompson A. L.; Martínez T. J. Time-Resolved Photoelectron Spectroscopy from First Principles: Excited State Dynamics of Benzene. Faraday Discuss. 2011, 150, 293–311. 10.1039/c1fd00003a. [DOI] [PubMed] [Google Scholar]
- Boggio-Pasqua M.; Groenhof G. On the Use of Reduced Active Space in CASSCF Calculations. Comput. Theor. Chem. 2014, 1040–1041, 6–13. 10.1016/j.comptc.2014.03.017. [DOI] [Google Scholar]
- Callomom J. H.; Dunn T. M.; Mills I I. M. Rotational Analysis of the 2600a Absorption System of Benzene. Philos. Trans. R. Soc. London, Ser. A 1966, 259, 499–532. 10.1098/rsta.1966.0023. [DOI] [Google Scholar]
- Callomon J. H.; Parkin J. E.; Lopez-Delgado R. Non-radiative Relaxation of the Excited à 1B2u State of Benzene. Chem. Phys. Lett. 1972, 13, 125–131. 10.1016/0009-2614(72)80059-9. [DOI] [Google Scholar]
- Riedle E.; Neusser H. J.; Schlag E. W. Sub-Doppler Spectroscopy of Benzene in the ‘Channel-three’ Region. Faraday Discuss. Chem. Soc. 1983, 75, 387–394. 10.1039/DC9837500387. [DOI] [Google Scholar]
- Riedle E.; Weber T.; Schubert U.; Neusser H. J.; Schlag E. W. Back to the Roots of ‘‘Channel Three’’: Rotationally Resolved Spectra of the 601103 band of C6H6. J. Chem. Phys. 1990, 93, 967–978. 10.1063/1.459123. [DOI] [Google Scholar]
- Riedle E.; Neusser H. J.; Schlag E. W.; Mills I. M.; Child M. S.; Marcus R. A. Pathways for Intramolecular Relaxation in S1 Benzene. Philos. Trans. R. Soc. London, Ser. A 1990, 332, 189–201. 10.1098/rsta.1990.0108. [DOI] [Google Scholar]
- Sobolewski A. L.; Woywod C.; Domcke W. Ab initio Investigation of Potential Energy Surfaces Involved in the Photophysics of Benzene and Pyrazine. J. Chem. Phys. 1993, 98, 5627–5641. 10.1063/1.464907. [DOI] [Google Scholar]
- Lasorne B.; Bearpark M. J.; Robb M. A.; Worth G. A. Controlling S1/S0 Decay and the Balance between Photochemistry and Photostability in Benzene: A Direct Quantum Dynamics Study. J. Phys. Chem. A 2008, 112, 13017–13027. 10.1021/jp803740a. [DOI] [PubMed] [Google Scholar]
- Parker D. S. N.; Minns R. S.; Penfold T. J.; Worth G. A.; Fielding H. H. Ultrafast Dynamics of the S1 Excited State of Benzene. Chem. Phys. Lett. 2009, 469, 43–47. 10.1016/j.cplett.2008.12.069. [DOI] [Google Scholar]
- Sobolewski A. L.; Domcke W. Photophysically Relevant Potential Energy Surface Functions of Low-Lying Singlet States of Benzene, Pyridine and Pyrazine: An Ab initio Study. Chem. Phys. Lett. 1991, 180, 381–386. 10.1016/0009-2614(91)90337-9. [DOI] [Google Scholar]
- Sobolewski A. L.; Woywod C.; Domcke W. Ab initio Investigation of Potential-Energy Surfaces Involved in the Photophysics of Benzene and Pyrazine. J. Chem. Phys. 1993, 98, 5627–5641. 10.1063/1.464907. [DOI] [Google Scholar]
- Su M.-D. Mechanistic Study of the Photochemical Isomerization Reactions of Silabenzene. Organometallics 2014, 33, 5231–5237. 10.1021/om500545a. [DOI] [Google Scholar]
- King R. A.; Lüthi H. P.; Schaefer H. F. III; Glarner F.; Burger U. The Photohydration of N-Alkylpyridinium Salts: Theory and Experiment. Chem. - Eur. J. 2001, 7, 1734–1742. 10.1002/1521-3765(20010417)7:8<1734::AID-CHEM17340>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Sowmiah S.; Esperanca J. M. S. S.; Rebelo L. P. N.; Afonso C. A. M. Pyridinium Salts: From Synthesis to Reactivity and Applications. Org. Chem. Front. 2018, 5, 453–493. 10.1039/C7QO00836H. [DOI] [Google Scholar]
- Damiano T.; Morton D.; Nelson A. Photochemical Transformations of Pyridinium Salts: Mechanistic Studies and Applications in Synthesis. Org. Biomol. Chem. 2007, 5, 2735–2752. 10.1039/b706244n. [DOI] [PubMed] [Google Scholar]
- Zou J.; Mariano P. S. The Synthetic Potential of Pyridinium Salt Photochemistry. Photochem. Photobiol. Sci. 2008, 7, 393–404. 10.1039/b801808c. [DOI] [PubMed] [Google Scholar]
- Delgado A. Recent Advances in the Chemistry of Aminocyclitols. Eur. J. Org. Chem. 2008, 2008, 3893–3906. 10.1002/ejoc.200800238. [DOI] [Google Scholar]
- Kaur N. Synthesis of Three-Membered and Four-Membered Heterocycles with the Assistance of Photochemical Reactions. J. Heterocycl. Chem. 2019, 56, 1141–1167. 10.1002/jhet.3491. [DOI] [Google Scholar]
- Wakita K.; Tokitoh N.; Okazaki R.; Takagi N.; Nagase S. Crystal Structure of a Stable Silabenzene and Its Photochemical Valence Isomerization into the Corresponding Silabenzvalene. J. Am. Chem. Soc. 2000, 122, 5648–5649. 10.1021/ja000309i. [DOI] [Google Scholar]
- Kaplan L.; Pavlik J. W.; Wilzbach K. E. Photohydration of Pyridinium Ions. J. Am. Chem. Soc. 1972, 94, 3283–3284. 10.1021/ja00764a089. [DOI] [Google Scholar]
- Yoon U. C.; Quillen S. L.; Mariano P. S.; Swanson R.; Stavinoha J. L.; Bay E. Exploratory and Mechanistic Aspects of the Electron-Transfer Photochemistry of Olefin-N-Heteroaromatic Cation Systems. J. Am. Chem. Soc. 1983, 105, 1204–1218. 10.1021/ja00343a022. [DOI] [Google Scholar]
- Ling R.; Yoshida M.; Mariano P. S. Exploratory Investigations Probing a Preparatively Versatile, Pyridinium Salt Photoelectrocyclization–Solvolytic Aziridine Ring Opening Sequence. J. Org. Chem. 1996, 61, 4439–4449. 10.1021/jo960316i. [DOI] [PubMed] [Google Scholar]
- Lewis G. N.; Kasha M. Phosphorescence in Fluid Media and the Reverse Process of Singlet-Triplet Absorption. J. Am. Chem. Soc. 1945, 67, 994–1003. 10.1021/ja01222a032. [DOI] [Google Scholar]
- Renner C. A.; Katz T. J.; Pouliquen J.; Turro N. J.; Waddell W. H. Energy Storage and Release. Direct and Sensitized Photoreactions of Benzvalene. Evidence for a Quantum Chain Process, an Adiabatic photorearrangement, a Degenerate Photovalence Isomerization, and Two Reactive Triplet States. J. Am. Chem. Soc. 1975, 97, 2568–2570. 10.1021/ja00842a059. [DOI] [Google Scholar]
- Carroll F. A.; Quina F. H. A New Method for the Determination of Intersystem Crossing Quantum Yields. Application to Benzene and Its Methyl Derivatives. J. Am. Chem. Soc. 1976, 98, 1–6. 10.1021/ja00417a001. [DOI] [Google Scholar]
- MacLeod P. J.; Pincock A. L.; Pincock J. A.; Thompson K. A. Photochemical Equilibration/Isomerization of p-, m-, and o-Methylbenzonitrile. J. Am. Chem. Soc. 1998, 120, 6443–6450. 10.1021/ja9808301. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.