

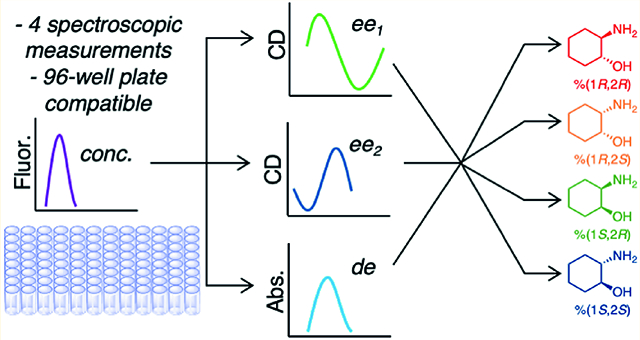
Abstract
Optical analysis of reaction parameters such as enantiomeric excess (ee), diastereomeric excess (de), and yield are becoming increasingly useful as assays for differing functional groups become available. These assays typically exploit reversible covalent or noncovalent assemblies that impart optical signals, commonly circular dichroism (CD), that are indicative of the stereochemistry and ee at a stereocenter proximal to the functional group of interest. Very few assays have been reported that determine ee and de when two stereocenters are present, and none have targeted two different functional groups that are vicinal and lack chromophores entirely. Using a CD assay that targets chiral secondary alcohols, a separate CD assay for chiral primary amines, a UV–vis assay for de, and a fluorescence assay for concentration, we demonstrate a work-flow for speciation of the enantiomers and diastereomers of 2-aminocyclohexanol as a test-bed analyte. Because of the fact the functional groups are vicinal, we found that the ee determination at the two stereocenters is influenced by the adjacent center, and this led us to implement a chemometric patterning approach, resulting in a 4% absolute error in full speciation of the four stereoisomers. The procedure presented herein would allow for the total speciation of around 96 reactions in 27 min using a high-throughput experimentation routine. While 2-aminocyclohexanol is used to demonstrate the methods, the general workflow should be amenable to analysis of other stereoisomers when two stereocenters are present.
Graphical Abstract
INTRODUCTION
Over the last few decades, asymmetric reaction discovery has been fueled by the advent of high-throughput experimentation (HTE).1,2 Many thousands of experimental conditions can be explored in a parallel fashion to identify the most efficient asymmetric transformations. However, a significant bottleneck is encountered with reaction analysis, due to the sheer number of reactions that can be conducted.3,4
Chiral chromatography is the most common method for characterizing the enantiomeric excess (ee) of the product of an asymmetric transformation. Recent developments in ultrafast chiral separations for high-throughput ee determination have made significant strides, with average analysis times on the 5–20 min time scale; however, in some cases, subminute separation times were achieved.5–7 Despite these remarkable advances, the serial nature of chromatography is not readily amenable to high-throughput screening (HTS) protocols. Parallelization of chromatographic techniques has been accomplished but requires specialized instrumentation and software.8,9 In an effort to overcome this bottleneck, optical methods have been developed for asymmetric reaction discovery.10–14 In contrast to chromatographic techniques, optical analyses can be accomplished in microwell plates, allowing for simple, fast, and cost-effective implementation in HTE protocols. To date, there have been numerous reports of optical methods such as UV–vis,15–18 fluorescence,19–23 and circular dichroism (CD)24–28 spectroscopy being successfully employed to determine the enantioselectivity of a given asymmetric transformation, as well as reaction yield.
While optical approaches to ee determination have had extensive success,29 few reports of optical assays for both enantiomers and diastereomers have been reported.23,30 Diastereomers are nonmirror image stereoisomers, and are possible whenever there are two or more stereocenters. Of course, the stereocenters can be far apart or proximal. In an asymmetric transformation that simultaneously sets more than one stereocenter, the stereocenters are commonly proximal and, in particular, vicinal.31–33 In such a scenario, the ee is typically determined within each diastereomeric set, for example, cis- and trans- (or threo and erythro) via chiral chromatography. Likewise, the most common technique to measure diastereomeric ratio (dr) is HPLC, but NMR and other spectroscopic methods can be used.34–36 These methods are again serial, and represent a bottleneck for rapid method development. Finally, while reaction yield is best defined by “isolated yield”, in HTE procedures it would be useful to have rapid optical methods to monitor percent conversion in situ, and such techniques have been created.37–39
In one report that has addressed the stereochemical determination of both ee and dr, Pu and co-workers utilized a pair of enantiomeric fluorescent sensors (1 and 2) that were both enantioselective and diastereoselective toward the four stereoisomers of N-carbobenzyloxy-threonine (3, Figure 1A).30 It was demonstrated that the fluorescence responses of the enantiomeric sensors at two emission wavelengths could be used to differentiate the four stereoisomers. On the basis of this observation, the authors stated that this system could be used to determine the relative concentration of each isomer in a mixture of four stereoisomers, but total speciation of the stereoisomers was not reported.
Figure 1.
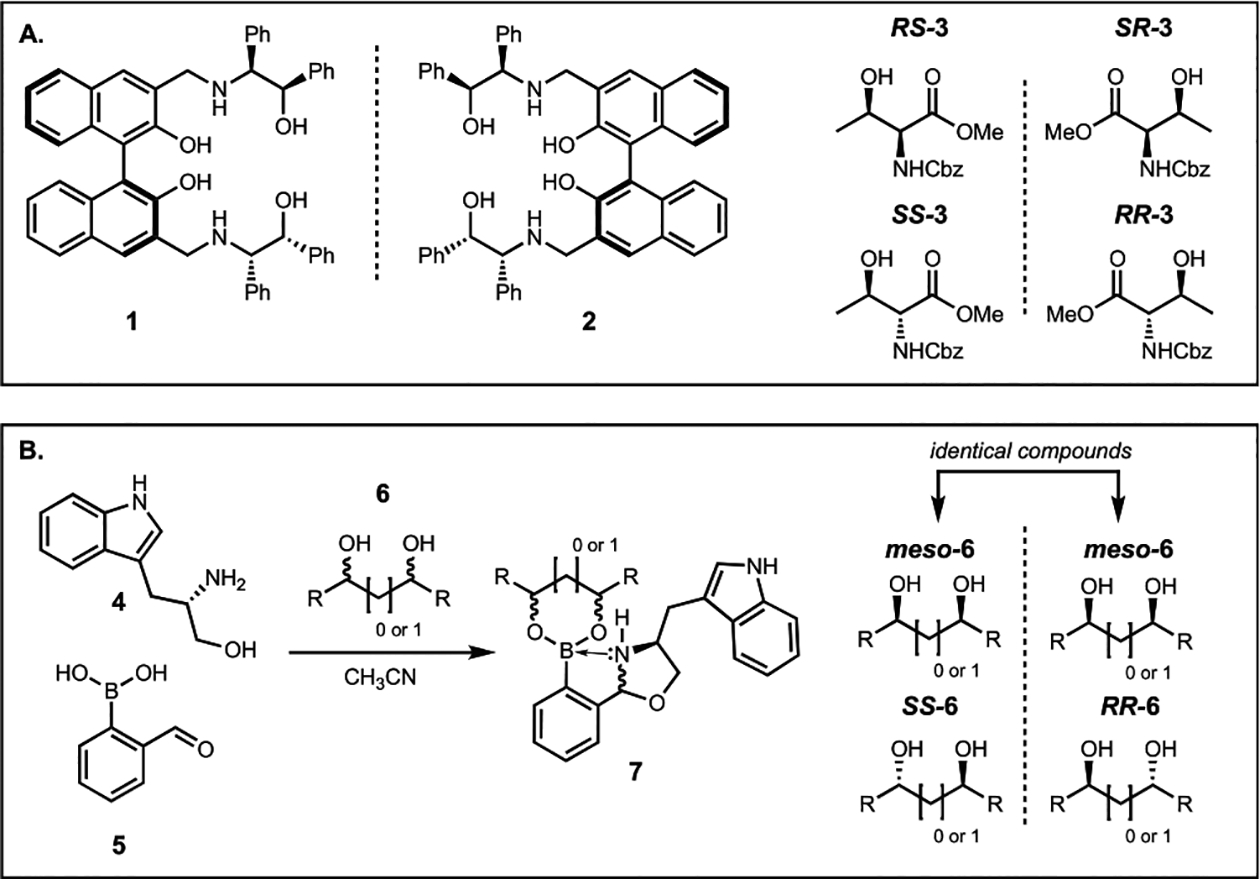
(A) A pair of enantiomeric BINOL sensors for differentiating the four stereoisomers of N-carboxybenzoyl threonine methyl ester. (B) A dynamic covalent multicomponent assembly for the differentiation of stereoisomeric diols, where there are only three possible stereoisomers due to the internal mirror plane present in the meso stereoisomer.
In another approach, Anzenbacher et al. utilized a dynamic covalent assembly of enantiopure tryptophanol (4) and oformyl phenyl boronic (5) acid for enantio- and diastereoselective fluorosensing of symmetrical chiral 1,2- or 1,3-diols (6) (Figure 1B).23 The stereochemistry at both –OH stereocenters imparts an effect on the fluorescence readout of complex 7, allowing for differentiation of three stereoisomers (RR, SS, and meso). It was demonstrated that an artificial neural network (ANN) analysis could be used to determine the ee of the stereoisomeric mixture and the total concentration of diol, but there was a caveat that only 5% of the meso form could be tolerated. Thus, diastereomeric excesses (de) were not determined, and full stereoisomeric speciation was not demonstrated. No group has yet reported using optical methods to perform a full speciation of stereoisomers when two stereocenters of differing chemical functionality are present. Achieving this was our fundamental goal, and we chose an amino-alcohol as a test-bed analyte.
Vicinal aminoalcohols are privileged scaffolds in organic synthesis in terms of their role in both chirality transfer processes and pharmacologically active natural products.40 As such, the synthesis of vicinal aminoalcohols remains a major thoroughfare of inquiry in the development of asymmetric methodologies. Several approaches have been demonstrated, including enantio- and diastereoselective reductive coupling of imino compounds with carbonyl compounds,41 diastereoselective addition of nucleophiles to enantiopure α-amino-carbonyl compounds,42 diastereoselective oxidations of allyl amines,43 and enantioselective ring opening of epoxides.44 In most cases, the absolute and relative configurations of synthetic vicinal aminoalcohols were determined for the N-derivatized substrates via chiral chromatography using racemic or enantiopure standards with chromatographic methods ranging from 12 to 25 min per run; however, it is noteworthy to mention that these approaches were not applicable to all vicinal aminoalcohols synthesized. For some substrates, the stereochemistry was determined via crystallographic methods where derivatization at the amine or alcohol stereocenter was often required to yield crystals suitable for crystallographic analysis.
With this in mind, we set out to create optical methods to determine ee, de, and reaction yield, and chose 2-aminocyclohexanol (8) as a model substrate. Not only are the stereocenters vicinal, but they contain different functionality, thus presenting the challenge of using two separate assays. Further, the structures do not possess chromophores, allowing the trivial characterization of ee values via CD spectropolarimetry on pure, underivatized samples. Here, we report on the utilization of dynamic covalent chemistries for the differentiation of the four stereoisomers of 2-aminocyclohexanol, and their concentrations (Figure 2). We discuss the complications encountered in doing such an analysis, and explore solutions to the complexities by using chemometric methods.
Figure 2.
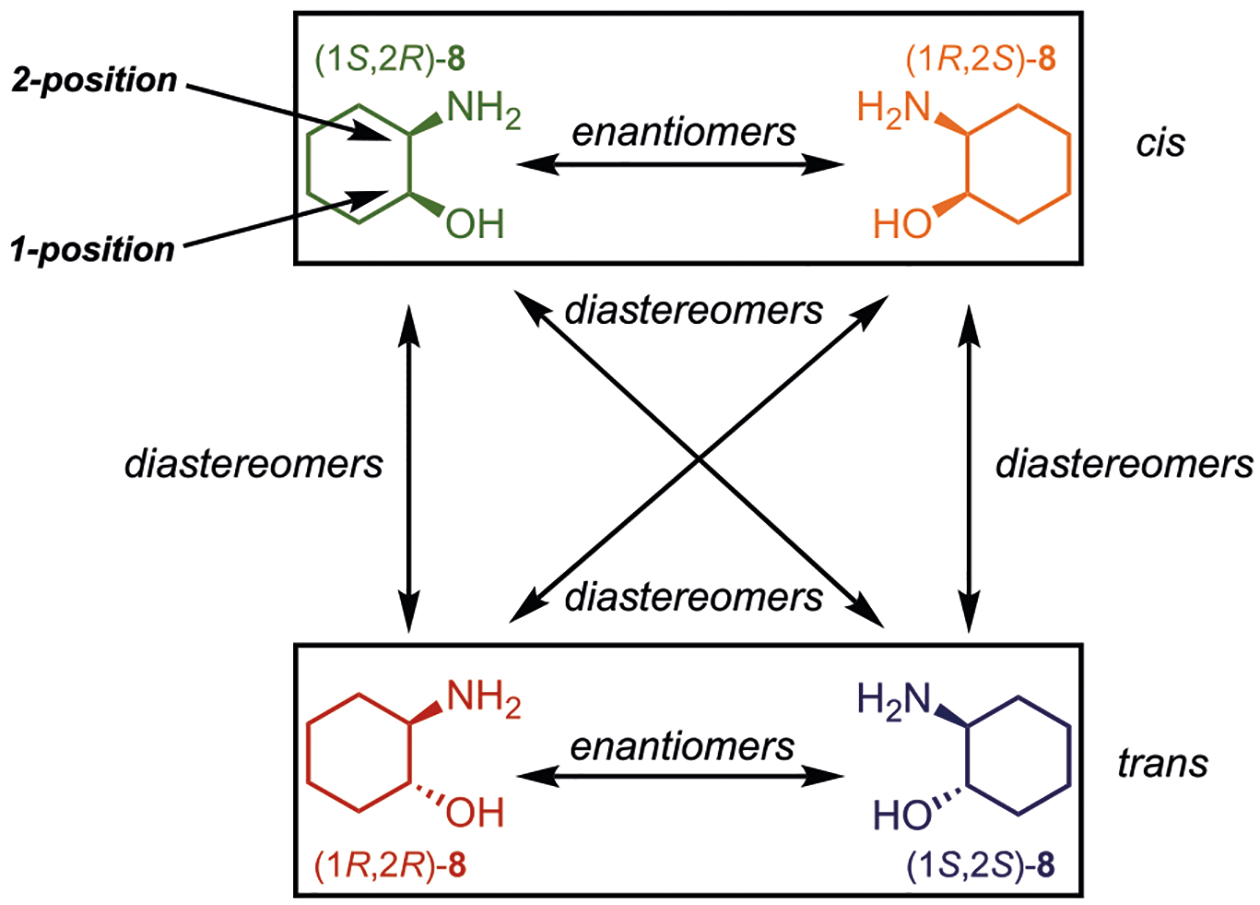
Four possible stereoisomers of 2-aminocyclohexanol (8), where the 1-position is the alcohol stereocenter and the 2-position is the amine stereocenter.
RESULTS AND DISCUSSION
Design Criteria and Strategy.
Optical assays for ee function by a chiral analyte interacting with a molecular sensor, inducing a spectroscopic readout that is dependent on the absolute configuration of the stereocenter being targeted. The current optical protocols for ee determination are designed for common functional groups, usually with a single α- (sometimes β- or γ-) stereocenter. Except for vibrational circular dichroism (VCD),45 optical methods for determining ee have focused primarily on molecules with only one stereocenter, and hence exist only as enantiomers. A problem arises when diastereomers exist. Because the optical methods assign R or S to individual stereocenters independently, the assignment of R or S is within both diastereomers. For example, an assay focused on stereocenter 1 would potentially report a 1S,2R stereoisomer the same as 1S,2S. Thus, we derived a mathematical relationship between the R and S configurations of two individual stereocenters with the ee of each set of diastereomers.46 As described in the previous report, one requires an assay for de (or dr) to perform a complete speciation of all four stereoisomers when two stereocenters are present (such as in Figure 2). When the optical signal for each individual stereocenter is not influenced by the additional stereocenter, an ee value for each stereocenter, and a de value for the four stereoisomers, allows for complete speciation of the four stereoisomers. Complete speciation allows one to calculate ee within each diastereomeric set.
Our group has reported chirality sensing systems for monoamines28,47,48 and secondary mono-ols49–54 via CD spectroscopy, and we anticipated the tandem use of these systems would be ideally suited for stereoisomeric differentiation of the 2-aminocyclohexanol isomers.
The chirality sensing system for primary-amines relies on the in situ generation of iron(II) complexes with exciton coupled circular dichroism (ECCD) active absorption bands in the UV region as the result of coupling between pyridyl chromophores (200–400 nm) and CD active absorption bands due to intense metal to ligand charge transfer (MLCT) bands in the visible region (400–700 nm). Imine formation between a chiral amine and 3-hydroxy-2-pyridinecarboxaldehyde (9) followed by 3:1 ligand complexation to an iron(II) center forms a stereoisomeric mixture of octahedral complexes (12) that possess helical chirality (Figure 3). These isomers are the result of helical isomerism (Λ- and Δ-), configurational isomerism (fac- and mer-), and the stereochemistry of the monoamines (R- and S-). This results in bisignate CD curves in the visible region due to (MLCT). The observed CD signal correlates to the helicity of the octahedral iron(II) complex and the stereochemistry of the chiral amine. Concentration-independent calibration curves are generated that correlate CD intensity to ee with an average error of ±5%. Importantly, this assay requires no synthesis; both 3-hydroxy-2-pyridinecarboxaldehyde and iron(II) triflate are commercially available reagents, making it a facile method for the ee determination of chiral primary-amines.
Figure 3.
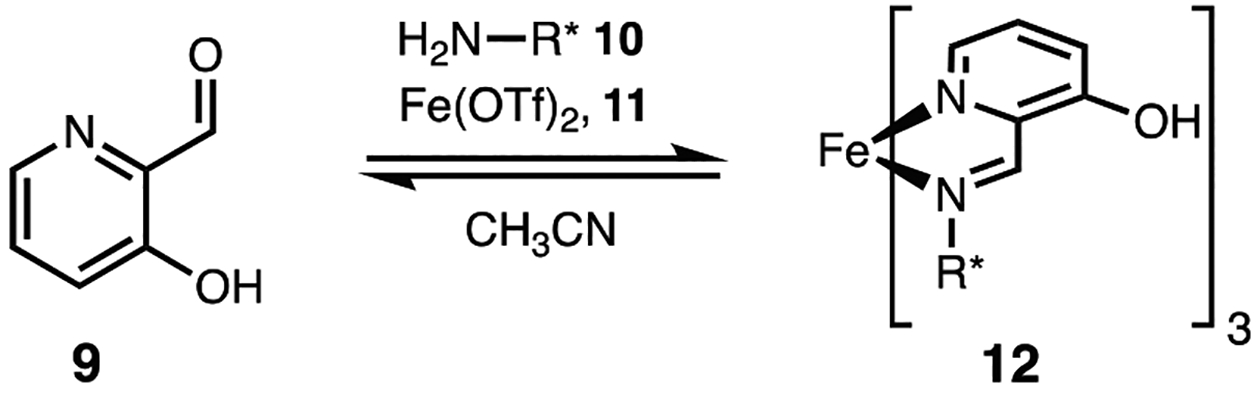
In situ-generated octahedral iron(II) complexes for the ee determination of chiral primary-amines.
An ECCD sensing system based on a dynamic multicomponent assembly process targets chiral secondary alcohols (Figure 4). Iminium formation between di(2-picolyl)amine (13) and 2-pyridinecarboxaldehyde (14) is followed by alcohol incorporation to yield a tren-like ligand (19). Complexation of the tris(pyridine) ligand to a zinc(II) center results in an ECCD-active trigonal bipyramidal complex as a result of the coupling between the pyridyl chromophores. The arrangement of the pyridinyl chromophores about the zinc(II) center is dependent on the stereochemistry of the hemiaminal ether stereocenter that is formed upon incorporation of the mono-ol into the tris(pyridine) ligand, whose stereochemistry in turn is dictated by the stereogenicity of the alcohol analyte. Thus, the sign of the Cotton effect is indicative of the stereochemistry of the chiral analyte. Concentration-independent calibration curves are generated that correlate CD intensity to ee to give extrapolated ee values with an average error of ±3%. As with the assay for chiral amines, all reagents are commercially available, making this a simple method for ee determination.
Figure 4.
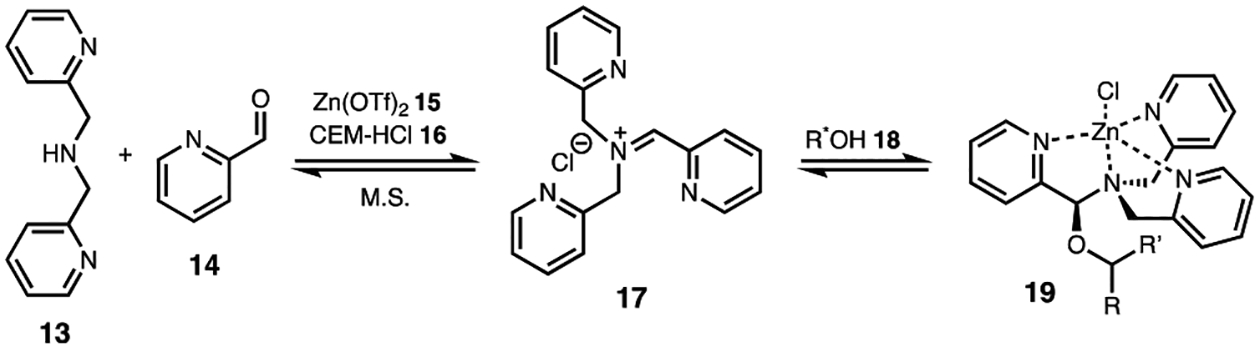
Dynamic covalent multicomponent assembly for the ee determination of chiral secondary alcohols, where CEM-HCl is 4-(2-chloroethyl)morpholine hydrochloride and M.S. is 4 Å molecular sieves.
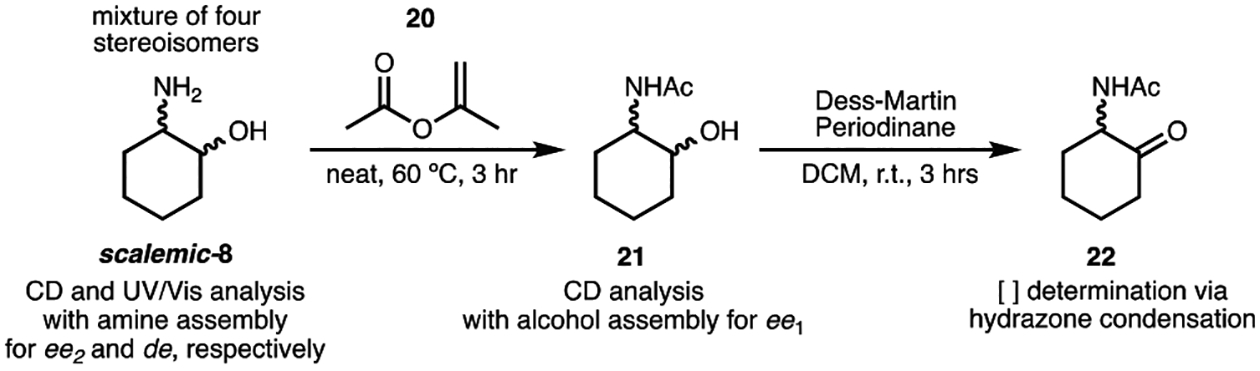
Our strategy for characterizing a stereoisomeric mixture of 2-aminocyclohexanol (8) is shown in Scheme 1. We hypothesized the total concentration of 8 could be determined via hydrazone formation of an oxidized form of the analyte and fluorescence spectroscopy (Scheme 1). With the concentration of the analyte known, both the amine and the alcohol sensing systems discussed above (Figures 3 and 4, respectively) could be conducted at concentrations above saturation to ensure that the CD signals indicative of ee were concentration-independent. The amine assay given in Figure 3 would also allow for the differentiation of the cis- and trans-1,2-aminoalcohols via UV–vis spectroscopy.
Scheme 1.
Workflow for Total Stereoisomeric Speciation
Stereoisomeric Speciation.
Concentration Determination.
As shown in Scheme 1, the total concentration of the four stereoisomers of 8 was determined with a simple three-step procedure that can be done in parallel reactions in plates: (1) in situ acetylation of the amino group with isopropenyl acetate (20), (2) in situ Dess–Martin oxidation of the resulting amidoalcohol to the corresponding amidoketone (22), and (3) addition of fluorescent 7-(diethylamino)coumarin-3-carbohydrazide (Figure S21). These transformations proceed quantitatively and require no chromatographic purifications. We found the oxidation to the ketone was advantageous for an additional reason besides yield determination. The oxidation removes diastereomeric differences, such that a mixture of four diastereomers become only enantiomers, which have the same emission.55 The products can be transferred from plates for future ee and/or de determination in parallel, via simple extractions, and concentration under centrifugal evaporation. In this workflow, it is necessary to first determine the concentration of the analyte to run the ee and de assays at the concentrations necessary for assembly formation.
Alcohol ee Determination.
To monitor the stereochemistry at the 1-position (alcohol), it was necessary for the amine moiety to be protected. This was accomplished by the quantitative acetylation procedure introduced above, releasing only an equivalent of acetone that is innocent in our assemblies (Scheme 1).56 All four stereoisomers of 21 were efficiently incorporated into the alcohol assembly, and characteristic Cotton effects were observed at 270 nm (Figure 5). The cis-diastereomers gave significantly higher CD signals (|CD|270 nm = 20 mdeg) as compared to trans- (|CD|270 nm = 7 mdeg).
Figure 5.
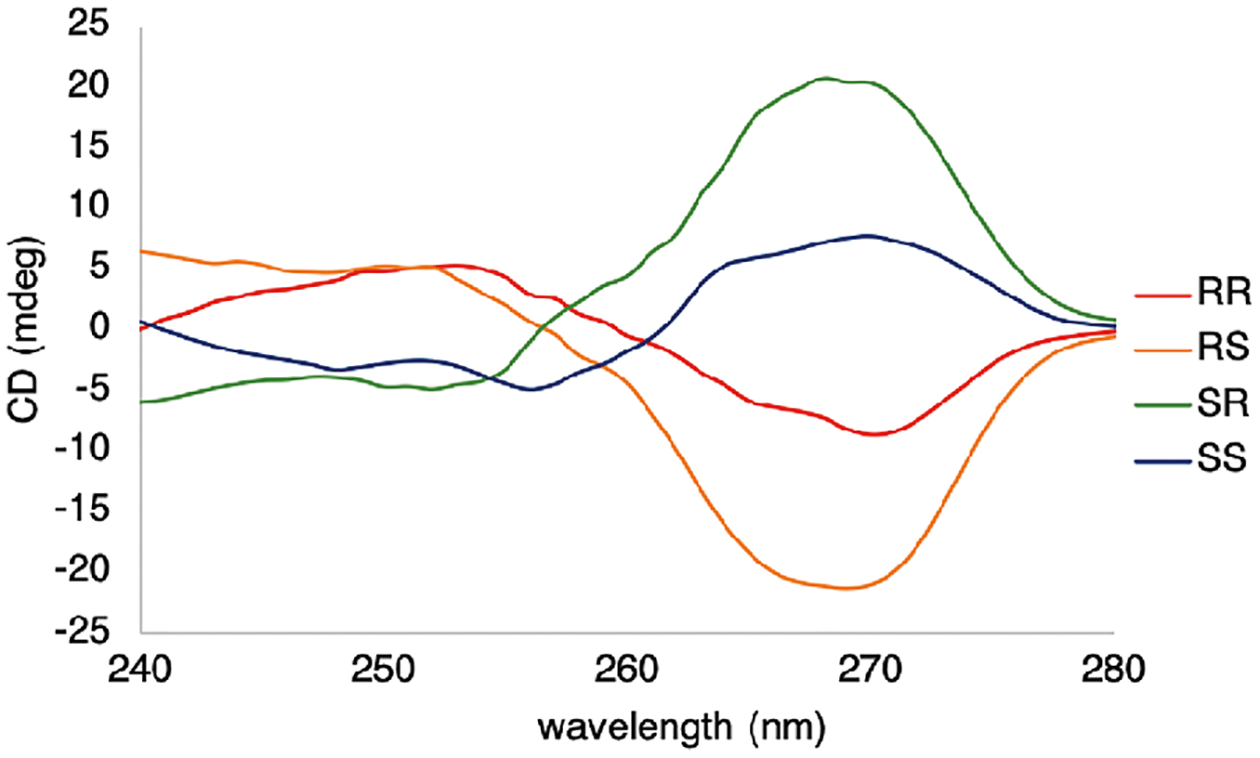
ECCD signals for the four stereoisomers N-(2-hydroxycyclohexyl)acetamide (21) with dipicolylamine, pyridine-2-carboxaldehyde, 4-(2-chloroethyl)morpholine hydrochloride, and Zn(OTf)2. The ECCD spectra were recorded in CH3CN at 25 °C (1.75 mM pyridine-2-carboxaldehyde, 8.75 mM 21, 1 mm cell).
The difference in the observed CD intensity at 270 nm for the diastereomers can be attributed to two possibilities. The stereochemistry at the 2-position could have an effect on the orientation of the pyridine rings in the tripodal zinc complex, or the diastereomers of 8 could be incorporated to different extents. In previous studies, we have determined that 5 equiv of alcohol relative to 2-pyridinecarboxaldehyde is needed to ensure efficient incorporation into the tris(pyridine) ligand. Thus, using 5 equiv of either diastereomer of 21, we found that the yield of the hemiaminal ether zinc complex (19) as determined via NMR integrals (Figures S1–S7) showed no significant differences in analyte incorporation. Hence, it was concluded that the difference in CD intensity at 270 nm was due to the adjacent stereocenter influencing the arrangement of the pyridine chromophores of zinc complex 19.
However, with the use of 5 equiv of alcohol, there is still the possibility that one diastereomer of 21 may preferentially be incorporated into the assembly (Scheme S2). If this is the case, the observed CD signal will instead reflect the preferred diastereomer of the analyte, and any resulting ee determinations will be incorrect. To verify a statistical distribution of incorporated alcohol, 1:1 diastereomeric mixtures were made with 5 equiv of alcohol, for example, 2.5 equiv:2.5 equiv (1R,2R):(1R,2S) and (1S,2S):(1S,2R), and subjected to the reaction conditions for alcohol assembly formation (Figure 4). As CD is a function of absorbance, it can be treated analogously as UV–vis spectra, which is simply additive. Thus, the CD of a solution with multiple components is the sum of the CD signals of the individual components. In our case, it was observed that the observed CD signal for the 1:1 diastereomeric mixtures was indeed additive. This was further confirmed by analyzing 1:1 diastereomeric mixtures that were made up from two enantiopure solutions, giving the same CD signals (Figure S12).
Amine ee Determination.
We found that the stereochemistry of the 2-position (amine) and the relative stereochemistry to the 1-position (alcohol) could be studied directly without prior derivatization of 2-aminocyclohexanol. Imine formation between 2-aminocyclohexanol and 3-hydroxypyridine-2-carboxaldehyde and subsequent complexation to an iron(II) center (Figure 3) gave intense MLCT bands with characteristic Cotton effects (Figure 6). The cis- and trans-8 gave CD signals with similar intensities (|CD|600 nm = 45 mdeg), albeit the (1S,2S) and (1S,2R) stereoisomers gave essentially the same CD spectra from 450–700 nm (see overlap of RR and RS in Figure 6; this coincidental overlap of CD spectra is not expected to be general and is not required for stereoisomeric differentiation). Thus, the arrangement of the pyridine chromophores around the iron center is primarily due to the configuration at the 1-position.
Figure 6.
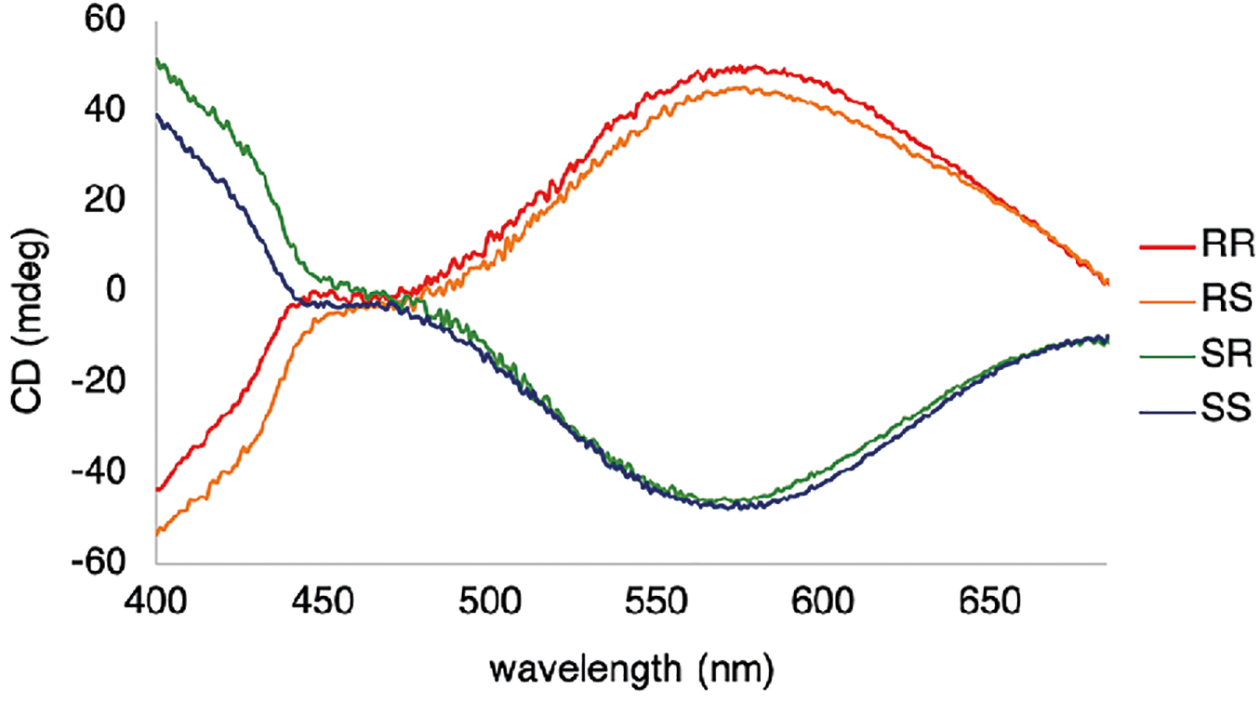
CD traces for four stereoisomers of 2-aminocyclohexanol with 3-hydroxy-pyridine-2-carboxaldehyde and Fe(OTf)2. The CD spectra were recorded in CH3CN at 25 °C (0.5 mM Fe(OTf)2, 1.5 mM 8, 1 cm cell).
In general, for 12, the imine ligands exist in the E-configuration and dock on the iron center with the bulkiest substituent oriented away from the iron(II) center.47 In the context of 2-aminocylohexanol, there is the added complexity that each stereoisomer can exist in two different chair flips (Figure 7). For the trans-diastereomeric set, the alcohol and amine functional groups can either both be axially or be equatorially oriented, whereas, in the cis-diastereomeric set, if one functional group is oriented axially, the other is necessarily oriented equatorially. Taking into consideration A-values (A for –NH2 ranges from 1.23 to 1.7, and A for –OH ranges from 0.6 to 1.04), the most stable chair for each stereoisomer is the conformer that has the amine group in the equatorial position, and thus the imine in the assembly of Figure 3 is similarly anticipated to be equatorial.57 Therefore, the alcohol is oriented axially in cis- and equatorially in the trans-, and this would give rise to a difference in the projection of the alcohol toward the pyridine rings in the octahedral Fe(II) complexes, thereby affecting the arrangement of the pyridyl rings around the Fe(II) center. Thus, the observed CD signals are strongly affected by the stereochemistry at the alcohol stereocenter.
Figure 7.
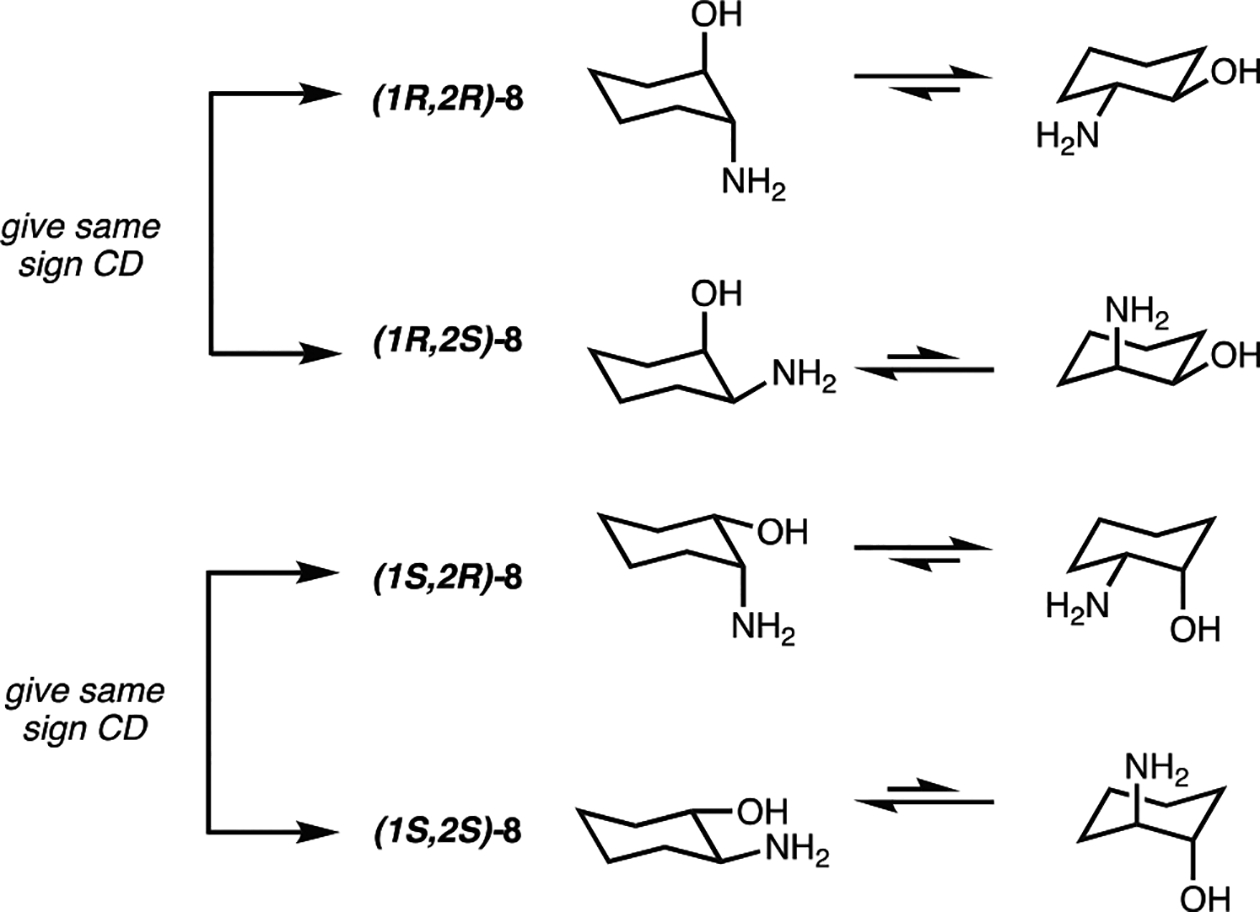
Chair-flip equilibria for the four stereoisomers of 2-aminocyclohexanol.
Aminoalcohol de Determination.
We anticipated the cis-and trans-diastereomers would be differentiated with the same octahedral iron complex via UV–vis spectroscopy. As anticipated, we observed a distinct difference in absorbance values between the cis- and trans-diastereomers of 12 (Figure 8). This difference in absorbance is likely due to the different orientation of ligands around the iron(II) between the cis- and trans-diastereomeric sets.58
Figure 8.
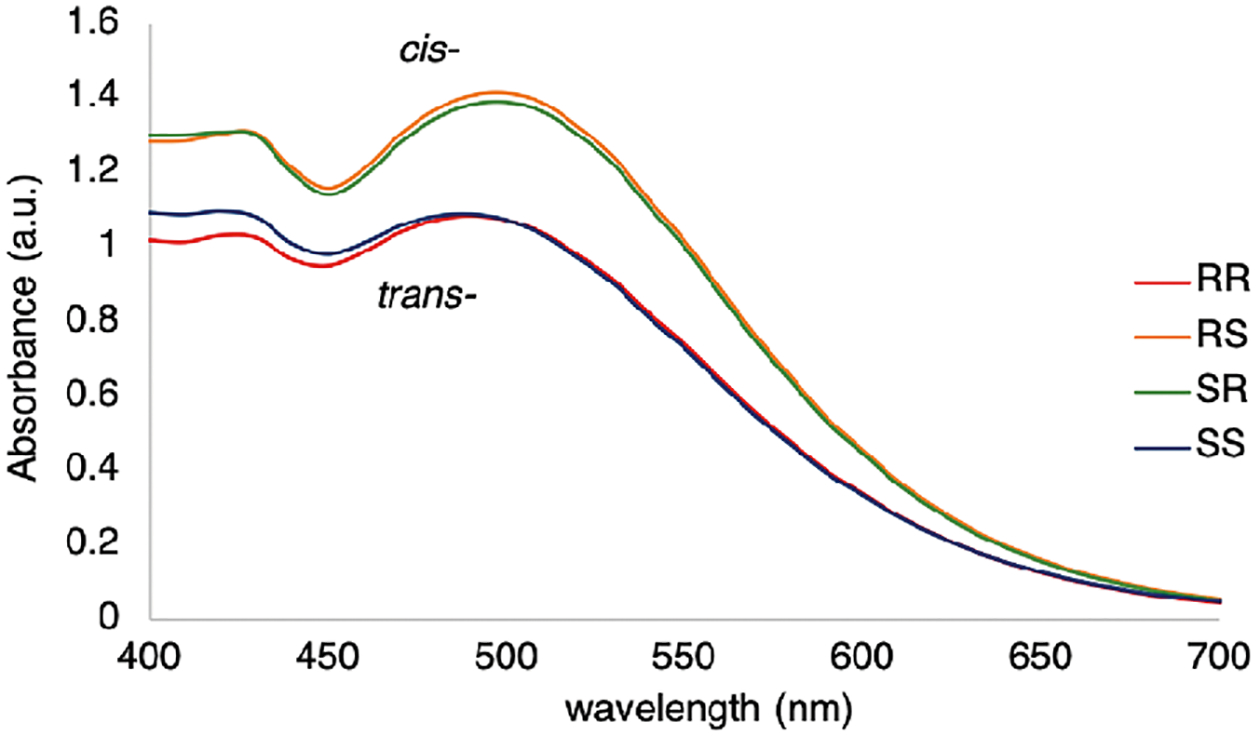
UV–vis spectra for the four stereoisomers of 2-aminocyclohexanol with 3-hydroxy-pyridine-2-carboxaldehyde and Fe(OTf)2. The UV–vis spectra were recorded in CH3CN at 25 °C (0.5 mM Fe(OTf)2, 1.5 mM 8, 1 cm cell).
A series of stereoisomeric solutions were made where the diastereomeric excesses (eq 1) were varied and monitored via UV–vis spectroscopy; a linear correlation between de and absorbance was observed (Figure 9). As expected, no change in absorbance was seen when the ee of the solution was varied (Figure S19).
| (1) |
The CD signals associated with the amine assembly are concentration-independent if the iron(II) center is fully saturated with chiral imine ligands; that is, the CD signal will reflect the ee of the chiral amine (Figure 2). For most substrates, a 3:3:1 ratio of chiral amine, 3-hydroxy-2-pyridinecarboxaldehdye, and iron triflate is suitable for complete assembly formation. The stereochemical complexity of the octahedral iron(II) complexes with a four-component stereoisomeric mixture led us to perform experiments similar to those discussed above for the 1-position and the alcohol assembly, where we checked if the analytes were statistically incorporated into the assemblies or if one diastereomer was preferred (Figure S20). A statistical distribution was verified, and the stereoisomeric complexity associated with the octahedral iron complexes was determined to not affect chiroptical analysis as the complexes rapidly interconvert in equilibria.
Figure 9.
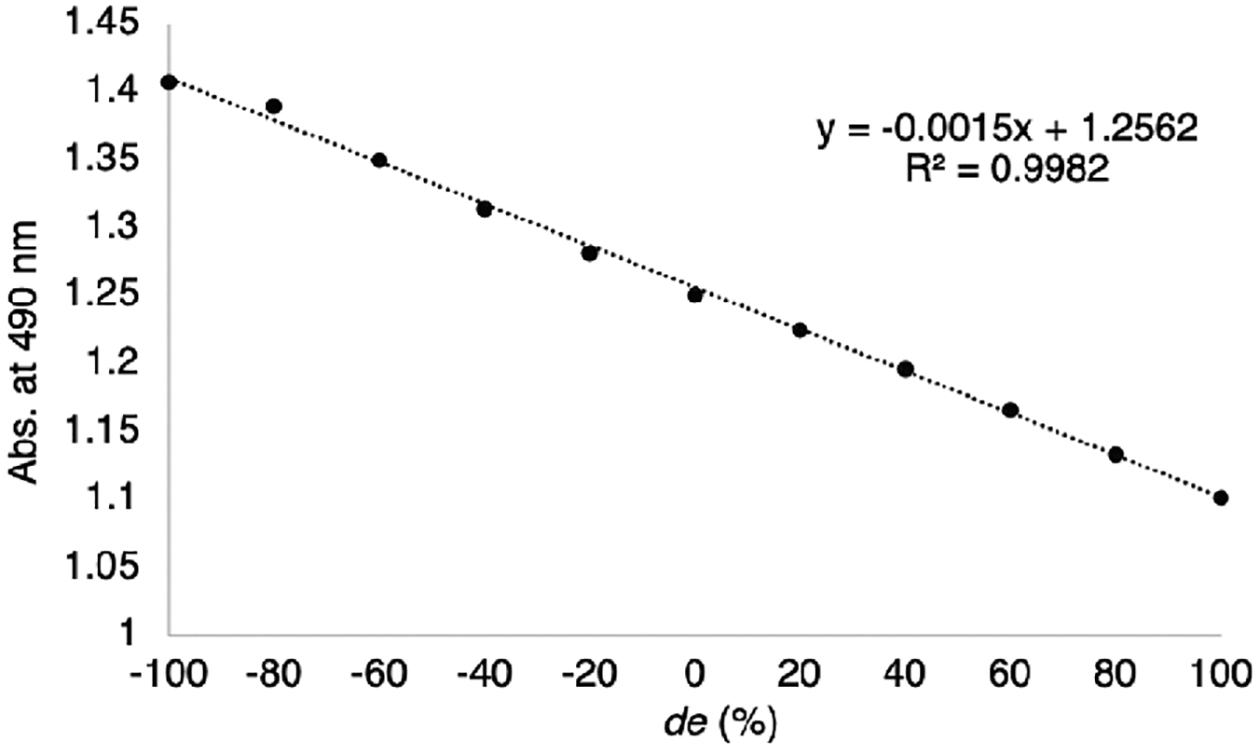
Calibration curve for determining de of a solution of 2-aminocyclohexanol.
Comined ee and de Analysis.
Having demonstrated enantiomeric differentiation of the four stereoisomers could be accomplished via CD spectroscopy of 12/19 and the relative configuration, that is, diastereomeric differentiation via UV–vis spectroscopy of 12, we turned our attention to determining the percent composition (speciation) for mixtures of the four stereoisomers. In a recent paper by our group, a mathematical relationship for the total speciation of a four-component stereoisomeric mixture using ee and de values was described.46 When discussing the success of an asymmetric transformation that forges two stereocenters, the enantioselectivity is typically characterized within the diastereomeric sets of enantiomers; that is, the ee is separately determined for the cis- and trans-isomers (eqs 2 and 3).
| (2) |
| (3) |
Yet, because our assays determine ee values at individual stereocenters, we defined the ee’s to be suited to our methods (eqs 4 and 5), while the traditional equation for diastereomeric excess remains unchanged (eq 6).
| (4) |
| (5) |
| (6) |
With these definitions, we aimed to individually home in on the absolute configuration at either the 1- or the 2-position without considering the stereochemistry of the adjacent stereocenter. Rearrangement of the three equations above and substitution gave us equations for determining the percent composition of the four stereoisomers based on two ee values and a de value (eqs 7–10).
| (7) |
| (8) |
| (9) |
| (10) |
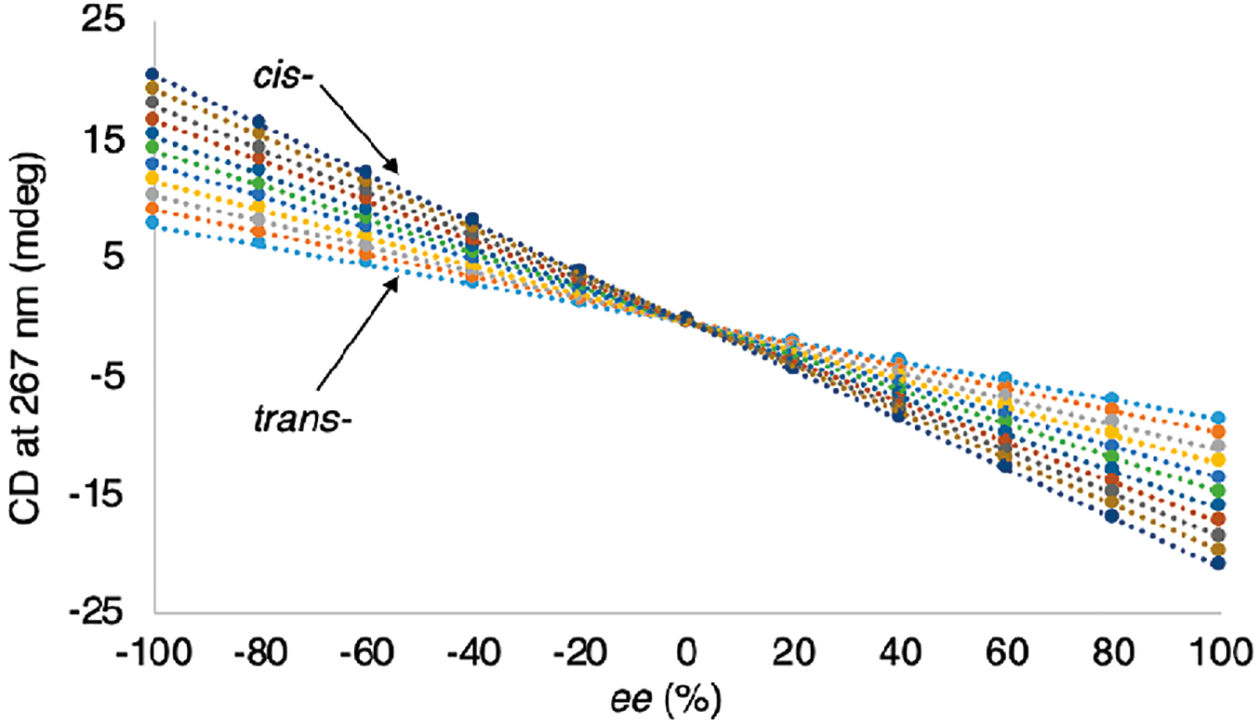
For the above equations to give accurate percent compositions, the optical measurements should be unaffected by the adjacent stereocenter. For example, when monitoring the 1-position, (1R,2R) and (1R,2S) alcohols should give the same CD signal at 267 nm. Yet, as discussed above, the adjacent stereocenter is imparting an effect on the spectroscopic signal. This can be clearly seen by considering the different intensities in CD magnitudes at the λmax of both the alcohol and the amine assemblies (Figures 10 and 11). These two plots show dramatic differences in the slopes of the ee versus CD signal for both the alcohol and the amine assemblies, and in fact the amine assembly slope switches from negative values for cis to positive values for trans.
Figure 10.
ECCD intensity of four component stereoisomeric mixtures of assembly 19 derived from the four stereoisomers of 21 plotted versus ee1 values. Each line corresponds to a single de value (−100, −80, −60, −40, −20, 0, 20, 40, 60, 80, 100) and 11 ee1 values (−100, −80, −60, −40, −20, 0, 20, 40, 60, 80, 100).
Figure 11.
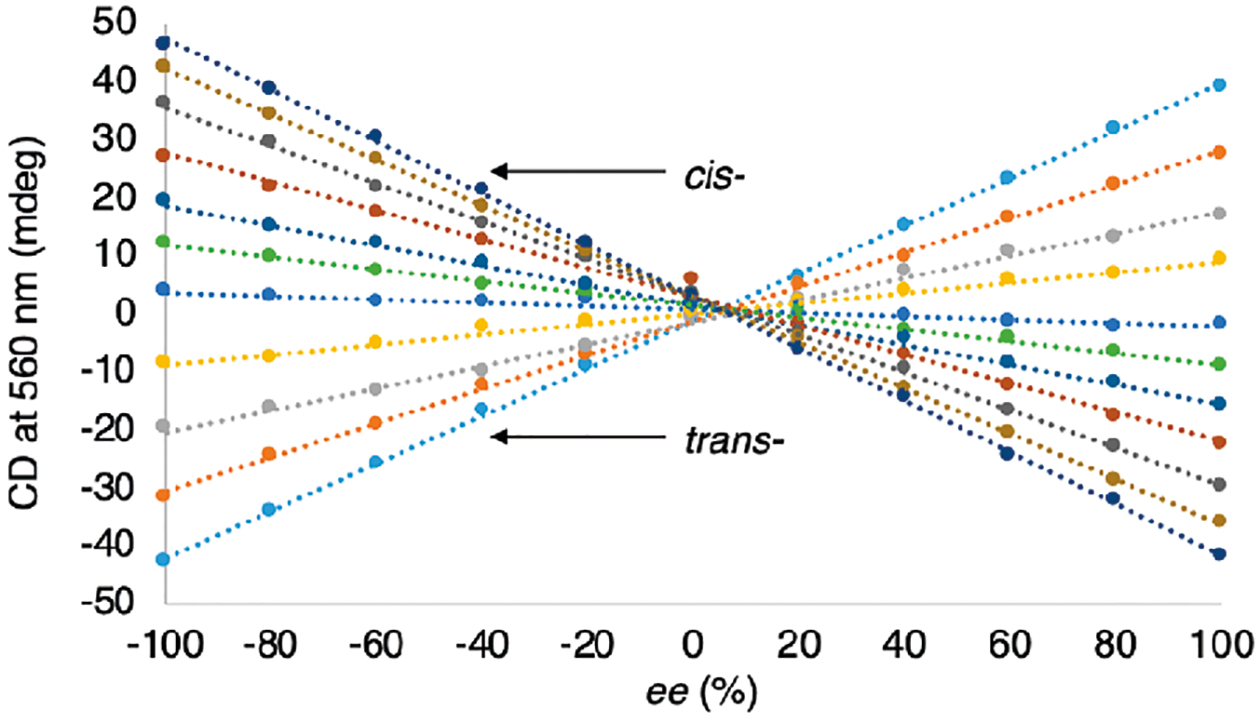
CD intensity of four component stereoisomeric mixtures of assembly 12 derived from the four stereoisomers of 8 plotted versus ee2 values. Each line corresponds to a single de value (−100, −80, −60, −40, −20, 0, 20, 40, 60, 80, 100) and 11 ee2 values (left to right −100, −80, −60, −40, −20, 0, 20, 40, 60, 80, 100).
This difference between stereoisomers that possess the same stereochemistry at the stereocenter being investigated results in the inability to accurately determine the ee at that stereocenter. For example, considering a hypothetical stereoisomeric mixture of unknown composition of 2-aminocyclohexanol, one might observe a CD intensity of 10 mdeg at the λmax of the alcohol assembly (Figure 10). Looking at Figure 10, a CD intensity of 10 mdeg corresponds to several different diastereomeric mixtures. At first glance, one might expect knowing the de of the mixture would allow for one to choose the line corresponding to the de in Figures 10 and 11 and determine ee1 and ee2; however, we have found that the mathematics do not permit this approach. We have found that only in a very special circumstance can one use this approach, and this is discussed extensively in ref 46. As a result, we were presented with the additional challenge of differentiating the stereochemistry of the adjacent stereocenter to accurately determine the ee values for the percent composition calculations using eqs 7–10. Thus, we turned to chemometrics as a means to pattern the optical responses.
Chemometric Analysis.
With the CD intensities for both the alcohol and the amine assemblies, and the UV–vis spectrum for the amine assembly as inputs, we employed a chemometric analysis (machine learning) to determine ee values at both stereocenters. We began with a training set of 70 samples and a test set of 10 samples and recorded the CD and absorbance spectra (Figures S22–S27). A visual representation of the space spanned by the training and test sets is shown in Figure 12. Note that we used values toward the high ends of ee and de for training, as these are the values that would be of most interest in a real screening scenario.
Figure 12.
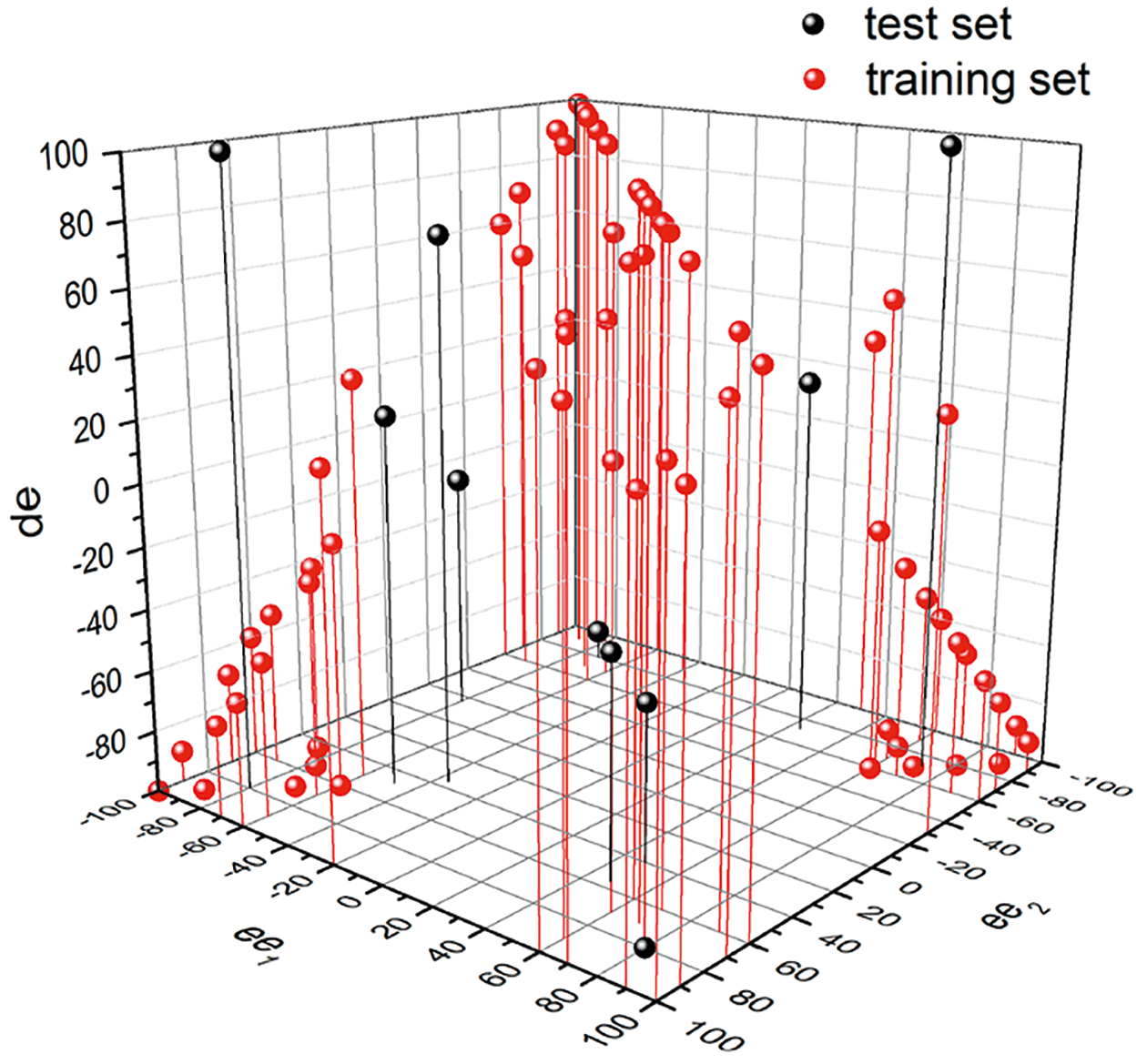
3D map of ee1, ee2, and de space spanned by training and test set, where the x-axis is ee1, y-axis is ee2, and the z-axis is de.
The three sets of measurements collected, that is, the CD intensities for 19 (XCD,alcohol) and for 12 (XCD,amine), and the UV–vis spectra of 12 (XUV–vis,amine), were used as predictors to build a linear regression model for the quantification of ee1 and ee2. In this framework, to take advantage of the multiblock nature of the data set, a novel multiblock regression method called sequential and orthogonalized covariance selection (SO-CovSel)59 was applied to find the best relationship between the measured variables (i.e., the spectral intensities) and the quantities to be predicted (ee1 and ee2). This method (which is explained in greater detail in section V of the Supporting Information) couples the search for the optimal regression parameter, leading to the most accurate predictions, with a highly effective variable selection strategy, which, leading to parsimonious models (i.e., models based only on a small number of predictors), are more robust, more easily interpretable, and also more suitable to be implemented in sensors. In the present study, the CD and UV–vis data measured on the calibration samples were used to build the regression model, that is, to select the best subset of variables (spectral intensities) to be used as predictors and to estimate their associated regression coefficients for the prediction of the responses (the values of ee1 and ee2). The optimal model (which was selected on the basis of a cross-validation procedure with seven cancelation groups) was the one built using 11 variables from the alcohol CD block (the spectral intensities at 240, 249, 252, 254, 256, 259, 262, 266, 269, 271, and 273 nm) and only one (ellipticity at 560 nm) and one (absorbance at 500 nm) for the amine CD and amine UV–vis blocks, respectively. These variables clearly would not have been chosen if one had simply assumed the λmax values would be optimal for variable selection. When applied to the training set, this model resulted in absolute errors of ±5.4% and ±10.7%, for the prediction of ee1 and ee2, respectively (Figure 13), and, in the validation stage, even better results were obtained on the test set (Table 1), ±6% for ee1 and ±5% for ee2, respectively.
Figure 13.
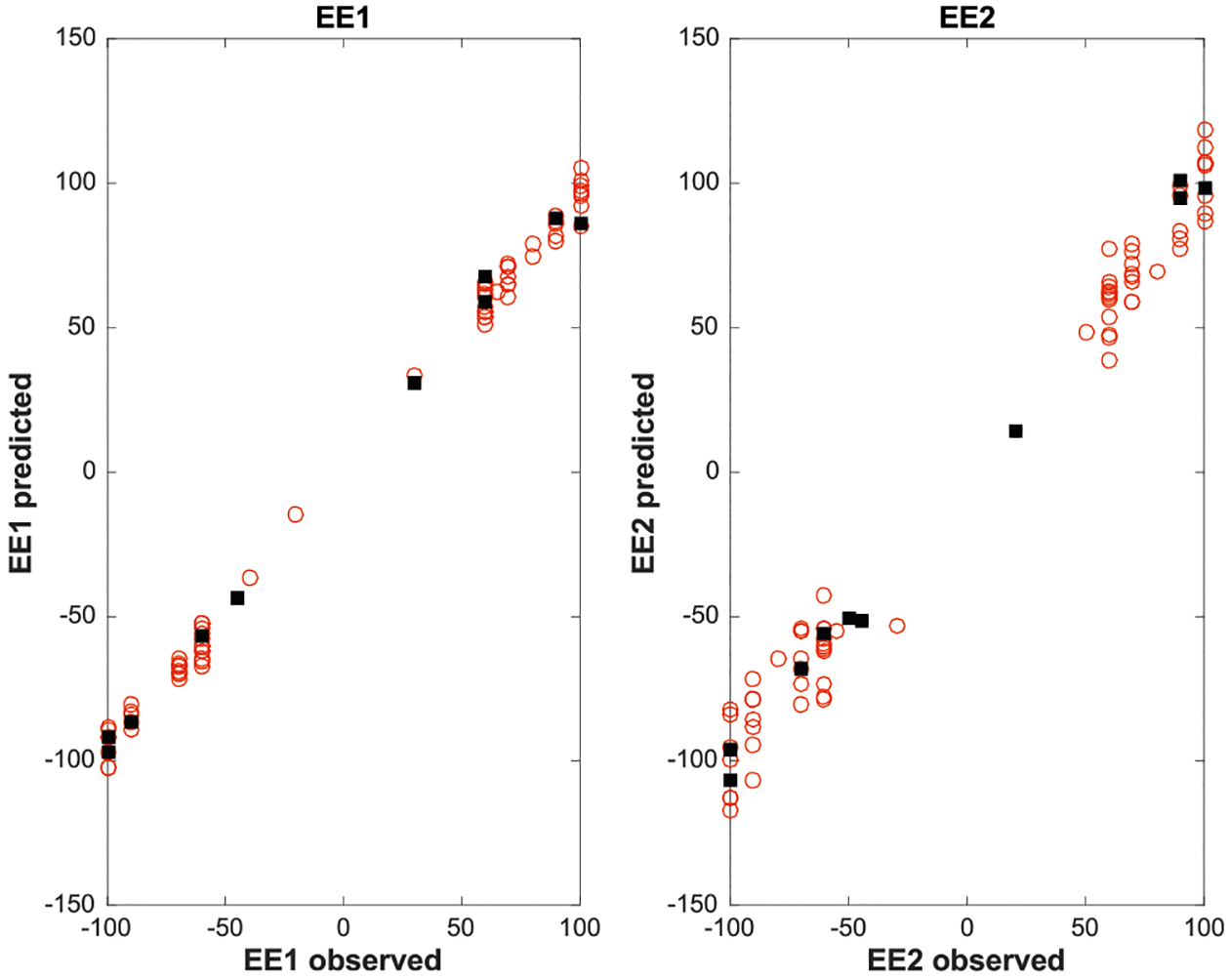
Plots of known ee (EE#,observed) versus ee predicted using the SO-CovSel regression method, where the red data points are the training set and the black data points are the test set.
Table 1.
Total Stereoisomeric Speciation with ee Values Predicted Using SO-CovSel and a de Value from Absorbance Dataa
| RR | SS | ||||
|---|---|---|---|---|---|
| calcd | actual | abs error | calcd | actual | abs error |
| 0.73 | 0 | 0.7 | 97.0 | 100 | −2.9 |
| 4.64 | 0 | 4.6 | 4.37 | 0 | 4.4 |
| 91.1 | 95 | −3.9 | −1.26 | 0 | −1.3 |
| −4.07 | 0 | −4.1 | −7.90 | 0 | −7.9 |
| 4.22 | 5.26 | −1.0 | 8.10 | 5.26 | 2.8 |
| 13.0 | 10 | 3.0 | 68.0 | 65 | 3.0 |
| 38.5 | 35 | 3.5 | 3.47 | 5 | −1.5 |
| 58.2 | 60 | −1.8 | 5.43 | 0 | 5.4 |
| 31.2 | 20 | 11.3 | 30.6 | 20 | 10.6 |
| 0.06 | 0 | 0.1 | 67.8 | 65 | 2.8 |
| RS | SR | ||||
| calcd | actual | abs error | calcd | actual | abs error |
| −3.90 | 0 | −3.9 | 6.08 | 0 | 6.1 |
| −2.78 | 0 | −2.8 | 93.8 | 100 | −6.2 |
| 8.16 | 5 | 3.2 | 2.02 | 0 | 2.0 |
| 102 | 95 | 6.7 | 10.3 | 5 | 5.3 |
| 20.2 | 21.05 | −0.9 | 67.5 | 68.42 | −0.9 |
| 11.6 | 15 | −3.4 | 7.42 | 10 | −2.6 |
| 62.1 | 60 | 2.1 | −4.13 | 0 | −4.1 |
| −1.11 | 0 | −1.1 | 37.5 | 40 | −2.5 |
| −9.19 | 0 | −9.2 | 47.3 | 60 | −12.7 |
| 15.9 | 15 | 0.9 | 16.34 | 20 | −3.7 |
All values are given in percentages.
With the ee1 and ee2 values predicted using SO-CovSel (Table S1) and de values determined directly from the absorbance at 490 nm of the octahedral iron complexes, eqs 7–10 were applied for total speciation and the percent composition of each stereoisomer (Table 1), resulting in an averaged absolute error of only ±4%. Finally, these percent compositions were expressed as the commonly used parameters of ee within each diastereomeric set (i.e., eecis and eetrans,Table 2) with similarly low absolute errors (±3%).
Table 2.
Calculated ee Values within Each Diastereomeric Seta
| eecis | eetrans | ||||
|---|---|---|---|---|---|
| calcd | actual | abs error | calcd | actual | abs error |
| −9.98 | 0 | −9.98 | −96.4 | −100 | 3.64 |
| −96.6 | −100 | 3.45 | 0.27 | 0 | 0.27 |
| 6.14 | 5 | 1.14 | 92.4 | 95 | −2.65 |
| 91.4 | 90 | 1.39 | 3.82 | 0 | 3.82 |
| −47.3 | −47.4 | 0.09 | −3.89 | 0 | −3.89 |
| 4.13 | 5 | −0.87 | −54.9 | −55 | 0.06 |
| 66.3 | 60 | 6.28 | 35.0 | 30 | 5.04 |
| −38.6 | −40 | 1.44 | 52.8 | 60 | −7.21 |
| −56.5 | −60 | 3.50 | 0.62 | 0 | 0.62 |
| −0.40 | −5 | 4.60 | −67.7 | −65 | −2.70 |
All values are given in percentages.
CONCLUSION
Rapid optical assays for the determination of reaction parameters in parallel synthesis routines targeted to optimizing ee and de values are becoming increasingly necessary as automation methods permeate reaction discovery. To explore methods for both ee and de analysis (as well as reaction yield), we turned our first study to a very challenging analyte: 2- aminocyclohexanol. Not only is there no chromophore in this analyte, but the stereocenters are vicinal, which led to the optical signal being influenced by both the targeted functional group and the neighboring functional group. Thus, a simple mathematical approach to achieve complete speciation was not possible, but instead a patterning technique (chemometrics) was implemented. This resulted in only a 4% absolute error on determining the percentages of each of the four stereoisomers in random mixtures. Further, optical analysis of 96 reactions can be accomplished in 27 min: 5 min for concentration determination via a single wavelength measurement, 5 min for simultaneous ee2/de determination via a single wavelength measurement, and 17 min for ee1 determination via 12 wavelength measurements. Because 2-aminocyclohexanol was simply chosen to be a challenging analyte for which to develop our methods, we are now turning toward using this general work-flow for screening reactions that simultaneously set two stereocenters in a single step with varying degrees of enantioand diastereoselectivities, such as the reduction of diketide thioesters via ketoreductases.31
Supplementary Material
ACKNOWLEDGMENTS
We thank Samuel D. Dahlhauser for his insightful comments and helpful critiques. We gratefully acknowledge financial support for this work from the National Institutes of Health (R01-GM077437 and 1 S10 OD021508-01) as well as the Welch Regents Chair to E.V.A. (F-0045).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b03844.
Detailed synthetic protocols, optical analyses, training set data, test set data, and predicted ee values using SOCovSel, as well as supporting text, supporting figures (Figures S1–S27), and a supporting table (Table S1) (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Krska SW; DiRocco DA; Dreher SD; Shevlin M The Evolution of Chemical High-Throughput Experimentation To Address Challenging Problems in Pharmaceutical Synthesis. Acc. Chem. Res 2017, 50 (12), 2976–2985. [DOI] [PubMed] [Google Scholar]
- (2).Shevlin M Practical High-Throughput Experimentation for Chemists. ACS Med. Chem. Lett 2017, 8 (6), 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Jagt RBC; Toullec PY; Schudde EP; De Vries JG; Feringa BL; Minnaard AJ Synthesis of Solution-Phase Phosphoramidite and Phosphite Ligand Libraries and Their In Situ Screening in the Rhodium-Catalyzed Asymmetric Addition of Arylboronic Acids. J. Comb. Chem 2007, 9 (3), 407–414. [DOI] [PubMed] [Google Scholar]
- (4).Burgess K; Lim H-J; Porte AM; Sulikowski GA New catalysts and conditions for a C-H insertion reaction identified by high throughput catalyst screening. Angew. Chem., Int. Ed. Engl 1996, 35 (2), 220–2. [Google Scholar]
- (5).Barhate CL; Joyce LA; Makarov AA; Zawatzky K; Bernardoni F; Schafer WA; Armstrong DW; Welch CJ; Regalado EL Ultrafast chiral separations for high throughput enantiopurity analysis. Chem. Commun 2017, 53 (3), 509–512. [DOI] [PubMed] [Google Scholar]
- (6).Patel DC; Wahab MF; Armstrong DW; Breitbach ZS Advances in high-throughput and high-efficiency chiral liquid chromatographic separations. J. Chromatogr. A 2016, 1467, 2–18. [DOI] [PubMed] [Google Scholar]
- (7).Barhate CL; Wahab MF; Breitbach ZS; Bell DS; Armstrong DW High efficiency, narrow particle size distribution, sub-2 μm based macrocyclic glycopeptide chiral stationary phases in HPLC and SFC. Anal. Chim. Acta 2015, 898, 128–137. [DOI] [PubMed] [Google Scholar]
- (8).Wahab MF; Dasgupta PK; Kadjo AF; Armstrong DW Sampling frequency, response times and embedded signal filtration in fast, high efficiency liquid chromatography: A tutorial. Anal. Chim. Acta 2016, 907, 31–44. [DOI] [PubMed] [Google Scholar]
- (9).Welch CJ; Sajonz P; Biba M; Gouker J; Fairchild J Comparison of Multiparallel Microfluidic HPLC Instruments for High Throughput Analyses in Support of Pharmaceutical Process Research. J. Liq. Chromatogr. Relat. Technol 2006, 29 (15), 2185–2200. [Google Scholar]
- (10).Leung D; Anslyn EV Rapid Determination of Enantiomeric Excess of α-Chiral Cyclohexanones Using Circular Dichroism Spectroscopy. Org. Lett 2011, 13 (9), 2298–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wolf C; Bentley KW Chirality sensing using stereodynamic probes with distinct electronic circular dichroism output. Chem. Soc. Rev 2013, 42 (12), 5408–5424. [DOI] [PubMed] [Google Scholar]
- (12).Pu L Fluorescence of Organic Molecules in Chiral Recognition. Chem. Rev 2004, 104 (3), 1687–1716. [DOI] [PubMed] [Google Scholar]
- (13).Jo HH; Lin C-Y; Anslyn EV Rapid Optical Methods for Enantiomeric Excess Analysis: From Enantioselective Indicator Displacement Assays to Exciton-Coupled Circular Dichroism. Acc. Chem. Res 2014, 47 (7), 2212–2221. [DOI] [PubMed] [Google Scholar]
- (14).Pu L Enantioselective Fluorescent Sensors: A Tale of BINOL. Acc. Chem. Res 2012, 45 (2), 150–163. [DOI] [PubMed] [Google Scholar]
- (15).Shabbir SH; Regan CJ; Anslyn EV A general protocol for creating high-throughput screening assays for reaction yield and enantiomeric excess applied to hydrobenzoin. Proc. Natl. Acad. Sci. U.S. A 2009, 106 (26), 10487–10492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhu L; Shabbir SH; Anslyn EV Two Methods for the Determination of Enantiomeric Excess and Concentration of a Chiral Sample with a Single Spectroscopic Measurement. Chem. - Eur. J 2007, 13 (1), 99–104. [DOI] [PubMed] [Google Scholar]
- (17).Dey S; Powell DR; Hu C; Berkowitz DB Cassette” In Situ Enzymatic Screening Identifies Complementary Chiral Scaffolds for Hydrolytic Kinetic Resolution Across a Range of Epoxides. Angew. Chem., Int. Ed 2007, 46 (37), 7010–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dey S; Karukurichi KR; Shen W; Berkowitz DB Double-Cuvette ISES: In Situ Estimation of Enantioselectivity and Relative Rate for Catalyst Screening. J. Am. Chem. Soc 2005, 127 (24), 8610–8611. [DOI] [PubMed] [Google Scholar]
- (19).Wen K; Yu S; Huang Z; Chen L; Xiao M; Yu X; Pu L Rational Design of a Fluorescent Sensor to Simultaneously Determine Both the Enantiomeric Composition and the Concentration of Chiral Functional Amines. J. Am. Chem. Soc 2015, 137 (13), 4517–4524. [DOI] [PubMed] [Google Scholar]
- (20).Yu S; Plunkett W; Kim M; Pu L Simultaneous Determination of Both the Enantiomeric Composition and Concentration of a Chiral Substrate with One Fluorescent Sensor. J. Am. Chem. Soc 2012, 134 (50), 20282–20285. [DOI] [PubMed] [Google Scholar]
- (21).Mei X; Wolf C Determination of Enantiomeric Excess and Concentration of Unprotected Amino Acids, Amines, Amino Alcohols, and Carboxylic Acids by Competitive Binding Assays with a Chiral Scandium Complex. J. Am. Chem. Soc 2006, 128 (41), 13326–13327. [DOI] [PubMed] [Google Scholar]
- (22).Liu S; Pestano JPC; Wolf C Enantioselective Fluorescence Sensing of Chiral α-Amino Alcohols. J. Org. Chem 2008, 73 (11), 4267–4270. [DOI] [PubMed] [Google Scholar]
- (23).Shcherbakova EG; Brega V; Lynch VM; James TD; Anzenbacher P Jr. High-Throughput Assay for Enantiomeric Excess Determination in 1,2- and 1,3-Diols and Direct Asymmetric Reaction Screening. Chem. - Eur. J 2017, 23 (42), 10222–10229. [DOI] [PubMed] [Google Scholar]
- (24).Nieto S; Lynch VM; Anslyn EV; Kim H; Chin J High-Throughput Screening of Identity, Enantiomeric Excess, and Concentration Using MLCT Transitions in CD Spectroscopy. J. Am. Chem. Soc 2008, 130 (29), 9232–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Nieto S; Dragna JM; Anslyn EV A Facile Circular Dichroism Protocol for Rapid Determination of Enantiomeric Excess and Concentration of Chiral Primary Amines. Chem. - Eur. J 2010, 16(1), 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bentley KW; Zhang P; Wolf C Miniature high-throughput chemosensing of yield, ee, and absolute configuration from crude reaction mixtures. Sci. Adv 2016, 2 (2), e1501162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).De los Santos ZA; Wolf C Chiroptical Asymmetric Reaction Screening via Multicomponent Self-Assembly. J. Am. Chem. Soc 2016, 138 (41), 13517–13520. [DOI] [PubMed] [Google Scholar]
- (28).Zhao Q; Wen J; Tan R; Huang K; Metola P; Wang R; Anslyn EV; Zhang X Rhodium-Catalyzed Asymmetric Hydrogenation of Unprotected NH Imines Assisted by a Thiourea. Angew. Chem., Int. Ed 2014, 53 (32), 8467–8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Herrera BT; Pilicer SL; Anslyn EV; Joyce LA; Wolf C Optical Analysis of Reaction Yield and Enantiomeric Excess: A New Paradigm Ready for Prime Time. J. Am. Chem. Soc 2018, 140 (33), 10385–10401. [DOI] [PubMed] [Google Scholar]
- (30).Yu SS; Pu L One enantiomeric fluorescent sensor pair to discriminate four stereoisomers of threonines. Sci. China: Chem 2013, 56 (3), 301–306. [Google Scholar]
- (31).Piasecki Shawn K.; Taylor Clint A.; Detelich Joshua F.; Liu J; Zheng J; Komsoukaniants A; Siegel Dionicio R.; Keatinge-Clay Adrian T. Employing Modular Polyketide Synthase Ketoreductases as Biocatalysts in the Preparative Chemoenzymatic Syntheses of Diketide Chiral Building Blocks. Chem. Bio 2011, 18 (10), 1331–1340. [DOI] [PubMed] [Google Scholar]
- (32).Tian X; Cassani C; Liu Y; Moran A; Urakawa A; Galzerano P; Arceo E; Melchiorre P Diastereodivergent Asymmetric Sulfa-Michael Additions of α-Branched Enones using a Single Chiral Organic Catalyst. J. Am. Chem. Soc 2011, 133 (44), 17934–17941. [DOI] [PubMed] [Google Scholar]
- (33).Li K; Shao X; Tseng L; Malcolmson SJ 2-Azadienes as Reagents for Preparing Chiral Amines: Synthesis of 1,2-Amino Tertiary Alcohols by Cu-Catalyzed Enantioselective Reductive Couplings with Ketones. J. Am. Chem. Soc 2018, 140 (2), 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ebinger K; Weller HN Comparison of chromatographic techniques for diastereomer separation of a diverse set of drug-like compounds. J. Chromatogr. A 2013, 1272, 150–154. [DOI] [PubMed] [Google Scholar]
- (35).Joshi N; Dhamarlapati B; Pillai A; Paulose J; Tan J; Blue LE; Tedrow J; Farrell B Separation and quantitation of eight isomers in a molecule with three stereogenic centers by normal phase liquid chromatography. J. Chromatogr. A 2018, 1538, 108–111. [DOI] [PubMed] [Google Scholar]
- (36).Chain WJ; Myers AG A Convenient, NMR-Based Method for the Analysis of Diastereomeric Mixtures of Pseudoephedrine Amides. Org. Lett 2007, 9 (2), 355–357. [DOI] [PubMed] [Google Scholar]
- (37).Guo H-M; Tanaka F A Fluorogenic Aldehyde Bearing a 1,2,3-Triazole Moiety for Monitoring the Progress of Aldol Reactions.J. Org. Chem 2009, 74 (6), 2417–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Matsumoto T; Urano Y; Takahashi Y; Mori Y; Terai T; Nagano T In Situ Evaluation of Kinetic Resolution Catalysts for Nitroaldol by Rationally Designed Fluorescence Probe. J. Org. Chem 2011, 76 (10), 3616–3625. [DOI] [PubMed] [Google Scholar]
- (39).Greene LE; Lincoln R; Krumova K; Cosa G Development of a Fluorogenic Reactivity Palette for the Study of Nucleophilic Addition Reactions Based on meso-Formyl BODIPY Dyes. ACS Omega 2017, 2 (12), 8618–8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Karjalainen OK; Koskinen AMP Diastereoselective synthesis of vicinal amino alcohols. Org. Biomol. Chem 2012, 10 (22), 4311–4326. [DOI] [PubMed] [Google Scholar]
- (41).Burchak ON; Py S Reductive cross-coupling reactions (RCCR) between CN and CO for β-amino alcohol synthesis. Tetrahedron 2009, 65 (36), 7333–7356. [Google Scholar]
- (42).Restorp P; Fischer A; Somfai P Stereoselective Synthesis of Functionalized Pyrrolidines via a [3 + 2]-Annulation of N-Ts-α-Amino Aldehydes and 1,3-Bis(silyl)propenes. J. Am. Chem. Soc 2006, 128 (39), 12646–12647. [DOI] [PubMed] [Google Scholar]
- (43).Clerc J; Schellenberg B; Groll M; Bachmann AS; Huber R; Dudler R; Kaiser M Convergent Synthesis and Biological Evaluation of Syringolin A and Derivatives as Eukaryotic 20S Proteasome Inhibitors. Eur. J. Org. Chem 2010, 2010 (21), 3991–4003. [Google Scholar]
- (44).Birrell JA; Jacobsen EN A Practical Method for the Synthesis of Highly Enantioenriched trans-1,2-Amino Alcohols. Org. Lett 2013, 15 (12), 2895–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sherer EC; Lee CH; Shpungin J; Cuff JF; Da C; Ball R; Bach R; Crespo A; Gong X; Welch CJ Systematic Approach to Conformational Sampling for Assigning Absolute Configuration Using Vibrational Circular Dichroism. J. Med. Chem 2014, 57 (2), 477–494. [DOI] [PubMed] [Google Scholar]
- (46).Herrera BT; Lin C-Y; Wright AM; Moor SR; Anslyn EV Mathematical Relationships of Individual Stereocenter er Values to dr Values. JOC 2019, 84 (9), 2922–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Dragna JM; Pescitelli G; Tran L; Lynch VM; Anslyn EV; Di Bari L In Situ Assembly of Octahedral Fe(II) Complexes for the Enantiomeric Excess Determination of Chiral Amines Using Circular Dichroism Spectroscopy. J. Am. Chem. Soc 2012, 134 (9), 4398–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Dragna JM; Gade AM; Tran L; Lynch VM; Anslyn EV Chiral Amine Enantiomeric Excess Determination Using Self-Assembled Octahedral Fe(II)-Imine Complexes. Chirality 2015, 27(4), 294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).You L; Pescitelli G; Anslyn EV; Di Bari L An Exciton-Coupled Circular Dichroism Protocol for the Determination of Identity, Chirality, and Enantiomeric Excess of Chiral Secondary Alcohols. J. Am. Chem. Soc 2012, 134 (16), 7117–7125. [DOI] [PubMed] [Google Scholar]
- (50).You L; Berman JS; Anslyn EV Dynamic multi-component covalent assembly for the reversible binding of secondary alcohols and chirality sensing. Nat. Chem 2011, 3 (12), 943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Giuliano MW; Lin CY; Romney DK; Miller SJ; Anslyn EV A Synergistic Combinatorial and Chiroptical Study of Peptide Catalysts for Asymmetric Baeyer-Villiger Oxidation. Adv. Synth. Catal 2015, 357 (10), 2301–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Jo HH; Gao X; You L; Anslyn EV; Krische MJ Application of a High-Throughput Enantiomeric Excess Optical Assay Involving a Dynamic Covalent Assembly: Parallel Asymmetric Allylation and Ee Sensing of Homoallylic Alcohols. Chem. Sci 2015, 6 (12), 6747–6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Lin C-Y; Giuliano MW; Ellis BD; Miller SJ; Anslyn EV From substituent effects to applications: enhancing the optical response of a four-component assembly for reporting ee values. Chem. Sci 2016, 7 (7), 4085–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Lin C-Y; Lim S; Anslyn EV Model Building Using Linear Free Energy Relationship Parameters–Eliminating Calibration Curves for Optical Analysis of Enantiomeric Excess. J. Am. Chem. Soc 2016, 138 (26), 8045–8047. [DOI] [PubMed] [Google Scholar]
- (55).If the concentration of analyte is known, the difference in physical properties among the diastereomers can be taken advantage of, and any of these assays could be used to characterize the diasteromeric excess of the mixture.
- (56).It has been demonstated that chiral monoamines, mono-ols, and monothiols can all be efficiently incorporated into this assembly; however, in the context of compounds with multiple compatible functional groups, the incorporation of analyte will be biased toward the more nucleophilic functional group.
- (57).Anslyn EV; Dougherty DA; Dougherty EV; Books US Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006. [Google Scholar]
- (58).Tamayo AB; Alleyne BD; Djurovich PI; Lamansky S; Tsyba I; Ho NN; Bau R; Thompson ME Synthesis and Characterization of Facial and Meridional Tris-cyclometalated Iridium(III) Complexes. J. Am. Chem. Soc 2003, 125 (24), 7377–7387. [DOI] [PubMed] [Google Scholar]
- (59).Biancolillo A; Marini F; Roger J-M SO-CovSel: A novel method for variable selection in a multiblock framework. J. Chemom 2019, e3120. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.