

Abstract

A regioselective protocol for the synthesis of substituted allylic chlorides, bromides, and fluorides has been established. Remarkably, the method can be applied to the enantioselective synthesis of challenging chiral allylic chlorides. When the allylic halides are treated with the base triazabicyclodecene as the catalyst, a [1,3]-proton shift takes place, giving the corresponding vinyl halides in excellent yields with excellent Z:E ratios. Furthermore, the [1,3]-proton shift takes place with an outstanding level of chirality transfer from chiral allylic alcohols (≤98%) to give chiral trifluoromethylated vinyl chlorides.
The direct transfer of a hydrogen atom across an allylic system by a [1,3]-sigmatropic shift is thermally forbidden, both supra- and antarafacially.1 Thus, when a catalyst promotes this process, the reaction must proceed in a stepwise manner via one or more intermediates.2 A relevant example of this class of reaction is the isomerization of allylic alcohols into carbonyl compounds, a synthetic transformation in organic chemistry.3 In recent decades, significant work has been put into the development of methods for performing this isomerization efficiently,4−6 in some instances even under mild reaction conditions, and with a large substrate scope. Different transition-metal catalysts have been used, in particular, ruthenium, rhodium, palladium, and iridium.6
The isomerization of allylic alcohols can be described as a formal [1,3]-hydrogen shift. Under catalytic conditions, we may understand that the reaction proceeds via a series of intermediates. These species could be harnessed and applied in further transformations, opening up new synthetic opportunities. Our group has shown that in the presence of a range of different electrophiles and even nucleophiles, α-substituted ketones are formed as single constitutional isomers.7,8
Because of the stepwise nature of the catalytic [1,3]-hydrogen shift reactions, stereospecific examples of this transformation are rare.9−11 In 2012, Cahard and co-workers reported an outstanding stereospecific isomerization of enantiopure β-trifluoromethylated allylic alcohols catalyzed by an achiral ruthenium complex.10a In 2016, we reported the stereospecific isomerization of electron-deficient allylic alcohols and ethers mediated by a simple guanidine-type base, triazabicyclodecene (TBD), thus, in the absence of a metal catalyst.11 This reaction showed excellent levels of stereospecificity (i.e., chirality transfer), yielding β-substituted ketones and enol ethers with excellent enantiomeric ratios. A similar interaction has also recently been proposed for the stereospecific isomerization of 3-substituted indenols using 1,4-diazabicyclo[2.2.2]octane (DABCO).12
For allylic systems, a stereospecific base-catalyzed [1,3]-hydrogen shift has been reported for only allylic alcohols and ethers.11,12 The application of this approach to other allylic systems would create new synthetic methods and provide access to new chiral scaffolds. For example, the stereospecific isomerization of allylic halides would yield chiral vinyl halides, which are very versatile building blocks in organic chemistry.13 Examples of the isomerization of allylic halides into vinyl halides are very rare,14 and no stereospecific examples have been reported. This is due to the difficulty in the synthesis of chiral allylic halides, as they are very prone to racemization.15
In this paper, we report a method for the regioselective synthesis of γ-trifluoromethylated allylic chlorides, bromides, and fluorides, and the isomerization of these compounds into vinyl halides using the bicyclic guanidine base TBD as a catalyst (Scheme 1a). Furthermore, we also report the regio- and enantioselective synthesis of allylic chlorides and the first stereospecific base-catalyzed isomerization of these compounds into chiral vinyl chlorides (Scheme 1b).
Scheme 1. (a) Regioselective Synthesis and [1,3]-Proton Shift of Allylic Chlorides, Bromides, and Fluorides and (b) Enantioselective Synthesis of Chiral Allylic Chlorides and Stereospecific [1,3]-Proton Shift.

Several protocols for the synthesis of allylic halides from the corresponding alcohols have been reported in the literature.16 However, these methods suffer from low regioselectivity due to competing SN1′ and SN2′ pathways.17 We thus started our investigations by developing a method for the regioselective synthesis of allylic chlorides. We used γ-trifluoromethylated allylic chloride 1a as a model substrate (Table 1). Treatment of 1a with SOCl2 (1 equiv) in THF gave a mixture of 2a, 2a′, and 2a″ in 78% combined yield (Table 1, entry 1).
Table 1. Regioselective Synthesis of 2aa.

| entry | Cl source | solvent | yield (%) | 2a:2a′:2a″ |
|---|---|---|---|---|
| 1 | SOCl2 | THF | 78 | 82:12:6 |
| 2 | SOCl2 | Et2O | 83 | 76:16:8 |
| 3 | SOCl2 | CH2Cl2 | 83 | 88:12:– |
| 4 | SOCl2b | CH2Cl2 | 47 | 80:14:6 |
| 5 | PCl3 | CH2Cl2 | 80 | 96:4:– |
| 6 | PCl5 | CH2Cl2 | 50 | 98:2:– |
| 7 | POCl3 | CH2Cl2 | 52 | 12:–:–c |
| 8 | NCS/PPh3d | CH2Cl2 | 40 | 94:6:– |
| 9 | NCS/PPh3d | THF | 67 | >99:–:– |
Unless otherwise noted, 1a (0.1 mmol, 0.1 M) and the chlorinating agent (0.1 mmol) at 0 °C overnight.
With 0.1 mmol of pyridine.
An unknown product was formed.
NCS (0.15 mmol) and PPh3 (0.15 mmol).
In an attempt to improve the regioselectivity and minimize any cis/trans isomerization, several reaction parameters were varied. Changing the solvent had little effect on the outcome of the transformation (Table 1, entries 2 and 3). HCl formed in the reaction could promote the formation of carbocationic intermediates, leading to 2a′ and 2a″. However, when a base was added to suppress the formation of HCl (Table 1, entry 4), significantly lower yields and low selectivity were obtained. Other chlorinating agents were then tested; PCl3 and PCl5 gave significantly better results in terms of selectivity (Table 1, entries 5 and 6, respectively), albeit with moderate yields. POCl3 gave similar results, but the reaction also gave an unidentified byproduct (Table 1, entry 7). When the reaction was carried out with NCS (N-chlorosuccinimide) and PPh3 in CH2Cl2, we obtained excellent selectivity [94:6 2a:2a′ (Table 1, entry 8)] and a moderate yield.18 Gratifyingly, when the reaction was performed in THF, 2a was formed 67% yield with complete regioselectivity (Table 1, entry 9). Moreover, similar conditions (Scheme 2a) using NBS (N-bromosuccinimide) gave allylic bromide 3a in 62% yield (Scheme 2a). Allylic fluoride 4a was also accessed using DAST (N,N-diethylaminosulfur trifluoride), with complete regioselectivity (Scheme 2b).19
Scheme 2. Regioselective Synthesis of γ-Trifluoromethylated Allylic Bromide 3a and Fluoride 4a.

The [1,3]-hydrogen shift of 2a to give vinyl chloride 5a was then investigated using various bases at 60 °C (Table 2). With NEt3 or Cs2CO3 in a stoichiometric amount, <5% of 5a was obtained (Table 2, entry 1 or 2, respectively). In contrast, with 1 equiv of the more basic DBU or TBD, full conversions were obtained, giving 5a in 71% or 46% yield, respectively (Table 2, entry 3 or 4, respectively). In both instances, though, decomposition products were detected. With a smaller amount of base (0.1 equiv), the level of decomposition also decreased (Table 2, entries 5 and 6). DBU gave a good yield of 86%, but TBD gave a >99% yield of 2a after the same reaction time (18 h). The use of 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene [MTBD (Table 2, entry 7)] afforded 5a in 81% yield, and full conversion was not achieved. It was also observed that more polar solvents such as CH2Cl2 and MeCN always gave lower yields due to the formation of decomposition products (Table 2, entries 8–11).
Table 2. Base-Catalyzed Isomerization of 2aa.

| entry | base (equiv) | solvent | conversionb (%) | yieldb,c (%) |
|---|---|---|---|---|
| 1 | NEt3 (1.0) | toluene | <5 | –d |
| 2 | Cs2CO3 (1.0) | toluene | <5 | –d |
| 3 | DBU (1.0) | toluene | >99 | 71e |
| 4 | TBD (1.0) | toluene | >99 | 46e |
| 5 | DBU (0.1) | toluene | 86 | 86e |
| 6 | TBD (0.1) | toluene | >99 | >99 |
| 7 | MTBD (0.1) | toluene | 90 | 81e |
| 8 | TBD (0.1) | THF | 90 | 84e |
| 9 | TBD (0.1) | CH2Cl2 | 67 | 60e |
| 10 | TBD (0.1) | dioxane | 78 | 78e |
| 11 | TBD (0.1) | MeCN | 38 | 20e |
2a (0.1 mmol, 0.1 M), base, overnight at 60 °C.
Determined by 19F NMR spectroscopy.
Against an internal standard.
>95% 2a recovered.
Decomposition observed.
Next, the scope was studied (Scheme 3) using the optimized reaction conditions (Table 2, entry 6). When an electron-withdrawing or electron-donating group was present at the para position of the aryl group at R1, excellent yields were obtained in all instances (5b–5f). Even when R1 was a naphthyl group, 5g was obtained in very good yield. The products were all formed with very good Z:E ratios, up to 96:4. Further study of the substituents at R1 revealed some limitations of the reaction. When R1 was CH3 or H, vinyl chlorides 5h and 5i were obtained in lower yields (30% and 51%, respectively). Moreover, the introduction of alkyl groups, aryl groups, or even H at R3 was very well tolerated, and vinyl chlorides 5j–5m were formed in very good yields. Importantly, the CF3 group at R4 could be replaced with a sulfonyl group, and 5n was obtained in excellent yield from both (E)-2n and (Z)-2n. Highly substituted allylic chloride 2o gave a good 45% yield of 5o with 20 mol % TBD.
Scheme 3. Scope of the Base-Catalyzed Isomerization of Allylic Halides.
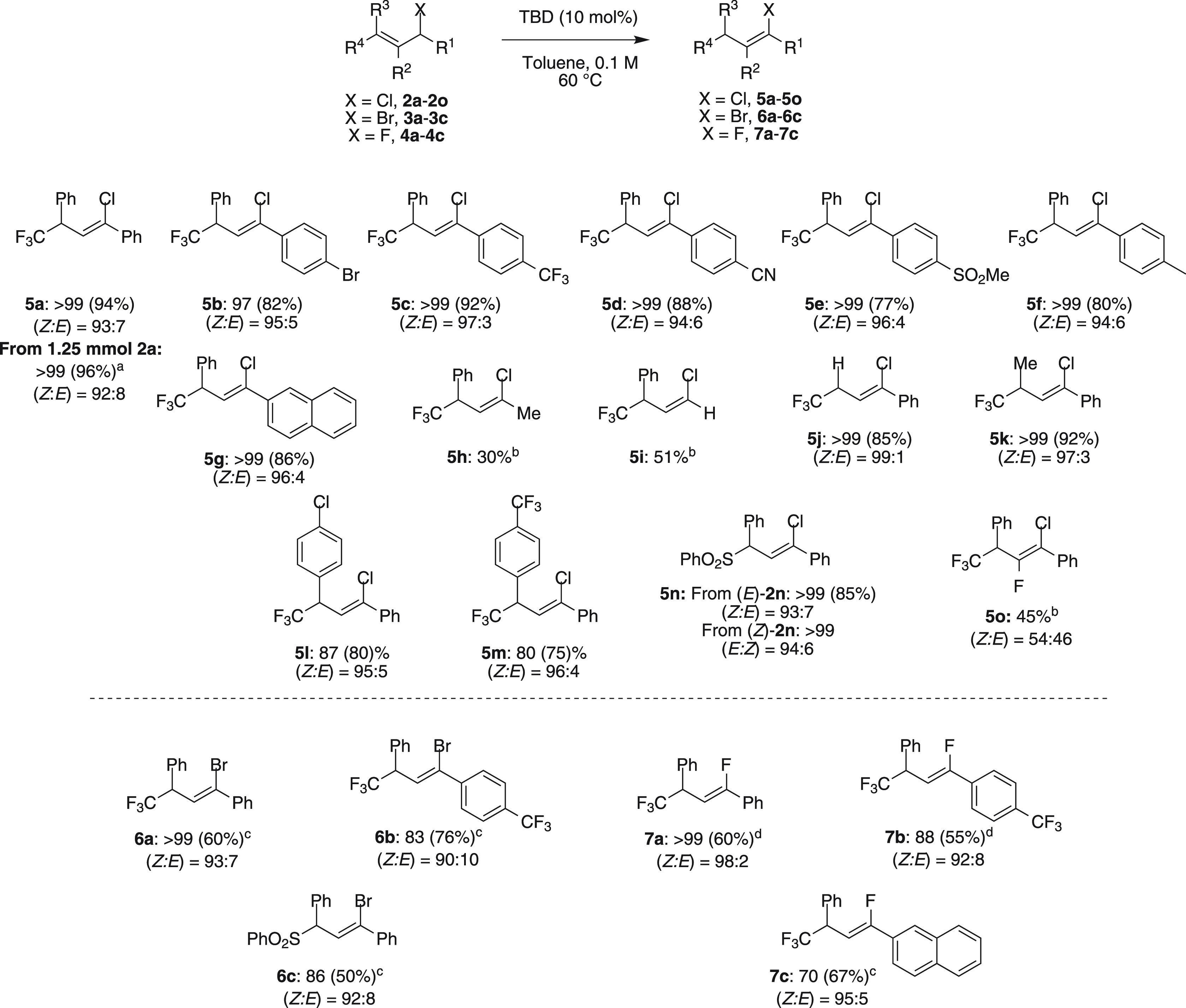
2a (1.25 mmol, 371 mg), TBD (0.125 mmol, 17 mg).
Conversion measured by 19F NMR.
TBD (30 mol %), 100 °C.
o-Xylene as a solvent at 145 °C.
Yields determined by 19F NMR spectroscopy (isolated yields in parentheses).
We also investigated the isomerization of allylic bromides 3 and fluorides 4. Both of these allylic substrates proved to be more challenging than the allylic chlorides. After a short optimization (see the Supporting Information), vinyl bromide 6a could be obtained in a good yield of 60% by using 30 mol % TBD at 100 °C. When an electron-withdrawing group was introduced into the aromatic group at R1 (3b), 83% was obtained with only 10 mol % TBD. Sulfonyl-substituted 3c isomerized well to 6c in 86% yield. Allylic fluorides 4a–4c showed lower reactivity, and a higher temperature of 145 °C was needed to reach full conversion. Under these conditions, vinyl fluorides 7a–7c were obtained with excellent Z:E ratios (≤98:2).
Next the stereospecificity of the reaction was investigated. To do this, a method for the enantioselective preparation of 2a had to be established.20 This is a challenging task, as benzyl and allyl groups can both stabilize transient cationic species that may form during the preparation of chiral allylic halides.21 As a result, SN1 pathways are favored and racemization occurs. The reaction of chiral (R)-1a (er of 99:1)10a gave allylic chloride (S)-2a regioselectively with an er of 77:23 (Table 3, entry 1, and Table S4). When the electron density of R1 was decreased, the racemization also decreased (Table 3, entries 2–5). This effect was more pronounced for the purely inductively electron-withdrawing CF3 group (1c, Table 3, entry 5); (S)-2c was obtained with an er of 94:6. In addition, a Me group at R3 also gave allyl chloride (S)-2m, with an er of 68:32 (Table 3, entry 6). Therefore, the best results are obtained for substrates with substituents that do not stabilize cationic species, preventing the chlorination from proceeding by SN1 pathways. Importantly, the isomerization of (S)-2a into (S)-5a took place with an excellent level of chirality transfer of 96% (Table 3, entry 1).
Table 3. Stereospecific Synthesis and Stereospecific 1,3-Proton Shift of Allylic Chloridesa.

| entry | (R)-1, R1/R3, er | (S)-2 er | (S)-5 erb | ssc (%) |
|---|---|---|---|---|
| 1 | 1a, Ph/Ph, 99:1 | 77:23 | 76:24 (99) | 96 |
| 2 | 1b, p-BrC6H4/Ph, 97:3 | 79:21 | 77:23 (99) | 95 |
| 3d | 1d, p-CNC6H4/Ph, 94:6 | 82:18 | 81:19 (92) | 98 |
| 4d | 1e, p-SO2MeC6H4/Ph, 93:7 | 87:13 | 76:24 (99) | 70 |
| 5e | 1c, p-CF3C6H4/Ph, 95:5 | 94:6 | 92:8 (99) | 95 |
| 6 | 1m, Ph/Me, 95:5 | 68:32 | 66:34 (99) | 94 |
(S)-2 prepared as in Table 1, entry 9, but with 1 equiv of NCS/PPh3 from (R)-1. Unless otherwise noted, 5 was obtained as in Table 2, entry 6. er is the enantiomeric ratio.
In parentheses, yields by 19F NMR spectroscopy.
Stereospecificity = ss = (ee of product 5/ee of 2) × 100.
5 prepared at 0 °C for 1 h.
5 prepared at room temperature for 1 h.
The absolute configuration of (S)-5a was determined by transforming it into a known β-trifluoromethylated ketone (see the Supporting Information). The stereochemistries of the other products were assigned by analogy. The exceptional transfer of chirality was also achieved for (S)-2b, (S)-2c, and (S)-2d (Table 3, entries 2, 3, and 5, respectively). An exception was (S)-2e, which bears a polar sulfonyl group; a minor loss of chirality was observed (Table 3, entry 4). Chloride (S)-5m was also obtained with a high stereosepecificity of 94% (Table 3, entry 6).
The mechanism is proposed to be a suprafacial [1,3]-proton shift (Figure 1), which likely is thermodynamically driven as previously observed in related suprafacial proton shifts.11,12 We carried out parallel isomerization reactions of 2a and 2a-d1 and observed a large kinetic isotope effect of 5.4 ± 0.6 (see the Supporting Information). This suggests that the deprotonation of C1 is the rate-determining step of the reaction. The high stereospecificity of the reaction indicates a highly efficient transfer of chirality from the starting chiral allylic chloride to the ion pair intermediate. Noncovalent chirality is induced on this ion pair, as interaction of the protonated base with the achiral planar allylic anion occurs exclusively from one specific side. As long as the ion pair remains tight, the chirality can be further transferred stereospecifically to the final product upon protonation of C3 (Figure 1). In conclusion, we have developed a method for the synthesis of allylic chlorides, bromides, and fluorides from allylic alcohols with full control of the regioselectivity. Furthermore, from enantiopure allylic alcohols, a new stereospecific chlorination reaction has been developed, giving access to γ-trifluoromethylated allylic chlorides in high enantiomeric ratios for the first time. The allylic halides were transformed in a single step into vinyl halides, important intermediates in synthetic organic chemistry. The reaction takes place by a stereospecific [1,3]-proton shift pathway catalyzed by the bicyclic guanidine base TBD. When applied to enantioenriched γ-trifluoromethylated allylic chlorides, the simple base catalyst can transfer the chirality affording vinyl chlorides with a stereogenic center at the allylic position, on a trifluoromethylated carbon. Formation of a very tight ion pair with induced noncovalent chirality, after a rate-determining deprotonation, is responsible for the outstanding levels of chirality transfer.
Figure 1.

Mechanism through a tight chiral ion pair.
Acknowledgments
The authors are grateful for support from the Swedish Research Council through Vetenskapsrådet and Formas, the Knut and Alice Wallenberg Foundation, and the Göran Gustafsson Foundation. This project was also funded by the European Union’s Horizon 2020 research and innovation programme under Grant Agreement 721223.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.0c01200.
Author Contributions
† S.M.-E. and V.G.-V. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Woodward R.; Hoffmann R. Stereochemistry of Electrocyclic Reactions. J. Am. Chem. Soc. 1965, 87, 395–397. 10.1021/ja01080a054. [DOI] [Google Scholar]; b Woodward R. B.; Hoffmann R.. The Conservation of Orbital Symmetry; Verlag Chemie: Weinheim, Germany, 1970. [Google Scholar]
- a Gauthier D.; Lindhardt A. T.; Olsen E. P. K.; Overgaard J.; Skrydstrup T. In Situ Generated Bulky Palladium Hydride Complexes as Catalysts for the Efficient Isomerization of Olefins. Selective Transformation of Terminal Alkenes to 2-Alkenes. J. Am. Chem. Soc. 2010, 132, 7998–8009. 10.1021/ja9108424. [DOI] [PubMed] [Google Scholar]; b Crossley S. W. M.; Barabé F.; Shenvi R. A. Simple, Chemoselective, Catalytic Olefin Isomerization. J. Am. Chem. Soc. 2014, 136, 16788–16791. 10.1021/ja5105602. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hassam M.; Taher A.; Arnott G. E.; Green I. R.; van Otterlo W. A. L. Isomerization of Allyl Benzenes. Chem. Rev. 2015, 115, 5462–5569. 10.1021/acs.chemrev.5b00052. [DOI] [PubMed] [Google Scholar]; d Kapat A.; Sperger T.; Guven S.; Schoenebeck F. E-Olefins through intramolecular radical relocation. Science 2019, 363, 391–396. 10.1126/science.aav1610. [DOI] [PubMed] [Google Scholar]; e Molleti N.; Martinez-Erro S.; Carretero Cerdán A.; Sanz-Marco A.; Gomez-Bengoa E.; Martín-Matute B. Base-Catalyzed [1, n]-Proton Shifts in Conjugated Polyenyl Alcohols and Ethers. ACS Catal. 2019, 9, 9134–9139. 10.1021/acscatal.9b02478. [DOI] [Google Scholar]
- a van der Drift R. C.; Bouwman E.; Drent E. Homogeneously catalyzed isomerization of allylic alcohols to carbonyl compounds. J. Organomet. Chem. 2002, 650, 1–24. 10.1016/S0022-328X(02)01150-6. [DOI] [Google Scholar]; b Trost B. M. The atom economy-a search for synthetic efficiency. Science 1991, 254, 1471–1477. 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- For reviews of transition-metal-catalyzed isomerization of allylic alcohols, see:; a Uma R.; Crevisy C.; Gree R. R. Transposition of Allylic Alcohols into Carbonyl Compounds Mediated by Transition Metal Complexes. Chem. Rev. 2003, 103, 27–52. 10.1021/cr0103165. [DOI] [PubMed] [Google Scholar]; b Cadierno V.; Crochet P.; Gimeno J. Ruthenium-Catalyzed Isomerizations of Allylic and Propargylic Alcohols in Aqueous and Organic Media: Applications in Synthesis. Synlett 2008, 8, 1105–1124. 10.1055/s-2008-1072593. [DOI] [Google Scholar]; c Lorenzo-Luis P.; Romerosa A.; Serrano-Ruiz M. Catalytic Isomerization of Allylic Alcohols in Water. ACS Catal. 2012, 2, 1079–1086. 10.1021/cs300092d. [DOI] [PubMed] [Google Scholar]
- For examples of the base-catalyzed isomerization of allyic alcohols, see:; a Johnston A. J. S.; McLaughlin M. G.; Reid J. P.; Cook M. J. NaH mediated isomerisation-allylation reaction of 1,3-substituted propenols. Org. Biomol. Chem. 2013, 11, 7662–7666. 10.1039/c3ob41857j. [DOI] [PubMed] [Google Scholar]; b Zheng H.-X.; Xiao Z.-F.; Yao C.-Z.; Li Q.-Q.; Ning X.-S.; Kang Y.-B.; Tang Y. Transition-Metal-Free Self-Hydrogen-Transferring Allylic Isomerization. Org. Lett. 2015, 17, 6102–6105. 10.1021/acs.orglett.5b03124. [DOI] [PubMed] [Google Scholar]; c Mondal K.; Mondal B.; Pan S. C. Organocatalytic Redox Isomerization of Electron-Deficient Allylic Alcohols: Synthesis of 1,4-Ketoaldehydes. J. Org. Chem. 2016, 81, 4835–4848. 10.1021/acs.joc.6b00243. [DOI] [PubMed] [Google Scholar]
- Selected examples:; a Ito M.; Kitahara S.; Ikariya T. Cp*Ru(PN) Complex-Catalyzed Isomerization of Allylic Alcohols and Its Application to the Asymmetric Synthesis of Muscone. J. Am. Chem. Soc. 2005, 127, 6172–6173. 10.1021/ja050770g. [DOI] [PubMed] [Google Scholar]; b Cadierno V.; García-Garrido S. E.; Gimeno J. A.; Varela-Álvarez A.; Sordo J. A. Bis(allyl)-Ruthenium(IV) Complexes as Highly Efficient Catalysts for the Redox Isomerization of Allylic Alcohols into Carbonyl Compounds in Organic and Aqueous Media: Scope, Limitations, and Theroretical Analysis of the Mechanism. J. Am. Chem. Soc. 2006, 128, 1360–1370. 10.1021/ja054827a. [DOI] [PubMed] [Google Scholar]; c Larionov E.; Lin L.; Guénée L.; Mazet C. Scope and Mechanism in Palladium-Catalyzed Isomerizations of Highly Substituted Allylic, Homoallylic, and Alkenyl Alcohols. J. Am. Chem. Soc. 2014, 136, 16882–16894. 10.1021/ja508736u. [DOI] [PubMed] [Google Scholar]; d Kress S.; Johnson T.; Weisshar F.; Lautens M. Synthetic and Mechanistic Studies on the Rhodium-Catalyzed Redox Isomerization of Cyclohexadienols. ACS Catal. 2016, 6, 747–750. 10.1021/acscatal.5b02387. [DOI] [Google Scholar]; e Erbing E.; Vázquez-Romero A.; Bermejo Gómez A.; Platero-Prats A. E.; Carson F.; Zou X.; Tolstoy P.; Martín-Matute B. General, Simple, and Chemoselective Catalysts for the Isomerization of Allylic Alcohols: The Importance of the Halide Ligand. Chem. - Eur. J. 2016, 22, 15659–15663. 10.1002/chem.201603825. [DOI] [PubMed] [Google Scholar]; f Spiegelberg B.; Dell’Acqua A.; Xia T.; Spannenberg A.; Tin S.; Hinze S.; de Vries J. G. Additive-Free Isomerization of Allylic Alcohols to Ketones with a Cobalt PNP Pincer Catalyst. Chem. - Eur. J. 2019, 25, 7820–7825. 10.1002/chem.201901148. [DOI] [PubMed] [Google Scholar]
- a Ahlsten N.; Bermejo Gómez A.; Martín-Matute B. Iridium-Catalyzed 1,3-Hydrogen Shift/Chlorination of Allylic Alcohols. Angew. Chem., Int. Ed. 2013, 52, 6273–6276. 10.1002/anie.201301013. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Martinez-Erro S.; Bermejo Gómez A.; Vázquez-Romero A.; Erbing E.; Martín-Matute B. 2,2-Diiododimedone: a mild electrophilic iodinating agent for the selective synthesis of α-iodoketones from allylic alcohols. Chem. Commun. 2017, 53, 9842–9845. 10.1039/C7CC04823H. [DOI] [PubMed] [Google Scholar]; c Sanz-Marco A.; Martinez-Erro S.; Martín-Matute B. Selective Synthesis of Unsymmetrical Aliphatic Acyloins through Oxidation of Iridium Enolates. Chem. - Eur. J. 2018, 24, 11564–11567. 10.1002/chem.201803117. [DOI] [PubMed] [Google Scholar]
- Sanz-Marco A.; Martinez-Erro S.; Pauze M.; Gómez-Bengoa E.; Martín-Matute B. An umpolung strategy to react catalytic enols with nucleophiles. Nat. Commun. 2019, 10, 5244–5253. 10.1038/s41467-019-13175-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Clark W. M.; Tickner-Eldridge A. M.; Huang G. K.; Pridgen L. N.; Olsen M. A.; Mills R. J.; Lantos I.; Baine N. H. A Catalytic Enantioselective Synthesis of the Endothelin Receptor Antagonists SB-209670 and SB-217242. A Base-Catalyzed Stereospecific Formal 1,3-Hydrogen Transfer of a Chiral 3-Arylindenol. J. Am. Chem. Soc. 1998, 120, 4550–4551. 10.1021/ja973882j. [DOI] [Google Scholar]; b Hedberg C.; Andersson P. G. Catalytic Asymmetric Total Synthesis of the Muscarinic Receptor Antagonist (R)-Tolterodine. Adv. Synth. Catal. 2005, 347, 662–666. 10.1002/adsc.200404234. [DOI] [Google Scholar]; c Dabrowski J. A.; Haeffner F.; Hoveyda A. H. Combining NHC-Cu and Brønsted Catalysis: Enantioselective Allylic Substitution/Conjugate Additions with Alkynylaluminium Reagents and Stereospecific Isomerization of the Products to Trisubstituted Allenes. Angew. Chem., Int. Ed. 2013, 52, 7694–7699. 10.1002/anie.201303501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bizet V.; Pannecoucke X.; Renaud J.-L.; Cahard D. Ruthenium-Catalyzed Redox Isomerization of Trifluoromethylated Allylic Alcohols: Mechanistic Evidence for an Enantiospeficic Pathway. Angew. Chem., Int. Ed. 2012, 51, 6467–6470. 10.1002/anie.201200827. [DOI] [PubMed] [Google Scholar]; b Bizet V.; Pannecoucke X.; Renaud J.-L.; Cahard D. J. Synthesis of β-CF3 Ketones from trifluoromethylated allylic alcohols by ruthenium catalyzed isomerization. J. Fluorine Chem. 2013, 152, 56–61. 10.1016/j.jfluchem.2013.01.004. [DOI] [Google Scholar]
- Martinez-Erro S.; Sanz-Marco A.; Bermejo Gómez A.; Vázquez-Romero A.; Ahlquist M. S. G.; Martín-Matute B. Base-Catalyzed Stereospecific Isomerization of Electron-Deficient Allylic Alcohols and Ethers through Ion-Pairing. J. Am. Chem. Soc. 2016, 138, 13408–13414. 10.1021/jacs.6b08350. [DOI] [PubMed] [Google Scholar]
- Ascough D. M. H.; Duarte F.; Paton R. S. Stereospecific 1,3-H Transfer of Indenols Proceeds via Persistent Ion-Pairs Anchored by NH···π Interactions. J. Am. Chem. Soc. 2018, 140, 16740–16748. 10.1021/jacs.8b09874. [DOI] [PubMed] [Google Scholar]
- a Shiers J. J.; Shipman M.; Hayes J. F.; Slawin A. M. Z. Rare Example of Nucleophilic Substitution at Vinylic Carbon with Inversion: Mechanism of Methyleneaziridine Formation by Sodium Amide Induced Ring Closure Revisited. J. Am. Chem. Soc. 2004, 126, 6868–6869. 10.1021/ja0482684. [DOI] [PubMed] [Google Scholar]; b Corsico E. F.; Rossi R. A. Sequential Reactions of Trimethylstannyl Anions with Vinyl Chlorides and Dichlorides by the SRN1 Mechanism Followed by Palladium-Catalyzed Cross-Coupliing Processes. J. Org. Chem. 2004, 69, 6427–6432. 10.1021/jo049287z. [DOI] [PubMed] [Google Scholar]; c Venkat Reddy C. R.; Urgaonkar S.; Verkade J. G. A Highyl Effective Catalyst System for the Pd-Catalyzed Amination of Vinyl Bromides and Chlorides. Org. Lett. 2005, 7, 4427–4430. 10.1021/ol051612x. [DOI] [PubMed] [Google Scholar]; d Ackermann L.; Gschrei C. J.; Althammer A.; Riederer M. Cross-coupling reactions of aryl and vinyl chlorides catalyzed by a palladium complex derived from an air-stable H-phosphonate. Chem. Commun. 2006, 13, 1419–1421. 10.1039/b518283b. [DOI] [PubMed] [Google Scholar]; e Poulsen T. B.; Bernardi L.; Bell M.; Jørgensen K. A. Organocatalytic enantioselective nucleophilic vinylic substitution. Angew. Chem., Int. Ed. 2006, 45, 6551–6554. 10.1002/anie.200602275. [DOI] [PubMed] [Google Scholar]; f Bernasconi C. F.; Rappoport Z. Recent Advances in Our Mechanistic Understanding for SNV Reactions. Acc. Chem. Res. 2009, 42, 993–1003. and references therein 10.1021/ar900048q. [DOI] [PubMed] [Google Scholar]
- a Oldendorf J.; Haufe G. Synthesis of Synthesis of γ-Fluoro-α,β-unsaturated Carboxylic Esters from Saturated α-Fluoro Aldehydes. J. Prakt. Chem. 2000, 342, 52–57. . [DOI] [Google Scholar]; b Guo R.; Huang J.; Zhao X. Organoselenium-Catalyzed Oxidative Allylic Fluorination with Electrophilic N–F Reagent. ACS Catal. 2018, 8, 926–930. 10.1021/acscatal.7b03829. [DOI] [Google Scholar]
- a Zhang Q.; Mixdorf J. C.; Reynders G. J. III; Nguyen H. M. Rhodium-catalyzed benzylic fluorination of trichloroacetimidates. Tetrahedron 2015, 71, 5932–5938. 10.1016/j.tet.2015.04.066. [DOI] [Google Scholar]; b Mixdorf J. C.; Sorlin A. M.; Zhang Q.; Nguyen H. M. Asymmetric Synthesis of Allyl Fluorides via Fluorination of Racemic Allylic Trichloroacetimidates Catalyzed by a Chiral Diene-Iridium Complex. ACS Catal. 2018, 8, 790–801. 10.1021/acscatal.7b03786. [DOI] [Google Scholar]
- a Appel R. Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P–N Linkage. Angew. Chem., Int. Ed. Engl. 1975, 14, 801–811. 10.1002/anie.197508011. [DOI] [Google Scholar]; b Huy P. H.; Hauch T.; Filbrich I. Lewis Base Catalyzed Nucleophilic Substitution of Alcohols. Synlett 2016, 27, 2631–2636. 10.1055/s-0036-1588633. [DOI] [Google Scholar]; c Larock R. C.; Zhang L.. Halogenation of Alcohols. In Comprehensive Organic Transformations; Larock R. C., Ed.; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- a Young W. G.; Caserio F. F.; Brandon D. D. Allylic Rearrangements. XLIX. The Controlled Conversion of α- and γ-Methylallyl Alcohols to Chlorides with Thionyl Chloride. J. Am. Chem. Soc. 1960, 82, 6163–6168. 10.1021/ja01508a047. [DOI] [Google Scholar]; b Snyder E. I. Conversion of allylic alcohols to chlorides without rearrangement. J. Org. Chem. 1972, 37, 1466–1466. 10.1021/jo00974a048. [DOI] [Google Scholar]; c Corey E.J.; Kim C.U.; Takeda M. New and highly effective method for the oxidation of primary and secondary alcohols to carbonyl compounds. Tetrahedron Lett. 1972, 4339–4344. 10.1016/S0040-4039(01)94310-2. [DOI] [Google Scholar]
- a Hanessian S.; Ponpipom M. M.; Lavallee P. Procedures for the direct replacement of primary hydroxyl groups in carbohydrates by halogen. Carbohydr. Res. 1972, 24, 45–56. 10.1016/S0008-6215(00)82258-2. [DOI] [Google Scholar]; b Bose A. K.; Lal B. A facile replacement of hydroxyl by halogen with inversion. Tetrahedron Lett. 1973, 3937–3940. 10.1016/S0040-4039(01)87077-5. [DOI] [Google Scholar]; c Jaseer E. A.; Naidu A. B.; Kumar S. S.; Rao R. K.; Thakur K. G.; Sekar G. Highly stereoselective chlorination of β-substituted cyclic alcohols using PPh3-NCS: factors that control the stereoselectivty. Chem. Commun. 2007, 867–869. 10.1039/B614512D. [DOI] [PubMed] [Google Scholar]
- Pacheco M. C.; Purser S.; Gouverneur V. The Chemistry of Propargylic and Allylic Fluorides. Chem. Rev. 2008, 108, 1943–1981. 10.1021/cr068410e. [DOI] [PubMed] [Google Scholar]
- Synthesis of chiral allylic substrates:Lumbroso A.; Cooke M. L.; Breit B. Catalytic Asymmetric Synthesis of Allylic Alcohols and Derivatives and their Applications in Organic Synthesis. Angew. Chem., Int. Ed. 2013, 52, 1890–1932. 10.1002/anie.201204579. [DOI] [PubMed] [Google Scholar]
- a Shandala M. Y.; Waight E. S.; Weinstock M. J. The rearrangement of 1-p-fluorophenylallyl, 1-m-tolylallyl, and optically active 1-phenylallyl chlorides in N,N-dimethylformamide. J. Chem. Soc. B 1966, 590–592. 10.1039/j29660000590. [DOI] [Google Scholar]; b Zhang Q.; Mixdorf J. C.; Reynders G. J. III; Nguyen H. M. Rhodium-Catalyzed Benzylic Fluorination of Trichloroacetimidates. Tetrahedron 2015, 71, 5932–5938. 10.1016/j.tet.2015.04.066. [DOI] [Google Scholar]; c Mixdorf J. C.; Sorlin A. M.; Zhang Q.; Nguyen H. M. Asymmetric Synthesis of Allylic Fluorides via Fluorination of Racemic Allylic Trichloroacetimidates Catalyzed by a Chiral Diene-Iridium Complex. ACS Catal. 2018, 8, 790–801. 10.1021/acscatal.7b03786. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.