

Abstract
Organofluorosilicon based 18F-radiolabeling is an efficient method for incorporating fluorine-18 into 18F-radiopharmaceuticals for positron emission tomography (PET) by 19F/18F isotopic exchange (IE). The first PET radiopharmaceutical, 18F-SiFAlin-TATE, radiolabeled with a silicon-based [18F]fluoride acceptor (SiFA), namely, a para-substituted di-tert-butyl[18F]fluorosilylbenzene, has entered clinical trials, and is paving the way for other potential [18F]SiFA-labeled radiopharmaceuticals for diagnostic use. In this study, we report the in vitro metabolism of an oxime-linked SiFA tetrazine (SiFA–Tz), a new PET-radiotracer candidate, recently evaluated for pretargeted PET imaging and macromolecule labeling. Metabolism of SiFA–Tz was studied in mouse liver microsomes (MLM) for elucidating its major biotransformation pathways. Nontargeted screening by ultrahigh performance liquid chromatography high-resolution mass spectrometry (UHPLC-HRMS) was utilized for detection of unknown metabolites. The oxime bond between the SiFA and Tz groups forms two geometric (E/Z) isomers, which underwent the same biotransformations, but unexpectedly with different kinetics. In total, nine proposed metabolites of SiFA–Tz from phase I and II reactions were detected, five of which were defluorinated in MLMs, elucidating the metabolic pathway leading to previously reported defluorination of [18F]SiFA–Tz in vivo. Based on the HRMS studies a biotransformation pathway is proposed: hydroxylation (+O) to tert-butyl group adjacent to the silicon, followed by oxidative defluorination (+OH/-F) cleaving the fluorine off the silicon. Interestingly, eight proposed metabolites of a reduced dihydrotetrazine analogue, SiFA–H2Tz, from phase I and II reactions were additionally detected. To the best of our knowledge, this is the first reported comprehensive investigation of enzyme mediated metabolic pathway of tetrazines and para-substituted di-tert-butylfluorosilylbenzene fluoride acceptors, providing novel structural information on the biotransformation and fragmentation patterns of radiotracers bearing these structural motifs. By investigating the metabolism preceding defluorination, structurally optimized new SiFA compounds can be designed for expanding the portfolio of efficient 19F/18F isotopic exchange labeling probes for PET imaging.
Keywords: defluorination, metabolism, positron emission tomography, silicon fluoride acceptor, tetrazine, geometric isomers
Introduction
Positron emission tomography (PET) is a nuclear imaging modality that utilizes pharmaceuticals radiolabeled with short-lived positron emitting radioisotopes administered to the subject for noninvasive diagnostics of physiological or pathological processes.1 As radiopharmaceuticals undergo metabolism in vivo, an important step in the development process of new PET radiopharmaceuticals is analysis of their metabolic stability in vitro and in vivo. The characteristics of radiometabolites, such as hydrophilicity of the formed metabolites and the percentage of intact radiolabeled parent compound remaining in tissues, can be investigated with various radiodetection methods, such as radio-high performance liquid chromatography (radio-HPLC), radio-thin layer chromatography (radio-TLC), and gamma counting.2,3 When coupled to mass spectrometry (MS) detectors, further analysis regarding potential biotransformation routes and proposed structures of radioactive and nonradioactive metabolites can be done.4 Radiotracers that demonstrate good in vivo stability and pharmacokinetics are generally utilized without detailed metabolite characterization by MS methods. When unexpected decomposition of the radiotracer occurs, a more detailed high-resolution mass spectrometry (HRMS) based investigation is often applied to understand the structural properties leading to radiolabel instability.4,5
Small molecule prosthetic groups, such as organophosphine fluoride acceptors6−8 or silicon-based fluoride acceptors9 (SiFA), are utilized for rapid, selective, and highly efficient radiofluorination of biomolecules via isotopic exchange. SiFAs are a new class of compounds for radiopharmaceutical chemistry where they are used for synthesizing new diagnostic tracer candidates for PET.9 SiFA chemistry is a radiofluorination strategy for fast and efficient radiolabeling of higher molecular weight constructs such as peptides, proteins, or nanoparticles that accumulate to tumors by enhanced permeability and retention (EPR) effect10,11 or by targeted strategies.12−14 Recently, a compound radiolabeled with SiFA chemistry, a somatostatin receptor-2 targeting peptide 18F-SiFAlin-TATE, has entered clinical trials.15−17 Main advantage of organophosphine fluoride acceptors or SiFA compounds lie in their tendency to undergo fast isotopic exchange (IE) of fluorine-19 to fluorine-18 (19F/18F), thus generating the 18F-PET-radiotracer in minutes at ambient temperature. Bulky substituents, such as two tert-butyl groups, have been incorporated into the structure to shield the fluorine from hydrolysis in vitro and in vivo leading to discovery of di-tert-butyl[18F]fluorosilylbenzyl moiety, the most widely used SiFA analogue for PET radiochemistry.18 Most SiFA analogues have demonstrated desirable in vitro and in vivo stability, with a few examples of in vivo defluorination reported to date.17,19,20 The vast majority of reported biodistribution studies are of peptide or macromolecular structures labeled with SiFA21 and only cautious extrapolation can be drawn from these structures to make conclusions about the stability of SiFA compounds of lower molecular weight.20 We recently reported biological evaluation of an oxime-linked SiFA-tetrazine (SiFA–Tz), [18F]-(E/Z)-SiFA–Tz (Figure 1) in healthy mice, which revealed rapid accumulation of radioactivity in both occipital bone and in tibia.22
Figure 1.

Chemical structure of SiFA–Tz (E/Z) and proposed structure of its reduced dihydrotetrazine form SiFA–H2Tz (E/Z).
In the light of the rapid defluorination in vivo, we concluded that the PET tracer candidate underwent metabolic transformation leading to the detachment of the radiolabel and subsequently high accumulation of radioactivity in the bone (Figure S1). Further investigation of the unforeseen defluorination and further elucidation of the biotransformation pathways of this new PET tracer candidate were consequently warranted. Defluorination is primarily a CYP2E1 mediated phase I transformation,23−26 although non-cytochrome P450 (CYP) mediated hydrolysis in solution has been reported for SiFAs with structural soft spots.17 For PET radiopharmaceuticals, metabolic or hydrolytic dissociation of [18F]fluorine from a compound is an unfavorable feature and is known to lead to rapid uptake of [18F]fluoride in the bone hydroxyapatite.27 This can be detrimental to the PET image quality and quantification. The high bone radioactivity lowers the region of interest-to-background contrast, and may cause spill-in of radioactivity into the region of interest, preventing accurate radioactivity quantification.28 Hence, designing the position and chemical environment of the fluorine atom in a radiotracer is crucial for ensuring sufficient in vivo stability.
Tetrazines are widely utilized as reactants in the chemoselective bioorthogonal inverse electron demand Diels–Alder (IEDDA) cycloaddition reaction, because of their remarkable reactivity with strained alkenes. Tetrazines are six-membered heterocyclic compounds, found as three structural isomers, containing a ring of four nitrogens and two carbons (Figure 2).29,30 The high reactivity of 1,2,4,5-tetrazine (s-tetrazine) is attributed to the extreme electron deficiency of the nitrogen-rich ring. The fast reaction kinetics of IEDDA between 1,2,4,5-tetrazines and trans-cyclooctenes makes it the most efficient bioorthogonal reaction to date.31,32 The discovery of IEDDA cycloaddition as a highly efficient method for pretargeted delivery of imaging and therapeutic radioisotopes to tumor sites in vivo has further accelerated its utility in bioconjugation.33 Conjugated s-tetrazines have revealed excellent stability in phosphate buffered saline (PBS) at ambient temperature and moderate stability in fetal bovine serum (FBS), with 15% decomposition after 15 h at 20 °C.34,35 Half-lives of up to 9 h in PBS and 100% of the tetrazine remaining intact in FBS up to 4 h have been reported.36 Unconjugated s-tetrazine, phenyl s-tetrazine, was shown to be 40% intact after 10 h incubation in FBS at 37 °C.37 Because of the high reactivity of s-tetrazines, they harbor the inherent ability to be reversibly reduced by accepting an electron, forming a stable anion in the absence of acids,38 and can accept a second electron forming 1,4-dihydrotetrazines, in both organic solvents and aqueous media.38−40 Reduction, if proceeding to completion, renders tetrazine inert to the IEDDA cycloaddition. However, the published literature to date indicates that detailed investigation of the metabolic profile of tetrazines is relatively limited.
Figure 2.

(A) Structural isomers of tetrazine and (B) reduction of 1,2,4,5-tetrazine (s-tetrazine) in aqueous and organic solutions.29,30,39,40
The oxime bond in SiFA–Tz adds an additional feature influencing its metabolism. Oximes are widely utilized in bioconjugation as well as in the synthesis of drugs and pesticides.41,42 The oxime bond formation is an acid catalyzed reaction between a hydroxylamine and an aldehyde or ketone. The oxime bond can form E- and Z-isomers, and the stereochemistry influences the geometry and pharmacology of the compounds. The major metabolic route reported for oximes is hydrolysis or N–O bond cleavage of the alkoxyamine.41 The metabolism can be specific to the compound, and recently a study reporting an antibody–drug conjugate studied in rats revealed that the oxime bond was left intact while a small molecule metabolite had cleaved at the terminal amide bond instead.43 Based on previous reports on the stability and metabolism of oximes, the oxime bond was presumed as a possible metabolic soft spot for SiFA–Tz.
To date, an extensive MS-based investigation of the in vivo stability and metabolic profile of low-molecular-weight compounds containing SiFA and tetrazine motifs is still lacking. Here, we report the investigation of the metabolic profile of SiFA–Tz and its proposed reduced analogue dihydrotetrazine SiFA–H2Tz (Figure 1) studied in mouse liver microsomes (MLMs) by using HRMS-based methods for elucidating the structural soft spots and defluorination detected previously in vivo. Additionally, a proposed metabolic pathway and elucidation of the major metabolites of this radiotracer are presented, in order to understand the structural features affecting the biological stability of SiFA and tetrazine containing compounds for the design of improved next-generation PET tracers of these motifs. To the best of our knowledge, detailed MS-based data on the biotransformation products of SiFA derivatives have not been previously presented.
Experimental Section
Chemicals and Reagents
All reagents and solvent were purchased from commercial providers and used as received without further purification. Ultrapure water (18.0 MΩ) was produced with a Milli-Q Integral Water Purification System (Merck Millipore, Burlington, MA, USA). Anhydrous acetonitrile (DNA synthesis quality, max. 10 ppm water) was purchased from Merck (Kenilworth, NJ, USA). CD-1 MLMs were purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). β-Nicotinamide adenine dinucleotide 2′-phosphate (NADPH) reduced tetrasodium salt hydrate, uridine 5′-diphosphoglucuronic acid (UDPGA) trisodium salt, and alamethicin were purchased from Sigma-Aldrich (St. Louis, Missouri, USA). Acetonitrile (ACN) and formic acid (FA) (both LC-MS grade) were purchased from Merck (LiChropur, Merck KGaA, Darmstadt, Germany). SiFA–Tz was synthesized in-house (purity >97%)22 and dissolved in ACN (DNA synthesis quality) for HRMS studies (= analytical standard).
Mouse Liver Sample Pretreatment
Livers of CD-1 mice (female, weight 25–33 g, 11 to 12 weeks, Charles River, n = 2) from a previously reported study were harvested after intravenous administration of [18F]SiFA–Tz into the lateral tail vein.22 The harvested organs were subjected to gamma counting (t = 1 min per organ), followed promptly by storage in freezer (−20 °C) until further processing. For the metabolite analysis, the harvested organs were taken to ambient temperature from a freezer (−20 °C), homogenized with a hand-held VDI 12 homogenizer (VWR, West Chester, PA, USA) in cold ACN–water solution (80:20), and centrifuged (10 000 g, 10 min), and the supernatants were filtered with 0.22 μm sterile syringe filter (Millex-GV, Merck Millipore Ltd., Tullagreen Carrigtwohill, Co. Cork Ireland). The animal experiment referenced in this study was conducted under a project license approved by the National Board of Animal Experimentation in Finland (license number ESAVI/12132/04.10.07/2017).22
In Vitro Metabolism of SiFA–Tz in CD-1 Mouse Liver Microsomes
Microsomal solutions for generating the in vitro metabolites were prepared following the manufacturer’s instructions and good practices in drug metabolism.44 The conditions used are presented in Supporting Information (Table S1). Briefly, for phase I in vitro samples 2 μL of SiFA–Tz (58 nmol) in ACN was diluted in 100 mM potassium phosphate buffer (183 μL, pH 7.4) and the solution was added into a suspension of MLMs (5 μL, 20 mg/mL solution). The suspension was incubated for 5 min at 37 °C, and the reaction was started by adding 20 mM NADPH (10 μL). For phase II in vitro samples, 2 μL of SiFA–Tz (58 nmol) in ACN was diluted in 100 mM potassium phosphate buffer containing 1 mM of MgCl2 (173 μL, pH 7.4), and the solution was added into a suspension of MLMs (5 μL, 20 mg/mL solution), followed by 10 μL of 20 mM UDPGA in phosphate buffer and 1 μL of alamethicin (5 mg/mL) in DMSO. The suspension was placed for 15 min in an ice bath, and the reaction was started by adding 10 or 15 μL of 20 mM NADPH (10 μL for t = 5 and 60 min, 15 μL for time points t = 120 and 240 min). The samples were incubated at 37 °C for the duration of selected time points (t = 5, 60, 120, and 240 min). The negative controls were prepared as follows: (a) incubation with all cofactors for 0 min (control 1), (b) without SiFA–Tz and UDPGA for 0 min (control 2), (c) without NADPH for 240 min (control 3), and (d) without microsomes for 240 min (control 4). The reactions were terminated with the addition of 200 μL of ice-cold ACN, the samples were vortexed and centrifuged (3000 rpm for 5 min), and thereafter the supernatants were separated from the pellets and stored at −80 °C prior to analysis. All samples were done in duplicate. The supernatants were defrosted and once at ambient temperature filtered (0.22 μm PVDF sterile filter) immediately before the UHPLC-HRMS analysis.
UHPLC-HRMS Method
The HRMS experiments were conducted with UHPLC Thermo Scientific Dionex Ultimate 3000 ultrahigh performance liquid chromatography (Germering, Germany) coupled to Thermo Scientific Orbitrap Fusion mass spectrometer (San Jose, USA). UHPLC separation was executed with Waters ACQUITY UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm) at 40 °C using gradient of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The gradient was run from 10% B at 0 min to 100% B at 5 min. After this, eluent B was kept at 100% for 1 min and at 5% for 2 min. The flow rate was 0.5 mL/min, and the injection volume was 2.5–5.0 μL. The ionization was done using heated electrospray ionization (HESI) in the positive ion mode. The instrument parameters were set to spray voltage 3500 V, source temperature 300 °C, ion transfer tube temperature 350 °C, sheath gas 40, auxiliary gas 10, and sweep gas 0. HRMS measurement was done with scan range of 70–1000 m/z using RF lens at 60% and quadrupole isolation at resolution of 120,000. Mass accuracy of the instrument using internal calibration was specified to be ≤3 ppm. MS/HRMS technique was applied for the structure elucidation of formed metabolites. Higher-energy collisional dissociation (HCD) was used for fragmentation of protonated molecules. Collision energies for different detected compounds are presented later in the text.
Data Analysis and Statistics
The data was acquired and processed with Xcalibur workstation (Thermo Fisher Scientific, Waltham, MA, USA). Compound Discoverer 3.0 and 3.1 by Thermo Fisher Scientific (Waltham, MA, USA) were used for the data filtering of most predominant phase I and phase II o-glucuronide-metabolites. MassFrontier 7.0 and 8.0 (Thermo Fisher Scientific, Waltham, MA, USA) were used for data filtering and elucidating fragmentation patterns from the data in MS/HRMS studies. The graphs were plotted and statistical calculations executed with GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA). The graphs with statistics are presented as mean ± standard deviation (SD).
Results and Discussion
We have previously reported the biological evaluation of [18F]SiFA–Tz in healthy CD-1 mice revealing defluorination of the tracer candidate in vivo.22 The fluorine-18 radiolabel detached from [18F]SiFA–Tz accumulated significantly in the bone within 1 h after administration (Figure S1). Therefore, in this study we decided to investigate this unexpected finding to further elucidate the biotransformation pathways of this proposed PET tracer to guide structural optimization. A more detailed study on the metabolism of (E/Z)-SiFA–Tz was necessary, and an in vitro experiment using MLM was conducted. The samples were analyzed with nontargeted HRMS method and the metabolites in addition to the isomers of SiFA–Tz and SiFA–H2Tz were screened with Xcalibur, Compound Discoverer, and MassFrontier softwares. The high mass accuracy measurements and the high resolving power provided by HRMS instrument enables the detection and identification of unknown metabolites from complex biological matrices, such as organ extracts and in vitro metabolism samples.
Identification of Geometric Isomers of SiFA–Tz and SiFA–H2Tz in Analytical Standard
SiFA–Tz in analytical standard was detected as two distinctly separated, but identical protonated molecule peaks at 4.37 min at m/z 509.24900 (Δ = −0.20865 ppm) and 4.64 min at m/z 509.24905 (Δ = 0.01860 ppm), with elemental composition C26H34FN6O2Si+, corresponding to its two geometric isomers; (Z)-SiFA–Tz and (E)-SiFA–Tz, as expected.42 The Z-isomer eluted before E-isomer indicating less retention on the C18 stationary phase, because of different geometric structures influencing the interaction with the column material. The proposed E- and Z-isomers were well separated and the percentage of the predominant E-isomer in the analytical standard was determined from two separate aliquots as 92 ± 0.7% (n = 2). Further structure elucidation for the detected isomers was performed by MS/HRMS using HCD 30%. The major fragment ions were detected at m/z 264.15716 (SiFA moiety) and at m/z 246.09795 (Tz moiety) by cleavage at the oxime bond. The MS/HRMS spectrum for (E)-SiFA–Tz, the proposed structures for fragment ions, and mass differences compared to theoretical values are presented in Supporting Information (Figure S2). Fragments at m/z 351.18915, m/z 246.09795, and m/z 171.06609 were determined as characteristic fragment ions important for identification of SiFA–Tz. In addition, two peaks at m/z 511.26478 were detected at 4.24 and 4.37 min corresponding to the elemental composition of C26H36FN6O2Si+ indicating two isomers and spontaneous reduction (+2 H) of SiFA–Tz in the analytical standard. The reduction is presumed to take place at the tetrazine ring as described in the literature.39,40 The reduction can be reversed sluggishly, but spontaneously, back to the original oxidized tetrazine form.38 The proposed reduced dihydrotetrazine SiFA–H2Tz was detected as two distinctly separated isomers with a percentage of (E)-SiFA–H2Tz 89 ± 7% (n = 2). MS/HRMS was used for further structure elucidation of SiFA–H2Tz and the fragmentation pattern is presented in Supporting Information (Figure S3). Characteristic fragments corresponding to ones observed for SiFA–Tz were detected for SiFA–H2Tz, which further corroborated the presence of the proposed reduced analogue of SiFA–Tz.
Identification of SiFA–Tz and SiFA–H2Tz in Ex Vivo Mouse Liver Samples
In full scan mode, two pairs of peaks at m/z 509.24890 (Δ = −0.40089 ppm, E-isomer) and at m/z 509.24820 (Δ = 0.84718 ppm, Z-isomer) corresponding to (E/Z)-SiFA–Tz, and peaks at m/z 511.26434 (Δ = −0.80647 ppm, E-isomer) and at m/z 511.26486 (Δ = 0.20827 ppm, Z-isomer) corresponding to (E/Z)-SiFA–H2Tz were detected in mouse liver extracts. Extracted ion chromatograms (EIC) for SiFA–Tz and SiFA–H2Tz detected in analytical standard and in mouse liver extracts are presented in Figure 3. The qualitative identification of detected compounds in mouse liver extracts was based on the European Union guidelines45 for LC-MS analysis regarding the retention time (tR) of the identified compound corresponding to the reference standard sample with tolerance of ±2.5%. As seen in Figure 3, the relative abundances indicated that the Z-isomers were metabolized slower than the E-isomers. The change in the ratio of E/Z from 92:8 to 48:52 (±7%, n = 2) for SiFA–Tz revealed a relatively rapid metabolism of the E-isomer and a prolonged biological half-life of the Z-isomer in vivo. Based on the HRMS studies of extracted liver samples, it was evident the E- and Z-isomers expressed different biological half-lives.
Figure 3.

EICs for E- and Z-isomers of (A) SiFA–Tz in analytical standard, (B) SiFA–Tz in liver extract of a mouse administered intravenously [18F]SiFA-Tz, (C) SiFA–H2Tz in analytical standard, and (D) SiFA–H2Tz in the liver extract.
Identification of SiFA–Tz and SiFA–H2Tz In Vitro in MLMs
The proposed reduced analogue SiFA–H2Tz (Figure 1) was additionally detected in vitro in MLM metabolite samples of SiFA–Tz. The quantitative comparison between the actual amounts of SiFA–Tz and SiFA–H2Tz in the samples was not possible because of the qualitative nature of the study, the inherent unique ionization efficiency of each compound,46 and the lack of synthesized analytical standard for SiFA–H2Tz. Nevertheless, SiFA–Tz and SiFA–H2Tz presented comparable relative abundances and similar metabolism kinetics throughout the in vitro MLM studies (Figure 4), further corroborating the presence of two extremely similar compounds subject to metabolism.
Figure 4.

Non-normalized relative abundances of (E)-SiFA–Tz and (E)-SiFA–H2Tz in the in vitro mouse liver microsome samples (n = 2).
As expected, the ratio of SiFA–H2Tz was observed to increase dramatically in the presence of the reducing agent NADPH and liver microsomes, by comparison of the relative abundances at different time points of SiFA–Tz and SiFA–H2Tz in the analytical standard (= ratio before incubation) and in vitro MLM samples (Figure S4). The fingerprint fragments containing the SiFA moiety at m/z 454 and at m/z 264 for SiFA–H2Tz corresponded to the fragments at m/z 454 and at m/z 264 for compound SiFA–Tz (Figures S2 and S3). Additionally, a fragment at m/z 191 containing the benzylacetamide was found for both SiFA–Tz and SiFA–H2Tz, indicating that the two added hydrogens did not attach to the SiFA or the benzylacetamide moieties. Furthermore, the tetrazine fragments with (+2 H) at m/z 248 and at m/z 173 for compound SiFA–H2Tz, and the analogous fragments at m/z 246 and at m/z 171 found for SiFA–Tz, verified that the reduction took place at the tetrazine ring. The MS/HRMS-fragmentation patterns revealed the presence of similar compounds, SiFA–Tz and SiFA–H2Tz, with identical major fragmentation patterns.
In Vitro Metabolite Screening
SiFA–Tz was incubated in MLMs with cofactors NADPH and UDPGA for the investigation of whether it undergoes other transformations than the hydrolytic defluorination discussed in detail in previously published studies.17,47 The in vitro samples were analyzed in full scan mode using nontargeted screening method. In total, 17 previously unknown compounds were detected in the in vitro MLM samples. The data were filtered with Compound Discoverer for phase I and phase II transformation reactions, which subsequently provided the retention times and biotransformations of the proposed major metabolites. For further structure elucidation, MS/HRMS technique was used. MS/HRMS and in silico data revealed the fragments leading to elucidation of proposed metabolites and sites of metabolism. The hypothesis regarding the presence of the reduced tetrazine in the solubilized samples was taken into account during the metabolite screening studies. In the in vitro MLM samples, the proposed Z-isomer metabolites were detected to form slower, and the (Z)-metabolites were apparently more resistant to further metabolism. The relative abundance of (Z)-SiFA–Tz remained almost constant; meanwhile, the (E)-SiFA–Tz declined throughout the study (Figure S5).
Phase I Metabolite Identification
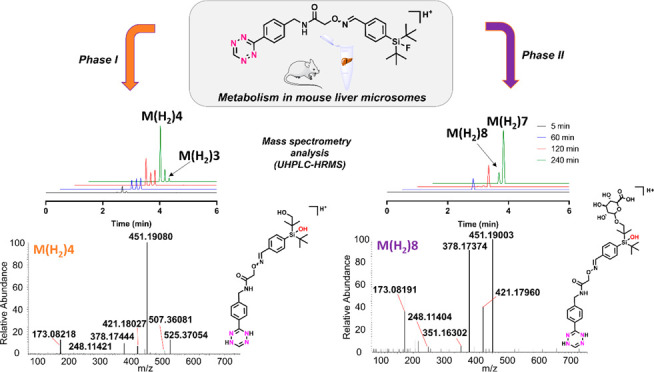
All proposed phase I metabolites, except for M3 (m/z 523) which was found as a single peak, were detected with another closely eluting peak with exactly the same mass and elemental composition. It was postulated that the pairs of peaks were isomers originating from the geometric E- and Z-isomers of SiFA–Tz. If the observed pairs of peaks were arising from, for example, hydroxylation to different positions, a more noticeable difference in the fragmentation patterns would be expected. The detected proposed major biotransformation products resulting from in vitro MLM samples were detected at earlier retention times on the reverse phase (C18) column, indicative of more hydrophilic compounds than SiFA–Tz. Proposed metabolite M1 at m/z 525.24402 (C26H34FN6O3Si+), at retention time of 3.68 min, and proposed metabolite M2 at m/z 525.24402 m/z (C26H34FN6O3Si+) at 3.64 min were detected, indicating SiFA–Tz underwent monohydroxylation. The proposed metabolites with detected masses and mass errors are presented in Supporting Information (Table S2). Additionally, three defluorinated proposed phase I metabolites (M3, M4, M5) of SiFA–Tz were detected. A monohydroxylated (+O) and oxidatively defluorinated (+OH/-F) proposed metabolite M3 (m/z 523.24836, C26H35N6O4Si+) eluted as a single peak at tR = 2.58 min. M4 (m/z 539.24237, C26H35N6O5Si+) at tR = 2.55 min and M5 (m/z 539.24240, C26H35N6O5Si+) at 2.27 min, presumed as isomers, were detected, indicating dihydroxylation and oxidative defluorination to SiFA–Tz. The same proposed metabolites were detected for the reduced analogue SiFA–H2Tz. The proposed major monohydroxylated metabolites of SiFA–H2Tz, eluted as two peaks presumed as isomers, M(H2)1 at m/z 527.25877 (C26H36FN6O3Si+, tR = 3.86 min) and M(H2)2 at m/z 527.25873 (C26H36FN6O3Si+, tR = 3.63 min) (Table S3). Two monohydroxylated and oxidatively defluorinated metabolites of SiFA–H2Tz were detected; M(H2)3 (m/z 525.26401, C26H37N6O4Si+, tR = 2.83 min) and M(H2)4 (m/z 525.26408, C26H37N6O4Si+, tR = 2.53 min). M(H2)4 continued to build up, while M(H2)3 was apparently subject to further metabolism (Figure S6). Proposed elemental compositions, measured masses, theoretical masses, mass differences and structures of detected proposed phase I metabolites are presented in Supporting Information (Tables S2, S3). None of the found proposed metabolites were detected in the negative control samples suggesting that all of the detected phase II metabolites were NADPH and UDPGA dependent.
Phase II Metabolite Identification
All proposed phase II metabolites were detected as pairs of peaks with same exact masses and elemental compositions presumed as isomers. Two proposed fluorine containing phase II metabolites of SiFA–Tz were detected, M6 at m/z 701.27497 (C32H42FN6O9Si+, tR = at 3.67 min) and M7 at m/z 701.27492 (C32H42FN6O9Si+, tR = 3.64 min). Based on measured masses, obtained elemental composition, and retention times, M6 and M7 were proposed to form after hydroxylation and addition of glucuronic acid to the OH after the NADPH-dependent oxidation (Table S4). Proposed metabolites M8 at m/z 699.28054 (C32H43N6O10Si+, tR = 2.96 min) and M9 at m/z 699.28039 (C32H43N6O10Si+, tR = 2.89 min) of SiFA–Tz were likely monohydroxylated and oxidatively defluorinated followed by o-glucuronidation. Similar analogous proposed metabolites of SiFA–H2Tz were detected; M(H2)7 at m/z 701.29471 (C32H45N6O10Si+, tR = 2.34 min) and M(H2)8 at m/z 701.29540 (C32H45N6O10Si+, tR = 2.20 min), which were likely monohydroxylated and oxidatively defluorinated followed by o-glucuronidation (Table S5). The proposed major phase II metabolites of SiFA–H2Tz, M(H2)5 and M(H2)6, formed likely after monohydroxylation and o-glucuronidation, and were detected at m/z 703.29099 (C32H44FN6O9Si+, tR = 2.73 min) and M(H2)6 at m/z 703.29058 (C32H44FN6O9Si+, tR = at 2.50 min), respectively. None of the found proposed metabolites were detected in the negative control samples indicating all of the detected phase II metabolites were NADPH and UDPGA dependent. The proposed hydroxylated metabolites, were rapidly o-glucuronidated (t = 5 min), and the hydroxylation was apparently the primary route followed by oxidative defluorination. The peak areas and the differences in the slopes of the formed metabolites (Figures S7, S8) indicated that the fluorine containing proposed hydroxylated biotransformation products formed early on and faster than the defluorinated analogues. This strongly indicated that the major metabolic route was most likely hydroxylation of SiFA–H2Tz followed by oxidative defluorination. A simultaneous and widely studied hydrolytic defluorination could not be totally ruled out as an additional mechanism, and could have participated to some extent to the cleavage of the fluoride. Minute defluorination (∼5%) of SiFA-Tz incubated in enzyme containing human plasma at 37 °C has been presented previously by our group.22
Structure Elucidation of SiFA–Tz Metabolites
For further structure elucidation of the detected proposed metabolites, MS/HRMS technique was applied using HCD for generating fragment ions of protonated molecules. Because SiFA–Tz has several possible oxidation sites, it was impossible to say with certainty to which exact carbon the biotransformation takes place. Previously reported findings on the metabolism of Finasteride48 and Docetaxel,49 both compounds containing a tert-butyl group and or aromatic ring, demonstrate that the tert-butyl undergoes oxidation at one sp3-carbon rather than an aromatic ring. The metabolism of 2,6-di-tert-butyl-p-cresol (BHT) demonstrated species-specific oxidation, with the aromatic ring bound methyl group being hydroxylated in rat, rabbit, and monkey, and tert-butyl groups in human.50 In all the aforementioned compounds, the sp3-carbon was oxidized in the presence of an aromatic ring, which was left intact. This supports the hypothesis that SiFA–Tz is likely oxidized from the tert-butyl groups adjacent to the silicon atom. The characteristic fragments at m/z 248 and m/z 173 detected for all four proposed metabolites M(H2)1, M(H2)3, M(H2)5, and M(H2)7 of SiFA–H2Tz (Table S9, Figures 5 and S10) indicated no biotransformations at the reduced tetrazine or the aromatic ring adjacent to it. Additionally, the cleavage of the glucuronic acid from M(H2)5 (m/z 703) to yield m/z 527 and o-glucuronic acid from M(H2)5 (m/z 703) to yield m/z 509 were detected as minor fragments (Figure S10). Fragments at m/z 451 for defluorinated M(H2)3 (m/z 525, phase I) and M(H2)7 (m/z 701.29, phase II), and the analogous fragments at m/z 453 for fluorine containing proposed metabolites M(H2)1 (m/z 527, phase I) and M(H2)5 (m/z 703, phase II) corroborated the hydroxylation site. The aforementioned fragments formed likely after simultaneous cleavage of one tert-butyl group, oxygen, and the glucuronic acid of proposed phase II metabolites. In this hypothesis, the tert-butyl group was hydroxylated, followed by o-glucuronidation, which was corroborated by the simultaneous cleavage of these two groups. The MS/HRMS experiments revealed fragmentation patterns with a slight difference in the relative abundances of the fragments, arising likely from the individual geometry of the isomers. The differences in the relative abundance, such as the 49% relative abundance at m/z 453 for Z-isomer compared with 100% for E-isomer, was attributed to the inaccessibility of the shielded tert-butyl group on the Z-isomer and the consequent lower probability of its cleavage. The E- and Z-isomers of proposed o-glucuronide-metabolites had the same predominant fragments with different relative abundances as indicated in Figure 5.
Figure 5.

Fragmentation patterns differentiating Z-isomer (A) M(H2)6 (tR = 2.50 min) from E-isomer (B) M(H2)5 (tR = 2.73 min) after proposed hydroxylation and o-glucuronidation using HCD 10%.
The tetrazine ring and its neighboring aromatic ring were intact and presented the characteristic fragment at m/z 246, indicating no metabolism to those functional groups. Corroborating this, a characteristic fragment at m/z 191 containing the benzylacetamide was found for M6, a proposed (E)-metabolite of SiFA–Tz (Figure S11). The spatial orientation of the isomers additionally supports the finding of the difference in metabolic speed at one of the tert-butyl groups, because the Z-isomer would be sterically highly hindered in compounds SiFA–Tz and SiFA–H2Tz, and their metabolites. Possible differences in metabolic rates of E/Z-isomers, due to the geometrical differences in their structures, is a well-known phenomenon and has been previously reported in the literature, for example, for the tricyclic antidepressant Doxepin.51 The increase in the relative abundance of proposed Z-isomer, probably due to intramolecular shielding from metabolism by steric hindrance, was present for all proposed formed phase I metabolites. It was hypothesized that in phase II metabolites, the buildup was slower for Z-isomer because of the impaired accessibility of the OH-group by the bulky glucuronic acid in the sterically hindered isomer. Nevertheless, the differences in geometry did not prevent the conjugation reaction from occurring and in total four proposed o-glucuronide conjugates were detected for both SiFA–Tz and SiFA–H2Tz. The influence of fluorine on the lipophilicity of a compound was demonstrated by the longer retention times for fluorine containing proposed metabolites of SiFA–H2Tz, compared to defluorinated proposed metabolites (Figure S12). Fluorine is known to increase the lipophilicity of a compound compared to its analogue with hydrogen or hydroxyl group.52
Proposed Metabolic Pathways
The proposed metabolic pathways for SiFA–Tz and its reduced analogue SiFA–H2Tz is shown in Scheme 1. It was hypothesized that first the compounds underwent hydroxylation to a tert-butyl group. Two propose metabolites (M4, M5) likely underwent a subsequent oxidation further to a carboxylic acid at the tert-butyl group. Additionally, oxidative defluorination took place, providing from these three steps in total nine proposed phase I metabolites of SiFA–Tz and SiFA–H2Tz. In the phase II metabolism, the proposed monohydroxylated biotransformation products were further o-glucuronidated forming in total eight proposed phase II metabolites. In phase I and phase II MLM samples, both fluorine-containing and defluorinated proposed metabolites were present. It was concluded that defluorination was likely a slower, mainly CYP enzyme mediated oxidative defluorination process in all proposed metabolites following an earlier hydroxylation biotransformation step to a tert-butyl carbon (Scheme 1).
Scheme 1. Proposed Major Metabolic Pathways of [19F/18F]SiFA–Tz and Its Reduced Form [19F/18F] SiFA–H2Tz In Vitro.

Conclusions
[18F]SiFA–Tz undergoes fast defluorination in vivo, in spite of displaying good stability in human plasma (∼95% intact) or PBS (>99% intact) in vitro. The defluorination for [19F/18F]SiFA–Tz was proven to be primarily a NADPH-dependent CYP enzyme mediated reaction originating from various biotransformation reactions, which were detected in both phase I and phase II in vitro samples. It was determined that one of the tert-butyl groups adjacent to the silicon atom was likely the major hydroxylation site, for both SiFA–Tz and its reduced analogue SiFA–H2Tz. SiFA–Tz was originally designed as a fast bioorthogonal synthon, and the tetrazine ring, in its oxidized form, plays a key role in the chemoselective reactions of the radiolabeled probe. In the IEDDA reaction, the electron-poor tetrazine ring reacts with strained alkenes efficiently in vitro and in vivo. After reduction of the tetrazine to dihydrotetrazine, this chemoselective IEDDA reaction is no longer favored. Even though the reduction of tetrazines is apparently kinetically relatively slow and might not be of major concern for the bioconjugation applications by IEDDA, the detection of the SiFA–H2Tz and its several metabolites during this investigation emphasizes the importance of this undermined phenomenon. Quantitative HRMS-based investigation is needed to estimate the actual amount of the reduced tetrazine SiFA–H2Tz in all of the samples compared to SiFA–Tz, because of the unique ionization efficiencies of the compounds. Nevertheless, the amount of reduced dihydrotetrazine was detected to increase 5 min after the start of incubation in the presence of NADPH and liver microsomes. It was determined that defluorination was primarily a secondary and slower biotransformation step preceded by hydroxylation proposed to take place at the tert-butyl carbon. Verifying the exact carbon undergoing hydroxylation would require further structural studies of synthesized metabolites aided by nuclear magnetic resonance spectroscopy, which fell beyond the scope of this work. Additionally, detection of the accurate masses of the fluorine-18 containing metabolites simultaneously with corresponding radiosignals, or collecting the radiolabeled metabolites and subsequently analyzing them with HRMS, could be conducted in future experiments to further corroborate the identity of the detected and proposed fluorine containing metabolites. In vitro liver microsome studies before in vivo studies in animal models can be utilized as a cost-effective and rapid approach to screen radiotracers for metabolic stability and defluorination, as well as to elucidate species differences in metabolism to optimize the choice of the model organism. Our earlier ex vivo findings indicated that the SiFA–Tz is susceptible to detrimental CYP-enzyme mediated biotransformation early on after its administration, and we were able to corroborate this finding in vitro in MLMs. Based on the proposed major metabolic site in SiFA–Tz and the biotransformation pathways preceding defluorination, structural optimizations can be carried out. These findings may be very useful for future development of more stable SiFA and tetrazine radiotracers, and in elucidating the reduction phenomenon capable of influencing the reactivity of tetrazines aimed at in vivo bioconjugation.
Acknowledgments
The authors would like to thank the Academy of Finland for the funding of this project (306239, 298481, and 278056). M.Sc. Surachet Imlimthan is acknowledged for his help with the animal studies referenced to in this study.
Glossary
Abbreviations
- ACN
acetonitrile
- CYP
cytochrome P450
- EIC
extracted ion chromatogram
- EPR
enhanced permeability and retention
- FA
formic acid
- FBS
fetal bovine serum
- HCD
higher-energy collisional dissociation
- HESI
heated electrospray ionization
- HPLC
radio-high performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- IE
isotopic exchange
- IEDDA
inverse-electron demand Diels–Alder
- LC-MS
liquid chromatography mass spectrometry
- MLM
mouse liver microsome
- MS
mass spectrometry
- NADPH
β-nicotinamide adenine dinucleotide 2′-phosphate
- PET
positron emission tomography
- SiFA
silicon fluoride-acceptor
- SiFA–H2Tz
silicon fluoride-acceptor dihydrotetrazine
- SiFA–Tz
silicon fluoride-acceptor tetrazine
- SD
standard deviation
- TLC
thin layer chromatography
- UDPGA
uridine 5′-diphosphoglucuronic acid
- UHPLC-HRMS
ultrahigh performance liquid chromatography high-resolution mass spectrometry
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.0c00523.
Biodistribution data, in vitro study conditions, MS/HRMS data of analytical standard and its relative abundances in in vitro samples; Elemental compositions, theoretical masses, measured masses, mass differences, structures and retention times of proposed in vitro metabolites; Formation speed of proposed phase II metabolites; Theoretical masses, retention times, biotransformations, and major fragments of proposed metabolites in in vitro samples; MS/HRMS of selected phase I and II metabolites; MS/MS/HRMS of a phase II metabolite; Extracted ion chromatogram of two phase II metabolites (PDF)
The authors declare no competing financial interest.
This paper was published ASAP on July 1, 2020, with an error in the Supporting Information. The corrected version was reposted on July 7, 2020.
Supplementary Material
References
- Vaquero J. J.; Kinahan P. Positron Emission Tomography: Current Challenges and Opportunities for Technological Advances in Clinical and Preclinical Imaging Systems. Annu. Rev. Biomed. Eng. 2015, 17, 385–414. 10.1146/annurev-bioeng-071114-040723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen A.; Hansen H. D.; Svarer C.; Lehel S.; Leth-Petersen S.; Kristensen J. L.; Gillings N.; Knudsen G. M. The importance of small polar radiometabolites in molecular neuroimaging: A PET study with [11C]Cimbi-36 labeled in two positions. J. Cereb. Blood Flow Metab. 2018, 38 (4), 659–668. 10.1177/0271678X17746179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao R.; Schou M.; Halldin C. Direct plasma metabolite analysis of positron emission tomography radioligands by micellar liquid chromatography with radiometric detection. Anal. Chem. 2012, 84 (7), 3222–30. 10.1021/ac2032657. [DOI] [PubMed] [Google Scholar]
- Keller J.; Baradat M.; Jouanin I.; Debrauwer L.; Gueraud F. ″Twin peaks″: searching for 4-hydroxynonenal urinary metabolites after oral administration in rats. Redox Biol. 2015, 4, 136–48. 10.1016/j.redox.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger B.; Bueschl C.; Neumann N.; Stuckler R.; Doppler M.; Chassy A. W.; Waterhouse A. L.; Rechthaler J.; Kampleitner N.; Thallinger G. G.; Adam G.; Krska R.; Schuhmacher R. Untargeted profiling of tracer-derived metabolites using stable isotopic labeling and fast polarity-switching LC-ESI-HRMS. Anal. Chem. 2014, 86 (23), 11533–7. 10.1021/ac503290j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Zhang L.; Xie F.; Zhuang R.; Jiang D.; Liu H.; Li J.; Yang H.; Zhang X.; Nie L.; Li Z. Rapid one-step 18F-radiolabeling of biomolecules in aqueous media by organophosphine fluoride acceptors. Nat. Commun. 2019, 10 (1), 989. 10.1038/s41467-019-08953-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Yang Y.; Sun M.; Cui M.; Fu Y.; Lin Y.; Li Z.; Nie L. Highly specific noninvasive photoacoustic and positron emission tomography of brain plaque with functionalized croconium dye labeled by a radiotracer. Chem. Sci. 2017, 8 (4), 2710–2716. 10.1039/C6SC04798J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q.; Huang S.; Wu Z.; Zheng J.; Chen X.; Nie L. Label-free Visualization of Early Cancer Hepatic Micrometastasis and Intraoperative Image-guided Surgery by Photoacoustic Imaging. J. Nucl. Med. 2019, jnumed.119.233155. 10.2967/jnumed.119.233155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard-Gauthier V.; Wängler C.; Schirrmacher E.; Kostikov A.; Jurkschat K.; Wängler B.; Schirrmacher R. 18F-labeled silicon-based fluoride acceptors: potential opportunities for novel positron emitting radiopharmaceuticals. BioMed Res. Int. 2014, 2014, 454503. 10.1155/2014/454503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J. Controlled Release 2012, 164 (2), 138–44. 10.1016/j.jconrel.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Wong A. D.; Ye M.; Ulmschneider M. B.; Searson P. C. Quantitative Analysis of the Enhanced Permeation and Retention (EPR) Effect. PLoS One 2015, 10 (5), e0123461. 10.1371/journal.pone.0123461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scodeller P.; Asciutto E. K. Targeting Tumors Using Peptides. Molecules 2020, 25 (4), 808. 10.3390/molecules25040808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W.; Rosenkrans Z. T.; Liu J.; Huang G.; Luo Q. Y.; Cai W. ImmunoPET: Concept, Design, and Applications. Chem. Rev. 2020, 120, 3787. 10.1021/acs.chemrev.9b00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke S.; Kampmann A. L.; Wuest M.; Bailey J. J.; Glowacki B.; Wuest F.; Jurkschat K.; Weberskirch R.; Schirrmacher R. 18F-Radiolabeling and In Vivo Analysis of SiFA-Derivatized Polymeric Core-Shell Nanoparticles. Bioconjugate Chem. 2018, 29 (1), 89–95. 10.1021/acs.bioconjchem.7b00630. [DOI] [PubMed] [Google Scholar]
- Ilhan H.; Lindner S.; Todica A.; Cyran C. C.; Tiling R.; Auernhammer C. J.; Spitzweg C.; Boeck S.; Unterrainer M.; Gildehaus F. J.; Boning G.; Jurkschat K.; Wängler C.; Wängler B.; Schirrmacher R.; Bartenstein P. Biodistribution and first clinical results of 18F-SiFAlin-TATE PET: a novel 18F-labeled somatostatin analog for imaging of neuroendocrine tumors. Eur. J. Nucl. Med. Mol. Imaging 2020, 47 (4), 870–880. 10.1007/s00259-019-04501-6. [DOI] [PubMed] [Google Scholar]
- Ilhan H.; Todica A.; Lindner S.; Boening G.; Gosewisch A.; Wängler C.; Wängler B.; Schirrmacher R.; Bartenstein P. First-in-human 18F-SiFAlin-TATE PET/CT for NET imaging and theranostics. Eur. J. Nucl. Med. Mol. Imaging 2019, 46 (11), 2400–2401. 10.1007/s00259-019-04448-8. [DOI] [PubMed] [Google Scholar]
- Dialer L. O.; Selivanova S. V.; Müller C. J.; Müller A.; Stellfeld T.; Graham K.; Dinkelborg L. M.; Kramer S. D.; Schibli R.; Reiher M.; Ametamey S. M. Studies toward the development of new silicon-containing building blocks for the direct 18F-labeling of peptides. J. Med. Chem. 2013, 56 (19), 7552–63. 10.1021/jm400857f. [DOI] [PubMed] [Google Scholar]
- Wängler C.; Kostikov A.; Zhu J.; Chin J.; Wängler B.; Schirrmacher R. Silicon-[F-18] Fluorine Radiochemistry: Basics, Applications and Challenges. Appl. Sci. 2012, 2 (2), 277–302. 10.3390/app2020277. [DOI] [Google Scholar]
- Lindner S.; Michler C.; Leidner S.; Rensch C.; Wängler C.; Schirrmacher R.; Bartenstein P.; Wängler B. Synthesis and in vitro and in vivo evaluation of SiFA-tagged bombesin and RGD peptides as tumor imaging probes for positron emission tomography. Bioconjugate Chem. 2014, 25 (4), 738–49. 10.1021/bc400588e. [DOI] [PubMed] [Google Scholar]
- Joyard Y.; Azzouz R.; Bischoff L.; Papamicael C.; Labar D.; Bol A.; Bol V.; Vera P.; Gregoire V.; Levacher V.; Bohn P. Synthesis of new 18F-radiolabeled silicon-based nitroimidazole compounds. Bioorg. Med. Chem. 2013, 21 (13), 3680–8. 10.1016/j.bmc.2013.04.029. [DOI] [PubMed] [Google Scholar]
- Schirrmacher R.; Bradtmoller G.; Schirrmacher E.; Thews O.; Tillmanns J.; Siessmeier T.; Buchholz H. G.; Bartenstein P.; Wängler B.; Niemeyer C. M.; Jurkschat K. 18F-labeling of peptides by means of an organosilicon-based fluoride acceptor. Angew. Chem., Int. Ed. 2006, 45 (36), 6047–50. 10.1002/anie.200600795. [DOI] [PubMed] [Google Scholar]
- Otaru S.; Imlimthan S.; Sarparanta M.; Helariutta K.; Wähälä K.; Airaksinen A. J. Evaluation of Organo [18F]Fluorosilicon Tetrazine as a Prosthetic Group for the Synthesis of PET Radiotracers. Molecules 2020, 25 (5), 1208. 10.3390/molecules25051208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchar M.; Mamat C. Methods to Increase the Metabolic Stability of 18F-Radiotracers. Molecules 2015, 20 (9), 16186–220. 10.3390/molecules200916186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharasch E. D.; Thummel K. E. Identification of cytochrome P450 2E1 as the predominant enzyme catalyzing human liver microsomal defluorination of sevoflurane, isoflurane, and methoxyflurane. Anesthesiology 1993, 79 (4), 795–807. 10.1097/00000542-199310000-00023. [DOI] [PubMed] [Google Scholar]
- Yin H.; Anders M. W.; Jones J. P. Metabolism of 1,2-dichloro-1-fluoroethane and 1-fluoro-1,2,2-trichloroethane: electronic factors govern the regioselectivity of cytochrome P450-dependent oxidation. Chem. Res. Toxicol. 1996, 9 (1), 50–7. 10.1021/tx950086n. [DOI] [PubMed] [Google Scholar]
- Bier D.; Holschbach M. H.; Wutz W.; Olsson R. A.; Coenen H. H. Metabolism of the A(1)1 adenosine receptor positron emission tomography ligand [18F]8-cyclopentyl-3-(3-fluoropropyl)-1-propylxanthine ([18F]CPFPX) in rodents and humans. Drug Metab. Dispos. 2006, 34 (4), 570–6. 10.1124/dmd.105.006411. [DOI] [PubMed] [Google Scholar]
- Piert M.; Zittel T. T.; Becker G. A.; Jahn M.; Stahlschmidt A.; Maier G.; Machulla H. J.; Bares R. Assessment of porcine bone metabolism by dynamic. J. Nucl. Med. 2001, 42 (7), 1091–100. [PubMed] [Google Scholar]
- Pan Y. The Dark Side of Fluorine. ACS Med. Chem. Lett. 2019, 10 (7), 1016–1019. 10.1021/acsmedchemlett.9b00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. C.; Boger D. L. Synthesis, Characterization, and Cycloaddition Reactivity of a Monocyclic Aromatic 1,2,3,5-Tetrazine. J. Am. Chem. Soc. 2019, 141 (41), 16388–16397. 10.1021/jacs.9b07744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin D.Y.; Alexandrou N.E. Reassignment of structures of the dihydro-v-tetrazines. Tetrahedron 1963, 19 (11), 1697–1703. 10.1016/S0040-4020(01)99243-9. [DOI] [Google Scholar]
- Wang M.; Svatunek D.; Rohlfing K.; Liu Y.; Wang H.; Giglio B.; Yuan H.; Wu Z.; Li Z.; Fox J. Conformationally Strained trans-Cyclooctene (sTCO) Enables the Rapid Construction of 18F-PET Probes via Tetrazine Ligation. Theranostics 2016, 6 (6), 887–95. 10.7150/thno.14742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc. 2008, 130 (41), 13518–9. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira B. L.; Guo Z.; Bernardes G. J. L. Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46 (16), 4895–4950. 10.1039/C7CS00184C. [DOI] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R.; Hilderbrand S. A. Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconjugate Chem. 2008, 19 (12), 2297–9. 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z.; Zhou L.; Li X.; Zhang J.; Dong S. Tetrazine-Containing Amino Acid for Peptide Modification and Live Cell Labeling. PLoS One 2015, 10 (11), e0141918. 10.1371/journal.pone.0141918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteegen R. M.; Rossin R.; ten Hoeve W.; Janssen H. M.; Robillard M. S. Click to release: instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem., Int. Ed. 2013, 52 (52), 14112–6. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Karver M. R.; Weissleder R.; Hilderbrand S. A. Synthesis and evaluation of a series of 1,2,4,5-tetrazines for bioorthogonal conjugation. Bioconjugate Chem. 2011, 22 (11), 2263–70. 10.1021/bc200295y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavier G.; Audebert P. s-Tetrazines as building blocks for new functional molecules and molecular materials. Chem. Rev. 2010, 110 (6), 3299–314. 10.1021/cr900357e. [DOI] [PubMed] [Google Scholar]
- Samanta S.; Ray S.; Ghosh A. B.; Biswas P. 3,6-Di(pyridin-2-yl)-1,2,4,5-tetrazine (pytz) mediated metal-free mild oxidation of thiols to disulfides in aqueous medium. RSC Adv. 2016, 6 (45), 39356–39363. 10.1039/C6RA01509C. [DOI] [Google Scholar]
- Fritea L.; Audebert P.; Galmiche L.; Gorgy K.; Le Goff A.; Villalonga R.; Sandulescu R.; Cosnier S. First Occurrence of Tetrazines in Aqueous Solution: Electrochemistry and Fluorescence. ChemPhysChem 2015, 16 (17), 3695–9. 10.1002/cphc.201500544. [DOI] [PubMed] [Google Scholar]
- Bohle D. S.; Chua Z.; Perepichka I.; Rosadiuk K. E/Z oxime isomerism in PhC(NOH)CN. Chem. - Eur. J. 2013, 19 (13), 4223–9. 10.1002/chem.201203357. [DOI] [PubMed] [Google Scholar]
- Kolmel D. K.; Kool E. T. Oximes and Hydrazones in Bioconjugation: Mechanism and Catalysis. Chem. Rev. 2017, 117 (15), 10358–10376. 10.1021/acs.chemrev.7b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder J. T.; Malinao M. C.; Dugal-Tessier J.; Atkinson J. E.; Anand B. S.; Okada A.; Mendelsohn B. A. Metabolism of an Oxime-Linked Antibody Drug Conjugate, AGS62P1, and Characterization of Its Identified Metabolite. Mol. Pharmaceutics 2018, 15 (6), 2384–2390. 10.1021/acs.molpharmaceut.8b00225. [DOI] [PubMed] [Google Scholar]
- Jia L.; Liu X. The conduct of drug metabolism studies considered good practice (II): in vitro experiments. Curr. Drug Metab. 2007, 8 (8), 822–9. 10.2174/138920007782798207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commission Decision 2002/657/EC implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02002D0657-20040110.
- Kruve A. Strategies for Drawing Quantitative Conclusions from Nontargeted Liquid Chromatography-High-Resolution Mass Spectrometry Analysis. Anal. Chem. 2020, 92 (7), 4691–4699. 10.1021/acs.analchem.9b03481. [DOI] [PubMed] [Google Scholar]
- Höhne A.; Yu L.; Mu L.; Reiher M.; Voigtmann U.; Klar U.; Graham K.; Schubiger P. A.; Ametamey S. M. Organofluorosilanes as model compounds for 18F-labeled silicon-based PET tracers and their hydrolytic stability: experimental data and theoretical calculations (PET = positron emission tomography). Chem. - Eur. J. 2009, 15 (15), 3736–43. 10.1002/chem.200802437. [DOI] [PubMed] [Google Scholar]
- Lundahl A.; Hedeland M.; Bondesson U.; Lennernas H. In vivo investigation in pigs of intestinal absorption, hepatobiliary disposition, and metabolism of the 5alpha-reductase inhibitor finasteride and the effects of coadministered ketoconazole. Drug Metab. Dispos. 2011, 39 (5), 847–57. 10.1124/dmd.110.035311. [DOI] [PubMed] [Google Scholar]
- Tang M. L.; Zhou L.; Chang J.; Hu Z. H.; Qin Y.; Sun X. Differential metabolism of 3FDT and docetaxel in RLMs, rats, and HLMs. Eur. J. Med. Chem. 2016, 113, 81–91. 10.1016/j.ejmech.2016.02.007. [DOI] [PubMed] [Google Scholar]
- Conning D. M.; Phillips J. C. Comparative metabolism of BHA, BHT and other phenolic antioxidants and its toxicological relevance. Food Chem. Toxicol. 1986, 24 (10–11), 1145–8. 10.1016/0278-6915(86)90300-5. [DOI] [PubMed] [Google Scholar]
- Yan J. H.; Hubbard J. W.; McKay G.; Midha K. K. Stereoselective in vivo and in vitro studies on the metabolism of doxepin and N-desmethyldoxepin. Xenobiotica 1997, 27 (12), 1245–57. 10.1080/004982597239831. [DOI] [PubMed] [Google Scholar]
- Park B. K.; Kitteringham N. R.; O’Neill P. M. Metabolism of fluorine-containing drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–70. 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.