

Conspectus
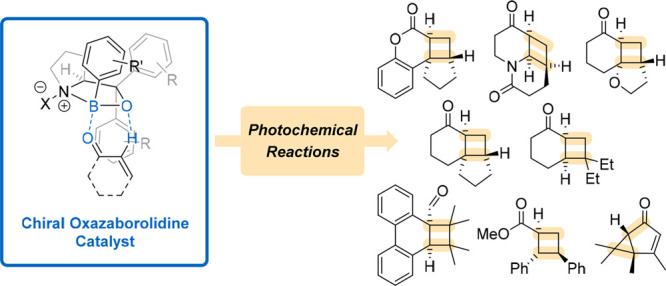
Asymmetric synthesis has posed a significant challenge to organic chemists for over a century. Several strategies have been developed to synthesize enantiomerically enriched compounds, which are ubiquitous in the pharmaceutical and agrochemical industries. While many organometallic and organic catalysts have been found to mediate thermal enantioselective reactions, the field of photochemistry lacks similar depth. Recently, chiral 1,3,2-oxazaborolidines have made the transition from Lewis acids that were exclusively applied to thermal reactions to catalysts for enantioselective photochemical reactions. Due to their modular structure, various 1,3,2-oxazaborolidines are readily available and can be easily fitted to a given chemical transformation. Their use holds great promise for future developments in photochemistry. This Account gives an overview of the substrate classes that are known to undergo enantioselective photochemical transformations in the presence of chiral 1,3,2-oxazaborolidines and touches on the catalytic mode of action, on the proposed enantiodifferentiation mechanism, as well as on recent computational studies.
Based on the discovery that the presence of Lewis acids enhances the efficiency of coumarin [2 + 2] photocycloadditions, chiral 1,3,2-oxazaborolidines were applied in 2010 for the first time to prepare enantiomerically enriched photoproducts. These Lewis acids were then successfully used in intramolecular [2 + 2] photocycloaddition reactions of 1-alkenoyl-5,6-dihydro-4-pyridones and 3-alkenyloxy-2-cycloalkenones. In the course of this work, it became evident that the chiral 1,3,2-oxazaborolidine must be tailored to the specific reaction; it was shown that both inter- and intramolecular [2 + 2] photocycloadditions of cyclic enones can be conducted enantioselectively, but the aryl rings of the chiral Lewis acids require different substitution patterns. In all [2 + 2] photocycloaddition reactions in which chiral 1,3,2-oxazaborolidines were used as catalysts, the catalyst loading could not be decreased below 50 mol % without sacrificing enantioselectivity due to competitive racemic background reactions. To overcome this constraint, substrates that reacted exclusively when bound to an oxazaborolidine were tested, notably phenanthrene-9-carboxaldehydes and cyclohexa-2,4-dienones. The former substrate class underwent an ortho photocycloaddition, the latter an oxadi-π-methane rearrangement. Several new 1,3,2-oxazaborolidines were designed, and the products were obtained in high enantioselectivity with only 10 mol % of catalyst. Recently, an iridium-based triplet sensitizer was employed to facilitate enantioselective [2 + 2] photocycloadditions of cinnamates with 25 mol % of chiral 1,3,2-oxazaborolidine. In this case, the relatively low catalyst loading was possible because the oxazaborolidine–substrate complex exhibits a lower triplet energy and an improved electronic coupling compared to the uncomplexed substrate, allowing for a selective energy transfer.
By synthetic and theoretical studies, it has become evident that chiral 1,3,2-oxazaborolidines are multifaceted catalysts: they change absorption behavior, alter energetic states, and induce chirality. While a diverse set of substrates has been shown to undergo enantioselective photochemical transformations in the presence of chiral 1,3,2-oxazaborolidines either through direct excitation or through triplet sensitization, these catalysts took on different roles for different substrates. Based on the studies presented in this Account, it can be assumed that there are still more photochemical reactions and substrate classes that could profit from chiral 1,3,2-oxazaborolidines.
Key References
Brimioulle, R.; Bauer, A.; Bach, T. Enantioselective Lewis Acid Catalysis in Intramolecular [2 + 2] Photocycloaddition Reactions: A Mechanistic Comparison between Representative Coumarin and Enone Substrates. J. Am. Chem. Soc. 2015, 137, 5170–5176.1 The mechanistic study revealed the major differences between two enantioselective [2 + 2] photocycloadditions discovered in prior work and helped to elucidate the mode of action of the chiral 1,3,2-oxazaborolidine Lewis acids.
Poplata, S.; Bach, T. Enantioselective Intermolecular [2 + 2] Photocycloaddition Reaction of Cyclic Enones and Its Application in a Synthesis of (−)-Grandisol. J. Am. Chem. Soc. 2018, 140, 3228–3231.2 Cyclic alkenones represent the most frequently used substrate class of [2 + 2] photocycloaddition chemistry. In this paper, a general solution was presented how these cycloaddition reactions can be performed enantioselectively.
Stegbauer, S.; Jandl, C.; Bach, T. Enantioselective Lewis Acid Catalyzed ortho Photocycloaddition of Olefins to Phenanthrene-9-carboxaldehydes. Angew. Chem. Int. Ed. 2018, 57, 14593–14596.3 The study revealed that catalyst loadings in Lewis acid-mediated photocycloaddition reactions can be significantly lowered when securing that Lewis acid coordination leads to an extensive bathochromic shift. Aldehydes were for the first time employed in a photochemical reaction catalyzed by a chiral 1,3,2-oxazaborolidine Lewis acid.
Leverenz, M.; Merten, C.; Dreuw, A.; Bach, T. Lewis Acid Catalyzed Enantioselective Photochemical Rearrangements on the Singlet Potential Energy Surface. J. Am. Chem. Soc. 2019, 141, 20053–20057.4 This work revealed that enantioselective photochemical reactions catalyzed by chiral Lewis acids are also feasible on the singlet hypersurface and that the application of Lewis acid catalysis in photochemistry goes beyond photocycloaddition chemistry.
1. Introduction
Five-membered heterocyclic compounds that consist of a consecutive set of oxygen, boron, nitrogen, and two carbon atoms are named 1,3,2-oxazaboroles. The history of 2,3-dihydro-1,3,2-oxazaboroles and tetrahydro-1,3,2-oxazaboroles (1,3,2-oxazaborolidines) dates back to the 1950s when the first representatives of this compound class were prepared. The synthesis of compound 1 (Figure 1) from ortho-aminophenol was described in 1958 by Sugihara and Bowman5 and by Dewar et al.6 The former authors performed the condensation with phenylboronic acid (reflux in acetone), and the latter authors used dichlorophenylboron as the boron source (reflux in benzene). To this day, the condensation of 1,2-aminoalcohols with alkyl- or arylboronic acids has remained the preferred method for the preparation of 1,3,2-oxazaborolidines. Compound 2 likely represents the first chiral 1,3,2-oxazaborolidine ever reported and was obtained by the condensation of (−)-ephedrine and phenylboronic acid in refluxing toluene.7 One of the first 2,3-dihydro-1,3,2-oxazaboroles in which a hydrogen atom was the substituent on the boron was compound 3, which was prepared by the reaction of 8-hydroxyquinoline with borane.8
Figure 1.

Structures of some historically relevant 1,3,2-oxazaborolidines 1–4.
The interest of synthetic organic chemists in dihydro- and tetrahydro-1,3,2-oxazaboroles was kindled by the report of Corey, Bakshi, and Shibata who discovered that the reductive power of a mixture of a (S)-diphenylprolinol and borane was linked to the intermediacy of 1,3,2-oxazaborolidine 4 (R = H).9 Prior work by Itsuno and coworkers had already revealed that chiral 1,2-aminoalcohols, like (S)-2-amino-3-methyl-1,1-diphenylbutan-1-ol, in combination with borane, can be used for the stoichiometric enantioselective reduction of aromatic ketones.10,11 A 1,3,2-oxazaborolidine had been proposed for the reaction of two equivalents of borane with (S)-valinol.12 Catalyst 4 (R = H, Me, Bu, aryl), which is known as the CBS (Corey–Bakshi–Shibata) catalyst, furnishes an enantioselective reduction of many prochiral ketones and has become one of the most frequently used catalysts for this transformation.13−17
Studies of the mechanistic mode of action of the CBS catalyst suggest that borane (BH3) coordinates to the nitrogen atom of the heterocyclic ring and thus increases the Lewis acidity of the 1,3,2-oxazaborolidine boron atom. Based on this insight, Corey and coworkers used 1,3,2-oxazaborolidines as chiral Lewis acidic catalysts upon activation by an appropriate acid. The Diels–Alder reaction of cyclopentadiene with methacrolein to product exo-5 served as one of the test reactions (Scheme 1). Initial work focused on the activation of l-proline-derived, triaryl-substituted 1,3,2-oxazaborolidines by trifluoromethanesulfonic acid (HOTf), and catalyst 6 was found to be the most active and selective Lewis acid furnishing the product exo-5 in a diastereomeric ratio (d.r. = exo:endo) of 91:9 and in 96% ee (enantiomeric excess).18
Scheme 1. Activation of Oxazaborolidines by Brønsted or Lewis Acids and the Use of Catalysts 6–8 in an Enantioselective Diels–Alder Reaction.
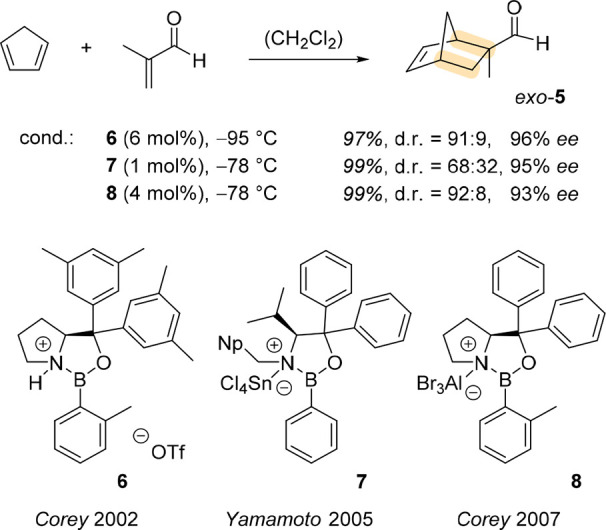
Futatsugi and Yamamoto showed that not only Brønsted but also Lewis acids can increase the Lewis acidity of 1,3,2-oxazaborolidines and render them useful chiral catalysts. Their preferred catalyst 7 was prepared from l-valine (Np = 1-naphthyl) and was activated by tin tetrachloride. In the Diels–Alder reaction to produce exo-5, the catalyst performed with high enantioselectivity but with reduced diastereoselectivity compared to 6.19 A final key discovery that is highly relevant for the work presented in this Account relates to the use of aluminum tribromide as the activating Lewis acid. Corey and coworkers presented catalyst 8 in which the boron atom of the 1,3,2-oxazaborolidine is more efficiently activated with Lewis than with Brønsted acid, making it a powerful catalyst for many enantioselective Diels–Alder reactions.20 Development of acid-activated 1,3,2-oxazaborolidines as chiral catalysts in thermal reactions has continued over the last ten years, and it is beyond the scope of this Account to summarize all the details. Excellent reviews are available that cover the developments in the field.21−24 Here, we focus on enantioselective reactions in photochemistry which pose the major challenge to unlock selective reaction pathways at electronically excited states more than 300 kJ mol–1 above the ground state. Given the extensive amount of energy a photon provides, enantioselective catalysis cannot rely on lowering transition states in a similar fashion as in thermal reactions.25
2. Discovery and Initial Applications
2.1. Coumarin [2 + 2] Photocycloaddition
Early examples of Lewis acid catalysis in photochemistry26 were reported by Lewis and coworkers. They studied, for example, the effects on the photochemical behavior of coumarin (9) in the presence of an acid.27,28 While the [2 + 2] photocycloaddition of an olefin to coumarin is inefficient in the absence of a catalyst, Lewis and Barancyk showed that upon addition of BF3·OEt2, photoproduct rac-10 was obtained in moderate yield (Scheme 2).28
Scheme 2. Lewis-Acid-Catalyzed [2 + 2] Photocycloaddition of 2,3-Dimethyl-2-butene and Coumarin 9, Furnishing rac-10.
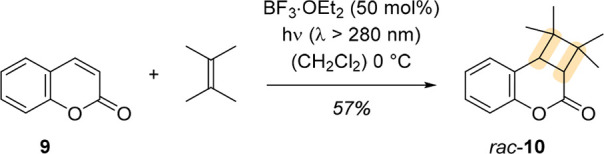
They attributed this observed increase in reactivity to a change of orbital energies in the coumarin–Lewis acid complex, which is made apparent by a bathochromic shift of the absorption band in UV/vis spectroscopy and an increased fluorescence of coumarin in the presence of Lewis acid. It was suggested that the energies of frontier orbitals, most profoundly the energy of the oxygen n-orbital (nO), are decreased. Consequently, the energy gap between the lowest unoccupied molecular orbital (LUMO) and the nO-orbital increases more than the energy gaps between the LUMO and the other occupied molecular orbitals. With a larger gap, the perturbation of ππ* and nπ* singlet states is reduced, which diminishes decay pathways and increases the lifetime of the singlet states. The increased lifetime explains the observed fluorescence of the coumarin–Lewis acid complex and presumably contributes to the increased quantum yield of the cycloaddition reaction that was suggested to occur via a triplet pathway. Upon sensitization with benzophenone, cycloaddition product rac-10 was also formed, but in the presence of BF3, higher conversions were achieved, and no side products were identified.
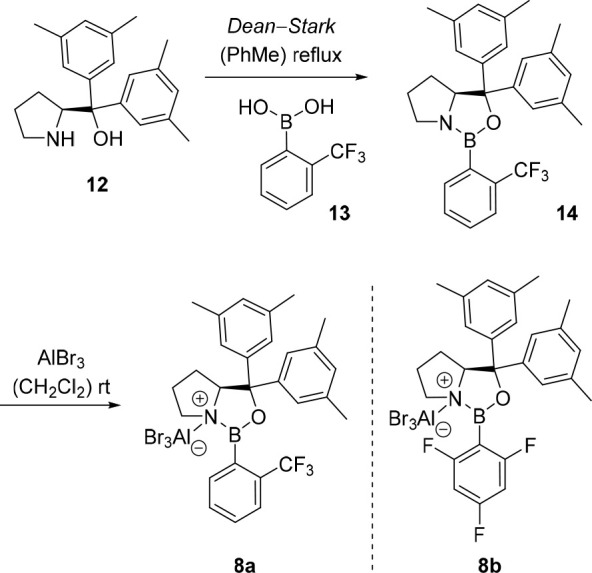
This finding inspired our group in 2010 to study the intramolecular [2 + 2] photocycloaddition of 4-(pent-4-enyl)coumarin (11a) with different chiral Lewis acids with the hope to achieve enantioselectivity through an unprecedented mode of enantiodifferentiation in photochemistry.29 After extensive screening, chiral 1,3,2-oxazaborolidine Lewis acids were the preferred catalysts (Scheme 3) for the desired transformation. Catalyst 8a and analogues like 8b were easily available from an l-prolinol derivative 12 and an arylboronic acid 13 through a condensation–activation procedure (via 14) as depicted in Scheme 3. Under the final reaction conditions, coumarin 11a underwent the desired photocycloaddition in the presence of oxazaborolidine catalyst 8a in high yield and high enantioselectivity (Scheme 4).
Scheme 3. Generation of Oxazaborolidine-Based Lewis Acid 8a by Condensation of l-Prolinol Derivative 12 and Boronic Acid 13 and Subsequent Activation of 14 by AlBr3 to Prepare Lewis Acid 8b.
Scheme 4. First Example of an Enantioselective [2 + 2] Photocycloaddition with a Chiral 1,3,2-Oxazaborolidine as Catalyst.
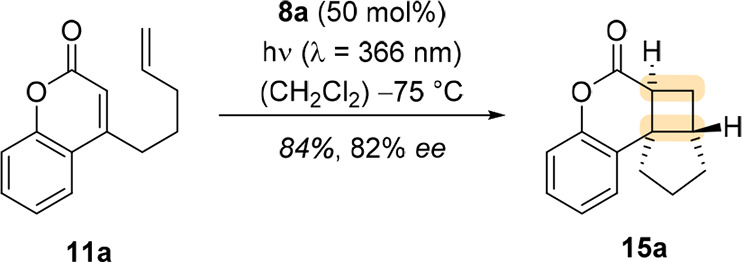
After screening multiple 1,3,2-oxazaborolidine catalysts, it became apparent that the substitution pattern on the aryl rings has a significant impact on the enantiomeric excess of 15a. The observed enantioselectivity was explained by a two-point binding of the 1,3,2-oxazaborolidine catalyst to the coumarin substrate, as had been suggested by Corey and others.24,30Figure 2a shows the potential structure of the coumarin–Lewis acid complex 11a·8a with a classical Lewis acid binding pattern of the boron atom to the carbonyl oxygen atom and a nonclassical binding of the oxazaborolidine oxygen atom to the olefinic α-hydrogen atom of the coumarin. An aryl ring of the prolinol backbone was proposed to shield one side of the coumarin, successfully differentiating the enantiotopic sides for a selective attack of the olefin tether upon irradiation. With the best catalyst 8a in hand, (E)- and (Z)-4-(hex-4-enyl)-coumarin (11b) were tested as substrates to investigate whether the reaction proceeds on the singlet or triplet hypersurface. Singlet reactions are expected to be concerted and so should be stereospecific. Because both (E)- and (Z)-11b gave predominantly the trans-product (15b, trans:cis = 77:23 and 62:38, respectively), it was suggested that the reaction likely proceeds via a triplet 1,4-diradical 16b that can undergo rotation around the C–C bond of the tether so that the favored conformation (Figure 2b) leads after ISC and ring closure to the major product.
Figure 2.
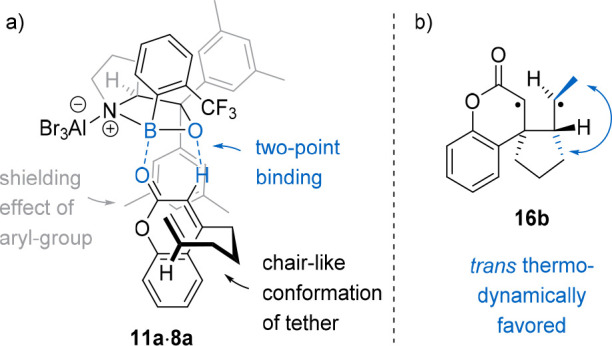
(a) Proposed conformation of the oxazaborolidine–substrate complex 11a·8a. (b) Intermediate 16b of the [2 + 2] photocycloaddition of 11b.
In a follow-up study, the established enantioselective [2 + 2] photocycloaddition was expanded to additional substrates.31 Representative photoproducts can be found in Figure 3. The products were obtained in very good yield and enantioselectivity, except for 15e, which bears a sulfur atom in its tether and likely underwent a cleavage reaction in the presence of the Lewis acid.
Figure 3.

Representative products 15 obtained by enantioselective [2 + 2] photocycloaddition of 4-substituted coumarins.
To understand the influence of Lewis acids on the coumarin [2 + 2] photocycloaddition, Brimioulle et al. conducted UV/vis and fluorescence spectroscopic studies.31 Upon addition of BF3, EtAlCl2, or AlBr3 as Lewis acids, the absorption maximum of substrate 11a was shifted bathochromically (Δλ < 10 nm), which favors the absorption at the chosen wavelength of λ = 366 nm but cannot explain the difference in reactivity of coumarins in the presence of Lewis acids. A notable difference, however, could be seen in fluorescence experiments. While there was no significant fluorescence without Lewis acid, an emission at λem ≈ 400 nm was observed in the presence of BF3 (λexc = 300 nm). This phenomenon can be explained by a reduced rate of internal conversion, i.e. radiation-less transition to the ground state, and an increased lifetime of the excited singlet state.27,28 Even though intersystem crossing (ISC) to the triplet state is slow, a significant number of molecules will undergo ISC when more molecules occupy the excited singlet state. Moreover, the presence of a Lewis acid favors the ISC to the reactive excited triplet state in comparison with the uncatalyzed pathway. In accordance with the previous findings of Lewis and Barancyk,28 the molecules can undergo a productive [2 + 2] photocycloaddition from the triplet state.
2.2. Intramolecular [2 + 2] Photocycloaddition of Enones
Once the enantioselective [2 + 2] photocycloaddition of coumarins was successfully established, cyclic enones were investigated as potential substrates. Not only do they undergo the reaction, but they also represent the most versatile and most useful substrate class of [2 + 2] photocycloaddition chemistry. Unlike coumarins, cyclic enones react very efficiently by direct excitation even at long wavelengths by nπ* excitation to their singlet state followed by rapid ISC to the triplet state. It was therefore surprising to note that oxazaborolidine 8a enabled the formation of photoproduct 18a from 5,6-dihydro-4-pyridone 17a in very good yield and enantioselectivity (Scheme 5).32 Other products were formed in equally good yield and selectivity, among them 18b–e, and the method was applied to the total synthesis of (+)-lupinine and the formal synthesis of (+)-thermopsine. The enantioface differentiation can be explained by an analogous transition state to that of the coumarins (cf. Figure 2a).
Scheme 5. Intramolecular [2 + 2] Photocycloaddition of 5,6-Dihydro-4-pyridones 17 in the Presence of Lewis Acid 8a.
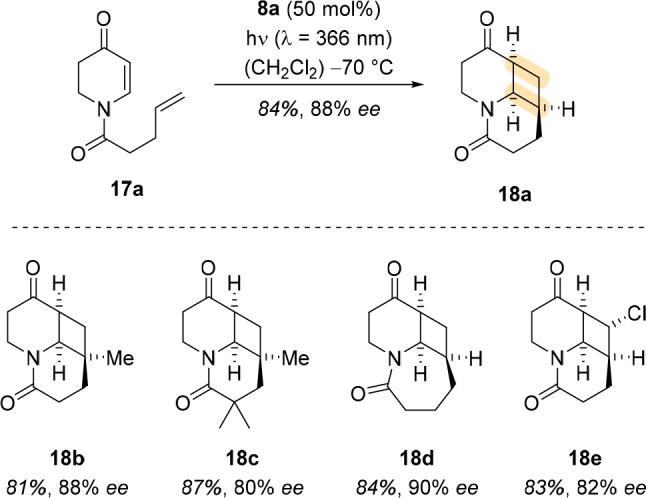
Notable differences between the 5,6-dihydro-4-pyridone substrates and the coumarin substrates (e.g., 17a vs 11a) are the absorption behavior and the mode of action.1 Upon addition of EtAlCl2, 5,6-dihydro-4-pyridones exhibit a significant bathochromic shift of Δλ > 50 nm of the allowed ππ* transition due to the coordination of the Lewis acid to the enone carbonyl moiety (Figure 4). After addition of Lewis acid, a new species was observed by 13C NMR spectroscopy. The signals related to the enone carbon atoms were significantly shifted, compared to the signal of the amide carbon atom, indicating coordination to the enone moiety. Because the extinction coefficient of the Lewis acid–substrate complex is more than 2 orders of magnitude higher (ε = 10 500 M–1 cm–1) than the extinction coefficient of the substrate (ε = 70 M–1 cm–1) at the chosen irradiation wavelength (λ = 366 nm), the racemic background reaction of the uncomplexed substrate is suppressed. Nevertheless, because the rate of intersystem crossing is reduced compared to the uncomplexed substrate due to the coordination of the Lewis acid, lowering the amount of catalyst diminishes the enantioselectivity as the background reaction becomes competitive.
Figure 4.
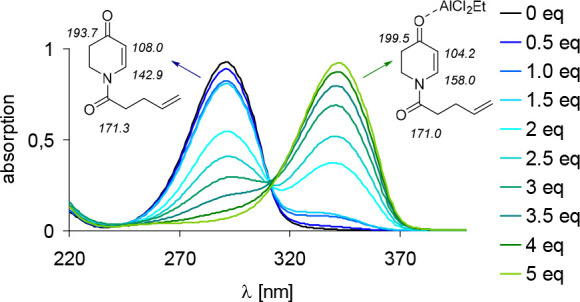
UV/vis spectra of 17a in the absence and presence of EtAlCl2 as the Lewis acid and corresponding species with selected 13C NMR signals. Reproduced with permission from ref (1). Copyright 2015 American Chemical Society.
After the 5,6-dihydro-4-pyridones, 3-alkenyloxy-2-cycloalkenones (19) were studied. It became apparent that they also undergo a [2 + 2] photocycloaddition in an enantioselective fashion in the presence of an 1,3,2-oxazaborolidine catalyst (Scheme 6).33 By screening different oxazaborolidines with 19a as substrate, Brimioulle et al. found that a 2,4,6-trifluoro-substituted boronic acid moiety resulted in a higher enantiomeric excess of photoproduct 20a. Multiple substrates yielded the corresponding products in good enantioselectivity with catalyst 8b; some of them are shown in Scheme 6. Similar to the 5,6-dihydro-4-pyridones, 3-alkenyloxy-2-cycloalkenones exhibit a bathochromic shift of Δλ ≈ 40 nm in the presence of Lewis acid, which explains the effectiveness of catalyst 8b. Products 20 are useful compounds that can undergo cyclobutane ring expansion reactions with Brønsted or Lewis acids.
Scheme 6. Intramolecular [2 + 2] Photocycloaddition of 3-Alkenyloxy-2-cycloalkenones 19 in the Presence of Lewis Acid 8b.

The success of the 1,3,2-oxazaborolidines as Lewis acid catalysts in the [2 + 2] photocycloadditions warranted an in-depth mechanistic study, which was conducted by Brimioulle et al. and focused on coumarin and 5,6-dihydro-4-pyridone substrates.1 In kinetic studies on the standard coumarin substrate 11a, the reaction proceeded substantially faster in the presence of catalyst 8a (50% conversion after 25 min) compared to the noncatalyzed reaction (11% after 5.0 h). In the case of 5,6-dihydro-4-pyridones substrate 17a, the opposite held true. The enantioselective reaction (50% conversion after 10 h) proceeded significantly slower than the reaction in the absence of a Lewis acid (full conversion within 1.0 h). While the catalyst helps to populate the triplet state and accelerate the photocycloaddition in the case of the coumarins by reducing the rate of internal conversion from the excited singlet state (vide supra), the catalyst reduces the rate of ISC in the case of the enones. The rate of ISC is high without Lewis acid (1nπ* → 3ππ*),34 whereby the effective rate of the enone [2 + 2] photocycloaddition is slowed. To corroborate the hypothesis that the reactions proceed on the triplet hypersurface, photocycloadditions with E- and Z-configured alkene tethers were conducted. A stereoconvergent reaction pathway indicates a triplet intermediate (see Figure 2b). For coumarins, the relative configuration of the tether in (E)- and (Z)-4-(hex-4-en-1-yl)-coumarin translated into the corresponding trans- and cis-photoproducts without catalyst 8a (stereospecific singlet reaction). When the reaction was performed with 8a, stereoconvergence was observed to give predominantly the trans-product. The studied (E)- and (Z)-1-(hex-4-enoyl)-5,6-dihydro-4-pyridones, on the other hand, yielded the corresponding photoproducts always in excellent diastereoselectivity of trans:cis = >95:5, irrespective of whether the reaction was catalyzed with 8a or not, indicating that they always react on the triplet hypersurface.
3. beyond Conventional Oxazaborolidines: New Catalysts and Reactions
3.1. [2 + 2] Photocycloaddition of Cycloalkenones
Coumarins, 5,6-dihydro-4-pyridones, and 3-alkenyloxy-2-cycloalkenones are not the only substrates suitable for enantioselective photochemical reactions with 1,3,2-oxazaborolidine Lewis acids. As discussed below, other substrates and reaction classes were investigated. This led to the discovery of new 1,3,2-oxazaborolidine catalysts 8c–8e (Figure 5), with which high enantioselectivities could be achieved in a variety of asymmetric photochemical transformations.
Figure 5.
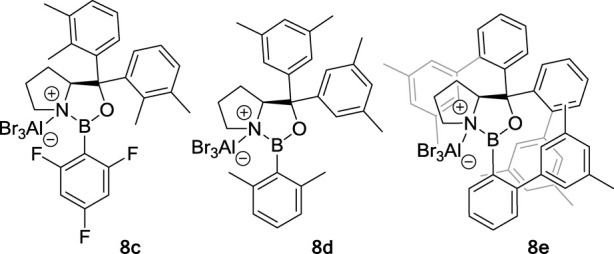
Structures of 1,3,2-oxazaborolidine Lewis acid catalysts 8c–8e.
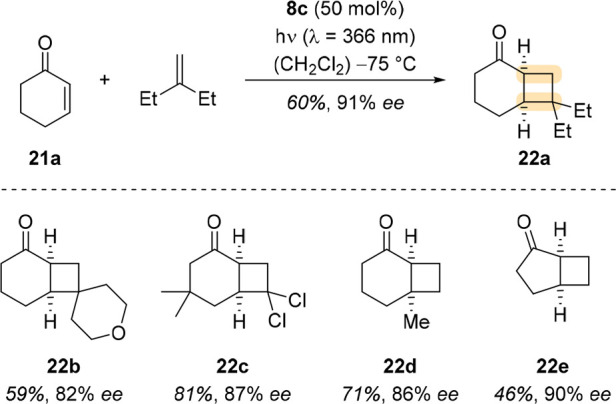
In 2018, Poplata and Bach demonstrated that cyclic alkenones such as 2-cyclohexenone (21a) and simple olefins reacted intermolecularly with high enantioselectivity (Scheme 7).2 While screening reaction conditions with 21a, it became evident that a different substitution pattern on the aryl groups of the l-prolinol backbone was needed to achieve high enantioselectivity. They presumed that in the case of 8b, a methyl substituent at the aryl group is oriented in a way that enables hydrogen abstraction of the excited enone from the catalyst, leading to its decomposition. By positioning the methyl groups differently, the design of catalyst 8c avoided any side reaction and allowed for a clean reaction and high enantioselectivity. The utility of the method was demonstrated by a short and enantioselective total synthesis of (−)-grandisol.
Scheme 7. Enantioselective Intermolecular [2 + 2] Photocycloadditions of Cyclic Enones with Simple Olefins, Catalyzed by Oxazaborolidine 8c.
Like the 5,6-dihydro-4-pyridone 17a, 2-cyclohexenone (21a) exhibits a bathochromic shift of the intense ππ* band in the presence of EtAlCl2, which explains the selective irradiation of substrate–8c complexes. The method provides a general access to cyclobutanes derived from cyclic alkenones, which in turn represent the most frequently used substrate class of [2 + 2] photocycloaddition chemistry.
The intramolecular variant of the same [2 + 2] photocycloaddition was successfully performed by Poplata et al. with catalyst 8c (Scheme 8).35 All products were isolated in good enantioselectivity, except for 24d, in which the terminal methyl groups presumably clashed with the oxazaborolidine catalyst.
Scheme 8. Enantioselective Intramolecular [2 + 2] Photocycloadditions of Cyclic 3-Substituted 2-Alkenones, Catalyzed by Oxazaborolidine 8c.
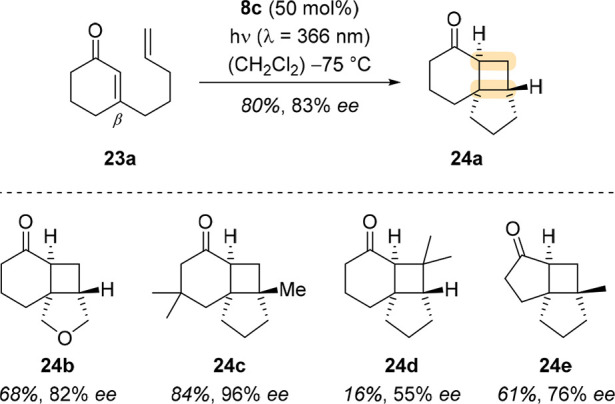
In contrast to the intermolecular case, the first C–C bond formation after excitation in the intramolecular case proceeds via a β-attack, as this furnishes the five-membered ring. The enantioselectivity is enabled through the proposed coordination of the oxazaborolidine, which was corroborated by density functional theory (DFT) calculations. Because both (E)- and (Z)-3-(hex-4-enyl)-cyclohex-2-enone gave predominantly the trans-configured product (vide supra), the intramolecular [2 + 2] photocycloaddition is also likely to proceed on the triplet hypersurface.
3.2. Overcoming the Constraints of Intersystem Crossing
All of the previously mentioned [2 + 2] photocycloaddition reactions of enones suffered from a background reaction that could only be sufficiently suppressed with a catalyst loading of at least 50 mol %. To overcome this limitation, substrates that exhibit an even more extensive bathochromic shift in the presence of Lewis acid were sought. It was hypothesized that in such a scenario only the Lewis acid–substrate complex and not the uncomplexed substrate would be excited at the chosen wavelength. Along these lines, Stegbauer et al. found that the bathochromic shift Δλ of phenanthrene-9-carboxaldehydes in the presence of EtAlCl2 was larger than 70 nm, reaching beyond the nπ*-absorption of the uncomplexed substrate (Figure 6).3
Figure 6.
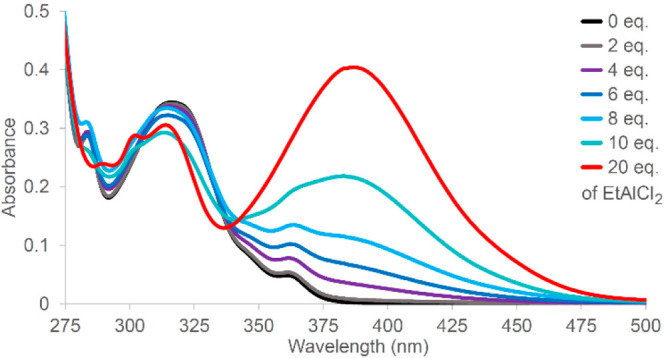
UV/vis spectra of phenanthrene-9-carboxaldehyde (25a, Scheme 9) in the absence and presence of EtAlCl2 as Lewis acid. Adapted with permission from ref (3). Copyright 2018 John Wiley and Sons.
Through elaborate screening, Stegbauer et al. showed that 10 mol % of oxazaborolidine 8d facilitated the formation of cyclobutane 26a, the product of an ortho photocycloaddition of 25a with 2,3-dimethylbut-2-ene, in excellent enantioselectivity (Scheme 9).3 Under the chosen reaction conditions with oxazaborolidine catalyst 8d, no side reactions from carbonyl photoreactivity, e.g. a Paternò–Büchi reaction,36 were observed.
Scheme 9. Enantioselective ortho Photocycloaddition of Phenanthrene-9-carboxaldehyde (25a) with 2,3-Dimethylbut-2-ene, Catalyzed by Oxazaborolidine 8d.
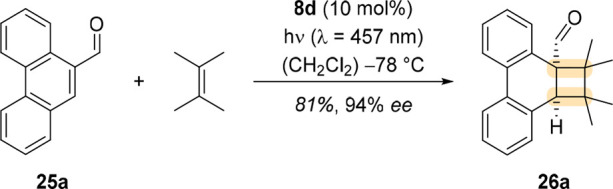
Several products were successfully prepared in excellent enantioselectivity when 20 mol % of 8d was employed as the catalyst. A subset thereof is shown in Figure 7. It is worth pointing out that the oxazaborolidine catalyst also enabled high regio- and diastereoselectivity as demonstrated by products 26d–f.
Figure 7.
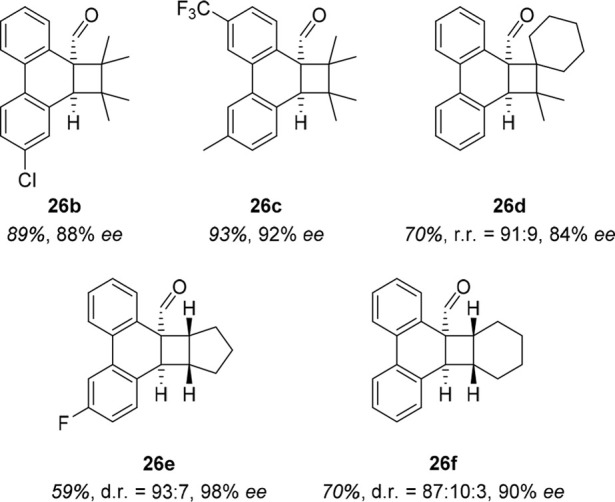
Exemplary products of the studied enantioselective ortho photocycloaddition.
In regards to the mechanism, Stegbauer et al. performed preliminary fluorescence studies in which they showed that the fluorescence of 25a in the presence of Lewis acid was quenched upon addition of 2,3-dimethylbut-2-ene. Because an energy transfer is not thermodynamically feasible due to the high singlet state energies of simple olefins, the authors tentatively suggested that the reactions proceed from the singlet state, which significantly reduces fluorescence as a decay pathway.
Yoon and coworkers found another way to reduce the amount of 1,3,2-oxazaborolidine catalyst in 2019.37 They used a dual catalysis approach38 and employed both a Lewis acid and a triplet sensitizer to enable the [2 + 2] photocycloaddition of methyl cinnamate (27a) and styrene (Scheme 10). Reasoning that a Lewis acid should lower the triplet energy of 27a upon complexation so that a sensitizer with a suitable triplet energy can only excite the complexed substrate, they found that 25 mol % of 8b′ and 1 mol % of [Ir(Fppy)2(dtbbpy)](PF6) resulted in product 28a in excellent yield and enantioselectivity. Yoon and coworkers applied these reaction conditions to several cinnamate esters, highlighting the usefulness of this general concept (Figure 8). They proved the usability of their findings through a concise synthesis of a norlignan natural product, for which they had to switch to a different iridium sensitizer so that the triplet energy of the specific cinnamate ester was suitable.
Scheme 10. Combining Oxazaborolidine Catalyst 8b′ (Activation with Tf2NH instead of AlBr3) and an Iridium Sensitizer to Accomplish an Enantioselective [2 + 2] Photocycloaddition of Cinnamate 27a and Styrene.

Figure 8.
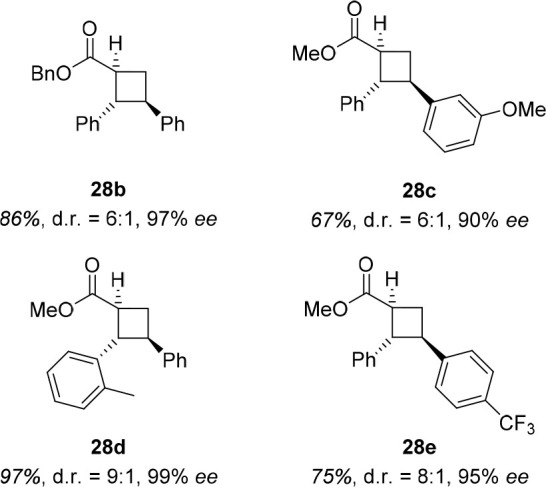
Exemplary products 28b–e of the iridium-sensitized [2 + 2] photocycloaddition of cinnamates.
Through detailed DFT studies, the authors demonstrated that even though the triplet energy of the cinnamate ester was lowered upon complexation with the oxazaborolidine catalyst 8b′, the impact on the triplet energy was not significant enough to explain the observed sensitization efficiency. Moreover, in the presence of another suitable Lewis acid the reaction was catalyzed even though no lowering of the triplet energy was observed in the Lewis acid-substrate complex. When Yoon and coworkers calculated the energies of the frontier molecular orbitals, they discovered that the oxazaborolidine Lewis acid significantly decreases the energies of the frontier orbitals. This makes these orbitals close in energy to the two singly occupied orbitals of the excited iridium sensitizer, which leads to greater electronic coupling and allows for rapid triplet energy transfer.
Cyclohexa-2,4-dienones (29) were found to exhibit a large bathochromic shift in the presence of Lewis acid, and their potential to undergo an enantioselective photochemical reaction was investigated by Leverenz et al.4 Upon irradiation, these substrates underwent an oxadi-π-methane rearrangement to bicyclic compounds. Leverenz et al. thoroughly studied this reaction and tested various 1,3,2-oxazaborolidine catalysts. Novel catalyst 8e with three 3′,5′-dimethyl-[1,1′-biphenyl]-2-yl groups successfully induced an enantioselective reaction course even though only 10 mol % of 8e was used (Scheme 11). Because the bathochromic shift is Δλ > 50 nm in the presence of Lewis acid and the absorption tails far into the visible light region, the solution could be irradiated with light of longer wavelength to avoid racemic background reactions.
Scheme 11. Photoinduced Oxadi-π-methane Rearrangement of Cyclohexa-2,4-dienone 29a to Bicyclo[3.1.0]hexenone 30a Catalyzed by Novel Oxazaborolidine Catalyst 8e.
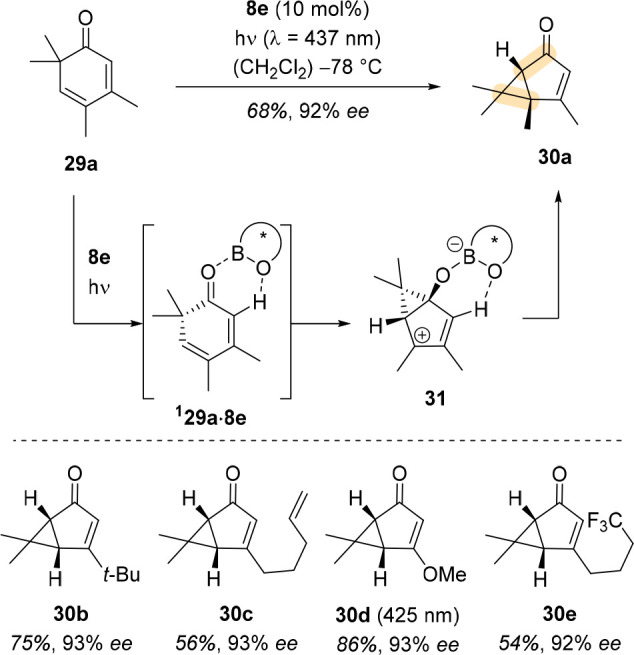
Leverenz et al. studied many substrates under these conditions and obtained the corresponding products in excellent enantioselectivity.4 Starting from photoproduct 30d, they were able to synthesize the monoterpene trans-chrysanthemic acid. The authors found no effect of the triplet quencher piperylene on the reaction course and therefore suggested a singlet reaction pathway. Computational studies corroborated their hypothesis and contributed an important detail regarding the enantiodifferentiation: the gem-dimethylated carbon atom of substrates 29 bends out of plane after excitation and, within the Lewis acid complex, the bending should occur away from the bulky aromatic group at the boron atom of the oxazaborolidine catalyst. Contrary to the other substrates, the opposite enantiotopic face of the molecule is shielded as the aryl group of the boronic acid, not an aryl group of the prolinol backbone, is responsible for successful face differentiation, which also becomes evident from the absolute configuration of the products 30.
Based on the examples of successfully developed enantioselective photochemical reactions, one can deduce that chiral 1,3,2-oxazaborolidines are suitable catalysts for many substrates. For all investigated reactions, careful screening of different oxazaborolidines was necessary and investigations into the mechanism of these reactions showed that the mode of action is more complex than meets the eye. Typical [2 + 2] photocycloaddition reactions through direct excitation have been thoroughly studied, and triplet sensitization has shown promising results. Recently, a 1,3,2-oxazaborolidine-catalyzed rearrangement reaction has been performed enantioselectively.4 Based on these findings, it stands to reason that the realm of enantioselective photochemical reactions with chiral 1,3,2-oxazaborolidines has a lot of dimensions yet to be explored.
4. Binding Modes and Computational Studies
4.1. Substrate Binding
Generally, chiral 1,3,2-oxazaborolidines as Lewis acids can bind to carbonyl groups and their aryl groups can differentiate the enantiotopic sides of the coordinated substrates. Nonclassical hydrogen bonding has been proposed with either coordination to the aldehyde hydrogen atom (as in the case of 2-substituted acroleins like 32) or coordination to the olefinic α-hydrogen atom (as in the cases of acrylic acid, enones, allenoates, or α,β-unsaturated esters like 33) as shown in Figure 9a.39,40 This coordination pattern was also observed in stable complexes of methyl cinnamate and (di)benzylidenacetone with BF3 as determined by X-ray single-crystal analysis (benzylidenacetone–BF3 complex 34 shown in Figure 9b).39 Nonclassical bonding is assumed to be an integral factor in effective catalyst complexation for multiple substrates.30 In their review article on nonclassical hydrogen bonding, Johnston and Cheong elaborate on the two-point binding mode and summarize that Corey and coworkers’ hypothesis has been corroborated by various computational studies.41 Over the years, many different substrates with a hydrogen available for nonclassical bonding have been employed in enantioselective 1,3,2-oxazaborolidine catalysis.
Figure 9.

(a) Two-point coordination of a generic 1,3,2-oxazaborolidine Lewis acid (8) to 2-methylacrolein (32) and ethyl acrylate (33). (b) Structure of isolated benzylidenacetone–BF3 complex 34.
4.2. Computational Studies
As mentioned, the two-point coordination model can explain the observed enantioselectivity in many reactions with 1,3,2-oxazaborolidines and has been corroborated by computational studies like those from Paddon-Row et al.,42 Paton,43 and Poplata et al.35 Notably, Paton stressed that the gem-diaryl moiety plays a crucial role in stereodifferentiation because it stabilizes the convex coordination of the substrate to the catalyst by attractive noncovalent interactions. However, there are instances in which a different transition state structure is favored because of other steric or electronic interactions.44 Therefore, detailed analysis for each individual reaction and substrate seems to be necessary. The role of aluminum bromide in the thermal catalysis of 1,3,2-oxazaborolidines was investigated through computational studies by Sakata and Fujimoto.45 They found that the coordination of the Lewis acid to the nitrogen atom of the oxazaborolidine is essential for its catalytic activity. Inherently, oxazaborolidine coordination to the substrate is disfavored due to destabilizing deformation. Upon activation with aluminum bromide, the B–N bond is polarized, which increases the Lewis acidity of the boron atom. Coordination of the substrate is then more energetically favorable.
Aluminum bromide also plays a significant role in the photochemical reactions through direct excitation with oxazaborolidine catalysts, as studied by Wang et al. (Figure 10).46,47 Based on calculations, they concluded that the heavy atoms of the AlBr3-activated oxazaborolidine catalyst facilitate spin–orbit coupling. This relativistic effect allows for efficient ISC, which is crucial for the successful enantioselective [2 + 2] photocycloaddition of coumarins and 5,6-dihydro-4-pyridones. In the case of 5,6-dihydro-4-pyridones (Figure 10a), after excitation to the 1nπ* state (λexc = 366 nm), the 1nπ* → 3ππ* transition is El-Sayed-allowed34 and exhibits high spin–orbit coupling. Therefore, the transition is fast, and once the 3ππ* state is reached, photocycloaddition is initiated. In the presence of an AlBr3-activated oxazaborolidine catalyst, the 1ππ* state is reached upon direct excitation. Even though the energy gap between 1ππ* and 3ππ* states is small, the 1ππ* → 3ππ* transition is El-Sayed-forbidden34 and slow. Here, the heavy atom effect increases the spin–orbit coupling and accelerates ISC. As the rate is still slower compared to the noncatalyzed pathway, the racemic background reaction must be suppressed by high catalyst loading.
Figure 10.
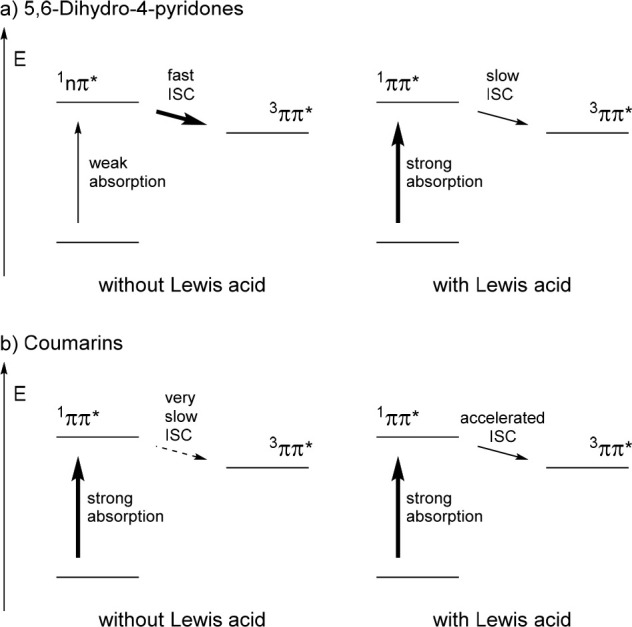
Qualitative, simplified energy diagrams highlighting calculated photophysical processes46,47 of (a) 5,6-dihydro-4-pyridones and (b) coumarins in the absence and presence of Lewis acid (energy levels not to scale).
In the case of the coumarins (Figure 10b), direct excitation to the 1ππ* was calculated to take place in both the uncatalyzed and catalyzed reaction.47 Because the 1ππ* → 3ππ* transition is slow, ISC is a minor pathway without catalyst, and the photocycloaddition reaction can, if at all, only follow a pathway on the singlet hypersurface. With the AlBr3-activated oxazaborolidine catalyst, spin–orbit coupling is again increased and leads to an accelerated ISC. A reaction channel on the triplet hypersurface is available, on which the photocycloaddition reaction occurs.
In summary, computational studies have corroborated the complex nature of 1,3,2-oxazaborolidines in both thermal and photochemical reactions. They have confirmed the role of a two-point binding mode including a nonclassical hydrogen bonding and have added nuances to our understanding of these reactions like the relativistic effect of aluminum bromide. Surely, further computational and mechanistic studies will enable scientists to advance the methodology of chiral 1,3,2-oxazaborolidine catalysis in photochemistry.
5. Conclusion
Throughout this Account, we have highlighted the variety and complexity of chiral 1,3,2-oxazaborolidines in photochemistry. As is often the case in synthetic chemistry, asymmetric synthesis is not a simple and straightforward endeavor. The mode of action of 1,3,2-oxazaborolidines transcends the standard substrate–catalyst binding and enantiotopic differentiation seen in many thermal reactions. Over the years, our understanding of their complex reactivity has increased through multiple studies, and it is now possible to transform a broad set of substrates into structurally intriguing products with excellent enantioselectivity. Continuous and elaborate scientific work has already paved the way, but it is surely not the end of the road for chiral 1,3,2-oxazaborolidine catalysts in enantioselective photochemical reactions. A major challenge is to overcome the still relatively high catalyst loadings for some of the reactions with possible solutions now being actively pursued. In addition, it needs to be explored whether Lewis acids can be meaningfully implemented in the production of chiral fine chemicals on a larger scale.
Acknowledgments
Our own research on this topic was generously supported by the European Research Council (ERC, Grant 665951-ELICOS), the Deutsche Forschungsgemeinschaft (DFG), the Alexander von Humboldt foundation, the Technical University of Munich, and the Fonds der Chemischen Industrie. T.B. gratefully acknowledges all colleagues and coworkers who have contributed to this project and who have been instrumental to its success. Their names are found in the reference section. It is due to their diligence, their dedication, and their intellectual input that this research topic has turned out to be so fruitful. D.P.S. is grateful to the Studienstiftung des Deutschen Volkes for a Ph.D. fellowship. The authors thank Camille Z. Rubel for her help in proof-reading the manuscript.
Biographies
Daniel P. Schwinger studied chemistry at the Technical University of Munich, where he obtained both his B.Sc. and M.Sc. During a research stay for his M.Sc. Thesis at the University of California, Berkeley with John F. Hartwig, he focused on the functionalization of aromatic C–H bonds. In 2019, he joined the group of Thorsten Bach and is currently working on enantioselective Lewis-acid-catalyzed photochemical reactions.
Thorsten Bach obtained his education at the University of Heidelberg and the University of Southern California, where he conducted his Diplom thesis with G. A. Olah. He received his Ph.D. in 1991 from the University of Marburg with M. T. Reetz and did postdoctoral work as a NATO fellow with D. A. Evans at Harvard University. He completed his Habilitation at the University of Münster in 1996, moved to the University of Marburg as an associate professor in 1997, and was appointed to the Chair of Organic Chemistry I at the Technische Universität München (TUM) in 2000. He has been an elected member of the German Academy of Sciences (Leopoldina) since 2006 and of the Bavarian Academy of Sciences since 2009.
The authors declare no competing financial interest.
References
- Brimioulle R.; Bauer A.; Bach T. Enantioselective Lewis Acid Catalysis in Intramolecular [2 + 2] Photocycloaddition Reactions: A Mechanistic Comparison between Representative Coumarin and Enone Substrates. J. Am. Chem. Soc. 2015, 137, 5170–5176. 10.1021/jacs.5b01740. [DOI] [PubMed] [Google Scholar]
- Poplata S.; Bach T. Enantioselective Intermolecular [2 + 2] Photocycloaddition Reaction of Cyclic Enones and Its Application in a Synthesis of (−)-Grandisol. J. Am. Chem. Soc. 2018, 140, 3228–3231. 10.1021/jacs.8b01011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegbauer S.; Jandl C.; Bach T. Enantioselective Lewis Acid Catalyzed ortho Photocycloaddition of Olefins to Phenanthrene-9-carboxaldehydes. Angew. Chem., Int. Ed. 2018, 57, 14593–14596. 10.1002/anie.201808919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverenz M.; Merten C.; Dreuw A.; Bach T. Lewis Acid Catalyzed Enantioselective Photochemical Rearrangements on the Singlet Potential Energy Surface. J. Am. Chem. Soc. 2019, 141, 20053–20057. 10.1021/jacs.9b12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara J. M.; Bowman C. M. Cyclic Benzeneboronate Esters. J. Am. Chem. Soc. 1958, 80, 2443–2446. 10.1021/ja01543a024. [DOI] [Google Scholar]
- Dewar M. J. S.; Kubba V. P. R.; Pettit R. New Heteroaromatic Compounds. Part II. Boron Compounds Isoconjugate with Indole, 2:3-Benzofuran, and Thionaphthen. J. Chem. Soc. 1958, 3076–3079. 10.1039/jr9580003076. [DOI] [Google Scholar]
- Pailer M.; Fenzl W. Borheterocyclen, 1. Monatsh. Chem. 1961, 92, 1294–1299. 10.1007/BF00914999. [DOI] [Google Scholar]
- Köster R.; Rotermund G. W. Borverbindungen, X1). Reaktionen von Organoboranen mit Chelatbildnern. Justus Liebigs Ann. Chem. 1965, 689, 40–64. 10.1002/jlac.19656890104. [DOI] [Google Scholar]
- Corey E. J.; Bakshi R. K.; Shibata S. Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. J. Am. Chem. Soc. 1987, 109, 5551–5553. 10.1021/ja00252a056. [DOI] [Google Scholar]
- Hirao A.; Itsuno S.; Nakahama S.; Yamazaki N. Asymmetric reduction of aromatic ketones with chiral alkoxy-amineborane complexes. J. Chem. Soc., Chem. Commun. 1981, 315. 10.1039/c39810000315. [DOI] [Google Scholar]
- Itsuno S.; Ito K.; Hirao A.; Nakahama S. Asymmetric reduction of aromatic ketones with the reagent prepared from (S)-(−)-2-amino-3-methyl-1,1-diphenylbutan-1-ol and borane. J. Chem. Soc., Chem. Commun. 1983, 469–470. 10.1039/C39830000469. [DOI] [Google Scholar]
- Itsuno S.; Hirao A.; Nakahama S.; Yamazaki N. Asymmetric synthesis using chirally modified borohydrides. Part 1. Enantioselective reduction of aromatic ketones with the reagent prepared from borane and (S)-valinol. J. Chem. Soc., Perkin Trans. 1 1983, 1673–1676. 10.1039/p19830001673. [DOI] [Google Scholar]
- Corey E. J.; Helal C. J. Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method. Angew. Chem., Int. Ed. 1998, 37, 1986–2012. . [DOI] [PubMed] [Google Scholar]
- Burkhardt E. R.; Matos K. Boron reagents in process chemistry: Excellent tools for selective reductions. Chem. Rev. 2006, 106, 2617–2650. 10.1021/cr0406918. [DOI] [PubMed] [Google Scholar]
- Cho B. T. Recent advances in the synthetic applications of the oxazaborolidine-mediated asymmetric reduction. Tetrahedron 2006, 62, 7621–7643. 10.1016/j.tet.2006.05.036. [DOI] [Google Scholar]
- Cho B. T. Recent development and improvement for boron hydride-based catalytic asymmetric reduction of unsymmetrical ketones. Chem. Soc. Rev. 2009, 38, 443–452. 10.1039/B811341F. [DOI] [PubMed] [Google Scholar]
- Shende V. S.; Singh P.; Bhanage B. M. Recent trends in organocatalyzed asymmetric reduction of prochiral ketones. Catal. Sci. Technol. 2018, 8, 955–969. 10.1039/C7CY02409F. [DOI] [Google Scholar]
- Corey E. J.; Shibata T.; Lee T. W. Asymmetric Diels–Alder reactions catalyzed by a triflic acid activated chiral oxazaborolidine. J. Am. Chem. Soc. 2002, 124, 3808–3809. 10.1021/ja025848x. [DOI] [PubMed] [Google Scholar]
- Futatsugi K.; Yamamoto H. Oxazaborolidine-derived Lewis acid assisted Lewis acid as a moisture-tolerant catalyst for enantioselective Diels–Alder reactions. Angew. Chem., Int. Ed. 2005, 44, 1484–1487. 10.1002/anie.200461319. [DOI] [PubMed] [Google Scholar]
- Liu D.; Canales E.; Corey E. J. Chiral oxazaborolidine–aluminum bromide complexes are unusually powerful and effective catalysts for enantioselective Diels–Alder reactions. J. Am. Chem. Soc. 2007, 129, 1498–1499. 10.1021/ja068637r. [DOI] [PubMed] [Google Scholar]
- Shim S. Y.; Ryu D. H. Enantioselective Carbonyl 1,2- or 1,4-Addition Reactions of Nucleophilic Silyl and Diazo Compounds Catalyzed by the Chiral Oxazaborolidinium Ion. Acc. Chem. Res. 2019, 52, 2349–2360. 10.1021/acs.accounts.9b00279. [DOI] [PubMed] [Google Scholar]
- Hughes D. L. Asymmetric Organocatalysis in Drug Development—Highlights of Recent Patent Literature. Org. Process Res. Dev. 2018, 22, 574–584. 10.1021/acs.oprd.8b00096. [DOI] [Google Scholar]
- Xu Y.; Conner M. L.; Brown M. K. Cyclobutane and cyclobutene synthesis: catalytic enantioselective [2 + 2] cycloadditions. Angew. Chem., Int. Ed. 2015, 54, 11918–11928. 10.1002/anie.201502815. [DOI] [PubMed] [Google Scholar]
- Corey E. J. Enantioselective catalysis based on cationic oxazaborolidines. Angew. Chem., Int. Ed. 2009, 48, 2100–2117. 10.1002/anie.200805374. [DOI] [PubMed] [Google Scholar]
- Brimioulle R.; Lenhart D.; Maturi M. M.; Bach T. Enantioselective Catalysis of Photochemical Reactions. Angew. Chem., Int. Ed. 2015, 54, 3872–3890. 10.1002/anie.201411409. [DOI] [PubMed] [Google Scholar]
- For a short review on the topic, see:Brenninger C.; Jolliffe J. D.; Bach T. Chromophor Activation of α,β-Unsaturated Carbonyl Compounds and its Application to Enantioselective Photochemical Reactions. Angew. Chem., Int. Ed. 2018, 57, 14338–14349. 10.1002/anie.201804006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis F. D.; Howard D. K.; Oxman J. D. Lewis acid catalysis of coumarin photodimerization. J. Am. Chem. Soc. 1983, 105, 3344–3345. 10.1021/ja00348a069. [DOI] [Google Scholar]
- Lewis F. D.; Barancyk S. V. Lewis acid catalysis of photochemical reactions. 8. Photodimerization and cross-cycloaddition of coumarin. J. Am. Chem. Soc. 1989, 111, 8653–8661. 10.1021/ja00205a015. [DOI] [Google Scholar]
- Guo H.; Herdtweck E.; Bach T. Enantioselective Lewis acid catalysis in intramolecular [2 + 2] photocycloaddition reactions of coumarins. Angew. Chem., Int. Ed. 2010, 49, 7782–7785. 10.1002/anie.201003619. [DOI] [PubMed] [Google Scholar]
- Ryu D. H.; Corey E. J. Triflimide activation of a chiral oxazaborolidine leads to a more general catalytic system for enantioselective Diels–Alder addition. J. Am. Chem. Soc. 2003, 125, 6388–6390. 10.1021/ja035393r. [DOI] [PubMed] [Google Scholar]
- Brimioulle R.; Guo H.; Bach T. Enantioselective intramolecular [2 + 2] photocycloaddition reactions of 4-substituted coumarins catalyzed by a chiral Lewis acid. Chem. - Eur. J. 2012, 18, 7552–7560. 10.1002/chem.201104032. [DOI] [PubMed] [Google Scholar]
- Brimioulle R.; Bach T. Enantioselective Lewis acid catalysis of intramolecular enone [2 + 2] photocycloaddition reactions. Science 2013, 342, 840–843. 10.1126/science.1244809. [DOI] [PubMed] [Google Scholar]
- Brimioulle R.; Bach T. [2 + 2] Photocycloaddition of 3-alkenyloxy-2-cycloalkenones: enantioselective Lewis acid catalysis and ring expansion. Angew. Chem., Int. Ed. 2014, 53, 12921–12924. 10.1002/anie.201407832. [DOI] [PubMed] [Google Scholar]
- El-Sayed M. A. Triplet state. Its radiative and nonradiative properties. Acc. Chem. Res. 1968, 1, 8–16. 10.1021/ar50001a002. [DOI] [Google Scholar]
- Poplata S.; Bauer A.; Storch G.; Bach T. Intramolecular [2 + 2] Photocycloaddition of Cyclic Enones: Selectivity Control by Lewis Acids and Mechanistic Implications. Chem. - Eur. J. 2019, 25, 8135–8148. 10.1002/chem.201901304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a related study, see:Stegbauer S.; Jeremias N.; Jandl C.; Bach T. Reversal of reaction type selectivity by Lewis acid coordination: The photocycloaddition of 1- and 2-naphthaldehyde. Chem. Sci. 2019, 10, 8566–8570. 10.1039/C9SC03315G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub M. E.; Jung H.; Lee B. J.; Won J.; Baik M.-H.; Yoon T. P. Enantioselective [2 + 2] Cycloadditions of Cinnamate Esters: Generalizing Lewis Acid Catalysis of Triplet Energy Transfer. J. Am. Chem. Soc. 2019, 141, 9543–9547. 10.1021/jacs.9b04643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skubi K. L.; Blum T. R.; Yoon T. P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D. H.; Lee T. W.; Corey E. J. Broad-spectrum enantioselective Diels–Alder catalysis by chiral, cationic oxazaborolidines. J. Am. Chem. Soc. 2002, 124, 9992–9993. 10.1021/ja027468h. [DOI] [PubMed] [Google Scholar]
- Wiest J. M.; Conner M. L.; Brown M. K. Allenoates in Enantioselective [2 + 2] Cycloadditions: From a Mechanistic Curiosity to a Stereospecific Transformation. J. Am. Chem. Soc. 2018, 140, 15943–15949. 10.1021/jacs.8b10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston R. C.; Cheong P. H.-Y. C-H···O non-classical hydrogen bonding in the stereomechanics of organic transformations: theory and recognition. Org. Biomol. Chem. 2013, 11, 5057–5064. 10.1039/c3ob40828k. [DOI] [PubMed] [Google Scholar]
- Paddon-Row M. N.; Anderson C. D.; Houk K. N. Computational evaluation of enantioselective Diels–Alder reactions mediated by Corey’s cationic oxazaborolidine catalysts. J. Org. Chem. 2009, 74, 861–868. 10.1021/jo802323p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton R. S. Dissecting non-covalent interactions in oxazaborolidinium catalyzed cycloadditions of maleimides. Org. Biomol. Chem. 2014, 12, 1717–1720. 10.1039/c4ob00009a. [DOI] [PubMed] [Google Scholar]
- Paddon-Row M. N.; Kwan L. C. H.; Willis A. C.; Sherburn M. S. Enantioselective oxazaborolidinium-catalyzed Diels-Alder reactions without CH···O hydrogen bonding. Angew. Chem., Int. Ed. 2008, 47, 7013–7017. 10.1002/anie.200802002. [DOI] [PubMed] [Google Scholar]
- Sakata K.; Fujimoto H. Quantum chemical study of Diels–Alder reactions catalyzed by Lewis acid activated oxazaborolidines. J. Org. Chem. 2013, 78, 3095–3103. 10.1021/jo400066h. [DOI] [PubMed] [Google Scholar]
- Wang H.; Cao X.; Chen X.; Fang W.; Dolg M. Regulatory Mechanism of the Enantioselective Intramolecular Enone [2 + 2] Photocycloaddition Reaction Mediated by a Chiral Lewis Acid Catalyst Containing Heavy Atoms. Angew. Chem., Int. Ed. 2015, 54, 14295–14298. 10.1002/anie.201505931. [DOI] [PubMed] [Google Scholar]
- Wang H.; Fang W.-H.; Chen X. Mechanism of the Enantioselective Intramolecular [2 + 2] Photocycloaddition Reaction of Coumarin Catalyzed by a Chiral Lewis Acid: Comparison with Enone Substrates. J. Org. Chem. 2016, 81, 7093–7101. 10.1021/acs.joc.6b00980. [DOI] [PubMed] [Google Scholar]