

Abstract
Transient assembled structures play an indispensable role in a wide variety of processes fundamental to living organisms including cellular transport, cell motility, and proliferation. Typically, the formation of these transient structures is driven by the consumption of molecular fuels via dissipative reaction networks. In these networks, building blocks are converted from inactive precursor states to active (assembling) states by (a set of) irreversible chemical reactions. Since the activated state is intrinsically unstable and can be maintained only in the presence of sufficient fuel, fuel depletion results in the spontaneous disintegration of the formed superstructures. Consequently, the properties and behavior of these assembled structures are governed by the kinetics of fuel consumption rather than by their thermodynamic stability. This fuel dependency endows biological systems with unprecedented spatiotemporal adaptability and inherent self-healing capabilities. Fascinated by these unique material characteristics, coupling the assembly behavior to molecular fuel or light-driven reaction networks was recently implemented in synthetic (supra)molecular systems. In this invited feature article, we discuss recent studies demonstrating that dissipative assembly is not limited to the molecular world but can also be translated to building blocks of colloidal dimensions. We highlight crucial guiding principles for the successful design of dissipative colloidal systems and illustrate these with the current state of the art. Finally, we present our vision on the future of the field and how marrying nonequilibrium self-assembly with the functional properties associated with colloidal building blocks presents a promising route for the development of next-generation materials.
Equilibrium versus Dissipative Assembly: Familiar versus the Unknown
Over the past few decades, the self-assembly of synthetic (molecular or colloidal) building blocks has proven to be an extremely powerful concept in fabricating materials with controllable (internal) structures in a bottom-up fashion. Typically, synthetic self-assembling systems are designed on the basis of thermodynamic principles. This implies that the targeted assembly resides in the global minimum of the free-energy landscape which is determined by thermodynamic parameters such as temperature and pressure. Naturally, this chemical thermodynamic definition of the free-energy minimum is operational and does indirectly imply that the (molecular) reactants and products do not decay into their stable chemical elements. Because the state associated with nonassembled building blocks is higher in free energy, the system will spontaneously evolve toward the assembled state (Figure 1a, left). The final ratio of assembled and nonassembled building blocks is thermodynamically determined by their free-energy difference, while the rates of structure formation and disintegration are governed by the energy barrier that connects the energy levels of the disassembled and assembled states. Once the assembled state is reached, the structure is rather insensitive to external thermal (or other) fluctuations and hence is considered to be stable. Structural stability does not imply that the superstructures are static. In a dynamic equilibrium, there is an exchange of building blocks between the assembled and nonassembled states, but the rates of exchange are equal. Consequently, no time-dependent structural evolution occurs. As the thermodynamic principles underlying assembly are (largely) understood and we are used to thinking about equilibrium processes, the design principles for equilibrium self-assembly are well-established. This resulted in an impressive set of self-assembled materials with controllable properties and/or functions.1−5
Figure 1.

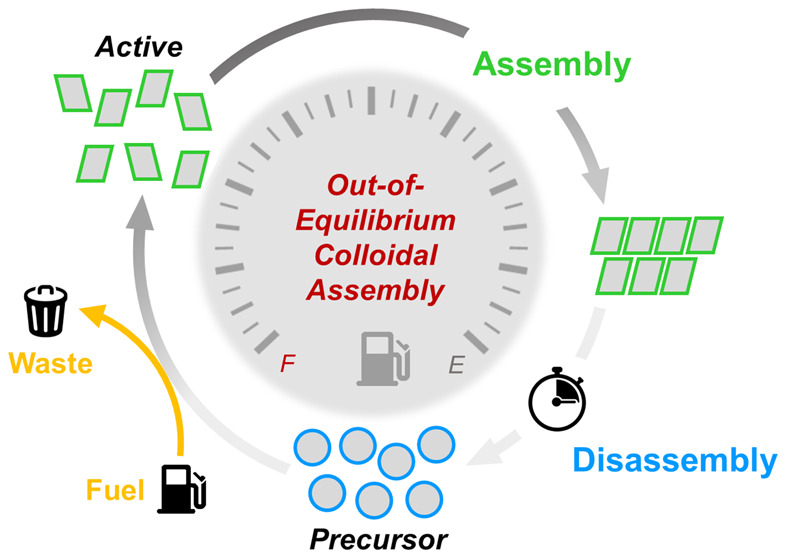
Pictorial representation of (a) equilibrium self-assembly (left, red) versus (b) dissipative assembly (right, blue/green). In equilibrium self-assembly, the assembled structure resides in a global minimum of the free-energy landscape. The assembled structure is therefore the thermodynamically favored state of the system, and a collection of individual building blocks spontaneously evolves to the assembled state (the rate of assembly is faster compared to disassembly). For dissipative assembly, building blocks in the precursor state (blue circles) are first promoted to their activated state (green rectangles) after fuel (yellow) consumption. Once in the activated state, assembly can commence. The assembled structure does not exist in a global minimum of the free-energy landscape and is therefore metastable. Once fuel is depleted, the activated state of the building blocks cannot be sustained, and the structure spontaneously disintegrates, thereby returning to its equilibrium state (well-dispersed precursors). At some point in the dissipative assembly cycle, energy is dissipated, and waste products are formed. In principle, waste formation can occur directly after fuel consumption or during the disassembly of the nonequilibrium superstructure.
However, there is more to life than equilibrium. Superstructures and materials can also exist out of equilibrium.6−8 Instead of reaching the thermodynamically favored global minimum in the free-energy landscape, systems can get trapped in local shallow minima giving rise to metastable states. In principle, these structures have a finite lifetime which depends on the height of the energy barriers surrounding the local energy minimum in which the structure resides. The possibility of structural evolution does imply that these systems are not in equilibrium. Typically, these metastable states can be found at energy levels slightly higher than the global minimum. A comprehensive review on metastable materials was recently published by Sorrenti et al.9 In this invited feature article, we focus on structures that form higher in the free-energy landscape. In contrast to their equilibrium or metastable analogues, these out-of-equilibrium systems rely on a net exchange of matter and energy with the environment to exist and are therefore typically referred to as dissipative systems. Only by a continuous consumption of energy (or matter), these out-of-equilibrium assembled structures can escape their thermodynamic fate and prevent degradation into equilibrium states. In fact, nonequilibrium systems in which energy is dissipated are intrinsic to life and the structures it comprises. The dependency on an influx of energy endows them with the capability to reconfigure, adapt, amplify signals, self-replicate, and self-heal in a highly dynamic fashion. Archetypical examples include microtubules and actin filaments, where energy-controlled assembly underlies cellular transport, cell motility, and proliferation.10−14
When inspecting these highly complex biological examples from a coarse-grained, physicochemical perspective, one will find that the assembly of these systems is regulated by direct coupling to an energy-providing reaction network.15−17 The connection between a reaction network and assembly is schematically depicted in Figure 1b. In equilibrium, the building blocks are in a nonassembling precursor state (Figure 1b, blue circles) and feel no thermodynamic propensity to undergo assembly. Upon reaction with an energy-rich molecule, typically referred to as a fuel, the precursors are activated to a higher free-energy state (Figure 1b, green rectangles). While in this activated state, the building blocks can undergo assembly, and analogous to the equilibrium case, this structuring is driven by a decrease in the total free-energy of the system. Crucial to employing chemical reaction networks to drive out-of-equilibrium assembly processes is a deactivation reaction that converts the activated building blocks back to their precursor state. During deactivation, the building blocks dissipate the energy they gained during the activation step into heat or waste products. Once deactivated, the assembly is no longer stable and spontaneously disintegrates into a collection of well-dispersed building blocks.
In this conventional example of dissipative systems, the building blocks are actively involved in the fuel-to-waste conversion. In principle, this is not required to drive a system away from equilibrium. For example, (molecular) fuels can take the role of transient templating agents18−21 or use their energy to alter solvent quality or ionic strength in a spatiotemporal manner to induce the assembly of building blocks.22,23 Systems that use fuel in this fashion are typically referred to as operating under dissipative conditions (Definitions and Terminology). Regardless of the exact nature of the underlying reaction network, the out-of-equilibrium superstructures can exist only in the presence of sufficient fuel and are therefore thermodynamically labile. When adding fuel in finite-sized quantities, the assembled state can be sustained as long as it takes for the system to consume the fuel. In the case where fuel is continuously supplied, an out-of-equilibrium steady state may form, governed by reaction-diffusion networks with emergent phenomena such as oscillations and bifurcations.7,24−28
Importantly, the (de)activation reactions underlying dissipative assembly have to be irreversible and are noninteracting or occur at sufficiently different rates. (Figure 1b, yellow and black arrows highlighting the fuel and waste symbols). Only when these criteria are met can energy be stored and dissipated, and structures can form in an out-of-equilibrium fashion. Processes underlying the conversion of the building blocks from their precursor state to the activated state are not subjected to any specific requirements and can include reactions involving the making or breaking of covalent bonds, supramolecular coordination processes, or conformational changes.38
In sharp contrast to equilibrium self-assembly where thermodynamics rules the final outcome of structural evolution, the assembly behavior of these out-of-equilibrium systems is governed by the kinetics of the (de)activation reactions.16 The direct coupling between reaction networks and assembly endows these systems with unique properties and facilitates spatiotemporal control by regulating the availability of the (chemical) fuel required for building block activation. Only at locations where sufficient fuel is present will assembly occur, and the lifetime of the dissipative state is directly linked to the ratio of the activation (fuel consumption) and deactivation rates. The formed superstructures are highly dependent on fluctuations in fuel (gradients) and are therefore capable of adapting to their environment by either growing, disintegrating, or a combination of both.39 This adaptability can also lead to truly self-healing properties.39−45 When the superstructures are damaged but the underlying reaction network remains intact, the system can autonomously recover itself and use available energy to prevent fatigue. At this point, we stress that equilibrium structures can in principle also respond and adapt to imposed stimuli. However, these triggered assembly processes are fundamentally different from dissipative assembly as no energy is stored in the system (Definitions and Terminology).
Finally, the transient nature of the assembled structures on the nanoscale can propagate across length scales to form dynamic patterns. Typically, feedback loops are required where the system influences its own or other chemical reaction networks to generate cascading and amplifying signals.46
Sparked by the unique properties of biological out-of-equilibrium systems, the quest for synthetic analogues that emulate these at a conceptual level to create emergent life-like behavior has attracted significant interest the last couple of years and had a distinct impact on the field of systems chemistry.17,47,48 The first examples included primarily supramolecular systems that form transient gels, vesicles (with supramolecular pores), micelles, or polymers using chemical fuels18−20,40,49−53 or light54−60 as an energy source. Various excellent review articles discussing these emergent materials appeared recently.16,38,39,61−64
The strategies applied to translate biologically inspired design criteria into synthetic molecular materials provide valuable input for translating key concepts of out-of-equilibrium assembly to the colloidal domain. As for equilibrium assembly, organizing building blocks of colloidal dimensions into (ordered) structures gives rise to materials with unique photonic, catalytic, magnetic, and mechanical properties originating not only from the positional order of the building blocks within the assembly but also from the chemical nature of the particle bulk (metallic, semiconductor, polymeric, inorganic, and minerals).65−68 Notable examples include the use of colloidal building blocks to fabricate optical (band gap) and electronic materials,69−71 lithographic masks,72,73 structural colorants,74−76 and catalytic scaffolds with tunable activity.77 Being able to marry these properties with the adaptability, responsiveness, and ability to self-heal and amplify signals in a spatiotemporal fashion will undoubtedly lead to new classes of dynamic functional materials. Additionally, colloids can serve as model systems to elucidate the physical concepts underlying (biological) out-of-equilibrium assembly, similar to the analogy between colloidal particles and atoms proposed more than a century ago.78
In this invited feature article, we provide an overview of experimental (synthetic) strategies applied to fabricate systems which display the dissipative assembly of colloidal building blocks. We will address the state of the art and present our vision on future applications and challenges within the field of reaction-network-driven colloidal assembly. Although this review is limited to systems coupled to reaction networks, we would like to emphasize that other fascinating out-of-equilibrium colloidal assembly routes have been reported. First, colloids can assemble into so-called arrested, glassy, or gel states. These amorphous, kinetically arrested states may have an (extremely) long lifetime and impede further structural evolution into a (desired) equilibrium structure. Although not ordered, the percolating colloidal structures endow these materials with novel mechanical load-bearing characteristics and potential use in conductive materials and photovoltaics. For an excellent description of the theoretical and experimental status of this field, we refer to the work of Sciortino, Foffi, and Dawson et al.79−82 Second, dynamic examples of out-of-equilibrium colloidal materials include structures that are formed under the influence of external magnetic or electric fields. Since these systems rely on field-induced polarization or magnetization for assembly, removal of the external fields will eliminate the driving force for assembly. Consequently, the system will relax back to its equilibrium state with well-dispersed individual building blocks. For a recent review of the current status of field-guided colloidal assembly, we refer the reader to a recent review of Liljeström et al.83 Third, suspensions of actively swimming colloids are inherently out of equilibrium. Directed (auto)phoretic motions are generated via the establishment of local molecular gradients.84,85 These gradients can be formed by the slow dissolution of colloids/colloidal droplets86−89 or by using Janus particles where only one face of the particle catalyzes a decomposition reaction90−94 or induces the local demixing of a binary dispersing medium driven by local heating.95 The nonequilibrium phoretic movements that these active colloids display can lead to higher-order structuring by analogy to the flocking of (micro)organisms.91,95−100
Definitions and Terminology
To emphasize the boundaries of this review and avoid confusing terminology within the multidisciplinary field of dissipative and out-of-equilibrium assembly, separate classes of (self-)assembly are defined. The definitions of dissipative systems are adapted from Ragazzon and Prins.16 Dissipative systems and systems operating under dissipative conditions using colloidal particles as primary building blocks will be discussed in this invited feature article.
Equilibrium Assembly
This is a self-assembly process in which the final outcome is governed by thermodynamic principles. Structure formation is driven by a lowering of the free-energy of the system. No fuel or (external) stimuli are required to drive structure formation. Examples include the crystallization of hard colloidal particles into close-packed crystals29 or the formation of micelles from amphiphilic building blocks.30,31
Triggered Assembly
This is an assembly process initiated by imposed triggers, e.g., changes in pH, temperature, or solvent quality.32 These systems in their final state cannot be classified as out-of-equilibrium, as the external trigger simply alters the free-energy landscape, forcing the assembly to evolve to a newly created free-energy minimum. Hence, returning to the initial state is not spontaneous, and a second trigger is required. In these processes, no energy is stored or dissipated. Notable examples include the assembly of colloids functionalized with sticky DNA,33,34 thermoresponsive polymers,35 or hydrogen-bonding tectons.36,37
Dissipative Assembly
This is an assembly process where energy is consumed via the conversion of a fuel through active participation of the building blocks. A part of this energy is used to drive assembly (stored energy), while a fraction of the energy is dissipated by waste formation. Assembled structures exist only in the presence of (sufficient) fuel and are therefore labeled as out-of-equilibrium. Upon fuel depletion and the absence of any substantial non-fuel-dependent attractive forces in the assembled state, the system evolves spontaneously back to its equilibrium state. (See Table 1 for examples.)
Table 1. Overview of Colloidal Systems Coupled to Reaction Networks.
| colloidal system | particle diameter (nm) | fuel | fuel-induced transformation | system classification | notes/applications | ref |
|---|---|---|---|---|---|---|
| gold/silver | 5 | UV light | azobenzene isomerization | dissipative | self-erasing images, temporary data storage | (110) |
| gold | 5 | UV light | azobenzene isomerization | dissipative | switchable catalytic activity | (111) |
| gold, silica, Fe3O4 | 6–20 | UV light | azobenzene isomerization | dissipative | capture and catalysis of organic compounds | (112) |
| Fe3O4 | 10 | UV light | azobenzene isomerization | dissipative | capture and transport of colloidal cargo | (126) |
| pSta | 5100 | UV light | azobenzene isomerization | dissipative control | reversible formation of 2D colloidal crystals | (23) |
| silica | 150 | UV light | spiropyran isomerization | dissipative | (128, 129) | |
| pMMA grafted silicab | 280–920 | UV light | spiropyran isomerization | dissipative | no spontaneous redispersion | (120) |
| gold | 6–10 | UV light | H+ generation via spiropyran isomerization | dissipative control | (22) | |
| gold | 6–12 | blue light | H+ generation via spiropyran isomerization | dissipative control | entrapment of organic compounds and self-erasing images | (114) |
| p(NIPAM-b-MMA) grafted pStc | 750 | DMSe | methylation of carboxylic acids | dissipative | (118) | |
| SiNCsd | 6 | EDCf | esterification of carboxylic acids | dissipative | delayed uptake by mammalian cells | (116) |
| gold, Fe3O4 | 4–15 | EDC | esterification of carboxylic acids | dissipative | experimental confirmation of pathway complexity | (115) |
| gold | 4 | estersg | H+ generation via ester hydrolysis | dissipative control | transient pH regulated by a combination of fast promotors and dormant deactivators | (117) |
| DNA-grafted pSt | 1000 | ATP | enzyme-mediated RNA production | dissipative control | transient clustering via enzymatic RNA production and degradation | (119) |
pSt = polystyrene.
pMMA = poly(methyl methacrylate).
p(NIPAM-b-MMA) = block copolymer of N-isopropylacrylamide and methyl methacrylate.
SiNCs = silicon nanocrystals.
DMS = dimethyl sulfate.
EDC = 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide.
Gluconic acid δ-lactone, methyl formate, β-butyrolactone, or ε-caprolactone.
Assembly under Dissipative Conditions
This is analogous to dissipative assembly, but the building blocks do not actively participate in the fuel-to-waste conversion. (See Table 1 for examples.)
Coupling Colloidal Assembly to Reaction Networks: Design Considerations
Conceptually, colloidal building blocks that assemble in a dissipative fashion are larger analogues to their supramolecular or biological counterparts. The underlying reaction network governs (de)activation of the building blocks, and assembly commences only when the concentration of building blocks in the activated state is sufficient. To rationally design colloidal out-of-equilibrium systems, a detailed understanding and a precise modulation of the (surface) forces that govern intercolloidal interactions are necessary. The effective potential (U) acting between colloidal objects is determined by an interplay between (surface) forces with varying strengths and ranges (Figure 2).101−104 An omnipresent source of attractive contributions to the interparticle potential are van der Waals forces (Figure 2, gray striped curve). These forces originate from a mismatch in polarizability between the colloidal particles and the surrounding medium and are strongly attractive at relatively short interparticle separations. van der Waals forces are significantly stronger for colloidal objects compared to molecular counterparts because the force scales with the particle size. To counteract these attractive forces and prevent the irreversible aggregation of colloidal particles, one can rely on steric and/or electrostatic stabilization (Figure 2, purple dotted curve) to generate an energy barrier that restricts the minimal interparticle distance (Figure 2, yellow circles).103
Figure 2.

Schematic representation of the net interparticle potentials (U) acting between (a) the particles in the precursor state (left, blue) and (b) the fuel-induced activated state (right, green or red). The net potentials are a sum of repulsive (dotted purple) and attractive (stripped gray) contributions. For stable dispersions, the repulsive contributions generate a potential energy barrier, preventing the colloids from accessing the primary potential energy minimum. Entering this minimum, where the interparticle potential is highly dominated by attractive van der Waals contributions, will result in an irreversible loss of colloidal stability. In the precursor state, the dominating repulsive contributions (highlighted by the yellow circle) at moderate interparticle distances (d) prevent colloids from assembling. In the activated state, the diminished repulsive contribution provides favorable conditions for assembly (top, green curve) as manifested by the formation of a secondary potential minimum. However, when the building blocks are activated to such an extent that the building blocks become too attractive, disordered and irreversible clusters are formed (bottom, red curve). Note that altering the range and absolute magnitude of the repulsive contributions can also be achieved by increasing the attraction strength between the particles.
For steric stabilization, the surface of the particles is decorated with a (dense) layer of small molecules or polymers. When these surfaces approach each other and the outer segments of the (polymeric) ligands start to deform and/or overlap, a disjoining force is generated. This force is essentially osmotic in nature and originates from the unfavorable entropy associated with compressing or confining the chains in between the surfaces.101,105 For electrostatic stabilization, the particles carry a net surface charge due to the dissociation of acidic or basic surface-tethered groups, isomorphic substitution, or the (covalent) attachment of ionic species. To ensure charge neutrality, the colloids are surrounded by an electrochemical double layer in which the concentration of counterions is higher than the concentration found in the bulk. When two charged colloidal objects approach, partial overlap of these double layers generates a repulsive osmotic force that drives the particles apart from each other again. By controlling the ionic strength of the medium, the significance of the electrostatic repulsion can be modulated both in strength and in range.102 In addition to van der Waals forces and steric and electrostatic repulsion, other contributions, e.g., hydrophobic,106 depletion,107 and Casimir forces,108 might have to be taken into account when considering the net interparticle potential.
Evidently, electrostatic and/or steric forces are required to safeguard colloidal stability prior to fuel-induced assembly. For subtle, fuel-induced changes to induce assembly, the overall interaction potential acting between the precursor building blocks should be sufficiently shallow at interparticle distances relevant to the assembled structure to generate a secondary potential energy minimum. In the deactivated state, repulsions should dominate the interparticle potentials to prevent clustering, while these repulsive contributions should be sufficiently weak in order for the fuel-induced attractions to overcome them once in the activated state (Figure 2b, top, green curve). Accessing the primary potential energy minimum in the activated state should be prevented, as the large attractive strength associated with the minimum will irrevocably result in an irreversible loss of colloidal stability (Figure 2b, red). Moreover, for the transient assembly processes considered here, limiting the magnitude of the attractive contributions in the activated state is necessary to promote spontaneous disintegration of the colloidal clusters after fuel depletion. Additionally, generating mildly attractive intercolloidal forces is pivotal when targeting assembled structures with high structural order and regularity. Only when the attractions are weak enough can particles reconfigure during assembly, ensuring that the building blocks find their optimum position within the growing structure. If attractions are too strong, then fractal(-like) aggregates are formed. In these clusters, the relative particle configurations are determined by the statistical diffusive processes that cause particles to find each other (Figure 2b, bottom, red curve). Because of the high attraction strength, particles irreversible stick to each other and no restructuring or reversibility is possible. Already for equilibrium self-assembling systems, fine-tuning of the potentials in terms of strength and range is a challenging task. Introducing the time-dependency of the potentials as additional design criterion increases the complexity of building block design even further.
In addition to interbuilding block forces that come into play when working with colloidal objects, the dimensions of the building blocks impose restrictions on the kinetics of the reaction networks that can be used for assembly. Compared to their molecular counterparts, the dynamics of colloids are significantly slower. The kinetics of the (de)activation reactions have to be matched to these slower dynamics in order to generate sufficiently high concentrations of activated building blocks and provide enough time for these activated colloids to find each other via diffusive processes. Naturally, this consideration becomes increasingly more important when larger colloids are used. Because of this reason, the vast majority of the dissipative colloidal systems described to date rely on relatively small (5–50 nm) nanoparticle-sized building blocks,22,32,109−117 while studies on (nearly) micrometer-sized particles are sparse.23,118−120
The dimensions of the colloidal particles can also impact the dynamics of the dissipative assembly when compared to their molecular counterparts. In fuel-driven molecular systems, (dis)assembly can be induced by (de)activating only a limited number of fuel-responsive moieties. The corresponding assembly process can therefore be a single-molecule event. This molecular control is, for example, elegantly used in actin tread milling.13,121 When using colloidal particles as elementary building blocks, this single-molecule character is lost. Colloids typically carry significantly more active sites than their molecular analogues. A fraction of these sites needs to be activated in order to affect the effective intercolloidal potentials and induce assembly. Similarly, for spontaneous disassembly sufficient sites have to be deactivated such that a threshold is crossed, after which the colloids are no longer attractive toward each other. This restriction limits the attainable dynamic behavior for colloidal particles unless, for example, very sharp fuel gradients, deactivation conditions, or fuel-mediated superselectivity122 can be realized.
Finally, as for molecular systems, the stability of colloidal systems sensitively depends on external conditions, e.g., salt concentration, pH, temperature, and solvent quality.101 Although these parameters can be used as experimental knobs to tune the interparticle potentials (in a transient fashion), this sensitivity introduces stringent limits on the types of reaction networks that can be used.
Through careful design and optimization of colloidal building blocks and associated reaction networks, the challenges underlying dissipative colloidal systems could be addressed, resulting in a number of elegant and promising dissipative colloidal systems. A wide variety of synthesis approaches were taken to imprint transient behavior to colloidal building blocks, highlighting the universality of the underlying kinetic concepts and the prospect of myriad future systems. In general, the systems reported so far can be divided according to the energy source used to fuel the reaction networks underlying particle assembly. Here, we differentiate between light- and chemical-fuel-driven systems. An overview of illustrative examples discussed in this invited feature article is given in Table 1.
Light-Mediated Out-of-Equilibrium Assembly
The use of light as an energy source has proven to be an attractive strategy to guide the self-assembly of colloidal systems in and out of equilibrium.37,109,123−126 Light can be switched on and off instantaneously and delivered with high spatial resolution. This facilitates running a large number of dissipative cycles without signs of fatigue or loss of reversibility. Typically, light sensitivity is imprinted on colloids by decorating the surface of (nano)particles with molecular switches that can undergo reversible isomerization reactions to alter their physical properties. On the basis of well-developed surface functionalization protocols, ligands containing the switches can be immobilized on the particles. Frequently employed switches are azobenzene- and spiropyran-based. In contrast to the use of molecular fuels (Fuel-Mediated Out-of-Equilibrium Assembly section), typically no waste products accumulate in light-driven systems as they rely on reversible conformational changes in molecular switches or the cyclic formation and consumption of protons (photoacids). Illustrative examples of incorporating molecular switches into reaction networks to drive colloidal assembly will be discussed below.
Azobenzene-Guided Colloidal Systems
Under the influence of UV light, surface-immobilized azobenzene-containing ligands can switch from their thermodynamically stable trans isomer to a metastable cis conformation. This conformational change is accompanied by generating a large dipole moment and hence an increase in hydrophilicity. Upon removal of the UV stimulus, the cis isomer has a limited lifetime under ambient conditions and will spontaneously convert back to the thermodynamically stable trans isomer. Effectively, particles with a transiently enhanced hydrophilicity are therefore obtained. By dispersing the colloidal objects in apolar environments, either solvents or organogels, this time-dependent hydrophilicity proved to be sufficient to drive the clustering of (nano)particles. In these particular cases, the UV light acts as an energy source that converts the trans-azobenzene-functionalized particles (precursor state) to the activated cis-azobenzene-functionalized particles. The energy stored by the systems is dissipated upon thermal relaxation of the azobenzene moieties.
Klajn et al. used this concept to prepare materials in which both the writing and self-erasure of images are controlled by the nonequilibrium aggregation of azobenzene-functionalized gold or silver nanoparticles (Figure 3).110 The generation of colored images with high spatial resolution was possible by irradiating a nanoparticle-containing gel through a photomask with UV light. In the illuminated areas, particle clustering commenced. By relying on the fact that the surface plasmon resonance of the metallic nanoparticles is cluster-size-dependent, a variety of colors could be generated. Due to the metastable character of these clusters, the images gradually self-erase. The erasure time could be controlled by the azobenzene ligand density and the fraction of these ligands in the cis conformation, which was in turn determined by the intensity of the UV light and illumination time. Multiple images could be written onto the same substrate either concurrently or after self-erasure. The smallest features that could be resolved were on the order of 10 μm. Within such small features, NPs diffuse into and out of the UV-irradiated regions on time scales comparable to typical irradiation times, leading to blurring or an inability to generate the desired feature. Improving the resolution by increasing the initial particle concentration or using higher irradiation powers could further facilitate the application of these transient images for storing sensitive information.
Figure 3.
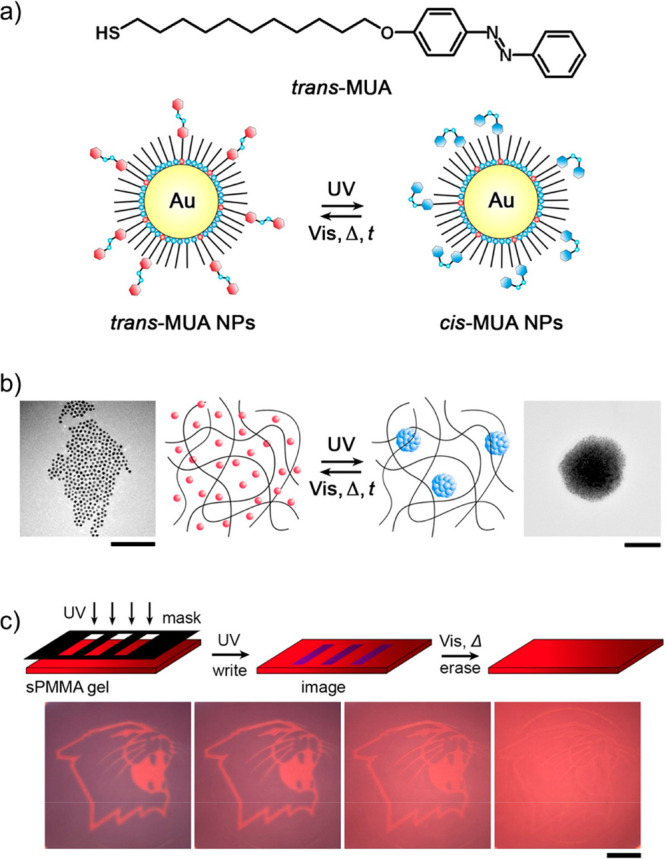
(a) Chemical structure of azobenzene-functionalized thiolated ligand (MUA). The azobenzene moiety is drawn in its thermodynamically stable trans conformation. Incorporating this functional thiol in the ligand corona on gold nanoparticles yields UV-responsive colloids able to reversibly convert the azobenzene fragments from their trans to cis conformations (trans/cis-MUA NPs). (b) Reversible aggregation of the colloids depicted in panel a under the influence of UV illumination in an organogel (scale bar on electron microscopy images = 100 nm). UV illumination forces the azobenzene moieties into the metastable cis conformation. The significant dipole moment associated with the cis conformation drives clustering in the apolar reaction environment. The cis conformation slowly reverts back to the thermodynamically stable trans form by thermal relaxation, resulting in cluster disintegration. (c) Writing self-erasable images by UV illumination through a mask. Only in the illuminated regions does clustering commence, leading to a red shift in the plasmon resonance. Macroscopically, this shift is observed as a transition from red (single particles) to blue (cluster). Since the clusters are transient, the images erase themselves (scale bar for all images = 1 cm). Adapted from ref (110). Copyright 2009, John Wiley and Sons.
An analogous approach was used by Grzybowski and co-workers to control the catalytic activity of nanoparticles.111 Also in this example gold nanoparticles were functionalized with thiolated azobenzene-containing ligands to transiently induce clusters upon UV illumination. In addition to the switchable ligands, the nanoparticle surface was decorated with mobile amine-containing ligands that allow for the gold surfaces to be accessible to molecules dissolved in the continuous phase. As the metallic surface is catalytically active for hydrosilylation reactions, the freely dispersed particles (equilibrium) show the conversion of model substrates with reasonable reaction rates. After UV-induced clustering (transient assembly), the catalytic activity decreased significantly due to a reduction in the available catalytic surface. Activity could be regained after thermal relaxation and hence redispersion of the catalytic colloids. Although the results are a neat demonstration of controlling catalytic activity in a temporal fashion, the number of photoswitchable catalytic cycles proved to be limited. Binding competition between the originally used surface ligands to cap the gold particles and employed hydrosilylation reactants prevented the complete switching off of the catalytic activity in the aggregated state.
An extension of using light-responsive nanoparticles to transiently control catalytic activity was reported by Klajn and co-workers.112 Instead of relying on the catalytic action of the particle surface, enhanced reaction kinetics for a variety of chemical transformations were driven by the nanoconfinement of the participating reactants (Figure 4a). Under appropriate conditions and UV illumination, azobenzene-functionalized particles (gold, silica, Fe3O4) assembled into ordered supracrystals containing well-defined tetrahedral interstitial cavities, in this work referred to as nanoflasks (Figure 4b,c). Polar additives present in the apolar dispersing medium could be preferentially captured during the formation of the supracrystals since the azobenzene ligands were forced by the UV light into the more polar cis conformation. Trapping efficiencies could be further enhanced by introducing ligands carrying specific recognition motifs for targeted substrates into the azobenzene-containing monolayer. Trapping increased the local concentration of reactants which translated into faster conversion. In addition to faster kinetics, it was neatly shown that the decoration of the particles with chiral ligands provides a means to control the stereochemistry of the products formed in the nanoflasks.
Figure 4.
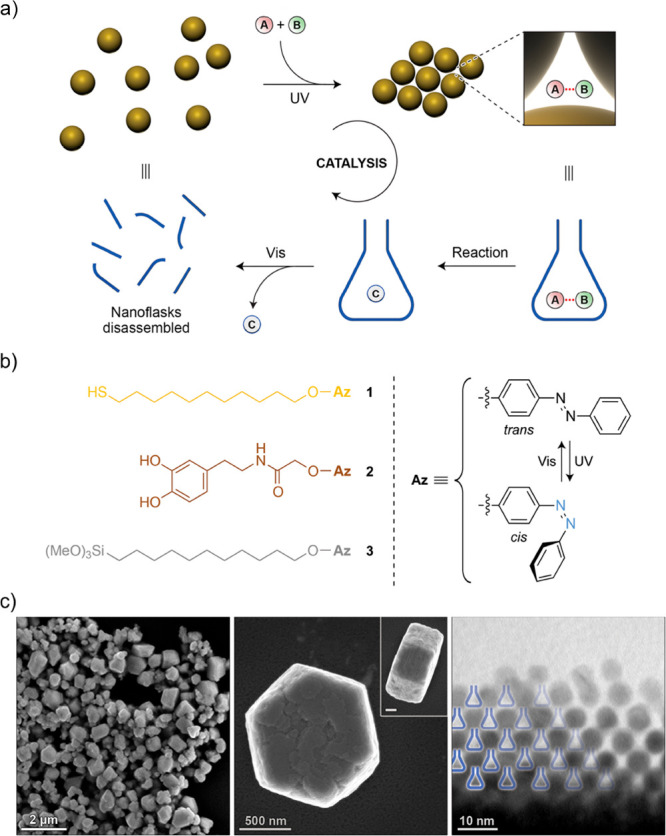
(a) Schematic representation of the reversible formation of nanoparticle-based crystals with confined interstitial voids (nanoflasks) in which polar reactants A and B are captured and subsequently coupled to form product C. The confined reaction volume created by the interstitial spaces enhances the local concentration of reactants and results in higher reaction rates. Crystals are transiently formed in apolar media under the influence of UV light, which forces the azobenzene-containing ligands into their more polar cis conformation (b, right). Thermal relaxation of the ligands to the trans conformation disintegrates the nanoflasks and liberates product C. (b) Employment of azobenzene-containing ligands to decorate gold (yellow), iron oxide (brown), and silica (gray) colloids. Nanoflasks could be generated for all three types of colloids. (c) Electron micrographs (at different magnifications) of the transient colloidal crystals with the nanoflasks highlighted in blue (right). Adapted from ref (112). Copyright 2015, Nature Publishing Group.
As the interparticle forces underlying nanoflask formation are transient, the reaction products could be easily released into the dispersing medium and the kinetics could be controlled in a temporal fashion. The authors speculate that the confined environments could be exploited to perform polymerizations of trapped monomers or enantioselective reactions, where the lengths of the polymer chains are determined by the dimensions of the nanoflasks or the enantiomeric excess of the product is controlled by the ligand chirality, respectively.
Chovnik et al. applied the principles discussed above for the out-of-equilibrium delivery of particulate cargo (Figure 5).127 In this work, diamagnetic particles could be manipulated using externally applied magnetic fields after the transient adsorption of azobenzene light-switchable/superparamagnetic Fe3O4 nanoparticles (Figure 5b). In a mixed dispersion of hydrophilic silica particles and the Fe3O4 colloids, the silica particles acted as a preferential adsorption site for the iron oxide particles after forcing the azobenzene ligands onto their surface in the more polar cis conformation. The resulting raspberry hybrid particles (Figure 5a, top middle) could subsequently be assembled into string-like clusters by applying an external magnetic field (Figure 5a, right). It is important to mention that the individual Fe3O4 particles were not susceptible to the applied magnetic field, as their magnetic Bjerrum length was smaller than their diameter. Only after multiple iron oxide particles were immobilized on a silica carrier, the combined magnetic dipoles were sufficiently strong to overcome thermal motion. The resulting elongated colloidal chains could subsequently be manipulated and navigated through space by controlling the direction of the magnetic field. As soon as the silica cargo reached the targeted destination, switching off the UV light released the adsorbed iron oxide nanoparticles (Figure 5a, bottom). This approach is an elegant way to transfer the transient characteristics of nanoparticles to significantly larger and otherwise inert objects.
Figure 5.
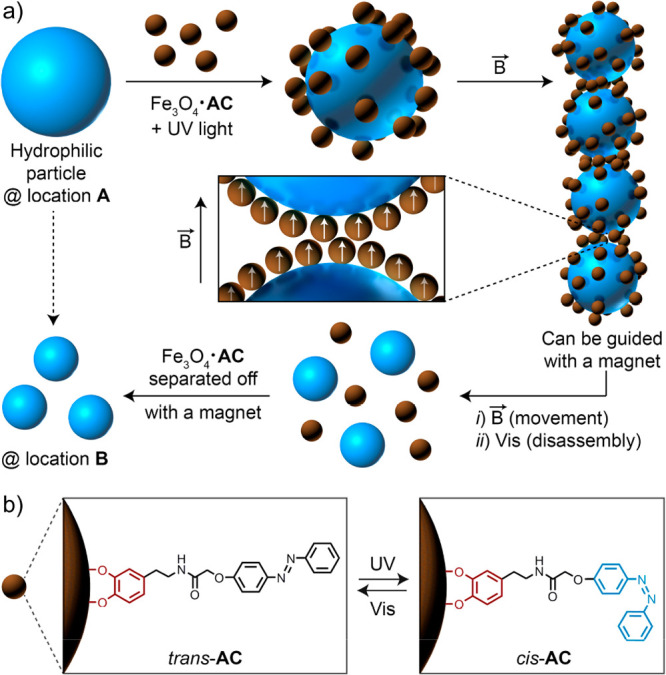
(a) Diamagnetic particles (blue) are transiently decorated with paramagnetic iron oxide (Fe3O4, brown) colloids via UV-light-mediated self-assembly. (b) The increased surface hydrophilicity caused by the conformational change between the trans and cis conformations of the azobenzene ligands immobilized on the Fe3O4 particles drives the heterocoagulation between the two colloid populations. The resulting raspberry-shaped hybrid colloids can subsequently be aligned and manipulated in a magnetic field to transport the diamagnetic cargo. After removal of the electric field and thermal relaxation of the azobenzene ligands, the hydrophilic cargo can be released. Adapted from ref (127). Copyright 2012, American Chemical Society.
Indirect manipulation of larger colloidal objects via coupling of the colloidal phase behavior to a light-driven azobenzene-based reaction system was also applied by Vialetto et al.23 In this particular example, which operates under dissipative conditions, energy input was used to realize the transient 2D crystallization of micrometer-sized colloidal beads confined at an air–water interface. The light responsiveness of the system originates from an azobenzene-containing surfactant (Figure 6a) that in equilibrium preferentially accumulates at the oil–water interface. The surfactant carries a positive charge by which it partially screens the electrostatic repulsion between the particles at the interface. In this state, disordered particle arrangements were observed (Figure 6b, left). Upon illumination with light, the surfactant switches to its more polar cis conformation and desorbs from the surface. The increased electrostatic repulsion between the colloids combined with the surface confinement induces the crystallization of particles into millimeter-sized highly ordered domains (Figure 6b, right). These ordered structures could be maintained only as long as the systems were illuminated. When illumination was stopped, isomerization of the surfactant followed by adsorption to the interface disrupted the crystals, and the systems returned to their disordered equilibrium states.
Figure 6.
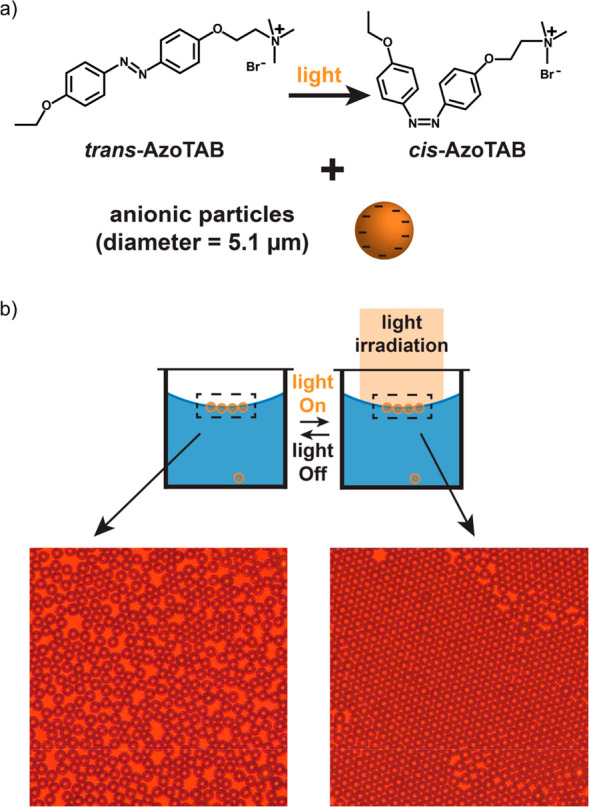
(a) Azobenzene-derived cationic surfactant in its trans and cis isomeric forms. In the equilibrium trans conformation, the surfactants preferentially partition at the air–water interface. In this situation, the surface charges of negatively charged particles pinned at the interface are partially screened, resulting in the formation of disordered 2D particle structures (panel b, left). Upon illumination with light, the surfactant is forced into its more polar cis form. This leads to desorption of the surfactant into the bulk water phase. The full electrostatic repulsion between the particles combined with confinement leads to crystallization (panel b, right). If illumination is stopped, then thermal equilibration of the surfactant and readsorption on the air–water interface randomizes the particle orientations again. Adapted from ref (23). Copyright 2019, John Wiley and Sons.
Spiropyran-Guided Colloidal Systems
In addition to azobenzene-based systems, spiropyran molecular switches have been applied to orchestrate colloidal assembly. Following the same strategy as described before, spiropyran-containing ligands can be coupled directly to the nanoparticle surface. Upon exposure to UV light, the spiropyran moiety undergoes ring-opening isomerization to generate its polar, zwitterionic merocyanine derivative (MCH+). This activated isomer is significantly more polar compared to the cis form of azobenzene, providing a larger driving force for particle assembly in organic media. This was experimentally verified for nanosized gold particles, where the spiropyran-functionalized systems were characterized by significantly faster (dis)assembly compared to their azobenzene counterparts.113 A detailed study on the effect of the spiropyran ligand density and size of the gold core provided control over the cluster size and lifetime. It is noteworthy that there are a limited number of dissipative cycles (∼30) that the system could go through, as well-known photoinduced degradation of the spiropyran ligands was observed.113 Degradation can be mainly attributed to bimolecular reactions involving the long-lived triplet excited state (dependent on the local spiropyran concentration) and oxidation of the photoswitch.128 Strategies to prevent fatigue are therefore used to minimize the stacking tendency by choosing highly apolar media or colloids with a lower spiropyran grafting density. Additionally, oxidation can be suppressed by performing photoswitching in the presence of antioxidants and/or in an airtight environment.
Motivated by the large dipole moment of the open MCH+ form, an analogous strategy was applied to significantly larger silica colloids (diameter = 150–920 nm) by Ueda et al.129,130 and Bell and Piech.120 Spiropyran fragments were immobilized by direct condensation onto the silica surface or by decorating the silica surface with a spiropyran-functionalized polymer brush (poly(methyl methacrylate), p(MMA)). When dispersed in highly apolar media (toluene, benzene, and xylene), aggregation could be induced by UV illumination. However, this aggregation proved to be (partially) irreversible. Despite the thermal relaxation of the zwitterionic MCH+ moieties, the input of external mechanical energy through sonication was required to disintegrate the clusters. Relatively strong van der Waals forces or physical entanglements between polymer brushes immobilized on different particles were proposed to explain the lack of complete reversibility. Technically, the requirement for additional energy to redisperse the particles makes this system not transient and illustrates the challenges involved with translating nanoscopic out-of-equilibrium systems across multiple length scales.
Another elegant use of spiropyran-mediated clustering of gold nanoparticles operating under dissipative conditions was recently introduced by Samanta and Klajn.22 On the basis of the pioneering work of Maity et al.,131 the spiropyran-derived switch functions as a transient photoacid to guide the assembly process (Figure 7). Instead of immobilizing the molecular switches on the surface of the particles, they were dissolved in the aqueous continuous phase of a dispersion of charge-stabilized gold nanoparticles. In equilibrium, the spiropyran is present in its open, protonated form (MCH+). Upon illumination with visible light, a ring-closed spiropyran is formed and a proton is released into the medium. The released protons lower the pH, leading to protonation of the charged carboxylic acids on the surface of the particles. By removing (part of) the surface charge, the electrostatic repulsion decreased and the particles aggregated under the influence of van der Waals forces. As the ring-closed spiropyrans proved to be metastable under the applied system conditions, they slowly convert back to their zwitterionic form, thereby extracting protons from the solution. This reverse process raises the pH again to its original level, reinstating the surface charges on the colloids which drives the dissociation of the colloidal clusters. Compared to the previous examples, this system operates in water which required careful optimization of the length of the carboxyl-containing thiol and ionic strength of the medium to find a window where the attractive forces were sufficiently strong to drive the clustering but weak enough to ensure cluster disintegration upon an increase in the pH.
Figure 7.
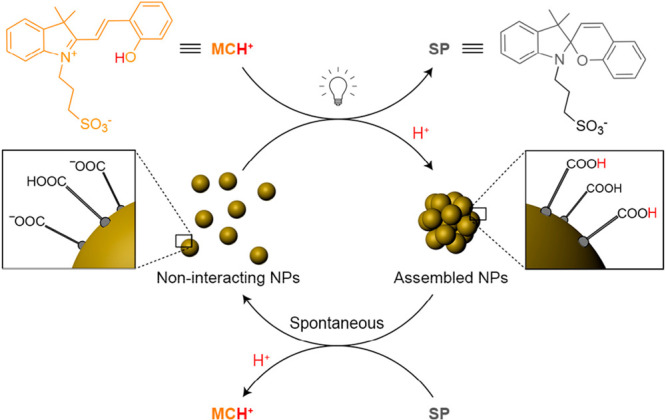
Visible-light-mediated assembly of gold nanoparticles functionalized with carboxylic-containing surface ligands dispersed in an aqueous medium operating under dissipative conditions. In the thermodynamic ground state, the colloids carry a net negative surface charge, safeguarding colloidal stability via electrostatic repulsion. Upon illumination with light, molecularly dissolved photoacids (MCH+, orange) undergo a ring-closure reaction, thereby releasing protons (H+). The associated decrease in pH results in protonation of the surface-immobilized carboxylic acids. The diminished surface charge is no longer sufficient to ensure colloidal stability resulting in the formation of clusters. As the ring-closed reaction product (SP, gray) is metastable under the imposed reaction conditions, it slowly reverts back to its zwitterionic form. Upon doing so, protons are taken up from the dispersing medium. This leads to an increase in pH and a reinstatement of the surface charges on the colloids. On a mesoscopic scale, this results in disintegration of the particle clusters. Adapted from ref (22). Copyright 2016, John Wiley and Sons.
A similar system was published by the same authors, where the particles were dispersed in an organic medium.114 Under these conditions, the particles were aggregated by the presence of surface charges in the absence of a light trigger. Upon illumination, protons were released into the medium and subsequent protonation of the surface-immobilized carboxylic acids caused transient disassembly of the nanoparticle clusters. In this example, not the disassembled but the assembled state resides in the global minimum of the free-energy landscape.
Fuel-Mediated Out-of-Equilibrium Assembly
In addition to (UV) light, energy-rich carrier molecules can be added to activate the precursor building blocks. These energy-rich compounds are typically referred to as (molecular) fuels and resemble the function of adenosine triphosphate (ATP) in biological systems. Compared to light-driven systems, molecular fuels can be added in excess to buffer the fuel demand of the system. Additionally, assembly (kinetics) can be controlled spatially by fuel diffusion, for example, from a high-concentration reservoir.119,132−134 Finally, the rate of fuel consumption and hence assembly kinetics can be regulated by relying on catalytic fuel-converting processes.135−138 However, the use of fuels is typically associated with an accumulation of waste products in the system (Figure 1). At sufficiently high concentrations, waste can impose limitations on the repeatability of the transient clustering by disrupting the reaction network or generating poisonous conditions that can result in fatigue and ultimately failure of the system. The additional constraint that the (de)activation reactions should be orthogonal and take place efficiently in a single reaction environment, makes the design of molecular fuel-driven systems a challenging task. An overview of recently reported systems will be discussed below.
Fully Synthetic-Fuel-Driven Systems
Van Ravensteijn and Hendriksen et al. contributed to the field of fuel-driven colloidal assembly by developing polymer-grafted particles capable of undergoing transient assembly upon addition of a strong methylating agent (dimethyl sulfate, DMS) as fuel.118 The chemistry underlying the assembly was previously developed for transient gelators and is schematically depicted in Figure 8. At the start of the clustering cycle, the particles were dispersed in an aqueous medium buffered with borate at a high pH (∼9). Consequently, the outer segments of the polymers grafted onto the colloidal core carried negatively charged carboxylic acids, ensuring colloidal stability. Upon fuel injection, the carboxylic acids were methylated with a significant increase in hydrophobicity as result. Once in this activated state, the particles aggregated into relatively large clusters. However, under the high pH conditions, the newly introduced methyl esters were marginally stable and slowly reverted back to the charged carboxylic acids. Redispersion of the colloids was promoted by using block copolymers of which only the outer segments were fuel responsive. The inner hydrophilic block prevented close contact between the colloidal cores and hence limited the magnitude of attractive van der Waals forces, which is key for the larger colloids used in this study (hydrodynamic diameter ≈ 750 nm). Once sufficient charges were reintroduced, cluster disassembly was observed. By the sequential addition of fuel, multiple assembly cycles could be performed. The acidity of the waste product (CH3SO4–) combined with the limited buffering capacity of the dispersing medium and the consumption of OH– in the hydrolysis step led to a loss of reversibility after three cycles.
Figure 8.
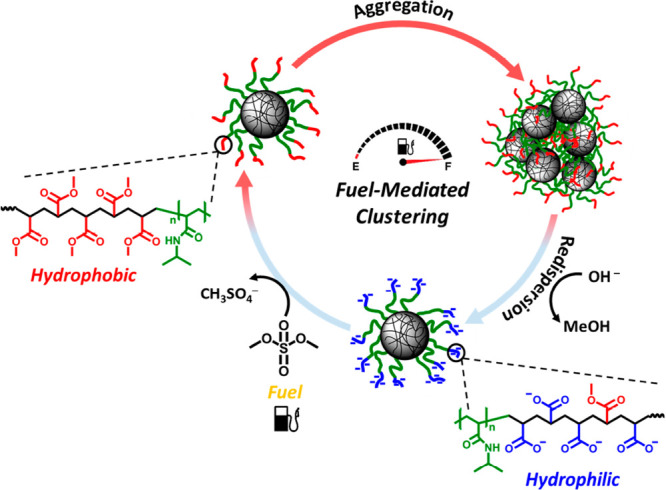
Molecular fuel-mediated clustering of colloidal building blocks. In the initial equilibrium situation, colloids are dispersed in a high-pH reaction medium. This causes the outer poly(methyl methacrylate) (p(MMA)) segments (blue) of the grafted polymers to be negatively charged due to the deprotonation of carboxylic acids (bottom). The negative charges provide colloidal stability through electrostatic repulsion. The addition of chemical fuel ((CH3)2SO4) removes these charges by methylation, thereby increasing the hydrophobicity of the polymer brush’s outer corona (left, red). The increased hydrophobicity triggers clustering between the colloidal particles (right). After fuel depletion, hydrolysis of the formed methyl esters becomes dominant. Over time, the charges are reintroduced on the grafted polymers, leading to the disintegration of the formed clusters and returning to the initial, well-dispersed state (bottom). Adapted from ref (118). Copyright 2017, American Chemical Society.
An alternative strategy for transiently converting surface-immobilized carboxylic acids based on the irreversible consumption of 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide (EDC) was introduced by Boekhoven and co-workers (Figure 9).116 Again, charged carboxylic acid-functionalized particles, in this case, photoluminescent silicon nanocrystals (SiN), served as precursors in the chemical reaction network. These precursors were activated by reaction with EDC to form N-hydroxysuccinimide (NHS) esters. Deactivation occurred via the spontaneous hydrolysis (pH 6.5) of the NHS esters to regain the carboxylate moieties and reinstate colloidal stability (Figure 9a). SiN clusters could therefore exist only in the presence of sufficient EDC. Again, the number of dissipative assembly cycles was limited to three by the disruptive effect that the waste product (1-[3-(dimethylamino)propyl]-3-ethylurea (EDU)) had on the reaction network. Interestingly, the transient clustering could be applied for the delayed uptake of SiN by mammalian cells under buffered artificial physiological conditions (1:1 phosphate-buffered saline (PBS)/2-(N-morpholino)ethanesulfonic acid (MES)), thereby showing the potential of these dynamic systems for the controlled delivery of therapeutics.
Figure 9.
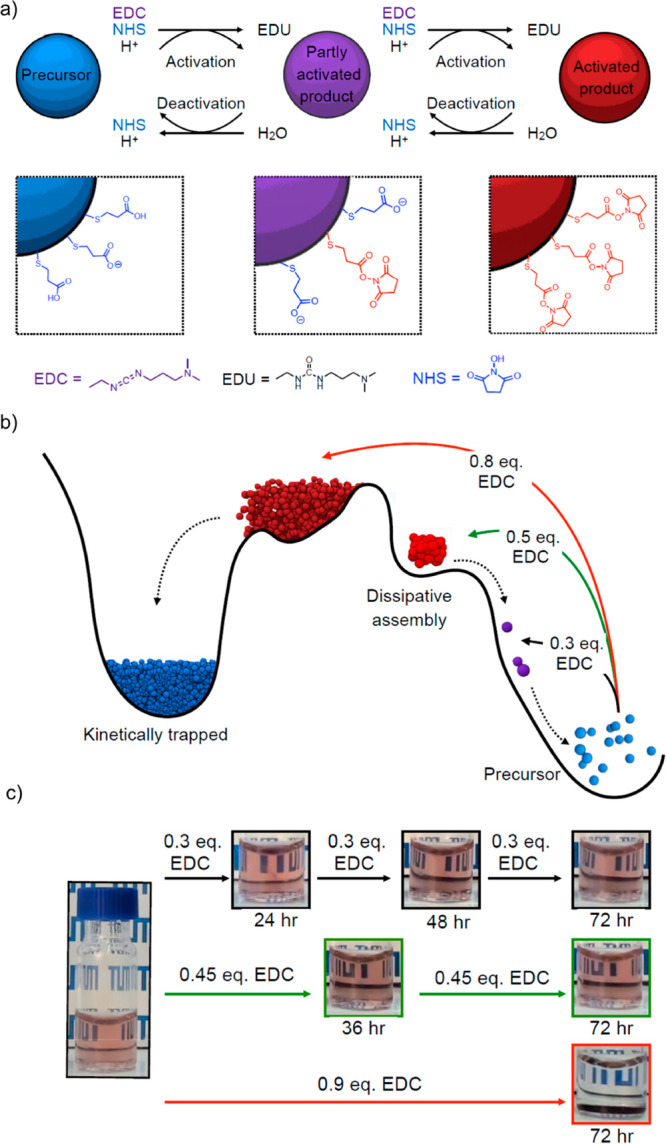
(a) Schematic representation of the transient activation of precursor building blocks via 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC)-mediated esterification of surface-immobilized carboxylic acid with N-hydroxysuccinimide (NHS). EDC acts as fuel in this system and is irreversibly consumed during esterification. Attachment of the NHS moieties to the nanoparticles decreases their surface charge, thereby generating a driving force for colloidal aggregation. The formed NHS esters are metastable under the employed reaction conditions. Hydrolysis causes the spontaneous reintroduction of the surface charges and hence disintegration of the aggregates. (b) Free-energy landscape of the EDC-driven transient clustering system depicted in panel a. Depending on the fuel (EDC) concentration, the particles can be partially activated (purple) without clustering, undergo transient clustering (light red), or get kinetically trapped (dark blue). The presence of a fuel-dependent kinetic trap indicates that the system is prone to pathway complexity, implying that the properties of the system depend on its preparative history. (c) Macroscopic manifestation of pathway complexity. Adding fuel in multiple small batches yields stable dispersions (homogeneous pink), while the addition of the same quantity of fuel in one shot results in (irreversible) clustering (colorless solution with sediment). Adapted from ref (115). Copyright 2019, American Chemical Society.
The importance of carefully optimizing reaction networks to promote reversible assembly was also illustrated for this chemical reaction cycle but with gold nanoparticles as colloidal building blocks.115 Only fuel concentrations within a certain concentration regime resulted in dissipative, transient assembly (Figure 9b). EDC concentrations that were too low led to incomplete activation and hence an insufficient driving force for particle clustering. On the contrary, the addition of too much fuel resulted in clusters trapped in a kinetically arrested state.115 The presence of a fuel-dependent kinetic trap implies that the behavior of the system is prone to pathway complexity; the final position of the system in the free-energy landscape depends on its preparative history. This finding was experimentally verified by showing that adding large quantities of fuel sequentially in small batches prevents the kinetic trap (Figure 9c). This behavior is reminiscent of the self-healing and proofreading capabilities of biological systems, which can recover from externally imposed damage as long as the underlying reaction network remains unaffected.41,42,45,139,140
Finally, Walther and colleagues introduced a conceptually different approach to coupling fuel-dependent reaction networks to the assembly of gold nanoparticles.117 Kinetic control over the temporal self-assembly was achieved by the simultaneous injection of rapid promoters and dormant deactivators (Figure 10). Evidently, the rapid promoters force building blocks from their inactive precursor states into activated states that undergo assembly. However, the deactivation of the building blocks is now governed by the reaction kinetics at which the dormant deactivator is converted into activated deactivators (ν*). The kinetic condition under which out-of-equilibrium superstructures can form in this situation is that the rate of activation (ν1) is significantly faster than the decay rate of the dormant deactivator. Since these two processes are decoupled, this kinetic requirement is significantly easier to fulfill than the situations in the examples discussed before, where the rates of activation (ν1) and competing deactivation (ν2) have to be delicately balanced.
Figure 10.
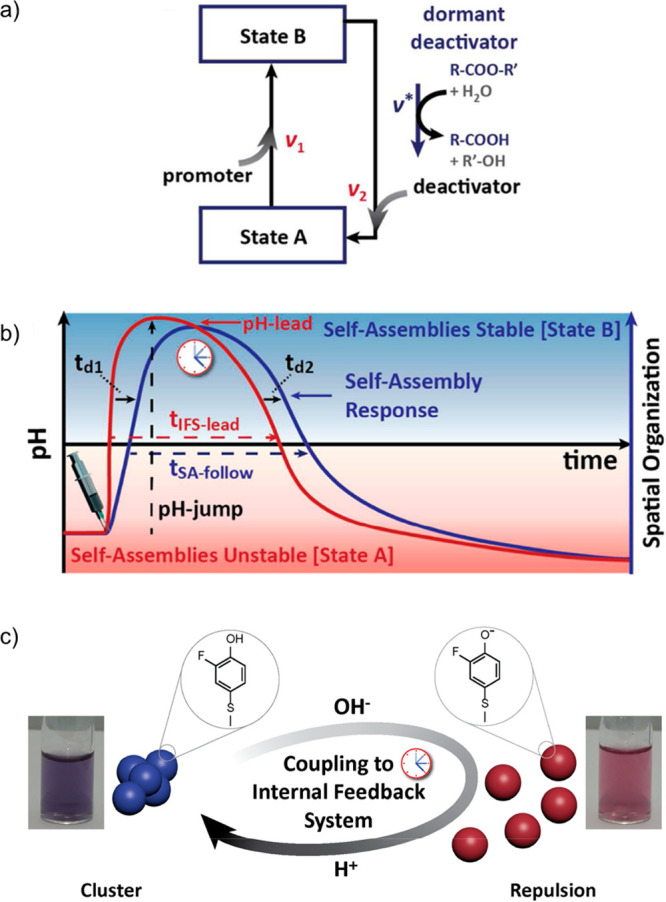
(a) Conceptual diagram of transient switching between states A and B by using a combination of rapid promoters and dormant deactivators. The presence of dormant deactivators lifts the stringent kinetic boundary ν1 ≫ ν2 to realize transient self-assembly. The relevant kinetic condition changes to ν1 ≫ ν* (the decay rate of the dormant deactivator into an active deactivator). The ratio between rate constants ν1 and ν2 is no longer leading, and the deactivation of state B to state A is ultimately determined by the time-dependent concentration of deactivator. (b) If promoter and dormant deactivators are added simultaneously, then a transient pH profile (red) is generated. The system follows this profile by undergoing (dis)assembly with a system-specific time offset. (c) Schematic representation and macroscopic appearance of gold nanoparticles undergoing transient assembly driven by the simultaneous action of rapid promoters and dormant deactivators. Adapted from ref (117). Copyright 2015, American Chemical Society.
The validity of this concept was shown by using molecular esters as dormant deactivators in combination with a strong base as a rapid promoter to transiently control the cluster state of 2-fluorophenol-functionalized gold nanoparticles in aqueous media. In equilibrium, the particles are clustered due to the absence of sufficient surface charges. The addition of the strong base deprotonates the ligands, leading to cluster disintegration driven by electrostatic repulsion. Simultaneously, the dormant esters slowly hydrolyze to liberate acidic species. In time, the pH of the medium decreases, which results in protonation of the phenol derivatives and hence a reinstatement of the clustered equilibrium state. By altering the chemical structure of the dormant deactivator, the hydrolysis rate and acidity of the corresponding acid can be tuned. Together with the absolute concentrations of dormant deactivators and strong promoter (base), the accessible transient pH window (5–9) and lifetime (minutes–days) of the activated state can be controlled. The robustness and versatility of this concept was emphasized by demonstrations of the transient assembly of colloidal particles, pH-responsive block copolymers into micellar structures, and coiled oligopeptides into nanofibrils.117
Biosynthetic Hybrid Systems
In addition to the fully synthetic examples discussed in the previous subsection, Dehne et al. coupled the transient clustering of micrometer-sized DNA coated colloids with a competing antagonistic enzymatic reaction of RNA polymerization and degradation.119 The system comprises two types of colloidal particles (Figure 11, red and green) both decorated with noncomplementary single-stranded DNA segments. In equilibrium, both types of colloids are mutually repelling due to the electrosteric repulsion of the DNA corona, resulting in a stable dispersion. The T7 Polymerase enzyme was used to synthesize complementary RNA linkers that induce specific clustering between the two types of colloids. The synthesis of this RNA linker from a double-stranded DNA template is driven by nucleoside triphosphate (NTP), which acts as the molecular fuel. Once in the aggregated state, cluster disintegration was realized by the presence of the RNaseH ribonuclease. This enzyme selectively degrades the RNA linkers once they are hybridized to the particle-immobilized DNA strands, generating RNA degradation products as waste. Transient clustering could be obtained by tuning the kinetics of the individual enzymatic reactions by varying the concentrations of enzymes and fuel. Following this strategy, clusters with controllable dimensions and lifetimes could be obtained. Spatial control over the clustering process could be obtained by localizing the RNA binder production. This was achieved by immobilizing the DNA template on confined regions in a microfluidic cell. RNA-linker diffusion from these regions coupled to the homogeneous degrading action of RNaseH led to the pulsating traveling of aggregation fronts.
Figure 11.

Schematic representation of transient colloidal aggregation coupled to a reaction network of two antagonistic enzymes. Clustering between single-stranded-DNA-coated particles is initiated by the production of complementary RNA linkers by T7 Polymerase. The RNA synthesis is templated by a double-stranded DNA template and is fueled by nucleoside triphosphate (NTP). Once in the hybridized state, the RNA is susceptible to degradation by RNaseH. This process leads to breaking of the colloidal bonds and hence disintegration of the clusters. By tuning the kinetics of the enzymatic reactions, clusters with a transient lifetime could be obtained. Adapted from ref (119). Copyright 2019, Nature Publishing Group.
Relying on the hybridization of DNA to assemble colloidal particles in conventional equilibrium self-assembly has been shown to be an extremely versatile approach.33,34 The high selectivity of DNA hybridization provides a means to accurately tune the colloidal interaction strength, temperature sensitivity, and directionality. Bringing these hallmarks to the out-of-equilibrium assembly domain will undoubtedly lead to new, biologically inspired, materials.
Future Prospects for Colloidal Out-of-Equilibrium Assembly
As illustrated by a variety of particle systems used for out-of-equilibrium or dissipative assembly, the concept has great potential for generating new classes of dynamic materials with spatiotemporal control and shows that the design principles exploited in (biological) supramolecular systems are extendable to the colloidal domain. The versatility of the colloidal systems that are capable of undergoing out-of-equilibrium structure formation is still limited compared to that of supramolecular systems. However, the field has slowly transcended its infancy status as particles with different dimensions, chemistries, and underlying activation mechanisms are being developed. Without exception, the dissipative systems developed thus far are extremely delicate and highly tailored. No off-the-shelf heuristic rules exist that can be used to easily fabricate new systems. Harnessing the full potential of out-of-equilibrium assembly to generate self-healing, highly adaptive, and communicative properties and create reaction-diffusion patterns has yet to be realized in colloidal systems. In most of the discussed examples, reversible association is used exclusively to showcase application potential. Future work will undoubtedly aim to further exploit this reversibility to realize spatio- or temporal control of the formed superstructures, including fuel-dependent feedback loops, and amplify fuel responses across multiple length scales.
As highlighted by Van Rossum et al. for (supra)molecular systems,38 the difficulty in designing dissipative systems is directly related to the complexity of selecting underlying reaction networks with suitable kinetics and finding reaction schemes with (completely) uncoupled activation and deactivation pathways. Kinetic considerations are important to ensuring that sufficiently high concentrations of building blocks can be accumulated into the activated state such that assembly can commence. In addition to the concentration of activated building blocks, the lifetime of the activated state should also be long enough to favor structure formation. In principle, these requirements can be fulfilled by having activation reactions which are faster compared to the deactivation pathways or by applying the dormant deactivator strategy introduced by Walther and co-workers.117 For colloids, these kinetic boundary conditions are extremely important to take into account, as the molecular reaction network operates on a different length scale than that for the particulate building blocks. Because of the slow colloidal dynamics (especially for particles approaching the micrometer size regime), reaction networks with slow deactivation pathways are a necessity. Potential strategies to realize these include tuning the chemical substitution patterns of photoswitches141,142 or, in case of hydrolysis-based deactivation pathways, regulating the stability and catalytic formation of the activated hydrolyzable bonds.117,138 Suppressing side reactions of fuel and nonassembling energy-dissipating pathways will become more challenging as the particles need to survive for longer in the activated state. Kinetic modeling and or a detailed analysis of reaction rate constants for fuel-dependent reactions could provide an important step forward for linking colloidal dynamics to underlying molecular processes.
These considerations are important if not only random aggregates but also ordered out-of-equilibrium clusters are desired. A majority of the transiently assembled systems realized to date lack structural (long-range) order and are best characterized as a collection of randomly associated particles. Naturally, being able to control the particle positions and relative orientations in complex (multicomponent) systems would be a pivotal step forward to transient materials with functional properties and open the potential application scope into the fields of responsive catalysis,111,112 optics,143,144 electronics, and sensors.145 To facilitate the formation of superstructures with controllable structural order, the fuel-induced activated state should not only survive long enough for the particles to find each other but also allow for subtle reconfigurations of the particles within the growing cluster to minimize the number of stacking faults (Coupling Colloidal Assembly to Reaction Networks: Design Considerations section). Balancing the attractive and repulsive contributions to the interparticle potential remains a great challenge in the colloid chemistry community, even for equilibrium systems. Additional restrictions associated with the transient nature of the attractions generated by reaction with fuels present an even greater challenge. To go beyond the trial-and-error methodology currently applied and find heuristic design rules for dissipative colloidal systems, measuring time-dependent intercolloidal forces during the dissipative cycle and relating this to (fuel-induced) chemical details of the building blocks are highly desired.
Once control over the strength and range of the fuel-dependent potentials is understood and programmable, more complex cluster morphologies could be targeted. Inspired by numerous examples of colloidal equilibrium self-assembly, higher degrees of structural order and directionality could be imprinted by, for example, relying on site-specific interactions by using Janus or patchy particles with well-defined fuel-responsive regions.1,2,5,66−68,146 Alternatively, the balance between the range of attractive and repulsive contributions can be altered upon activation to shift the preferred cluster morphology.147−152
Recent simulation studies showed that even for isotropic colloids out-of-equilibrium assembly pathways can lead to transient open-crystalline lattices which are extremely hard or even impossible to fabricate in equilibrium.153−155 These low-packing-density crystals are formed if the particles interact with each other via oscillating potentials that switch between a net repulsive and attractive state. Evidently, this switching could be driven by (molecular) fuels. A similar oscillating interparticle potential approach was used to anneal out stacking faults in growing colloidal crystals, providing a means to fabricate truly long-range-ordered superstructures. Alternatively, the local and time-dependent fuel concentration can be used as an additional design parameter to realize adaptive materials. For example, one could envision materials with temporal gradients in mechanical or optical properties due to spatiotemporal changes in the shape138 and/or local ordering of the colloidal building blocks.
Conclusions
Perfected in biology and increasingly more applied to synthetic supramolecular systems, the concepts of dissipative assembly to force systems out-of-equilibrium and imprint transient properties to materials are slowly but surely permeating the colloidal world. The wide variety of dissipative (supra)molecular building blocks and ever-growing library of fuel- or light-driven reaction networks serves as great inspiration for the colloid and physical chemical community. Coupling the assembly behavior of colloidal particles to molecular networks presents significant challenges caused by nonmatching time and length scales. Even more importantly than for equilibrium assembly are strategies to prevent irreversible assembly and the formation of kinetically arrested states. To rationally synthesize particles that balance fuel-induced attractions and repulsion successfully, insight into how molecular details of the colloidal particles propagate into time-dependent interparticle potentials is essential. Despite the fact that the field is still very much in its infancy, an impressive set of colloidal dissipative systems were reported in the last couple of years. The myriad chemical strategies used to fabricate out-of-equilibrium colloidal superstructures show the prospect of the large parameter space that can be explored in the pursuit of colloidal materials that truly mimic the self-healing and highly adaptive properties seen in biology. Additionally, dissipative colloidal systems could serve as next-generation model systems for out-of-equilibrium biological processes. Analogous to the role that hard colloidal particles played in elucidating the physics underlying atomic crystallization, dissipative colloidal systems might shed light on the fundamentals of (bio)molecular out-of-equilibrium processes.
Biographies

Bas G. P. van Ravensteijn completed his Ph.D. on the synthesis and physical–chemical characterization of colloidal model systems for self-assembly at the Van’t Hoff Laboratory for Physical and Colloid Chemistry (Utrecht University, The Netherlands) in 2015. He then moved to the University of California—Santa Barbara to work as a postdoctoral researcher on the development of new synthesis procedures for well-defined star polymers and their application as next-generation lubricant additives. As a next step, he continued his scientific career at The Netherlands Organisation for Applied Scientific Research. Here, he employed fundamental polymer and colloid science for application-driven research. Currently, he is a Marie Skłodowska-Curie fellow at the Eindhoven University of Technology where his research revolves around using state-of-the-art controlled polymerization techniques to control the assembly behavior of colloids in and out of equilibrium.

Ilja K. Voets studied molecular sciences at Wageningen University in Wageningen, The Netherlands. She performed her Ph.D. research on complex coacervate core micelles assembled from oppositely charged copolymers at the same university, after which she moved to the Adolphe Merkle Institute in Fribourg, Switzerland for a postdoctoral research stay to investigate the phase behavior of concentrated protein mixtures. In 2011, she returned to The Netherlands to develop her own line of research in physical chemistry as a university lecturer in the Department of Chemical Engineering and Chemistry and the Institute for Complex Molecular Systems at TU/e. Here, she was appointed associate professor in 2015 and then full professor in the field of self-organizing soft matter in 2018. In her research on (biological) soft matter, Voets seeks to understand the simple design rules that orchestrate how complex functionalities emerge from hierarchical self-organization processes and to translate these into rational design strategies for innovative functional materials.

Willem K. Kegel is a professor in the Van’t Hoff Laboratory at Utrecht University. With his group he works on a broad range of subjects in the field of soft matter and biophysics, in particular, self-organization in colloids, (block) polymers, and (virus) proteins, and, more recently, the statistical physics of transcription regulation.

Rienk Eelkema is an associate professor at TU Delft in The Netherlands. He obtained his Ph.D. in chemistry (cum laude) with Prof. Ben Feringa at the University of Groningen. After postdoctoral work at the University of Oxford with Prof. Harry Anderson, he joined the TU Delft faculty in 2008 (tenured in 2013). His main research interests include the use of chemical reactivity to control interactive soft materials and the design and synthesis of new molecules and materials with advanced functions for applications in physics, biology, and biomedical engineering.
This work was financially supported by the Marie Curie Research Grants Scheme, Grant 838585 – STAR Polymers (B.G.P.v.R). I.K.V. acknowledges The Netherlands Organisation for Scientific Research (NWO VIDI Grant 723.014.006) for financial support.
The authors declare no competing financial interest.
References
- Gerth M.; Voets I. K. Molecular Control over Colloidal Assembly. Chem. Commun. 2017, 53 (32), 4414–4428. 10.1039/C6CC09985H. [DOI] [PubMed] [Google Scholar]
- Elacqua E.; Zheng X.; Shillingford C.; Liu M.; Weck M. Molecular Recognition in the Colloidal World. Acc. Chem. Res. 2017, 50 (11), 2756–2766. 10.1021/acs.accounts.7b00370. [DOI] [PubMed] [Google Scholar]
- Boles M. A.; Engel M.; Talapin D. V. Self-Assembly of Colloidal Nanocrystals: From Intricate Structures to Functional Materials. Chem. Rev. 2016, 116 (18), 11220–11289. 10.1021/acs.chemrev.6b00196. [DOI] [PubMed] [Google Scholar]
- Pawar A. B.; Kretzschmar I. Fabrication, Assembly, and Application of Patchy Particles. Macromol. Rapid Commun. 2010, 31 (2), 150–168. 10.1002/marc.200900614. [DOI] [PubMed] [Google Scholar]
- Duguet É.; Hubert C.; Chomette C.; Perro A.; Ravaine S. Patchy Colloidal Particles for Programmed Self-Assembly. C. R. Chim. 2016, 19 (1–2), 173–182. 10.1016/j.crci.2015.11.013. [DOI] [Google Scholar]
- Fialkowski M.; Bishop K. J. M.; Klajn R.; Smoukov S. K.; Campbell C. J.; Grzybowski B. A. Principles and Implementations of Dissipative (Dynamic) Self-Assembly. J. Phys. Chem. B 2006, 110 (6), 2482–2496. 10.1021/jp054153q. [DOI] [PubMed] [Google Scholar]
- Nicolis G.; Prigogine I.. Self-Organization in Nonequilibrium Systems; John Wiley & Sons, 1977. [Google Scholar]
- de Groot S. R.; Mazur P.. Non-Equilibrium Thermodynamics; Dover Publications, 1984. [Google Scholar]
- Sorrenti A.; Leira-Iglesias J.; Markvoort A. J.; De Greef T. F. A.; Hermans T. M. Non-Equilibrium Supramolecular Polymerization. Chem. Soc. Rev. 2017, 46 (18), 5476–5490. 10.1039/C7CS00121E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A.; Mitchison T. J. Microtuble Polymerization Dynamics. Annu. Rev. Cell Dev. Biol. 1997, 13, 83–117. 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- Mitchison T.; Kirschner M. Dynamic Instability of Microtubule Growth. Nature 1984, 312 (5991), 237–242. 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- Karsenti E. Self-Organization in Cell Biology: A Brief History. Nat. Rev. Mol. Cell Biol. 2008, 9 (3), 255–262. 10.1038/nrm2357. [DOI] [PubMed] [Google Scholar]
- Blanchoin L.; Boujemaa-Paterski R.; Sykes C.; Plastino J. Actin Dynamics, Architecture, and Mechanics in Cell Motility. Physiol. Rev. 2014, 94 (1), 235–263. 10.1152/physrev.00018.2013. [DOI] [PubMed] [Google Scholar]
- Hess H.; Ross J. L. Non-Equilibrium Assembly of Microtubules: From Molecules to Autonomous Chemical Robots. Chem. Soc. Rev. 2017, 46 (18), 5570–5587. 10.1039/C7CS00030H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N.; Formon G. J. M.; De Piccoli S.; Hermans T. M. Devising Synthetic Reaction Cycles for Dissipative Nonequilibrium Self-Assembly. Adv. Mater. 2020, 32, 1906834. 10.1002/adma.201906834. [DOI] [PubMed] [Google Scholar]
- Ragazzon G.; Prins L. J. Energy Consumption in Chemical Fuel-Driven Self-Assembly. Nat. Nanotechnol. 2018, 13 (10), 882–889. 10.1038/s41565-018-0250-8. [DOI] [PubMed] [Google Scholar]
- Ashkenasy G.; Hermans T. M.; Otto S.; Taylor A. F. Systems Chemistry. Chem. Soc. Rev. 2017, 46 (9), 2543–2554. 10.1039/C7CS00117G. [DOI] [PubMed] [Google Scholar]
- Maiti S.; Fortunati I.; Ferrante C.; Scrimin P.; Prins L. J. Dissipative Self-Assembly of Vesicular Nanoreactors. Nat. Chem. 2016, 8 (7), 725–731. 10.1038/nchem.2511. [DOI] [PubMed] [Google Scholar]
- Dhiman S.; Jain A.; Kumar M.; George S. J. Adenosine-Phosphate-Fueled, Temporally Programmed Supramolecular Polymers with Multiple Transient States. J. Am. Chem. Soc. 2017, 139 (46), 16568–16575. 10.1021/jacs.7b07469. [DOI] [PubMed] [Google Scholar]
- Mishra A.; Korlepara D. B.; Kumar M.; Jain A.; Jonnalagadda N.; Bejagam K. K.; Balasubramanian S.; George S. J. Biomimetic Temporal Self-Assembly via Fuel-Driven Controlled Supramolecular Polymerization. Nat. Commun. 2018, 9 (1), 1–9. 10.1038/s41467-018-03542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- della Sala F.; Maiti S.; Bonanni A.; Scrimin P.; Prins L. J. Fuel-Selective Transient Activation of Nanosystems for Signal Generation. Angew. Chem., Int. Ed. 2018, 57 (6), 1611–1615. 10.1002/anie.201711964. [DOI] [PubMed] [Google Scholar]
- Samanta D.; Klajn R. Aqueous Light-Controlled Self-Assembly of Nanoparticles. Adv. Opt. Mater. 2016, 4 (9), 1373–1377. 10.1002/adom.201600364. [DOI] [Google Scholar]
- Vialetto J.; Anyfantakis M.; Rudiuk S.; Morel M.; Baigl D. Photoswitchable Dissipative Two-Dimensional Colloidal Crystals. Angew. Chem., Int. Ed. 2019, 58 (27), 9145–9149. 10.1002/anie.201904093. [DOI] [PubMed] [Google Scholar]
- Lebon G.; Jou D.; Casa-Vázquez J.. Understanding Non-Equilibrium Thermodynamics; Springer, 2008. [Google Scholar]
- Lagzi I.; Kowalczyk B.; Wang D.; Grzybowski B. A. Nanoparticle Oscillations and Fronts. Angew. Chem., Int. Ed. 2010, 49 (46), 8616–8619. 10.1002/anie.201004231. [DOI] [PubMed] [Google Scholar]
- Nabika H.; Oikawa T.; Iwasaki K.; Murakoshi K.; Unoura K. Dynamics of Gold Nanoparticle Assembly and Disassembly Induced by pH Oscillations. J. Phys. Chem. C 2012, 116 (10), 6153–6158. 10.1021/jp300650c. [DOI] [Google Scholar]
- Szalai I.; Cuiñas D.; Takács N.; Horváth J.; De Kepper P. Chemical Morphogenesis: Recent Experimental Advances in Reaction-Diffusion System Design and Control. Interface Focus 2012, 2 (4), 417–432. 10.1098/rsfs.2012.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein I. R.; Xu B. Reaction-Diffusion Processes at the Nano- and Microscales. Nat. Nanotechnol. 2016, 11 (4), 312–319. 10.1038/nnano.2016.41. [DOI] [PubMed] [Google Scholar]
- Pusey P. N.; van Megen W. Phase Behaviour of Concentrated Suspensions of Nearly Colloidal Spheres. Nature 1986, 320, 340. 10.1038/320340a0. [DOI] [Google Scholar]
- Riess G. Micellization of Block Copolymers. Prog. Polym. Sci. 2003, 28 (7), 1107–1170. 10.1016/S0079-6700(03)00015-7. [DOI] [Google Scholar]
- Tanford C. Thermodynamics of Micelle Formation: Prediction of Micelle Size and Size Distribution. Proc. Natl. Acad. Sci. U. S. A. 1974, 71 (5), 1811–1815. 10.1073/pnas.71.5.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzelczak M.; Liz-Marzán L. M.; Klajn R. Stimuli-Responsive Self-Assembly of Nanoparticles. Chem. Soc. Rev. 2019, 48 (5), 1342–1361. 10.1039/C8CS00787J. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wang Y.; Zheng X.; Ducrot É.; Yodh J. S.; Weck M.; Pine D. J. Crystallization of DNA-Coated Colloids. Nat. Commun. 2015, 6, 7253. 10.1038/ncomms8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J. S.; Lee S.; Glotzer S. C.; Yi G. R.; Pine D. J. Colloidal Fibers and Rings by Cooperative Assembly. Nat. Commun. 2019, 10, 3936. 10.1038/s41467-019-11915-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson M. I.; O’reilly R. K. To Aggregate, or Not to Aggregate? Considerations in the Design and Application of Polymeric Thermally-Responsive Nanoparticles. Chem. Soc. Rev. 2013, 42 (17), 7204–7213. 10.1039/C3CS60035A. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Santos P. J.; Kubiak J. M.; Guo X.; Lee M. S.; Macfarlane R. J. Multistimuli Responsive Nanocomposite Tectons for Pathway Dependent Self-Assembly and Acceleration of Covalent Bond Formation. J. Am. Chem. Soc. 2019, 141 (33), 13234–13243. 10.1021/jacs.9b06695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Feijter I.; Albertazzi L.; Palmans A. R. A.; Voets I. K. Stimuli-Responsive Colloidal Assembly Driven by Surface-Grafted Supramolecular Moieties. Langmuir 2015, 31 (1), 57–64. 10.1021/la5031872. [DOI] [PubMed] [Google Scholar]
- Van Rossum S. A. P.; Tena-Solsona M.; Van Esch J. H.; Eelkema R.; Boekhoven J. Dissipative Out-of-Equilibrium Assembly of Man-Made Supramolecular Materials. Chem. Soc. Rev. 2017, 46 (18), 5519–5535. 10.1039/C7CS00246G. [DOI] [PubMed] [Google Scholar]
- Arango-Restrepo A.; Barragán D.; Rubi J. M. Self-Assembling Outside Equilibrium: Emergence of Structures Mediated by Dissipation. Phys. Chem. Chem. Phys. 2019, 21 (32), 17475–17493. 10.1039/C9CP01088B. [DOI] [PubMed] [Google Scholar]
- Boekhoven J.; Hendriksen W. E.; Koper G. J. M.; Eelkema R.; Van Esch J. H. Transient Assembly of Active Materials Fueled by a Chemical Reaction. Science 2015, 349, 1075–1079. 10.1126/science.aac6103. [DOI] [PubMed] [Google Scholar]
- Schaedel L.; John K.; Gaillard J.; Nachury M. V.; Blanchoin L.; Thery M. Microtubules Self-Repair in Response to Mechanical Stress. Nat. Mater. 2015, 14 (11), 1156–1163. 10.1038/nmat4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck O.; Speck T. An Overview of Bioinspired and Biomimetic Self-Repairing Materials. Biomimetics 2019, 4 (1), 26. 10.3390/biomimetics4010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A.; Lindsey-Boltz L. A.; Ünsal-Kaçmaz K.; Linn S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73 (1), 39–85. 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Tang S. K. Y.; Marshall W. F. Self-Repairing Cells: How Single Cells Heal Membrane Ruptures and Restore Lost Structures. Science 2017, 356, 1022–1025. 10.1126/science.aam6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V.; Sharma P. K.; Sharma N.; Pal O. P.; Malviya R.. Biological Archetypes for Self-Healing Materials. Self-healing Materials; Advances in Polymer Science; Springer: Cham, 2015; Vol. 273, pp 307–344 10.1007/12_2015_334. [DOI] [Google Scholar]
- Lim W. A.; Lee C. M.; Tang C. Design Principles of Regulatory Networks: Searching for the Molecular Algorithms of the Cell. Mol. Cell 2013, 49 (2), 202–212. 10.1016/j.molcel.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto S. Dynamic Molecular Networks: From Synthetic Receptors to Self-Replicators. Acc. Chem. Res. 2012, 45 (12), 2200–2210. 10.1021/ar200246j. [DOI] [PubMed] [Google Scholar]
- Ludlow R. F.; Otto S. Systems Chemistry. Chem. Soc. Rev. 2008, 37 (1), 101–108. 10.1039/B611921M. [DOI] [PubMed] [Google Scholar]
- Che H.; Cao S.; Van Hest J. C. M. Feedback-Induced Temporal Control of “Breathing” Polymersomes to Create Self-Adaptive Nanoreactors. J. Am. Chem. Soc. 2018, 140 (16), 5356–5359. 10.1021/jacs.8b02387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tena-Solsona M.; Rieß B.; Grötsch R. K.; Löhrer F. C.; Wanzke C.; Käsdorf B.; Bausch A. R.; Müller-Buschbaum P.; Lieleg O.; Boekhoven J. Non-Equilibrium Dissipative Supramolecular Materials with a Tunable Lifetime. Nat. Commun. 2017, 8, 15895. 10.1038/ncomms15895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A.; Dhiman S.; Dhayani A.; Vemula P. K.; George S. J. Chemical Fuel-Driven Living and Transient Supramolecular Polymerization. Nat. Commun. 2019, 10, 450. 10.1038/s41467-019-08308-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden W. A.; Guan Z. Redox Chemical-Fueled Dissipative Self-Assembly of Active Materials. ChemSystemsChem. 2020, 2, e1900030. 10.1002/syst.201900030. [DOI] [Google Scholar]
- Boekhoven J.; Brizard A. M.; Kowlgi K. N. K.; Koper G. J. M.; Eelkema R.; Van Esch J. H. Dissipative Self-Assembly of a Molecular Gelator by Using a Chemical Fuel. Angew. Chem., Int. Ed. 2010, 49 (28), 4825–4828. 10.1002/anie.201001511. [DOI] [PubMed] [Google Scholar]
- Cissé N.; Kudernac T. Light-Fuelled Self-Assembly of Cyclic Peptides into Supramolecular Tubules. ChemSystemsChem. 2020, 10.1002/syst.202000012. [DOI] [Google Scholar]
- Endo M.; Fukui T.; Jung S. H.; Yagai S.; Takeuchi M.; Sugiyasu K. Photoregulated Living Supramolecular Polymerization Established by Combining Energy Landscapes of Photoisomerization and Nucleation-Elongation Processes. J. Am. Chem. Soc. 2016, 138 (43), 14347–14353. 10.1021/jacs.6b08145. [DOI] [PubMed] [Google Scholar]
- De Jong J. J. D.; Hania P. R.; Pugžlys A.; Lucas L. N.; De Loos M.; Kellogg R. M.; Feringa B. L.; Duppen K.; Van Esch J. H. Light-Driven Dynamic Pattern Formation. Angew. Chem., Int. Ed. 2005, 44 (16), 2373–2376. 10.1002/anie.200462500. [DOI] [PubMed] [Google Scholar]
- Rakotondradany F.; Whitehead M. A.; Lebuis A. M.; Sleiman H. F. Photoresponsive Supramolecular Systems: Self-Assembly of Azodibenzoic Acid Linear Tapes and Cyclic Tetramers. Chem. - Eur. J. 2003, 9 (19), 4771–4780. 10.1002/chem.200304864. [DOI] [PubMed] [Google Scholar]
- Xie X.; Wang L.; Liu X.; Du Z.; Li Y.; Li B.; Wu L.; Li W. Light-Powered and Transient Peptide Two-Dimensional Assembly Driven by: Trans -to- Cis Isomerization of Azobenzene Side Chains. Chem. Commun. 2020, 56 (12), 1867–1870. 10.1039/C9CC09448B. [DOI] [PubMed] [Google Scholar]
- Ragazzon G.; Baroncini M.; Silvi S.; Venturi M.; Credi A. Light-Powered Autonomous and Directional Molecular Motion of a Dissipative Self-Assembling System. Nat. Nanotechnol. 2015, 10 (1), 70–75. 10.1038/nnano.2014.260. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Song G.; Jiao Y.; Zheng P.; Xu J.-F.; Zhang X. Dissipative Supramolecular Polymerization Powered by Light. CCS Chem. 2019, 1 (4), 335–342. 10.31635/ccschem.019.20190013. [DOI] [Google Scholar]
- De S.; Klajn R. Dissipative Self-Assembly Driven by the Consumption of Chemical Fuels. Adv. Mater. 2018, 30 (41), 1706750. 10.1002/adma.201706750. [DOI] [PubMed] [Google Scholar]
- Wang G.; Liu S. Strategies to Construct a Chemical-Fuel-Driven Self-Assembly. ChemSystemsChem. 2020, 2, e1900046. 10.1002/syst.201900046. [DOI] [Google Scholar]
- Grzybowski B. A.; Fitzner K.; Paczesny J.; Granick S. From Dynamic Self-Assembly to Networked Chemical Systems. Chem. Soc. Rev. 2017, 46 (18), 5647–5678. 10.1039/C7CS00089H. [DOI] [PubMed] [Google Scholar]
- Merindol R.; Walther A. Materials Learning from Life: Concepts for Active, Adaptive and Autonomous Molecular Systems. Chem. Soc. Rev. 2017, 46 (18), 5588–5619. 10.1039/C6CS00738D. [DOI] [PubMed] [Google Scholar]
- Rogers W. B.; Shih W. M.; Manoharan V. N. Using DNA to Program the Self-Assembly of Colloidal Nanoparticles and Microparticles. Nat. Rev. Mater. 2016, 1, 16008. 10.1038/natrevmats.2016.8. [DOI] [Google Scholar]
- Vogel N.; Retsch M.; Fustin C. A.; Del Campo A.; Jonas U. Advances in Colloidal Assembly: The Design of Structure and Hierarchy in Two and Three Dimensions. Chem. Rev. 2015, 115 (13), 6265–6311. 10.1021/cr400081d. [DOI] [PubMed] [Google Scholar]
- Yi G. R.; Pine D. J.; Sacanna S. Recent Progress on Patchy Colloids and Their Self-Assembly. J. Phys.: Condens. Matter 2013, 25, 193101. 10.1088/0953-8984/25/19/193101. [DOI] [PubMed] [Google Scholar]
- Glotzer S. C.; Solomon M. J.; Kotov N. A. Self-Assembly: From Nanoscale to Microscale Colloids. AIChE J. 2004, 50 (12), 2978–2985. 10.1002/aic.10413. [DOI] [Google Scholar]
- Vanmaekelbergh D. Self-Assembly of Colloidal Nanocrystals as Route to Novel Classes of Nanostructured Materials. Nano Today 2011, 6 (4), 419–437. 10.1016/j.nantod.2011.06.005. [DOI] [Google Scholar]
- Kim S. H.; Lee S. Y.; Yang S. M.; Yi G. R. Self-Assembled Colloidal Structures for Photonics. NPG Asia Mater. 2011, 3 (1), 25–33. 10.1038/asiamat.2010.192. [DOI] [Google Scholar]
- Zhu J.; Hersam M. C. Assembly and Electronic Applications of Colloidal Nanomaterials. Adv. Mater. 2017, 29, 1603895. 10.1002/adma.201603895. [DOI] [PubMed] [Google Scholar]
- Shao L.; Zheng J. Fabrication of Plasmonic Nanostructures by Hole-Mask Colloidal Lithography: Recent Development. Appl. Mater. Today 2019, 15, 6–17. 10.1016/j.apmt.2018.12.014. [DOI] [Google Scholar]
- Wang Y.; Zhang M.; Lai Y.; Chi L. Advanced Colloidal Lithography: From Patterning to Applications. Nano Today 2018, 22, 36–61. 10.1016/j.nantod.2018.08.010. [DOI] [Google Scholar]
- Colvin V. L. From Opals to Optics: Colloidal Photonic Crystals. MRS Bull. 2001, 26 (8), 637–641. 10.1557/mrs2001.159. [DOI] [Google Scholar]
- Kim J. B.; Lee S. Y.; Lee J. M.; Kim S. H. Designing Structural-Color Patterns Composed of Colloidal Arrays. ACS Appl. Mater. Interfaces 2019, 11 (16), 14485–14509. 10.1021/acsami.8b21276. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Xie Z.; Gu H.; Zhu C.; Gu Z. Bio-Inspired Variable Structural Color Materials. Chem. Soc. Rev. 2012, 41 (8), 3297–3317. 10.1039/c2cs15267c. [DOI] [PubMed] [Google Scholar]
- Cargnello M. Colloidal Nanocrystals as Building Blocks for Well-Defined Heterogeneous Catalysts. Chem. Mater. 2019, 31 (3), 576–596. 10.1021/acs.chemmater.8b04533. [DOI] [Google Scholar]
- Poon W. Colloids as Big Atoms. Science 2004, 304, 830. 10.1126/science.1097964. [DOI] [PubMed] [Google Scholar]
- Di Michele L.; Varrato F.; Kotar J.; Nathan S. H.; Foffi G.; Eiser E. Multistep Kinetic Self-Assembly of DNA-Coated Colloids. Nat. Commun. 2013, 4, 2007. 10.1038/ncomms3007. [DOI] [PubMed] [Google Scholar]
- Varrato F.; Di Michele L.; Belushkin M.; Dorsaz N.; Nathan S. H.; Eiser E.; Foffi G. Arrested Demixing Opens Route to Bigels. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (47), 19155–19160. 10.1073/pnas.1214971109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciortino F.; Tartaglia P. Glassy Colloidal Systems. Adv. Phys. 2005, 54 (6–7), 471–524. 10.1080/00018730500414570. [DOI] [Google Scholar]
- Dawson K. A. The Glass Paradigm for Colloidal Glasses, Gels, and Other Arrested States Driven by Attractive Interactions. Curr. Opin. Colloid Interface Sci. 2002, 7 (3–4), 218–227. 10.1016/S1359-0294(02)00052-3. [DOI] [Google Scholar]
- Liljeström V.; Chen C.; Dommersnes P.; Fossum J. O.; Gröschel A. H. Active Structuring of Colloids through Field-Driven Self-Assembly. Curr. Opin. Colloid Interface Sci. 2019, 40, 25–41. 10.1016/j.cocis.2018.10.008. [DOI] [Google Scholar]
- Illien P.; Golestanian R.; Sen A. Fuelled” Motion: Phoretic Motility and Collective Behaviour of Active Colloids. Chem. Soc. Rev. 2017, 46 (18), 5508–5518. 10.1039/C7CS00087A. [DOI] [PubMed] [Google Scholar]
- Paxton W. F.; Sundararajan S.; Mallouk T. E.; Sen A. Chemical Locomotion. Angew. Chem., Int. Ed. 2006, 45 (33), 5420–5429. 10.1002/anie.200600060. [DOI] [PubMed] [Google Scholar]
- Moerman P. G.; Moyses H. W.; Van Der Wee E. B.; Grier D. G.; Van Blaaderen A.; Kegel W. K.; Groenewold J.; Brujic J. Solute-Mediated Interactions between Active Droplets. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 2017, 96, 032607 10.1103/PhysRevE.96.032607. [DOI] [PubMed] [Google Scholar]
- Maass C. C.; Krüger C.; Herminghaus S.; Bahr C. Swimming Droplets. Annu. Rev. Condens. Matter Phys. 2016, 7 (1), 171–193. 10.1146/annurev-conmatphys-031115-011517. [DOI] [Google Scholar]
- Jin C.; Kru-ger C.; Maass C. C. Chemotaxis and Autochemotaxis of Self-Propelling Droplet Swimmers. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (20), 5089–5094. 10.1073/pnas.1619783114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibele M.; Mallouk T. E.; Sen A. Schooling Behavior of Light-Powered Autonomous Micromotors in Water. Angew. Chem., Int. Ed. 2009, 48 (18), 3308–3312. 10.1002/anie.200804704. [DOI] [PubMed] [Google Scholar]
- Sanchez S.; Soler L.; Katuri J. Chemically Powered Micro- and Nanomotors. Angew. Chem., Int. Ed. 2015, 54 (5), 1414–1444. 10.1002/anie.201406096. [DOI] [PubMed] [Google Scholar]
- Liebchen B.; Löwen H. Synthetic Chemotaxis and Collective Behavior in Active Matter. Acc. Chem. Res. 2018, 51 (12), 2982–2990. 10.1021/acs.accounts.8b00215. [DOI] [PubMed] [Google Scholar]
- Ebbens S. J.; Gregory D. A. Catalytic Janus Colloids: Controlling Trajectories of Chemical Microswimmers. Acc. Chem. Res. 2018, 51 (9), 1931–1939. 10.1021/acs.accounts.8b00243. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Blackman N. M. K.; Kopp N. D.; Sen A.; Velegol D. Chemotaxis of Nonbiological Colloidal Rods. Phys. Rev. Lett. 2007, 99, 178103. 10.1103/PhysRevLett.99.178103. [DOI] [PubMed] [Google Scholar]
- Morgan A. R.; Dawson A. B.; McKenzie H. S.; Skelhon T. S.; Beanland R.; Franks H. P. W.; Bon S. A. F. Chemotaxis of Catalytic Silica-Manganese Oxide “Matchstick” Particles. Mater. Horiz. 2014, 1 (1), 65–68. 10.1039/C3MH00003F. [DOI] [Google Scholar]
- Buttinoni I.; Bialké J.; Kümmel F.; Löwen H.; Bechinger C.; Speck T. Dynamical Clustering and Phase Separation in Suspensions of Self-Propelled Colloidal Particles. Phys. Rev. Lett. 2013, 110, 238301. 10.1103/PhysRevLett.110.238301. [DOI] [PubMed] [Google Scholar]
- Palacci J.; Sacanna S.; Steinberg A. P.; Pine D. J.; Chaikin P. M. Colloidal Surfers. Science 2013, 339, 936–939. 10.1126/science.1230020. [DOI] [PubMed] [Google Scholar]
- Wang W.; Duan W.; Ahmed S.; Sen A.; Mallouk T. E. From One to Many: Dynamic Assembly and Collective Behavior of Self-Propelled Colloidal Motors. Acc. Chem. Res. 2015, 48 (7), 1938–1946. 10.1021/acs.accounts.5b00025. [DOI] [PubMed] [Google Scholar]
- Bialké J.; Speck T.; Löwen H. Active Colloidal Suspensions: Clustering and Phase Behavior. J. Non-Cryst. Solids 2015, 407, 367–375. 10.1016/j.jnoncrysol.2014.08.011. [DOI] [Google Scholar]
- Lin Z.; Gao C.; Chen M.; Lin X.; He Q. Collective Motion and Dynamic Self-Assembly of Colloid Motors. Curr. Opin. Colloid Interface Sci. 2018, 35, 51–58. 10.1016/j.cocis.2018.01.006. [DOI] [Google Scholar]
- Kagan D.; Balasubramanian S.; Wang J. Chemically Triggered Swarming of Gold Microparticles. Angew. Chem., Int. Ed. 2011, 50 (2), 503–506. 10.1002/anie.201005078. [DOI] [PubMed] [Google Scholar]
- Israelachvili J. N.Intermolecular and Surface Forces, 3rd ed.; Elsevier, 2011. [Google Scholar]
- Verwey E. J. W.; Overbeek J. T. G.. Theory of the Stability of Lyophobic Colloids; Elsevier, 1948. [DOI] [PubMed] [Google Scholar]
- Theodoor J.; Overbeek G. Strong and Weak Points in the Interpretation of Colloid Stability. Adv. Colloid Interface Sci. 1982, 16 (1), 17–30. 10.1016/0001-8686(82)85003-3. [DOI] [Google Scholar]
- Evertt C. H.Basic Principles of Colloid Science; Royal Society of Chemistry, 1988. [Google Scholar]
- Zhulina E. B.; Borisov O. V.; Priamitsyn V. A. Theory of Steric Stabilization of Colloid Dispersions by Grafted Polymers. J. Colloid Interface Sci. 1990, 137 (2), 495–511. 10.1016/0021-9797(90)90423-L. [DOI] [Google Scholar]
- Israelachvili J.; Pashley R. The Hydrophobic Interaction Is Long Range, Decaying Exponentially with Distance. Nature 1982, 300 (5890), 341–342. 10.1038/300341a0. [DOI] [PubMed] [Google Scholar]
- Lekkerkerker H. N. W.; Tuinier R.. Colloids and the Depletion Interaction; Springer, 2011. [Google Scholar]
- Bonn D.; Otwinowski J.; Sacanna S.; Guo H.; Wegdam G.; Schall P. Direct Observation of Colloidal Aggregation by Critical Casimir Forces. Phys. Rev. Lett. 2009, 103, 156101. 10.1103/PhysRevLett.103.156101. [DOI] [PubMed] [Google Scholar]
- Bian T.; Chu Z.; Klajn R. The Many Ways to Assemble Nanoparticles Using Light. Adv. Mater. 2020, 32, 1905866. 10.1002/adma.201905866. [DOI] [PubMed] [Google Scholar]
- Klajn R.; Wesson P. J.; Bishop K. J. M.; Grzybowski B. A. Writing Self-Erasing Images Using Metastable Nanoparticle “Inks. Angew. Chem., Int. Ed. 2009, 48 (38), 7035–7039. 10.1002/anie.200901119. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Han S.; Kim J.; Soh S.; Grzybowski B. A. Photoswitchable Catalysis Mediated by Dynamic Aggregation of Nanoparticles. J. Am. Chem. Soc. 2010, 132 (32), 11018–11020. 10.1021/ja104260n. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Sen S.; Udayabhaskararao T.; Sawczyk M.; Kucanda K.; Manna D.; Kundu P. K.; Lee J. W.; Král P.; Klajn R. Reversible Trapping and Reaction Acceleration within Dynamically Self-Assembling Nanoflasks. Nat. Nanotechnol. 2016, 11 (1), 82–88. 10.1038/nnano.2015.256. [DOI] [PubMed] [Google Scholar]
- Kundu P. K.; Das S.; Ahrens J.; Klajn R. Controlling the Lifetimes of Dynamic Nanoparticle Aggregates by Spiropyran Functionalization. Nanoscale 2016, 8 (46), 19280–19286. 10.1039/C6NR05959G. [DOI] [PubMed] [Google Scholar]
- Kundu P. K.; Samanta D.; Leizrowice R.; Margulis B.; Zhao H.; Börner M.; Udayabhaskararao T.; Manna D.; Klajn R. Light-Controlled Self-Assembly of Non-Photoresponsive Nanoparticles. Nat. Chem. 2015, 7 (8), 646–652. 10.1038/nchem.2303. [DOI] [PubMed] [Google Scholar]
- Grötsch R. K.; Wanzke C.; Speckbacher M.; Angl A.; Rieger B.; Boekhoven J. Pathway Dependence in the Fuel-Driven Dissipative Self-Assembly of Nanoparticles. J. Am. Chem. Soc. 2019, 141 (25), 9872–9878. 10.1021/jacs.9b02004. [DOI] [PubMed] [Google Scholar]
- Grötsch R. K.; Angı A.; Mideksa Y. G.; Wanzke C.; Tena-Solsona M.; Feige M. J.; Rieger B.; Boekhoven J. Dissipative Self-Assembly of Photoluminescent Silicon Nanocrystals. Angew. Chem., Int. Ed. 2018, 57 (44), 14608–14612. 10.1002/anie.201807937. [DOI] [PubMed] [Google Scholar]
- Heuser T.; Steppert A. K.; Molano Lopez C.; Zhu B.; Walther A. Generic Concept to Program the Time Domain of Self-Assemblies with a Self-Regulation Mechanism. Nano Lett. 2015, 15 (4), 2213–2219. 10.1021/nl5039506. [DOI] [PubMed] [Google Scholar]
- Van Ravensteijn B. G. P.; Hendriksen W. E.; Eelkema R.; Van Esch J. H.; Kegel W. K. Fuel-Mediated Transient Clustering of Colloidal Building Blocks. J. Am. Chem. Soc. 2017, 139 (29), 9763–9766. 10.1021/jacs.7b03263. [DOI] [PubMed] [Google Scholar]
- Dehne H.; Reitenbach A.; Bausch A. R. Transient Self-Organisation of DNA Coated Colloids Directed by Enzymatic Reactions. Sci. Rep. 2019, 9, 7350. 10.1038/s41598-019-43720-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell N. S.; Piech M. Photophysical Effects between Spirobenzopyran-Methyl Methacrylate- Functionalized Colloidal Particles. Langmuir 2006, 22 (4), 1420–1427. 10.1021/la0516375. [DOI] [PubMed] [Google Scholar]
- Pantaloni D.; Le Clainche C.; Carlier M. Mechanism of Actin-Based Motility. Science 2001, 292, 1502–1507. 10.1126/science.1059975. [DOI] [PubMed] [Google Scholar]
- Martinez-Veracoechea F. J.; Frenkel D. Designing Super Selectivity in Multivalent Nano-Particle Binding. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (27), 10963–10968. 10.1073/pnas.1105351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elacqua E.; Zheng X.; Weck M. Light-Mediated Reversible Assembly of Polymeric Colloids. ACS Macro Lett. 2017, 6 (10), 1060–1065. 10.1021/acsmacrolett.7b00539. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Lin H.; Kim Y.; Yang M.; Skakuj K.; Du J. S.; Lee B.; Schatz G. C.; Van Duyne R. P.; Mirkin C. A. Light-Responsive Colloidal Crystals Engineered with DNA. Adv. Mater. 2020, 32, 1906600. 10.1002/adma.201906600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plunkett K. N.; Mohraz A.; Haasch R. T.; Lewis J. A.; Moore J. S. Light-Regulated Electrostatic Interactions in Colloidal Suspensions. J. Am. Chem. Soc. 2005, 127 (42), 14574–14575. 10.1021/ja054666a. [DOI] [PubMed] [Google Scholar]
- Kathan M.; Hecht S. Photoswitchable Molecules as Key Ingredients to Drive Systems Away from the Global Thermodynamic Minimum. Chem. Soc. Rev. 2017, 46 (18), 5536–5550. 10.1039/C7CS00112F. [DOI] [PubMed] [Google Scholar]
- Chovnik O.; Balgley R.; Goldman J. R.; Klajn R. Dynamically Self-Assembling Carriers Enable Guiding of Diamagnetic Particles by Weak Magnets. J. Am. Chem. Soc. 2012, 134 (48), 19564–19567. 10.1021/ja309633v. [DOI] [PubMed] [Google Scholar]
- Klajn R. Spiropyran-Based Dynamic Materials. Chem. Soc. Rev. 2014, 43 (1), 148–184. 10.1039/C3CS60181A. [DOI] [PubMed] [Google Scholar]
- Ueda M.; Kudo K.; Ichimura K. Photochromic Behaviour of a Spirobenzopyran Chemisorbed on a Colloidal Silica Surface. J. Mater. Chem. 1995, 5 (7), 1007–1011. 10.1039/jm9950501007. [DOI] [Google Scholar]
- Ueda M.; Kim H. B.; Ichimura K. Photocontrolled Aggregation of Colloidal Silica. J. Mater. Chem. 1994, 4 (6), 883–889. 10.1039/jm9940400883. [DOI] [Google Scholar]
- Maity C.; Hendriksen W. E.; Van Esch J. H.; Eelkema R. Spatial Structuring of a Supramolecular Hydrogel by Using a Visible- Light Triggered Catalyst. Angew. Chem., Int. Ed. 2015, 54, 998–1001. 10.1002/anie.201409198. [DOI] [PubMed] [Google Scholar]
- Kubota R.; Makuta M.; Suzuki R.; Hamachi I. Force Generation by a Propagating Wave of Supramolecular Nanofibers. Nat. Commun. 2020, 11, 3541. 10.1038/s41467-020-17394-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Brinke E.; Groen J.; Herrmann A.; Heus H. A.; Rivas G.; Spruijt E.; Huck W. T. S. Dissipative Adaptation in Driven Self-Assembly Leading to Self-Dividing Fibrils. Nat. Nanotechnol. 2018, 13, 849–855. 10.1038/s41565-018-0192-1. [DOI] [PubMed] [Google Scholar]
- Sevim S.; Sorrenti A.; Franco C.; Furukawa S.; Pane S.; deMello A. J.; Puigmarti-Luis J. Self-Assembled Materials and Supramolecular Chemistry within Microfluidic Environments: From Common Thermodynamic States to Non-Equilibrium Structures. Chem. Soc. Rev. 2018, 47, 3788–3803. 10.1039/C8CS00025E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eelkema R.; Van Esch J. H. Catalytic Control over the Formation of Supramolecular Materials. Org. Biomol. Chem. 2014, 12 (33), 6292–6296. 10.1039/C4OB01108B. [DOI] [PubMed] [Google Scholar]
- Solís Muñana P.; Ragazzon G.; Dupont J.; Ren C. Z. J.; Prins L. J.; Chen J. L. Y. Substrate-Induced Self-Assembly of Cooperative Catalysts. Angew. Chem., Int. Ed. 2018, 57 (50), 16469–16474. 10.1002/anie.201810891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal S.; Das K.; Ahmed S.; Das D. Chemically Fueled Dissipative Self-Assembly That Exploits Cooperative Catalysis. Angew. Chem., Int. Ed. 2019, 58 (1), 244–247. 10.1002/anie.201811749. [DOI] [PubMed] [Google Scholar]
- van der Helm M. P.; Wang C.; Fan B.; Macchione M.; Mendes E.; Eelkema R.. Organocatalytic Control over a Fuel-driven Transient Esterification Network. Angew. Chem., Int. Ed. 2020, 10.1002/anie.202008921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninio J. Kinetic Amplification of Enzyme Discrimination. Biochimie 1975, 57 (5), 587–595. 10.1016/S0300-9084(75)80139-8. [DOI] [PubMed] [Google Scholar]
- Hopfield J. J. Kinetic Proofreading: A New Mechanism for Reducing Errors in Biosynthetic Processes Requiring High Specificity. Proc. Natl. Acad. Sci. U. S. A. 1974, 71 (10), 4135–4139. 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calbo J.; Weston C. E.; White A. J. P.; Rzepa H. S.; Contreras-García J.; Fuchter M. J. Tuning Azoheteroarene Photoswitch Performance through Heteroaryl Design. J. Am. Chem. Soc. 2017, 139 (3), 1261–1274. 10.1021/jacs.6b11626. [DOI] [PubMed] [Google Scholar]
- Simeth N. A.; Crespi S.; Fagnoni M.; König B. Tuning the Thermal Isomerization of Phenylazoindole Photoswitches from Days to Nanoseconds. J. Am. Chem. Soc. 2018, 140 (8), 2940–2946. 10.1021/jacs.7b12871. [DOI] [PubMed] [Google Scholar]
- Sawczyk M.; Klajn R. Out-of-Equilibrium Aggregates and Coatings during Seeded Growth of Metallic Nanoparticles. J. Am. Chem. Soc. 2017, 139 (49), 17973–17978. 10.1021/jacs.7b09111. [DOI] [PubMed] [Google Scholar]
- Jha P. K.; Kuzovkov V.; Grzybowski B. A.; Olvera de la Cruz M. Dynamic Self-Assembly of Photo-Switchable Nanoparticles. Soft Matter 2012, 8 (1), 227–234. 10.1039/C1SM06662E. [DOI] [Google Scholar]
- Klajn R.; Browne K. P.; Soh S.; Grzybowski B. A. Nanoparticles That “Remember” Temperature. Small 2010, 6 (13), 1385–1387. 10.1002/smll.200902272. [DOI] [PubMed] [Google Scholar]
- Li W.; Palis H.; Mérindol R.; Majimel J.; Ravaine S.; Duguet E. Colloidal Molecules and Patchy Particles: Complementary Concepts, Synthesis and Self-Assembly. Chem. Soc. Rev. 2020, 49 (6), 1955–1976. 10.1039/C9CS00804G. [DOI] [PubMed] [Google Scholar]
- Mani E.; Lechner W.; Kegel W. K.; Bolhuis P. G. Equilibrium and Non-Equilibrium Cluster Phases in Colloids with Competing Interactions. Soft Matter 2014, 10 (25), 4479–4486. 10.1039/C3SM53058B. [DOI] [PubMed] [Google Scholar]
- Zhang T. H.; Klok J.; Hans Tromp R.; Groenewold J.; Kegel W. K. Non-Equilibrium Cluster States in Colloids with Competing Interactions. Soft Matter 2012, 8 (3), 667–672. 10.1039/C1SM06570J. [DOI] [Google Scholar]
- Mossa S.; Sciortino F.; Tartaglia P.; Zaccarelli E. Ground-State Clusters for Short-Range Attractive and Long-Range Repulsive Potentials. Langmuir 2004, 20 (24), 10756–10763. 10.1021/la048554t. [DOI] [PubMed] [Google Scholar]
- Campbell A. I.; Anderson V. J.; Van Duijneveldt J. S.; Bartlett P. Dynamical Arrest in Attractive Colloids: The Effect of Long-Range Repulsion. Phys. Rev. Lett. 2005, 94, 208301. 10.1103/PhysRevLett.94.208301. [DOI] [PubMed] [Google Scholar]
- Groenewold J.; Kegel W. K. Anomalously Large Equilibrium Clusters of Colloids. J. Phys. Chem. B 2001, 105 (47), 11702–11709. 10.1021/jp011646w. [DOI] [Google Scholar]
- Guo Y.; Van Ravensteijn B. G. P.; Kegel W. K. Self-Assembly of Isotropic Colloids into Colloidal Strings, Bernal Spiral-like, and Tubular Clusters. Chem. Commun. 2020, 56, 6309–6312. 10.1039/D0CC00948B. [DOI] [PubMed] [Google Scholar]
- Tagliazucchi M.; Weiss E. A.; Szleifer I. Dissipative Self-Assembly of Particles Interacting through Time-Oscillatory Potentials. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (27), 9751–9756. 10.1073/pnas.1406122111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman Z. M.; Swan J. W. Dynamic, Directed Self-Assembly of Nanoparticles via Toggled Interactions. ACS Nano 2016, 10 (5), 5260–5271. 10.1021/acsnano.6b01050. [DOI] [PubMed] [Google Scholar]
- Long C.; Lei Q. L.; Ren C. L.; Ma Y. Q. Three-Dimensional Non-Close-Packed Structures of Oppositely Charged Colloids Driven by pH Oscillation. J. Phys. Chem. B 2018, 122 (12), 3196–3201. 10.1021/acs.jpcb.8b00441. [DOI] [PubMed] [Google Scholar]