

Abstract
We describe an integrated approach of using hydrogen deuterium exchange mass spectrometry (HDX-MS), chemical crosslinking mass spectrometry (XL-MS), and molecular docking to characterize the binding interface and to predict the three-dimensional quaternary structure of a protein-protein complex in solution. Interleukin 7 (IL-7) and its α-receptor, IL-7Rα, serving as essential mediators in the immune system, are the model system. HDX kinetics report widespread protection on IL-7Rα but show no differential evidence of binding-induced protection or remote conformational change. Crosslinking with reagents that differ in spacer lengths and targeting residues increases the spatial resolution. Using five cross-links as distance restraints for protein-protein docking, we generated a high-confidence model of the IL-7/IL-7Rα complex. Both the predicted binding interface and regions with direct contacts agree well with those in the solid-state structure, as confirmed by previous X-ray crystallography. An additional binding region was revealed to be the C-terminus of helix B of IL-7, highlighting the value of solution-based characterization. To generalize the integrated approach, protein-protein docking was executed with a different number of cross-links. Combining cluster analysis and HDX kinetics adjudication, we found that two intermolecular crosslink-derived restraints are sufficient to generate a high-confidence model with root mean square distance (r.m.s.d.) value of all alpha carbons below 2.0 Å relative to the crystal structure. The remarkable results of binding-interface determination and quaternary structure prediction highlight the effectiveness and capability of the integrated approach, which will allow more efficient and comprehensive analysis of inter-protein interactions with broad applications in the multiple stages of design, implementation, and evaluation for protein therapeutics.
Introduction
Precisely regulated but complex protein-protein interactions are paramount to safeguard homeostasis in the immune system1. Protein-binding events initiate vast arrays of signal transduction, communicating critical information to orchestrate multiple health-related immune responses2. Comprehensive characterization and impartial understanding of the binding interfaces at high spatial resolution are imperative for both academic and pharmaceutical research. A traditional approach to obtain binding information is X-ray crystallography3. The need for fastidious crystallization and long growth times can limit its application. In addition, the delivered solid-state information can raise questions about its relevance to the physiological environment. Nuclear magnetic resonance (NMR), on the other hand, provides a liquid-phase characterization but at the expense of large amounts of isotopically-labelled sample, demanding signal averaging, and complicated data interpretation4. Mass spectrometry (MS)-based methods are exceptionally appealing in this context given their low detection limit, fast throughput, and compelling use of native proteins.
HDX-MS5,6 is a robust analytical technique for characterizing protein-protein interactions,7 protein-ligand binding, higher-order structure8, and conformational dynamics.9–11 Conformational changes induced by protein-protein binding lead to changes in deuterium uptake kinetics, enabling comparisons between different states (e.g., bound and unbound), thus permitting the determination of binding interfaces and changes in dynamics. Spatial resolution exceeding the peptide-level can be achieved by multi-protease digestion for overlapping peptides, by ETD/ECD fragmentation12–14, or by combining HDX results with other characterization approaches (e.g., chemical cross-linking and docking, as in this study).
Chemical crosslinking with subsequent MS analysis has considerable value in the structural biology toolbox.15–16 It can deliver topological information of protein complexes by bridging the neighboring domains in close proximity, defined by characteristic spacer lengths on cross-linkers. Advanced analysis software17–19 and new crosslinking reagents and protocols20 have greatly escalated its application. The obtained information is even more propitious to uncover higher-order quaternary structure when coupled with other methods (e.g., macromolecular docking).
Complementing X-ray crystallography and NMR, protein-protein docking is a computational methodology that also yields complete three-dimensional structural information for protein complexes. Despite rapid and regular increases in computer speed, parallel processing, acceleration of computation through the use of GPUs and the development of improved algorithms and scoring functions, accurate modeling of protein complexes remains challenging, even starting with high-quality structural models (e.g., X-ray crystal structures) of the binding partners21. This is due in part to the large number of potential complex structures that must be evaluated in the absence of additional information (sampling problem)22 and to limitations in the ability to score accurately the models generated (scoring problem)23–24. An integrated platform25–28 that utilizes experiment-derived information (i.e., HDX-MS and XL-MS data) to define restraints that guide the sampling stage of protein-protein docking has the potential to provide models with high accuracy and resolution.
In this study, we chose interleukin 7 (IL-7) as a test case to demonstrate the integrated characterization platform. IL-7 is a representative member of the cytokine family, modulating immune cell physiology through receptor recognition2, 29. IL-7 sequentially binds to IL-7Rα, its α-receptor, and γc through its extracellular domain to form a ternary-complex that activates the Janus kinase (JAK) and enables phosphorylation of the signal transducer and activator of transcription (STAT).30 Downstream signal transduction switches on transcription of the anti-apoptotic genes, promotes cell survival, and causes proliferation of both naïve and memory T cells.31 Many investigators consider recombinant human IL-7 an ideal treatment agent in cancer immunotherapy. 32 It can promote immune reconstitution33–34 through peripheral T cell expansion, increase the efficacy of tumor regression,35 and antagonize the immunosuppressive network.36 Understanding the critical role of regulating immune functions requires characterization of the involved binding interface, further motivating the choice of IL-7/IL-7Rα as a model system. The approach, once established, will doubtlessly benefit other protein systems (e.g., other cytokine complexes, antigen/antibody complexes). Deeper understanding will aid many steps in biomedical design37 including protein interface engineering2 and epitope-based preventive vaccines38 that are crucial to maintain homeostasis and physiological well-being.
Our goal is to implement and test an analytical approach that is comprised of HDX, chemical crosslinking and protein-protein docking to characterize the binding interface of IL-7/IL-7Rα. With the integrated methods, we generated an accurate three-dimensional model that allows detailed description on the direct inter-protein contacts in the binding region. Furthermore, we investigated how HDX kinetics can adjudicate candidate docking models to an ultimate high-confidence complex structure. A previous X-ray crystallographic study39 of IL-7/IL-7Rα is available for final comparison purposes.
The binding regions assigned by using the approach are in good accord with the published crystal structure and, moreover, indicate another involvement (i.e., C-terminus of helix B in IL-7). Docking results generated with different numbers of cross-links further demonstrate that, with HDX kinetics adjudication, a high-confidence model can be delivered by using only two intermolecular cross-links. Although HDX-MS has been employed for complementary information in some biological studies40–44 along with XL-MS, the two methods are usually treated individually. Other studies27–28 that incorporated XL-based docking have shown that HDX kinetics can be utilized to examine the constructed 3D model; however, none has deployed the process in a systematic way. The integration and evaluation of the three approaches convincingly shows the combination can yield high-order structural information and provide basic and practical insights for further applications.
Experimental
Material
A description of the materials used in this study is given in SI Material and Method.
Hydrogen-Deuterium Exchange
The HDX experiments were performed with three states: IL-7 unbound state, IL-7Rα unbound state, and IL-7/IL-7Rα bound state (details see SI Material and Method). Data were acquired on a custom-built HDX platform coupled with Linear Ion Trap-Fourier Transform mass spectrometer (Thermo Fisher, Waltham, MA) and analyzed by HDExaminer (Sierra Analytics, Modesto, CA). Per residue deuterium uptake difference for each time point was calculated and exported from the software based on available overlapping peptides. The cumulative uptake differences and the associated propagation error were calculated manually. Three times propagation error was considered to give 99.7% certainty.
BS3 and BS2G Crosslinking
Prior to any experiments, IL-7 and IL-7Rα were incubated as 1:1 ratio for 1 h in 10 mM PBS buffer (2.7 mM KCl, 137 mM NaCl, pH = 7.4). The BS3-h12/d12 crosslinker (1 mg) was dissolved in PBS buffer to make a 25 mM stock solution. The IL-7/ IL-7Rα complex was then mixed with the fresh BS3-h12/d12 solution at 1:25, 1:50 and 1:100 ratio, where the concentration of IL-7/ IL-7Rα was 10 μM. The reaction mixture was kept at 25 °C for 45 min. Tris-HCl solution (1 M, pH = 7.4) was added at a final concentration of 50 mM and incubated for 15 mins to stop the crosslinking chemistry. For the BS2G crosslinking, the experimental procedure was identical except that BS2G-d4 and BS2G-h4 stock solutions were prepared separately at 25 mM and manually mixed later at 1:1 ratio prior to the XL reactions. Cross-linking experiments were in triplicates for each condition.
EDC Crosslinking
IL-7 and IL-7Rα were incubated in 35 mM MES buffer (15 mM KCl, pH = 6.5) with 1:1 ratio for 1 h. A freshly made EDC/sulfo-NHS stock solution (400 mM EDC/sulfo-NHS in MES buffer, pH = 6.5) was added to the IL-7/IL-7Rα solution for 1 h at 25 °C to allow the XL reactions; the protein concentration was 17 μM and EDC/sulfo-NHS was 5-20 mM in different trials. To quench the reaction, 2 M Tris-HCl (pH = 7.4) was added to a final concentration of 200 mM, followed by 15 mins incubation. Cross-linking experiments were in triplicates for each condition.
Enzymatic In-solution Digestion and LC-MS/MS Analysis
The protein mixture was desalted with a Zeba column and denatured by RapiGest. After reduction and alkylation of disulfate bonds with DTT and IAM, the cross-linked sample was further digested and deglycosylated wih trypsin and PNGase F. Peptides mixture was further desalted by C18 NuTip and submitted for LC-MS/MS analysis. Data was acquired on a Thermo W Exactive Plus orbitrap mass spectrometer coupled with a Nano Ultimate 3000 Rapid Separation system (Dionex, Co). A detailed description is given SI Material and Method.
Identification of Crosslinked Products
The raw LC-MS/MS files were analyzed by pLink19, 45 (ver. 2.3.1, Institute of Computing Technology, Chinese Academy of Sciences, Beijing, China) and Protein Prospector (ver. 5.22.1, UCSF MS Facility). IL-7 and IL-7Rα sequence were added manually to the search database. XL information, including monoisotopic linker mass of light and heavy form of BS3 (156.079 Da and 168.154 Da) and BS2G (118.057 Da and 114.032 Da), linked sites and composition were all required in pConfig. Trypsin/chymotrypsin digestion was manually defined and added to the library. Search parameters in pLink were: enzyme: trypsin/chymotrypsin; missed cleavage: 3; precursor tolerance: 10 ppm; fragment tolerance: 30 ppm; variable modification: oxidation of M, deamidation of N, Q and N-terminus; fixed modification: carbamidomethyl of C; minimal peptide length: 6 aa; maximal peptide length: 60 aa; minimal peptide mass: 600 Da; maximal peptide mass: 6000 Da. The crosslinked peptides were examined in pLabel to give corresponding summary reports. The identified XL peptides were equal or smaller than a 5% false discovery rate at spectral level with a 10 ppm MS1 filter tolerance. Isotopic doublets were manually confirmed in the raw file, and product ion (MS/MS) spectra were further validated and compared with the calculated product ions.
Protein-protein Docking
IL-7/IL-7Rα protein-protein docking analyses were performed with the Rosetta (v. 3.846–48) docking_protocol (RosettaDock) code49–50 on a cluster of multi-processor Linux servers. For the docking analyses, the A chain from the X-ray crystal structure of unbound IL-7R extracellular domain (PDB ID: 3UP1) was used as the protein model for IL-7R51. Unfortunately, no crystal structure of unbound IL-7 was available either in-house or in the public domain. Thus, the A chain from the IL-7/IL-7R complex crystal structure (PDB ID: 3DI2) provided the model for IL-739. The protein models were prepared for docking in a two-stage process starting with the Rosetta score_jd2 program which added residue atoms that were not present in the PDB files, added hydrogen atoms, and removed non-protein residues. The second preparation step utilized the Rosetta relax program to perform energy minimization of the structures output by score_jd2 within the context of the Rosetta energy model. With the relax application, 10 models were generated for each structure using the “constrain_relax_to_start_coords” and “coord_constrain_sidechains” options to limit structural changes, and the model of each protein with the lowest Rosetta total energy score was selected.
The IL-7 and IL-7R models were imported into Maestro (Schrödinger Release 2019-1: Maestro, Schrödinger, LLC, New York, NY, 2019.) and manually displaced relative to each other to arbitrary extents and then merged into a single two-chain model that was written to a PDB format file as input for the subsequent protein-protein docking analyses. The details of docking study utilizing different number of cross-links as the distance restraints is given in SI Material and Method. All data are available upon request.
Results and Discussion
Hydrogen Deuterium Exchange (HDX).
We conducted HDX on the unbound IL-7 and the bound-state in presence of IL-7Rα (Figure S1) and collected kinetic plots with 94% sequence coverage and 48 unique peptides (see Figure 1A for representative HDX kinetic plots). Regions 17-24 (a), 58-66 (c) and 81-91 (e) of IL-7 show less deuterium uptake across the incubation time up to 2 h, indicating their involvement in the binding interaction. On the other hand, region 35-42 (b) takes up more deuterium, showing exposure when bound to IL-7Rα. Another region, represented by peptide 71-76 (d), is an example not affected by binding, serving as a negative control. Statistical analysis of the cumulative deuterium uptake differences further supports these observations sometimes up to near residue-level (Figure 1B). We consider the difference as significant when they are greater than three times the propagated error, giving 99.7% confidence. We mapped the cumulative HDX differences on the IL-7 structure (Figure 1C) to show that the most protected regions lie on helix A (Region a) and helix C (Region e). The C-terminus of helix B (Region c) also shows moderate protection, whereas the loop region (Region b) between helix A and B becomes more flexible upon binding.
Figure 1.
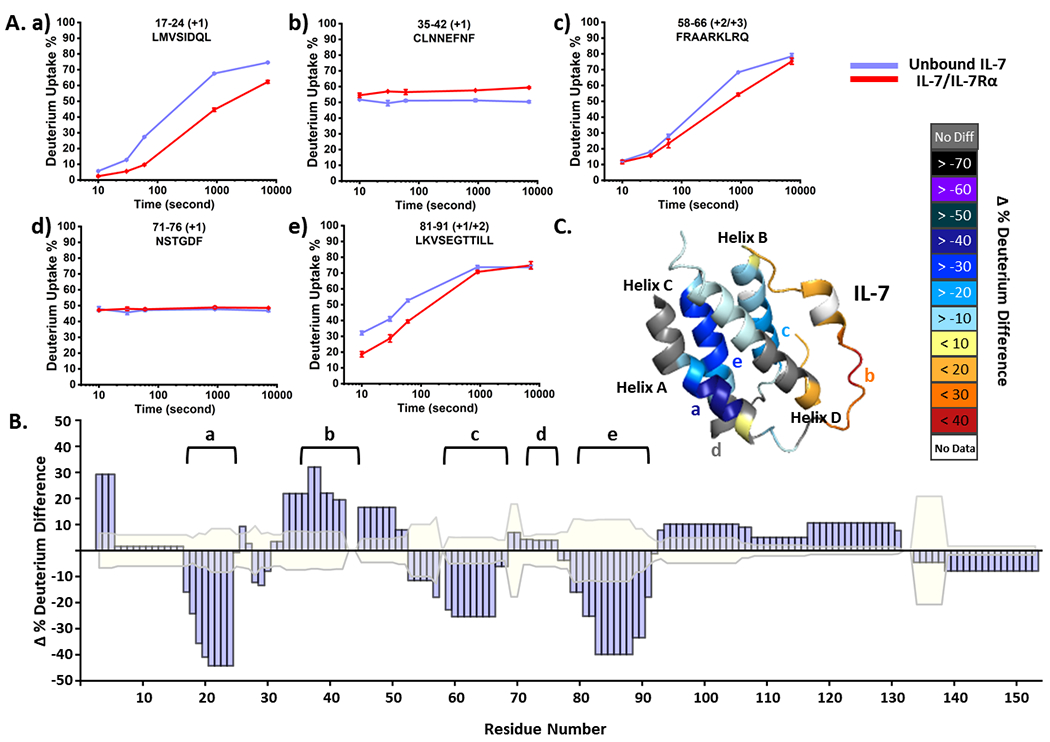
(A) Representative HDX kinetics of unbound IL-7 (purple) and of bound with IL-7Rα (red). (B) Statistical analysis of cumulative HDX difference for each residue. Residues are considered being affected upon binding with IL-7Rα when the difference is greater than three times the propagated error (shaded in faint yellow) of all time points. (C) The cumulative HDX difference of each residue mapped onto the crystal structure of IL-7 (PDB:3DI2).
To map the binding interface of IL-7Rα, we compared HDX kinetics of its bound and unbound states (Figure S2A and Figure S3) and obtained 80% sequence coverage and 45 unique peptides. The N-terminal peptide (Region a) shows identical uptake kinetics between the two states. All other peptides, however, present various extents of protection in the HDX profiles, as is clearly demonstrated in the statistical analysis of cumulative deuterium uptake difference (Figure S2B). The most protected residues, colored black in the IL-7Rα structure (Figure S2C, region c), comprise the elbow region on domain 1 (D1), indicating high probability to be the binding interface. Other regions with pronounced protection are on the other elbow region (Region e) and on one beta-strand (Region d) of domain 2 (D2). The widespread protection indicates that IL-7Rα exhibits a more structured and compact conformation upon binding; however, there is no differential evidence for being the binding interface or undergoing remote conformational changes with the stand-alone HDX. To increase the resolution, to reveal the associated peptide regions in IL-7/ IL-7Rα, and to provide distance restraints needed for docking, we applied crosslinking coupled with MS to obtain complementary information.
Cross-linking of IL-7/IL-7Rα Complex.
To address the interacting regions in the IL-7/ IL-7Rα complex, we chose isotope-encoded BS3-H12/D12 (11 Å), BS2G-H4/D4 (8 Å) and EDC/NHS (“zero-length crosslinker”) as the cross-linking reagents. Their spacer lengths cover a range of distance length restraints, and their use combined with that of BS3 allows interrogation of not only lysine (K) and the N-terminal -NH2 groups but also glutamic (D) and aspartic acids (E), affording broad coverage. We incubated IL-7Rα and IL-7 at a 1:1 molar ratio to allow complex formation and then mixed them with the cross-linkers at various excess concentrations in separate experiments. We monitored the yields with gel electrophoresis (Figure S4). The cross-linked complexes present clearly at approximately 64 kDa, and the yields are estimated accordingly to the intensities of the bands. We found that 100-fold excess BS3 and BS2G (1 mM) gives more abundant cross-linked IL-7/ IL-7Rα than does an excess of 25 (0.25 mM) and 50-fold (0.5 mM). For EDC/NHS crosslinking (Figure S4B), use of 10 mM and 20 mM of reagent results in product bands of nearly equal intensity. We submitted the crosslinked IL-7/ IL-7Rα from individual trials to in-solution digestion followed by LC-MS/MS analysis, in which the “light” and “heavy” encoding aids the identification of cross-links (corresponding cross-links are shown in Figure S5–S14). We manually validated the isotope-encoded doublets and the product ions formed in MS/MS from raw files, establishing that the experimental masses agree with their theoretical values.
In total, we identified 16 intermolecular cross-links. Six of them contain one cross-linked site on a long and flexible loop between helix C and D of IL-7, suggesting the flexibility of the loop region but providing no evidence of the binding interface. This loop can be ruled out as a binding interface, even in the absence of an X-ray structure, because it does not show protection in HDX upon binding. Of the remaining 10 inter cross-links (Table 1), those on IL-7 are K11 on helix A (cross-link 1 and 2), D75 on helix C (cross-link 3), K69 on the C-terminus of helix B (cross-link 4) further support the previously assigned protected regions in the HDX kinetics study. In addition, we observed several cross-links on a series of residues on the N-terminus (crosslink 6 to 10) and one on helix D, all of which deliver topological information about the binding complexes.
Table 1.
Identified inter cross-links with BS3, BS2G and EDC cross-linking.
| IL-7 | IL-7Rα | Cross-linker | Reported Cross-linkable Cα- Cα Distance, Å | Measured Cα- Cα Distance in the Generated Model, Å | |
|---|---|---|---|---|---|
| 1 | K11 | K84 | BS3 | 9-3052 | 14.3 |
| 2 | K11 | K141 | BS3 | 9-3052 | 13.9 |
| 3 | D75 | K77 | EDC | 6-1653 | 9.8 |
| 4 | K69 | K78 | BS3 | 9-3052 | 17.2 |
| 5 | K152 | K141 | BS3 | 9-3052 | 24.2 |
| 6 | K8 | K84 | BS2G / BS3 | 6-26/9-3052 | 12.3a |
| 7 | N-term | K84 | BS2G / BS3 | 6-26/9-3052 | - |
| 8 | D2 | K84 | EDC | 6-1653 | - |
| 9 | D4 | K84 | EDC | 6-1653 | - |
| 10 | E6 | K84 | EDC | 6-1653 | - |
Cross-linked distance is measured at the Cα of D9 instead of K8 in crystal structure (PDB: 3DI2) because the N-terminal region from M1-K8 is missing in the X-ray structure.
For the IL-7Rα, there are four cross-linked residues; three of them, K77, K78 (cross-link 3 and 4) and K84 (cross-link 1 and cross-link 6 to 10) are on the elbow region of D1, and the fourth, K141 (cross-link 2 and 5) is located on the elbow loop of D2, showing discriminating evidence that the two elbow regions constitute the binding interface of IL-7Rα rather than allosteric regions. To delineate the interacting regions and the three-dimensional structure of the binding complexes, we turned to protein-protein docking based on crosslinking distance restraints to elucidate the quaternary structure.
Restraint-based protein-protein docking.
Protein-protein docking calculations were performed with the RosettaDock program to discern the likely structure of the IL-7/IL-7Rα complex in aqueous solution. Five representative intermolecular cross-links (i.e., crosslinks 1-5 in Table 1) were selected to define distance restraints. The other five cross-links involve N-terminal residues of IL-7 that are not resolved in the input X-ray crystal structure and, thus, are not included as docking restraints. Initial protein-protein docking experiments with RosettaDock utilized all five crosslink-derived restraints simultaneously. Clustering of the top-scoring 100 IL-7/IL-7Rα complex models (RosettaDock total score) was performed with the Rosetta cluster program with a 2.0 Å cluster radius. The largest cluster contained 53 similar models. We chose one representative model (Figure 2, IL-7 in purple and IL-7Rα in green) and overlaid it with the X-ray crystal structure (Figure 2, black). The r.m.s.d. difference across all alpha carbons is 1.7 Å which is less than 2.0 Å cutoff used to define a high-confidence model.
Figure 2.
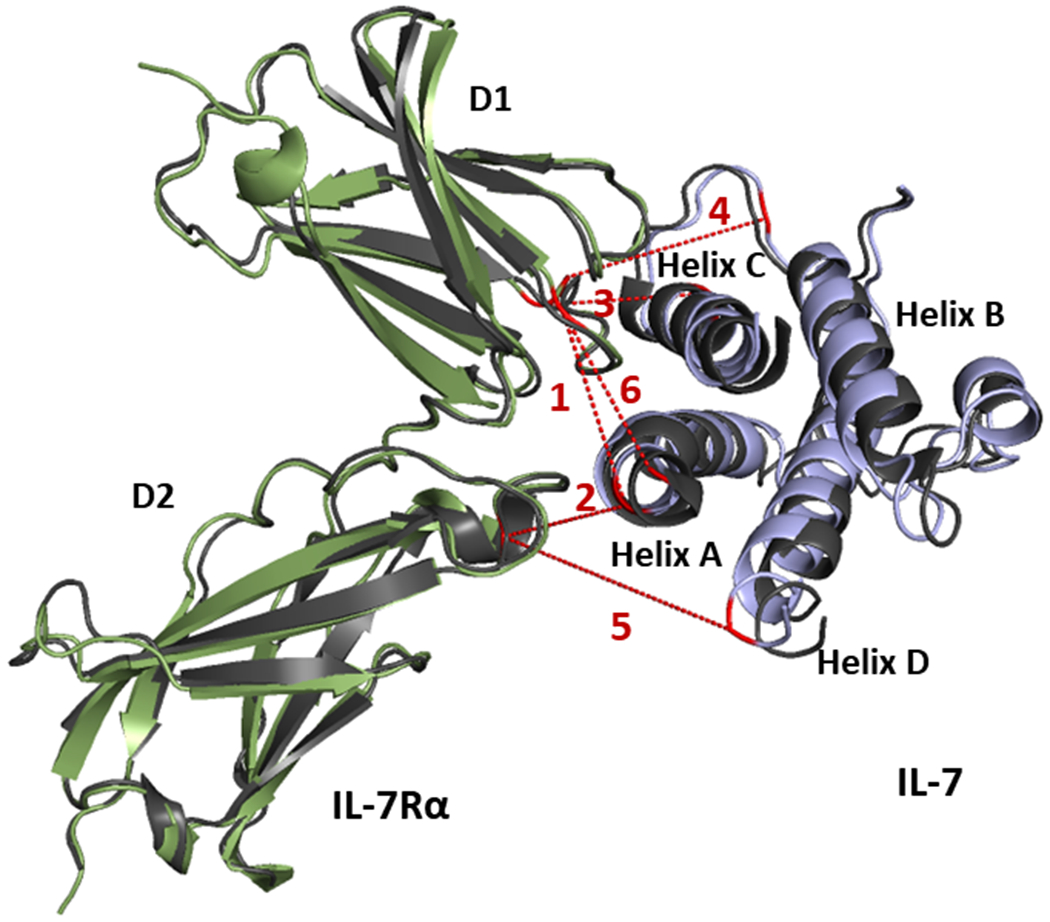
X-ray crystal structure of the IL-7/IL-7Rα (black, PDB: 3DI2) overlaid with generated docking models (green for IL-7Rα and purple for IL-7) that were generated using five intermolecular cross-links (1-5) as restraints. Cross-linked residues are in red, and dashed lines depict the corresponding crosslinked spans. The adjoining number corresponds to cross-links shown in Table 1.
We further mapped cross-links 1-6 onto the generated model (Figure 2). Although the N-terminal region from M1 to K8 in IL-7 is not resolved, cross-link 6 is located more closely than cross-links 7-10 to the resolved residue D9, allowing its Cα-Cα distance to be estimated and taken for mapping (Figure 2). Cross-links 7-10, therefore, are not mapped in the complex model. All of the measured Euclidean Cα- Cα distances are within the reported range52,53 for each specific cross-linker (Table 1). The excellent accord between the model and the X-ray crystal structure demonstrates the validity of solution-based data and the feasibility of using an integrative approach to obtain an accurate structure. In contrast, clustering of the top-scoring 100 models from an unrestrained docking exercise where more extensive sampling was performed (100K models) resulted in identification of only four two-membered clusters, none of which contained models that recapitulated the X-ray structure of the complex. The success of generating an accurate model provides insight about the binding interface, especially direct contacting regions that cannot be assigned by either stand-alone crosslinking or HDX.
Binding interface for IL-7 and IL-7Rα.
On D1 of IL-7Rα, the elbow region (Figure S2–c) is heavily protected upon binding and considered to interact primarily with helix C of the IL7. Not only the adjacent positions of these two domains, from directly viewing the docking model, but also a short EDC cross-link 3 further support their proximity. The observation is consistent with the contacting residues resolved from the crystal structure39, where K77 on the D1 elbow region of IL-7Rα forms H-bonds with D75 and K82 on helix C of IL-7. Furthermore, HDX kinetics reveals another participating region on the C-terminus of helix B, evidenced by the low deuterium uptake (Figure 1A–c) and cross-link 4. In addition, additional short cross-links (i.e., cross-links 8-10) indicate a third direct contacting region, the N-terminus of helix A, with D1 elbow region; the information is not found in the crystallographic study due to the partially unresolved structure. The other elbow region (Figure S2–e) on D2 of IL-7Rα is bridged to helix A of IL-7 by cross-link 2, closely locating those regions in the generated model. The cross-linked residue, K11, is the nearest linkable residue to Q23, which contributes to H-bond formation with the paired residue K138 in domain 239. The C-terminus of helix D adjoins the D2 elbow loop, as represented by cross-link 5. The less than 10% cumulative deuterium uptake difference in region 138-152 (Figure 1B) and distant location in the docking model, however, suggest involvement but not prominent interaction. Cross-link 1 and cross-links 6 and 7 obtained with BS3 span the D1 domain and helix A, delivering topological information about the binding complex and further show the limited information from stand-alone cross-linking chemistry. Those long cross-links are not highly effective restraints in terms of defining the interface unless coupled with other orthogonal methods. The integrated platform of using multiple cross-linkers, especially shorter ones, and of incorporating protein-protein docking significantly increases the certainty and defines better the directly contacting regions.
The number of intermolecular cross-links needed for a high-confidence model.
Excellent results were obtained in the protein-protein docking exercise that utilized five crosslink-derived restraints. However, in the context of the integrated approach, it is possible that accurate models might be obtained with fewer restraints. To examine this possibility, we conducted docking runs with all possible combinations of using 1 to 5 cross-links as distance restraints. For each restraint combination, we identified the top-scoring 100 IL-7/IL-7Rα docking models with RosettaDock total score and clustered them with the Rosetta cluster program by using the aforementioned criteria (Cα atom r.m.s.d. ≤ 2.0). For an unknown system, the clusters with the largest size are likely to be the most promising ones to choose. Thus, we summarized and evaluated all the largest clusters for each of the combinations (Figure 3). When using a restraint combination, multiple clusters of equal size are possible (e.g., for cross-link 1 and the combination of cross-links 4 and 5). Docking with larger number of restraints generally leads to larger cluster sizes; however, the size also depends on the cross-links chosen. We found that shorter cross-links are more demanding and lead to larger clusters. Supporting this contention is that the largest clusters in each category (e.g., one restraint, two restraints) all contain cross-link 3, the EDC cross-link. This observation further emphasizes the importance of employing a multiple-cross-linker strategy to provide both tight requirements for crosslinking and, at the same time, relatively loose restraints that allow many cross-links to form, thus aiding the downstream analysis.
Figure 3.
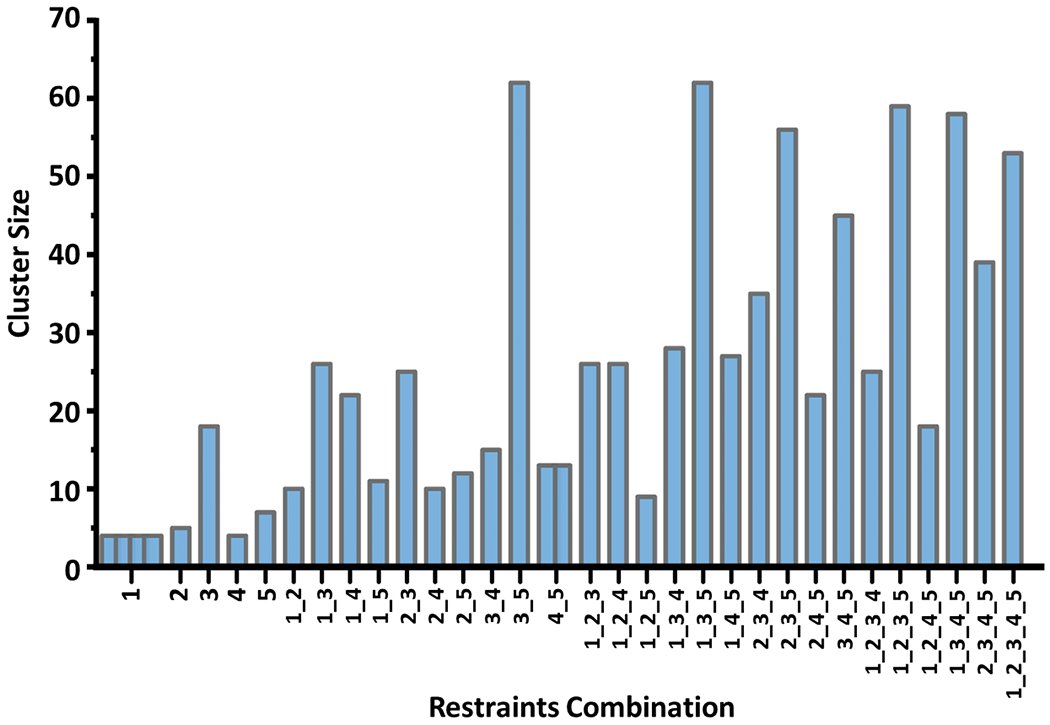
Cluster sizes for largest clusters of IL-7/IL-7Rα models generated with each possible combination of 1-5 crosslink-derived restraints from protein-protein docking. Several clusters with nearly equal members are formed for restraint combinations of cross-link 1 and combinations of cross-links 4 and 5.
Protein-protein docking with 10 different combinations of 5 crosslink-derived restraints taken two at a time yielded 11 largest clusters in total (Figure 3 and Table 2). Based on the structural similarity, we grouped and overlaid them into three different types (Figure 4). Our approach utilizes HDX results at this stage to evaluate the accuracy of the models. The omnipresent protection of IL-7Rα upon binding make IL-7Rα a poor choice to adjudicate the models. Instead, we chose only IL-7 HDX kinetics for the evaluation and mapped the residue-level cumulative deuterium uptake difference onto its structure as shown before (Figure 1). To place the comparison on a quantitative basis, we calculated the solvent accessible area (SASA) of the two most protected regions, 19VSIDQL24 and 83VSEGTTIL90, as indicated by HDX, for each generated model (Table 2). In the type 1 models, which are the most populated models given by seven different combinations (Table 2), the averaged SASA is 143 ± 12 Å and 212 ± 6 Å for 19VSIDQL24 and 83VSEGTTIL90, respectively. The values are significantly smaller than 230 Å and 319 Å in the unbound IL-7, suggesting there would be less deuterium uptake in the bound IL-7/IL-7Rα, as is seen in the HDX kinetics.
Table 2.
Summary of representative models from the largest clusters by using any two cross-links as docking restraints. The solvent accessible area (SASA) of two most protected regions in IL-7, as indicated by HDX kinetics, are calculated for each docking model and for the unbound-state IL-7 in Pymol.
| Crosslink-based Restraints | Size of Largest Cluster | Model Category | SASA of Peptide in IL-7, Å | Matching with HDX (Y/N) | Compared to X-ray Structure, r.m.s.d, Å | ||||
|---|---|---|---|---|---|---|---|---|---|
| 19VSIDQL24 | 83VSEGTTIL90 | ||||||||
| Bound IL-7 in Models | Unbound IL-7 | Bound IL-7 in Models | Unbound IL-7 | ||||||
| 1_2 | 10 | Type 3 | 230 | 230 | 268 | 319 | N | 12.3 | |
| 1_3 | 26 | Type 1 | 133 | 220 | Y | 1.7 | |||
| 1_4 | 22 | Type 2 | 230 | 247 | N | 11.0 | |||
| 1_5 | 11 | Type 1 | 141 | 211 | Y | 1.4 | |||
| 2_3 | 25 | Type 1 | 158 | 213 | Y | 1.9 | |||
| 2_4 | 10 | Type 2 | 230 | 269 | N | 10.9 | |||
| 2_5 | 12 | Type 1 | 137 | 209 | Y | 1.5 | |||
| 3_4 | 15 | Type 1 | 142 | 214 | Y | 1.7 | |||
| 3_5 | 62 | Type 1 | 160 | 201 | Y | 1.7 | |||
| 4_5 | 4_5.1 | 13 | Type 1 | 127 | 213 | Y | 1.3 | ||
| 4_5.2 | 13 | Type 2 | 230 | 253 | N | 10.9 | |||
Figure 4.
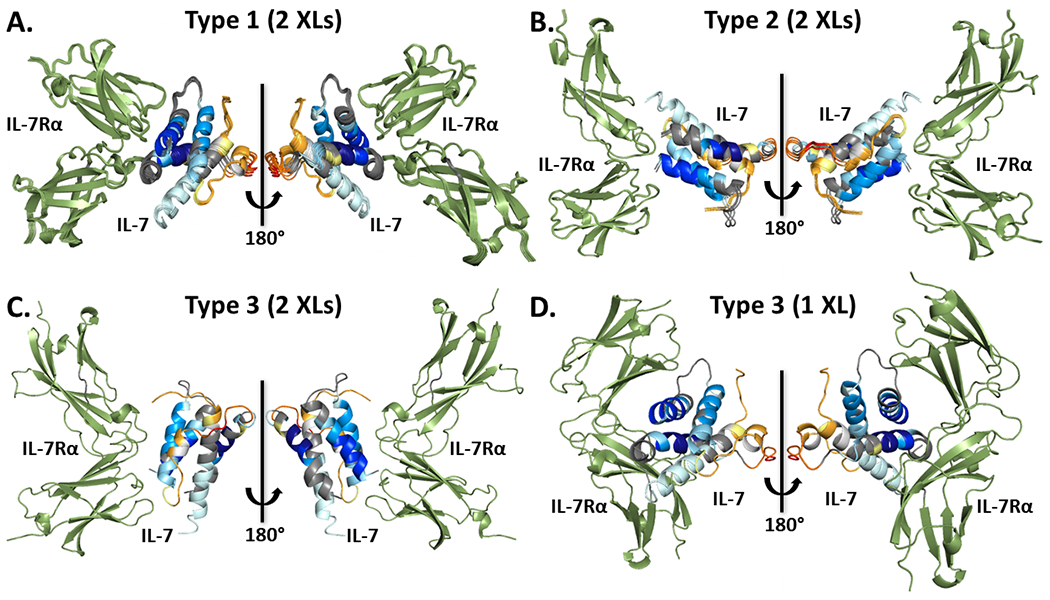
Different IL-7/IL-7Rα model-types, including type 1 (A), type 2 (B) and type 3 (C), generated from protein-protein docking with different combinations of two crosslink-derived restraints. IL-7 is mapped with the cumulative HDX uptake difference whereas IL-7Rα is colored in green. Similar models in each type are overlaid. (D) Type 3 IL-7/IL-7Rα model generated using only cross-link 1.
For the type 2 models (Figure 4B), however, the two regions are outside the contacting interface and nearly entirely exposed, evidenced explicitly by a SASA for the region 19VSIDQL24 of 230 ± 1 Å that is identical to the SASA of the unbound IL-7. This identity indicates no binding-induced protection on this region, in contradistinction to the HDX results. This inconsistency is further shown in other regions, for example the loops that connect different helices. Although type 2 models suggest the loops are the binding interface, HDX reports they either become more exposed upon binding or show no differences between two states (Figure 1B). The conflicting conclusions from HDX and docking forcefully decrease the likelihood of type 2 models to be correct. From similar reasoning, the type 3 model (Figure 4C) is also inconsistent with the HDX kinetics. Although the SASA of 83VSEGTTIL90 is 268 Å, smaller than that of unbound IL-7, the SASA of the other most protected region, 19VSIDQL24, is the same as that of unbound, indicating no involvement in the IL-7/IL-7Rα interaction. Instead, the suggested binding interface is consisted of a mini-helix and a flexible loop, which shows more deuterium uptake (Figure 1A–b) in HDX upon binding. Guidance from the HDX data allows us to reasonably rule out dubious docking models.
A subsequent evaluation between the remaining models, i.e. type 1 models, after HDX adjudication and comparison with the published crystal structure reveals only subtle structural differences (Table 2), all smaller than 2 Å r.m.s.d for all alpha carbons. These latter models meet the criteria promoted in this study to define a high-confidence model. Using this integrated platform, we can conclude that docking restraints based on two intermolecular cross-links are adequate to identify an accurate quaternary model for this binding complex.
Using three or four inter cross-links as restraints leads to better populated clusters and more well-defined IL-7/IL-7Rα models. Only two types of models (Figure S15) are generated and the second type can be excluded by incorporating the HDX results in the decision, where not the SASA values of both two peptides show differences between the bound and unbound IL-7 (Table S1). The remaining models are all considered as high-confidence models upon comparison to the X-ray structure. Although using more than two cross-links also gives an accurate model with no surprise, the successful exercise provides confidence for utilizing integrated methods to obtain HOS information.
When using only one cross-link as the docking restraint, however, seven different model-types are generated (Figure S16 and Figure 4D). Although some of these can survive HDX adjudication, they still deliver erroneous structures. Type 3 model (Figure 4D) is an example, showing the two most protected regions interacting with the IL-7Rα. The SASA value for 19VSIDQL24 and 83VSEGTTIL90 are 65 Å and 182.3 Å, respectively, all smaller than that of the unbound IL-7, suggesting consistent protection behavior observed from HDX kinetics. However, due to the mis-orientation of IL-7Rα, the r.m.s.d. difference in comparison with the crystal structure is 14.8 Å (Table S2), far exceeding the 2 Å threshold for a good model. A similar situation also applies to type 5 and type 7 models. Serving as a comparison control, one cross-link is insufficient to assign an accurate quaternary structure, thus the minimal number of cross-links to fulfill this goal is two.
Conclusion
This work highlights the effectiveness of the integrated approach to deliver quaternary structural information. Although the generality of the number “two cross-links” may vary from system to system, the prospect of generating a high-confidence model based on a few cross-links is clearly demonstrated. More crosslinks-derived restraints increase the likelihood of generating accurate models, but only modestly, and extensive efforts to identify large numbers of cross-links may not be justified for some protein-protein complexes.
The study also demonstrates successful utilization of HDX results in combination with XL-enabled docking studies to identify the IL-7/IL-7Rα binding interface and to predict the quaternary structure of the complex in solution. Use of multiple cross-linkers spanning diverse distances and targeting different amino acids increases the chances of identifying inter-protein contacting regions and the spatial-resolution; restraints derived from shorter crosslinking reagents more effectively focus the docking results towards native-like models. The excellent results achieved for the IL-7/IL-7Rα system may be due in part to the rich concentration of lysine residues along the binding interface, but for interfaces that are not lysine-rich, employing non-selective crosslinkers, (e.g., sulfo-NHS-SDA), can overcome that limitation.
Implementation of several complementary methods significantly enriches the structural information from tertiary to quaternary, compared to any stand-alone method. The delivery of a high-confidence model with only two experimental cross-links as restraints demonstrate the capability of an integrated platform. It can not only categorize changes arising from either binding-induced protection or remote structural changes but also define the binding interface more precisely with modeling. For many other protein complexes, which require considerable effort to crystalize or don’t crystallize at all, the approach is an intriguing alternative to obtain structural information. That information applies to the proteins in solution and can fill in regions that do not diffract well. The established approach will certainly aid the design of protein therapeutics.
Supplementary Material
Acknowledgement:
The authors thank Dr. Daniel A Weisz, from department of biology in WUSTL, for his generous help and Dr. Dina Schneidman, Hebrew University of Jerusalem, for helpful discussion. This work was supported by a Research Collaboration with Bristol-Myers Squibb and by the NIH 2P41GM103422. The authors thank Dr. Olafur Gudmundsson and Dr. Lois Lehman-McKeeman from Bristol-Myers Squibb for their support of this project.
Footnotes
Supporting Information
Material, experimental Details of Hydrogen-deuterium exchange, enzymatic in-solution digestion, LC-MS/MS analysis, protein-protein docking; HDX kinetics of bound/unbound IL-7; representative HDX kinetics of bound/unbound IL-7α with statistical analysis of cumulative HDX difference on each residue; HDX kinetics of bound/unbound of bound/unbound IL-7α; gel picture of cross-linked IL-7/IL-7α by BS3 and BS2G and EDC; mass spectrum of obtained cross-links; docking models generated using any three and four cross-links; summary of representative models from the largest clusters using any three and four cross-links; docking models generated using one cross-link; summary of representative models from the largest clusters using one cross-link (PDF).
The authors declare no competing financial interest.
References
- 1.Rebsamen M; Kandasamy RK; Superti-Furga G, Protein interaction networks in innate immunity. Trends in Immunology 2013, 34 (12), 610–9. [DOI] [PubMed] [Google Scholar]
- 2.Krohl PJ; Ludwig SD; Spangler JB, Emerging technologies in protein interface engineering for biomedical applications. Current Opinion Biotechnology 2019, 60, 82–88. [DOI] [PubMed] [Google Scholar]
- 3.Amit AG; Mariuzza RA; Phillips SE; Poljak RJ, Three-dimensional structure of an antigen-antibody complex at 2.8 A resolution. Science 1986, 233 (4765), 747–53. [DOI] [PubMed] [Google Scholar]
- 4.Mayer M; Meyer B, Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew Chemie International Edition in English 1999, 38 (12), 1784–1788. [DOI] [PubMed] [Google Scholar]
- 5.Weis DD, Hydrogen Exchange Mass Spectrometry of Proteins. Wiley Online Library: 2015. [Google Scholar]
- 6.Cummins DJ; Espada A; Novick SJ; Molina-Martin M; Stites RE; Espinosa JF; Broughton H; Goswami D; Pascal BD; Dodge JA; Chalmers MJ; Griffin PR, Two-Site Evaluation of the Repeatability and Precision of an Automated Dual-Column Hydrogen/Deuterium Exchange Mass Spectrometry Platform. Analytical Chemistry 2016, 88 (12), 6607–14. [DOI] [PubMed] [Google Scholar]
- 7.Wang HL; Shu Q; Rempel DL; Frieden C; Gross ML, Understanding curli amyloid-protein aggregation by hydrogen-deuterium exchange and mass spectrometry. International Journal of Mass Spectrometry 2017, 420, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang RY; Chen G, Higher order structure characterization of protein therapeutics by hydrogen/deuterium exchange mass spectrometry. Analytical andn Bioanalytical Chemistry 2014, 406 (26), 6541–58. [DOI] [PubMed] [Google Scholar]
- 9.Wang H; Shu Q; Rempel DL; Frieden C; Gross ML, Understanding Curli Amyloid-Protein Aggregation by Hydrogen-Deuterium Exchange and Mass Spectrometry. International Journal of Mass Spectrometry 2017, 420, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J; Wei H; Krystek SR Jr.; Bond D; Brender TM; Cohen D; Feiner J; Hamacher N; Harshman J; Huang RY; Julien SH; Lin Z; Moore K; Mueller L; Noriega C; Sejwal P; Sheppard P; Stevens B; Chen G; Tymiak AA; Gross ML; Schneeweis LA, Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Analytical Chemistry 2017, 89 (4), 2250–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chik JK; Schriemer DC, Hydrogen/deuterium exchange mass spectrometry of actin in various biochemical contexts. Journal of Molecular Biology 2003, 334 (3), 373–385. [DOI] [PubMed] [Google Scholar]
- 12.Pan JX; Han J; Borchers CH; Konermann L, Characterizing Short-Lived Protein Folding Intermediates by Top-Down Hydrogen Exchange Mass Spectrometry. Analytical Chemistry 2010, 82 (20), 8591–8597. [DOI] [PubMed] [Google Scholar]
- 13.Sterling HJ; Williams ER, Real-time hydrogen/deuterium exchange kinetics via supercharged electrospray ionization tandem mass spectrometry. Analytical Chemistry 2010, 82 (21), 9050–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang RY; Krystek SR Jr.; Felix N; Graziano RF; Srinivasan M; Pashine A; Chen G, Hydrogen/deuterium exchange mass spectrometry and computational modeling reveal a discontinuous epitope of an antibody/TL1A Interaction. MAbs 2018, 10 (1), 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinz A, Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrometry Reviews 2006, 25 (4), 663–682. [DOI] [PubMed] [Google Scholar]
- 16.Yu C; Huang L, Cross-Linking Mass Spectrometry: An Emerging Technology for Interactomics and Structural Biology. Analytical Chemistry 2018, 90 (1), 144–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gotze M; Pettelkau J; Schaks S; Bosse K; Ihling CH; Krauth F; Fritzsche R; Kuhn U; Sinz A, StavroX-A Software for Analyzing Crosslinked Products in Protein Interaction Studies. Journal of the American Society Mass Spectrometry 2012, 23 (1), 76–87. [DOI] [PubMed] [Google Scholar]
- 18.Rey M; Sarpe V; Burns KM; Buse J; Baker CAH; van Dijk M; Wordeman L; Bonvin AMJJ; Schriemer DC, Mass Spec Studio for Integrative Structural Biology. Structure 2014, 22 (10), 1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang B; Wu YJ; Zhu M; Fan SB; Lin JZ; Zhang K; Li S; Chi H; Li YX; Chen HF; Luo SK; Ding YH; Wang LH; Hao ZQ; Xiu LY; Chen S; Ye KQ; He SM; Dong MQ, Identification of cross-linked peptides from complex samples. Nature Methods 2012, 9 (9), 904-+. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt C; Robinson CV, A comparative cross-linking strategy to probe conformational changes in protein complexes. Nature Protocol 2014, 9 (9), 2224–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q; Feng T; Xu L; Sun H; Pan P; Li Y; Li D; Hou T, Recent Advances in Protein-Protein Docking. Current Drug Targets 2016, 17 (14), 1586–1594. [DOI] [PubMed] [Google Scholar]
- 22.Park H; Lee H; Seok C, High-resolution protein–protein docking by global optimization: recent advances and future challenges. Current Opinion in Structural Biology 2015, 35, 24–31. [DOI] [PubMed] [Google Scholar]
- 23.Gromiha MM; Yugandhar K; Jemimah S, Protein–protein interactions: scoring schemes and binding affinity. Current Opinion in Structural Biology 2017, 44, 31–38. [DOI] [PubMed] [Google Scholar]
- 24.Moal IH; Moretti R; Baker D; Fernandez-Recio J, Scoring functions for protein–protein interactions. Current Opinion in Structural Biology 2013, 23 (6), 862–867. [DOI] [PubMed] [Google Scholar]
- 25.Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH, Solving protein structures using short-distance cross-linking constraints as a guide for discrete molecular dynamics simulations. Science Advances 2017, 3 (7), e1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rampler E; Stranzl T; Orban-Nemeth Z; Hollenstein DM; Hudecz O; Schlögelhofer P.; Mechtler K, Comprehensive cross-linking mass spectrometry reveals parallel orientation and flexible conformations of plant HOP2–MND1. Journal of Proteome Research 2015, 14 (12), 5048–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin S-J; Chen Y-F; Hsu K-C; Chen Y-L; Ko T-P; Lo C-F; Wang H-C; Wang H-C, Structural Insights to the Heterotetrameric Interaction between the Vibrio parahaemolyticus PirAvp and PirBvp Toxins and Activation of the Cry-Like Pore-Forming Domain. Toxins 2019, 11 (4), 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Komolov KE; Du Y; Duc NM; Betz RM; Rodrigues JP; Leib RD; Patra D; Skiniotis G; Adams CM; Dror RO, Structural and functional analysis of a β2-adrenergic receptor complex with GRK5. Cell 2017, 169 (3), 407–421. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elenkov IJ; Iezzoni DG; Daly A; Harris AG; Chrousos GP, Cytokine dysregulation, inflammation and well-being. Neuroimmunomodulation 2005, 12 (5), 255–69. [DOI] [PubMed] [Google Scholar]
- 30.Walsh STR, Structural insights into the common gamma-chain family of cytokines and receptors from the interleukin-7 pathway. Immunological Review 2012, 250, 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kittipatarin C; Khaled AR, Interlinking interleukin-7. Cytokine 2007, 39 (1), 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao JB; Zhao LT; Wan YSY; Zhu B, Mechanism of Action of IL-7 and Its Potential Applications and Limitations in Cancer Immunotherapy. International J Molecular Sciences 2015, 16 (5), 10267–10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.ElKassar N; Gress RE, An overview of IL-7 biology and its use in immunotherapy. Journal of Immunotoxicology 2010, 7 (1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lundstrom W; Fewkes NM; Mackall CL, IL-7 in human health and disease. Seminars in Immunology 2012, 24 (3), 218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Habibi M; Kmieciak M; Graham L; Morales JK; Bear HD; Manjili MH, Radiofrequency thermal ablation of breast tumors combined with intralesional administration of IL-7 and IL-15 augments anti-tumor immune responses and inhibits tumor development and metastasis. Breast Cancer Research and Treatment 2009, 114 (3), 423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pellegrini M; Calzascia T; Elford AR; Shahinian A; Lin AE; Dissanayake D; Dhanji S; Nguyen LT; Gronski MA; Morre M; Assouline B; Lahl K; Sparwasser T; Ohashi PS; Mak TW, Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies (vol 15, pg 528, 2009). Nature Medicine 2009, 15 (7), 819–819. [DOI] [PubMed] [Google Scholar]
- 37.Mackall CL; Fry TJ; Gress RE, Harnessing the biology of IL-7 for therapeutic application. Nature Review Immunology 2011, 11 (5), 330–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gershoni JM; Roitburd-Berman A; Siman-Tov DD; Tarnovitski Freund N; Weiss Y, Epitope mapping: the first step in developing epitope-based vaccines. BioDrugs 2007, 21 (3), 145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McElroy CA; Dohm JA; Walsh STR, Structural and Biophysical Studies of the Human IL-7/IL-7R alpha Complex. Structure 2009, 17 (1), 54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu WH; Roemer SC; Zhou Y; Shen Z-J; Dennehey BK; Balsbaugh JL; Liddle JC; Nemkov T; Ahn NG; Hansen KC, The Cac1 subunit of histone chaperone CAF-1 organizes CAF-1-H3/H4 architecture and tetramerizes histones. Elife 2016, 5, e18023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mysling S; Kristensen KK; Larsson M; Beigneux AP; Gårdsvoll H; Fong LG; Bensadouen A; Jørgensen TJ; Young SG; Ploug M, The acidic domain of the endothelial membrane protein GPIHBP1 stabilizes lipoprotein lipase activity by preventing unfolding of its catalytic domain. Elife 2016, 5, e12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng J; Corzo C; Chang MR; Shang J; Lam VQ; Brust R; Blayo A-L; Bruning JB; Kamenecka TM; Kojetin DJ, Chemical crosslinking mass spectrometry reveals the conformational landscape of the activation helix of PPARγ; a model for ligand-dependent antagonism. Structure 2018, 26 (11), 1431–1439. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rimmer MA; Nadeau OW; Artigues A; Carlson GM, Structural characterization of the catalytic γ and regulatory β subunits of phosphorylase kinase in the context of the hexadecameric enzyme complex. Protein Science 2018, 27 (2), 485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhat JY; Miličić G; Thieulin-Pardo G; Bracher A; Maxwell A; Ciniawsky S; Mueller-Cajar O; Engen JR; Hartl FU; Wendler P, Mechanism of enzyme repair by the AAA+ chaperone Rubisco activase. Molecular Cell 2017, 67 (5), 744–756. e6. [DOI] [PubMed] [Google Scholar]
- 45.Lu S; Fan SB; Yang B; Li YX; Meng JM; Wu L; Li P; Zhang K; Zhang MJ; Fu Y; Luo JC; Sun RX; He SM; Dong MQ, Mapping native disulfide bonds at a proteome scale. Nature Methods 2015, 12 (4), 329–U73. [DOI] [PubMed] [Google Scholar]
- 46.Bender BJ; Cisneros III A; Duran AM; Finn JA; Fu D; Lokits AD; Mueller BK; Sangha AK; Sauer MF; Sevy AM, Protocols for molecular modeling with Rosetta3 and RosettaScripts. Biochemistry 2016, 55 (34), 4748–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simons KT; Kooperberg C; Huang E; Baker D, Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. Journal of Molecular Biology 1997, 268 (1), 209–225. [DOI] [PubMed] [Google Scholar]
- 48.Simons KT; Ruczinski I; Kooperberg C; Fox BA; Bystroff C; Baker D, Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins: Structure, Function, and Bioinformatics 1999, 34 (1), 82–95. [DOI] [PubMed] [Google Scholar]
- 49.Gray JJ; Moughon S; Wang C; Schueler-Furman O; Kuhlman B; Rohl CA; Baker D, Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. Journal of Molecular Biology 2003, 331 (1), 281–299. [DOI] [PubMed] [Google Scholar]
- 50.Chaudhury S; Berrondo M; Weitzner BD; Muthu P; Bergman H; Gray JJ, Benchmarking and analysis of protein docking performance in Rosetta v3. 2. PloS One 2011, 6 (8), e22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McElroy CA; Holland PJ; Zhao P; Lim JM; Wells L; Eisenstein E; Walsh ST, Structural reorganization of the interleukin-7 signaling complex. Proceedings of the National Academy of Sciences of the United States of America 2012, 109 (7), 2503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leitner A; Faini M; Stengel F; Aebersold R, Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines. Trends Biochemical Sciences 2016, 41 (1), 20–32. [DOI] [PubMed] [Google Scholar]
- 53.Leitner A; Joachimiak LA; Unverdorben P; Walzthoeni T; Frydman J; Forster F; Aebersold R, Chemical cross-linking/mass spectrometry targeting acidic residues in proteins and protein complexes. Proceedings of the National Academy of Sciences of the United States of America 2014, 111 (26), 9455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.