

Abstract
We report an improved and scalable synthesis of MIDD0301, a positive GABAA receptor modulator that is under development as oral and inhaled treatments for asthma. In contrast to other benzodiazepines in clinical use, MIDD0301 is a chiral compound that has limited brain absorption. The starting material to generate MIDD0301 is 2-amino-5-bromo-2’-fluorobenzophenone, which has a non-basic nitrogen due to electron withdrawing substituents in the ortho and para positions, reducing its reactivity towards activated carboxylic acids. Investigations of peptide coupling reagents on multigram scale resulted in moderate yields due to incomplete conversions. Secondly, basic conditions used for the formation of the seven-membered 1,4-diazepine ring resulted in racemization of the chiral center. We found that neutral conditions comparable to the pKa of the primary amine were sufficient to support the formation of the intramolecular imine but did not enable the simultaneous removal of the protecting group. Both difficulties were overcome with the application of the N-carboxyanhydride of D-alanine. Activated in the presence of acid, this compound reacted with non-basic 2-amino-5-bromo-2’-fluorobenzophenone and formed the 1,4-diazepine upon neutralization with triethylamine. Carefully designed workup procedures and divergent solubility of the synthetic intermediates in solvents and solvent combinations were utilized to eliminate the need for column chromatography. To improve compatibility with large scale reactors, temperature-controlled slow addition of reagents generated the imidazodiazepine at −20 °C. All intermediates were isolated with a purity of >97% and impurities were identified and quantified. After the final hydrolysis step, MIDD0301 was isolated in a 44% overall yield and purity of 98.9% after recrystallization. The enantiomeric excess was greater than 99.0%.
Keywords: GABAA receptor, asthma, imidazodiazepine, amino acid N-carboxyanhydride
Graphical Abstract

Introduction
The synthesis of 6-phenyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylic acid derivatives was patented in 19761 and two years later published by Walser et al.2 Three different benzodiazepines were used as starting material with the following leaving groups in the 2 position: N-nitrosomethylamino,3 dimorpholinylphosphinyloxy4 and diethyl phosphate. However, the synthesis included 5–6 steps in an overall yield of 10–14%. A significant improvement was accomplished in the same year using a phenyl stabilized nitrone5 under basic conditions that generated only 1-phenyl substituted ethyl 8-chloro-6-(2-fluorophenyl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylates in one step with yields as high as 95%.6 An alternative route was developed starting from quinazoline 3-oxides using carbanions that gave substituted 6-phenyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylates in 45–55% yield using four sequential reactions.7 The first general one-pot procedure to convert readily accessible 5-(2-fluorophenyl)-7-nitro-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one to ethyl 6-(2-fluorophenyl)-8-nitro-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate using diethyl chlorophosphate and ethyl isocyanoacetate was published in 1978 but no yields were reported.8 The reaction was carried out with the starting material in tetrahydrofuran (THF) at 0 °C and the addition of potassium t-butoxide (t-BuOK) followed by sequential addition of diethyl chlorophosphate and a THF solution of ethyl isocyanate and t-BuOK. More than a decade later Watjen et al.9 used a similar method by reacting diazepines with sodium hydride (NaH) in dimethylformamide (DMF), followed by the sequential addition of diethyl chlorophosphate at −20 °C and a THF solution of lithium isopropyl amide and ethyl isocyanate at −78 °C. The only yield reported was 47% for ethyl 8-chloro-6-oxo-5,6-dihydro-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate. A 44% yield was reported for ethyl 8-nitro-6-(4-chlorophenyl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate when the starting material and t-BuOK were stirred in THF at 0 °C, followed by the sequential addition of diethyl chlorophosphate, ethyl isocyanate and t-BuOK.10 Using ethyl (E)-2-(((dimethylamino)methylene)amino)acetate instead of ethyl isocyanoacetate did not improve the yield of this reaction.11 An improved yield of 58% for ethyl 8-bromo-6-(2-fluorophenyl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate was achieved when the starting material was treated with NaH in THF, followed by the dropwise addition of diethyl chlorophosphate and a slow addition of ethyl isocyanate and NaH.12 A systematic analysis of this reaction identified t-BuOK as the best base used at 1.1 equiv. for the reaction of the starting material and diethyl chlorophosphate (1.3 equiv.) at 0 °C followed by the slow sequential addition of ethyl isocyanate (1.1 equiv.) and NaH (1.1 equiv.) at −35 °C or below.13 A combined yield of 87% was reported for ethyl 8-chloro-6-(2-phenyl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate when precipitated in diethyl ether and the remainder isolated from the mother liquor using column chromatography. Several subsequent publications have used these conditions but the necessity for column chromatography to achieve moderate to good yields precluded multigram scale production.14–18 Herein, we describe an improved four step synthesis of (R)-8-bromo-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylic acid (MIDD0301) that enabled us to manufacture 88 g of this chiral compound without racemization. MIDD0301 is a clinical drug candidate for asthma that binds allosterically to the gamma amino butyric acid type A receptor (GABAAR).19 Several in vivo studies have shown that inhaled and orally administered MIDD0301 reduced airway smooth muscle constriction and lung inflammation without toxicity.20–22
Results and Discussion
The synthesis of many benzodiazepines used clinically involves the reaction of substituted 2-aminobenzophenones and activated carboxylic acids or acid halides. Our published procedure for MIDD0301 involved the addition of N,N’-dicyclohexylcarbodiimide (DCC) as coupling reagent to react 1 and Boc-D-alanine (Table 1, entry 1),12 but incomplete conversion of 1 was observed for a 204 mmol scale reaction resulting in 58.6% yield of 2. A longer reaction time (48 h) did not appreciably increase the yield (Table 1, entry 2), nor did a reaction temperature of 40 °C (Table 1, entry 3) or the addition of more DCC (2 equiv.) and Boc-D-alanine (2 equiv.) (Table 1, entry 4). The addition of Boc-D-alanine to a solution of 1 and DCC in dichloromethane resulted in a yield similar to adding DCC to a solution of Boc-D-alanine and 1 (Table 1, entry 5).
Table 1:
Reaction between 1 and Boc-D-alanine.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Coupling Reagent | Boc-D-Alanine | Additive | Conditions | Solvent | Yield (%) |
| 1a | DCC, 1.2 eq | 1.0 eq | None | 24 hr, rtg | CH2Cl2 | 59 |
| 2b | DCC, 1.2 eq | 1.0 eq | None | 48 hr, rtg | CH2Cl2 | 60 |
| 3b | DCC, 1.2 eq | 1.0 eq | None | 24 hrg, 40 ºC | CH2Cl2 | 55 |
| 4 b | DCC, 2.0 eq | 2.0 eq | None | 24 hr, rtg | CH2Cl2 | 62 |
| 5b,c | DCC, 1.2 eq | 1.0 eq | None | 24 hr, rtg | CH2Cl2 | 57 |
| 6b,d | DCC, 1.2 eq | 1.0 eq | None | 24 hr, rtg | CH2Cl2 | 10 |
| 7b | DCC, 1.2 eq | 1.0 eq | DMAP, 0.1 eq | 24 hr, rtg | CH2Cl2 | 53 |
| 8b | DCC, 1.2 eq | 1.0 eq | DMAP, 1.0 eq | 24 hr, rtg | CH2Cl2 | 50 |
| 9b | DCC, 1.2 eq | 1.0 eq | None | 24 hr, rtg | CH3CN | 9 |
| 10e | EDCI, 1.2 eq | 1.0 eq | TEA, 1.2 eq | 24 hr, rtg | CH2Cl2 | 10 |
| 11e | EDCI, 1.2 eq | 1.0 eq | TEA, 1.2 eq DMAP, 1.2 eq |
24 hr, rtg | CH2Cl2 | 39 |
| 12f | Methyl chloroformate | 1.5 eq | NMM, 3 eq | 24 hr, rtg | THF | 50 |
| 13f | Ethyl chloroformate | 1.5 eq | NMM, 3 eq | 24 hr, rtg | THF | 25 |
| 14f | Isobutyl chloroformate | 1.5 eq | NMM, 3 eq | 24 hr, rtg | THF | 22 |
204 mmol scale
68 mmol scal
Addition of Boc-D-alanine to a solution of 1 and DCC
Addition of 1 to a solution of DCC and Boc-D-alanine
22.1 mmol scale
21.6 mmol scale
rt = room temperature
However, adding 1 to a solution of Boc-D-alanine and DCC reduced the yield of 2 to 9.6% (Table 1, entry 6), probably due to the formation of an N-acylurea species proposed by Valuer and Bradlely.23 Additives such as 4-dimethylaminopyridine (DMAP) were first reported by Neises and Steglich24 for amide coupling reactions as an acyl transfer agent to circumvent the 1,3-rearrangement problem of the O-acylisourea intermediate by rapidly converting it into a more stable DMAP complex. However, the addition of DMAP in catalytic or stoichiometric amounts did not improve the yield of 2 (Table 1, entries 7 and 8). The application of acetonitrile instead of dichloromethane (CH2Cl2) reduced the yield to 8.7% for this reaction (Table 1, entry 9). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) was investigated instead of DCC as coupling reagent in the presence of triethylamine (TEA) (Table 1, entry 10) or DMAP (Table 1, entry 11). Both reactions gave lower yields in comparison with the DCC mediated reaction. Next, the mixed carbonic anhydride approach was explored to form the amide bond25 using methyl-, ethyl-, and isobutylchloroformates (Table 1, entries 12–14). These conditions were reported by Anderson, et al.26 and later used in the synthesis of similar benzodiazepine analogs by Reddy, et al.27 The reaction using methyl chloroformate in the presence of N-methylmorpholine (NMM) gave the best yield for 2 with 50.0%. From our observation of similar yields using different activation reagents for Boc-D-alanine, it can be concluded that the nucleophilicity of 1 is the controlling aspect of this reaction. The pKa of 1 is −1.12 in comparison to 4.61 for aniline.28 This significant change is based on the electron withdrawing character of the acetophenone and bromine substituents in the ortho and para position, respectively. Although the pKa is a measurement of basicity, it is directly related to nucleophilicity as both parameters define the electronic nature of the nitrogen atom. Another important parameter of an amide bond formation reaction is steric hindrance. Therefore, we employed highly reactive acyl chlorides for our next approach as reported for synthesis of achiral benzodiazepines (Scheme 1).29
Scheme 1.

Synthesis of 5 using (9H-fluoren-9-yl)methyl (R)-(1-chloro-1-oxopropan-2-yl)carbamate.
The procedure reported by Carpino et al.30 and Prabhu et al.31 was followed to synthesize 147.5 g of 3 with 93% yield. The reaction between 1 and 3 was carried out at room temperature and complete conversion was observed after 30 min on a 0.4 M scale. Six equiv. of piperidine were added slowly to the reaction mixture to form compound 4. The crude product was dissolved in methanol, followed by the addition of 1.6 M aqueous sodium hydroxide to enable the formation of 5 after 12 h at room temperature with 68% yield. Despite the increased yield, racemization was observed for this process. For similar achiral compounds such as nitrazepam32 and nordiazepam,28 the amide hydrogen is relative acidic with a pKa of 10.8 and 11.6, respectively. Resonance structures can be employed to show racemization of the α-carbon atom under strongly basic conditions. This is supported by the fact that the same reaction in the absence of the aqueous sodium hydroxide produced 5 with an enantiomeric excess of 76.9% when performed on a 0.01 M scale. Further analysis confirmed that traces of piperidine were still present during the cyclization step due to insufficient removal by multiple washes with water and aqueous bicarbonate during the workup. Because basic conditions induce racemization, acid labile protecting groups such as t-butyl carbamates were investigated for the formation of 5 on multigram scale.
In contrast to Fmoc, Boc groups can be removed in the presence of acid, which has been reported for 2 using HCl gas. Alternatively, HCl in dioxane was used for multigram scale (Table, 2, entries 2–4). After neutralizing aqueous workup, cyclization of the crude product 4 was achieved in methanol:water at pH 8.5 using 0.1 equiv. of sodium hydroxide. The product formed for this reaction was optically pure (Table 2, entry 1). 12
Table 2:
1,4-Diazepine formation reaction with 2.
 | ||||
|---|---|---|---|---|
| Entry | a) | b) | Yield % | % ee |
| 1a | HCl (g), saturated, rtd, 18 h | MeOH / H2O (9 : 1), 0.1 eq NaOH, rtd, 10 h | 8212 | 99.512 |
| 2b | HCl in dioxane, 4 eq, rtd, 4 h | MeOH / H2O (9 : 1), 0.1 eq NaOH, rtd, 48 h | 84 | 53.9 |
| 3c | HCl in dioxane, 4 eq, rtd, 4 h | MeOH / H2O (9 : 1), 1 eq NaOH, rtd, 12 h | 50 | 0 |
| 4c | HCl in dioxane, 4 eq, rtd, 4 h | Toluene, 60 °C, 4 h | 48 | 99.2 |
60 mM scale
116 mM scale
122 mM scale
rt = room temperature
Longer reaction times were required for complete conversion of 4 and scaling the reaction from 60 mM to 116 mM led to racemization resulting in an enantiomeric excess of 53.9% for 5 (Table 2, entry 2). To reduce the reaction time, one equiv. of sodium hydroxide was used instead of 0.1 equiv. Full conversion was achieved in 12 h but a racemic product was obtained (Table 2, entries 3). In an attempt to circumvent the use of base, crude product 4 was heated in toluene at 60 °C for 4 h, which gave 5 in a yield of 50% and an enantiomeric excess of 99.2% (Table 2, entry 4).
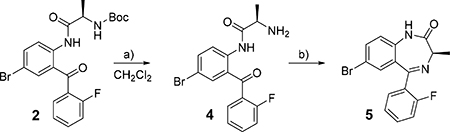
To overcome the moderate yield of 2 mentioned earlier and racemization encountered in both approaches, we followed a procedure developed by Fier and Whitakker33 using amino acid N-carboxyanhydrides (NCAs) to synthesize optically pure benzodiazepines. Compound 6 was synthesized using triphosgene and D-alanine, as reported,34, 35 however a semisolid product was obtained in less than 50% yield. Employing Boc-D-alanine instead D-alanine, 6 was synthesized using triphosgene and triethylamine using a procedure developed by Wilder and Mobashery.36 On a 0.53M scale, 35 g of pure crystalline 6 (58% yield) was obtained from a dichloromethane solution triturated with hexanes.
As reported for natural NCAs, 6 reacted with 1 in the presence of 2 equiv. of trifluoroacetic acid (TFA) releasing carbon dioxide and forming the salt form of 4 (Scheme 2). Two equiv. of triethylamine were added to the reaction vessel to neutralize the acid and to liberate amine 4 enabling the formation of imine 5 and water. In contrast to the reported procedure, the reaction was carried out at 50 °C with longer reactions times. Upon reaction completion, an aqueous work-up removed the triethylammonium trifluoroacetate and 5 was triturated in 10% ethyl acetate in heptane to remove traces of 1 and oligomeric impurities providing 127 g (77% yield). The purity (absolute area %) of 5 was 97.3% and the enantiomeric excess was 98.8%. Impurities found were compound 5a + 1 (0.7 %) and 5b (2.0 %) (Figure 1 and Supporting Information).
Scheme 2.

Improved scale-up synthesis of MIDD0301.
Figure 1.

Purity determination of each synthesis intermediate for manufactured MIDD0301
The third step of the MIDD0301 synthesis installs the imidazole ring to produce imidazodiazepine 7. The established procedure13 involved the formation of an iminophosphate in the presence of base (potassium t-butoxide), followed by addition of diethylchlorophosphate. The subsequent reaction with the enolate of ethyl isocyanoacetate yielded the desired imidazobenzodiazepine. Earlier trial reactions monitored by thin layer chromatography (TLC) showed that the iminophosphate was not formed until a reaction temperature of −20 °C was reached. Similarly, it was observed that the reaction onset temperature of the iminophosphate with the enolate of ethyl isocyanoacetate was above −30 °C. Therefore, 5 in THF was cooled to −20 °C and the reagents adjusted to room temperature were added dropwise sequentially ensuring a reaction temperature of −20 °C throughout the process with the use of a dry ice/isopropanol bath. After a 30 min addition of t-BuOK (1.3 equiv.) in THF (1.56M), the reaction mixture was stirred for an additional 30 min to ensure complete deprotonation of the amide starting material. Next, diethylchlorophosphate (1.4 equiv.) was added dropwise over 15 minutes, followed by 2 h of stirring at −20 °C to complete the formation of iminophosphate as indicated by TLC. Then, ethyl isocyanoacetate (1.3 equiv.) was added dropwise over 15 min followed directly by a 30 min addition of t-BuOK (1.3 equiv.) in THF (1.56M) at −20 °C. After the addition, the cooling bath was removed and the reaction mixture stirred for an additional 1 h once room temperature was reached, at which point full conversion was observed by TLC. After aqueous workup, the crude product was treated with t-butyl methyl ether at 55 °C for 30 min to dissolve impurities. After stirring at room temperature for 2 h, 7 was collected by filtration and washed with t-butyl methyl ether. The synthesis provided 96.6 g (61% yield) of 7 with a purity of 97.2%. Identified impurities were the t-butyl ester of MIDD0301 (7a, 1.4 %) and the 6H isomer 7b (1.4 %) (Figure 1 and Supporting Information). The transesterification with t-BuOK and formation of the 6H isomer of imidazobenzodiazepines under these reaction conditions have been reported.37, 38 The enantiomeric excess of 7 was 99.0 %. Using our published hydrolysis conditions to scale-up synthesis of MIDD0301 (8 equiv. of sodium hydroxide in ethanol) an insoluble and gel-like MIDD0301 sodium salt was observed. To overcome this problem a 1:4 ratio of water and THF was employed, which remained as a biphasic solution throughout the process. The initial 8 equiv. of sodium hydroxide were reduced to 4 equiv. and the reaction temperature lowered from 78 °C to 50 °C, achieving full conversion of the starting material within 4 h. Our group recently reported the pH dependent equilibrium between a closed seven-membered ring and the open-ring acyclic benzophenone structure of MIDD0301.20 Although both compounds are interconvertible without racemization, they exhibited different aqueous solubility. The lowest aqueous solubility of MIDD0301 was observed a pH 3–6. Therefore, acetic acid was used to adjust the pH to 5.0 after dilution with water. The isolated yield was 98% with a purity of 97.2%. One impurity of this product was 7a (Figure 1 and Supporting Information), which was removed by recrystallization in ethanol. The final yield after recrystallization was 94% with a purity of 98.9 %. The 6H isomer MIDD0301a (1.14%) (Figure 1 and Supporting Information) was the only impurity. The enantiomeric excess of MIDD0301 was >99.0%.
Conclusion
We conclude that NCAs are superior amino acid derivatives for reaction with non-basic nitrogen atoms. Furthermore, these compounds obviate the need for protecting groups and therefore enable a one pot peptide coupling reaction and imine formation without racemization. The careful adjustment of solvent mixtures can avoid column chromatography by simply dissolving byproducts and precipitating the product. Furthermore, slow addition of reagents to form the imidazole ring can significantly reduce that need for low reaction temperatures if products like imidazodiazepines are thermodynamically stable. Other important achievements reported are multiple reactions carried out in the same reaction vessel and the synthesis of MIDD0301 in three steps. Classified metal salts and class 1 solvents are not employed and optically pure MIDD0301 was reliably generated at intermediate scale.
Experimental Procedure
All reactions were performed in round-bottom flasks with overhead mechanical stirrers under an argon atmosphere. Chemicals were purchased from either Millipore Sigma, Oakwood Chemical, Alfa Aesar, Matrix Scientific, or Acros Organic and used as received. Reaction progress was monitored by silica gel TLC (Dynamic Adsorbents Inc.) with fluorescence indicator. 1H, 13C and 19F-NMR spectra were obtained on Bruker 300 MHz and 500 MHz instruments with the chemical shifts in δ (ppm) reported by reference to the deuterated solvents as an internal standard DMSO-D6: δ = 2.50 ppm (1H-NMR) and δ = 39.52 ppm (13C-NMR) and CDCl3: δ = 7.20 ppm (1H-NMR) and δ = 77.00 ppm (13C-NMR) (see Supporting Information for NMR spectra). HRMS spectral data were recorded using a LCMS-IT-TOF spectrometer (Shimadzu). High performance liquid chromatography (Shimadzu Nexara series HPLC) coupled with a Photo Diode Array detector (PDA, Shimidzu SPD-M30A) and a single quadrupole mass analyzer (LCMS 2020, Shimadzu, Kyoto, Japan) was used for purity analysis (absolute area %). Analytes were separated using a Restek Pinnacle-C18 (4.6 mm x 50 mm, 5 μm particle size) column with gradient elution of water and methanol (0.1% formic acid) at a flow rate of 0.8 mL/min. Optical purity was determined with an Agilent 1100 HPLC system using a DAD detector. The mobile phase consisted of HPLC grade ethanol and n-hexane and the stationary phase was a Pirkle Whelk-01 column (4.6 mm x 25 cm, 5 μm particle size) to separate 5 and 7 and a Chiralpak IB-N3 column (4.6 mm x 15 cm, 3 μm) for MIDD0301. Trifluoroacetic acid (0.1%) was used as modifier for the analysis of MIDD0301.
Tert-Butyl-(R)-(1-((4-bromo-2-(2-fluorobenzoyl)phenyl)amino)-1-oxopropan-2-yl) carbamate (2) (Table 1, entries 1–9)
2-Amino-5-bromo-2’fluorobenzophenone (1) (60 g, 204 mmol) and Boc-D-alanine (38.6 g, 204 mmol) were dissolved in anhydrous dichloromethane (300 mL) and stirred at 0 °C. Dicyclohexylcarbodiimide (51.4 g, 248.9 mmol) was dissolved in anhydrous dichloromethane (200 mL) to form a homogenous solution, which was added to the former mixture dropwise over a 30 min period at 0 °C. The solution formed a white precipitate and was allowed to stir for 22 h at room temperature, at which point no further conversion of the starting materials was detected (TLC, 50% ethyl acetate in hexanes, Rf (1) = 0.5 and Rf (Boc-D alanine) = 0.4). The formed dicyclohexyl urea byproduct was removed by filtration and washed with dichloromethane until the solid was colorless. The organic layers were combined and concentrated under reduced pressure. The residue, was triturated with hexanes (200 mL) to yield a pale-yellow solid. The solid was filtered and washed with hexanes (50 mL x 3) followed by reflux in hexanes (200 mL) for 30 min. After cooling to room temperature, the white solid 2 was filtered and washed with hexanes (50 mL x 3) and dried under vacuum at 40 °C (55.3 g, 59% yield). 1H NMR (300 MHz, CDCl3) δ 11.68 (s, 1H), 8.71 (d, J = 9.0 Hz, 1H), 7.69 (dd, J = 9.0, 2.3 Hz, 1H), 7.64 – 7.53 (m, 2H), 7.51 – 7.42 (m, 1H), 7.35 – 7.27 (m, 1H), 7.21 (t, J = 9.1 Hz, 1H), 5.13 (s, 1H), 4.37 (s, 1H), 1.52 (d, J = 7.2 Hz, 3H), 1.45 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 195.47 (s), 172.42 (s), 159.56 (d, J = 253.2 Hz), 155.28 (s), 139.68 (s), 137.97 (s), 135.97 (d, J = 1.9 Hz), 133.62 (d, J = 8.4 Hz), 130.31 (d, J = 2.4 Hz), 126.77 (d, J = 14.5 Hz), 124.51 (s), 124.46 (s), 122.63 (s), 116.55 (d, J = 21.4 Hz), 114.95 (s), 80.26 (s), 51.86 (s), 28.26 (s), 18.58 (s); HRMS (ESI/IT-TOF) m/z: [M + H]+ Calcd for C21H23BrFN2O4 465.0820; found 465.0845.
Tert-Butyl-(R)-(1-((4-bromo-2-(2-fluorobenzoyl)phenyl)amino)-1-oxopropan-2-yl) carbamate (2) (Table 1, entries 10–11)
2-Amino-5-bromo-2’-fluorobenzophenone (6.5 g, 22.1 mmol), Boc-D-alanine (4.6 g, 24.3 mmol), triethylamine (2.9 g, 28.7 mmol) in anhydrous dichloromethane (50 mL) was stirred at room temperature for 30 min. The solution was cooled to 0 °C and N-(3-dimethylaminopropyl)-N’-ethylcarbodimide hydrochloride (EDC, 5.5 g, 28.7 mmol) added in one portion. The yellow solution was allowed to warm to room temperature and stirred for 24 h, at which point TLC analysis (50% ethyl acetate in hexanes, Rf (1) = 0.5, Rf (Boc-D-alanine) = 0.4) showed no further conversion. The reaction mixture was diluted in water (50 mL) and the biphasic mixture was separated and washed with 25% aqueous potassium carbonate (50 mL) and 10% aqueous sodium chloride (50 mL). The organic layer was dried over sodium sulfate, evaporated under reduced pressure, and triturated with 10% ethyl acetate in hexanes (100 mL). The white solid 2 was collected by filtration, washed with hexanes (10 mL x 2), and dried under vacuum at 40 °C (1.0 g, 10% yield).
Tert-Butyl-(R)-(1-((4-bromo-2-(2-fluorobenzoyl)phenyl)amino)-1-oxopropan-2-yl) carbamate (2) (Table 1, entries 12–14)
Methyl chloroformate (1.7 mL, 32.3 mmol) was added dropwise over 20 min to a solution of Boc-D-alanine (4.1 g, 21.6 mmol) and N-methylmorpholine (2.7 mL, 32.3 mmol) in THF (100 mL) at −20 °C. To the white slurry, a solution of 2-amino-5-bromo-2’-fluorobenzophenone (6.3 g, 21.6 mmol), N-methylmorpholine (2.7 mL, 32.3 mmol) in THF (50 mL) was added dropwise over 15 min at −20 °C. The mixture was allowed to warm to room temperature and stirred for 24 h, at which point analysis of the reaction progress by TLC indicated a 50/50 mixture of product to starting material (50% ethyl acetate in hexanes, Rf (1) = 0.5, Rf (Boc-D-alanine) = 0.4). The solvent was removed under reduced pressure and the residue dissolved in dichloromethane (100 mL) followed by the addition of 25% aqueous sodium chloride (100 mL). The biphasic mixture was separated, washed twice with 25% aqueous potassium carbonate (100 mL x 2), and dried over sodium sulfate. The solvents were removed under reduced pressure and the residue was triturated with 10% ethyl acetate in hexanes (40 mL). A white solid 2 was filtered, washed with hexanes (10 mL x 2), and dried under vacuum at 40 °C (4.9 g, 50% yield).
(9H-fluoren-9-yl)methyl (R)-(1-chloro-1-oxopropan-2-yl)carbamate (3)
To a mixture of Fmoc-D-alanine (150 g, 481.8 mmol), N,N-dimethylformamide (3.7 mL, 48.2 mmol), and anhydrous dichloromethane (3000 mL) was added thionyl chloride (351.4 mL, 4818.0 mmol) dropwise over 60 min while keeping the temperature between 25 – 30 °C. The resulting SO2(g) and HCl(g) was trapped with a methanolic scrubber system. The solution was stirred at room temperature for 1 h followed by 2 h at 40 °C. The completion of the reaction was determined by converting an aliquot with methanol in the presence of trimethylamine. The starting material was eluted by TLC using 25% methanol in dichloromethane with 1% trimethylamine. The solvents and excess thionyl chloride were removed under reduced pressure. The residual thionyl chloride was removed by drying the solid residue under reduced pressure at 40 °C, followed by dissolution in dichloromethane (350 mL) and addition of heptane (3150 mL) over 1 h with vigorous stirring to precipitate the product. The stirring was continued for 2 h. The solid was filtered and washed with 10 % dichloromethane in heptane (250 mL x 2). The solid was dried under vacuum at room temperature to yield 3 as an off-white solid (147.5 g, 93%): 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 7.6 Hz, 2H), 7.65 – 7.53 (m, 2H), 7.48 – 7.40 (m, 2H), 7.35 (td, J = 7.4, 1.2 Hz, 2H), 5.37 – 5.22 (m, 1H), 4.64 (p, J = 7.4 Hz, 1H), 4.53 (dd, J = 10.6, 6.6 Hz, 1H), 4.45 (dd, J = 10.8, 6.9 Hz, 1H), 4.25 (t, J = 6.8 Hz, 1H), 1.57 (d, J = 7.3 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 175.55, 155.48, 143.54, 141.36, 127.83, 127.15, 125.02, 124.95, 120.07, 67.34, 58.67, 47.09, 17.28.
(R)-7-bromo-5-(2-fluorophenyl)-3-methyl-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one (5) (Scheme 1)
2-Amino-5-bromo-2’-fluorobenzophenone (1) (120 g, 408.0 mmol) was dissolved in anhydrous dichloromethane (1200 mL) and a solution of (9H-fluoren-9-yl)methyl (R)-(1-chloro-1-oxopropan-2-yl)carbamate (3) (148 g, 448.8 mmol) in anhydrous dichloromethane (1000 mL) added dropwise over 60 min and keeping the reaction temperature between 25 – 30 °C. After continuous stirring at room temperature for 1 h, the reaction was deemed complete by TLC analysis (silica gel, 50% ethyl acetate in hexanes). Piperidine (241.8 mL, 2448 mmol) was added dropwise to the reaction mixture over 30 min while maintaining the temperature at 25 – 30 °C. Stirring at room temperature was continued for 12 h resulting disappearance of the intermediate as analyzed by TLC (silica gel, 50% ethyl acetate in hexanes). The reaction mixture was quenched with 5% aqueous sodium bicarbonate (1200 mL) and separated. The aqueous layer was extracted with dichloromethane (1200 mL) and the combined organic layers washed with 5% aqueous sodium bicarbonate solution (1200 mL) and then twice with deionized water (1200 mL x 2). The solvents were removed under reduced pressure and the residue was dissolved in dichloromethane (1200 mL) and methanol (1200 mL). A solution of sodium hydroxide (16.3 g, 408.0 mmol) and deionized water (240 mL) was added dropwise to the reaction mixture over 15 min and the reaction mixture was stirred for an additional 12 h at room temperature. Upon completion of conversion (TLC analysis: silica gel, 50% ethyl acetate in hexanes), solvents were removed under reduced pressure. The resulting semi-solid was dissolved in dichloromethane (1200 mL) and treated with 10% aqueous sodium chloride (1200 mL). The biphasic mixture was separated and the aqueous layer was extracted with dichloromethane (1200 mL) followed by the combination of the organic layers and a wash with 10% aqueous sodium chloride (1200 mL). The organic layer was dried over Na2SO4, evaporated under reduced pressure, and triturated in methanol (1600 mL) at 50 °C for 30 min. After stirring for 3 h at room temperature, the solid byproduct (1-((9H-fluoren-9-yl)methyl)piperidine) was removed by filtration and washed with methanol (200 mL x 2). The combined methanol layers were evaporated under reduced pressure and the residue was stripped with 10% ethyl acetate in heptane (200 mL x 2). The solid was triturated in 10% ethyl acetate in heptane (1600 mL) at 60 °C for 30 min to dissolve 1. The mixture was cooled to room temperature and stirred for 2 h. The solid material was collected by filtration and washed with 10% ethyl acetate in heptane (50 mL x 2) and then with heptane (50 mL x 2). The solid was dried under vacuum at 35 °C to afford 5 as an off-white solid (97.3 g, 69%) with a purity of 99.8%. 1H NMR (500 MHz, CDCl3) δ 9.69 (s, 1H), 7.63 – 7.59 (m, 1H), 7.59 (dd, J = 8.5, 2.3 Hz, 1H), 7.47 (dddd, J = 8.2, 7.0, 5.0, 1.8 Hz, 1H), 7.36 (d, J = 2.2 Hz, 1H), 7.26 (td, J = 7.5, 1.1 Hz, 1H), 7.12 (d, J = 8.6 Hz, 1H), 7.08 (ddd, J = 10.2, 8.3, 1.1 Hz, 1H), 3.79 (q, J = 6.5 Hz, 1H), 1.78 (d, J = 6.5 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 172.39 (s), 164.53 (s), 160.45 (d, 1JCF = 251.9 Hz), 136.47 (s), 134.74 (s), 132.19 (d, 3JCF = 8.3 Hz), 132.03 (d, JCF = 1.5 Hz, NOE coupling), 131.56 (d, 3JCF = 2.2 Hz), 130.15 (s), 127.13 (d, 2JCF = 12.4 Hz), 124.46 (d, 4JCF = 3.6 Hz), 122.98 (s), 116.54 (s), 116.29 (d, 2JCF = 21.5 Hz), 58.85 (s), 16.93 (s); HRMS (ESI/IT-TOF): m/z [M + H]+ calcd for C16H13BrFN2O: 347.0190; found: 347.0181.
(R)-7-bromo-5-(2-fluorophenyl)-3-methyl-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one (5) (Table 2, entry 2–4)
Compound 2 (55 g, 118.2 mmol) in anhydrous dichloromethane (500 mL) was cooled to 0 °C. A solution of 4.0M hydrogen chloride in dioxane (118 mL, 472.8 mmol) was added dropwise to the reaction mixture at 0 °C over a period of 60 min. The reaction mixture was stirred for 4 h at 0 °C, at which point the starting material was converted (TLC analysis, 50% ethyl acetate in hexanes). The reaction mixture was diluted with 10% aq sodium bicarbonate (500 mL) and the biphasic mixture was separated. The aqueous layer was extracted with dichloromethane (500 mL) and the combined organic layers were washed with 5% aqueous sodium bicarbonate (500 mL) and twice with deionized water (500 mL x 2). The solvents were removed under reduced pressure and the residue was dissolved in methanol (500 mL). A solution of sodium hydroxide (0.5 g, 11.8 mmol) and deionized water (50 mL) was added dropwise to the reaction mixture over 15 min and stirred at room temperature for 48 h, at which point the reaction was deemed complete by TLC analysis (silica gel and 50% ethyl acetate in hexanes). The solvent was removed under reduced pressure and the residue was dissolved in dichloromethane (500 mL) and treated with 10% aqueous sodium chloride (500 mL). The biphasic mixture was separated and the aqueous layer was extracted with dichloromethane (500 mL). The combined organic layers were washed with 10% aqueous sodium chloride (500 mL) and dried with Na2SO4. The solvent was removed under reduced pressure and the residue was stripped with 10% ethyl acetate in heptane (200 mL x 2) and triturated with 10% ethyl acetate in heptane (500 mL) at 60 °C for 30 min. Upon stirring at room temperature for 2 h, the solid product was collected by filtration and washed with 10% ethyl acetate in heptane (50 mL x 2) followed by heptane (50 mL x 2). The solid was dried under vacuum at 40 °C to yield 5 as an off-white solid (34.6 g, 84%): 1H NMR (500 MHz, CDCl3) δ 9.69 (s, 1H), 7.63 – 7.59 (m, 1H), 7.59 (dd, J = 8.5, 2.3 Hz, 1H), 7.47 (dddd, J = 8.2, 7.0, 5.0, 1.8 Hz, 1H), 7.36 (d, J = 2.2 Hz, 1H), 7.26 (td, J = 7.5, 1.1 Hz, 1H), 7.12 (d, J = 8.6 Hz, 1H), 7.08 (ddd, J = 10.2, 8.3, 1.1 Hz, 1H), 3.79 (q, J = 6.5 Hz, 1H), 1.78 (d, J = 6.5 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 172.39 (s), 164.53 (s), 160.45 (d, 1JCF = 251.9 Hz), 136.47 (s), 134.74 (s), 132.19 (d, 3JCF = 8.3 Hz), 132.03 (d, JCF = 1.5 Hz, NOE coupling), 131.56 (d, JCF = 2.2 Hz), 130.15 (s), 127.13 (d, JCF = 12.4 Hz), 124.46 (d, JCF = 3.6 Hz), 122.98 (s), 116.54 (s), 116.29 (d, 2JCF = 21.5 Hz), 58.85 (s), 16.93 (s); HRMS (ESI/IT-TOF): m/z [M + H]+ calcd for C16H13BrFN2O: 347.0190; found: 347.0181; HPLC Purity: 97.1%; Optical Purity: 53.9%.
(R)-4-methyloxazolidine-2,5-dione (6)
To a solution of Boc-D-Alanine (100 g, 528.5 mmol), triphosgene (62.7g, 211.4 mmol) in anhydrous ethyl acetate (4000 mL) was added triethylamine (81.0 mL, 581.4 mmol) dropwise over 60 min while keeping the temperature below 30 °C. The formed CO2, HCl, and residual phosgene was trapped by a methanolic scrubber system. The reaction mixture was stirred for 1 h at room temperature followed by 2 h at reflux (65–70 °C), at which point the gas evolution ceased and the Boc-D-alanine was converted (TLC, 50% ethyl acetate in hexanes, Rf = 0.5). The reaction mixture was cooled to room temperature and a white solid (TEA-HCl and residual D-Alanine) was removed by filtration to yield a pale-yellow solution. The solid was washed with ethyl acetate (2 × 100 mL) and the organic layers were combined and evaporated under reduced pressure. The semi-solid was dissolved in dichloromethane (400 mL) and hexanes was added dropwise (400mL) over 1 h with vigorous stirring to precipitate the product. After 12 h at −20 °C, the solid was filtered, washed with hexanes (250 mL x 2), and dried under vacuum at room temperature to afford 6 as an off-white solid (35.2 g, 58%): 1H NMR (500 MHz, D6-DMSO) δ 9.01 (s, 1H), 4.48 (qd, J = 7.0, 1.1 Hz, 1H), 1.34 (d, J = 7.1 Hz, 3H); 13C NMR (126 MHz,D6-DMSO) δ 172.88, 152.15, 53.28, 17.23.
(R)-7-bromo-5-(2-fluorophenyl)-3-methyl-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one (5)
A mixture of 2-amino-5-bromo-2’-fluorobenzophenone (1) (140.7 g, 478.4 mmol) and trifluoroacetic acid (73.3 mL, 956.7 mmol) in anhydrous toluene (2200 mL) was stirred at room temperature for 30 min to form a solution. N-carboxy-D-alanine anhydride (66.0 g, 574.0 mmol) was added and the reaction mixture was heated for 1 h at 50 °C. After disappearance of 1 (TLC, 50% ethyl acetate in hexanes), triethylamine (133.3 mL, 956.7 mmol) was added dropwise to the reaction mixture over 30 min, while maintaining the temperature at 50 °C. After 2 h at 50 °C, the intermediate 4 was not detected (TLC, 50% ethyl acetate in hexanes). Upon cooling the reaction mixture to room temperature, solvents were removed under reduced pressure and the residue was dissolved in ethyl acetate (1500 mL) and water (1500 mL). The resulting biphasic mixture was separated and the organic layer was washed with 5% aqueous sodium bicarbonate solution (1500 mL) followed by 10% aqueous sodium chloride solution (1500 mL). The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The residue was stripped with 10% ethyl acetate/heptane (25 mL x 2) and slurried with 10% ethyl acetate in heptane (1700 mL) at 60 °C for 30 min to dissolve unreacted starting material 1. The reaction mixture was cooled to room temperature and stirred for an additional 2 h before the product was collected by filtration and washed with 10% ethyl acetate in heptane (50 mL x 2) followed by heptane (50 mL x 2). The solid was dried under vacuum at 40 °C to afford 5 as an off-white solid (127.0 g, 77%): 1H NMR (500 MHz, CDCl3) δ 9.69 (s, 1H), 7.63 – 7.59 (m, 1H), 7.59 (dd, J = 8.5, 2.3 Hz, 1H), 7.47 (dddd, J = 8.2, 7.0, 5.0, 1.8 Hz, 1H), 7.36 (d, J = 2.2 Hz, 1H), 7.26 (td, J = 7.5, 1.1 Hz, 1H), 7.12 (d, J = 8.6 Hz, 1H), 7.08 (ddd, J = 10.2, 8.3, 1.1 Hz, 1H), 3.79 (q, J = 6.5 Hz, 1H), 1.78 (d, J = 6.5 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 172.39 (s), 164.53 (s), 160.45 (d, 1JCF = 251.9 Hz), 136.47 (s), 134.74 (s), 132.19 (d, 3JCF = 8.3 Hz), 132.03 (d, JCF = 1.5 Hz, NOE coupling), 131.56 (d, 3JCF = 2.2 Hz), 130.15 (s), 127.13 (d, 2JCF = 12.4 Hz), 124.46 (d, 4JCF = 3.6 Hz), 122.98 (s), 116.54 (s), 116.29 (d, 2JCF = 21.5 Hz), 58.85 (s), 16.93 (s); 19F NMR (471 MHz, CDCl3) δ −112.53; HRMS (ESI/IT-TOF): m/z [M + H]+ calcd for C16H13BrFN2O: 347.0190; found: 347.0181; HPLC Purity: 97.3%; Optical Purity: 98.8% ee.
Ethyl (R)-8-bromo-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a] [1,4]diazepine-3-carboxylate (7)
Compound 5 (125.0 g, 360.0 mmol) in anhydrous tetrahydrofuran (2000 mL) was cooled to −20 °C using a dry ice/IPA bath. A solution of t-BuOK (52.5 g, 468.0 mmol) in tetrahydrofuran (300 mL) was added dropwise to the reaction mixture over 30 min, while maintaining a temperature of −20 °C. Upon completion of the addition, the reaction mixture was allowed to stir for an additional 60 min at −20 °C. Diethyl chlorophosphate (72.8 mL, 504.1 mmol) was then added dropwise to the reaction mixture over 15 min at −20 °C. After 2 h at −20 °C the starting material was converted (TLC, 100% ethyl acetate). Ethyl isocyanoacetate (51.2 mL, 468.0 mmol) was added dropwise over 15 min while maintaining −20 °C, followed by the dropwise addition of a solution of t-BuOK (52.5 g, 468.0 mmol) in tetrahydrofuran (300 mL) over 30 min at −20 °C. Upon completion of the addition, the reaction mixture was allowed to warm to room temperature and stir for an additional 1 h, at which point the intermediate was fully converted (TLC, 100% ethyl acetate). The reaction mixture was diluted with 5% aqueous sodium bicarbonate (2000 mL) and ethyl acetate (2000 mL). The resulting emulsion was cleared by filtration and separated after 30 min. The aqueous layer was extracted with ethyl acetate (2000 mL) and the combined organic layers were washed with 10% aqueous sodium bicarbonate solution (2000 mL) and 20% aqueous sodium chloride solution (2000 mL). The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The residue was stripped with t-butyl methyl ether (200 mL x 2) and slurried with t-butyl methyl ether (1000 mL) at 55 °C for 30 min. The mixture was stirred for 12 h at room temperature followed by filtration and washed with t-butyl methyl ether (100 mL x 4). The solid was dried under vacuum at 40 °C to yield 7 as a white powder (96.6 g, 61%): 1H NMR (500 MHz, CDCl3) δ 7.92 (s, 1H), 7.73 (dd, J = 8.5, 2.2 Hz, 1H), 7.60 (dt, J = 7.3, 3.9 Hz, 1H), 7.48 (d, J = 8.6 Hz, 1H), 7.50 – 7.42 (m, 1H), 7.42 (d, J = 2.2 Hz, 1H), 7.26 (td, J = 7.5, 1.1 Hz, 1H), 7.10 – 7.02 (m, 1H), 6.71 (q, J = 7.3 Hz, 1H), 4.54 – 4.28 (m, 2H), 1.42 (t, J = 7.1 Hz, 3H), 1.29 (d, J = 7.4 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 162.93 (s), 162.67 (s), 160.10 (d, 1JCF = 250.7 Hz), 141.59 (s), 134.85 (s), 134.75 (s), 133.68 (s), 133.05 (s), 132.12 (d, 3JCF = 8.2 Hz), 131.17 (s), 129.59 (s), 128.42 (d, 2JCF = 12.3 Hz), 124.57 (d, 4JCF = 3.3 Hz), 123.65 (s), 120.96 (s), 116.25 (d, 2JCF = 21.4 Hz), 60.82 (s), 50.12 (s), 14.87 (s), 14.43 (s); 19F NMR (471 MHz, CDCl3) δ −112.36; HRMS (ESI/IT-TOF): m/z [M + H]+ calcd for C21H18BrFN3O2: 442.0561; found: 442.0563; HPLC Purity: 97.2%; Optical Purity: 99.0% ee.
(R)-8-bromo-6-(2-fluorophenyl)-4-methyl-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylic acid [MIDD0301]
To a solution of 7 (96.0 g, 217.0 mmol) in tetrahydrofuran (1500 mL),500 mL of a 1.74M aqueous solution of sodium hydroxide was added dropwise over 15 min while keeping the temperature at 30 °C. The reaction mixture was heated for 4 h at 55 °C to convert the starting material (TLC, 100% ethyl acetate). The reaction mixture was then cooled to room temperature followed by the addition of water (1000 mL) and acetic acid (74.5 mL, 1302.3 mmol) dropwise over 15 min while maintaining the temperature at 25 °C. Tetrahydrofuran was removed under reduced pressure and methanol (750 mL) was added to the mixture and heated for 30 min at 60 °C. After cooling to room temperature, the mixture was stirred for 12 h. The solid was filtered and washed with water (200 mL x 4). The solid was dried under vacuum at 40 °C to yield MIDD0301 as an off-white powder (87.7 g, 97.5%). The purity was 97.2% by HPLC. Recrystallization in ethanol gave 94% yield with a 98.9% purity. The optical purity: >99.0% ee. 1H NMR (500 MHz, D6-DMSO) δ 12.83 (s, 1H), 8.42 (s, 1H), 8.09 – 7.92 (m, 1H), 7.89 (d, J = 8.7 Hz, 1H), 7.59 (t, J = 5.5 Hz, 1H), 7.55 (dtd, J = 7.5, 5.4, 2.5 Hz, 1H), 7.36 – 7.30 (m, 2H), 7.23 (dd, J = 10.7, 8.2 Hz, 1H), 6.52 (q, J = 7.3 Hz, 1H), 1.17 (d, J = 7.3 Hz, 3H); 13C NMR (126 MHz, D6-DMSO) δ 164.68 (s), 162.37 (s), 159.85 (d, 1JCF = 248.3 Hz), 140.86 (s), 136.64 (s), 135.49 (s), 134.02 (s), 132.74 (d, 3JCF = 7.9 Hz), 132.38 (s), 131.93 (s), 130.89 (s), 129.58 (s), 128.70 (d, 2JCF = 11.7 Hz), 125.64 (s), 125.18 (d, 4JCF = 2.8 Hz), 120.26 (s), 116.43 (d, 2JCF = 21.1 Hz), 49.83 (s), 15.05 (s); 19F NMR (471 MHz, DMSO) δ −114.15; HRMS (ESI/IT-TOF): m/z [M + H]+ calcd for C19H14BrFN3O2: 414.0248; found: 414.0246.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (USA) R41HL147658 (L.A.A. and D.C.S.), R01NS076517 (J.M.C. and L.A.A.) and R01HL118561 (J.M.C., L.A.A., and D.C.S.), as well as the University of Wisconsin-Milwaukee, University of Wisconsin-Milwaukee Research Foundation (Catalyst grant), the Lynde and Harry Bradley Foundation, and the Richard and Ethel Herzfeld Foundation. In addition, this work was supported by grant CHE-1625735 from the National Science Foundation, Division of Chemistry.
Abbreviations
- THF
tetrahydrofuran
- t-BuOK
potassium t-butoxide
- DMF
dimethylformamide
- NaH
sodium hydride
- GABAAR
gamma amino butyric acid type A receptor
- DCC
N,N’-dicyclohexylcarbodiimide
- Boc-D-alanine
tert-butoxycarbonyl-D-alanine
- DMAP
4-dimethylaminopyridine
- CH2Cl2
dichloromethane
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- TEA
trimethylamine
- NMM
N-methyl morpholine
- NCA
N-carboxyanhydride
- Fmoc
((9H-fluoren-9-yl)methoxy)carbonyl)
- TLC
thin layer chromatography
- NMR
nuclear magnetic resonance
- LC-MS
liquid chromatography-mass spectrometry
- DMSO
dimethyl sulfoxide
- TLC
thin layer chromatography
- HRMS
high resolution mass spectrometry
- ESI
electron spray ionization
- IT-TOF
ion trap-time of flight
- HSQC
heteronuclear single quantum coherence
Footnotes
The authors declare the following competing financial interest(s): Drs. Stafford, Cook and Arnold are inventors of patent application WO2018035246A1, Gaba(A) receptor modulators and methods to control airway hyperresponsiveness and inflammation in asthma. Stafford and Arnold have equity interests in Pantherics Incorporated, which has certain intellectual property rights to these patents.
Supporting Information
The Supporting Information is available free of charge at: https://pubs.acs.org
Detailed HPLC methods for purity and optical purity analysis, HPLC, MS, 1H, 13C and 2D NMR spectra of compounds.
References
- [1].Fryer RI, and Walser A Diazepine derivatives. Patent DE2540522, 1976. [Google Scholar]
- [2].Walser A, Flynn T, and Fryer RI Quinazolines and 1,4-benzodiazepines. LXXXV. Syntheses of 3-substituted imidazo[1,5-a][1,4]benzodiazepines. J. Heterocycl. Chem 1978, 15, 577–583. [Google Scholar]
- [3].Walser A, Fryer RI, Sternbach LH, and Archer MC Quinazolines and 1,4‐benzodiazepines. LXV some transformations of chlordiazepoxide. J. Heterocycl. Chem 1974, 11, 619–621. [Google Scholar]
- [4].Ning RY, Fryer RI, Madan PB, and Sluboski BC Quinazolines and 1,4-Benzodiazepines. 74. Phosphorylation of Ambident Anions. Preparation of Some Di-4-morpholinylphosphinyloxy Imines via -Phosphorylation of Anions of Lactams. J. Org. Chem 1976, 41, 2720–2724. [Google Scholar]
- [5].Buehler E, and Brown GB A general synthesis of N-hydroxyamino acids. J. Org. Chem 1967, 32, 265–267. [Google Scholar]
- [6].Walser A, Lauer RF, and Fryer RI Quinazolines and 1,4-Benzodiazepines. LXXXVII. Synthesis of 1- and 3-Phenylimidazo [1,5-a] [ 1,4] benzodiazepines. J. Heterocycl. Chem 1978, 15, 855–858. [Google Scholar]
- [7].Walser A, Flynn T, Mason C, and Fryer RI Quinazolines and 1,4-Benzodiazepines. XCV. Synthesis of 1,4-Benzodiazepines by Ring Expansion of 2-Chloromethylquinazolines with Carbanions. J. Heterocycl. Chem 1986, 23, 1303–1314. [Google Scholar]
- [8].Walser A Synthesis of imidazo[1,5-a]diazepine-3-carboxylates Patent US 4118386, 1978. [Google Scholar]
- [9].Watjen F, Baker R, Engelstoff M, Herbert R, MacLeod A, Knight A, Merchant K, Moseley J, Saunders J, Swain CJ, and et al. Novel benzodiazepine receptor partial agonists: oxadiazolylimidazobenzodiazepines. J. Med. Chem 1989, 32, 2282–2291. [DOI] [PubMed] [Google Scholar]
- [10].Fryer RI, Gu ZQ, and Wang CG Synthesis of Novel, Substituted 4h-Imidazo[1,5-a][1,4]Benzodiazepines. J. Heterocycl. Chem 1991, 28, 1661–1669. [Google Scholar]
- [11].Fryer RI, Kudzma LV, Gu ZQ, Lin KY, and Rafalko PW Addition of Glycinate Enolate Equivalents to 1,4-Benzodiazepine Imino Phosphates - Preparation of Synthetically Useful 2-(Ethyl Glycinat-Alpha-Ylidene)-1,4-Benzodiazepines and Related Derivatives. J. Org. Chem 1991, 56, 3715–3719. [Google Scholar]
- [12].Cook JM, Zhou H, Huang S, Sarma PV, and Zhang C Stereospecific anxiolytic and anticonvulsant agents with reduced muscle relaxant, sedative-hypnotic and ataxic effects. Patent US7618958 2006. [Google Scholar]
- [13].Yang J, Teng Y, Ara S, Rallapalli S, and Cook JM An Improved Process for the Synthesis of 4H-Imidazo[1,5-a][1,4]benzodiazepines. Synthesis-Stuttgart 2009, 1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li GG, Golani LK, Jahan R, Rashid F, and Cook JM Improved Synthesis of Anxiolytic, Anticonvulsant, and Antinociceptive alpha2/alpha3-GABA(A)-ergic Receptor Subtype Selective Ligands as Promising Agents to Treat Anxiety, Epilepsy, and Neuropathic. Synthesis-Stuttgart 2018, 50, 4124–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Taghizadeh MJ, Malakpouri GR, and Javidan A Improved and scalable methods for the synthesis of midazolam drug and its analogues using isocyanide reagents. J Iran Chem Soc 2019, 16, 785–794. [Google Scholar]
- [16].Cook JM, Huang S, Edwankar R, Namjoshi OA, Wang Z-J, Huang Q, He X, Li X, Yu J, and Han D Synthesis of GABAA receptor subunit selective benzodiazepine derivatives for pain suppression. Patent US8835424B2, 2010. [Google Scholar]
- [17].Cook JM, Edwankar RV, Edwankar CR, Huang S, Jain HD, Yang J, Rivas FM, and Zhou H Selective anticonvulsant agents and their uses Patent US20100261711A1, 2010. [Google Scholar]
- [18].Anzini M, Valenti S, Braile C, Cappelli A, Vomero S, Acaro S, Ortuso F, Marinelli L, Limongelli V, Novellino E, Betti L, Giannaccini G, Lucacchini A, Daniele S, Martini C, Ghelardini C, Mannelli LD, Giorgi G, Mascia MP, and Biggio G New Insight into the Central Benzodiazepine Receptor-Ligand Interactions: Design, Synthesis, Biological Evaluation, and Molecular Modeling of 3-Substituted 6-Phenyl-4H-imidazo[1,5-a][1,4]benzodiazepines and Related Compounds. J. Med. Chem 2011, 54, 5694–5711. [DOI] [PubMed] [Google Scholar]
- [19].Forkuo GS, Nieman AN, Kodali R, Zahn NM, Li G, Rashid Roni MS, Stephen MR, Harris TW, Jahan R, Guthrie ML, Yu OB, Fisher JL, Yocum GT, Emala CW, Steeber DA, Stafford DC, Cook JM, and Arnold LA A Novel Orally Available Asthma Drug Candidate That Reduces Smooth Muscle Constriction and Inflammation by Targeting GABAA Receptors in the Lung. Mol. Pharm 2018, 15, 1766–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Roni MSR, Li G, Mikulsky BN, Knutson DE, Mian MY, Zahn NM, Cook JM, Stafford DC, and Arnold LA The Effects of pH on the Structure and Bioavailability of Imidazobenzodiazepine-3-Carboxylate MIDD0301. Mol. Pharm 2020, 17, 1182–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zahn NM, Huber AT, Mikulsky BN, Stepanski ME, Kehoe AS, Li G, Schussman M, Rashid Roni MS, Kodali R, Cook JM, Stafford DC, Steeber DA, and Arnold LA MIDD0301 - A first-in-class anti-inflammatory asthma drug targets GABAA receptors without causing systemic immune suppression. Basic. Clin. Pharmacol. Toxicol 2019, 125, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yocum GT, Perez-Zoghbi JF, Danielsson J, Kuforiji AS, Zhang Y, Li G, Rashid Roni MS, Kodali R, Stafford DC, Arnold LA, Cook JM, and Emala CW Sr. A novel GABAA receptor ligand MIDD0301 with limited blood-brain barrier penetration relaxes airway smooth muscle ex vivo and in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol 2019, 316, L385–L390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Valeur E, and Bradley M Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev 2009, 38, 606–631. [DOI] [PubMed] [Google Scholar]
- [24].Neises B, and Steglich W Einfaches Verfahren zur Veresterung von Carbonsäuren. Angew. Chem 1978, 90, 556–557. [Google Scholar]
- [25].Meienhofer J The Mixed Carbonic Anhydride Method of Peptide Synthesis. Peptides 1979, 1, 263–314. [Google Scholar]
- [26].Anderson GW, Zimmerman JE, and Callahan FM A reinvestigation of the mixed carbonic anhydride method of peptide synthesis. J. Am. Chem. Soc 1967, 89, 5012–5017. [DOI] [PubMed] [Google Scholar]
- [27].Reddy DR, Ballante F, Zhou NJ, and Marshall GR Design and synthesis of benzodiazepine analogs as isoform-selective human lysine deacetylase inhibitors. Eur. J. Med. Chem 2017, 127, 531–553. [DOI] [PubMed] [Google Scholar]
- [28].Calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02 (© 1994–2020. ACD/Labs). [Google Scholar]
- [29].Cook J, Li G, Golani L, Jahan R, and Rashid F Improved Synthesis of Anxiolytic, Anticonvulsant, and Antinociceptive α2/α3-GABA(A)-ergic Receptor Subtype Selective Ligands as Promising Agents to Treat Anxiety, Epilepsy, and Neuropathic Pain. Synthesis 2018, 50, 4124–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Carpino LA, Cohen BJ, Stephens KE, Sadat-Aalaee SY, Tien JH, and Langridge DC (Fluoren-9-ylmethoxy)carbonyl (Fmoc) amino acid chlorides. Synthesis, characterization, and application to the rapid synthesis of short peptide segments. J. Org. Chem 1986, 51, 3732–3734. [Google Scholar]
- [31].Prabhu G, Basavaprabhu Narendra, N., Vishwanatha TM, and Sureshbabu VV Amino acid chlorides: a journey from instability and racemization toward broader utility in organic synthesis including peptides and their mimetics. Tetrahedron 2015, 71, 2785–2832. [Google Scholar]
- [32].Clifford JM Polarographic Study of the Acid-Base Equilibria Existing in Aqueous Solutions of the 1,4-Benzodiazepines. Z. Anal. Chem 1973, 264, 149–153. [Google Scholar]
- [33].Fier PS, and Whittaker AM An Atom-Economical Method To Prepare Enantiopure Benzodiazepines with N-Carboxyanhydrides. Org. Lett 2017, 19, 1454–1457. [DOI] [PubMed] [Google Scholar]
- [34].Daly WH, and Poché D The preparation of N-carboxyanhydrides of α-amino acids using bis(trichloromethyl)carbonate. Tetrahedron Lett 1988, 29, 5859–5862. [Google Scholar]
- [35].Van Dijk-Wolthuis WNE, van de Water L, van de Wetering P, Van Steenbergen MJ, Kettenes-van den Bosch JJ, Schuyl WJW, and Hennink WE Synthesis and characterization of poly-L-lysine with controlled low molecular weight. Macromol. Chem. Phy 1997, 198, 3893–3906. [Google Scholar]
- [36].Wilder R, and Mobashery S The use of triphosgene in preparation of N-carboxy .alpha.-amino acid anhydrides. J. Org. Chem 1992, 57, 2755–2756. [Google Scholar]
- [37].Vasin VA, and Razin VV Mild and Efficient Method for Preparation of tert-Butyl Esters. Synlett 2001, 5, 658–660. [Google Scholar]
- [38].Walser A, Benjamin LE, Flynn T, Mason C, Schwartz R, and Fryer RI Quinazolines and 1,4-Benzodiazepines. 84. Synthesis and Reactions of Imidazo[l,5-a][l,4]benzodiazepines. J. Org. Chem 1978, 43, 936–944. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.