

Structure-function characterization of AF9 complexes with BCOR and CBX8 identifies BCOR-Eya1-Myc axis in MLL-AF9-driven leukemogenesis.
Abstract
MLL is a target of chromosomal translocations in acute leukemias with poor prognosis. The common MLL fusion partner AF9 (MLLT3) can directly bind to AF4, DOT1L, BCOR, and CBX8. To delineate the relevance of BCOR and CBX8 binding to MLL-AF9 for leukemogenesis, here we determine protein structures of AF9 complexes with CBX8 and BCOR, and show that binding of all four partners to AF9 is mutually exclusive. Using the structural analyses, we identify point mutations that selectively disrupt AF9 interactions with BCOR and CBX8. In bone marrow stem/progenitor cells expressing point mutant CBX8 or point mutant MLL-AF9, we show that disruption of direct CBX8/MLL-AF9 binding does not impact in vitro cell proliferation, whereas loss of direct BCOR/MLL-AF9 binding causes partial differentiation and increased proliferation. Strikingly, loss of MLL-AF9/BCOR binding abrogated its leukemogenic potential in a mouse model. The MLL-AF9 mutant deficient for BCOR binding reduces the expression of the EYA1 phosphatase and the protein level of c-Myc. Reduction in BCOR binding to MLL-AF9 alters a MYC-driven gene expression program, as well as altering expression of SIX-regulated genes, likely contributing to the observed reduction in the leukemia-initiating cell population.
Significance:
Direct recruitment of BCOR to MLL-AF9 is essential for leukemia via EYA1 phosphatase regulation, altering MYC and SIX gene expression programs. Specific partner binding (AF4, DOT1L, and BCOR) contributes in distinct ways to MLL leukemia. This may provide a rationale for combination DOT1L and EYA1 inhibition for MLL fusion leukemia treatment.
This article is highlighted in the In This Issue feature, p. 127
Introduction
The mixed lineage leukemia (MLL) protein is a histone methyltransferase that writes the histone H3 lysine 4 trimethyl (H3K4me3) mark at the promoters of target genes such as HOXA9 and MEIS1. MLL is the target of chromosomal translocations that fuse it in frame to one of over 90 partners, leading to acute myeloid and lymphoid leukemias (AML and ALL, respectively) characterized by poor prognoses (1). MLL fusions activate transcription by recruiting the AF4 family/ENL family/P-TEFb (AEP) complex and the DOT1L–AF10 family–ENL family complex (DOT1L complex, or DotCom; refs. 2, 3). Transcriptional activation via AF4 recruitment and transcriptional maintenance via DOT1L recruitment are required for MLL leukemias (2, 4, 5).
Despite the large number of fusion partners, members of the AEP complex account for nearly 70% of MLL rearrangements (6). These fusions constitutively activate MLL targets (3) by bypassing regulated recruitment via ENL (MLLT1) and AF9 (MLLT3) YEATS domain binding to crotonylated or acetylated histone H3 (7, 8). The AF9 ANC1 homology domain (AHD), retained in MLL fusions, is intrinsically disordered but undergoes coupled folding and binding upon interaction with its binding proteins (9). The AHD recruits AF4 and DOT1L, which support transcriptional elongation, as well as the BCL6 corepressor (BCOR) and chromobox homolog 8 (CBX8), which are implicated in transcriptional repression. BCOR and CBX8 are members of variant PRC1 complexes distinguished by their polycomb group RING finger (PCGF) components.
CBX8 (HPC3) is a mammalian ortholog of Drosophila polycomb that binds trimethylated histone H3 lysine 9 and 27 (H3K9me3 and H3K27me3) with variable affinity and is a component of the canonical PRC1 complex (10, 11, 12). A Cbx7-containing polycomb repressive complex 1 (PRC1) maintains pluripotency of mouse embryonic stem cells (ESC) by repressing targets including Cbx8 (13, 14). Replacement of Cbx7 with Cbx8 is required for ESC differentiation and the accompanying transition to gene activation (15). Previous reports indicate that CBX8 is required for MLL-AF9 and MLL-ENL leukemias (16, 17).
BCOR is a transcriptional corepressor that augments BCL6-mediated repression (18, 19) and is a component of the noncanonical PRC1.1 complex (10, 12). The BCL6 POZ domain forms a ternary complex with BCOR and SMRT, repressing targets via recruitment of PRC1.1 and HDAC3 (20). BCOR translocations and mutations have been found in a range of cancers. Although it is broadly expressed throughout the hematopoietic system (BloodSpot; ref. 21), little is known about BCOR function in hematopoiesis. BCOR has been implicated in regulation of myeloid cell proliferation and differentiation, and is necessary for MLL-AF9 leukemogenesis (22, 23).
While the roles of the direct MLL-AF9/AF4 and MLL-AF9/DOT1L interactions have been the subject of previous structural and functional studies (4, 5, 24), the roles of the direct interactions of MLL-AF9 with CBX8 and BCOR remain uncharacterized. Here, we present the structures of the AF9 AHD–CBX8 and AF9 AHD–BCOR complexes. We developed structure-guided point mutants to modulate affinity of CBX8 for AF9. Decreased affinity had no effect on colony-forming ability or differentiation of MLL-AF9–transformed cells. A point mutant was also developed to selectively disrupt BCOR binding to AF9. In the context of MLL-AF9, this mutant increases in vitro proliferative ability without an effect on colony formation yet, strikingly, is unable to cause leukemia in vivo. RNA-sequencing (RNA-seq) analysis shows that EYA1 is the most downregulated direct target gene upon loss of BCOR binding to MLL-AF9 and that its interaction partner SIX1 is also downregulated. Consistent with a documented role for EYA1 in enhancing the stability of the MYC protein, we observe a reduction in MYC protein and a MYC signature in our RNA-seq data. In addition, we observe altered expression of numerous genes known to be regulated by SIX1 and SIX2, to which EYA1 is known to bind. These changes in gene expression lead to a reduction in the leukemia-initiating cell (LIC) population, an effect that is partially abrogated by forced EYA1 expression in MLL-AF9–mutant cells.
Results
Structure of the CBX8–AF9 AHD Complex
The interaction of CBX8 with AF9 and ENL has previously been localized roughly to CBX8 residues 250–349 (16, 27). We used a nuclear magnetic resonance (NMR)–based method that can indicate regions of flexibility in the polypeptide backbone [15N-1H heteronuclear NOE (hetNOE)] to guide the construction of several constructs to coexpress different length CBX8 peptides with the AF9 AHD. Two-dimensional NMR spectra (15N-1H HSQC spectra) were evaluated for peak quality and dispersion, ultimately narrowing the minimal interacting region to CBX8 residues 326–349.
The solution NMR structure of the CBX8–AF9 complex was solved using dihedral angle and NOE restraints without significant violations (PDB: 2N4Q; Supplementary Table S1). An ensemble of the 10 lowest energy conformations derived from the structure calculations shows a well-formed complex of mixed alpha–beta structure (Fig. 1A). The AF9 binding motif comprises a β strand of alternating hydrophobic residues whose side chains are buried in a hydrophobic pocket on the AF9 surface. The interaction is mediated by an antiparallel β sheet formed by this strand and an AF9 β hairpin. The remaining AF9 residues pack around the β sheet in three α helices, making additional contacts to CBX8. The structure is highly similar to the AF4– and DOT1L–AF9 complexes we have described previously (refs. 5, 9; Fig. 1B). As we observed for the AF4–AF9 complex we previously characterized (9), NMR measurements of the backbone 15N relaxation rates indicate significant conformational exchange for the complex (Supplementary Fig. S1A–S1C).
Figure 1.

Structure and binding properties of the CBX8–AF9 AHD complex. A, Ensemble of the 10 lowest energy conformers from the structure calculations with CBX8 in orange and AF9 in purple. B, Alignment of the CBX8–AF9 and DOT1L–AF9 (PDB: 2MV7) structures with CBX8 in orange, its AF9 in purple, DOT1L in gold, and its AF9 in cyan. Backbone RMSD for AF9 residues 502–562 between CBX8–AF9 and the other AF9 complexes is shown in the table on the right. See also Supplementary Table S1 and Supplementary Fig. S1. C, Results of FA assays for binding of MBP-AF9 to fluorescein-labeled CBX8 WT (blue) and A335V (black) peptides. Error bars indicate SEM for three replicates. D, Top, sequence alignment of the AF9 binding motif. Conserved hydrophobic residues are in red. Bottom-comparison of the binding affinities of fluoresceinated peptides for all binding partners for MBP-ENL to affinities for MBP-AF9 measured by FA. Fold change is calculated as AF9 KD/ENL KD. Error bars, SEM for three replicates. E, Results of FA assays for competition of a fluorescein-labeled DOT1L site 3 peptide off of MBP-AF9 by unlabeled CBX8 A335V (black), WT (blue), and R336D (red) peptides. Error bars, SEM for three replicates. For details, see Supplementary Fig. S2.
AF9 Binding to CBX8 Is Enhanced by an Ala to Val Mutation and Weakened by an Arg to Asp Mutation
To characterize the CBX8–AF9 interaction, binding of a fluorescein-labeled CBX8 peptide (FITC-CBX8) to MBP-AF9 was measured by fluorescence anisotropy (FA). Unlike the other binding proteins, the affinity of CBX8 for AF9 is weak, so binding cannot be saturated in this assay (Fig. 1C), which is limited in the upper concentration that can be used by the propensity for MBP-AF9 to aggregate. The AF9 binding site in each partner consists of alternating hydrophobic residues roughly conforming to an LXVXIXLXXL motif (Fig. 1D). While AF4 and BCOR fit this pattern exactly and DOT1L differs in only the last position, CBX8 departs from the motif more significantly. We hypothesized that CBX8 A335 may not pack into the hydrophobic pocket of the AF9 binding site as well as the valine at this position in the other binding proteins, thereby weakening the interaction. To test this, binding of an FITC-CBX8 A335V peptide to MBP-AF9 was measured. The A to V mutation significantly increased CBX8 affinity for AF9, with a KD of 12 ± 1 nmol/L (Fig. 1C). A second mutation was made with the intention of weakening CBX8 affinity for AF9. Because AF9 D544 forms a salt bridge with CBX8 R336, a charge reversal to D was investigated for its AF9-binding ability. The weak nature of WT CBX8 binding makes it impossible to measure the affinity of this interaction in our typical FA assay. However, the relative affinity can be delineated by competing another peptide off AF9. To this end, unlabeled CBX8 WT, A335V, and R336D peptides were titrated into FITC-DOT1L site 3 peptide/MBP-AF9 at a concentration three times the DOT1L site 3-AF9 KD (Fig. 1E). As expected, CBX8 A335V was the most effective competitor, with an IC50 of 96 ± 16 nmol/L. WT CBX8 was much less effective, with an IC50 of 1876 ± 202 nmol/L, while CBX8 R336D was so ineffective at displacing DOT1L that it was unable to do so completely even at a concentration of 20 μmol/L.
The ENL AHD is a close AF9 AHD homolog (80% identical) and is known to associate with AF4, DOT1L, and CBX8. Binding of fluorescein-labeled AF4, DOT1L site 3, DOT1L site 2, BCOR (1176–1206), BCOR (1176–1225), and CBX8 peptides to MBP-ENL was measured by FA. Interestingly, while the affinity of the AF4 and DOT1L peptides for ENL was comparable with their affinity for AF9, there was a substantial enhancement in binding of CBX8 and BCOR (Fig. 1D; Supplementary Fig. S2A–S2F). The binding of BCOR (aa 1176–1225) was 9-fold tighter, and CBX8, whose AF9 binding was too weak to saturate, bound to ENL with a KD of 112 ± 13 nmol/L.
CBX8 A335V Mutation Inhibits MLL-AF9 Colony Formation, whereas CBX8 R336D Has No Effect
The fact that all the AF9 binding partners bind to the same site on AF9 makes it difficult to design a mutation that affects the binding of only one partner. In the absence of an AF9 mutation selective for CBX8 binding, we studied the role of the direct CBX8–AF9 interaction in the context of MLL leukemia by knockout and complementation. This involved the use of the A335V and R336D mutations described above. MLL-AF9–transformed BM progenitor cells from conditional Cbx8 knockout mice (17) were transduced with either WT or mutant CBX8 and assessed for colony formation. Knockout of endogenous Cbx8 was effective and led to a marked decrease in colony formation and cell number compared with the control (Fig. 2A and B), as has been reported previously (17). Visualization of individual colonies shows that Cbx8 knockout colonies are distinctly smaller and more diffuse than their Cbx8-expressing counterparts (Fig. 2C). Exogenous CBX8 expression levels were similar for the WT and both mutants when measured relative to vehicle control (Fig. 2D). Compared with addback of WT CBX8, the A335V mutant saw a significant decrease in both number of colonies and cell number (Fig. 2E, panels 2 and 4). This effect was magnified by the knockout of endogenous Cbx8 (Fig. 2E, panels 1 and 3). Addback of the R336D mutant was not significantly different from the WT addback (Fig. 2E). Colonies expressing CBX8 A335V are more diffuse than WT and R336D colonies, and Wright–Giemsa staining shows that these cells are more differentiated compared with the more blast-like untreated, WT, and R336D cells (Fig. 2F and G).
Figure 2.

CBX8 A335V inhibits colony formation, and CBX8 R336D has no effect. MLL-AF9–transformed mouse BM cells from Cbx8fl/fl (flox/flox) and conditional Cbx8 knockout (Cbx8KO) mice after methylcellulose culture 7 days with vehicle or 4-hydroxytamoxifen (4-OHT). For A, B, D, and E, data are from two experiments, each in triplicate, and are represented as mean ± SEM. Statistical significance was determined using a Student two-tailed t test with a confidence interval of 95%. A, Quantification of endogenous Cbx8 RNA expression, normalized to Polr2a by qRT-PCR. B, Quantification of colony and cell number relative to vehicle control. C, Bright-field images of colonies treated with vehicle (top) or 4-OHT (bottom). D, Quantification of exogenous CBX8 WT (gray), A335V (red), and R336D (blue) RNA expression, normalized to Polr2a by qRT-PCR. E, Number of colonies (panels 1 and 2) and cells (panels 3 and 4) produced by conditional Cbx8 knockout mouse BM cells treated with 4-OHT (panels 1 and 3) or vehicle (panels 2 and 4) and expressing exogenous CBX8 WT (gray), A335V (red), or R336D (blue). F, Bright-field images of conditional Cbx8 knockout mouse BM cells expressing CBX8 WT (left), A335V (middle), or R336D (right) and treated with vehicle (top) or 4-OHT (bottom). G, Images of Wright–Giemsa stained flox/flox (top) and conditional Cbx8 knockout (bottom) mouse BM cells treated with vehicle (panel 1) or 4-OHT and expressing exogenous CBX8 WT (panel 2), A335V (panel 3), or R336D (panel 4).
Defining BCOR Binding Motif for Interaction with AF9 AHD
Of the four BCOR isoforms expressed in mice and humans, only those which avoid use of an alternative splice site at the 5′ end of exon 8 are able to bind AF9 (28, 29). The difference in isoforms encompasses 34 amino acids that were presumed to include residues required for AF9 binding, including a 7–amino acid stretch of alternating hydrophobic residues consistent with the AF9 binding motifs of AF4 and DOT1L. Similar to our approach with the CBX8–AF9 complex, the minimally interacting region was identified by evaluating the dispersion and quality of two-dimensional NMR spectra (15N HSQC spectra) of several BCOR constructs of varying length coexpressed with the AF9 AHD. A construct expressing BCOR residues 1176–1228 bound to AF9 [BCOR (1176–1228)–AF9] produced good-quality spectra, but resonances for roughly 20 amino acids near the C-terminus were not observable by NMR, suggesting they were broadened out by conformational exchange resulting in a lack of signal intensity. Spectra of a construct expressing BCOR residues 1176–1207 in complex with AF9 [BCOR (1176–1207)–AF9] showed good dispersion, and all the resonances could be assigned to specific amino acids, so this construct was selected for structural studies.
Structure of the BCOR–AF9 AHD Complex
The solution NMR structure of the BCOR–AF9 complex was solved using NOE and dihedral angle restraints without significant violations (PDB: 6B7G; Supplementary Table S2). As in the CBX8–AF9 complex, an ensemble of the 10 lowest energy structures from the structure calculations shows an interaction site involving a β strand of alternating hydrophobic BCOR residues that forms an antiparallel β sheet with a β hairpin in AF9 (Fig. 3A). The remaining AF9 residues form three helices that fold around the binding site, making additional contacts with the BCOR peptide and burying the hydrophobic side chains of the β sheet. This structure is similar to those of the AF4–, DOT1L–, and CBX8–AF9 complexes, with backbone root mean square deviations (RMSDs) of 1.9, 1.4, and 1.6 Å, respectively, for structured AF9 residues (Fig. 3B). Importantly, the structures with all four partners show that they bind to the same site on AF9, indicating their binding is mutually exclusive with one another. As we observed for the AF4–AF9 complex we previously characterized (9), as well as the CBX8–AF9 complex, NMR measurements of the backbone 15N relaxation rates indicate significant conformational exchange for the complex (Supplementary Fig. S1D–S1F).
Figure 3.

Structure and binding properties of the BCOR–AF9 AHD complex. A, Ensemble of the 10 lowest energy conformers from the structure calculations, with BCOR in red and AF9 in blue. Unstructured BCOR residues 1176–1191 have been omitted for clarity. B, Alignment of the BCOR–AF9 and DOT1L–AF9 structures, with BCOR in red and its AF9 in blue, and DOT1L in gold and its AF9 in cyan. Backbone RMSD for AF9 residues 502–562 between BCOR–AF9 and the other AF9 complexes is shown in the table on the right. Unstructured BCOR residues 1176–1191 and DOT1L residues 893–900 have been omitted for clarity. See also Supplementary Table S2 and Supplementary Fig. S1. C, Results of FA assays for binding of MBP-AF9 to fluorescein-labeled BCOR (1176–1206) (blue) and BCOR (1176–1225) (black) peptides. Error bars, SEM for three replicates. D, Weighted chemical shift difference for AF9 residues in 15N-1H HSQC spectra of BCOR (1176–1228)–AF9 versus BCOR (1175–1207)–AF9 complexes calculated using |Δδ15N|/4.69 + |Δδ1HN|. AF9 E531 is shown in red. E, Top, results of FA assays for binding of MBP-AF9 E531R (blue) and MBP-AF9 (black) to a fluorescein-labeled BCOR (1176–1225) peptide. Error bars, SEM for three replicates. Bottom, effect of the AF9 E531R mutation on the binding of fluoresceinated peptides of all binding partners measured by FA. Fold change is calculated as E531R KD/WT KD. See also Supplementary Fig. S2. F, Immunoprecipitation (IP) of endogenous BCOR with FLAG-tagged AF9 or AF9 E531R from HEK293T cells. One representative immunoblot and summary from three independent experiments quantified relative to input MLLT3 levels.
Structure-Based AF9 E531R Mutation Specifically Disrupts BCOR Binding
To probe the functional role of the BCOR–AF9 interaction, we sought a mutation in AF9 that would abrogate binding to BCOR without affecting the other binding proteins. This is challenging due to the structural similarity of the AF9 AHD complexes. A shorter BCOR peptide, BCOR (aa 1176–1206)-FITC, binds MBP-AF9 too weakly to be saturated (Fig. 3C). However, a longer BCOR peptide, BCOR (aa 1176–1225)-FITC, binds much more tightly, with a KD of 18 ± 2 nmol/L (Fig. 3C). The significantly tighter binding of the longer BCOR peptide to AF9 makes its C-terminal residues (not present in the shorter BCOR peptide) an attractive target for selectively disrupting the BCOR–AF9 interaction. Candidate residues were identified by quantifying the chemical shift differences between 15N-1H HSQC NMR spectra of BCOR (aa 1176–1207)–AF9 and BCOR (aa 1176–1228)–AF9 complexes (Fig. 3D). The largest difference was residue E531 in AF9. Because the C-terminus of BCOR (aa 1176–1225) contains several positively charged residues, we hypothesized that AF9 E531 may be involved in an electrostatic interaction. On the basis of this, we developed a charge reversal mutation, AF9 E531R. This mutation greatly weakened the binding of the longer BCOR peptide, recapitulating the binding of the shorter BCOR peptide (Fig. 3E). Furthermore, we have shown by coimmunoprecipitation using full-length AF9 and AF9 (E531R) that introduction of the E531R mutation disrupts binding of BCOR in cells (Fig. 3F). Importantly, the binding of AF4, DOT1L sites 2 and 3, and CBX8 A335V saw little to no change (Fig. 3E; Supplementary Fig. S2G–S2J). It should be noted that the minor changes observed in the binding of AF9 E531R to AF4, DOT1L, and CBX8 are much smaller than we observed previously for an AF9 D544R mutation that retains colony-forming ability (5), so we do not believe these minor changes are biologically relevant.
MLL-AF9 E531R Does Not Impact Colony-Forming Ability but Does Enhance Cell Proliferation
The robust reduction in BCOR affinity for AF9 E531R as well as its demonstrated specificity make this mutation an excellent tool for probing the functional effects of this interaction in cells. To that end, WT MLL-AF9, MLL-AF9 E531R, and MLL-AF9 D546R (which abrogates DOT1L binding; ref. 5) or empty vector (MSCVneo) were transduced into mouse BM stem/progenitor cells and serially replated to measure colony formation ability. Both mutants were efficiently expressed with D546R 2-fold higher and E531R 6-fold higher than WT (Supplementary Fig. S3). While the D546R mutant severely inhibited colony formation as we have previously reported (5), the E531R mutant had no effect on the number of colonies (Fig. 4A). However, the colonies were larger (Fig. 4B) and had a greater number of cells per colony at weeks 2 to 4, with an increase in mean Feret diameter compared with WT colonies (Fig. 4C and D). Wright–Giemsa staining revealed a partial differentiation phenotype of MLL-AF9 E531R cells as compared with MLL-AF9 (Fig. 4E). As we previously observed, MLL-AF9 D546R cells with abrogated Dot1l interaction were the most differentiated (5). Surface marker staining reinforced this observation, with decreased c-Kit expression and increased, albeit not statistically significant, expression of CD11b in MLL-AF9(E531R) cells (Fig. 4F).
Figure 4.

MLL-AF9 E531R shows increased cell proliferation and prevents leukemogenesis in vivo. A, Quantification of methylcellulose colony formation from serial replating of MLL-AF9– and MLL-AF9 E531R– or D546R–transduced mouse BM cells. Data are from six experiments, each in triplicate, and are represented as mean ± SEM. B, Bright-field images of MLL-AF9 WT and E531R colonies. C, Quantification of cells per colony formed from serial replating of transduced mouse BM cells as in A. Data are from six experiments, each in triplicate, and are represented as mean ± SEM. Statistical significance was determined using a Student two-tailed t test with a confidence interval of 95%. D, Feret diameter box plots for MLL-AF9 WT (n = 115) and MLL-AF9 E531R (n = 88) colonies. Box indicates 1st and 3rd quartiles, line indicates median, and whiskers represent max and min. Statistical significance was determined using a Student two-tailed t test with a confidence interval of 95%. E, Images of Wright–Giemsa-stained MLL-AF9–, MLL-AF9 E531R–, and MLL-AF9 D546R–expressing mouse BM cells following the third plating in methylcellulose (scale bar, 10 μm). F, Summary of immunofluorescence staining and one representative flow cytometry plot of MLL-AF9 and MLL-AF9 E531R cells from E for hematopoietic stem and differentiation surface markers. Relative mean fluorescence intensity (MFI) of staining is shown. Data are from five independent experiments; **, P < 0.01. G, Survival curves for sublethally irradiated mouse recipients (n = 10 for each cohort) of BM cells transformed with MLL-AF9 WT (blue) or MLL-AF9 E531R (red). Statistical significance was determined using a log-rank (Mantel–Cox) test. For details, see also Supplementary Fig. S3.
The MLL-AF9/BCOR Interaction Is Required for MLL-AF9 In Vivo Leukemogenesis
To evaluate whether the disruption of the BCOR–AF9 interaction affects leukemogenesis in vivo, we transplanted retrovirally transformed BM cells into sublethally irradiated mice. Mice expressing WT MLL-AF9 developed AML as expected under these conditions (small number of cells transplanted, sublethal irradiation), whereas none of the MLL-AF9 E531R–expressing mice developed leukemia (Fig. 4G).
The MLL-AF9/BCOR Interaction Targets a Unique Set of Genes
RNA-seq was performed on mouse BM cells transformed with either MLL-AF9 or MLL-AF9 E531R to further probe the importance of the interaction with BCOR (GEO: GSE105767). The E531R mutation resulted in altered expression of 1,002 genes by >2-fold, with a false discovery rate (FDR) < 0.05 (Fig. 5A and B). Of these, 516 were upregulated and 486 were downregulated. Of 139 previously identified MLL-AF9 chromatin immunoprecipitation (ChIP) targets (30), a very small number were affected by the MLL-AF9 E531R mutation (Fig. 5C). Only four were downregulated by >2-fold, while none were upregulated at this level, although a total of 18 were significantly altered (Padj < 0.05; Fig. 5A). We have shown previously that a subset of MLL-AF9 targets exhibit a loss of H3K79 methylation upon loss of MLL-AF9/DOT1L binding (5). Genes affected by loss of MLL-AF9/BCOR binding show very little overlap with this subset, with only two of 37 genes altered by >2-fold. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway–associated terms for altered genes include hematopoietic cell lineage and cell adhesion molecules (CAM; Supplementary Fig. S4A). Gene ontology (GO) analysis indicates significant changes in genes associated with immune response and inflammation, in line with known BCL6 function, and positive regulation of the ERK1 and ERK2 cascade, which regulates multiple cell processes, including proliferation, differentiation, and survival. ChIP Enrichment Analysis (ChEA) 2016 shows significant enrichment of 36 ChIP datasets representing 27 unique transcription factors at the sites of altered genes (Supplementary Fig. S4B; Supplementary Table S3). Among these are components of PRC1 and PRC2 (e.g., RING1B and EZH2) and transcription factors with critical roles in hematopoiesis [e.g., GATA2, GFI1, and MYB, all of which are downregulated in MLL-AF9 (E531R) cells].
Figure 5.

The BCOR/MLL-AF9 interaction regulates a unique set of genes. A, Left, volcano plot showing differential RNA expression in MLL-AF9 E531R samples compared with WT. Genes with FDR < 0.05 and altered >2-fold are shown in orange. Those with FDR < 0.05 and altered <2-fold are shown in red. Genes selected for qPCR are shown in blue. Ccnd1–3 are indicated in green. Right, MLL-AF9 E531R–altered genes that are direct MLL-AF9 targets. Log2 (fold change) is reported relative to WT. Genes labeled in C are highlighted in blue. B, Heatmap of genes with >2-fold expression change between MLL-AF9 WT and MLL-AF9 (E531R), with increased expression in red and decreased in blue. C, Venn diagram showing overlap of genes altered >2-fold by MLL-AF9 E531R (pink) with MLL-AF9 ChIP targets (blue; ref. 30) and genes showing loss of H3K79me2 (yellow) or H3K79me3 (green) in MLL-AF9 D546R samples (5). D, GSEA analysis identified “HALLMARK_MYC_TARETS_V1” signature based on comparison of MLL-AF9 and MLL-AF9 (E531R) RNA-seq data. E, Western blot analysis for Myc protein in MLL-AF9– and MLL–AF9 (E531R)–expressing cells. Myc protein level was quantified relative to actin from three independent experiments using the Fiji distribution of ImageJ.
To directly compare the effect of specifically modulating binding of BCOR, CBX8, AF4, or DOT1L to MLL-AF9 via direct AF9 interaction, gene expression of select direct MLL-AF9 target genes was assessed in BM stem/progenitor cells transduced with the different MLL-AF9 point mutants (E531R, D544R, or D546R) or transduced with CBX8 A335V following Cbx8 knockdown. Interestingly, target genes were differentially affected by the various mutants (Supplementary Fig. S4C–S4F). For example, decreased binding of DOT1L showed significantly decreased expression of Hoxa7, Hoxa9, Hoxa10, whereas abrogated BCOR binding showed increased Hoxa7, no significant effect on Hoxa9, and decreased Hoxa10 expression. Higher binding affinity of CBX8, which will displace other binding partners due to the site of binding being the same for all binding partners, significantly increased Hoxa10 expression but decreased Meis1 and Myb expression. In contrast, all mutations caused decreased expression of Eya1, with the caveat that the D544R and D546R mutations decrease binding of DOT1L as well as BCOR (5). Gfi1 expression was decreased in MLL-AF9 E531R–transformed cells, in contrast to the RNA-seq results. This was verified with three primer sets (Supplementary Fig. S4G) and could be due to variance in amplification during sample preparation and sequencing.
Loss of MLL-AF9/BCOR Binding Alters an MYC-Regulated Set of Genes
Gene set enrichment analysis (GSEA) (31, 32) of our gene set identified an MYC molecular signature (Fig. 5D; Supplementary Fig. S4H). The phosphatase EYA1 is the direct MLL-AF9 target gene with the largest reduction in expression in our gene set (∼9-fold). EYA1 has been shown to decrease the proteasomal degradation of MYC via dephosphorylation of T58 (33, 34), leading to increased MYC levels. Consistent with the reduced EYA1 level in MLL-AF9 E531R cells, we observe decreased MYC protein in E531R cells relative to WT cells (Fig. 5E).
Loss of BCOR Binding to MLL-AF9 Alters a Set of Genes That Is Critical for MLL-AF9–Transformed Long-term Hematopoietic Stem Cells
The observation that MLL-AF9 (E531R) induced increased proliferation in the colony assay but a complete loss of leukemia development in vivo suggested a possible reduction in the LIC population upon loss of BCOR binding. Stavropoulou and colleagues (35) identified a leukemia-initiating population when MLL-AF9 was expressed in long-term hematopoietic stem cells (LT-HSC), but not in GMPs. As shown in Fig. 6, we observe that approximately 10% of the genes identified by Stavropoulou and colleagues as increased in LT-HSCs versus GMPs upon expression of MLL-AF9 were reduced in expression in MLL-AF9 (E531R) cells. This set of genes includes a number of genes with well-documented roles in leukemia, including Bcl2, Flt3, Fyn, and Gata2 (identified in ChEA analysis of RNA-seq data; Supplementary Table S3), Maml3, and Sox4. As also shown in Fig. 6, approximately 10% of the genes identified as increased in GMPs versus LT-HSCs upon expression of MLL-AF9 were increased in expression in MLL-AF9 (E531R) cells. Similarly, comparison to a leukemia stem cell (LSC) gene signature identified by Dick and colleagues (36) as well as to a gene set comparing MLL-AF9 in hematopoietic stem cells (HSC) and GMPs from Armstrong and colleagues (37) also showed overlap (Supplementary Fig. S5A and S5B).
Figure 6.
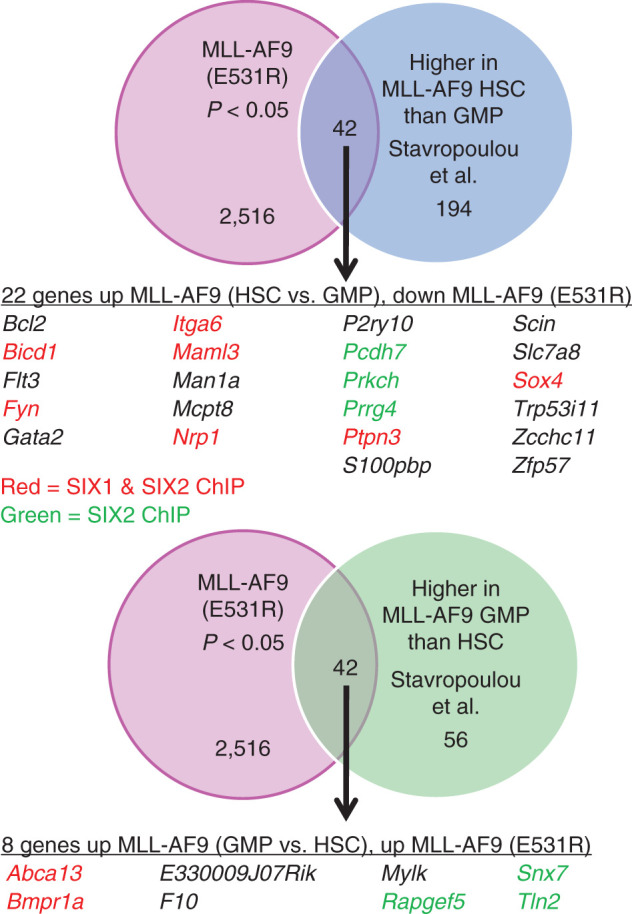
Comparison of MLL-AF9 E531R (disruption of BCOR binding) RNA-seq data to RNA-seq data from Stavropolou and colleagues (35), identifying genes with increased expression in MLL-AF9 HSCs versus MLL-AF9 GMPs. Top, Venn diagram showing overlap of genes altered in the MLL-AF9 E531R gene set and increased in MLL-AF9–expressing HSCs versus MLL-AF9–expressing GMPs. The table lists all the genes in the overlap set that are increased in MLL-AF9 HSCs versus GMPs and that are down in the MLL-AF9 E531R gene set. The genes in the table that have been shown by ChIP-seq to be either SIX1 and SIX2 targets or solely SIX2 targets are colored in red and green, respectively. Bottom, Venn diagram showing overlap of genes altered in the MLL-AF9 E531R gene set and increased in MLL-AF9–expressing GMPs versus MLL-AF9–expressing HSCs. The table lists all the genes in the overlap set that are increased in MLL-AF9 GMPs versus HSCs and increased in the MLL-AF9 E531R gene set. The genes in the table are colored as above.
As noted above, the most downregulated gene among direct MLL-AF9 targets (30) in MLL-AF9 (E531R) cells was the phosphatase Eya1 (down ∼9-fold). In addition, the transcription factor Six1, a known binding partner of Eya1 (38, 39) as well as a direct MLL-AF9 target, was also downregulated. This suggested that the changes in gene expression observed may be driven to a substantial extent by alteration of Eya1/Six–regulated genes. We compared our gene expression data to a recent study that used ChIP sequencing (ChIP-seq) to identify genes regulated by both SIX1 and SIX2, only by SIX2, and only by SIX1 in nephron progenitors (40). Of the 2,558 genes showing altered expression with P < 0.05 in our gene set, 771 (30%) contain binding sites for SIX1 and/or SIX2 (Supplementary Fig. S6A–S6C). As Eya1 and Six2 are also known binding partners (38, 39), the data suggest that the changes in gene expression observed in MLL-AF9 (E531R) cells are driven to a meaningful extent by alteration of an Eya1/Six1/Six2–regulated set of genes.
Loss of BCOR/MLL-AF9 Binding Reduces the LIC Population, and Addback of Eya1 Partially Rescues This Effect
To test whether loss of BCOR/MLL-AF9 binding alters the LIC population, we compared the survival of mice receiving MLL-AF9 cells, MLL-AF9(E531R) cells, 10-fold more MLL-AF9(E531R) cells (10×), and MLL-AF9(E531R) cells with forced expression of Eya1 (Fig. 7A). Whereas MLL-AF9(E531R) cells resulted in 100% survival at 120 days, transplant of 10× MLL-AF9(E531R) cells resulted in 0% survival, confirming a reduction in the LIC population upon loss of BCOR binding. BM cells transplanted into mice immediately after infection with MLL-AF9(E531R), as we have done previously (25), also did not cause leukemia (Fig. 7A). Forced expression of Eya1 alone did not cause leukemia (Fig. 7A), whereas expression of Eya1 in MLL-AF9(E531R) cells resulted in 0% survival at 120 days, confirming a critical role for decreased Eya1 in reducing the LIC population in MLL-AF9 (E531R) cells. Leukemia cells from mice with forced Eya1 expression in MLL-AF9(E531R) had a more homogeneous blast-like morphology compared with those from 10× MLL-AF9 (E531R), suggesting qualitative differences in the leukemias derived from these cells (Supplementary Fig. S7A).
Figure 7.

EYA1/SIX1 partially rescues phenotype from abrogated MLL-AF9/BCOR binding and schematic of BCOR/MLL-AF9–dependent gene regulation. A, Survival curves for sublethally irradiated mouse recipients (n = 5–10 per cohort) of BM cells transformed with MLL-AF9 WT (blue), MLL-AF9 E531R (1×; red), MLL-AF9 E531R (NC; red, cross), MLL-AF9 E531R (10×; maroon), MLL-AF9 E531R + Eya1 (green, triangle), or Eya1 alone (green, open diamond). All mice received the same number of transduced cells except those indicated (10×) which received 10-fold more cells. All mice received cells that had been cultured one week in methylcellulose prior to transplant except those indicated as (NC) that had no in vitro culture prior to transplant. Statistical significance compared with MLL-AF9 E531R was determined using a log-rank (Mantel–Cox) test. B, Images of Wright–Giemsa-stained mouse BM cells expressing MLL-AF9, MLL-AF9 E531R, and MLL-AF9 E531R coexpressing Eya1, SIX1, or both Eya1 and SIX1, following the third plating in methylcellulose (scale bar, 10 μm). C, Summary of immunofluorescence staining of MLL-AF9 E531R cells from B for relevant hematopoietic stem and differentiation surface markers. Relative mean fluorescence intensity (MFI) of staining is shown. Data are from three independent experiments; *, P < 0.05. D, Schematic of key effects of loss of BCOR/MLL-AF9 interaction. The levels of expression of Eya1, Six1, Hoxa7, and Cyclins D1–3 are regulated by the interaction of BCOR with WT MLL-AF9. In the absence of the MLL-AF9/BCOR interaction, levels of Eya1 and Six1 are decreased, as is the level of Myc protein, reducing the LIC population and resulting in a lack of in vivo leukemogenesis. The levels of Hoxa7 and Cyclins D1–3 are increased, resulting in increased proliferation as seen in the colony-forming assay.
To further examine the extent to which loss of Eya1 and Six1 drive the observed effects, we expressed Eya1 and/or SIX1 in the MLL-AF9(E531R) cells (Supplementary Fig. S7B). As shown in Fig. 7B, addback of Eya1, but not SIX1, changes the in vitro cellular morphology to more resemble the blast-like morphology seen with WT MLL-AF9. Similar results were obtained with addback of Eya1 + SIX1. To probe this further, we have assessed surface marker expression. Previous studies have shown that loss of MLL-AF9 leads to differentiation with decreased surface c-kit (35, 41). As shown in Fig. 7C and Supplementary Fig. S7D, addback of Eya1 or Eya1 + SIX1 to MLL-AF9(E531R) cells increased c-kit and decreased CD11b, a marker present on monocytic cells. As shown in Supplementary Fig. S7C, addback of Eya1 and SIX1 had minimal effects on the expression of Hoxa7, Hoxa9, and Meis1 but increased expression of Hoxa10 almost 4-fold, which corresponds roughly to the fold change in its expression upon introduction of the E531R mutation in MLL-AF9. Thus, the change in expression of Hoxa10 is largely driven by changes in Eya1 and Six1 expression. Addback of Eya1 increased the expression of Six1, whereas addition of human SIX1 decreased expression of the mouse Six1, suggesting a negative feedback loop for Six1 on its own expression.
Discussion
Whereas the need for CBX8 and BCOR, integral components of the canonical PRC1 and the noncanonical PRC1.1 complexes, respectively, to be expressed in order for MLL-AF9 to induce leukemia has been demonstrated (16, 17, 22), the functional role of their direct binding to MLL-AF9 has not been established. We show that the CBX8– and BCOR–AF9 complexes adopt mixed alpha–beta structures highly similar to the AF4– and DOT1L–AF9 complexes we described previously (5, 9). This demonstrates that all of the interactions of the AF9 AHD are mutually exclusive with one another, as described for AF4 and DOT1L and suggested for CBX8 and BCOR (3, 9, 42). Of the AF9 binding proteins, CBX8 is distinct in its relatively weak AF9 binding (Fig. 1C). Intriguingly, CBX8 and BCOR both interact much more strongly with ENL (Fig. 1D) despite little to no difference in the binding of AF4 or DOT1L. This provides potential insight into the phenotypic differences observed between MLL-AF9 and MLL-ENL leukemias. Cbx8 knockout in mouse BM cells harboring MLL-AF9 severely limits colony formation, as reported previously (17), which can be rescued by addback of exogenous CBX8 (Fig. 2E and F). Addback of the CBX8 A335V mutant, which has increased affinity for AF9, reduces the number of colonies and cells and results in a more differentiated phenotype (Fig. 2E–G). This is true even in the presence of endogenous Cbx8, as the tighter-binding mutant likely outcompetes the weakly bound WT protein for AF9 interaction. As all four binding partners bind to the same site on AF9, increased binding of Cbx8 will necessitate decreased binding of all the other interaction partners. As we have previously shown that loss of AF4 or DOT1L binding leads to reduced colony formation, the observed reduced colony formation with CBX8(A335V) is almost certainly due to the displacement of AF4 and DOT1L. The R336D mutation was designed to weaken CBX8 binding to AF9 by disrupting an electrostatic interaction between CBX8 R336 and AF9 D544, a residue that is also critical for AF4–AF9 binding (4, 9). Addback of this mutant results in no significant change in colony formation despite its weaker affinity for AF9 (Figs. 1E and 2E), indicating that the direct CBX8/MLL-AF9 interaction is not essential for MLL-AF9–driven leukemia.
Previous studies showed that Cbx8 knockdown resulted in loss of Hoxa9 expression and suggested that loss of CBX8 binding to MLL-AF9 also reduced Hoxa9 expression, indicating that CBX8 could play an activating role in this context (17). However, we have previously noted that the mutations used in those studies also affect AF4 and DOT1L binding (5). In addition, AF9 T542A causes a significant reduction in BCOR binding, making the results difficult to interpret and strongly suggesting that the observed effects derived from reduced recruitment of binding partners other than CBX8.
Our structural studies made it possible to identify an AF9 point mutation, which selectively disrupts BCOR binding, thus avoiding the challenges associated with affecting other binding proteins. The AF9 E531R mutant drastically reduces BCOR binding affinity with little effect on AF4, DOT1L, and CBX8 binding (Fig. 3E and F; Supplementary Fig. S2G–S2J). Introduction of this mutation into MLL-AF9 completely abrogates the ability of MLL-AF9 to induce leukemia in vivo (Fig. 4G), showing that direct BCOR recruitment is essential. As loss of BCOR binding likely results in increased occupancy of other binding partners, it is unclear whether departure of BCOR or increases in occupancy of other partner proteins drives the phenotypic effects observed. There is a reduction in the LIC population with loss of BCOR binding as transplant of at least 10× more cells was required to develop leukemia, albeit with a longer latency and altered morphology (Fig. 7A; Supplementary Fig. S7A). This suggests both a qualitative and quantitative difference in LICs with abrogated BCOR binding. Analysis of RNA-seq data comparing WT and E531R-mutant MLL-AF9 cells showed the phosphatase Eya1 was the most downregulated direct MLL-AF9 target gene and that one of its interaction partners, the transcription factor Six1, is also downregulated (Fig. 5). Importantly, forced expression of Eya1 in E531R-mutant MLL-AF9 rescued leukemia-initiating activity, but with delayed latency (Fig. 7A). Interestingly, genes altered by the E531R mutation overlap minimally with those affected by abrogation of MLL-AF9/DOT1L binding, indicating that BCOR recruitment to MLL-AF9 regulates a distinct subset of genes (Fig. 5C).
EYA phosphatase family members (1, 2, 3, 4) bind to the transcription factors SIX1 and SIX2 to regulate gene expression (38), with the phosphatase activity of the EYA partner being critical for gene activation (43). Indeed, either SIX proteins alone or EYA1 alone show moderate or low levels of activation of reporter constructs, whereas cotransfection of both produces synergistic activation (44). As Eya1 is the most downregulated direct MLL-AF9 target gene in MLL-AF9 (E531R) cells and Six1 is also downregulated, the data suggested that a meaningful portion of the changes in gene expression may arise from altered Eya1/Six regulation of target genes. Comparison of the MLL-AF9 (E531R) expression data to ChIP-seq data for SIX1 and SIX2 in nephron progenitors (40) indeed shows 30% of our altered genes are direct targets of SIX1 and/or SIX2 (Supplementary Fig. S6A–S6C), confirming the importance of Eya1/Six–regulated genes in the effects seen with loss of BCOR binding to MLL-AF9.
Recent studies have shown that altered expression of the retinal determination gene network (RDGN) components DACH, EYA, and SIX can drive carcinogenesis with DACH acting as a tumor suppressor in opposition to EYA and SIX acting as oncogenes (38). Consistent with this, in cancers, EYA and SIX genes are upregulated, while DACH genes are downregulated (45). Importantly for our studies, components of the RDGN have been demonstrated to regulate the balance between self-renewal and differentiation of cancer stem cells (46, 47). Indeed, Eya1 overexpression immortalizes hematopoietic progenitor cells, and heterodimerization with Six1 augments this effect (26). Furthermore, two recent reports demonstrate a reduced LIC population in Six1 knockout or knockdown mouse cells expressing MLL-AF9 versus in a Six1 WT background (48, 49). The unexpected observation that we see increased proliferation in the colony formation assay with MLL-AF9(E531R) could in part be due to increased expression of Hoxa7 (Fig. 5D; Supplementary Fig. S4C), which contributes to hematopoietic stem/progenitor cell proliferative capacity (50). The Eya1/Six1/Dach1 axis likely contributes as well. Reduced Eya1/Six1 levels enable Dach1-dependent increases in Ccnd1 and Ccnd3 in myeloid cells (51), consistent with the increased expression of Ccnd1, Ccnd2, and Ccnd3 in our gene expression data (Figs. 5A and 7D).
In addition to its role in regulating SIX transcription factor function, EYA1 has been shown to decrease the proteasomal degradation of MYC via dephosphorylation of T58 (33, 34). Consistent with the reduced Eya1 level in MLL-AF9 E531R cells, we observe decreased Myc protein in E531R cells relative to WT cells (Fig. 5E). GSEA analysis of our RNA-seq data identified a MYC signature (Fig. 5D; Supplementary Fig. S4H). MYC overexpression, via a variety of mechanisms, is observed in 30% of all human cancers and is correlated with increased risk of relapse and poor outcome (52). A critical role for MYC has been demonstrated in multiple types of leukemia, including MLL-AF9 leukemia (53, 54, 55, 56). The decreased Myc protein level observed in our MLL-AF9 (E531R) cells undoubtedly contributes to the reduction in LICs we observe. In human cells, EYA1 levels are lower in more differentiated myeloid lineages (CMP, GMP, and MEP) than in HSCs (BloodSpot– HemaExplorer dataset), suggesting higher levels of EYA1 are needed for maintenance of the HSC phenotype. Furthermore, EYA1 levels are consistently similar to or higher than that seen in HSCs across almost all subtypes of AML, including t(11q23)/MLL (BloodSpot–Merged AML dataset), consistent with a potential need for an increased level of EYA1 to maintain self-renewal. It is important to point out that as we only observe an effect on leukemia in vivo but no reduction, rather an increase, in proliferation ex vivo, EYA1 may not show up in genome-wide screens for dependencies such as the Cancer Dependency Map (depmap.org) that rely solely on effects on proliferation/survival ex vivo as their readout.
Our data indicate that the reduction in Eya1 and Six1 with loss of BCOR binding drives a loss of LICs, resulting in a lack of in vivo leukemogenesis (Fig. 4G). In support of this, we compared gene expression data from MLL-AF9 (E531R) with data from Stavropoulou and colleagues (35), which identified genes specifically upregulated or downregulated in LT-HSCs versus GMPs expressing MLL-AF9. MLL-AF9–expressing LT-HSCs, as compared with GMPs, had increased putative LICs, and generated a more aggressive population of LICs with increased migratory capacity and Ara-C resistance. The latter result is consistent with that of Armstrong and colleagues, who also showed an increased LIC population for MLL-AF9 in HSCs versus GMPs as well as increased chemoresistance (37). As shown in Fig. 6, comparison of our RNA-seq data for MLL-AF9 (E531R) identified genes that are increased with MLL-AF9 expressed in LT-HSCs versus GMPs but decreased with MLL-AF9 (E531R), as well as the converse. These 30 genes likely contribute to the observed reduction in LICs. Among the genes identified are numerous well-characterized regulators of LSC and/or HSC function that are down in MLL-AF9 (E531R) cells, including Bcl2, Flt3, Fyn, Gata2, Maml3, Sox4.
Here we have demonstrated the structural basis of CBX8 and BCOR binding to AF9. We have established that direct BCOR recruitment to MLL-AF9 is essential for leukemia, whereas direct CBX8 recruitment is dispensable. Furthermore, we show that the genes regulated by direct BCOR recruitment are distinct from those regulated by direct AF4 and DOT1L recruitment and are driven to a large extent by EYA1/SIX and MYC regulation of gene expression. This set of altered genes upon loss of BCOR binding includes genes that play an important role in MLL-AF9 LIC function. The importance of the BCOR interaction for MLL-AF9 leukemia has implications for development of efficacious targeted therapies for this devastating disease, particularly in light of the limited efficacy seen in initial clinical testing of enzymatic inhibitors of DOT1L. On the basis of our results, it is likely necessary to inhibit all the protein–protein interactions of the AF9 (or ENL) portions of MLL-AF9 (or MLL-ENL) rather than targeting selected partners to achieve efficacy. Furthermore, our data strongly support the likely therapeutic utility of targeting the phosphatase activity of Eya1.
Methods
Structure Determination
Standard NMR methods were used to determine the CBX8–AF9 and BCOR–AF9 complex structures. Details are provided in Supplementary Information.
Fluorescence Polarization–Binding Assays
Standard methods were employed for the fluorescence polarization (FP) measurements of binding. Details are provided in Supplementary Information.
Serial Replating Assays
All studies involving mice were approved by Loyola University's Institutional Animal Care and Use Committee, according to standards set forth in the NIH Guidelines. MLL-AF9 wild-type (WT) and MLL-AF9–mutant serial replating assays were conducted with murine bone marrow (BM) c-kit+ cells transduced with MSCVneo, MSCVneo-MLL-AF9 (WT), MSCVneo-MLL-AF9 (D546R), or MSCVneo-MLL-AF9 (E531R) retroviruses. Cells were plated in methylcellulose medium with cytokines and G418, as we have described previously (25). Colonies and cells were enumerated, and cells were serially replated after 7 days for each of 4 weeks. Colony size was quantified using the Fiji distribution of ImageJ analysis software. The arithmetic mean of the individual mean Feret diameters was then calculated for each experimental group. Statistical significance was determined using Student two-tailed t test with a confidence interval of 95%. Colony assays were conducted in duplicate and repeated n = 6 for all of the constructs.
For CBX8 genetic complementation methylcellulose assays, full-length CBX8, either WT or containing different mutations, was cloned into an MSCV-mCherry retroviral vector (kindly provided by Dr. Jiwang Zhang, Loyola University Chicago, Chicago, IL) and verified by sequencing. MLL-AF9–transformed BM progenitor cells from Cbx8fl/fl-Cre-ERT2 mice, which were previously published (kindly provided by Dr. Hess, Indiana University, Indianapolis, IN), were infected with retroviruses expressing mCherry-CBX8 (WT or mutant). After 20 to 24 hours, mCherry-positive cells were sorted by FACS and cultured in methylcellulose as described above, but containing 4-hydroxytamoxifen (Sigma; H-7904) solubilized in 100% EtOH to induce endogenous Cbx8 deletion or EtOH as vehicle control. After 1 week, colonies were counted and cells processed for cytospin, RNA, and genomic DNA. PCR was used to determine the status of the endogenous Cbx8 floxed allele using primers as described previously (17).
For Eya1 and SIX1 complementation assays, MLL-AF9 E531R–transformed cells were infected with MSCVneo-Eya1 and/or MSCVpuro-SIX1 retroviruses (kindly provided by Dr. Michael Thirman, University of Chicago, Chicago, IL; described in ref. 26) and plated in methylcellulose medium with cytokines and puromycin or G418 as described above.
Flow Cytometry
Flow cytometry was carried out using standard approaches as we have done previously. Details are provided in Supplementary Information.
In Vivo Transplantation
All studies involving mice were approved by Loyola University's Institutional Animal Care and Use Committee, according to standards set forth in the NIH Guidelines. Transformed BM cells (25,000) were transplanted into sublethally irradiated (450 cGy, Radsource, RS-2000) 7- to 9-week-old C57Bl/6 mice (both males and females) via tail vein injection. Male and female mice were equally distributed and randomly assigned to receive MLL-AF9– or MLL-AF9(E531R)– or MLL-AF9(E531R) + Eya1– or Eya1 alone–transformed cells. For 10X MLL-AF9(E531R) experiments, mice received 250,000 cells. Additional mice received 100,000 freshly infected MLL-AF9(E531R) BM cells without prior culture in methylcellulose as previously described (25). Mice were group housed in sterile rodent microisolator caging with filtered cage tops and provided with water and chow ad libitum. Mice were prophylactically treated with Baytril (Enrofloxacin, Sigma, #17849) at 0.1 mg/mL in water ad libitum for 14 days postirradiation. Mice were monitored for leukemia development by observing for lethargy, significant weight loss, or ruffled fur. Leukemia was verified by peripheral blood CBC (complete blood count) analysis (Hemavet 950FS) and after sacrifice by examining the BM, peripheral blood, and spleen.
RNA-seq
RNA was isolated from three MLL-AF9 WT and three MLL-AF9E531R samples by HudsonAlpha Genome Services Lab using the miRNeasy Mini Kit (Qiagen) and the standard protocol with on-column DNase treatment. Isolated RNA was quantified by Qubit (Invitrogen) and underwent quality control with a 2100 Bioanalyzer (Agilent). Samples were normalized to 500 ng of input in 50 μL, and mRNAs were enriched by poly(A) selection. Resultant RNA was used for directional first-strand synthesis and cDNA library synthesis. Libraries were sequenced using a 100PE lane on a HiSeq 2500 (Illumina). Details of the analysis of the RNA-seq data are provided in Supplementary Information.
Quantitative RT-PCR
qPCR was carried out using standard approaches as we have done previously with expression normalized to Polr2a. Details are provided in Supplementary Information. Primers used are listed in Supplementary Table S4.
Western Blot Analysis for Myc Protein Level
Whole-cell lysates were prepared from MLL-AF9 and MLL-AF9(E531R)–transformed primary BM cells using standard approaches. Details are provided in Supplementary Information.
Accession Numbers
The Protein Data Bank (PDB) IDs for the CBX8– and BCOR–AF9 structures are 2N4Q and 6B7G, respectively. The Gene Expression Omnibus (GEO) accession number for RNA-seq data is GSE105767.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors' Contributions
C.R. Schmidt: Investigation, writing–original draft, writing–review and editing. N.J. Achille: Investigation. A. Kuntimaddi: Investigation. A.M. Boulton: Investigation. B.I. Leach: Investigation. S. Zhang: Investigation. N.J. Zeleznik-Le: Conceptualization, resources, writing–original draft, funding acquisition, writing review and editing. J.H. Bushweller: Conceptualization, formal analysis, funding acquisition, writing–review and editing.
Supplementary Material
Acknowledgments
This work was supported by grants from the NCI (R01 CA155328 and R01 CA233749; to N.J. Zeleznik-Le and J.H. Bushweller) and an ASH Bridge Grant (to N.J. Zeleznik-Le). The 800 MHz NMR spectrometer at the University of Virginia used in these studies was purchased with funds from NIH S10RR023035. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grant P41GM103399 [National Institute of General Medical Sciences (NIGMS); old number: P41RR002301]. The authors also acknowledge HudsonAlpha for their work in RNA isolation and performance of RNA-seq and Dr. Jay Hess for MLL-AF9–transformed conditional Cbx8 knockout cells.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Blood Cancer Discovery Online (https://bloodcancerdiscov.aacrjournals.org/).
Current address for B.I. Leach: Department of Radiology, University of California, San Diego, La Jolla, California.
Blood Cancer Discov 2020;1:162–77
References
- 1. Meyer C, Schneider B, Jakob S, Strehl S, Attarbaschi A, Schnittger S, et al. The MLL recombinome of acute leukemias. Leukemia 2006;20:777–84. [DOI] [PubMed] [Google Scholar]
- 2. Okuda H, Stanojevic B, Kanai A, Kawamura T, Takahashi S, Matsui H, et al. Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J Clin Invest 2017;127:1918–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 2010;17:198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lokken AA, Achille NJ, Chang MJ, Lin JJ, Kuntimaddi A, Leach BI, et al. Importance of a specific amino acid pairing for murine MLL leukemias driven by MLLT1/3 or AFF1/4. Leuk Res 2014;38:1309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuntimaddi A, Achille NJ, Thorpe J, Lokken AA, Singh R, Hemenway CS, et al. Degree of recruitment of DOT1L to MLL-AF9 defines level of H3K79 Di- and tri-methylation on target genes and transformation potential. Cell Rep 2015;11:808–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meyer C, Burmeister T, Groger D, Tsaur G, Fechina L, Renneville A, et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018;32:273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 2014;159:558–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, et al. Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol Cell 2016;62:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leach BI, Kuntimaddi A, Schmidt CR, Cierpicki T, Johnson SA, Bushweller JH. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure 2013;21:176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Di Carlo V, Mocavini I, Di Croce L. Polycomb complexes in normal and malignant hematopoiesis. J Cell Biol 2019;218:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaustov L, Ouyang H, Amaya M, Lemak A, Nady N, Duan S, et al. Recognition and specificity determinants of the human Cbx chromodomains. J Biol Chem 2011;286:521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, et al. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell 2012;45:344–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morey L, Pascual G, Cozzuto L, Roma G, Wutz A, Benitah SA, et al. Nonoverlapping functions of the Polycomb group Cbx family of proteins in embryonic stem cells. Cell Stem Cell 2012;10:47–62. [DOI] [PubMed] [Google Scholar]
- 14. O'Loghlen A, Munoz-Cabello AM, Gaspar-Maia A, Wu HA, Banito A, Kunowska N, et al. MicroRNA regulation of Cbx7 mediates a switch of Polycomb orthologs during ESC differentiation. Cell Stem Cell 2012;10:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Creppe C, Palau A, Malinverni R, Valero V, Buschbeck M. A Cbx8-containing polycomb complex facilitates the transition to gene activation during ES cell differentiation. PLoS Genet 2014;10:e1004851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maethner E, Garcia-Cuellar MP, Breitinger C, Takacova S, Divoky V, Hess JL, et al. MLL-ENL inhibits polycomb repressive complex 1 to achieve efficient transformation of hematopoietic cells. Cell Rep 2013;3:1553–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tan J, Jones M, Koseki H, Nakayama M, Muntean AG, Maillard I, et al. CBX8, a polycomb group protein, is essential for MLL-AF9-induced leukemogenesis. Cancer Cell 2011;20:563–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahmad KF, Melnick A, Lax S, Bouchard D, Liu J, Kiang CL, et al. Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol Cell 2003;12:1551–64. [DOI] [PubMed] [Google Scholar]
- 19. Huynh KD, Fischle W, Verdin E, Bardwell VJ. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev 2000;14:1810–23. [PMC free article] [PubMed] [Google Scholar]
- 20. Hatzi K, Jiang Y, Huang C, Garrett-Bakelman F, Gearhart MD, Giannopoulou EG, et al. A hybrid mechanism of action for BCL6 in B cells defined by formation of functionally distinct complexes at enhancers and promoters. Cell Rep 2013;4:578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bagger FO, Sasivarevic D, Sohi SH, Laursen LG, Pundhir S, Sonderby CK, et al. BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res 2016;44:D917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van den Boom V, Maat H, Geugien M, Rodriguez Lopez A, Sotoca AM, Jaques J, et al. Non-canonical PRC1.1 targets active genes independent of H3K27me3 and is essential for leukemogenesis. Cell Rep 2016;14:332–46. [DOI] [PubMed] [Google Scholar]
- 23. Cao Q, Gearhart MD, Gery S, Shojaee S, Yang H, Sun H, et al. BCOR regulates myeloid cell proliferation and differentiation. Leukemia 2016;30:1155–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood 2011;117:6912–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cierpicki T, Risner LE, Grembecka J, Lukasik SM, Popovic R, Omonkowska M, et al. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat Struct Mol Biol 2010;17:62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang QF, Wu G, Mi S, He F, Wu J, Dong J, et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood 2011;117:6895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hemenway CS, de Erkenez AC, Gould GC. The polycomb protein MPc3 interacts with AF9, an MLL fusion partner in t(9;11)(p22;q23) acute leukemias. Oncogene 2001;20:3798–805. [DOI] [PubMed] [Google Scholar]
- 28. Srinivasan RS, de Erkenez AC, Hemenway CS. The mixed lineage leukemia fusion partner AF9 binds specific isoforms of the BCL-6 corepressor. Oncogene 2003;22:3395–406. [DOI] [PubMed] [Google Scholar]
- 29. Wamstad JA, Bardwell VJ. Characterization of Bcor expression in mouse development. Gene Expr Patterns 2007;7:550–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011;20:66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34:267–73. [DOI] [PubMed] [Google Scholar]
- 33. Li J, Rodriguez Y, Cheng C, Zeng L, Wong EYM, Xu CY, et al. EYA1's conformation specificity in dephosphorylating phosphothreonine in Myc and its activity on Myc stabilization in breast cancer. Mol Cell Biol 2017;37:e00499–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu J, Wong EY, Cheng C, Li J, Sharkar MT, Xu CY, et al. Eya1 interacts with Six2 and Myc to regulate expansion of the nephron progenitor pool during nephrogenesis. Dev Cell 2014;31:434–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stavropoulou V, Kaspar S, Brault L, Sanders MA, Juge S, Morettini S, et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell 2016;30:43–58. [DOI] [PubMed] [Google Scholar]
- 36. Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016;540:433–7. [DOI] [PubMed] [Google Scholar]
- 37. Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia 2013;27:852–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu Y, Han N, Zhou S, Zhou R, Yuan X, Xu H, et al. The DACH/EYA/SIX gene network and its role in tumor initiation and progression. Int J Cancer 2016;138:1067–75. [DOI] [PubMed] [Google Scholar]
- 39. Jemc J, Rebay I. The Eyes absent family of phosphotyrosine phosphatases: properties and roles in developmental regulation of transcription. Annu Rev Biochem 2007;76:513–38. [DOI] [PubMed] [Google Scholar]
- 40. O'Brien LL, Guo Q, Lee Y, Tran T, Benazet JD, Whitney PH, et al. Differential regulation of mouse and human nephron progenitors by the Six family of transcriptional regulators. Development 2016;143:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006;10:257–68. [DOI] [PubMed] [Google Scholar]
- 42. Biswas D, Milne TA, Basrur V, Kim J, Elenitoba-Johnson KS, Allis CD, et al. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc Natl Acad Sci U S A 2011;108:15751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 2003;426:247–54. [DOI] [PubMed] [Google Scholar]
- 44. Silver SJ, Davies EL, Doyon L, Rebay I. Functional dissection of Eyes absent reveals new modes of regulation within the retinal determination gene network. Mol Cell Biol 2003;23:5989–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Miller SJ, Lan ZD, Hardiman A, Wu J, Kordich JJ, Patmore DM, et al. Inhibition of Eyes Absent Homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene 2010;29:368–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Farabaugh SM, Micalizzi DS, Jedlicka P, Zhao R, Ford HL. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-beta signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene 2012;31:552–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCoy EL, Iwanaga R, Jedlicka P, Abbey NS, Chodosh LA, Heichman KA, et al. Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J Clin Invest 2009;119:2663–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang LS, Kang X, Lu J, Zhang Y, Wu X, Wu G, et al. Installation of a cancer promoting WNT/SIX1 signaling axis by the oncofusion protein MLL-AF9. EBioMedicine 2019;39:145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chu Y, Chen Y, Li M, Shi D, Wang B, Lian Y, et al. Six1 regulates leukemia stem cell maintenance in acute myeloid leukemia. Cancer Sci 2019;110:2200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fisher JB, Peterson J, Reimer M, Stelloh C, Pulakanti K, Gerbec ZJ, et al. The cohesin subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic repression of Hoxa7 and Hoxa9. Leukemia 2017;31:712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee JW, Kim HS, Kim S, Hwang J, Kim YH, Lim GY, et al. DACH1 regulates cell cycle progression of myeloid cells through the control of cyclin D, Cdk 4/6 and p21Cip1. Biochem Biophys Res Commun 2012;420:91–5. [DOI] [PubMed] [Google Scholar]
- 52. Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene 1999;18:3004–16. [DOI] [PubMed] [Google Scholar]
- 53. Pulikkan JA, Hegde M, Ahmad HM, Belaghzal H, Illendula A, Yu J, et al. CBFb-SMMHC inhibition triggers apoptosis by disrupting MYC chromatin dynamics in acute myeloid leukemia. Cell 2018;174:172–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018;553:515–20. [DOI] [PubMed] [Google Scholar]
- 55. Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev 2013;27:2648–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 2014;20:1130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.