

Summary
Background
In the ongoing phase 3 CheckMate 214 trial, nivolumab plus ipilimumab (NIVO+IPI) demonstrated superior efficacy over sunitinib (SUN) in patients with previously untreated IMDC intermediate/poor-risk advanced renal cell carcinoma (aRCC) with a manageable safety profile. We assessed efficacy and safety after extended follow-up to inform the long-term clinical value of NIVO+IPI versus SUN in this setting.
Methods
In the phase 3, randomised, controlled CheckMate 214 trial, patients aged 18 years and older with previously untreated, advanced or metastatic renal cell carcinoma with a clear-cell component were recruited from 175 hospitals and cancer centres in 28 countries. Patients were categorised by IMDC risk status into favourable, intermediate, and poor risk subgroups and randomly assigned (1:1) to open-label NIVO 3 mg/kg IV plus IPI 1 mg/kg IV every 3 weeks for four doses, followed by NIVO 3 mg/kg IV every 2 weeks; or SUN 50 mg orally once daily for 4 weeks (6-week cycle). Randomisation was done with a block size of four and stratified by risk status and region. Here, we report overall survival (OS), investigator-assessed PFS, the proportion of patients achieving an investigator-assessed objective response per RECIST v1.1, characterisation of response, and safety in all patients (ITT and across risk groups) with extended follow-up. The co-primary endpoints of the trial reported previously were OS, proportion of patients achieving an objective response per IRRC, and progression-free survival (PFS) per IRRC in intermediate/poor-risk patients. This study is registered with ClinicalTrials.gov, number NCT02231749, and is ongoing but is now closed to recruitment.
Findings
Between October 16, 2014, and February 23, 2016, of 1390 patients screened, 1096 (79%) were randomised to NIVO+IPI versus SUN (550 vs 546 ITT; 425 vs 422 intermediate/poor risk, 125 vs 124 favourable risk). Efficacy outcomes were assessed in all randomised patients; safety outcomes were assessed in all treated patients. Median follow-up was 32·4 months (IQR 13·4–36·3). In intermediate/poor-risk patients, improved OS (HR 0·66; 95% CI 0·54–0·80; p<0·0001; median not reached [95% CI 35·6-not estimable] vs 26·6 [95% CI 22·1–33·4] months), PFS (HR 0·77; 95% CI 0·65–0·90; p<0·01; median 8·2 [95% CI 6·9–10·0] vs 8·3 [95% CI 7·0–8·8] months), and proportion of patients achieving objective response (178 [42%] of 425 vs 124 [29%] of 422; p<0·01, Δresponse = 12·8% [95% CI 6·5–19·2]) were maintained with NIVO+IPI versus SUN, respectively. Similarly in ITT patients, longer OS (HR 0·71; 95% CI 0·59–0·86; p<0.01; median not reached [95% CI not estimable] vs 37·9 [95% CI 32·2-not estimable] months), longer PFS (HR 0·85; 95% CI 0·73–0·98; p=0·03; median 9·7 [95% CI 8·1–11·1] vs 9·7 [95% CI 8·3–11·1] months), and a higher proportion of patients achieving objective response (227 [41%] of 550 vs 186 [34%] of 546; p=0·02, Δresponse = 7·6% [95% CI 2·0–13·2]) were observed with NIVO+IPI versus SUN, respectively.Of all treated patients (547 NIVO+IPI; 535, SUN), the most common grade 3–4 treatment-related AEs were increased lipase (57 [10%]), amylase (31 [6%], and alanine aminotransferase (28 [5%]) with NIVO+IPI vs hypertension (90 [17%]), fatigue (51 [10%]), and palmar-plantar erythrodysaesthesia (49 [9%]) with SUN. Eight deaths in the NIVO+IPI arm (one each due to pneumonitis, pneumonia and aplastic anaemia, immune-mediated bronchitis, lower gastrointestinal haemorrhage, hemophagocytic syndrome, sudden death, liver toxic effects, and lung infection) and four deaths in the SUN arm (two cardiac arrests and one each of heart failure and multiple organ failure) were reported as treatment-related.
Interpretation
These results suggest that the superior benefit-risk profile of NIVO+IPI over SUN were maintained in intermediate/poor-risk and ITT patients with extended follow-up and demonstrate the long-term benefits of NIVO+IPI in patients with previously untreated aRCC across all risk categories.
Keywords: nivolumab, ipilimumab, sunitinib, first-line, treatment-naïve, advanced renal cell carcinoma, RCC, metastatic renal cell carcinoma, characterisation of response, IMDC risk, combination immunotherapy
Introduction
The immunotherapy combination of nivolumab (NIVO; a programmed death 1 immune checkpoint inhibitor antibody) plus ipilimumab (IPI; a cytotoxic T-lymphocyte antigen 4 antibody) is a new standard-of-care option for first-line treatment of intermediate/poor-risk patients with advanced renal cell carcinoma (aRCC), based on superior overall survival (OS) and a higher proportion of patients achieving an objective response compared with sunitinib (SUN; a tyrosine kinase inhibitor [TKI]) demonstrated in the randomised, phase 3 CheckMate 214 trial.1–5
At a prespecified interim analysis with a minimum follow-up of 17·5 months, CheckMate 214 met two of three co-primary endpoints, with NIVO+IPI demonstrating superior OS and a higher objective response per independent radiology review committee (IRRC) over SUN in the primary efficacy population of International Metastatic RCC Database Consortium (IMDC) intermediate/poor-risk aRCC. Median OS was not reached (NR) versus 26·0 months, respectively; hazard ratio [HR] for death was 0·63 (p<0·01). Higher proportions of intermediate/poor-risk patients achieved confirmed objective responses (42% vs 27% [p<0·01]) and complete responses (9% vs 1% [p<0·01]) with NIVO+IPI versus SUN. Median progression-free survival (PFS) was 11·6 versus 8·4 months for NIVO+IPI versus SUN (p=0·03), a clinically meaningful improvement that did not meet the alpha level (p<0·009) for statistical significance (HR for disease progression or death, 0·82; 99·1% confidence interval [CI] 0·64–1·04). Among the secondary endpoints in the intention-to-treat (ITT) population, including both intermediate/poor- and favourable-risk patients, NIVO+IPI demonstrated longer OS (HR for death 0·68; 99·8% CI 0·49–0·95; p<0·01), numerically higher objective response (39% vs 32%; p=0·02), and similar PFS versus SUN (HR for disease progression or death, 0·98; 99·1% CI, 0·79–1·23; p=0·85). In the exploratory favourable-risk subpopulation, objective response was higher (29% vs 52%; p<0·01), and PFS (HR for disease progression or death, 2·18; 99·1% CI, 1·29–3·68; p<0·01) and OS were longer with SUN versus NIVO+IPI (HR for death 1·45; 99·8% CI, 0·51–4·12; p=0·27); however, only 37 deaths had occurred as of this database lock.1 Health-related quality of life was consistently better with NIVO+IPI than SUN in both ITT and intermediate/poor-risk patients.6
Long-term updates critically inform the value of immunotherapy-based regimens. Targeted agents have demonstrated improved short-term endpoints such as objective response and PFS,7 but immunotherapy combinations are better judged with endpoints indicative of durable response and long-term survival. Here, we report expanded CheckMate 214 efficacy and safety analyses with extended follow-up including OS, a post-hoc multivariable model of OS to inform future prognostic models, investigator-assessed PFS and objective response, characterisation of response, treatment-related select adverse event (AE) and common treatment-related AE incidence, together with high-grade treatment-related AEs over time.
Methods
Study design and participants
CheckMate 214 was a randomised, open-label, phase 3 trial comparing NIVO+IPI induction followed by NIVO maintenance versus SUN monotherapy in patients with previously untreated aRCC, recruited from 175 hospitals and cancer centres in 28 countries. Detailed methodology was reported previously.1 Eligible patients were aged 18 years or older with histological confirmation of advanced or metastatic renal cell carcinoma with a clear cell component with no previous systemic therapy for their disease, with the exception of one previous adjuvant or neoadjuvant therapy (not including VEGF-targeted agents) for completely resectable renal cell carcinoma if recurrence occurred 6 months or more after the last dose. Further inclusion criteria were a Karnofsky performance status of 70% or more, and measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Key exclusion criteria were history of or current central nervous system metastases and active or recent history of autoimmune disease or use of systemic corticosteroids within 14 days of group assignment. Additionally, patients with any of the following laboratory test findings were deemed ineligible: white blood cells <2000/mm3, neutrophils <1500/mm3, platelets < 100,000/mm3, aspartate aminotransferase or alanine aminotransferase >3× upper limit of normal (ULN; >5× ULN if liver metastases are present), total bilirubin >1·5× ULN (except subjects with Gilbert syndrome, who could have total bilirubin <3·0 mg/dL), serum creatinine >1·5× ULN, or creatinine clearance <40 mL/min. Some patients were not randomised post enrolment due to AEs, withdrawal of consent, non-compliance, no longer meeting study criteria, or death. CheckMate 214 was approved by the institutional review board or ethics committee at each site and conducted in accordance with Good Clinical Practice guidelines per the International Conference on Harmonisation. All patients provided written informed consent per Declaration of Helsinki principles. The protocol is available in the appendix (pp 13–144). Protocol deviations considered relevant were collected by treatment group and overall in all randomised patients (data not shown).
Randomisation and masking
Patients were randomised in a 1:1 ratio to NIVO+IPI or SUN through an Interactive Voice Response System (IVRS). Randomisation procedures were carried out via permuted blocks within each stratum, with a block size of four and stratified by IMDC risk score (favourable [0] vs intermediate [1–2] vs poor [3–6]) and by geographical region (United States vs Canada and Europe vs rest of the world).1,8 Allocation and implementation was managed via IVRS. Patients and investigators were not masked to treatment assignment as this was an open-label trial.
Procedures
The treatment regimen was administered in cycles of 6 weeks. NIVO 3 mg/kg and IPI 1 mg/kg were administered intravenously over approximately 60 and 30 minutes, respectively, every 3 weeks for four doses (induction), followed by NIVO 3 mg/kg every 2 weeks (maintenance). SUN 50 mg was administered orally once daily for 4 weeks on and 2 weeks off in each 6-week cycle. Treatment continued until disease progression or unacceptable toxicity. No dose reductions were allowed in the NIVO +IPI arm, whereas two dose reductions were permitted for SUN in 12·5-mg increments for grade 3–4 toxic side-effects.9 Patients could continue study treatment after initial investigator-assessed progression per RECIST if the investigator thought the patient tolerated and benefited from treatment despite initial evidence of disease progression. As of a November 13, 2017, protocol amendment, NIVO+IPI arm patients could discontinue after 2 years of study treatment even in the absence of disease progression or unacceptable toxicity at the discretion of the patient and/or investigator; NIVO+IPI patients who were receiving NIVO monotherapy could also switch to a flat dose of NIVO 240 mg every 2 weeks; and among intermediate/poor-risk patients, crossover from the SUN arm to the NIVO+IPI arm was allowed. Disease assessments were done with CT or MRI at baseline, and treated patients were evaluated for response according to RECIST v1.1 guidelines beginning 12 (±1) weeks after randomisation and continuing every 6 (±1) weeks for the first 13 months, then every 12 (±1) weeks until disease progression or treatment discontinuation, whichever occurred later. AEs were assessed continuously during treatment visits, then at follow-up visits 1 and 2 and during OS follow-up, graded per National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v4.0,10 and reported from the first dose and up to and including 30 days after the last study treatment dose. Blood chemistry assessments were done on day 1 weeks 1 and 4 during cycles 1 and 2; day 1 of weeks 1, 3, and 5 during subsequent cycles; and at follow-up visits 1 and 2.
Outcomes
The co-primary endpoints of CheckMate 214 were OS, PFS per IRRC, and the proportion achieving objective response per IRRC in intermediate/poor-risk patients. Secondary endpoints were OS, PFS per IRRC, the proportion achieving objective response per IRRC in the ITT population, and incidence of AEs in all treated patients. Exploratory endpoints included efficacy in favourable-risk patients; all were reported previously.1 In the present prespecified follow-up analysis after co-primary endpoints were met, OS was analysed as reported previously, however, progression and incidence of objective response (including time to response and duration of response) were assessed per investigator using RECIST v1.1 instead of IRRC. OS was defined as the time from randomisation to the date of death. For patients who were alive, their survival time was censored at the date of last contact; OS was censored for patients at the date of randomisation if they were randomised but had no follow-up. Objective response rate was defined as the proportion of randomised patients who achieved a best response of complete response or partial response using RECIST v1.1 based on investigator assessment. The primary definition of PFS was specified as the time between the date of randomisation and the first date of documented progression, based on investigator assessments (per RECIST v1.1), or death due to any cause, whichever occurred first. Patients who died without a reported progression were considered to have progressed on the date of death. Prespecified safety outcomes included treatment-related AEs and treatment-related AEs leading to discontinuation or death, per CTCAE, v4.0.10 Incidence of these AEs was defined as the proportion of patients with any-grade AEs among patients treated in each treatment arm. Treatment-related select AEs (prespecified and defined as events that may be immune-mediated, differ from those caused by non-immunotherapeutic agents, may require immunosuppression for management, and whose early recognition may mitigate severe toxicity) were also reported, including skin, gastrointestinal, endocrine, hepatic, pulmonary, or renal system events. Additional methodology details for outcomes are reported in the appendix (pp 6).
Statistical analyses
Details of the statistical analyses for the primary and secondary endpoints have been previously reported; as the first planned interim OS analysis met the pre-specified statistical significance boundary for the coprimary endpoint of OS, this was considered the final primary analysis per protocol.1 Briefly, 1070 participants were estimated to be required to enrol 820 (eg, the number needed for robust statistical analysis) who were at intermediate or poor risk. The overall alpha level of 0·05 was split among the coprimary endpoints (0·001 for the proportion of patients who achieved an objective response, 0·009 with 80% power for PFS, and 0·04 with 90% power for OS) in the primary analysis. A formal comparison of OS in ITT patients was conducted using a two-sided stratified log-rank test at the same alpha level at the time of the final analysis of co-primary endpoints based on a hierarchical testing procedure. Alpha allocations inform the sample size determination to adequately power the study and are no longer relevant in this analysis. We include p-values for descriptive purposes to confirm consistency with primary outcomes as appropriate. Efficacy outcomes were assessed in all randomised patients (ITT population and by risk) and safety outcomes were assessed in all treated patients. The prespecified analyses of OS, PFS, duration of study therapy, and duration of response were estimated per Kaplan-Meier methodology.11 Stratified Cox proportional HRs and 95% CIs were calculated between treatment arms for OS and PFS, and as an exploratory measure of duration of response. Post-hoc analysis of the effects of clinically relevant baseline features (tumour programmed death ligand 1 [PD-L1] expression, neutrophil-to-lymphocyte ratio, tumour burden, prior nephrectomy, individual IMDC risk factors,8 and individual Memorial Sloan Kettering Cancer Center risk factors12) on OS was performed using univariable and multivariable models for each ITT treatment arm separately to distinguish factors relevant to NIVO+IPI versus SUN. Individual factors first underwent univariable analysis to preclude introducing collinearity into the model. A biologically plausible multivariable model was built and verified, as baseline factors associated with OS at p<0·1 in the univariable model were entered into a full Cox proportional hazards multivariable regression model. Parsimonious (reduced) multivariable OS models included all baseline factors associated with OS at a nominal p<0·05. The proportion of patients achieving investigator-assessed objective response and the exact two-sided 95% CI were computed per Clopper-Pearson methodology,13 with strata-adjusted objective response differences between treatment arms, based on DerSimonian-Laird methods,14 included as an exploratory measure. Additional post-hoc efficacy analyses in responders included baseline characteristics, disposition, depth of response, additional response kinetics, and treatment-free survival (defined as the time between protocol therapy discontinuation and subsequent systemic anticancer treatment initiation or death, whichever occurred first, in patients who discontinued protocol therapy for any reason).9 Prevalence of grade 3–4 treatment-related AEs of clinical interest was analysed post hoc in all treated patients and graphed by system organ class using density plots summing vectors over time for each treatment arm, where each vector represents an individual patient’s time to onset through resolution of the AE; if multiple events were reported for a patient in the same organ category, the earliest and the latest to resolve in the category were used. All statistical analyses were conducted using SAS v8.2 (SAS Institute, North Carolina, USA) or East v5.4. (Cytel, Cambridge, Massachusetts, USA). This study is registered with ClinicalTrials.gov, number NCT02231749.
Role of the funding source
The funders (Bristol-Myers Squibb and ONO Pharmaceutical) contributed to the study design, data analysis, and data interpretation in collaboration with the authors. The funders had no role in data collection. Financial support for editorial and writing assistance was provided by the funders. A data confidentiality agreement was in place between Bristol-Myers Squibb and the investigators. All authors vouch for the completeness and accuracy of the data and analyses and for the adherence of the trial to the protocol. All authors had full access to all of the data included in the study, and all authors contributed to drafting the manuscript and provided final approval to submit the manuscript. The corresponding author had full access to all of the data and final responsibility to submit for publication.
Results
CheckMate 214 enrolled patients from October 16, 2014, through February 23, 2016; 1390 patients were assessed and 1096 (79%) were randomised (240 [17%] of 1390 did not meet inclusion criteria, 24 [2%] withdrew consent, and 30 [2%] were not randomised for other reasons; figure 1). The ITT population was analysed for efficacy and comprised 550 patients randomised to NIVO+IPI (425 intermediate/poor, 125 favourable risk) and 546 patients randomised to SUN (422 intermediate/poor, 124 favourable risk); 547 NIVO+IPI arm and 535 SUN arm patients received treatment and were included in the safety analysis. Baseline characteristics were similar in the two treatment arms, and across intermediate/poor-risk, ITT, and favourable-risk patients (table S1, pp 7). However, baseline tumour PD-L1 expression level was lower in both treatment arms among favourable-risk versus intermediate/poor-risk and ITT patients.1 The data cutoff for this analysis with extended follow-up was August 6, 2018. At a minimum OS follow-up of 30 months (median 32·4 [IQR 13·4–36·3]), 82 of 547 patients (15%) continued therapy in the NIVO+IPI arm and 49 of 535 (9%) continued in the SUN arm (figure 1).
Figure 1: CONSORT diagram for patient disposition.
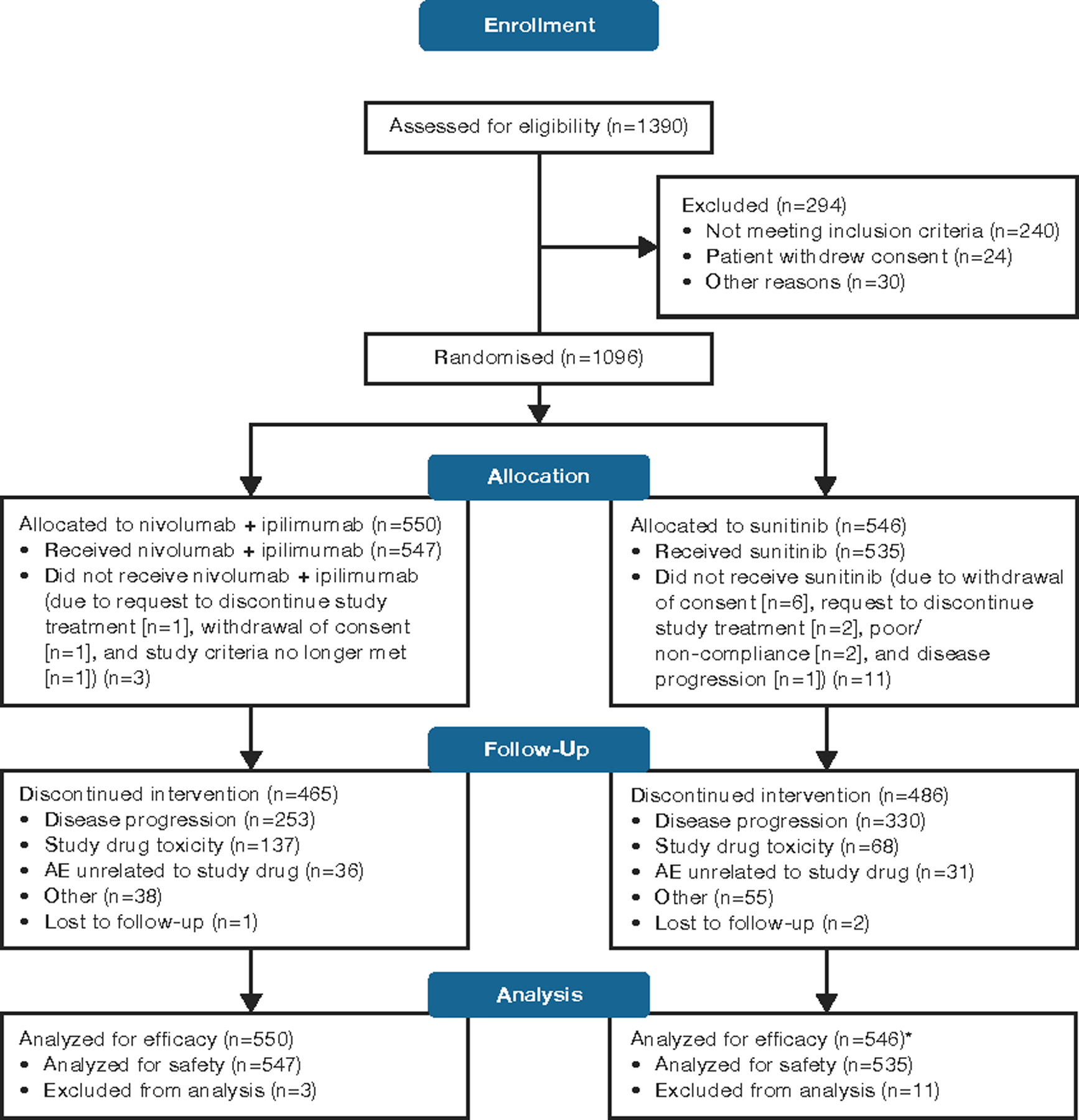
*11 intermediate/poor-risk patients in the sunitinib arm crossed over in the nivolumab + ipilimumab (NIVO+IPI) arm, but were not analysed as part of the NIVO+IPI efficacy or safety population.
Median (95% CI) duration of treatment was 7·9 months (6·5–8·4) in the NIVO+IPI arm and 7·8 months (6·4–8·6) in the SUN arm. Treated patients in the NIVO+IPI arm received a median (interquartile range [IQR] of 14·0 (4·0–43·0) doses of NIVO and 4·0 (4·0–4·0) doses of IPI. The median average daily dose of SUN received was 46·4 mg (IQR 37·0–53·6) over the 28-day cycle. Dose reductions occurred in 286 (53%) of the 535 patients treated with SUN.
In the primary efficacy population of intermediate/poor-risk patients, the OS benefit was maintained with NIVO+IPI versus SUN with longer follow-up. The HR was 0·66 (95% CI 0·54–0·80; p<0·0001), which remains in favour of NIVO+IPI. The 30-month OS probability (95% CI) was 60% (55–64) with NIVO+IPI versus 47% (43–52) with SUN (figure 2A). Among intermediate/poor-risk patients, 182 (43%) of 425 versus 227 (54%) of 422 patients died in the NIVO+IPI versus SUN arms. In the ITT (secondary efficacy) population, the OS benefit was also maintained with NIVO+IPI over SUN (HR 0·71; 95% CI 0·59–0·86; p<0·01). The 30-month OS probability (95% CI) was 64% (60–68) with NIVO+IPI versus 56% (52–60) with SUN (figure 2B). Among ITT patients, 214 (39%) of 550 versus 254 (47%) of 546 patients died in the NIVO+IPI versus SUN arm. In favourable-risk patients (exploratory efficacy population), OS was similar in the two arms (HR 1·22; 95% CI 0·73–2·04; p=0·44), with comparable 30-month OS probabilities (80% [72–86] with NIVO+IPI vs 85% [77–90] with SUN) (figure 2C). Among favourable-risk patients, 32 (26%) of 125 versus 27 (22%) of 124 patients died in the NIVO+IPI versus SUN arm.
Figure 2: Overall survival in IMDC intermediate/poor-risk patients.

(A), in the ITT population (B), and in IMDC favourable-risk patients (C) CI = confidence interval; HR = hazard ratio; IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; ITT = intention-to-treat; NE = not evaluable; NIVO+IPI = nivolumab plus ipilimumab; NR = not reached; SUN = sunitinib.
A multivariable model was used to assess the impact of baseline clinical features on OS. Baseline factors were first analysed individually in the univariable analysis to preclude introducing collinearity into the model (table S2, pp 8–9). In the reduced multivariable models we observed a similar significant negative prognostic effect of the following baseline clinical risk factors on OS among ITT patients in both the NIVO+IPI and SUN arms (table 1): lower Karnofsky performance status (KPS ≤70%), higher lactate dehydrogenase (>1·5× ULN), higher neutrophil-to-lymphocyte ratio (≥2·9), and higher sum of reference diameters of target lesions (≥ median, eg, greater tumour burden). Higher corrected calcium (>10 mg/dL) only significantly impacted OS in the NIVO+IPI arm, whereas lower haemoglobin (< lower limit of normal), higher tumour PD-L1 expression (≥1%), and no prior nephrectomy only significantly impacted OS in the SUN arm (table 1).
Table 1:
Multivariable analysis of effect of baseline clinical features on probability of overall survival in the ITT population in the NIVO+IPI and in the SUN arms
| Arm | Baseline characteristic | n | HR (95% CI) | p value | Median OS (95% CI), months | 24-month OS, % | 30-month OS, % | |
|---|---|---|---|---|---|---|---|---|
| NIVO+IPI | KPS | 2·80 (1·93–4·06) | <0·0001 | |||||
| >70% | 495 | NR (NE) | 75 | 68 | ||||
| Corrected calcium | 0·63 (0·46–0·86) | <0·01 | ||||||
| >10 mg/dL | 95 | 29·9 (24·3–NE) | 61 | 49 | ||||
| LDH | 2·02 (1·22–3·34) | <0·01 | ||||||
| ≤1·5× ULN | 515 | NR (NE) | 72 | 66 | ||||
| NLR | 1·64 (1·24–2·17) | <0·01 | ||||||
| <2·9 | 274 | NR (NE) | 79 | 73 | ||||
| Median sum of reference diameters of target lesions | 0·55 (0·41–0·75) | <0·01 | ||||||
| ≥64·0 mm | 282 | 32·8 (28·2‒NE) | 61 | 54 | ||||
| SUN | KPS | 1·98 (1·35–2·92) | <0·01 | |||||
| >70% | 496 | NR (33·4‒NE) | 63 | 58 | ||||
| Haemoglobin | 1·78 (1·35–2·34) | <0·0001 | ||||||
| ≥LLN | 294 | NR (NE) | 73 | 69 | ||||
| LDH | 4·02 (2·45–6·59) | <0·0001 | ||||||
| ≤1·5× ULN | 521 | NR (34·3‒NE) | 63 | 58 | ||||
| NLR | 1·82 (1·38–2·39) | <0·0001 | ||||||
| <2·9 | 271 | NR (NE) | 73 | 69 | ||||
| PD-L1 status | 0·70 (0·52–0·93) | 0·01 | ||||||
| ≥1% | 127 | 23·9 (15·8–34·4) | 49 | 44 | ||||
| Prior nephrectomy | 1·44 (1·06–1·96) | 0·02 | ||||||
| Yes | 437 | NR (39·7‒NE) | 65 | 61 | ||||
| Median sum of reference diameters of target lesions | 0·69 (0·52–0·91) | <0·01 | ||||||
| ≥64·0 mm | 269 | 24 (19·2–30·1) | 50 | 44 | ||||
CI = confidence interval; HR = hazard ratio; ITT = intention-to-treat; KPS = Karnofsky performance status; LDH = lactate dehydrogenase; LLN = lower limit of normal; NE = not estimable; NLR = neutrophil-to-lymphocyte ratio; NR = not reached; NIVO+IPI = nivolumab plus ipilimumab; OS, overall survival; PD-L1 = programmed death ligand 1; SUN = sunitinib; ULN = upper limit of normal.
In intermediate/poor-risk patients (primary efficacy population), NIVO+IPI demonstrated a delayed PFS benefit, with a separation in favour of NIVO+IPI in the tails of the curves (HR 0·77; 95% CI 0·65–0·90; p<0·01), and the 30-month PFS probability (95% CI) was 28% (23–33) in the NIVO+IPI arm versus 12% (8–16) in the SUN arm (figure 3A). Among intermediate/poor-risk patients, 275 (65%) of 425 versus 304 (72%) of 422 experienced a progression event with NIVO+IPI versus SUN. In the ITT population (secondary efficacy population), NIVO+IPI demonstrated a similar late benefit in investigator-assessed PFS, with Kaplan-Meier curves separating after 12 months. A plateau emerged at 30 months with NIVO+IPI, and the tails of these curves showed ≥10% separation between arms (HR 0·85; 95% CI 0·73–0·98; p=0·03). The 30-month PFS probability (95% CI) was 28% (24–32) with NIVO+IPI versus 18% (14–22) with SUN (figure 3B). Among ITT patients, 357 (65%) of 550 versus 385 (71%) of 546 experienced a progression event with NIVO+IPI versus SUN. In favourable-risk patients (exploratory efficacy population), PFS was numerically shorter with NIVO+IPI versus SUN although the difference between arms was not statistically significant (HR 1·23; 95% CI 0·90–1·69; p=0·19). The 30-month PFS probability (95% CI) was 29% (21–38) with NIVO+IPI versus 35% (26–44) with SUN (figure 3C). Among favourable-risk patients, 82 [66%] of 125 versus 81 [65%] of 124 experienced a progression event with NIVO+IPI versus SUN.
Figure 3: Progression-free survival per investigator in IMDC intermediate/poor-risk patients.

(A), in the ITT population (B), and in IMDC favourable-risk patients (C) CI = confidence interval; HR = hazard ratio; IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; NIVO+IPI = nivolumab plus ipilimumab; PFS = progression-free survival; SUN = sunitinib.
The proportion of patients achieving an investigator-assessed confirmed objective response with NIVO+IPI was consistent across risk groups (178 [42%] of 425 in intermediate/poor-risk [primary efficacy population], 227 [41%] of 550 in ITT [secondary efficacy population], and 49 [39%] of 125 in favourable-risk patients [exploratory efficacy population]); whereas the proportion achieving an objective response with SUN was increasingly lower among patients with worse IMDC prognosis (intermediate/poor-risk, 124 [29%] of 422; ITT, 186 [34%] of 546; favourable-risk patients, 62 [50%] of 124; table 2). Depth of response was greater with NIVO+IPI versus SUN, as seen in the proportion of ITT patients who achieved ≥50% best tumour burden reduction; 185 (34%) of 550 versus 114 (21%) of 546 respectively. Additionally, the proportion of patients achieving a complete response was consistently higher with NIVO+IPI versus SUN across all risk categories: 48 (11%) of 425 versus 5 (1%) of 422 in intermediate/poor-risk, 58 (11%) of 550 versus 10 (2%) of 546 in ITT, and 10 (8%) of 125 versus 5 (4%) of 124 in favourable-risk patients (table 2). The proportions of patients achieving a partial response were similar between arms, with the exception of favourable-risk patients in the SUN arm: 130 (31%) of 425 versus 119 (28%) of 422 in intermediate/poor-risk, 169 (31%) of 550 versus 176 (32%) of 546 in ITT, and 39 (31%) of 125 versus 57 (46%) of 124 in favourable-risk patients (table 2). Strata-adjusted differences in proportion of patients achieving an objective response (95% CI) were 12·8% (6·5–19·2) in intermediate/poor-risk, 7·6% (2·0–13·2) in ITT, and –11·0% (–12·0 to 1·0) in favourable-risk patients.
Table 2:
Investigator-assessed objective response rates by RECIST v1.1
| IMDC intermediate/poor risk | ITT population | IMDC favourable risk | ||||
|---|---|---|---|---|---|---|
| Variable | NIVO+IPI (N=425) | SUN (N=422) | NIVO+IPI (N=550) | SUN (N=546) | NIVO+IPI (N=125) | SUN (N=124) |
| Objective response rate, % (95% CI) | 42 (37–47) | 29 (25–34) | 41 (37–46) | 34 (30–38) | 39 (31–48) | 50 (41–59) |
| p<0·01 | p=0·02 | p=0·14 | ||||
| Unable to determine/not reported | 31 (7) | 44 (10) | 37 (7) | 52 (10) | 6 (5) | 8 (6) |
| Time to confirmed objective response, median (IQR), months* | N=176 2·8 (2·7–3·1) |
N=124 4·0 (2·8–5·5) |
N=225 2·8 (2·7–4·0) |
N=186 4·0 (2·8–6·0) |
N=49 2·9 (2·7–5·5) |
N=62 4·2 (2·8–6·9) |
| Time to confirmed complete response, median (IQR), months | N=48 5·8 (2·9‒10·5) |
NC | N=58 7·6 (3·8–11·2) |
NC | N=10 10·5 (7·0–14·0) |
NC |
| Patients with duration of response ≥18 months, n/N (%)* |
92/176 (52) | 35/124 (28) | 120/225 (53) | 72/186 (39) | 28/49 (57) | 37/62 (60) |
| Patients with ongoing response, n/N (%)* | 104/176 (59) | 43/124 (35) | 131/225 (58) | 69/186 (37) | 27/49 (55) | 26/62 (42) |
| Patients with ongoing complete response, n/N (%) | 42/48 (88) | 4/5 (80) | 51/58 (88) | 6/10 (60) | 9/10 (90) | 2/5 (40) |
Characterisation of response kinetics could not be calculated for two intermediate/poor-risk partial responders to NIVO+IPI due to missing date of partial response.
CI = confidence interval; IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; IQR = interquartile range; ITT = intention-to-treat; NC = not calculated; NIVO+IPI = nivolumab plus ipilimumab; RECIST = Response Evaluation Criteria in Solid Tumors; SUN = sunitinib.
Among ITT patients (secondary efficacy population) in the NIVO+IPI arm, baseline characteristics for responders (N=227), complete responders (N=58), and non-responders (N=323) were largely similar (table S3, pp 10). However, responders and complete responders to NIVO+IPI tended to have lower disease burdens, including fewer sites with lesions, less bone and kidney involvement, and smaller target lesions at baseline versus non-responders. Additionally, responders and complete responders to NIVO+IPI both had higher baseline tumour PD-L1 expression than non-responders (table S3, pp 10).
Median (IQR) time to confirmed objective response was 2·8 months (2·7–4·0) for NIVO+IPI responders versus 4·0 months (2·8–6·0) for SUN responders among ITT patients (secondary efficacy population); 169 (75%) of 225 responders to NIVO+IPI achieved response by 4 months (table 2). Among all complete responders to NIVO+IPI, 3 (5·2%) of 58 achieved complete response at the first scan, while most converted from partial response (44 [75·9%] of 58 at a median of 6·9 months (IQR 3·2–9·1) or from stable disease (11 [19·0%] of 58 at 11·3 months [IQR 3·8–15·4]). In the NIVO+IPI arm, there were six additional complete responses since the previous database lock in 2017, and 44 (75%) of 58 complete responses were reached by 11·2 months.
Duration of response was longer with NIVO+IPI versus SUN in ITT patients, with increasing separation between the curves in favour of NIVO+IPI with extended follow-up (HR 0·51, 95% CI 0·38–0·68; figure 4A). The Kaplan-Meier duration of response curve reached a plateau above 50% in the NIVO+IPI arm, whereas the probability of maintained response with SUN dropped over time. A higher proportion of all responders to NIVO+IPI versus SUN achieved durable response lasting at least 18 months (120 [53%] of 225 vs 72 [39%] of 186; table 2). Regardless of risk, a higher proportion of NIVO+IPI versus SUN responders had ongoing response as of the database lock, including 51 (88%) of 58 versus 6 (60%) of 10 complete responders, respectively (table 2).
Figure 4: Duration of response in the NIVO+IPI and SUN arms.

(A), treatment duration and treatment-free survival in responders in the NIVO+IPI arm (B), and in the SUN arm (C) NE = not evaluable; NIVO+IPI = nivolumab plus ipilimumab; NR = not reached; SUN = sunitinib.
The median (95% CI) duration of treatment among responders was 20·6 months (17·7–23·2) in the NIVO+IPI arm versus 21·2 months (18·9–24·4) in the SUN arm, longer durations than seen in all treated patients per arm, respectively. Among responders who discontinued study therapy across all risk categories, higher proportions in the NIVO+IPI versus the SUN arm did not continue on to subsequent therapy, including more NIVO+IPI versus SUN responders with ongoing treatment-free survival (68 [38%] of 178 vs 32 [26%] of 124 intermediate/poor-risk and 18 [37%] of 49 vs 12 [19%] of 62 favourable-risk patients, respectively; figure 4B, 4C).
Among ITT patients, 264 (48%) of 550 in the NIVO+IPI arm and 334 (61%) of 546 in the SUN arm received subsequent systemic therapy; most commonly SUN (120 [22%] of 550), pazopanib (95 [17%] of 550), axitinib (86 [16%] of 550), and cabozantinib (61 [11%] of 550) in the NIVO+IPI arm, and NIVO (192 [35%] of 546), axitinib (117 [21%] of 546), SUN (65 [12%] of 546), and everolimus (60 [11%] of 546) in the SUN arm. Although we did not collect reasons for subsequent therapies, the proportion of patients in the SUN arm who received SUN as a subsequent therapy may not have been able to tolerate the protocol-specific 4:2 schedule and went off study to switch to the 2:1 schedule. As of the database lock, 11 intermediate/poor-risk patients crossed over from SUN to NIVO+IPI, per a protocol amendment that provided this option for second-line NIVO+IPI treatment as a part of the study.
No new safety signals emerged with longer follow-up. Similar proportions of patients experienced treatment-related AEs of any grade in the NIVO+IPI versus the SUN arms (513 [94%] of 547 vs 521 [97%] of 535 patients). Fewer grade 3 or 4 treatment-related AEs occurred with NIVO+IPI versus SUN (255 [47%] of 547 vs 342 [64%] of 535). Any-grade treatment-related AEs occurring in >15% of patients in either arm with treatment-related grade 3–4 AEs are reported in figure 5A. Tracking the most common organ classes of high-grade treatment-related AEs over time, differences in early and chronic toxicity burden between arms were observed, with SUN toxicity continuing despite dose adjustments, while the overall toxicity burden in the NIVO+IPI arm was low (figure 5B and 5C). More grade 3 or 4 treatment-related AEs occurred during the combination induction versus the maintenance phase in the NIVO+IPI arm.
Figure 5: Any-grade treatment-related AEs occurring in >15% of patients in either arm with treatment-related grade 3–4 AEs (all treated patients).

(A) and proportion of patients with treatment-related grade 3–4 AEs by common system organ class over time in the NIVO+IPI arm (B) and in the SUN arm (C) *Additional patients reported common any-grade treatment-related AEs with longer follow-up compared with the primary database lock (NIVO+IPI arm: diarrhoea [n=9], pruritis, rash [both n=6], fatigue [n=5]; hypothyroidism [n=4]; asthenia, increased lipase, anaemia [all n=2]; PPE, nausea, mucosal inflammation, stomatitis, decreased appetite, vomiting, dyspepsia, thrombocytopenia [all n=1] vs SUN arm: vomiting, hypothyroidism [both n=5]; diarrhoea [n=4]; increased lipase, hypertension, nausea [all n=3]; fatigue, rash, PPE, mucosal inflammation, decreased appetite [all n=2]; asthenia, dysgeusia, stomatitis, dyspepsia [all n=1]; †< 1% reported grade 3–4 treatment-related AE; ‡No patients reported a grade 3–4 treatment-related AE.
NIVO+IPI = nivolumab plus ipilimumab; PPE = palmoplantar erythrodysesthesia; SUN = sunitinib
Treatment-related AEs leading to discontinuation occurred in 119 (22%) of 547 patients in the NIVO+IPI arm, most commonly due to increased alanine aminotransferase (15 [2·7%] of 547), diarrhoea (14 [2·6%] of 547), and increased aspartate aminotransferase (12 [2·2%] of 547); the majority of these patients discontinued after completing the NIVO+IPI induction phase. Treatment-related AEs leading to discontinuation occurred in 66 (12%) of 535 patients in the SUN arm, most commonly fatigue (7 [1·3%] of 535), increased alanine aminotransferase (5 [0·9%] of 535), diarrhoea, pancreatitis, and thrombocytopenia (all 4 [0·7%] of 535). Per protocol, patients who were required to discontinue NIVO+IPI could not continue on NIVO monotherapy. No additional treatment-related deaths were reported since the primary analysis: eight (1%) of 547 treatment-related deaths in the NIVO+IPI arm (one each due to pneumonitis, pneumonia and aplastic anaemia, immune-mediated bronchitis, lower gastrointestinal haemorrhage, haemophagocytic syndrome, sudden death, liver toxic effects, and lung infection) and four (1%) of 535 in the SUN arm (two cardiac arrests and one each of heart failure and multiple organ failure).1 Among all treated patients, 212 (39%) of 547 NIVO+IPI arm patients died of any cause (disease, 173 [32%] of 547; study drug toxicity, 8 [1%] of 547; unknown/other, 31 [6%] of 547), and 251 (47%) of 535 SUN arm patients died of any cause (disease, 221 [41%] of 535; study drug toxicity, 4 [1%] of 535; unknown/other, 26 [5%] of 535).
Treatment-related select AEs (potentially immune-mediated) of any grade were reported in 443 (81%) of 547 versus 443 (83%) of 535 patients treated with NIVO+IPI versus SUN within 30 days of the last dose (table S4, pp 11–12), the majority of which were grade 1 or 2 in both arms (table 3). The same preferred terms were reported for SUN arm patients, although the mechanism driving those AEs may be different from NIVO+IPI. The majority of treatment-related select AEs with NIVO+IPI resolved, apart from some select endocrine treatment-related AEs, which were managed with appropriate hormonal therapies (table S4, pp 11–12). Overall, 157 (28·7%) of 547 NIVO+IPI treated patients received ≥40 mg prednisone daily or equivalent to manage select treatment-related AEs; 53 (9·7%) of 547 received ≥40 mg prednisone daily or equivalent continuously for ≥30 days and 102 (18·6%) of 547 patients received ≥40 mg prednisone daily or equivalent continuously for ≥2 weeks.
Table 3:
Treatment-related select AEs (any grade, reported in ≥1% of all treated patients in either arm)
| System* Preferred term | NIVO+IPI (N=547) | SUN (N=535) | ||||
|---|---|---|---|---|---|---|
| Grade 1 or 2, n (%) | Grade 3, n (%) | Grade 4, n (%) | Grade 1 or 2, n (%) | Grade 3, n (%) | Grade 4, n (%) | |
| Skin | 252 (46·1) | 21 (3·8) | 0 | 251 (46·9) | 53 (9·9) | 1 (<1) |
| Pruritis | 157 (8·7) | 3 (<1) | 0 | 49 (9·2) | 0 | 0 |
| Rash | 115 (21·0) | 9 (1·6) | 0 | 69 (12·9) | 0 | 0 |
| Rash (maculo-papular) | 43 (7·9) | 8 (1·5) | 0 | 21 (3·9) | 1 (<1) | 0 |
| Erythema | 16 (2·9) | 0 | 0 | 5 (<1) | 0 | 0 |
| Rash (macular) | 8 (1·5) | 0 | 0 | 2 (<1) | 0 | 0 |
| Pruritis generalised | 8 (1·5) | 1 (<1) | 0 | 2 (<1) | 0 | 0 |
| Urticaria | 7 (1·3) | 1 (<1) | 0 | 2 (<1) | 0 | 0 |
| Rash (pruritic) | 6 (1·1) | 1 (<1) | 0 | 1 (<1) | 0 | 0 |
| Palmar-plantar erythrodysaesthesia syndrome | 5 (<1) | 1 (<1) | 0 | 184 (34·4) | 48 (9·0) | 1 (<1) |
| Skin exfoliation | 3 (<1) | 0 | 0 | 14 (2·6) | 0 | 0 |
| Blister | 2 (<1) | 0 | 0 | 7 (1·3) | 3 (<1) | 0 |
| Eczema | 2 (<1) | 0 | 0 | 6 (1·1) | 0 | 0 |
| Skin hypopigmentation | 2 (<1) | 0 | 0 | 15 (2·8) | 0 | 0 |
| Endocrine | 142 (26·0) | 31 (5·7) | 7 (1·3) | 165 (30·8) | 1 (<1) | 0 |
| Hypothyroidism | 87 (15·9) | 2 (<1) | 0 | 138 (25·8) | 1 (<1) | 0 |
| Hyperthyroidism | 57 (10·4) | 2 (<1) | 0 | 13 (2·4) | 0 | 0 |
| Adrenal insufficiency | 17 (3·1) | 9 (1·6) | 2 (<1) | 0 | 0 | 0 |
| Thyroiditis | 15 (2·7) | 1 (<1) | 0 | 0 | 0 | 0 |
| Blood thyroid stimulating hormone increased | 12 (2·2) | 0 | 0 | 29 (5·4) | 0 | 0 |
| Hypophysitis | 7 (1·3) | 13 (2·4) | 2 (<1) | 0 | 0 | 0 |
| Diabetes mellitus | 4 (<1) | 0 | 2 (<1) | 0 | 0 | 0 |
| Gastrointestinal | 135 (24·7) | 27 (4·9) | 0 | 251 (46·9) | 30 (5·6) | 1 (<1) |
| Diarrhoea | 133 (24·3) | 21 (3·8) | 0 | 251 (46·9) | 30 (5·6) | 1 (<1) |
| Colitis | 7 (1·3) | 11 (2·0) | 0 | 2 (<1) | 0 | 0 |
| Hepatic | 56 (10·2) | 38 (6·9) | 9 (1·6) | 59 (11·0) | 20 (3·7) | 0 |
| AST increased | 39 (7·1) | 17 (3·1) | 3 (<1) | 44 (8·2) | 7 (1·3) | 0 |
| ALT increased | 33 (6·0) | 23 (4·2) | 5 (<1) | 42 (7·9) | 9 (1·7) | 0 |
| Blood alkaline phosphatase increased | 16 (2·9) | 9 (1·6) | 0 | 11 (2·1) | 1 (<1) | 0 |
| Blood bilirubin increased | 12 (2·2) | 1 (<1) | 0 | 15 (2·8) | 3 (<1) | 0 |
| Transaminases increased | 9 (1·6) | 3 (<1) | 1 (<1) | 4 (<1) | 1 (<1) | 0 |
| Gamma-glutamyltransferase increased | 7 (1·3) | 4 (<1) | 1 (<1) | 3 (<1) | 4 (<1) | 0 |
| Hepatic enzyme increased | 4 (<1) | 1 (<1) | 1 (<1) | 0 | 0 | 0 |
| Renal | 48 (8·8) | 4 (<1) | 3 (<1) | 41 (7·7) | 6 (1·1) | 0 |
| Blood creatinine increased | 39 (7·1) | 0 | 1 (<1) | 34 (6·4) | 2 (<1) | 0 |
| Acute kidney injury | 8 (1·5) | 3 (<1) | 1 (<1) | 6 (1·1) | 3 (<1) | 0 |
| Pulmonary | 28 (5·1) | 6 (1·1) | 0 | 1 (<1) | 0 | 0 |
| Pneumonitis | 26 (4·8) | 6 (1·1) | 0 | 0 | 0 | 0 |
Includes events reported after the first dose and within 30 days of last dose of study therapy
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; NIVO+IPI = nivolumab + ipilimumab; Select = immune-mediated; SUN = sunitinib.
Discussion
With extended follow-up in CheckMate 214, significant OS benefit was maintained with NIVO+IPI versus SUN with an early and consistent separation of Kaplan-Meier curves in both the intermediate/poor-risk and ITT populations. The ITT group included patients with IMDC favourable, intermediate, and poor-risk disease, while the primary objective of the study was to assess OS and other efficacy endpoints in the intermediate/poor-risk population. The 30-month survival probability was 60% versus 47% in intermediate/poor-risk patients, and 64% versus 56% in ITT patients. A late PFS benefit was observed in intermediate/poor-risk and ITT patients, with the Kaplan-Meier curves separating after 9 and 12 months and ≥10% separation in favour of NIVO+IPI was maintained at the tails of both curves. The 30-month PFS probability was higher with NIVO+IPI versus SUN among both intermediate/poor-risk (28% vs 12%) and ITT patients (28% vs 18%), and was consistent with NIVO+IPI regardless of risk category. In the ITT population, median time to confirmed response was less than 3 months, the proportion of patients achieving an objective response was higher, and more patients had an ongoing response with NIVO+IPI versus SUN (58% vs 37%). These observations suggest that NIVO+IPI resulted in rapid disease volume control. Of note, more responses to NIVO+IPI were complete (11%) and durable (88% ongoing complete responses; 58% ongoing responses) versus SUN. A high proportion of responders who discontinued NIVO+IPI still derived treatment-free survival benefit (ie, remained progression-free or were alive) without subsequent systemic therapy, regardless of risk category.
Historically, favourable-risk patients have less-aggressive disease and respond well to TKIs, and many patients can be managed at diagnosis with surveillance followed by delayed therapeutic intervention.15 While the 30-month probability was slightly numerically higher with SUN, survival probabilities between arms were similar. Compared with the results from the primary analysis,1 the magnitude of difference between arms in OS in the favourable-risk subgroup decreased with longer follow-up, and as median OS was not reached in either treatment arm, the OS data could still be considered immature in this group. While updated response was measured per investigator instead of per IRRC assessment, the responses with NIVO+IPI in favourable-risk patients were durable. In this relatively small exploratory group, nine of ten patients in the NIVO+IPI arm versus only two of five patients in the SUN arm have ongoing complete response. Available data in favourable-risk patients suggest that further follow-up will continue to inform the benefit-risk ratio for NIVO+IPI versus SUN in this group of patients.
The exploratory multivariable analysis showed that the IMDC baseline risk factors anaemia, neutrophilia, thrombocytosis, and time from diagnosis to treatment were not prognostic of OS with NIVO+IPI. Interestingly, these results suggest that baseline tumour PD-L1 expression ≥1% predicts poor OS with SUN but not with NIVO+IPI in aRCC. Neutrophil-to-lymphocyte ratio (a parameter indicating relative lymphocyte counts and marker of systemic inflammation) has shown previous prognostic value in aRCC with immune checkpoint inhibitors, and was an independent prognostic parameter in both arms in the present analysis.16,17 In the current era of immune checkpoint inhibitor-based therapies, these findings highlight a need for improved prognostic models based on understanding the host response and underlying tumour biology.18
No new safety signals emerged in the NIVO+IPI arm with longer follow-up, and no treatment-related deaths occurred since the primary analysis data cutoff. Rates of discontinuation of NIVO+IPI in part reflected the protocol design, which specified that patients who required treatment discontinuation due to AEs arising during NIVO+IPI induction were not allowed to continue study treatment with NIVO monotherapy. Interestingly, in a recent exploratory analysis of CheckMate 214, comparable efficacy outcomes were observed in all patients who discontinued due to treatment-related AEs relative to the overall NIVO+IPI efficacy population.19
The toxicity profiles for NIVO+IPI and SUN were expected to differ based on disparate underlying mechanisms of action. When comparing treatment arms, common grade 3 or 4 treatment-related AEs in the NIVO+IPI arm arose early and generally resolved within the first 4–6 months of treatment. In contrast, both early and chronic toxicity were apparent in the SUN arm despite dose adjustments. Chronic toxicities commonly observed with TKIs, including hypertension and palmar-plantar erythrodysaesthesia, were both evident in this analysis with SUN and in recent reports with TKI/immunotherapy combinations.20,21
Most treatment-related select AEs occurring within 30 days of last dose in the NIVO+IPI arm were low-grade, and the majority resolved and were manageable using established algorithms. However, vigilance for safety events by the health care team throughout and after immunotherapy treatment completion is recommended. There remains a lack of standardised reporting of corticosteroid use for the immune checkpoint drug class as a whole. The initial CheckMate 214 presentation reported higher systemic corticosteroid use with NIVO+IPI22; that figure reflected a very broad definition of any systemic corticosteroid use from all-cause AEs, and included corticosteroid use for AEs related and unrelated to study treatment. The present analysis reported use of corticosteroids (≥40 mg prednisone daily or equivalent) to manage treatment-related select AEs occurring within 30 days of last dose with an appropriate denominator of all treated patients (28·7% of 547 NIVO+IPI-treated patients). This definition is more clinically meaningful and in line with corticosteroid-use definitions used in recent PD-(L)1/VEGFR combination trials.20,23 Additionally, in a separate analysis of CheckMate 214 based on the primary database lock, health-related quality of life was maintained or significantly improved from baseline with NIVO+IPI versus SUN in ITT patients,6 further supporting the favourable benefit-risk profile of NIVO+IPI versus SUN.6,24
Limitations should be considered when interpreting the results. Outcomes in the relatively small subset of favourable-risk patients characterised by wide 95% CIs should be considered exploratory. To this point, the favourable-risk patients were included in the ITT population along with intermediate- and poor-risk patients, and efficacy endpoints of objective response, PFS, and OS were formally assessed as secondary endpoints for the trial. Additionally, the present response outcomes were analysed per investigator versus IRRC and minor differences between outcomes based on these different methodologies could be expected.
Ongoing work and future directions include exploring the potential role for NIVO+IPI in treating novel patient populations including the adjuvant RCC setting in patients with high risk of relapse after nephrectomy (CheckMate 914, NCT03138512), and aRCC subpopulations with unmet need. A post-hoc, exploratory analysis of CheckMate 214 showed improved OS and PFS benefits with NIVO+IPI (N=60) versus SUN (N=52) in intermediate/poor-risk patients with sarcomatoid features, with a higher proportion of patients achieving an objective response (56·7% vs 19·2%; p<0·0001) and a higher proportion of patients achieving a complete response (18·3% vs 0%); the magnitude of benefit from NIVO+IPI versus SUN was greater in these sarcomatoid-positive patients compared with the overall intermediate/poor-risk population, and detailed analyses are ongoing.24 Furthermore, a prospective phase 3b/4 study of NIVO+IPI in treatment-naïve aRCC patients with clear cell disease, non-clear cell disease, or non-active brain metastases (all with KPS ≥70%), or any histology/non-active brain metastasis with KPS 50–60% (CheckMate 920; NCT02982954), and a phase 2 study of NIVO+IPI versus SUN in patients with non-clear cell aRCC (SUNIFORECAST; NCT03075423) are ongoing. The updated CheckMate 214 results support ongoing studies combining NIVO+IPI with novel agents (eg, with NKTR-214 in PIVOT-02; NCT02983045). Ongoing molecular and genomic analyses of CheckMate 214 and other studies may illuminate predictive biomarkers of response or prognostic factors to NIVO+IPI. Additional CheckMate 214 analyses outside the current scope, such as outcomes in patients who discontinued therapy before finishing NIVO+IPI induction, updated health-related quality of life after extended follow-up, and further details of treatment-related AE kinetics, could be performed in future investigations.
In summary, with extended follow-up, improved OS was maintained with NIVO+IPI versus SUN in both the entire ITT population and the intermediate/poor-risk populations, and OS was similar between arms in the relatively small favourable-risk subgroup in CheckMate 214. An improvement was also observed in PFS with NIVO+IPI over SUN with longer follow-up, with a notable proportion of ITT patients remaining progression-free at 30 months. Complete and durable responses were observed with NIVO+IPI regardless of IMDC-risk based prognosis.
Supplementary Material
Research in Context Panel.
Evidence before this study
A search of PubMed for clinical trial reports published with no restrictions on article type or language, from database inception before May 2019, using the terms “nivolumab,” “nivolumab AND ipilimumab,” “sunitinib,” “renal cell carcinoma,” “RCC,” “kidney cancer”, “advanced and metastatic RCC”, “advanced and metastatic renal cell carcinoma,” and “renal cell carcinoma” with specific attention to randomised phase 3 trials with any following comparators: VEGF inhibitors or immuno-oncology therapeutics. The only randomised, open-label, phase 3 trial we found in first-line therapy was CheckMate 214, which compared nivolumab plus ipilimumab (NIVO+IPI) with sunitinib (SUN) in patients with previously untreated, advanced or metastatic renal cell carcinoma. Our search parameters also revealed multiple published randomised studies of relevance in patients with previously treated and untreated clear cell advanced renal cell carcinoma. Various targeted therapies, including antiangiogenic drugs targeting vascular endothelial growth factor (VEGF) and its receptors and mTOR inhibitors, have improved outcomes for patients with advanced renal cell carcinoma (aRCC) over the past decade. Sunitinib (a VEGF tyrosine kinase inhibitor [TKI]) has been considered a gold standard first-line option for patients with aRCC. Yet, durable complete responses are rare with targeted therapies, most patients eventually progress, and overall survival (OS) and toxicity profiles are suboptimal. Single-agent anti–PD-1/PD-L1 immunotherapies (I-O) proved efficacious in pre-treated patients with aRCC, and subsequently investigative I-O regimens (including I-O/I-O, and I-O/TKI combinations) showed notable antitumour activity in first-line aRCC, however, unfavourable toxicity was reported with some I-O/TKI regimens. The combination I-O/I-O regimen of nivolumab (monoclonal antibody against PD-1) plus ipilimumab (anti–cytotoxic T-lymphocyte–associated antigen 4 antibody) demonstrated manageable safety and notable antitumour activity in the phase 1 CheckMate 016 trial of patients with aRCC. Building upon these results, the phase 3 CheckMate 214 trial demonstrated a significant OS and objective response benefit with NIVO+IPI over SUN in the first-line treatment of IMDC intermediate- or poor-risk patients with aRCC with a clear cell component. To our knowledge, CheckMate 214 was the first study to investigate the efficacy and safety of first-line NIVO+IPI and the first to demonstrate a statistically significant OS benefit over SUN. On the basis of these results, the combination of NIVO+IPI is approved by the European Commission and the US Food and Drug Administration for the first-line treatment of patients with intermediate- and poor-risk aRCC.
Added value of this study
Long-term updates critically inform the value of immunotherapy-based regimens. As such, we report updated and key additional efficacy and safety data with longer follow-up. The superior long-term OS, late progression-free survival (PFS) benefit, proportion of patients achieving complete response, and durability of response observed in the current analysis are encouraging and further inform the multifaceted benefits of NIVO+IPI in clinical practice.
Implications of all the available evidence
Extended follow-up of first-line combination treatment with NIVO+IPI in patients with aRCC continues to demonstrate impressive antitumour activity over sunitinib in CheckMate 214. Improved OS was maintained with NIVO+IPI versus sunitinib in both the intermediate/poor-risk and the intention-to-treat (ITT) populations, and OS was similar between arms in the smaller favourable-risk subgroup. An improvement was also observed in PFS with NIVO+IPI over sunitinib with longer follow-up, with 28% of ITT patients remaining progression-free at 30 months. Complete and durable responses were observed with NIVO+IPI regardless of IMDC risk–based prognosis. No new safety signals emerged with NIVO+IPI, and the adverse event profile continued to be manageable. These results demonstrate the long-term benefits of NIVO+IPI in patients with previously untreated aRCC, and support future analyses exploring the role for NIVO+IPI in treating novel patient populations and ongoing studies combining NIVO+IPI with novel agents to further improve outcomes for patients with aRCC.
Acknowledgments
CheckMate 214 was funded by Bristol-Myers Squibb and ONO Pharmaceutical. We thank the patients and their families who are making this study possible; the late Paul Gagnier, the initial medical monitor; Jennifer McCarthy, the CheckMate 214 protocol manager; and Dako, an Agilent Technologies, Inc. company, for collaborative development of the PD-L1 IHC 28-8 pharmDx assay. Professional medical writing and editorial assistance were provided by Jennifer Tyson, PhD, and Lawrence Hargett of Parexel, funded by Bristol-Myers Squibb. Patients treated at the Memorial Sloan Kettering Cancer Center were supported in part by Memorial Sloan Kettering Cancer Center Support Grant/Core Grant (P30 CA008748).
Research support: This study was sponsored by Bristol-Myers Squibb and ONO Pharmaceutical Company Limited. Authors received no financial support or compensation for publication of this manuscript.
Funding Bristol-Myers Squibb and ONO Pharmaceutical Company Ltd.
Footnotes
Data Sharing
Bristol-Myers Squibb’s policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html. Deidentified and anonymised datasets of clinical trial information, including patient-level data, will be shared with external researchers for proposals that are complete, for which the scientific request is valid and the data are available, consistent with safeguarding patient privacy and informed consent. Upon execution of an agreement, the deidentified and anonymised data sets can be accessed via a secured portal that provides an environment for statistical programming with R as the programming language. The trial protocol and statistical analysis plan will also be available. Data will be available for 2 years from the study completion or termination of the program (August 2024).
Presented, in part, at the European Society of Medical Oncology, Madrid, Spain; September 8–12, 2017; and at the 17th International Kidney Cancer Symposium; November 2–3, 2018; Miami, FL, USA
Declaration of interests
RJM reports grants from Bristol-Myers Squibb during the conduct of the study; and grants and personal fees from Pfizer, Novartis, Eisai, Exelixis, and Genentech/Roche, personal fees from Merck, grants and personal fees from Eisai and Pfizer, and personal fees from Novartis, outside the submitted work.
BIR reports grants and personal fees from Bristol-Myers Squibb, outside submitted work.
DFM reports personal fees from Exelixis, Array BioPharma, Bristol-Myers Squibb, Merck, Genentech, Novartis, Pfizer, and Eisai, grants from Prometheus, Bristol-Myers Squibb, Merck, Genentech/Roche, Pfizer, Exelixis, Novartis, and X4 Pharma, and personal fees from Alkermes, Inc., outside the submitted work.
OAF reports personal fees from Roche, Tecnofarma, Novartis, and Bristol-Myers Squibb, and other from Pfizer, outside the submitted work.
HJH reports grants and personal fees from Bristol-Myers Squibb, personal fees from Pfizer and Exelixis, grants and personal fees from Merck, and personal fees from Armo Biosciences and Novartis, during the conduct of the study.
MAC reports personal fees from Astellas Pharma, AbbVie, Roche/Genentech, Pfizer, and Foundation Medicine, grants from Bristol-Myers Squibb, AstraZeneca, Gilead Sciences, EMD Serono, and Effector, outside the submitted work.
PS has nothing to disclose.
BE reports honoraria from Ipsen, Novartis, Pfizer, Bristol-Myers Squibb, Exelixis, EUSA, Aveo, and Roche.
BB reports grants and personal fees from Pfizer and Bristol-Myers Squibb, and personal fees from Ipsen, outside the submitted work.
AA reports grants, personal fees, and non-financial support from Bristol-Myers Squibb and Merck, grants and personal fees from Dynavax, personal fees and non-financial support from Pfizer, Exelixis, Bioarray, and Novartis, outside the submitted work.
CP reports personal fees from Bristol-Myers Squibb and MSD, grants and personal fees from Pfizer, personal fees from Novartis, Ipsen, EUSA, Eisai, Janssen, AstraZeneca, and General Electric, outside submitted work.
SG reports personal fees from AstraZeneca, grants and personal fees from Bayer, Bristol-Myers Squibb and Novartis, personal fees from Exelixis and Janssen, grants and personal fees from Corvus, personal fees from Genentech and Sanofi/Genzyme, grants and personal fees from Pfizer, grants from Acceleron, Merck, Agensys, and Eisai, and personal fees from EMD Serono, outside the submitted work.
VN has nothing to disclose.
SB reports non-financial support from Novartis, personal fees, non-financial support and other from Astellas, personal fees and non-financial support from Janssen, personal fees, non-financial support and other from Pfizer, non-financial support and other from Bristol-Myers Squibb, non-financial support and other from Roche, personal fees and non-financial support from MSD, non-financial support from Exelixis, and non-financial support from AstraZeneca, outside the submitted work.
SST reports clinical trial support received on behalf of his institution from Bristol-Myers Squibb, non-financial support from Bristol-Myers Squibb, during the conduct of the study; clinical trial support received on behalf of his institution from Peloton Therapeutics, Merck, Nektar Therapeutics, and Calithera Biosciences, personal fees from Calithera Biosciences, clinical trial support received on behalf of his institution from Jounce Therapeutics, Pfizer, Genentech, and Prometheus Laboratories, personal fees from Prometheus Laboratories, and clinical trial support received on behalf of his institution from ARGOS Therapeutics, outside the submitted work.
PB reports advisory board funding from Bristol-Myers Squibb, Pfizer, Roche, Ipsen, MSD, Janssen Cilag, and Novartis, outside the submitted work.
RLA reports honoraria from Bristol-Myers Squibb, MSD, Roche, Isotopia, and Bayer, advisory role travel grant from Janssen, advisory role from Sanofi, advisory role travel grant from Pfizer, and advisory role from Astellas, outside the submitted work.
ERP reports grants from Bristol-Myers Squibb, consulting fees from Bristol-Myers Squibb, and travel fees from Bristol-Myers Squibb during the conduct of the study; consultant fees from AstraZeneca, Bristol-Myers Squibb, Genentech/Roche, Merck, Novartis, Pfizer, Eli Lilly, SynerGene Therapeutics, Inovio, Clovis, Horizon Pharma, Exelixis, funds for development of educational presentations from Bristol-Myers Squibb, Merck, Roche, and Novartis; and grants to institution for conduct of clinical trial(s) from AstraZeneca, Bristol-Myers Squibb, Merck, Peloton, Pfizer, and Astellas. In addition, Dr. Plimack has a patent U.S. Patent Application No.: 14/588,503 issued, and a patent US Patent Application No: 15/226,474 issued.
SFO reports non-financial support from Bristol-Myers Squibb, during the conduct of the study; grants from Celldex, and grants from Novartis, outside the submitted work.
BR has nothing to disclose.
BM reports personal fees and honoraria for advisory boards, speeches, and travel support from Bristol-Myers Squibb, Roche, MSD, Merck Serono, and Novartis, personal fees from Pfizer, Ipsen, Sanofi, Astellas, Janssen, Eisai, AstraZeneca, and Amgen, outside the submitted work.
TP reports grants from AstraZeneca, Roche, Novartis, Merck, Bristol-Myers Squibb, Pfizer, Roche, Ipsen, Novartis, and Exelixis, outside the submitted work.
PN reports board membership, manuscript preparation, and travel fees from Bristol-Myers Squibb, outside the submitted work.
SO reports grants, personal fees and non-financial support from Bristol-Myers Squibb, Pfizer, Novartis, Eisai, and Bayer, outside the submitted work.
DP reports research funding from Bristol-Myers Squibb, during the conduct of the study; personal fees and advisory board fees from Bristol-Myers Squibb, and personal fees, research funding, and advisory board funding from Pfizer, outside the submitted work.
TKC reports grants, personal fees, and non-financial support from Pfizer and Exelixis during the conduct of the study; grants and personal fees from AstraZeneca, Bayer, Bristol-Myers Squibb, Cerulean, Eisai, Foundation Medicine Inc., Exelixis, Genentech, Roche, GlaxoSmithKline, Merck, Novartis, Peloton, Pfizer, Prometheus Labs, Corvus, and Ipsen; grants from Tracon; personal fees from Alligent, Up-to-Date, NCCN, Analysis Group, Michael J. Hennessy (MJH) Associates, Inc. (Healthcare Communications Company and several brands such as OnClive and PER), L-path, Kidney Cancer Journal, Clinical Care Options, Platform Q, Navinata Healthcare, Harborside Press, American Society of Medical Oncology, New England Journal of Medicine, and Lancet Oncology; and grants from Calithera and Takeda, outside the submitted work.
FD reports grants from Novartis, Pfizer, and Ipsen, outside the submitted work.
MOG reports treatment fees from Bristol-Myers Squibb for participation in CheckMate 214 during the conduct of this trial; grants and personal fees from Novartis and Bristol-Myers Squibb; and personal fees from Pfizer, Bayer HealthCare, Astellas, Intuitive Surgical, Sanofi Aventis, Hexal, Apogepha, Amgen, AstraZeneca, MSD, Janssen Cilag, ONO Pharmaceutical, Ipsen Pharma, and Medac, outside submitted work.
HG reports personal fees from Bristol-Myers Squibb, during the conduct of the study; and personal fees from Pfizer, Astellas, Ipsen, and Roche, outside the submitted work.
DYCH reports personal fees from Bristol-Myers Squibb, Pfizer, and Novartis, outside the submitted work.
CKK reports personal fees from Bristol-Myers Squibb, Pfizer, Eisai, Ipsen, Roche, and AstraZeneca, outside the submitted work.
MRH reports grants from Pfizer, during the conduct of the study; grants from Argos, Bristol-Myers Squibb, Exelixis, and Genentech; personal fees from Argos, Exelixis, Genentech, and Bristol-Myers Squibb; consulting fees from Argos, Exelixis, and Pfizer; and speakers bureau fees from Exelixis and Genentech, outside the submitted work.
YT reports grants from Astellas, AstraZeneca, ONO, Pfizer, and Chugai; and personal fees from Astellas, Bristol-Myers Squibb, Novartis, ONO, Pfizer, and Taiho, outside the submitted work.
ID reports personal fees from Bristol-Myers Squibb and Novartis, personal fees and non-financial support from Ipsen, grants, personal fees, and non-financial support from Roche Genentech and AstraZeneca, and personal fees from Sanofi, Bayer, Pharmacyclics, Jansen, and MSD, outside the submitted work.
VG reports grants and personal fees from Pfizer, grants, personal fees, and non-financial support from Bristol-Myers Squibb, personal fees and non-financial support from Roche, Novartis, and Eisai, grants, personal fees, and non-financial support from Ipsen, and grants and personal fees from EUSA Pharma, during the conduct of the study; grants, personal fees, non-financial support and stock holdings in MSD, grants from Novartis, grants, personal fees, and stock holdings in AstraZeneca, grants, personal fees, non-financial support, and stock holdings in Bristol-Myers Squibb, personal fees and non-financial support from Merck Serono, personal fees from Roche, Pfizer, and Lilly, and personal fees and non-financial support from PharmaMar, outside the submitted work.
MBM reports personal fees, and employment and stock holdings in Bristol-Myers Squibb, outside the submitted work.
SM reports personal fees from and employment by Bristol-Myers Squibb, outside the submitted work.
NMT reports grants and personal fees from Bristol-Myers Squibb, Pfizer, and Novartis, personal fees from Nektar Therapeutics and Oncorena, grants and personal fees from Exelixis, Inc, personal fees from Eisai Medical Research, grants from Calithera Biosciences, and personal fees from ONO Pharmaceutical, outside the submitted work.
References
- 1.Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 2018; 378: 1277–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.OPDIVO (nivolumab) [package insert]. Bristol-Myers Squibb Company, Princeton, NJ, USA; 2018. [Google Scholar]
- 3.YERVOY (ipilimumab) [package insert]. Bristol-Myers Squibb Company, Princeton, NJ, USA; 2018. [Google Scholar]
- 4.Jonasch E Updates to the management of kidney cancer. J Natl Compr Canc Netw 2018; 16: 639–41. [DOI] [PubMed] [Google Scholar]
- 5.European Medicines Agency. European Public Assessment Report (EPAR), Opdivo https://www.ema.europa.eu/en/medicines/human/EPAR/opdivo (accessed March 20, 2019), 2019.
- 6.Cella D, Grünwald V, Escudier B, et al. Patient-reported outcomes of patients with advanced renal cell carcinoma treated with nivolumab plus ipilimumab versus sunitinib (CheckMate 214): a randomised, phase 3 trial. Lancet Oncol 2019; 20: 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anagnostou V, Yarchoan M, Hansen AR, et al. Immuno-oncology trial endpoints: Capturing clinically meaningful activity. Clin Cancer Res 2017; 23: 4959–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heng DY, Xie W, Regan MM, et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009; 27: 5794–9. [DOI] [PubMed] [Google Scholar]
- 9.McDermott DF, Rini BI, Motzer RJ, et al. Treatment-free survival (TFS) after discontinuation of first-line nivolumab (NIVO) plus ipilimumab (IPI) or sunitinib (SUN) in intention-to-treat (ITT) and IMDC favorable-risk patients (pts) with advanced renal cell carcinoma (aRCC) from CheckMate 214. Presented at: 2019 Genitourinary Cancers Symposium, February 14–16, 2019; San Francisco, CA, USA Abstract. [Google Scholar]
- 10.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40 (accessed September 25, 2017).
- 11.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc 1958; 53: 457–81. [Google Scholar]
- 12.Motzer RJ, Bacik J, Murphy BA, Russo P, Mazumdar M. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 2002; 20: 289–96. [DOI] [PubMed] [Google Scholar]
- 13.Clopper CJ, Pearson ES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika 1934; 26: 404–13. [Google Scholar]
- 14.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials 1986; 7: 177–88. [DOI] [PubMed] [Google Scholar]
- 15.Atkins MB, Tannir NM. Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat Rev 2018; 70: 127–37. [DOI] [PubMed] [Google Scholar]
- 16.Lalani AA, Xie W, Martini DJ, et al. Change in neutrophil-to-lymphocyte ratio (NLR) in response to immune checkpoint blockade for metastatic renal cell carcinoma. J Immunother Cancer 2018; 6: 5. doi: 10.1186/s40425-018-0315-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zahoor H, Barata PC, Jia X, et al. Patterns, predictors and subsequent outcomes of disease progression in metastatic renal cell carcinoma patients treated with nivolumab. J Immunother Cancer 2018; 6: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDermott DF, Huseni MA, Atkins MB, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 2018; 24: 749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tannir NM, Motzer RJ, Plimack ER, et al. Outcomes in patients (pts) with advanced renal cell carcinoma (aRCC) who discontinued (DC) first-line nivolumab + ipilimumab (N+I) or sunitinib (S) due to treatment-related adverse events (TRAEs) in CheckMate 214. Presented at: 2019 Genitourinary Cancers Symposium, February 14–16, 2019; San Francisco, CA, USA Abstract. [Google Scholar]
- 20.Motzer RJ, Penkov K, Haanen J, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019; 380: 1103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019. [DOI] [PubMed] [Google Scholar]
- 22.Escudier B, Tannir NM, McDermott DF, et al. CheckMate 214: Efficacy and safety of nivolumab plus ipilimumab vs sunitinib for treatment-naïve advanced or metastatic renal cell carcinoma, including IMDC risk and PD-L1 expression subgroups. Presented at: European Society of Medical Oncology 2017, September 8–12, 2017; Madrid, Spain Oral presentation LBA5. [Google Scholar]
- 23.Motzer RJ, Powles T, Atkins MB, et al. IMmotion151: A randomized phase III study of atezolizumab plus bevacizumab vs sunitinib in untreated metastatic renal cell carcinoma (mRCC) Presented at: 2018 Genitourinary Cancers Symposium, February 8–10, 2018; San Francisco, CA, USA Poster 578. [Google Scholar]
- 24.McDermott DF, Motzer RJ, Rini BI, et al. CheckMate 214 retrospective analyses of nivolumab plus ipilimumab or sunitinib in IMDC intermediate/poor-risk patients with previously untreated advanced renal cell carcinoma with sarcomatoid features. Presented at: 17th International Kidney Cancer Symposium, November 2–3, 2018; Miami, FL, USA Poster. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.