

Supplemental Digital Content is available in the text.
Keywords: angiotensin-converting enzyme 2; endothelium; MDM2 protein, human; pulmonary arterial hypertension; vascular remodeling
Abstract
Background:
Angiotensin-converting enzyme 2 (ACE2) converts angiotensin II, a potent vasoconstrictor, to angiotensin-(1–7) and is also a membrane protein that enables coronavirus disease 2019 (COVID-19) infectivity. AMP-activated protein kinase (AMPK) phosphorylation of ACE2 enhances ACE2 stability. This mode of posttranslational modification of ACE2 in vascular endothelial cells is causative of a pulmonary hypertension (PH)–protective phenotype. The oncoprotein MDM2 (murine double minute 2) is an E3 ligase that ubiquitinates its substrates to cause their degradation. In this study, we investigated whether MDM2 is involved in the posttranslational modification of ACE2 through its ubiquitination of ACE2, and whether an AMPK and MDM2 crosstalk regulates the pathogenesis of PH.
Methods:
Bioinformatic analyses were used to explore E3 ligase that ubiquitinates ACE2. Cultured endothelial cells, mouse models, and specimens from patients with idiopathic pulmonary arterial hypertension were used to investigate the crosstalk between AMPK and MDM2 in regulating ACE2 phosphorylation and ubiquitination in the context of PH.
Results:
Levels of MDM2 were increased and those of ACE2 decreased in lung tissues or pulmonary arterial endothelial cells from patients with idiopathic pulmonary arterial hypertension and rodent models of experimental PH. MDM2 inhibition by JNJ-165 reversed the SU5416/hypoxia-induced PH in C57BL/6 mice. ACE2-S680L mice (dephosphorylation at S680) showed PH susceptibility, and ectopic expression of ACE2-S680L/K788R (deubiquitination at K788) reduced experimental PH. Moreover, ACE2-K788R overexpression in mice with endothelial cell–specific AMPKα2 knockout mitigated PH.
Conclusions:
Maladapted posttranslational modification (phosphorylation and ubiquitination) of ACE2 at Ser-680 and Lys-788 is involved in the pathogenesis of pulmonary arterial hypertension and experimental PH. Thus, a combined intervention of AMPK and MDM2 in the pulmonary endothelium might be therapeutically effective in PH treatment.
Clinical Perspective.
What Is New?
MDM2 (murine double minute 2) expression is increased in lung tissues from patients with idiopathic pulmonary arterial hypertension and animals with experimental pulmonary hypertension.
MDM2, an E3 ligase of angiotensin-converting enzyme 2 (ACE2), ubiquitinates ACE2 at K788. AMP-activated protein kinase phosphorylates ACE2 at S680 and inhibits MDM2-mediated ubiquitination of ACE2 at K788.
Functionally, ACE2 phosphorylation and deubiquitination increase the endothelial nitric oxide synthase–mediated nitric oxide bioavailability in endothelial cells.
What Are the Clinical Implications?
Inhibition of MDM2 (eg, by JNJ-165) has great therapeutic potential for pulmonary arterial hypertension/pulmonary hypertension by stabilizing ACE2.
Posttranslational modification of ACE2 is a novel strategy to develop new therapies for pulmonary arterial hypertension/pulmonary hypertension.
The renin-angiotensin system (RAS) regulates blood pressure, fluid and electrolyte balance, and vascular resistance.1 Sustained pulmonary vasoconstriction, concentric pulmonary vascular remodeling, and in situ thrombosis are major causes for the elevated pulmonary vascular resistance and pulmonary arterial pressure in patients with pulmonary arterial hypertension (PAH).2,3 Within the RAS system, the angiotensin-converting enzyme 2 (ACE2)/angiotensin (Ang)-(1–7)/Mas receptor axis counterposes the ACE1/Ang II/AT1 receptor axis. With a homologous catalytic domain as for ACE1, ACE2 competes with ACE1 to convert Ang II to Ang-(1–7), which provides antivasoconstrictive, anti-inflammatory, antihypertrophic, and antifibrotic effects on various vascular beds, including the pulmonary vasculature.4,5 Thus, inhibiting the canonical RAS (ie, ACE1) and activating the counterbalancing ACE2/Ang-(1–7)/Mas axis have complementary efficacies in cardiovascular diseases, including pulmonary hypertension (PH).6–8
Posttranslational modifications (PTMs) play an essential role in regulating protein expression, activity, and subcellular distribution. As 2 major modes of PTMs, protein phosphorylation and ubiquitination are critical for cellular responses to physiological and pathophysiological stimuli.9,10 We have previously shown that AMP-activated protein kinase (AMPK) phosphorylates ACE2 Ser-680 (S680) in vascular endothelial cells (ECs) and thereby inhibits the proteasome-mediated ubiquitination of ACE2.8 Consequently, ACE2 stability is increased under conditions in which AMPK is activated (eg, metformin treatment).11,12 Activation of the AMPK-ACE2 axis is beneficial to the pulmonary endothelium, as supported by the PH-resistant phenotype seen in a mouse line harboring the phosphomimetic form of ACE2 (ie, ACE2-S680D).8 However, the molecular basis underlying ACE2 ubiquitination leading to its degradation and its correlation with the onset of PAH remains elusive.
MDM2 (murine double minute 2), or HDM2 in humans, is an oncoprotein, in part, through its negative regulation of p53.13–15 Functioning as an E3 ubiquitin ligase, MDM2 ubiquitinates p53 and induces p53 protein degradation.16–18 MDM2 is upregulated in diverse cancer cell types, including but not limited to, colorectal adenocarcinoma, breast carcinomas, lung cancer, and osteogenic sarcoma.13,19 JNJ-26854165 (JNJ-165), an oral MDM2 inhibitor, can induce apoptosis and decrease proliferation in various tumor models.20,21 Dysregulated MDM2 is also implicated in cardiovascular impairments. For example, MDM2 overexpression enhances vascular calcification by ubiquitinating histone deacetylase 1.22 Nutlin-3a, a small-molecule antagonist of MDM2, mitigates both PH and retinal angiogenesis by decreasing the proliferation of pulmonary artery smooth muscle cells and ECs, respectively.23,24
In this study, we explored the role of MDM2 and its crosstalk with AMPK in regulating the expression of ACE2 in the pulmonary endothelium. Our results showed that AMPK phosphorylation of ACE2 at S680 hindered MDM2 ubiquitination of ACE2 at K788 in ECs. In concert, these 2 PTM-associated mechanisms regulated the stability of ACE2. A maladapted PTM of ACE2 is implicated in the pulmonary endothelium in patients with idiopathic pulmonary arterial hypertension (IPAH) and several animal models of experimental PH.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Cell Cultures and Reagents
Human pulmonary artery ECs (PAECs) were obtained from the Pulmonary Hypertension Breakthrough Initiative. Demographic and clinical data of controls and patients with IPAH are in Table I in the Data Supplement. On the basis of the Pulmonary Hypertension Breakthrough Initiative facility’s protocol,25 the cells were isolated from lung tissues of health controls and patients with IPAH and cultured in endothelial cell growth medium (EGM-2; Lonza) on fibronectin-coated plates. Cells were passaged at 70% to 80% confluence by dissociation from plates using Trypsin-EDTA. Primary cultures of passage 5 to 8 were used in experiments. PAEC authentication was performed by using antibodies against the EC markers, CD31, von Willebrand factor, and VE-cadherin, and fluorescence-activated cell sorting/immunocytochemistry. The percentage positively stained with these EC markers was >93%. Human umbilical vein endothelial cells were cultured in M199 medium (Sigma-Aldrich). Human embryonic kidney 293 (HEK 293) cells and bovine aortic endothelial cells were obtained from ATCC and maintained in Dulbecco's Modified Eagle Medium (Life Technologies). All cell cultures were maintained at 37°C in 95% air and 5% CO2.
The reagents used and their commercial sources were as follows: SU5416 (Cayman); cycloheximide, MG132, and monocrotaline (Sigma-Aldrich); JNJ26854165 (Selleckchem); Ang-(1–7) ELISA kit (Peninsula Laboratories International); Microfil MV-122, MV-Diluent, and MV curing agent (Flow Tech); anti-ACE2 and anti-ACE1 (Abcam); anti-Ub, anti–endothelial nitric oxide synthase (eNOS), and anti–α-tubulin (Cell Signaling Technology); anti-MDM2, anti–β-actin, and anti-GAPDH (Santa Cruz Biotechnology); anti-peNOS (S1177) and anti-peNOS (T495) (BD Biosciences). Expression plasmids encoding ACE2, ubiquitin, eNOS, MDM2, and MDM2 C464A were purchased from Addgene. ACE2 Ser-680 was mutated to Asp (D) or Leu (L) and Lys-788 to Arg (R) by using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies).
Human Lung Samples
Human lung specimens were from healthy donor lungs or explanted lungs from subjects with IPAH undergoing lung transplantation at the University of California, San Diego. The study was approved by the institutional review board at the University of California, San Diego; written informed consent was obtained from all participants. Demographic and clinical data of controls and patients with IPAH are in Table I in the Data Supplement.
Cell Transfection and Virus Infection
MDM2 small interfering RNA or scramble control RNA (Qiagen) was transfected into various cell types at a concentration of 20 nmol/L with the use of Lipofectamine RNAi Max (Invitrogen). Adenoviruses overexpressing ACE2 wild type (WT), S680D, S680L, or S680L/K788R were created by using the ViraPower Adenoviral Expression System (Invitrogen), with protocols provided by the manufacturer. Adenovirus-driven ectopic expressing ACE2 (WT) and the various ACE2 mutants in cells were achieved by infecting cultured cells at 10 multiplicity of infection.
In Silico Method
UbiBrowser (http://ubibrowser.ncpsb.org) was used to predict the E3 ligases. Combining confidence and evidence modes, a list of putative MDM2 E3 ligases is shown in Table II in the Data Supplement. For the heat map shown in Figure 1C, we analyzed a GEO RNA-sequencing data set (GSE79613) generated from the induced pluripotent stem cell–derived ECs from patients with PAH26 with the online tool GENE_E (Morpheus) used to produce the presented heat map.
Figure 1.
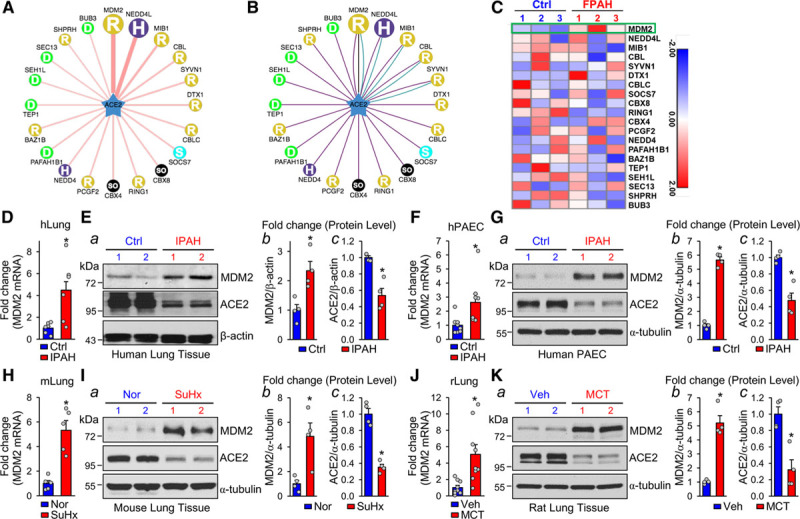
MDM2 level is elevated in PH. A and B, In the network view, a node is positioned in the center of the canvas showing the putative substrates, surrounded by nodes revealing predicted E3 ligases. The node colors and characters denote the E3 ligase type with the edge width and node size representing the confidence score. In the confidence mode (A), both edge width and node size are positively correlated with the UbiBrowser score. In the evidence mode (B), the edge of each predicted E3-substrate interaction consists of multiple colored lines, with distinct colors representing different types of supporting evidence, including E3-recognizing motif (blue), network loops (black), and enriched gene ontology pair (purple). C, RNA-sequencing data for predicted E3 ligases in induced pluripotent stem cell–derived endothelial cells. The heat map shows the expression of genes involved in patients with familial PAH (FPAH) with BMPR2 mutation versus controls. D and E, qPCR quantification of MDM2 mRNA levels and Western blot analysis of MDM2 and ACE2 levels in lung tissue from patients with IPAH (n=6) and controls without PAH (n=6). F and G, qPCR quantification of MDM2 mRNA level in PAECs isolated from IPAH (n=8) and control lungs (n=8) and Western blot analysis of MDM2 and ACE2 levels in PAECs isolated from IPAH (n=4) and control lungs (n=4). H through K, qPCR quantification of MDM2 mRNA level and Western blot analysis of MDM2 and ACE2 levels in the lung tissues of SuHx-induced PH mice (n=5) and control mice (n=5) (H and I) and MCT-induced PH rat models (n=9) versus controls (n=9) (J and K). Data are mean±SEM. For data with normal distribution (D, H, and J), statistical significance was determined by 2-tailed Student t test with Welch correction between 2 indicated groups. Nonnormally distributed data (E through G, I, and K) were analyzed using the Mann-Whitney U test between 2 indicated groups. *P<0.05 versus control patients (Ctrl) or Normoxia (Nor) or vehicle (Veh) controls. ACE2 indicates angiotensin-converting enzyme 2; IPAH, idiopathic pulmonary arterial hypertension; MCT, monocrotaline; MDM2, murine double minute 2; PAEC, pulmonary artery endothelial cells; PH, pulmonary hypertension; qPCR, quantitative polymerase chain reaction; and SuHx, SU5416/hypoxia.
Western Blot Analysis, Immunoprecipitation, and Quantitative Real-Time Polymerase Chain Reaction
Cells or tissue was lysed in the EBC buffer supplemented with protease inhibitors (Thermo Scientific), phosphatase inhibitors (Roche), and ubiquitinase inhibitors (Sigma-Aldrich). Lysates were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunoblotted with antibodies as indicated. For immunoprecipitation, the lysates were incubated with the described antibodies for 4 hours at 4°C, followed by a 2-hour incubation with Dynabeads Protein G (Life Technologies). Immunoprecipitated products were washed with the NETN buffer before resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunoblotting with the indicated antibodies. For quantitative real-time polymerase chain reaction, RNA was extracted from homogenized tissue or cells by using TRIzol reagent (Invitrogen). The cDNA was synthesized by using Takara (Clontech Laboratories). Quantitative polymerase chain reaction was performed using SYBR Green Realtime PCR Master Mix (Bio-Red) with GAPDH or β-actin as internal controls. The relative gene expression was quantified by the ΔΔCq method. Primer sequences used for quantitative polymerase chain reaction are listed in Table III in the Data Supplement.
Animal Studies, Hemodynamics, Histology, and Pulmonary Angiogram
Animal experiments were approved by Institutional Animal Care and Use Committee of University of California, San Diego and the Institutional Animal Ethics Committee of Xi’an Jiaotong University. Adult male mice at 8 weeks old were given SU5416 (20 mg/kg) once a week and placed under 10% hypoxia for 5 weeks (12-hour light/dark cycle). JNJ-165 was administered daily through oral gavage for the last 2 weeks, and adenovirus (Ad-null, Ad-ACE2L K788R, Ad-ACE2-WT, and ACE2-K788R) (5×108 plaque-forming units) was delivered intratracheally (once). Right ventricular systolic pressure (RVSP) and systemic systolic blood pressure (were measured with standardized protocols. Fulton index (right ventricular hypertrophy) was determined as the ratio of the right ventricular (RV) weight to those of the left ventricle (LV)+septum (S) weight. For investigating vessel wall remodeling, formaldehyde-fixed, paraffin-embedded lung tissue sections were stained with hematoxylin and eosin and observed under a NanoZoomer Slide Scanner. For pulmonary angiography, mice were anesthetized by pentobarbital sodium (120 mg/kg) through intraperitoneal injection. The chest was opened by median sternotomy, and the thymus and adipose tissue were carefully excised. Heparin (20 IU) was immediately injected into the right ventricle to prevent blood from coagulation. A catheter composed of PE-20 tubing was inserted into the main pulmonary artery through the right ventricle, which was ligated together with the ascending aorta using a 6-0 black silk suture. Mouse lung was initially perfused with phosphate-buffered saline 0.05 mL/min for 3 minutes using PE-20 tubing until the lung turned white and then was perfused with Microfil, a casting agent at 0.05 mL/min. Microfil composition consists of 600 µL of Microfil and 750 µL of diluent. Just before microfil perfusion, 67.5 µL of curing agent was added. The working time of the Microfil solution is 15 minutes. Lungs were left overnight at 4°C covered with wet paper to avoid drying. The next day, the lungs were separated carefully and placed in phosphate-buffered saline and kept gently shaking for 15 minutes at room temperature. Phosphate-buffered saline was removed and lungs underwent gradient dehydration using ethyl alcohol (50%, 70%, 80%, 95%, and 100% twice) per hour to digest lung tissue. After dehydration, lungs were immersed in methyl salicylate to view the lung vasculature. Lungs were photographed with a camera (MU1000, FMA050, Amscope). The periphery of lung was selected with Photoshop and converted to binary images using National Institutes of Health Image J to quantitate the total branch length, number of branches, and number of junctions normalized to area used for analysis.
Statistics
All statistical analyses were performed with SPSS 14.0 or GraphPad Prism 7 software. The numbers in figure legends indicate biological replicates performed in each experiment. Initially, the data sets were analyzed for normality using the Shapiro-Wilk test (P<0.05) and equal variance using the F test (P>0.05). For data with normal distribution, 2-tailed Student t test was used to compare 2 groups, and 1-way ANOVA with Bonferroni post hoc test was used to compare multiple groups. Two-way ANOVA was used in Figure 2D and 2E and Figure 3D for comparison with 2 independent variables. For nonnormally distributed data or experiments with small sample size (n<5), the Mann-Whitney U test was used to compare 2 groups, and the Kruskal-Wallis test was used for multiple groups. Data are expressed as mean±SEM and P<0.05 was considered statistically significant.
Figure 2.
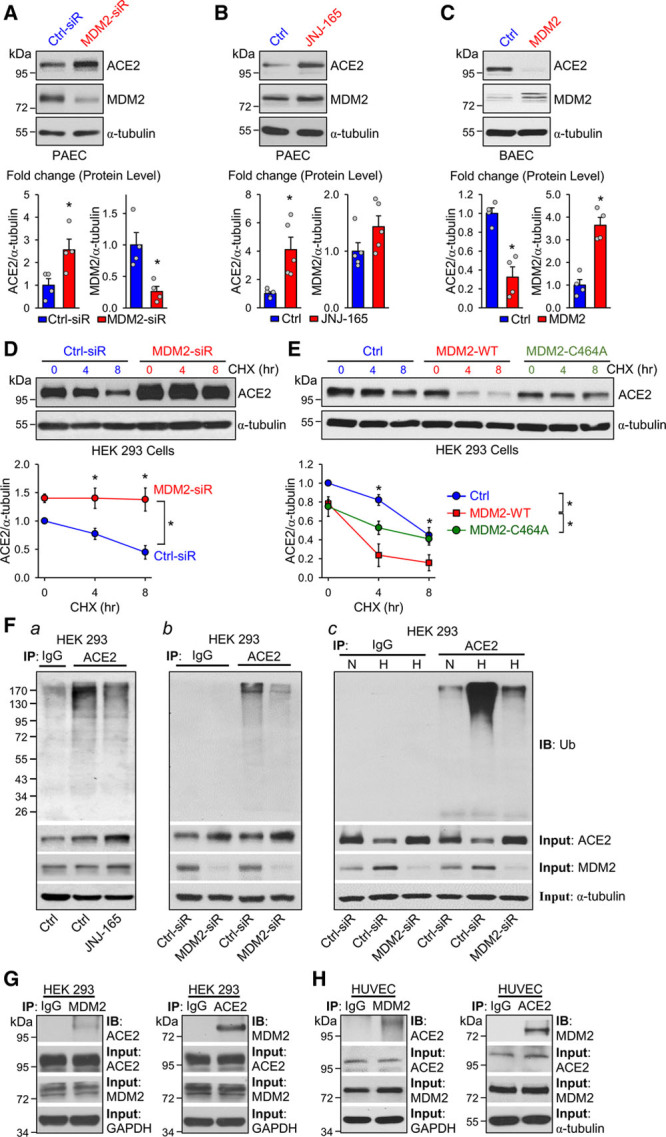
MDM2 decreases ACE2 stability through ubiquitination. A through C, Western blot analysis of ACE2 and MDM2 levels in PAECs transfected with MDM2 or control siRNA (20 nmol/L) for 48 hours (A); JNJ-165 (10 μmol/L) or vehicle for 16 hours (B); and MDM2 overexpression or control plasmid (1 μg/mL) in BAECs for 48 hours (C). D and E, Western blot analysis of ACE2 in HEK 293 cells transfected with MDM2 siRNA (20 nmol/L), MDM2 overexpression plasmids (MDM2-WT), or MDM2 RING domain mutation plasmids (MDM2-C464A) together with ACE2 overexpression plasmids (1 μg/mL), then treated with CHX (100 μg/mL) for up to 8 hours. F, HEK 293 cells were cotransfected with ACE2 and ubquitin (Ub) overexpression plasmids (1 μg/mL) and were treated with MG132 (20 μmol/L) 10 hours before harvesting the cells. Separately, HEK 293 cells were treated with JNJ-165 (10 μmol/L) or vehicle for 16 hours before harvesting (a). HEK 293 cells were treated with MDM2 or control siRNA (20 nmol/L) and cultured under a normal or hypoxia condition (b and c) for 24 hours. Cell extracts were immunoprecipitated with anti-ACE2 or IgG and immunoblotted with anti-Ub. The input proteins were immunoblotted with antibodies against ACE2, MDM2, or α-tubulin. G, HEK 293 cells were transfected with plasmids overexpressing ACE2 or MDM2 (1 μg/mL). H, HUVECs were treated with MG132 (20 μmol/L) for 10 hours. ACE2 or MDM2 was immunoprecipitated. The input protein was immunoblotted with antibodies against ACE2, MDM2, α-tubulin, or GAPDH. Data are mean±SEM from 3 to 5 independent experiments. Normally distributed data (B) were analyzed by 2-tailed Student t test with Welch correction between 2 indicated groups and normally distributed data (D and E) were analyzed by 2-way ANOVA test between multiple groups. Nonnormally distributed data (A and C) were analyzed using the Mann-Whitney U test between 2 indicated groups. *P<0.05 versus controls (Ctrl) or control-siRNA (Ctrl-siR). ACE2 indicates angiotensin-converting enzyme 2; BAEC, bovine aortic endothelial cell; CHX, cycloheximide; HEK 293, human embryonic kidney 293; HUVEC, human umbilical vein endothelial cell; IB, immunoblotted; IgG, immunoglobulin G; IP, immunoprecipitation; MDM2, murine double minute 2; PAEC, pulmonary artery endothelial cells; siR, siRNA, small interfering RNA; and WT, wild type.
Results
MDM2 Is Correlated With ACE2 in PH
We used UbiBrowser (http://ubibrowser.ncpsb.org) to explore the putative E3 ligases that can ubiquitinate ACE2.27 Combined results of the confidence mode (Figure 1A) and evidence mode (Figure 1B) suggested that MDM2, NEDD4L (neural precursor cell expressed developmentally downregulated gene 4-like), and MIB1 (mindbomb E3 ubiquitin protein ligase 1) had the highest scores as E3 ligases to ubiquitinate ACE2. We then mined a GEO RNA-sequencing data set (GSE79613; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE79613) generated from the induced pluripotent stem cell–derived ECs from patients with familial PAH. The mRNA level of MDM2, but not that of NEDD4L, MIB1, and other E3 ligases was elevated in induced pluripotent stem cells-derived ECs from patients with familial PAH (heat map in Figure 1C). We validated these in silico results by examining the MDM2 and ACE2 expression levels in lung tissues from patients with IPAH. The protein and mRNA levels of MDM2 were both increased, but the protein level of ACE2 was decreased in lung tissues from patients who had IPAH versus healthy controls (Figure 1D and 1E), which was verified in PAECs from patients with IPAH (Figure 1F and 1G) and lung tissues from mice with SU5416/hypoxia (SuHx)-induced PH (Figure 1H and 1I) and rats with monocrotaline-induced PH (Figure 1J and 1K).
MDM2 Is an E3 Ligase of ACE2
In light of augmented MDM2 level and decreased ACE2 level in PAECs from patients with IPAH and lung tissues from rodents with experimental PH, we next explored whether MDM2 regulates ACE2 stability through its ubiquitination of ACE2 and hence affects the expression level of ACE2. MDM2 knockdown by small interfering RNA or inhibition by JNJ-165, an agent targeting the MDM2 ubiquitinase catalytic domain (RING domain), increased ACE2 expression in PAECs (Figure 2A and 2B). By contrast, MDM2 overexpression in ECs decreased ACE2 level (Figure 2C). We performed a cycloheximide pulse-chase experiment to determine whether MDM2 affected ACE2 stability. MDM2 knockdown prolonged, but MDM2 overexpression shortened ACE2 half-life in HEK 293 cells (Figure 2D and 2E). However, overexpression of the loss-of-function mutant MDM2 C464A (C464 in the RING domain is mutated to A)17 did not affect the protein half-life of ACE2. We next investigated whether MDM2 ubiquitinates ACE2 and therefore affects ACE2 stability. ACE2 ubiquitination was significantly attenuated in HEK 293 cells treated with JNJ-165 or transfected with MDM2 small interfering RNA (Figure 2Fa and 2Fb). Hypoxia induced ACE2 ubiquitination in HEK 293 cells, which was significantly attenuated by MDM2 knockdown (Figure 2Fc). We also explored the interaction between MDM2 and ACE2. As shown in Figure 2G, exogenously expressed MDM2 and ACE2 in HEK 293 cells were reciprocally immunoprecipitated. The interaction between MDM2 and ACE2 was further confirmed by the endogenously expressed MDM2 and ACE2 in ECs (Figure 2H). Together, these results suggest that MDM2 ubiquitinated ACE2 leading to ACE2 degradation.
MDM2 Ubiquitination of ACE2 at K788
By using UbPred (http://www.ubpred.org), we predicted that ACE2-K788 is a putative ubiquitination site (Figure 3A). K788 is located 108 amino acids downstream of S680 which is phosphorylated by AMPK.8 Given that AMPK increases ACE2 stability, we hypothesized that AMPK phosphorylation of S680 inhibits MDM2 ubiquitination of ACE2-K788. To test this hypothesis, we created ACE2-S680L, a dephosphomimetic of ACE2 (ubiquitination-prone mutant), and ACE2-S680L/K788R, its ubiquitination-resistant counterpart. To examine the crosstalk between AMPK and MDM2 in regulating ACE2 expression level, we transfected HEK 293 cells with ACE2-S680D (phosphomimetic), ACE2-S680L, or ACE2-S680L/K788R. Level of ubiquitination was higher for ACE2-S680L than for ACE2-S680D. However, ACE2-S680L/K788R reversed the ubiquitination of ACE2 (Figure 3B). Under hypoxia, the level of ubiquitination was lower for ACE2-S680L/K788R than for ACE2-S680L (Figure 3C). Cycloheximide pulse-chase experiments showed remitted stability of ACE2-S680L/K788R (Figure 3D). Functionally, ACE2-S680L/K788R increased the eNOS-mediated nitric oxide (NO) bioavailability (ie, increased S1177 phosphorylation and decreased T495 dephosphorylation) to a level similar to that of ACE2-S680D in both HEK 293 cells and PAECs. In contrast, ACE1 expression level was lower in S680L/K788R-transfected cells than ACE2-S680L cells (Figure 3E and 3F). As the major proteolytic product of ACE2, Ang-(1–7) level was elevated by ≈3-fold in PAECs overexpressing ACE2-S680D or ACE2-S680L/K788R in comparison with ACE2-S680L (Figure 3G).
Figure 3.
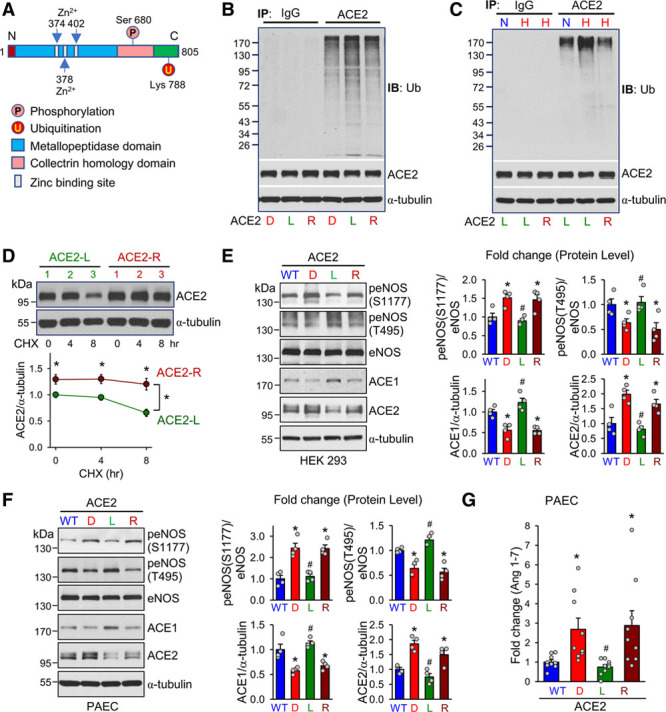
MDM2 ubiquitinates ACE2 at K788. A, Schematic diagram showing the functional domains, and S680 and K788, as well, of ACE2, which are conserved among human, mouse, and rat. B, HEK 293 cells were transfected with expression plasmids encoding ACE2-S680D, ACE2-S680L, or ACE2-S680L/K788R (ACE2-R) together with Ub plasmids (1 μg/mL) and MG132 (20 μmol/L) treatment in the last 10 hours. Cell lysates were immunoprecipitated by anti-ACE2 or IgG, then immunoblotted with anti-Ub. The input proteins were immunoblotted with anti-ACE2 or anti–α-tubulin. C, HEK 293 cells were transfected with ACE2-S680L or ACE2-S680L/K788R together with Ub plasmids (1 μg/mL) under hypoxia or normoxia for 24 hours with MG132 (20 μmol/L) treatment in the last 10 hours. Ubiquitination of various forms of ACE2 was analyzed. D, Western blot analysis of ACE2 and α-tubulin in HEK 293 cells transfected with ACE2-S680L or ACE2-S680L/K788R (ACE2-R) plasmids, then treated with CHX for 0, 4, and 8 hours. E, HEK 293 cells were transfected with ACE2-WT, ACE2-S680D, ACE2-S680L, or ACE2-S680L/K788R (ACE2-R), together with eNOS expression plasmids (1 μg/mL). Cell lysates were immunoblotted with antibodies against peNOS (S1177), peNOS (T495), eNOS, ACE2, ACE1, or α-tubulin. F, PAECs were infected with ACE2-WT, ACE2-S680D, ACE2-S680L, and ACE2-S680L/K788R adenovirus (10 MOI). Cell lysates were immunoblotted with antibodies against peNOS (S1177), peNOS (T495), eNOS, ACE2, ACE1, or α-tubulin. G, ELISA assay of Ang-(1–7) in PAECs infected with ACE2-WT, ACE2-S680D, ACE2-S680L, and ACE2-S680L/K788R adenovirus (10 MOI). Data are mean±SEM from 3 to 9 independent experiments. Normally distributed data (G) were analyzed by 1-way ANOVA test between multiple groups, and normally distributed data (D) were analyzed by 2-way ANOVA test between multiple groups. Nonnormally distributed data (E and F) were analyzed using the Kruskal-Wallis test between multiple groups. *P<0.05 versus WT, #P<0.05 versus D and R. ACE2 indicates angiotensin-converting enzyme 2; Ang, angiotensin; CHX, cycloheximide; eNOS, endothelial nitric oxide synthase; HEK 293, human embryonic kidney 293; IB, immunoblotted; IgG, immunoglobulin G; IP, immunoprecipitation; MDM2, murine double minute 2; MOI, multiplicity of infection; PAEC, pulmonary artery endothelial cells; Ub, ubiquitin; and WT, wild type.
MDM2 Inhibition Mitigates PH in Mice
We used 3 mouse models to infer the involvement of the AMPK/MDM2/ACE2 axis in PH. First, we compared the SuHx-induced PH in C57BL/6 mice administered with JNJ-165 or vehicle (Figure 4A). RVSP and Fulton index (RV/LV+S weight) were elevated in mice with 5-week SuHx treatment (Figure 4B and 4C). JNJ-165 administration at the beginning of the 4th week ameliorated the SuHx-increased RVSP and RV hypertrophy (Figure 4B and 4C). Consistent with changes in hemodynamics, mice receiving JNJ-165 showed abrogated pulmonary artery remodeling, as shown by the hematoxylin and eosin staining (Figure 4D). Moreover, angiogram revealed better-preserved lung vasculature in mice receiving JNJ-165 (Figure 4E). At the molecular level, lung tissues from mice administered JNJ-165 showed lower levels of ACE2 ubiquitination and eNOS T494 phosphorylation, and higher levels of eNOS S1176 phosphorylation and ACE2, as well (Figure 4F). Furthermore, we compared the mRNA levels of genes implicated in the development of PH, including EDN1 (endothelin-1), ANGPT1 (angiopoietin 1), PTGIS (prostacyclin synthase), and BMPR2 (bone morphogenetic protein receptor type 2) in mouse lung tissues. JNJ-165 decreased the mRNA levels of EDN1 and ANGPT1 by ≈0.3- and 0.5-fold, respectively, but increased those of PTGIS and BMPR2 by ≈2- and 3-fold, respectively, in lung tissues from SuHx-PH mice (Figure 4G). However, JNJ-165 decreased the expression of genes involved in EC proliferation and inflammation, namely, vascular cell adhesion molecule 1, vascular endothelial growth factor, platelet-derived growth factor subunit A, interleukin 6, MCP-1 (monocyte chemoattractant protein-1), and interleukin 1β in lung tissues in SuHx-PH mice (Figure 4H). We have also performed an additional set of experiments in which JNJ-165 was administered to C57BL/6 mice at the beginning of the 5-week period. As shown in Figure I in the Data Supplement, PH was significantly prevented in mice receiving JNJ-165 along with SuHx treatment. Collectively, Figure 4 and Figure I in the Data Supplement suggest the effect of MDM2 inhibition in reducing experimental PH, which, at least in part, was mediated through the rectified ACE2 expression.
Figure 4.
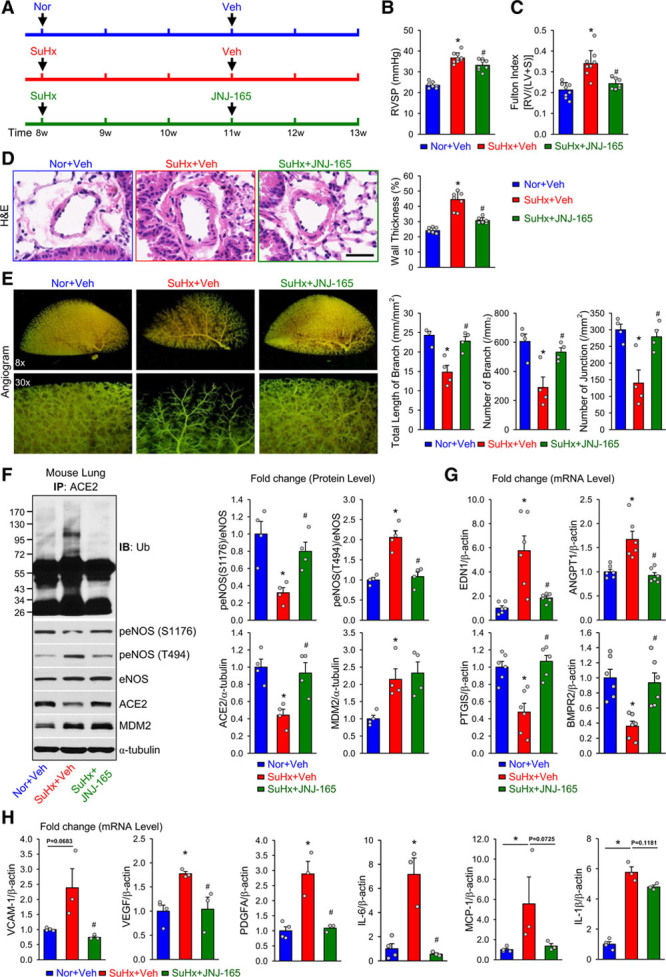
MDM2 inhibition alleviates experimental pulmonary hypertension in mice. A, Eight-week-old C57BL/6 mice were subjected to normoxia (Nor) or SuHx (10% O2 with subcutaneous injection of SU5416 at 20 mg·kg–1·wk–1) for 5 weeks. Nor and SuHx mice were given vehicle (Veh) or JNJ-165 (20 mg·kg–1·d–1) during the last 2 weeks by oral gavage. B and C, Summarized data (mean±SEM) showing right ventricular systolic pressure (RVSP, B) and Fulton index, the ratio of the weight of right ventricle (RV) to the weight of the left ventricle (LV) and septum (S) (RV/[LV+S], C) in Nor and SuHx mice treated with Veh or JNJ-165. D, H&E staining of pulmonary arteries (Left) and summarized data (mean±SEM, Right) showing the pulmonary arterial wall thickness from Nor+Veh, SuHx+Veh, and SuHx+JNJ-165 mice. Scale bar = 20 µm. E, Representative lung angiogram (Left) of the left lungs at 8× (Upper) and 30× (Lower) magnification from Nor+Veh, SuHx+Veh, and SuHx+JNJ-165 mice. Summarized data (mean±SEM, n=4 lungs, Right) showing the total length of branches, number of branches, and number of junctions of the left lungs from Nor+Veh, SuHx+Veh, and SuHx+JNJ-165 mice. F, Western blot analysis (Left) of eNOS phosphorylation at S1176 and T494, eNOS, ACE2, and MDM2, along with summarized data (mean±SEM, Right) showing the expression levels of peNOS (S1176), peNOS (T494), ACE2, and MDM2, in lung tissues from Nor+Veh, SuHx+Veh, or SuHx+JNJ-165 mice. G and H, Quantitative polymerase chain reaction analysis of endothelin-1 (END1), angiopoietin 1 (ANGPT1), prostacyclin synthase (PTGIS), and bone morphogenetic protein receptor II (BMPR2) mRNAs (G); vascular cell adhesion molecule 1 (VCAM-1), vascular endothelial growth factor (VEGF), platelet derived growth factor subunit A (PDGFA), interleukin 6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), and interleukin 1β (IL-1β) (H) in lung tissues from Nor+Veh, SuHx+Veh, and SuHx+JNJ-165 mice. Data are mean±SEM (7–9 mice per group). Normally distributed data (B through D, and G) were analyzed by 1-way ANOVA test between multiple groups. Nonnormally distributed data (E and F) were analyzed using Kruskal-Wallis test between multiple groups. *P<0.05 versus Nor+Veh; #P<0.05 versus SuHx+Veh. ACE2 indicates angiotensin-converting enzyme 2; eNOS, endothelial nitric oxide synthase; H&E, hematoxylin and eosin; IB, immunoblotted; IP, immunoprecipitation; MDM2, murine double minute 2; and Ub, ubiquitin.
ACE2-S680L/K788R Attenuates PH in ACE2-S680L Mice
We previously found that ACE2-S680D (mimicking constitutively phosphorylated ACE2-S680 leading to increased ACE2 expression) mice showed reduced PH.8 To substantiate the crosstalk between AMPK and MDM2 in regulating ACE2 in terms of PAH, we created an ACE2-S680L (mimicking constitutively dephosphorylated ACE2-S680 leading to decreased ACE2 expression) mouse line. In comparison with their ACE2-S680D counterparts, SuHx treatment caused aggravated PH in ACE2-S680L mice, as demonstrated by elevated RVSP, increased Fulton index, and vascular remodeling (Figure 5A through 5D). The enhanced PH in ACE2-S680L mice was reminiscent of EC-AMPKα2–/– and ACE2–/– mice.8 To test whether inhibition of ACE2-K788 ubiquitination attenuates PH, we administered ACE2-S680L mice an adenovirus expressing ACE2-S680L/K788R (Ad-ACE2-S680L/K788R) through intratracheal injection. Overexpression of the exogenous ACE2-S680L/K788R (mimicking constitutively dephosphorylated ACE2-S680 and deubiquitinated K788 leading to increased ACE2 expression) mitigated PH in ACE2-S680L mice (Figure 5B through 5D). However, systolic blood pressure was not significantly different between these groups (Figure II in the Data Supplement). As expected, ACE2 level was lower in the lung of ACE2-S680L than ACE2-S680D mice under SuHx treatment, which was consistent with decreased NO bioavailability (Figure 5E). Regarding PH-related genes, mice overexpressing ACE2-S680L/K788R showed reduced mRNA levels of EDN1 (≈0.3-fold) and ANGPT1 (≈0.3-fold) and increased mRNA levels of PTGIS (≈5-fold) and BMPR2 (≈3-fold) (Figure 5F). Collectively, results in Figure 5 suggest that dephosphorylation of ACE2-S680 aggravates, but deubiquitination of ACE2-K788 mitigates, experimental PH.
Figure 5.
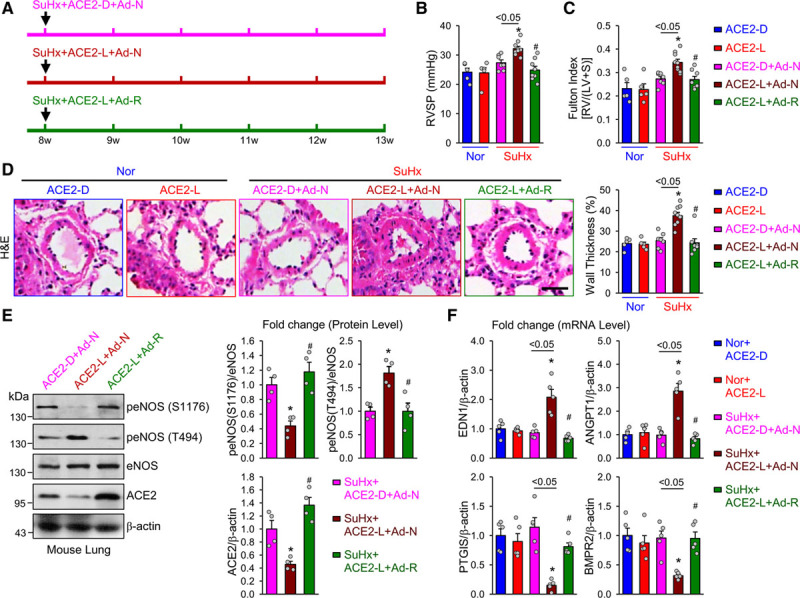
ACE2-K788R alleviates pulmonary hypertension in ACE2-S680L mice. A, Eight-week-old ACE2-S680D (ACE2-D) and ACE2-S680L (ACE2-L) mice were subjected to normoxia or SuHx for 5 weeks. The SuHx-treated mice were administrated adenoviral-null (Ad-N) or adenoviral-ACE2-S680L/K788R (Ad-R) intratracheally at the beginning of the experiments. B and C, Summarized data (mean±SEM) showing RVSP (B) and Fulton index, the ratio of the weight of right ventricle (RV) to the weight of the left ventricle (LV) and septum (S) (RV/[LV+S], C) in normoxic (Nor) ACE2-D and ACE2-L mice and SuHx-treated ACE2-D and ACE2-L mice receiving Ad-N or Ad-R. D, Histological images with H&E staining of pulmonary arteries (Left) and summarized data (mean±SEM, Right) showing the pulmonary arterial wall thickness from Nor+ACE2-D, Nor+ACE2-L, SuHx+ACE2-D+Ad-N, SuHx+ACE2-L+Ad-N, or SuHx+ACE2-L+Ad-R mice. Scale bar = 20 µm. E, Western blot analysis (Left) of peNOS S1176 or T494, eNOS, ACE2, and MDM2, along with summarized data (mean±SEM, Right) showing the expression levels of peNOS S1176, peNOS T494, ACE2, and MDM2, in lung tissues from SuHx+ ACE2-D+Ad-N, SuHx+ACE2-L+Ad-N, or SuHx+ACE2-L+Ad-R mice. F, Quantitative polymerase chain reaction analysis of END1, ANGPT1, PTGIS, and BMPR2 mRNA in lung tissues from Nor+ACE2-D, Nor+ACE2-L, SuHx+ACE2-D+Ad-N, SuHx+ACE2-L+Ad-N, or SuHx+ACE2-L+Ad-R mice. Data are mean±SEM (5–9 mice per group). Normally distributed data (B through D and F) were analyzed by 1-way ANOVA test between multiple groups. Nonnormally distributed data (E) were analyzed using the Kruskal-Wallis test between multiple groups. *P<0.05 versus Nor+ACE2-L; #P<0.05 versus SuHx+ACE2-L+Ad-N; *P<0.05 between SuHx+ACE2-D+Ad-N and SuHx+ACE2-L+Ad-N as indicated. ACE2 indicates angiotensin-converting enzyme 2; ANGPT1, angiopoietin 1; BMPR2, bone morphogenetic protein receptor II; END1, endothelin-1; eNOS, endothelial nitric oxide synthase; H&E, hematoxylin and eosin; MDM2, murine double minute 2; PTGIS, prostacyclin synthase; RVSP, right ventricular systolic pressure; and SuHx, SU5416/hypoxia.
ACE2-K788R Improves PH in EC-AMPKα2–/– Mice
To further decipher the role of MDM2 in modulating ACE2 through interacting with AMPK, we investigated whether ectopic expression of ACE2-K788R is sufficient to rescue SuHx-mediated PH in EC-AMPKα2–/– mice.8 To this end, EC-AMPKα2–/– mice were under SuHx for 3 weeks before adenovirus-null (Ad-null) and adenovirus-expressing wild type ACE2 (Ad-ACE2-WT) or ACE2-K788R (Ad-ACE2-K788R) was introduced intratracheally. Two weeks later, we examined the effects of Ad-null, Ad-ACE2-WT, or Ad-ACE2-K788R on SuHx-induced PH in EC-AMPKα2–/– mice. SuHx treatment significantly increased RVSP, mean pulmonary arterial pressure (mPAP; estimated according to the equation: mPAP=0.61 RVSP+2 mm Hg)28 and RV contractility (RV ±dP/dt) in EC-AMPKα2–/– mice (Figure 6A). Overexpression of ACE2-K788R in pulmonary endothelium was confirmed by Western blot revealing increased ACE2 level in lung ECs isolated from mice receiving Ad-ACE2-K788R (Figure III in the Data Supplement). Increased RVSP/mPAP was associated with significant pulmonary vascular remodeling as revealed by lung histology (Figure 6B), angiogram (Figure 6C), and RV hypertrophy (Figure 6D). Administration of Ad-ACE2-K788R significantly attenuated SuHx-induced PH. However, EC-AMPKα2–/– mice receiving Ad-ACE2-K788R exhibited fewer increases in RVSP/mPAP (Figure 6A), pulmonary artery wall thickness (Figure 6B), Fulton index (Figure 6D), and preserved pulmonary artery branches (Figure 6C) than EC-AMPKα2–/– mice receiving Ad-null or Ad-ACE2-WT (Figure 6A through 6D). In line with the in vivo hemodynamic and ex vivo histological data, ACE2 ubiquitination was decreased and its expression was increased in SuHx-treated EC-AMPKα2–/– mice infected with Ad-ACE2-K788R in comparison with those infected with Ad-ACE2-WT (Figure 6E and 6F). Furthermore, eNOS S1176 phosphorylation was increased (≈2.5-fold) and eNOS T494 phosphorylation was decreased by ≈0.5-fold (Figure 6E); levels of EDN1 and ANGPT1 mRNA were decreased by ≈0.5- and 0.7-fold, respectively, and those of PTGIS and BMPR2 were increased by ≈1.5- and 2-fold, respectively, in SuHx-treated EC-AMPKα2–/– mice with Ad-ACE2-K788R in comparison with Ad-null or Ad-ACE-WT (Figure 6F). Ad-ACE2-WT overexpression in EC-AMPKα2–/– mice did not significantly increase ACE2 level, whereas ACE2 ubiquitination level was much higher with SuHx, which indicates that omission of AMPK phosphorylation at S680 enhanced ACE2 ubiquitination. Together, these results suggest that AMPK phosphorylation at S680 was necessary for ACE2 deubiquitination at K788, which endowed a PH-resistant phenotype.
Figure 6.
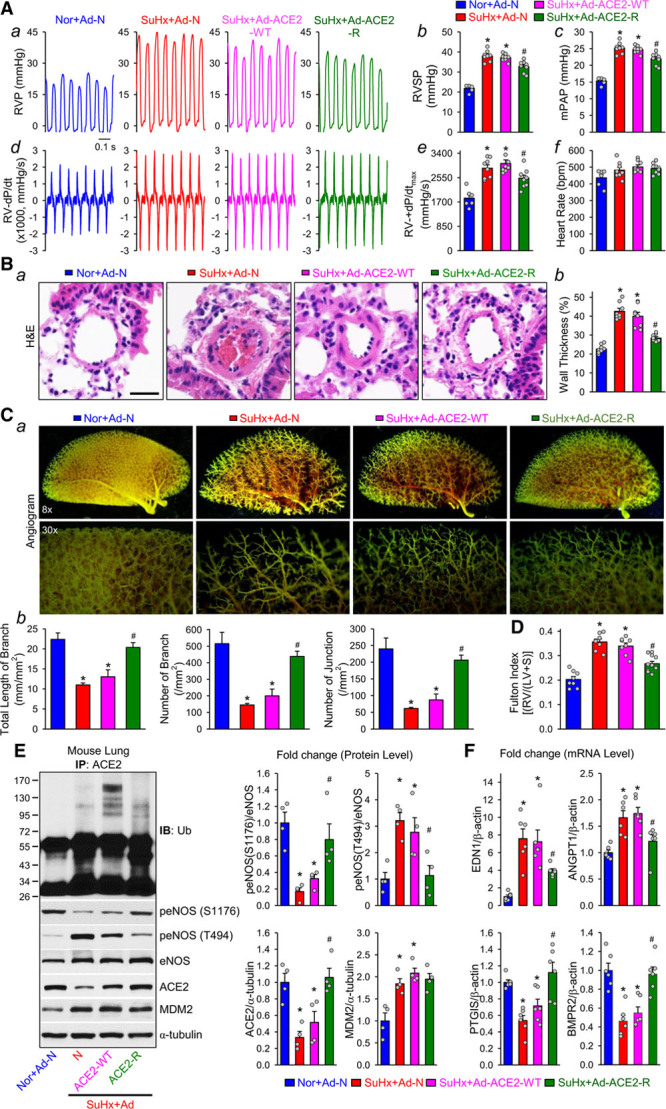
ACE2-K788R ameliorates pulmonary hypertension in EC-AMPKα2–/– mice. Eight-week-old EC-AMPKα2–/– mice were subcutaneously injected with SU5416 and exposed to hypoxia (10% O2) for 3 weeks before adenoviral infection was introduced intratracheally; the hemodynamic, histological, and angiogram measurements were conducted in the mice 2 weeks after adenoviral infection. A, Representative records of right ventricular pressure (RVP) (a), summarized RVSP (b), and estimated mean pulmonary arterial pressure (mPAP, calculated by the equation: mPAP=0.6 RVSP+2 mm Hg, (c) in normoxic mice receiving adenovirus null (Nor-Ad-N) and SU5416/Hypoxia (SuHx)–treated mice with adenoviral null (SuHx+Ad-N) or adenoviral infection of wild-type ACE2 (SuHx-ACE2-WT) and ACE2-K788R (SuHx+ACE2-R). Representative RV ±dP/dt (d) and summarized values of RV +dp/dtmax (e, mean±SEM) from Nor-Ad-N, SuHx+Ad-N, SuHx+ACE2-WT, and SuHx+ACE2-K788R mice are also shown. f shows averaged heart rate in Nor+Ad-N, SuHx+Ad-N, SuHx+ACE2-WT, and SuHx+ACE2-K788R mice. B, Histological images with H&E staining of pulmonary arteries (a) and summarized data (mean±SEM) showing the pulmonary arterial wall thickness (b) from Nor+Ad-N, SuHx+Ad-N, SuHx+ACE2-WT, and SuHx+ACE2-K788R mice. Scale bar = 20 μm. C, Representative lung angiogram (a) of the left lung at 8× (Upper) and 30× (Lower) magnification from Nor+Ad-N, SuHx+Ad-N, SuHx+ACE2-WT, and SuHx+ACE2-K788R mice. Summarized data (mean±SEM, n=5 mouse lungs, b) showing the total length of branches (Left), number of branches (Middle), and number of junctions (Right). D, Fulton index, the ratio of the weight of right ventricle (RV) to the weight of the left ventricle (LV) and septum (S) ([RV/LV+S]), in Nor+Ad-N, SuHx+Ad-N, SuHx+ACE2-WT and SuHx-ACE2-K788R mice. E, Immunoprecipitation (IP) and Western blot analyses (Left) on Ub, peNOS at S1176 and T494, eNOS, ACE2, and MDM2, along with summarized data (mean±SEM) showing the expression levels of peNOS S1176, peNOS T494, ACE2, and MDM2, in lung tissues from Nor+Ad-N, SuHx+Ad-N, SuHx+ACE2-WT, and SuHx+ACE2-R mice. F, qPCR analysis of END1, ANGPT1, PTGIS, and BMPR2 transcripts in lung tissues from Nor+Ad-N, SuHx+Ad-N, SuHx+ACE2-WT, and SuHx+ACE2-R mice. Data are mean±SEM (6–10 mice per group). Normally distributed data (A, B, and D) were analyzed by 1-way ANOVA test between multiple groups. Nonnormally distributed data (C, E, and F) were analyzed using the Kruskal-Wallis test between multiple groups. *P<0.05 versus Nor+Ad-N; #P<0.05 versus SuHx+Ad-N and SuHx+ACE2-WT. ACE2 indicates angiotensin-converting enzyme 2; ANGPT1, angiopoietin 1; BMPR2, bone morphogenetic protein receptor II; END1, endothelin-1; eNOS, endothelial nitric oxide synthase; H&E, hematoxylin and eosin; MDM2, murine double minute 2; PTGIS, prostacyclin synthase; RVSP, right ventricular systolic pressure; and Ub, ubiquitin.
Discussion
In this study, we demonstrated that the expression level of ACE2 in ECs was coregulated by a PTM mechanism involving AMPK and MDM2. The implication of this mechanism in PH builds on the observations that: (1) MDM2 level is increased and ACE2 level decreased in lung tissues and PAECs from patients with IPAH and in lung tissues from animals with experimental PH and (2) pharmacological inhibition of MDM2 (which increased ACE2) or genetic manipulation of ACE2 K788 (which rendered ACE2 more resistant to MDM2-mediated ubiquitination and degradation) attenuated the development of PH and partially reversed established PH in mice. Mechanistically, we demonstrated that an interplay between the AMPK-mediated phosphorylation of ACE2 at S680 and the MDM2-mediated ubiquitination of ACE2 at K788 played an important role in determining the level of ACE2. In mice with experimental PH, inhibition of ACE2 phosphorylation at S680 and upregulation of MDM2 expression resulted in ACE2 ubiquitination at K788 and, consequently, ACE2 degradation (summarized in Figure 7).
Figure 7.
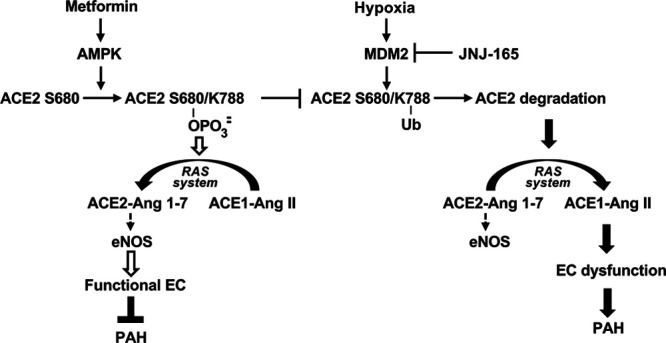
Schematic illustration of AMPK and MDM2-modulated the phosphorylation and ubiquitination of ACE2 in PAH. In functional pulmonary endothelial cells or those under metformin therapy, AMPK phosphorylates ACE2 at S680 to increase its stability to maintain the eNOS-derived nitric oxide bioavailability. As an E3 ligase of ACE2, MDM2 ubiquitinates ACE2 at K788, resulting in its degradation under hypoxia, thereby inducing disorder of RAS system and dysfunction of ECs, eventually lead to PAH. Such pathological modulation may be reversed by JNJ-165 therapy, which inhibits the MDM2 E3 function. ACE2 indicates angiotensin-converting enzyme 2; AMPK, AMP-activated protein kinase; Ang, angiotensin; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; MDM2, murine double minute 2; PAH, pulmonary arterial hypertension; RAS, renin-angiotensin system; and Ub, ubiquitin.
The RAS is intricately balanced between the detrimental ACE1-Ang II-AT1R axis and the beneficial ACE2-Ang-(1–7)-Mas axis. Suppressed ACE2 or aggravated ACE1 have been implicated in PH.29 Herein, the decreased level of ACE2 in lung tissues of animals with experimental PH and patients with IPAH, more specifically, in PAECs from these patients (Figure 1D through 1K) are in line with the previous reports. Although there might be various mechanisms involved in the regulation of the 2 axes, alone or in concert, the data from the current study indicated that MDM2 ubiquitination of ACE2 would decrease the ratio of ACE2 to ACE1. Consequently, impaired EC functions became evident, as suggested by the impaired eNOS-derived NO bioavailability (Figure 3F). Because ACE1 in the lung endothelium plays a critical role in regulating RAS in the cardiovascular system, the dysregulated MDM2-ACE2 might also potentiate the ACE1-Ang II-AT1R axis at the system level, which agrees with the increased serum level of Ang I and Ang II in patients with IPAH.30
Sustained pulmonary vasoconstriction and excessive pulmonary vascular remodeling attributable to increased proliferation of PAECs, pulmonary artery smooth muscle cells, and fibroblasts are the major causes of the elevated pulmonary vascular resistance in patients with PAH.31 The ACE1-Ang II-AT1R arm promotes vasoconstriction, proliferation, and growth, which would be counteracted by the ACE2-Ang-(1–7)-Mas arm.32 We found that ACE2-S680 phosphorylation and ACE2 K788 deubiquitination increased eNOS-mediated NO production and Ang-(1–7) in PAECs, which may account for the decreased RVSP seen in ACE2-S680D mice and S680L mice infected with ACE2-S680L/K788R adenovirus (Figure 5).
PAH has emerged as a cancer-like cardiovascular disease.33,34 This hypothetical notion is supported by the efficacy of anticancer drugs (eg, imatinib [tyrosine kinase inhibitor], everolimus [mammalian target of rapamycin inhibitor], and dichloroacetate [pyruvate dehydrogenase kinase inhibitor]) in their use in clinical trials of PAH.35 Related to the current work, given that MDM2 inhibitors are being considered in preclinical experiments and clinical trials for solid malignancies, these drugs might be useful in treating PAH. One of these agents, JNJ-165 (also known as serdemetan) has been suggested to exert both p53-dependent and -independent effects on killing tumor cells.20,36 The therapeutic effect of MDM2 inhibitors on PAH, if any, would be attributable, at least in part, to their inhibition of the MDM2-ACE2 axis. Besides MDM2, other E3 ligases (eg, NEDD4L and MIB1) are likely to be involved in ACE2 ubiquitination through their ubiquitination of K788 or other Lys residues. Thus, during the pathophysiological course of PAH, a number of E3 ligases in the pulmonary endothelium, including MDM2, are activated by the cancer-like milieu, which results in ACE2 ubiquitination leading to its degradation.
Data shown in Figures 5 and 6 suggest that ACE2-S680 phosphorylation is necessary and sufficient for K788 deubiquitination. In contrast, S680 dephosphorylation might potentiate K788 ubiquitination. Both S680 and K788, located at the extracellular domain of ACE2 and PTM of these 2 residues, might modify the 3-dimensional conformation of ACE2, which will either hinder or favor the catalysis by MDM2. Of note, the binding of severe acute respiratory syndrome coronavirus 2 Spike glycoprotein (S-protein) to ACE2 is required for viral infection and development of COVID-19–associated lung diseases.37–40 Although knowledge is lacking whether COVID-19 aggravates PH, the present study leads us to speculate that COVID-19 is likely associated with EC dysfunction through dysregulated ACE2. Recombinant human ACE2 can inhibit severe acute respiratory syndrome coronavirus 2 infection of engineered blood vessels.41 Because ACE2-S680D/K788R has a longer half-life than the WT ACE2, recombinant human ACE2-S680D/K788R might potentially be a better therapeutic strategy.
In summary, this study demonstrates a novel crosstalk between the metabolic regulator AMPK and the oncoprotein MDM2 by which ACE2 in the lung vasculature is decreased to contribute to the development of pulmonary arterial hypertension. Under normal or physiological conditions, AMPK-mediated ACE2 phosphorylation at S680 hinders MDM2-mediated ACE2 ubiquitination at K788; thus, the ACE2 stability is maintained and ACE2-mediated conversion of Ang II to Ang-(1–7), and, eNOS-mediated NO production, as well, is enhanced in lung ECs. A maladapted PTM modification of ACE2 results in the decrease of ACE2 stability attributable to MDM2-mediated ubiquitination and degradation, which then causes increased Ang II and decreased NO in lung ECs, contributing to the development and progression of PAH. Metformin, an activator of AMPK, induces AMPK-mediated ACE2 phosphorylation at S680 and protects ACE2 from MDM2-mediated ubiquitination, thereby exerting a therapeutic effect on PAH. In addition, given the specific binding of severe acute respiratory syndrome coronavirus 2 to ACE2, the AMPK/MDM2-mediated PTM of ACE2 may also be implicated in severe acute respiratory syndrome coronavirus 2–associated acute and chronic lung injury and acute respiratory distress syndrome.
Acknowledgments
H. Shen and Drs Zhang, Thistlethwaite, Wang, Z.-Y. Yuan, J.X.-J. Yuan, and Shyy designed research; H. Shen, C. Wang, Drs Jain, Xiong, and Shi, Y.-Y. Lei, S.-S. Chen, and Dr Yin performed research; H. Shen, C. Wang, Drs Jain, Xiong, J.X.-J. Yuan, and Shyy analyzed data; H. Shen, Drs Zhang, Gong, Z.-Y. Yuan, J.X.-J. Yuan, and Shyy wrote and edited the article. The authors acknowledge Drs Wang, Kang, Babicheva, Dong, and Moya, Y. Zhang and Y. Zhang at the University of California, San Diego; Drs Wang, Lai, and Bai, L. Chen and J. Zhang at Xi’an Jiaotong University, Xi’an, China, for their technical assistance. The authors also thank Drs He, Gongol, and Powell at the University of California, San Diego; Drs Liu and Zhou at the Xi’an Jiaotong University, Xi’an, China; Dr Lee at The Chinese University of Hong Kong, Shenzhen, China, for their consultation and useful discussion. The authors are grateful to the Pulmonary Hypertension Breakthrough Initiative for providing human lung cells for this study. The Pulmonary Hypertension Breakthrough Initiative is funded by Cardiovascular Medical Research and Education Fund.
Sources of Funding
This work was supported by the NIH grant R01HL108735 (to J.Y.-J.S.); National Natural Science Foundation of China grants 81941005 (to Dr Z.-Y. Yuan); 81970225 (to Dr Gong); 81800328 (to Dr Zhang); the National Key Research and Development Program grant 2018YFC1311500 (to Dr Z.-Y. Yuan); the Clinical Research Award of the First Affiliated Hospital of Xi’an Jiaotong University, China, grant XJTU1AF-CRF-2016-004 (to Dr Z.-Y. Yuan); Xi’an Jiaotong University Financial support; the Yangzhou University International Academic Exchange Fund no 20190224 (to H. Shen); and Jiangsu Key R&D Project in Social Development grant no. BE2015663 (Dr Gong).
Disclosures
None.
Supplemental Materials
Expanded Methods
Data Supplement Tables I–III
Data Supplement Figures I–III
Supplementary Material
Footnotes
H. Shen, Dr Zhang, and C. Wang contributed equally.
Sources of Funding, see page 1202
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCULATIONAHA.120.048191.
References
- 1.Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res. 2015;116:960–975. doi: 10.1161/CIRCRESAHA.116.303587 [DOI] [PubMed] [Google Scholar]
- 2.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:1 supplS20–S31. doi: 10.1016/j.jacc.2009.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ, Nicolls MR. Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur Respir J. 2012;40:1555–1565. doi: 10.1183/09031936.00046612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, Campagnole-Santos MJ. The ACE2/angiotensin-(1–7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1–7). Physiol Rev. 2018;98:505–553. doi: 10.1152/physrev.00023.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oudit GY, Crackower MA, Backx PH, Penninger JM. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc Med. 2003;13:93–101. doi: 10.1016/s1050-1738(02)00233-5 [DOI] [PubMed] [Google Scholar]
- 6.Jiang F, Yang J, Zhang Y, Dong M, Wang S, Zhang Q, Liu FF, Zhang K, Zhang C. Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat Rev Cardiol. 2014;11:413–426. doi: 10.1038/nrcardio.2014.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrell NW, Atochina EN, Morris KG, Danilov SM, Stenmark KR. Angiotensin converting enzyme expression is increased in small pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. J Clin Invest. 1995;96:1823–1833. doi: 10.1172/JCI118228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Dong J, Martin M, He M, Gongol B, Marin TL, Chen L, Shi X, Yin Y, Shang F, et al. AMP-activated protein kinase phosphorylation of angiotensin-converting enzyme 2 in endothelium mitigates pulmonary hypertension. Am J Respir Crit Care Med. 2018;198:509–520. doi: 10.1164/rccm.201712-2570OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019 [DOI] [PubMed] [Google Scholar]
- 10.Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan NJ, Villén J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat Methods. 2013;10:676–682. doi: 10.1038/nmeth.2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420 [DOI] [PubMed] [Google Scholar]
- 12.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0 [DOI] [PubMed] [Google Scholar]
- 14.Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res. 2008;14:5318–5324. doi: 10.1158/1078-0432.CCR-07-5136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res. 2003;1:993–1000 [PubMed] [Google Scholar]
- 16.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0 [DOI] [PubMed] [Google Scholar]
- 17.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4 [DOI] [PubMed] [Google Scholar]
- 18.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0 [DOI] [PubMed] [Google Scholar]
- 19.Oliner JD, Saiki AY, Caenepeel S. The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb Perspect Med. 2016;6:a026336 doi: 10.1101/cshperspect.a026336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kojima K, Burks JK, Arts J, Andreeff M. The novel tryptamine derivative JNJ-26854165 induces wild-type p53- and E2F1-mediated apoptosis in acute myeloid and lymphoid leukemias. Mol Cancer Ther. 2010;9:2545–2557. doi: 10.1158/1535-7163.MCT-10-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You L, Liu H, Huang J, Xie W, Wei J, Ye X, Qian W. The novel anticancer agent JNJ-26854165 is active in chronic myeloid leukemic cells with unmutated BCR/ABL and T315I mutant BCR/ABL through promoting proteosomal degradation of BCR/ABL proteins. Oncotarget. 2017;8:7777–7790. doi: 10.18632/oncotarget.13951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwon DH, Eom GH, Ko JH, Shin S, Joung H, Choe N, Nam YS, Min HK, Kook T, Yoon S, et al. MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat Commun. 2016;7:10492 doi: 10.1038/ncomms10492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mouraret N, Marcos E, Abid S, Gary-Bobo G, Saker M, Houssaini A, Dubois-Rande JL, Boyer L, Boczkowski J, Derumeaux G, et al. Activation of lung p53 by Nutlin-3a prevents and reverses experimental pulmonary hypertension. Circulation. 2013;127:1664–1676. doi: 10.1161/CIRCULATIONAHA.113.002434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chavala SH, Kim Y, Tudisco L, Cicatiello V, Milde T, Kerur N, Claros N, Yanni S, Guaiquil VH, Hauswirth WW, et al. Retinal angiogenesis suppression through small molecule activation of p53. J Clin Invest. 2013;123:4170–4181. doi: 10.1172/JCI67315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104:1342–1347. doi: 10.1073/pnas.0605080104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu M, Shao NY, Sa S, Li D, Termglinchan V, Ameen M, Karakikes I, Sosa G, Grubert F, Lee J, et al. Patient-specific iPSC-derived endothelial cells uncover pathways that protect against pulmonary hypertension in BMPR2 mutation carriers. Cell Stem Cell. 2017;20:490–504.e5. doi: 10.1016/j.stem.2016.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Xie P, Lu L, Wang J, Diao L, Liu Z, Guo F, He Y, Liu Y, Huang Q, et al. An integrated bioinformatics platform for investigating the human E3 ubiquitin ligase-substrate interaction network. Nat Commun. 2017;8:347 doi: 10.1038/s41467-017-00299-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chemla D, Castelain V, Humbert M, Hébert JL, Simonneau G, Lecarpentier Y, Hervé P. New formula for predicting mean pulmonary artery pressure using systolic pulmonary artery pressure. Chest. 2004;126:1313–1317. doi: 10.1378/chest.126.4.1313 [DOI] [PubMed] [Google Scholar]
- 29.Ferreira AJ, Shenoy V, Yamazato Y, Sriramula S, Francis J, Yuan L, Castellano RK, Ostrov DA, Oh SP, Katovich MJ, et al. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179:1048–1054. doi: 10.1164/rccm.200811-1678OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, François C, Schalij I, Dorfmüller P, Simonneau G, Fadel E, et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:780–789. doi: 10.1164/rccm.201203-0411OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54:1 supplS10–S19. doi: 10.1016/j.jacc.2009.04.006 [DOI] [PubMed] [Google Scholar]
- 32.Bradford CN, Ely DR, Raizada MK. Targeting the vasoprotective axis of the renin-angiotensin system: a novel strategic approach to pulmonary hypertensive therapy. Curr Hypertens Rep. 2010;12:212–219. doi: 10.1007/s11906-010-0122-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rai PR, Cool CD, King JA, Stevens T, Burns N, Winn RA, Kasper M, Voelkel NF. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:558–564. doi: 10.1164/rccm.200709-1369PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guignabert C, Tu L, Le Hiress M, Ricard N, Sattler C, Seferian A, Huertas A, Humbert M, Montani D. Pathogenesis of pulmonary arterial hypertension: lessons from cancer. Eur Respir Rev. 2013;22:543–551. doi: 10.1183/09059180.00007513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boucherat O, Vitry G, Trinh I, Paulin R, Provencher S, Bonnet S. The cancer theory of pulmonary arterial hypertension. Pulm Circ. 2017;7:285–299. doi: 10.1177/2045893217701438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tabernero J, Dirix L, Schöffski P, Cervantes A, Lopez-Martin JA, Capdevila J, van Beijsterveldt L, Platero S, Hall B, Yuan Z, et al. A phase I first-in-human pharmacokinetic and pharmacodynamic study of serdemetan in patients with advanced solid tumors. Clin Cancer Res. 2011;17:6313–6321. doi: 10.1158/1078-0432.CCR-11-1101 [DOI] [PubMed] [Google Scholar]
- 37.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, Raizada MK, Grant MB, Oudit GY. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res. 2020;126:1456–1474. doi: 10.1161/CIRCRESAHA.120.317015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, Leopoldi A, Garreta E, Hurtado Del Pozo C, Prosper F, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181:905–913.e7. doi: 10.1016/j.cell.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.