

Abstract
Patients with inflammatory bowel disease (IBD) have an increased risk of colorectal cancer, particularly in ulcerative colitis (UC) when the majority of colon epithelial cells may be exposed to inflammation-associated mutagenesis. In addition to mutagenesis generated by oxidative stress, inflammation can induce activation-induced cytidine deaminase (Aicda), a mutator enzyme in the APOBEC family, within colon epithelial cells. This study tested the hypothesis that deletion of the Aicda gene could protect against the development of inflammation-associated colorectal cancers, using a model of UC-like colitis in “T/I” mice deficient in TNF and IL10. Results showed that T/I mice that were additionally Aicda-deficient (“TIA” mice) spontaneously developed moderate to severe UC-like colitis soon after weaning, with histologic features and colon inflammation severity scores similar those in T/I mice. Although the mean survival of TIA mice was decreased compared to T/I mice, multivariable analysis that adjusted for age when neoplasia was ascertained showed a decreased numbers of neoplastic colorectal lesions in TIA mice, with a trend toward decreased incidence of neoplasia. Aicda deficiency increased serum IL1α and slightly decreased IL12p40 and M-CSF, as compared with T/I mice, and led to undetectable levels of IgA, IgG1, IgG2a, IgG2b, and IgG3. Taken together, these studies show that Aicda deficiency can decrease the number of neoplastic lesions but is not sufficient to prevent the risk of inflammation-associated colorectal neoplasia in the setting of severe UC-like inflammation. The TIA model may also be useful for assessing the roles of antibody class-switch recombination deficiency and somatic hypermutation on regulation of microbiota and inflammation in the small intestine and colon, as well as the pathogenesis of colitis associated with hyper-IgM syndrome in humans. Further studies will be required to determine the mechanisms that drive early mortality in TIA mice.
Introduction
Cancer is known to result from accumulation of non-lethal mutations and changes in gene regulation that affect cell growth and genomic stability and allow continuous self-renewal. General risk factors for colorectal cancer include older age, family history of colon cancer, smoking, alcohol consumption, being overweight, and not exercising. A personal history of inflammatory bowel disease (IBD) further increases colorectal cancer risk beyond that of the general population, although reported relative risks have varied widely depending on the populations studied [1–4]. Cancer risk in IBD appears to be proportional to the area of colon that is inflamed, with a lower risk in Crohn disease (CD) where inflammation is typically focal and higher in ulcerative colitis (UC) where inflammation is characteristically geographically continuous such that more colon epithelial cells are potentially exposed to inflammation-associated mutagenesis [4]. Although the oxidative stress generated by inflammation within the colon can clearly enhance the rate of mutation accumulation, this alone appears to be insufficient to explain the increased risk of colorectal cancer in IBD patients.
The apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) family mutator enzyme called activation-induced cytidine deaminase (gene = Aicda; protein = AID) is normally expressed during B cell development to facilitate the mutations and DNA rearrangements that enhance antibody diversity [5]. However, a variety of pro-inflammatory cytokines have also been found to induce Aicda in colon epithelial cell lines. AID also appears to be highly expressed in vivo by colon epithelial cells from humans with IBD [6]. Importantly, forced expression of AID in colon epithelial cells in vitro was shown to enhance acquisition of inactivating mutations in the Tp53 gene after as little as 6 weeks of culture [6]. Unrelated studies showed that AID expression facilitated induction of the OCT4 and NANOG genes that are associated with self-renewal (“stemness”) [7], perhaps via its contribution to methylation/ demethylation reactions that regulate gene expression [8]. Changes in methylation and inappropriate expression of stem cell genes can contribute to the continuous self-renewal that is a hallmark of cancer. Together, these observations suggest that inflammation-induced induction of endogenous Aicda in colon epithelium may drive the development of colorectal cancers by enhancing survival and self-renewal, in addition to enhancing mutagenesis & decreasing genomic stability. This study tested the hypothesis that deletion of the Aicda gene could protect against the development of inflammation-associated colorectal cancers driven by the above mechanisms.
Testing this hypothesis requires an appropriate animal model. Gene variants for IL-10 and its receptor increase risk for developing both CD [9] and UC in humans [10, 11]. Il10-/- mice on the C57BL/6 background readily develop moderate to severe colitis when their mucosal barrier is compromised by exogenous triggers such as infection with Helicobacter species [12, 13] or exposure to non-steroidal anti-inflammatory drugs (NSAIDs) [14, 15]. Colitis in Il10-/- mice is characterized by colonic inflammation that is transmural but discontinuous, forming “skip lesions” similar to what is observed in humans with CD. Il10-/- mice with colitis also have an increased risk of dysplasia and invasive colon cancer relative to control mice [12, 13, 16–18]. Aicda deficiency was previously reported to decrease inflammation-associated colon cancer in mice, based on the observation that adenocarcinoma had developed in 6 of 22 Il10-/- mice vs. 1 of 23 Il10-/- Aicda-/- mice by ~1 year of age (p = 0.05; [19]). However, Aicda deficiency did not affect the percentage of mice with precursor dysplastic lesions in that model (90.9% for Il10-/- and 90.3% for Il10-/- Aicda-/- mice) [19]. Furthermore, the percentage of the colonic area involved by inflammation and thus at risk for inflammation-associated neoplasia was not reported in this CD-like model.
We previously reported a novel murine model of colitis that closely resembles human UC rather than CD [20]. Il10-/- mice that are also TNF-deficient (Tnf -/- Il10-/-; termed “T/I” mice) uniformly develop moderate to severe colitis by age 4–6 weeks without the need for exogenous triggering [20]. Inflammation in these mice always involves the rectum and continues proximally in a linear fashion, typically affecting the entire colon (“pancolitis”). The lamina propria of inflamed tissues is packed with inflammatory cells. Crypt abcesses and ulceration are common, but inflammation generally does not extend below the muscularis mucosae. This clinical and histologic pattern closely resembles what is typically seen in human UC. Like humans with UC, T/I mice develop non-polypoid (“flat”) colonic neoplasia with high penetrance (63% incidence by age 28 weeks, with a mean of 2 lesions/mouse) [20]. Based on its extremely close clinical and histologic similarities to human UC, the T/I model may more accurately reproduce mechanisms of inflammation-associated carcinogenesis that are relevant in humans with UC compared with CD-like murine models.
The purpose of this study was to determine how AID presence or absence affected inflammation severity and the incidence of colorectal neoplasia in the T/I mouse model of inflammation-associated colorectal cancer.
Materials and methods
Animal studies
C57BL/6 Tnf -/- Il10-/- (“T/I”) mice were created by crossing founder mice obtained from Jackson Laboratories, Bar Harbor, ME (strain name = B6.129P2-Il10tm1Cgn/J; stock # 002251 and strain name = B6.129S6-Tnftm1Gkl/J; stock # 005540). The same parent lines were crossed with Aicda-/- mice that had been extensively back-crossed onto the C57BL/6 background (obtained from their creator, Dr. Tasuku Honjo; [21]) to create mice that were homozygous for TNF, IL-10, and AID deficiency (Tnf-/-, Il10-/-, Aicda-/-; termed “TIA” mice). Since mice deficient in TNF and IL10 develop UC-like disease at or soon after weaning and reproduce poorly [22], these lines were typically maintained by breeding T/I, Tnf-/- Il10+/- (T/Ihet), or Tnf-/- Il10+/- Aicda-/- (TIhetA) males to T/Ihet or TIhetA females. We showed previously that T/Ihet mice did not develop colitis [22]; therefore T/Ihet mice produced as littermates to and co-housed with T/I mice were used for biomarker studies as non-colitis controls.
Mice were housed in polycarbonate micro-isolator cages in individually ventilated racks under barrier conditions that excluded helicobacter and norovirus as well as other known mouse pathogens. Mice had ad libitum access to PicoLab Mouse Diet 20/5058 (LabDiet, St. Louis, MO, USA) and water. Sentinel mice exposed repetitively to dirty bedding from the mice used in this study were negative for parasites by microscopic examination, negative for Citrobacter rodentium by fecal culture, negative for infection with Helicobacter species by PCR of feces and negative by serology for a panel of 22 murine protozoal, bacterial and viral pathogens, including murine parvovirus, murine hepatitis virus, and murine norovirus (S1 Table).
Mice were euthanized by CO2 asphyxiation for assessment of colitis severity and neoplasia when they either reached the pre-defined study endpoint of 28 weeks of age or met humane endpoints of >15% loss of body weight, rectal prolapse, or other clinical signs of pain and distress, including hunching, shivering, minimal movement on gentle prodding, or grimace [23]. Once respiration ceased, death was assured by exsanguination, harvest of vital organs, and/or decapitation. Although colitis has the potential to produce pain and distress, analgesics were not employed in these studies, since non-steroidal anti-inflammatory agents (NSAIDs) can exacerbate colitis and opioids affect intestinal motility and function. Monitoring, including determination of body weight, generally occurred daily, but was always provided at least 3 times weekly. Suffering was minimized and unattended deaths were prevented by providing euthanasia as soon as possible (≤ 2 hours) for mice that met humane endpoints. All mice involved in this study were euthanized, either at defined time points or when they met humane endpoints, which defined the mortality described in this study. There were no unattended natural deaths.
Tissue analysis
After euthanasia, the entire digestive tract was dissected out from stomach to anus. A segment of mid-jejunum and the entire colon were routinely submitted for histologic examination for all mice. The lower stomach/duodenum and mesenteric lymph nodes from TIA mice were also routinely sampled for histologic examination. Five colors of permanent tissue marking dye (Bradley Products, Bloomington, MN) were used to identify specific colonic regions (cecum, proximal, mid-, and distal colon, and terminal colon/rectum). Intact, unopened intestinal segments were fixed in Carnoy’s solution for 2–4 hrs then embedded in paraffin. This method is ideal for identification of flat neoplastic lesions, since it allows examination of 2 sides of the colon and the presence of feces prevents tissue curling during fixation but does not interfere with most slide-based assays. Hematoxylin and eosin (H&E)-stained sections were evaluated by a board-certified pathologist who was blinded to tissue identity/genotype. The severity of inflammation was scored as previously described [15], using a scale from 0–75 that takes into account mucosal changes such as hyperplasia and ulceration, degree of inflammation, and percentage of each bowel segment affected by these changes. Using this scale, a score 0–12 indicates the absence of colitis, 13–24 indicates mild, and ≥ 25 indicates moderate to severe colitis. Animals with histologic scores that fall into the moderate to severe range typically have either scattered severe inflammatory lesions or extensive disease involving the mucosa. Sections were also scored for non-invasive or invasive neoplasia [24]. Gastrointestinal intraepithelial neoplasia (synonymous with atypical hyperplasia, microadenoma, carcinoma in situ) and adenoma were considered to be non-invasive lesions. A diagnosis of invasive carcinoma required the presence of a desmoplastic response (formation of an abundant collagenous stroma) to differentiate invasion from mucosal herniation or pseudoinvasion. Regions of neoplasia that were separated by regions of normal mucosa were scored as separate lesions.
Serum biomarker levels
Serum was obtained from eight to 10 week old T/I, TIA, and T/Ihet mice immediately following euthanasia. Serum biomarker profiling (cytokines, chemokines, immunoglobulins) was performed in the Duke Regional Biocontainment Laboratory (RBL) Immunology Unit (Durham, NC) under the direction of Dr. Gregory D. Sempowski, using Luminex bead-based multiplex immunoassays (BioRad), according to the manufacturer’s instructions.
Statistical analysis
Statistical comparison of histologic scores was performed using the Mann Whitney U test. Serum cytokine/chemokine measurements from T/I, TIA, and control T/Ihet mice were log-transformed and compared via 2-way ANOVA with Tukey’s test for multiple comparisons (GraphPad Prism, version 8.4.3). Categorical data was compared via Fisher’s exact test. Survival rates were determined by Kaplan-Meier analysis, with p-values calculated using the log rank test (MediCalc Software, Version 13.3, Mariakerke, Belgium). A Poisson regression model was used to determine the effect of genotype on number of neoplastic lesions, while adjusting for age (R statistical program). A p value ≤ 0.05 was considered to represent a significant difference between groups.
Ethics statement
All animal studies were approved (protocol numbers A151-09-05, A093-12-04, or A43-15-02) by the Institutional Animal Care and Use Committee of Duke University, an institution accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), International.
Results
Spontaneous colon inflammation is rare in Aicda-deficient mice
Mice that were deficient in Aicda alone demonstrated no grossly discernable phenotype over the course of this study. 100% of a cohort of 16 Aicda-/- mice survived to the experimental endpoint of 28 weeks. Most of the mice in the cohort had no colon inflammation (S1 Fig). However, one mouse in this group demonstrated focal mild to moderate mucosal hyperplasia and inflammation that scored as mild colitis (histologic score = 16). This led to a mean ± SD histologic score = 3 ± 4 for this genotype (n = 16).
Aicda deficiency does not prevent the development of UC-like colitis in TIA mice
We previously reported that T/I mice spontaneously developed moderate to severe UC-like colitis soon after weaning [20]. All T/I mice that were additionally Aidca-deficient (TIA mice) also spontaneously developed moderate to severe UC-like colitis (mean ± SD histologic score = 49 ± 9; n = 77). As in human UC, inflammation involved the entire colon (“pan-colitis”), including the cecum (S2 Fig). Severe colitis was uniformly present in all 6–7 week old TIA mice examined (mean ± SD histologic score = 53 ± 6; n = 11), consistent with onset soon after weaning. UC-like colitis in TIA mice (Fig 1) was qualitatively indistinguishable from that seen in T/I mice (S3 Fig and [20]). For both genotypes, the lamina propria of all inflamed regions was packed with inflammatory cells with frequent crypt abscesses and ulceration and inflammation generally was confined to the mucosal layer and did not extend deep into the muscle wall (Fig 1). Squamous metaplasia of the rectum was common, appearing in 36 of 69 (52%) evaluable TIA mice (Fig 2A).
Fig 1. Histology of colitis in TIA mice.
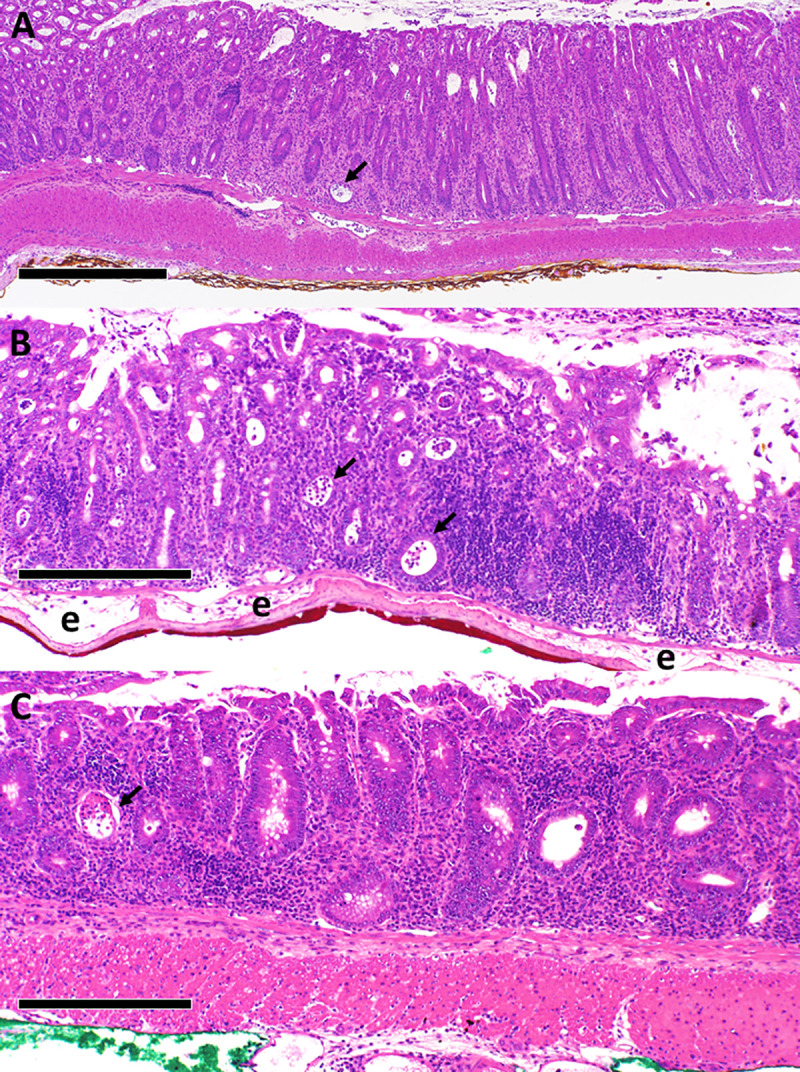
TIA mice generally presented with moderate to severe pan-colitis, primarily involving the mucosa, as shown in these representative fields derived from the terminal colon/rectum (A), distal colon (B), and mid-colon (C). The lamina propria is packed with inflammatory cells. Representative crypt abscesses are indicated by arrows. “e” in panel B denotes areas of edema in the submucosa. Scale bar represents 500 μm in A and 250 μm in B and C.
Fig 2. Squamous metaplasia and neoplasia in TIA mice.
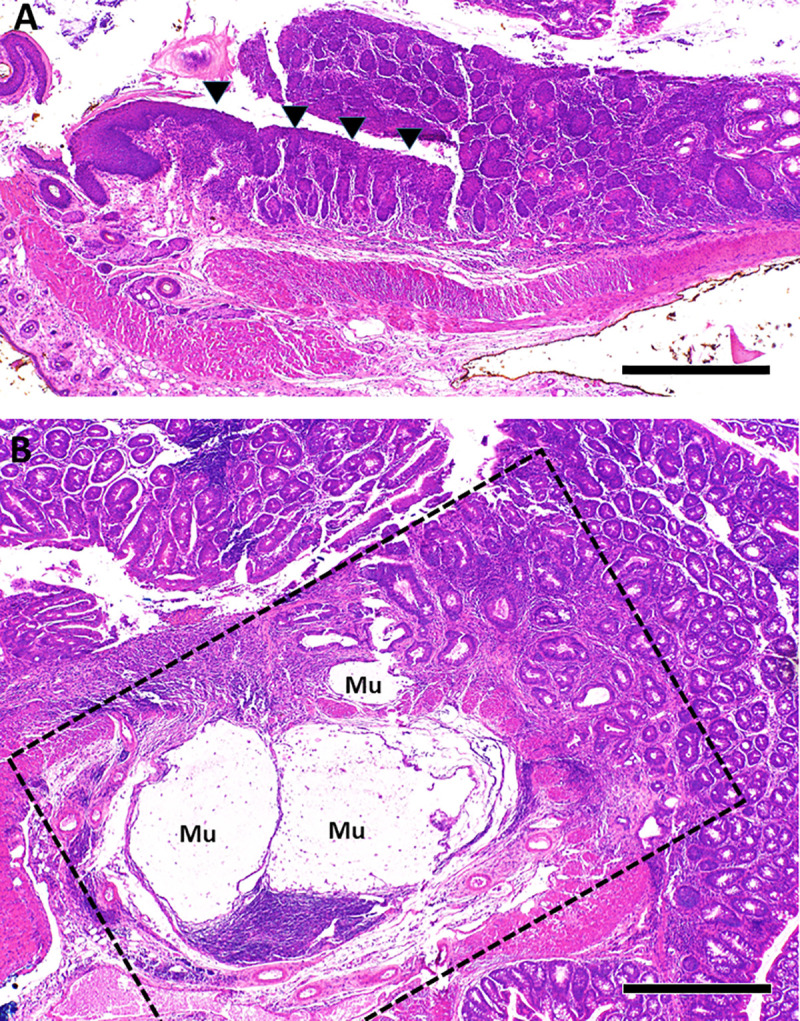
A. Squamous metaplasia (arrowheads) was common in the rectum of TIA mice, shown here in a 9 week old. B. The dotted lines highlight a focus of invasive mucinous adenocarcinoma identified in the proximal colon of a 15 week old TIA mouse with severe pan-colitis (histologic score = 55). “Mu” indicates mucin lakes associated with invasive carcinoma. Scale bar in A and B represents 500 μm.
To begin to identify potential biomarkers that could distinguish colitis in T/I versus TIA mice, concentrations of a panel of 28 serum cytokines and chemokines were measured in serum from 8–10 week old T/I (n = 28) and TIA mice (n = 17) (Table 1). Eight to 10 week old T/Ihet mice (n = 13) were also studied as non-colitis controls. Results showed that TIA mice had increased serum IL1α and decreased IL12p40 and M-CSF compared with T/I mice. Levels of 25 other cytokines and chemokines tested (Table 1) were similar in T/I and TIA mice. Serum levels of GM-CSF, IFN-γ, IL6, IL17, IP10, MCP-1, MIP1α, and MIG were higher and serum levels of IL1α, IL5, IL12(p70), MCP1, and MIP1β were lower in T/I mice compared to their non-inflamed T/Ihet littermates (Table 1). Similar differences were seen between TIA and control T/Ihet mice for most of these biomarkers, with the exception that serum levels of IL12(p70) and MCP-1 in TIA mice remained similar to control (Table 1).
Table 1. Serum cytokine and chemokine levels in T/I, TIA, and control T/Ihet micea.
| T/I mice (n = 28) | TIA mice (n = 17) | Control T/Ihet mice (n = 13) | |
|---|---|---|---|
| Eotaxin | 918 (471–1607) | 928 (559–1388) | 954 (412–1760) |
| GM-CSF | 16 (8–30) | 16 (8–37) | 4 (2–38)b,c |
| IFN-γ | 38 (4–524) | 43 (2–236) | 4 (2–11) b,c |
| IL1α | 112 (28–1883) | 157 (23–1683)* | 192 (5–1605) b,c |
| IL1β | 2 (2–12) | 2 (2–265)* | 4 (1–10) |
| IL2 | 5 (2–8) | 2 (2–9) | 6 (2–11) |
| IL3 | 2 (2–4) | 2 (2–8) | 2 (2–22) |
| IL4 | 2 (2–11) | 2 (2–42) | 2 (2–2) |
| IL5 | 2 (2–11) | 2 (2–79) | 12 (2–30) b,c |
| IL6 | 19 (2–44) | 36 (4–104) | 2 (2–8) b,c |
| IL7 | 2 (2–84) | 2 (2–26) | 2 (2–14) |
| IL9 | 47 (10–487) | 69 (10–245) | 46 (46–106) |
| IL12p40 | 12 (2–98) | 7 (2–14)* | 9 (2–30) |
| IL12p70 | 4 (1–156) | 8 (1–211) | 8 (7–45) b |
| IL13 | 154 (9–350) | 186 (69–913) | 91 (58–206) |
| IL15 | 29 (8–1378) | 27 (8–1909) | 54 (22–135) |
| IL17 | 26 (4–159) | 60 (7–146) | 2 (2–7) b,c |
| LIF | 2 (2–16) | 2 (2–17) | 2 (2–2) |
| LIX | 4634 (2397–7787) | 3900 (809–7336) | 5930 (544–7824) |
| IP10 | 292 (120–670) | 355 (150–916) | 140 (27–334) b,c |
| KC | 175 (44–765) | 189 (16–1226) | 122 (68–211) |
| MCP1 | 29 (9–58) | 27 (9–479) | 46 (24–60) b |
| MIP1α | 31 (10–62) | 29 (10–258) | 24 (1–33) b |
| MIP1β | 9 (9–28) | 9 (9–22) | 36 (9–45) b,c |
| M-CSF | 20 (7–517) | 11 (6–44)* | 12 (6–33) |
| MIP2 | 80 (20–606) | 77 (37–126) | 87 (32–132) |
| MIG | 302 (68–2448) | 368 (51–907) | 49 (30–138) b,c |
| RANTES | 36 (8–328) | 21 (6–84)* | 29 (16–65) |
a Values shown represent the median (range) of cytokine/chemokine in pg/ml in cohorts of 8–10 week old mice.
b Indicates p < 0.05 for comparison of control T/Ihet versus T/I mice.
c Indicates p < 0.05 for comparison of control T/Ihet versus TIA mice.
* Indicates p < 0.05 for comparison of T/I versus TIA mice.
To further assess immune system function, serum immunoglobulins were measured in this same cohort. Total serum immunoglobulin (Ig) was decreased in TIA mice (mean ± SEM = 6.8 ± 1.0 mg/ml) compared with T/I mice (12.1 ± 1.1 mg/ml; p = 0.0005). However, IgM levels were increased in TIA mice relative to T/I mice (p = 0.006) and TIA mice had undetectable levels of IgA, IgG1, IgG2a, IgG2b, and IgG3 (Fig 3). This phenotypic pattern of Ig expression results from a deficiency of Ig class switching and is diagnostic of hyper-IgM syndrome [25]. Levels of all immunoglobulin isotypes were increased in T/I mice relative to their non-inflamed T/Ihet littermates (p values from 0.002 for IgG1 to 1.3 x 10−7 for IgG2a; Fig 3).
Fig 3. Serum immunoglobulin concentrations in TIA, T/I, and control T/Ihet mice.
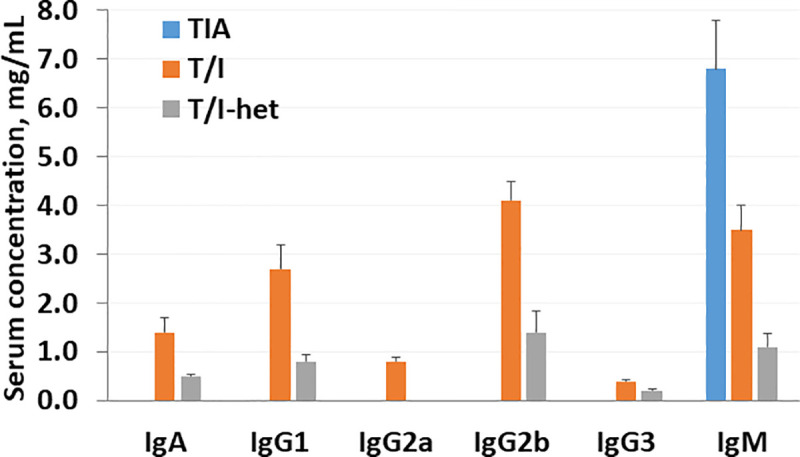
The serum concentration of various immunoglobulin isotypes are shown for 8–10 week old TIA (n = 17), T/I (n = 28), and control T/Ihet mice (n = 13). Values shown represent the mean ± standard error of the mean (SEM). Values obtained for TIA mice were statistically different than for T/I mice for all isotypes tested, with p values ranging from p = 0.006 for IgM to 7 x 10−12 for IgG3 (Student’s t-test).
Aicda deficiency increases mortality in T/I mice with UC-like colitis
We next assessed the effect of Aicda deficiency on mortality by comparing survival of T/I versus TIA mice. As shown in Fig 4, the mean survival of TIA mice was 13 weeks (median 12 wks; 95% CI, 10–14 weeks; n = 90), while the mean survival of T/I mice was 21 weeks (median 21 wks; 95% CI, 18–24 weeks; n = 109) (p < 0.0001, log rank test). Excess mortality of TIA mice was observed beginning at 8 weeks of age and typically manifested as weight loss or other markers of pain or distress rather than as rectal prolapse.
Fig 4. Survival of T/I and TIA mice as a function of age.
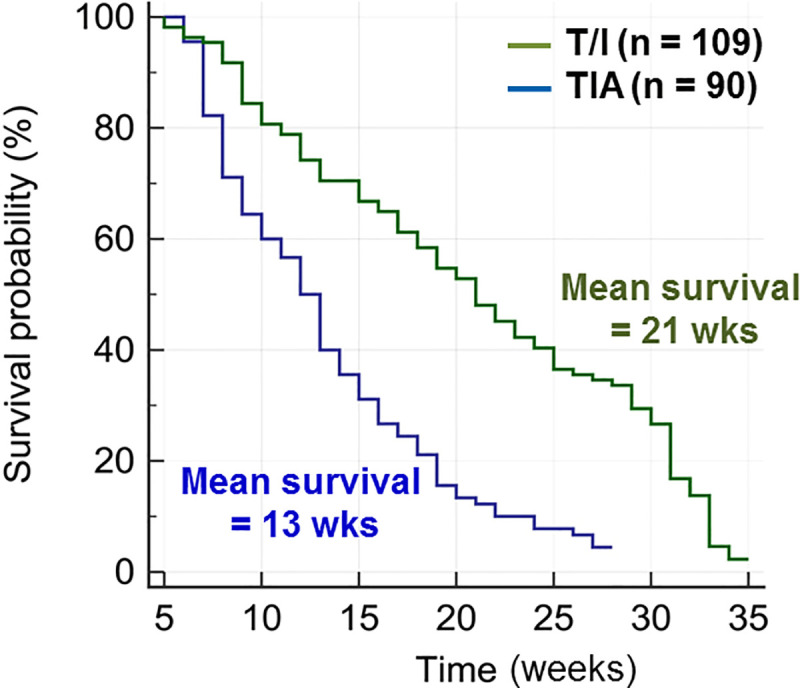
To prevent unnecessary suffering, morbidity that triggered humane endpoints was used as a surrogate for mortality, as described in Materials and Methods. Kaplan-Meier analysis demonstrates that TIA mice have a significantly shorter mean survival (13 weeks) than T/I mice (21 weeks; p = 0.0001, log-rank test).
Effect of Aicda deficiency on colorectal neoplasia in mice with UC-like colitis
To assess the effect of Aicda deficiency on the incidence of colorectal neoplasia, cohorts of T/I (n = 19) and TIA mice (n = 41) were closely monitored between ages of 12 and 28 weeks and euthanized at age 28 weeks or when they met humane endpoints, whichever came first. Of the 19 T/I mice studied, eleven (58%) had colorectal neoplasia, with a mean of 2 neoplastic lesions/mouse with neoplasia (range = 1–4) (Fig 5). Of the 41 TIA mice studied, fourteen (34%) exhibited colorectal neoplasia (p = 0.10 vs. T/I, Fisher’s exact test), with a mean of 1 neoplastic lesion/mouse with neoplasia (range = 1–2; p = 0.06 vs. T/I) (Fig 5). However, since the TIA cohort was younger than the T/I cohort at the time of study (TIA = 19 ± 5 weeks vs T/I = 21 ± 3 weeks; p = 0.03), it was important to rule out that the trends toward decreased incidence and multiplicity of neoplasia in TIA mice were due to insufficient time for neoplasia to become evident before euthanasia was required. Multivariable analysis that adjusted for age when neoplasia was ascertained showed a significant decrease in the numbers of neoplastic lesions in TIA compared with T/I mice (p = 0.017; Poisson regression analysis).
Fig 5. Incidence and multiplicity of colonic neoplasia in T/I versus TIA mice.
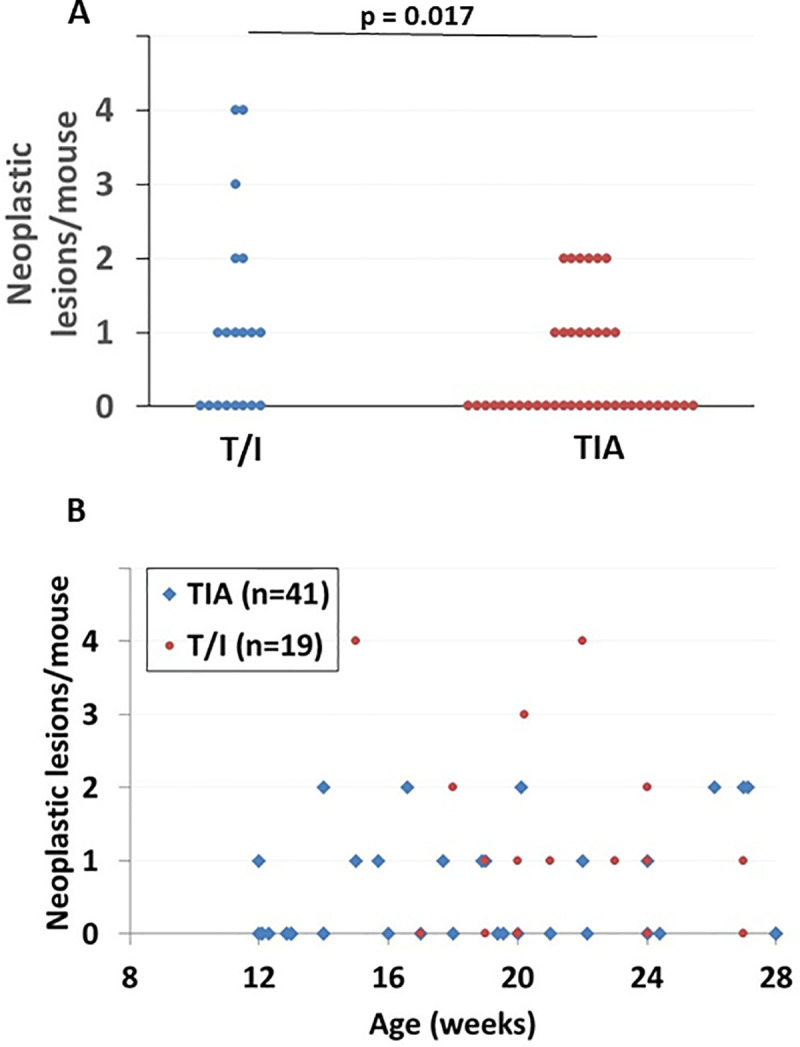
A. The number of tumors per mouse is shown for T/I (n = 19) and TIA mice (n = 41). Each point represents a single mouse. The p value shown is the result of a multivariable analysis that adjusted for age when neoplasia was ascertained (Poisson regression analysis). B. The number of tumors per mouse is shown as a function of age at euthanasia for the same cohort illustrated in panel A. Each point represents a single mouse, however some points are not visible due to multiple mice with the same age and tumor count.
Neoplastic lesions in TIA mice ranged from gastrointestinal intraepithelial neoplasia to adenocarcinoma that invaded through the serosa (Fig 2B). No evidence of metastasis was found grossly in the liver or in any of the lymph nodes examined histologically. All TIA mice in this cohort demonstrated moderate to severe colitis (S4 Fig), with a trend toward higher histologic scores (mean ± SD = 48 ± 10; n = 41), compared with 41 ± 13 (n = 19) in the T/I group (p = 0.06).
Other histologic findings in TIA mice
In addition to colitis with or without colonic neoplasia as described above, TIA mice frequently exhibited grossly evident swelling at the gastroduodenal junction. Histologic examination of this area revealed a change in the epithelium of the duodenal surface mucosa and submucosal Brunner’s glands to resemble colonic epithelial cells (“colonic metaplasia”; Fig 6) in 42 of 66 evaluable TIA mice (64%). This metaplastic change was typically associated with acute and chronic mucosal inflammation that was limited to the short segment of duodenum. Random sampling showed no evidence of inflammation in the mid-jejunum of these animals. Inflammation was also occasionally seen in the gastric glands and in the lamina propria of the glandular stomach in a subset of TIA mice, with a histology that resembled the crypt abscesses seen in the colon (Fig 7). Mesenteric lymph nodes were often massively enlarged, consistent with the severe colon inflammation observed, but the protruding lymphoid follicular structures previously described to be present on the antemesenteric side of duodenal and jejunal segments in singly Aicda-deficient mice [26] were not typically seen along the small intestine of TIA mice.
Fig 6. Colonic metaplasia of Brunner’s glands and duodenal inflammation in TIA mice.
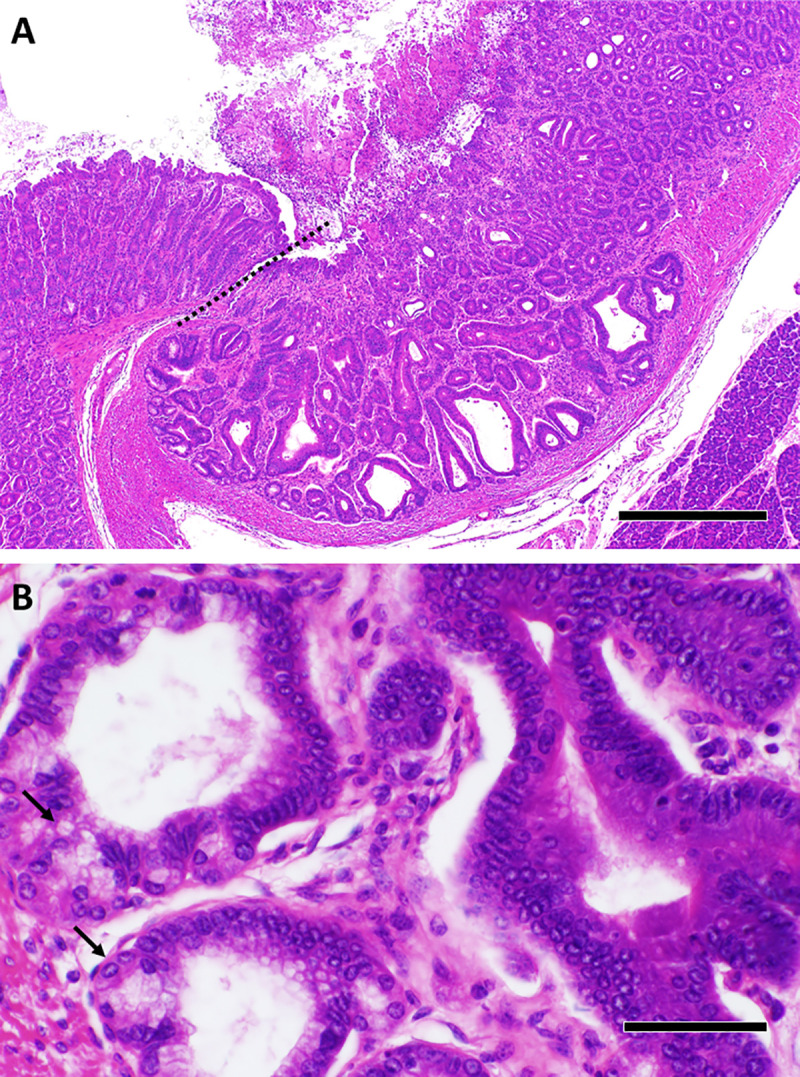
A. The pyloric region of the glandular stomach (left of dashed line) and the duodenum (right of dashed line) are shown. The duodenal mucosa demonstrates mucosal epithelium and Brunner’s glands with loss of villous architecture and histology similar to that seen in the colon (colonic metaplasia). B. A higher magnification views demonstrates the metaplastic epithelium and surrounding lamina propria and submucosa with marked chronic and active inflammation, similar to what was observed in the colon of these mice. Images shown are from a 9 week mouse, where some residual cells (arrows) exhibit morphology typical of Brunner’s glands. Older mice with such metaplasia typically lacked residual identifiable Brunner’s glands. Scale bar represents 500 μm in A and 50 μm in B.
Fig 7. Stomach inflammation in TIA mice.
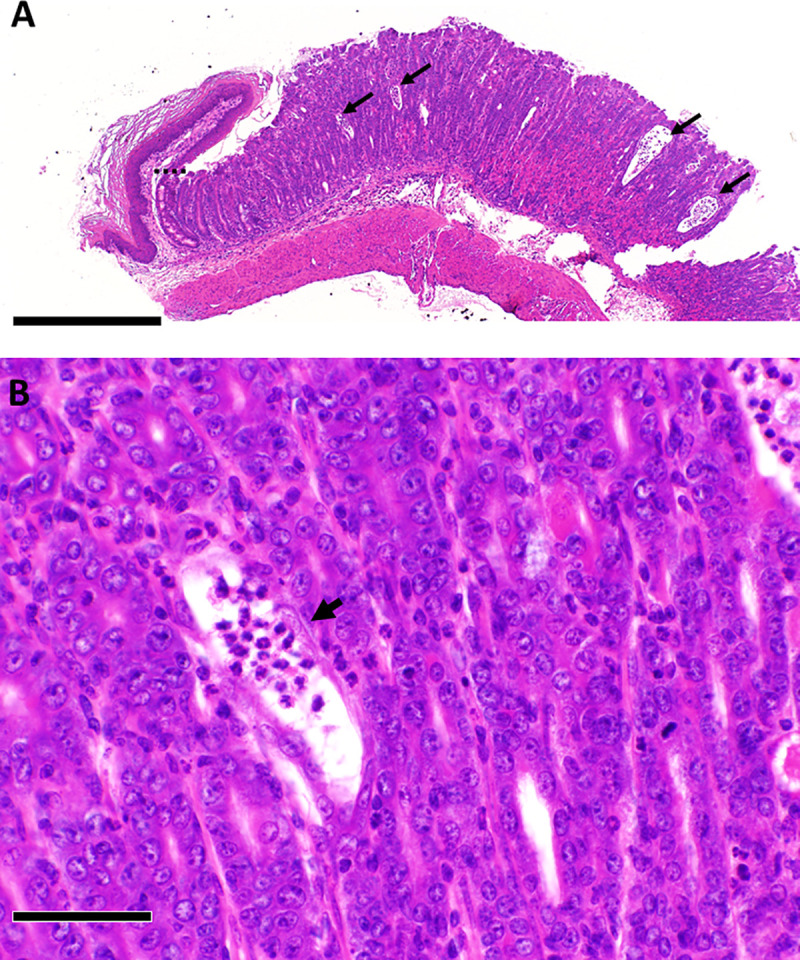
The dashed line in A represents the boundary between the forestomach and the glandular stomach. A higher magnification view in B shows inflamed gastric glands that resemble crypt abscesses, with thinning of glandular epithelium and neutrophils present within the dilated lumens, as indicated by arrows. Scale bar represents 500 μm in A and 50 μm in B.
Discussion
The studies reported here show that TIA mice that are triply deficient in Il10, Tnf, and the APOBEC family mutator enzyme Aicda develop severe UC-like colitis soon after weaning, similar to T/I mice that are deficient in Tnf and Il10 alone. Multivariable analysis that adjusted for age when neoplasia was ascertained showed that deletion of the Aicda gene decreased the development of inflammation-associated colorectal neoplasia, with decreased numbers of neoplastic colorectal lesions and a trend toward decreased incidence of neoplasia in TIA mice. Inflammation severity was statistically similar in T/I and TIA mice, with a trend toward slightly increased histologic scores in TIA mice. Serum biomarker analysis revealed that TIA mice had defective Ig class-switching that resulted in hyper-IgM syndrome, as well as increased serum IL1α and slightly decreased IL12(p40) and M-CSF compared with T/I mice. Taken together, these studies show that Aicda deficiency can decrease but is not sufficient to prevent inflammation-associated colon neoplasia in the setting of severe UC-like inflammation. The generally similar cytokine profiles in TIA and T/I mice, combined with similar histologic severity and extent of inflammation in the colon, strongly suggest that the decreased colorectal neoplasia observed in TIA relative to T/I mice is due to direct or indirect effects of Aicda deficiency that affect mechanisms of carcinogenesis, rather than simply affecting inflammation severity.
Previous research showed that the expression of the mutation-inducing enzyme encoded by Aicda is induced by colon inflammation and can enhance the acquisition of p53 mutations by colon epithelial cells [6, 19]. This is important since, in contrast to what is observed in sporadic colon cancers, p53 mutations occur early in UC, with higher frequencies in inflamed vs. non-inflamed regions of the colon [27, 28]. Consistent with the lack of early WNT-APC mutations that stimulate polypoid growth (reviewed in [27, 29]), the colorectal cancers that occur in IBD patients typically develop from non-polypoid (“flat”) mucosa and progress rapidly. Takai et al. previously showed that although Aicda deficiency did not affect inflammation severity based on cytokine production in Il10-/- mice, it decreased the frequency of nucleotide alterations in the Trp53 gene [19]. However, Aicda deficiency was not sufficient to prevent oncogenic mutagenesis in the setting of ongoing inflammation in their model, since although it decreased the percentage of Il10-/- mice that developed invasive colon cancers, it did not affect the percentage of Il10-/- mice with dysplastic precursor lesions such as adenomas at one year of age [19]. Similarly, we found that the severe inflammation present throughout the colon in our UC-like model continued to drive the development of neoplasia by 28 weeks of age or earlier, even in the setting of Aicda deficiency. The more limited anti-neoplastic effects of Aicda deficiency observed in our study compared with Takai et al [19] may reflect the increased proportion of colon epithelial cells that were exposed to inflammation-associated mutagenesis in our geographically continuous UC-like colitis models compared to the focal CD-like colon inflammation typically characteristic of mice deficient in IL10 alone. Other murine models of carcinogenesis, such as the azoxymethane plus dextran sulfate sodium model, may be useful for further elucidating mechanisms by which Aicda deficiency may decrease inflammation-associated colon carcinogenesis.
The absence of Aicda in all cells of the TIA mice we studied led to hyper-IgM syndrome, similar to when humans have bi-allelic lack-of-function mutations in AICDA [25]. Hyper-IgM syndrome is characterized by defective immunoglobulin class-switch recombination as well as defective somatic hypermutation of B cell antigen receptors. Interestingly, although the single knockout Aicda-/- mice housed in our specific pathogen-free facility showed no evidence of colitis, IBD has been reported in humans with AICDA mutations [30]. Aicda-/- and Aicda-mutant mice were previously shown to develop grossly evident hyperplasia of small intestinal lymphoid follicles, along with expansion of microbiota in the small intestine [26, 31]. However, in our study, mice singly deficient in Aicda showed no evidence of small intestinal lymphoid follicular hyperplasia and minimal to no colitis. This lack of phenotype may be due to the relative cleanliness of our specific pathogen-free environment (S1 Table), which we have also shown to be sufficient to prevent the development of colitis in mice deficient in Il10-/- alone, in the absence of specific triggering [13, 32]. TIA mice demonstrated enlarged mesenteric lymph nodes consistent with their severe colitis, but did not exhibit the small intestinal follicular hyperplasia reported by Fagarson et al [26]. Instead, many TIA mice exhibited colonic metaplasia in the duodenum that often developed inflammation similar to what was seen in the colon of these mice. The mechanisms underlying this pathology will require additional study, but may include local changes in abundance and type of microbiota, changes in gut permeability related to lack of IgG or IgA, the lack of somatic hypermutation to modify antibody repertoire to shape the relative composition and distribution of microbiota, systemic invasion of gut microbes, and/or other mechanisms. However, the generally similar severity of colitis in T/I and TIA mice, as evidenced by similar histologic scores of inflammation severity and similar levels of key pro-inflammatory cytokines and chemokines definitively shows that the abilities to carry out somatic hypermutation and immunoglobulin class-switching to produce IgG and IgA do not protect against the development of colitis in the setting of TNF and IL10 deficiency.
Although most aspects of inflammation seen in T/I and TIA mice very closely resemble those seen in human UC, the colonic metaplasia of Brunner’s glands and the duodenal inflammation we observed have not previously been reported in humans with UC. Additional studies will be required to fully assess the importance of these observations and the responsible mechanisms. The squamous metaplasia that was frequently observed in the rectum of TIA mice may be related to their lack of TNF, since squamous metaplasia of the rectum has also been observed in other strains of mice that are genetically TNF-deficient [20] as well as in wild type mice that were treated with an inhibitor of TNF transcription [33].
Since the experiments reported here used mice globally deficient in Aicda, it is not possible to determine whether the phenotypic differences between T/I and TIA mice are directly caused by AID deficiency in the gut epithelium, are a consequence of inefficient humoral adaptive immune system due to the effects of AID deficiency on B cells that severely alter immunoglubulin levels and/or repertoires in both the Peyer's patches and the lumen of the gut, and/or are due to other mechanisms. Follow-up studies using mice with targeted tissue-specific knockout of Aicda will be required to address these issues. Determining which cells must be AID-deficient to decrease the frequency of inflammation-associated colorectal neoplasia has important clinical implications, since this may allow the development of therapeutic interventions that decrease the incidence of colorectal neoplasia without causing undesirable changes in serum antibodies and gut microbiota.
In summary, the studies reported here show that Aicda deficiency can decrease, but is not sufficient to prevent, the risk of inflammation-associated colorectal neoplasia in the setting of severe UC-like inflammation. The TIA model may be useful for assessing the roles of antibody class-switch recombination deficiency and somatic hypermutation on regulation of microbiota in the small intestine and colon, as well as the pathogenesis of colitis associated with hyper-IgM syndrome in humans. Further studies will be required to determine the mechanisms that drive early mortality in TIA mice.
Supporting information
Fields shown are from the terminal colon/rectum (A), distal colon (B), and mid-colon (C). Scale bar represents 100 μm.
(TIF)
The severity of inflammation was scored as previously described [15], using a scale that takes into account mucosal architectural changes (M), degree of inflammation (I), and percentage of each bowel segment affected by any and severe changes (E1, E2). The bars indicate the mean ± SD score for the 5 colon segments examined in each mouse: terminal colon/rectum, distal colon, mid-colon, proximal colon, and cecum. The scores for each segment are summed to provide the overall histologic score of 49 ± 9 observed for TIA mice (n = 77), as described in Results. For the M score, 0 = no significant lesions, 1 = mild epithelial hyperplasia, 2 = moderate epithelial hyperplasia, and 3 = severe epithelial hyperplasia, with crypt branching or herniation. For the I score, 0 = no inflammation, 1 = mild inflammation limited to the mucosa, 2 = moderate inflammation present in mucosa and submucosa, 3 = severe inflammation with obliteration of normal architecture, erosions, and/or crypt abscesses, and 4 = level 3 changes plus ulceration. The E1 score is derived from the percent of the segment affected in any manner. The E2 score is derived the percent of the segment with level 3 or 4 changes. For the E1 and E2 scores, 1 = <5% of segment affected, 2 = 5–30% of segment affected, 3 = 31–60% of segment affected, and 4 = >60% of segment affected. Since the total histologic score is derived from summing (M + I + E1 +E2) scores from the 5 segments examined, the maximum score is 75.
(TIF)
T/I mice generally developed moderate to severe mucosal inflammation involving cecum to rectum (“pan-colitis”) soon after weaning, as described in [20]. The representative fields shown are from the terminal colon/rectum (A), distal colon (B), and mid-colon (C). Marked epithelial hyperplasia is present and the lamina propria is packed with inflammatory cells. Representative crypt abscesses are indicated by arrows. Scale bar represents 250 μm in panels A and B and 500 μm in panel C.
(TIF)
Graph shows histologic scores for T/I (n = 19) and TIA mice (n = 41) between the ages of 12 and 28 weeks who were euthanized for determination of neoplasia, either when they met humane endpoints or at the experimental endpoint of 28 weeks. Each point represents a single mouse. p = 0.06 (Student’s t-test).
(TIF)
(DOCX)
Acknowledgments
The author would like to thank Paula Greer and Drs. Eniko Nagy and Amelia Karlsson for developing and maintaining the colonies of mice used in these studies, and Drs. Andrew Macintyre and Ivo Shterev for assistance with data analysis.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was supported by the United States National Institutes of Health (http://www.nih.gov), grant number CA-170069 to LPH. The funder had no role in the study design, data collection and analysis, decision to publish, or preparation of this manuscript.
References
- 1.Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. New Engl J Med. 1990;323: 1228–1233. 10.1056/NEJM199011013231802 [DOI] [PubMed] [Google Scholar]
- 2.Munkholm P. Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment Pharmacol Ther. 2003;18: 1–5. [DOI] [PubMed] [Google Scholar]
- 3.Nordenholtz KE, Stowe SP, Stormont JM, Stowe MM, Chessin LN, Shah AN, et al. The cause of death in inflammatory bowel disease: a comparison of death certificates and hospital charts in Rochester, New York. Am J Gastroenterol. 1995;90: 927–932. [PubMed] [Google Scholar]
- 4.Soderlund S, Brandt L, Lapidus A, Karlen P, Brostrom O, Lofberg R, et al. Gastroenterol. 2009;136: 1561–1567. [DOI] [PubMed] [Google Scholar]
- 5.Upton DC, Gregory BL, Arya R, Unniraman S. AID: a riddle wrapped in a mystery inside an enigma. Immunol Res. 2011;49: 14–24. 10.1007/s12026-010-8190-x [DOI] [PubMed] [Google Scholar]
- 6.Endo Y, Marusawa H, Kou T, Nakase H, Fuji S, Fujimori T, et al. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterol. 2008;135: 889–898. [DOI] [PubMed] [Google Scholar]
- 7.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature 2010;463: 1042–1047. 10.1038/nature08752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nabel CS, Kohli RM. Demystifying DNA methylation. Science 2011;333: 1229–1230. 10.1126/science.1211917 [DOI] [PubMed] [Google Scholar]
- 9.Glocker E-O, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. New Engl J Med. 2009;361: 2033–2045. 10.1056/NEJMoa0907206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franke A, Balschun T, Karlsen TH, Sventoraityte J, Nikolaus S, Mayr G, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nature Genet. 2008;40: 1319–1323. 10.1038/ng.221 [DOI] [PubMed] [Google Scholar]
- 11.Imielinski M, Baldassano RN, Griffiths A, Russell RK, Annese V, Dubinsky M, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nature Genet. 2009;41: 1335–1340. 10.1038/ng.489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hale LP, Perrera D, Gottfried MR, Maggio-Price L. Srinivasan S, Marchuk DA. Neonatal infection with Helicobacter species markedly accelerates the development of inflammation-associated colonic neoplasia in IL-10-/- mice. Helicobacter 2007;12: 598–604. 10.1111/j.1523-5378.2007.00552.x [DOI] [PubMed] [Google Scholar]
- 13.Chichlowski M, Sharp JM, Vanderford DA, Myles MH, Hale LP. Helicobacter typhlonius and H. rodentium differentially affect the severity of colon inflammation and inflammation-associated neoplasia in IL-10-deficient mice. Comp Med. 2008;58: 534–541. [PMC free article] [PubMed] [Google Scholar]
- 14.Berg DJ, Zhang J, Weinstock JV, Ismail HF, Earle KA, Alila H, et al. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterol. 2002;123: 1527–1542. [DOI] [PubMed] [Google Scholar]
- 15.Hale LP, Greer PK, Trinh CT, Gottfried MR. Treatment with oral bromelain decreases colonic inflammation in the IL-10-deficient murine model of inflammatory bowel disease. Clin Immunol. 2005;116: 135–142. 10.1016/j.clim.2005.04.011 [DOI] [PubMed] [Google Scholar]
- 16.Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4+ Th1-like responses. J Clin Invest. 1996;98: 1010–1020. 10.1172/JCI118861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sturlan S, Oberhuber G, Beinhauer BG, Tichy B, Kappel S, Wang J, et al. Interleukin-10-deficient mice and inflammatory bowel disease associated cancer development. Carcinogenesis 2001;22: 665–671. 10.1093/carcin/22.4.665 [DOI] [PubMed] [Google Scholar]
- 18.Hale LP, Chichlowski M, Trinh CT, Greer PK. Dietary supplementation with fresh pineapple juice decreases inflammation and colonic neoplasia in IL-10-deficient mice with colitis. Inflamm Bowel Dis. 2010;16: 2012–2021. 10.1002/ibd.21320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takai A, Marusawa H, Minaki Y, Watanabe T, Nakase H, Kinoshita K, et al. Targeting activation-induced cytidine deaminase prevents colon cancer development despite persistent colonic inflammation. Oncogene 2012;31: 1733–1742. 10.1038/onc.2011.352 [DOI] [PubMed] [Google Scholar]
- 20.Hale LP, Greer PK. A novel murine model of inflammatory bowel disease and inflammation-associated colon cancer with ulcerative colitis-like features. PLOS One 2012;7: e41797 10.1371/journal.pone.0041797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102: 553–563. 10.1016/s0092-8674(00)00078-7 [DOI] [PubMed] [Google Scholar]
- 22.Nagy E, Rodriguiz RM, Wetsel WC, MacIver NJ, Hale LP. Reproduction and growth in a murine model of early life-onset inflammatory bowel disease. PLOS One. 2016;11: e0152764 10.1371/journal.pone.0152764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langford DJ, Bailey AL, Chanda ML, Clarke SE, Drummond TE, Echols S, et al. Coding of facial expressions of pain in the laboratory mouse. Nature Methods 2010;7: 447–449. 10.1038/nmeth.1455 [DOI] [PubMed] [Google Scholar]
- 24.Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, et al. Pathology of mouse models of intestinal cancer: Consensus report and recommendations. Gastroenterol. 2003;124: 762–777 [DOI] [PubMed] [Google Scholar]
- 25.Imai K, Catalan N, Plebani A, Marodi L, Sanal O, Kumaki S, et al. Hyper-IgM syndrome type 4 with a B lymphocyte-intrinsic selective deficiency in Ig class-switch recombination. J Clin Invest. 2003:112: 136–142. 10.1172/JCI18161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fagarsan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science 2002;298: 1424–1427. 10.1126/science.1077336 [DOI] [PubMed] [Google Scholar]
- 27.Hussain SP, Amstad P, Raja K, Ambs S, Nagashima M, Bennett WP, et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res. 2000;60: 3333–3337. [PubMed] [Google Scholar]
- 28.Chen R, Rabinovitch PS, Crispin DA, Emond MJ, Koprowicz KM, Bronner MP, et al. DNA fingerprinting abnormalities can distinguish ulcerative colitis patients with dysplasia and cancer from those who are dysplasia/cancer-free. Amer J Pathol. 2003;162: 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. New Engl J Med. 2015;372: 1441–1452. 10.1056/NEJMra1403718 [DOI] [PubMed] [Google Scholar]
- 30.Quartier P, Bustamante J, Sanal O, Plebani A, Debre´ M, Deville A, et al. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to Activation-Induced Cytidine Deaminase deficiency. Clinical Immunol. 2004;110: 22–29. [DOI] [PubMed] [Google Scholar]
- 31.Wei M, Shinkura R, Doi Y, Maruya M, Fagarasan S, Honjo T. Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nature Immunol. 2011;12: 264–270. [DOI] [PubMed] [Google Scholar]
- 32.Chichlowski M, Westwood GS, Abraham SN, Hale LP. Role of mast cells in inflammatory bowel disease and inflammation-associated colorectal neoplasia. PLOS One 2010;5: e12220 10.1371/journal.pone.0012220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hale LP, Cianciolo G. Treatment of experimental colitis in mice with LMP-420, an inhibitor of TNF transcription. J Inflamm. 2008;5: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fields shown are from the terminal colon/rectum (A), distal colon (B), and mid-colon (C). Scale bar represents 100 μm.
(TIF)
The severity of inflammation was scored as previously described [15], using a scale that takes into account mucosal architectural changes (M), degree of inflammation (I), and percentage of each bowel segment affected by any and severe changes (E1, E2). The bars indicate the mean ± SD score for the 5 colon segments examined in each mouse: terminal colon/rectum, distal colon, mid-colon, proximal colon, and cecum. The scores for each segment are summed to provide the overall histologic score of 49 ± 9 observed for TIA mice (n = 77), as described in Results. For the M score, 0 = no significant lesions, 1 = mild epithelial hyperplasia, 2 = moderate epithelial hyperplasia, and 3 = severe epithelial hyperplasia, with crypt branching or herniation. For the I score, 0 = no inflammation, 1 = mild inflammation limited to the mucosa, 2 = moderate inflammation present in mucosa and submucosa, 3 = severe inflammation with obliteration of normal architecture, erosions, and/or crypt abscesses, and 4 = level 3 changes plus ulceration. The E1 score is derived from the percent of the segment affected in any manner. The E2 score is derived the percent of the segment with level 3 or 4 changes. For the E1 and E2 scores, 1 = <5% of segment affected, 2 = 5–30% of segment affected, 3 = 31–60% of segment affected, and 4 = >60% of segment affected. Since the total histologic score is derived from summing (M + I + E1 +E2) scores from the 5 segments examined, the maximum score is 75.
(TIF)
T/I mice generally developed moderate to severe mucosal inflammation involving cecum to rectum (“pan-colitis”) soon after weaning, as described in [20]. The representative fields shown are from the terminal colon/rectum (A), distal colon (B), and mid-colon (C). Marked epithelial hyperplasia is present and the lamina propria is packed with inflammatory cells. Representative crypt abscesses are indicated by arrows. Scale bar represents 250 μm in panels A and B and 500 μm in panel C.
(TIF)
Graph shows histologic scores for T/I (n = 19) and TIA mice (n = 41) between the ages of 12 and 28 weeks who were euthanized for determination of neoplasia, either when they met humane endpoints or at the experimental endpoint of 28 weeks. Each point represents a single mouse. p = 0.06 (Student’s t-test).
(TIF)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.