

Abstract

A general, scalable two-step regio- and diastereoselective method has been described for the synthesis of versatile alkaloid-type azetidines from simple building blocks with excellent overall yields. In the kinetically controlled reaction, only the formation of the strained four-membered ring can be achieved instead of the thermodynamically favorable five-membered rings under appropriate conditions. Remarkable functional group tolerance has also been demonstrated. In this paper, we give a new scope of Baldwin’s rules by density functional theory (DFT) calculations with an explicit solvent model, confirming the proposed reaction mechanisms and the role of kinetic controls in the stereochemical outcome of the reported transition-metal-free carbon–carbon bond formation reactions.
Introduction
Despite the irrefutable importance of azetidines as bioactive compounds and pharmaceutical building blocks, they have received moderate attention compared to larger-ring-sized pyrrolidines and piperidines.1−3 The most important strategy for the synthesis of saturated N-heterocycles relies on unimolecular cyclization reactions by nucleophilic substitution. This approach results in the efficient formation of three-, five-, and six-membered heterocycles but often fails to result in four-membered heterocycles. In general, azetidines are considered the most difficult of all to form.4 Azetidines have excellent physicochemical properties, bioavailability and metabolic stability.5−8 A wide variety of antibiotics,9−13 numerous anticancer agents14,15 containing azetidines, and other drug molecules have been developed over the last decade.13,16−21 They have a significant role as synthetic building blocks of foldamers,22,23N-heterocycles,24−28 and polymers.29 It has also been demonstrated, that the introduction of these strained rings improves enormously the fluorescent properties of rhodamines30−32 and coumarins33 and the efficacy of homogenous catalysts as ligands,34,35 which makes them an attractive and challenging synthetic topic for chemists nowadays. Compared to their widespread uses, there are relatively few synthetic methods available, although they cover a wide range of structural variability.3,36−38 In conclusion, there are no general methods for the synthesis of azetidines, with a wide variety of functional groups.39 Only a few methods for the preparation of druglike azetidines have been reported.40−46
2-Arylazetidines also have a huge potential in synthetic and medicinal chemistry47−51 (Scheme 1), even though no general synthetic method is known. Only a few approaches have been developed for the synthesis of 2-phenylazetidines. N-Protected 2-arylazetidines can be synthesized by stereospecific cross-coupling reactions,52 by selective intermolecular sp3-C–H amination,53 or by [2+2] photocycloaddition.1 A few efficient synthetic methods of diversely substituted N-aryl-2-cyanoazetidines have also been published, based on an anionic ring-closure reaction, which requires the presence of an electron-withdrawing group (EWG) in the starting material.54 A similar reaction promoted by an uncommonly used base has been reported by our group few years ago.55
Scheme 1. Chemical Skeleton of 2-Arylazetidine-3-ylmethanol Derivatives (1) and Selected Examples for Bioactive 2-Arylazetidines.

(LG: leaving group.).
Our goal was to enrich the available chemical library with trans-3-(hydroxymethyl)-2-arylazetidines (1 in Scheme 1) for fragment-based drug discovery by a novel and versatile chemical synthesis from simple and readily available oxiranylmethyl-substituted benzylamines. This method is inspired by and designed according to Baldwin’s rules.56 While some theoretical explanation has been published in the last decade,57−59 the quantum chemical background has not been established to date.
Results and Discussion
Our synthetic method to prepare 2-arylazetidines 1 consists of only two or four simple synthetic steps from commercially available starting materials (substituted oxiranes and N-alkyl-benzylamines), providing a large range for functionalization. The key intermediates 2 were synthesized from N-substituted benzylamines (3) and epichlorohydrin (4) or readily prepared 2-substituted-(oxiran-2-yl)methyl 4-toluenesulfonates (5–7) (Scheme 2). As was demonstrated earlier, these versatile epoxide intermediates could be synthesized easily in a two-step procedure from commercially available allyl alcohol derivatives (8).60,61
Scheme 2. Synthesis and isolated Yields of the Key Intermediate Oxiranylmethyl-benzylamine Derivatives (2) from Epichlorohydrine (4) or Tosylates (5–7).

The key intermediates 2 (0.5 mmol) were treated with the mixture of lithium diisopropylamide and potassium tert-butoxide (LiDA-KOR superbase) in tetrahydrofuran at −78 °C. The four-membered ring was formed regio- and diastereoselectively.55 Noteworthily, compound 2 reacted exclusively on its benzylic position (Scheme 3), excluding the formation of alternative cyclic products. Spectroscopic investigations (J in 1H NMR, ROESY, NOESY) of products confirmed that the substituents were situated in a trans geometry around the azetidine ring in positions 2 and 3.The application of other bases did not yield product 1. To test the feasibility of the reaction, the metalation of 2d (in structure 2 R1 = Pr, R2 = H, R3 = Me, R4 = H) was investigated by different types of organometallic bases used in analogue cases under similar conditions.62 Using 3 equiv of lithium diisopropyl amide (LDA) or lithium 2,2,6,6-tetramethyl piperidide (LiTMP) or KHMDS (potassium hexamethyldisilazide), only unreacted 2d oxirane was isolated. When the reaction was performed by BuLi or LiC-KOR (a mixture of butyl lithium and potassium tert-butoxide), a complex mixture was obtained with low conversion and azetidine 1d was detected only in traces.
Scheme 3. Scope of the LiDA-KOR-Induced Azetidine Formation.

Isolated yields of the products are also given. See Scheme 4.
Other products were also formed; see the Experimental Section.
This reaction was proved to be a scalable process as 1k was prepared in 20 mmol scale with similar yields of the 0.5 mmol reaction.
In general, the isolated yields varied from moderate to good values with rather a good substituent tolerance, except a few examples. Compared to alternate literature protocols, this two-step synthetic procedure has significantly higher overall production and efficacy. In the cases when the R1/R2 group was H, Pr, or Ph, the azetidine products were obtained in moderate to good yields without a significant appearance of any side product.
In contrast, the expected yields were slightly reduced for R2 = CH2OTrt products, as some unidentified side products were formed in small amounts.
Various R3 alkyl groups typically do not influence the good yields of the reactions. However, benzyl substitution allows the formation of certain byproducts through alternative deprotonation, lowering the overall yields. In the case of the product 1g, two diastereomers formed in 5/1 ratio (liquid chromatography–mass spectrometry, LC–MS), which were separated by common column chromatography. The N-boc protection at the N atom allows the parallel deprotonation at the C atom in ca. 1/1 ratio and the formation of an allylic-type byproduct (9, Scheme 4), which competes with the main ring-opening mechanism.
Scheme 4. Formation of the Allylic Side Product (9) in the Case of N-Boc Protection of the Amine.

The electron donating group (EDG) substituents (tBu, OMe) and F at the Ph ring (R4) had a beneficial effect on the yields of the reaction. In contrast, the strong EWG CF3 group at para and ortho positions (see Supporting Information (SI)) could stabilize the benzyl anion and decreased its nucleophilic character to inhibit the ring opening. Benzylamine moiety-containing bicyclic compounds were also tested. The N-oxiranylmethyl isoindole did not afford the desired fused heterocyclic product under the conditions applied. However, it should be emphasized that the reactions of the very similar tetrahydroisoquinoline derivatives have different routes, yielding completely different bridged heterocyclic systems.63
Reaction Mechanism Study
It is well known for ring-closure reactions that generally five-membered heterocycles are formed more commonly than four-membered products. In general, the relative reaction rate of cyclization steps can be 2 orders of magnitude larger for five-membered rings.4
Theoretical calculations were performed at the M06-2X/6-31G(d,p) level of theory64 with the implementation of an implicit-explicit solvation model65 (ε = 12.2) by G16.66 The explicit model67 includes one Li+ ion and one K+ ion, bound to the O– and the Ph ring, respectively, as shown in Figure 1. Moreover, both cations were solvated by two explicit THF molecules each, to mimic the surrounding media in the best way. The calculated values are given in the SI.
Figure 1.
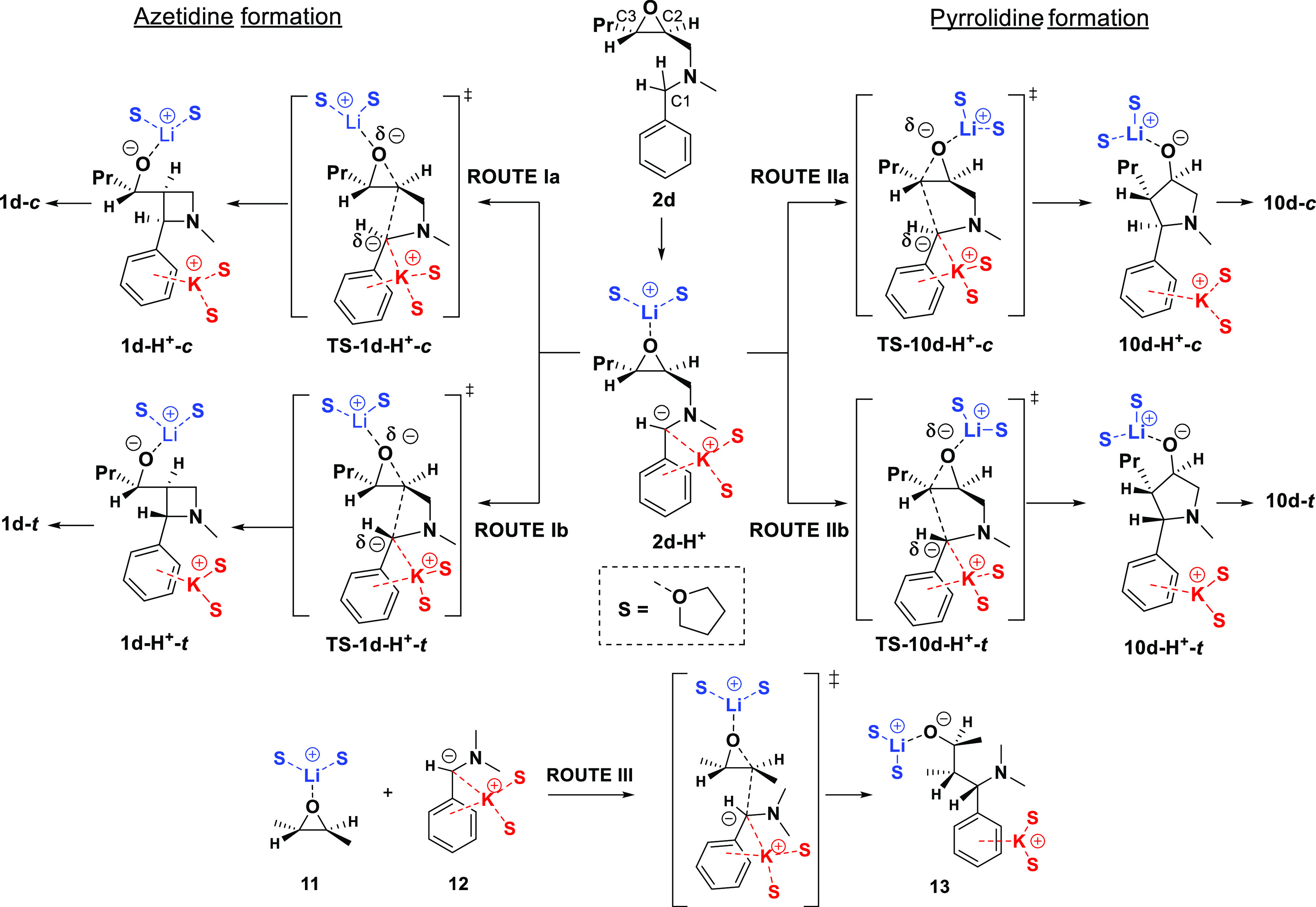
Conversion of the epoxide (2d-H+) into the four possible products (1d and 10d) via the four TSs (above), together with the bimolecular reaction (below). For the corresponding thermodynamic values, see Table 1.
From a thermodynamic point of view, there are five deprotonation sites at compound 2. However, the enthalpically and kinetically more favorable deprotonation was found at the benzylic position (C1), in agreement with the experiment. During these reaction mechanism studies, we focused only on the ring-closure steps from the metalation, leading to products for one selected molecule as an example (2d → 1d), including all of the possible routes (Ia, Ib, IIa, and IIb, see Table 1 and Figure 1). The ring closure (Figure 1) of the metalated benzylaminomethyloxirane 2d-H+ may theoretically result in both pyrrolidine (10d-H+-c, 10d-H+-t) and azetidine products (1d-c, 1d-t), but in our experiments, the trans four-membered heterocycle was formed regio- (up to 90%) and diastereoselectively.
Table 1. Conversion of the Epoxides (2d-H+) into the Four Possible Products (1d and 10d, t = trans, c = cis) and Their Reaction Enthalpy (ΔH), Gibbs Free Energy (ΔG) in kJ mol–1, and Entropy (ΔS) in J mol–1 K–1 a.
| TS |
product-H+ |
|||||
|---|---|---|---|---|---|---|
| 2d-H+→ | ΔH‡ | ΔG‡ | ΔS‡ | ΔH | ΔG | ΔS |
| 1d-H+-t | 50.5 | 58.0 | –25.2 | –160.2 | –152.4 | –26.1 |
| 1d-H+-c | 65.6 | 67.7 | –6.8 | –128.8 | –120.8 | –27.1 |
| 10d-H+-t | 70.8 | 79.4 | –28.7 | –267.1 | –245.1 | –73.6 |
| 10d-H+-c | 77.1 | 80.8 | –12.4 | –215.3 | –205.9 | –31.8 |
| TS-13 | 13 | |||||
| 11 → 13 | 60.2 | 70.8 | –72.1 | –197.0 | –167.4 | –99.3 |
The resulting negatively charged nucleophilic carbon atom (C1) can react with both electrophilic carbon atoms in the oxirane (C2 and C3) from two faces (routes Ia, Ib, IIa, and IIb), as shown in Figure 1, leading to the two cis–trans product pairs (1d-c/1d-t and 10d-c/10d-t), allowed by Baldwin’s rules. In terms of the calculated thermodynamic stability of the resulting products, the two five-membered pyrrolidine derivatives are more stable than the more stressed four-membered azetidine derivatives, regardless of their cis (10d-c → 1d-c: 80.2 kJ mol–1) or trans (10d-t → 1d-t: 63.0 kJ mol–1) arrangements. In general, the trans geometries are always more stable than cis (1d-c → 1d-t: 25.3 kJ mol–1; 10d-c → 10d-t: 8.1 kJ mol–1), which is due to the larger steric hindrance of the neighboring substituents (Table 1). Since the isolated products were azetidine derivatives in all of the cases, these thermodynamic data allowed us to conclude that the reaction was controlled kinetically.
In the next section, we sought to answer the question of why the formation of the thermodynamically unfavorable four-membered azetidines is more advantageous, in agreement with Baldwin’s rules. The two lowest TSs undoubtedly belong to the formation of the two azetidines 1d-H+-t and 1d-H+-c, preferring the formation of trans products, in contrast to the formation of pyrrolidines (10d-H+-c, 10d-H+-t). The enthalpy (ΔH‡) and Gibbs free energy (ΔG‡) difference between the two lowest gaps is only about 10 kJ mol–1. Due to the low reaction temperature applied, this difference provides sufficient diastereoselectivity under kinetic control, in good agreement with the experiments.
Although the computed TS values confirm the experimental findings, they did not give a deep explanation for the exclusive formation of the azetidine derivatives. Moreover, Baldwin’s rules also provide only a superficial and phenomenological interpretation. To reveal more details about the mechanism, a bimolecular model reaction ((11+12) → TS-13 → 13, route III in Figure 1) was also carried out under the same condition as a concerted SN2-type reaction, which mimics a nonrestricted alternative of the previous reaction (Figure 1). Here, the deprotonated dimethyl benzylamide anion (12) reacts with trans oxiranes (11). Surprisingly, the computed activation parameters (ΔH‡ and ΔG‡) are significantly higher compared to those in the formation of the stretched four-membered product and somewhat lower than in the case of the five-membered product. On this basis, we suppose that the azetidine formation is superbeneficial.
In the ideal transition state for SN2 reactions, the direction of the electrophile attack should be around 180° with the central atom and the leaving group, which provides the highest overlap between the nucleophilic highest occupied molecular orbital (HOMO) and the electrophilic lowest unoccupied molecular orbital (LUMO). In this ring-closure reaction, the central atom (C2 or C3), the attacking carbanion (C1), and the leaving oxygen atom (O) cannot be in this optimal linear arrangement in either case due to the bent bond angles in the oxirane-ring.
Analyzing the geometries of the transition states leading to the five- (TS-10d-H+-c and TS-10d-H+-t) and four-membered rings (TS-1d-H+-c, TS-1d-H+-t; Figure 2), as well as the TS-13, we found that the angle of the attack enclosed by the C1–C2–O atoms is most favorable for the formation of the four-membered trans and cis isomers (trans: 166.2° and cis: 163.9°) than five-membered cases (C–C3–O, trans: 133.2°, cis: 127.5°). Moreover, the nonrestricted model also exhibits lower angles than TS-1d-H+ (150.6°). The special preferred arrangement of the C1–C2–O in the TS-1d-H+ is due to the stretched double bicyclic transition structure, which provides the optimal arrangement for the orbital overlap and can be illustrated by the Δα value (=180° – α, blue angles in Figures 2 and 3). In the case of route II, pyrrolidine is already distorted in the opposite direction, thereby significantly reducing the overlap.
Figure 2.
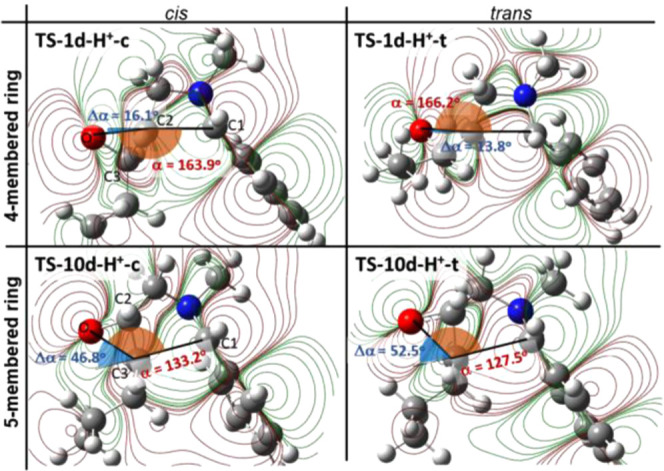
Transition states (TS-1d-H+ and TS-10d-H+) of the four- and five-membered rings (without explicit ions and molecules), representing their HOMOs leading to the azetidine (1d) and pyrrolidine derivatives (10d) derived from the oxirane derivative 2d-H+. Black lines show the bond angles between the C1–C2–O and C1–C3–O atoms, corresponding to α (orange). Δα = 180° – α (blue). The carbon, hydrogen, nitrogen, and oxygen atoms are marked in gray, white, blue, and red, respectively.
Figure 3.
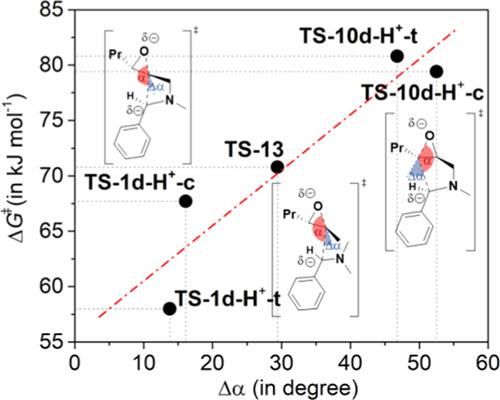
Correlation between the Δα (Δα = 180° – α.) (blue) of TS-1d-H+, TS-10d-H+, and TS-13 and the related ΔG‡ values.
This overlap between the HOMO of 12 and the LUMO of 13 is significantly less than that of the TS-1-H+, thanks to the dominance of the steric hindrance in the absence of the supporting ring stretch. Consequently, this may be the reason for the lowest activation ΔG‡ of the four-membered trans azetidine derivative.
Conclusions
In summary, an efficient, scalable, and stereo- and diastereo-specific method was developed for the preparation of 2-arylazetidines (1), using strong alkali amide-type bases. The results of the quantum chemical investigation of the mechanism are consistent with the experimental findings and shed light on the details of the regio- and stereoselectivity of the azetidine formation reaction. Thus, we were able to find the quantum mechanical and structural explanation of Baldwin’s rules for the ring opening of oxiranes, controlled by the balance between the ring stretch and overlap.
Experimental Section
General Remarks
All commercial starting materials were purchased from Sigma-Aldrich Kft., Hungary, and were used without further purification. All organometallic reactions were conducted under a dry nitrogen atmosphere using the Schlenk-technique. Solvents were freshly distilled and dried over molecular sieves.
1H NMR and 13C {1H} NMR spectra were recorded at 500/300 and 126/75 MHz on Bruker Avance 500 or 300 spectrometers. All 1H NMR and 13C chemical shifts were referenced to the tetramethylsilane (TMS). All chemical shifts are quoted in parts per million (ppm), measured from the center of the signal, except in the case of multiplets, which are quoted as a range. Coupling constants are quoted to the nearest 0.1 Hz. Splitting patterns are abbreviated as follows: singlet (s), doublet (d), triplet (t), quartet (q), quintet (quin), sextet (sxt), multiplet (m), broad singlet (br. s), and combinations thereof. The assignment of spectra was aided by DEPT 135 and 1D (NOESY) and 2D NMR spectroscopy (NOESY, ROESY, HSQC). For the assignment, see SI NMR spectra.
HRMS-EI+ data were obtained using either electrospray ionization (ESI) or electron impact (EI) techniques. High-resolution ESI analyses were performed on an Agilent 6230 TOF LC/MS spectrometer (ion trap; analyzed using Excalibur). High-resolution EI analysis was performed on an Autospec spectrometer (magnetic sector; analyzed using MassLynx).
Thin-layer chromatography (TLC) was performed on commercially available precoated TLC plates (Merck Silica gel 60 F254 aluminum sheets or Merck aluminum oxide 60 F254 plates). Visualization was achieved either under UV light at 254 nm or by exposure to iodine or the aqueous solution of (NH4)6Mo7O24, Ce(SO4)2, and sulfuric acid.
Flash column chromatography was performed by a CombiFlash Rf 150 (Teledyne ISCO) apparatus using gradient elution in normal (silica column; hexane–ethyl acetate as the eluent) phase mode. Gradient elution preparative high-performance liquid chromatography (HPLC) was performed (HPLC Gilson 333 instrument, UV detector 220 nm) on a Phenomenex Gemini C18 (250 mm × 50.00 mm; 10 μm, 110 A) column using 0.4 g of NH4HCO3 in 1 L of water and acetonitrile (A/B) or 10 mL of trifluoroacetic acid in 1 L of water and acetonitrile (C/B) as the two solvents.
General Procedures
General Procedure A for the Preparation of Trialkyl Amines from Epichlorohydrin
To a solution of amines (30.0 mmol, 1.0 equiv) in EtOH (4 mL) and water (2 mL) was added epichlorohydrin (30.0 mmol, 2.35 mL, 1.0 equiv) at 0 °C using an ice bath. The mixture was stirred for 5 h at room temperature and then cooled to 0 °C in an ice bath. Toluene (3 mL) and NaOH (0.054 mmol, 2.16 g) were added and then stirred at 25 °C for 16 h. The mixture was concentrated under reduced pressure, and then water (20.0 mL) was added to it. The organic compounds were extracted with dichloromethane (4 × 30 mL). The combined organic layers were washed with brine (10 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude mixture was purified by column chromatography.
General Procedure B for the Preparation of Trialkyl Amines from Tosylates
Tosylate (10.0 mmol, 1.0 equiv) was dissolved in dry N,N-dimethylformamide (DMF, 10 mL) under a dry nitrogen atmosphere, and potassium iodide (5.00 mmol, 0.5 equiv) was added into it. The solution was cooled to 0 °C in an ice bath, and the secondary amine (HNRR’, 21.00 mmol, 2.1 equiv) was added into the solution. The reaction mixture was stirred for 24 h at 40 °C and heated by an oil bath, and then it was poured into a mixture of ice (100 g), saturated sodium hydrogen carbonate solution (200 mL), and diethyl ether (50 mL). Then, the phases were separated and the aqueous mixture was extracted with diethyl ether (4 × 60 mL). The combined organic layers were washed with brine (50 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude mixture was purified by column chromatography.
General Procedure C for the Preparation of N-Boc Aminomethyloxiranes
Sodium hydride (55% in mineral oil, 1.8 mmol, 72.2 mg, 1.5 equiv) was washed with dry hexane (3 × 3 mL) under a dry nitrogen atmosphere. Then, it was dried in vacuo followed by the addition of dry DMF (2 mL) and cooled (0 °C in an ice bath). A dimethylformamide solution (7 mL) of N-boc-benzylamine derivative68 (1.20 mmol, 1.0 equiv) was added dropwise to the cold (0 °C) suspension of sodium hydride and DMF, and the mixture was stirred for 2 h at room temperature. The obtained yellowish solution was slowly added into a dry DMF solution (7 mL) of tosylate (1.2 mmol, 1.0 equiv) at 5 °C and cooled in an ice bath under a nitrogen atmosphere. The reaction mixture was stirred for 24 h, and then it was poured into a mixture of ice (15 g) and saturated sodium hydrogen carbonate solution (10 mL). The aqueous mixture was extracted with diethyl ether (5 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude mixture was purified by column chromatography.
General Procedure D for the Preparation of Azetidines via Superbase-Induced Reactions
Potassium tert-butoxide in tetrahydrofuran (THF, 1.0 mmol, 1 mL in 1 M THF solution) was cooled to −78 °C in a cold bath, using dry ice in acetone, in a Schlenk tube under a nitrogen atmosphere and diluted with 1 mL of absolute THF. Diisopropylamine (1.0 mmol, 0.10 g, 0.14 mL, 2.0 equiv) and a 1.59 M hexane solution of butyllithium (1.5 mmol, 0.94 mL, 3.0 equiv) were added dropwise into the solution. The reaction mixture was stirred for 20 min at −78 °C. Oxirane (2, 0.5 mmol, 1.0 equiv) in absolute THF (2 mL) was added dropwise, and the mixture was stirred at −78 °C for 2 h. Water (10.0 mL) and diethyl ether (5 mL) were added to the cold mixture, and then it was allowed to warm up to room temperature. The phases were separated, and the aqueous phase was extracted with diethyl ether (3 × 5 mL). The combined organic layers were washed with brine (1 × 10 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude mixture was purified by column chromatography or in some cases by preparative HPLC.
Synthesis of Benzylaminomethyl-oxiranes
N-Benzyl-N-methyl-1-(oxiran-2-yl)methanamine (2a)
It was prepared according to general procedure A using N-methylbenzylamine (30.0 mmol, 3.64 g, 3.87 mL) and epichlorohydrin (30.0 mmol, 2.35 mL). Purification by column chromatography on silica gel (10–40% EtOAc in hexane) afforded the title compound as a yellowish oil (2a, 3.16 g, 18.0 mmol, 60%). 1H NMR (CDCl3, 300 MHz) δH: 7.37–7.21 (5H, m, C[7,8,9]-H), 3.64 (1H, AB d, J = 13.1, Hz, C[5]-H), 3.50 (1H, AB d, J = 13.1, Hz, C[5]-H), 3.15–3.07 (1H, m, C[2]-H), 2.81–2.68 (2H, m, 2 × C[1]-H), 2.49–2.41 (1H, m, C[3]-H), 2.38–2.32 (4H, m, C[3]-H, 3 × C[4]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 138.7 (C[6]), 129.0 (C[7]), 128.3 (C[8]), 127.1 (C[9]), 62.6 (C[5]), 59.7 (C[3]), 50.8 (C[2]), 45.0 (C[1]), 42.9 (C[4]). HRMS (ESI) m/z: [M + H]+ calcd for C11H16NO 178.1226; found 178.1219.
N-Benzyl-N-ethyl-1-(oxiran-2-yl)methanamine (2b)
It was prepared according to general procedure A using N-ethylbenzylamine (30.0 mmol, 4.06 g, 4.46 mL) and epichlorohydrin (30.0 mmol, 2.35 mL). Purification by column chromatography on silica gel (0–30% EtOAc in hexane) afforded 2b as a yellowish oil (3.10 g, 16.2 mmol, 54%). 1H NMR (CDCl3, 300 MHz) δH: 7.37–7.21 (5H, m, C[8,9,10]-H), 3.76 (1H, AB d, J = 13.7, Hz, C[6]-H), 3.57 (1H, AB d, J = 13.7, Hz, C[6]-H), 3.09–3.00 (1H, m, C[2]-H), 2.81–2.52 (4H, m, 2 × C[1]-H, 2 × C[4]-H), 2.52–2.37 (2H, m, C[3]-H), 1.08 (3H, t, J = 7.1 Hz, 3 × C[5]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 139.5 (C[7]), 128.9 (C[8]), 128.2 (C[9]), 126.9 (C[10]), 58.5 (C[6]), 55.8 (C[3]), 51.1 (C[2]), 48.2 (C[4]), 45.2 (C[1]), 11.9 (C[5]). HRMS (ESI) m/z: [M + H]+ calcd for C12H18NO 192.1383; found 192.1382.
N,N-Dibenzyl-1-(oxiran-2-yl)methanamine (2c)
It was prepared according to general procedure A using dibenzylamine (30.0 mmol, 5.92 g, 5.77 mL) and epichlorohydrin (30.0 mmol, 2.35 mL). Purification by column chromatography on silica gel (0–15% EtOAc in hexane) afforded 2c as a yellowish oil (1.98 g, 7.80 mmol, 26%). 1H NMR (CDCl3, 300 MHz) δH: 7.38 (4H, d, J = 7.2 Hz, 4 × C[6]-H), 7.30 (4H, t, J = 7.2 Hz, 4 × C[7]-H), 7.26–7.20 (2H, m, Hz, 2 × C[8]-H), 3.80 (2H, AB d, J = 13.7, Hz, 2 × C[4]-H), 3.56 (1H, AB d, J = 13.7, Hz, 2 × C[4]-H), 3.06 (1H, td, J = 6.2 Hz, 3.4 Hz, C[2]-H), 2.76 (1H, dd, J = 13.8 Hz, 3.4 Hz, C[3]-H) 2.65 (1H, t, J = 4.5 Hz, C[1]-H), 2.46–2.37 (2H, m, C[1]-H, C[3]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 139.3 (C[5]), 128.8 (C[6]), 128.2 (C[7]), 126.9 (C[8]), 58.9 (C[4]), 55.8 (C[3]), 51.0 (C[2]), 45.0 (C[1]). HRMS (ESI) m/z: [M + H]+ calcd for C17H20NO 254.1539; found 254.1539.
N-Benzyl-N-methyl-1-(3-propyloxiran-2-yl)methanamine (2d)
It was prepared according to general procedure B using N-methylbenzylamine (16.6 mmol, 2.01 g, 2.15 mL) and (3-propyloxiran-2-yl)methyl 4-methylbenzenesulfonate (6.67 mmol, 1.80 g). Purification by column chromatography on silica gel (0–25% EtOAc in hexane) afforded the title compound (2d) as a yellowish oil (1.26 g, 5.75 mmol, 86%). 1H NMR (CDCl3, 300 MHz) δH: 7.34–7.24 (5H, m, C[7,8,9]-H), 3.63 (1H, AB d, J = 13.0 Hz, C[5]-H), 3.50 (1H, d, J = 13.0 Hz, C[5]-H), 2.91–2.85 (1H, m, C[2]-H), 2.71–2.64 (2H, m C[1]-H, C[3]-H), 2.40 (1H, AB dd, J = 13.2 Hz, 6.2 Hz, C[3]-H) 2.30 (3H, s, 3 × C[4]-H), 1.53–1.44 (4H, m, 2 × C[10]-H, 2 × C[11]-H), 0.95 (3H, t, J = 6.9 Hz, 3 × C[12]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 138.7 (C[6]), 129.0 (C[7]), 128.3 (C[8]), 127.1 (C[9]), 62.6 (C[5]), 59.3 (C[3]), 57.0 (C[1/2]), 56.8 (C[1/2]), 42.9 (C[4]), 33.9 (C[10]), 19.3 (C[11]), 13.9 (C[12]). HRMS (ESI) m/z: [M + H]+ calcd for C14H22NO 220.1701; found 220.1703.
N-Benzyl-N-ethyl-1-(3-propyloxiran-2-yl)methanamine (2e)
It was prepared according to general procedure B using N-ethylbenzylamine (3.88 mmol, 0.525 g, 0.57 mL) and (3-propyloxiran-2-yl)methyl 4-methylbenzenesulfonate (1.85 mmol, 0.500 g). Purification by column chromatography on silica gel (0–30% EtOAc in hexane) afforded the title compound as a yellowish oil (2e, 0.363 g, 1.55 mmol, 84%). 1H NMR (CDCl3, 300 MHz) δH: 7.36–7.22 (5H, m, C[8,9,10]-H), 3.73 (1H, AB d, J = 13.6 Hz, C[6]-H), 3.73 (1H, AB d, J = 13.6 Hz, C[6]-H), 2.86–2.80 (1H, m, C[2]-H), 2.72–2.46 (5H, m, C[1]-H, 2 × C[4]-H, 2 × C[6]-H) 1.52–1.39 (4H, m, 2 × C[11]-H, 2 × C[12]-H) 1.07 (3H, t, J = 7.2 Hz, 3 × C[5]-H), 0.94 (3H, t, J = 6.8 Hz, 2 × C[13]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 139.6 (C[7]), 128.9 (C[8]), 128.2 (C[9]), 126.9 (C[10]), 58.5 (C[6]), 57.3 (C[1/2]), 57.0 (C[1/2]), 55.4 (C[3]), 48.1 (C[4]), 33.9 (C[12]), 19.3 (C[13]), 13.9 (C[14]), 11.8 (C[5]). HRMS (ESI) m/z: [M + H]+ calcd for C15H24NO 234.1852; found 234.1861.
N,N-Dibenzyl-1-(3-propyloxiran-2-yl)methanamine (2f)
It was prepared according to general procedure B using dibenzylamine (3.88 mmol, 0.765 g, 0.745 mL) and (3-propyloxiran-2-yl)methyl 4-methylbenzenesulfonate (1.85 mmol, 0.500 g). Purification by column chromatography on silica gel (0–12% EtOAc in hexane) afforded the title compound as a yellowish oil (0.535 g, 1.81 mmol, 93%). 1H NMR (CDCl3, 300 MHz) δH: 7.38 (4H, d, J = 7.2 Hz, 4 × C[6]-H), 7.31 (4H, t, J = 7.2 Hz, 4 × C[7]-H), 7.26–7.19 (2H, m, Hz, 2 × C[8]-H), 3.76 (2H, AB d, J = 13.5 Hz, 2 × C[4]-H), 3.56 (2H, AB d, J = 13.5 Hz, 2 × C[4]-H), 2.86–2.84 (1H, m, C[1]-H), 2.68 (1H, dd, J = 13.5 Hz, 4.1 Hz, C[3]-H), 2.62 (1H, m, C[2]-H), 2.49 (1H, dd, J = 13.5 Hz, 5.7 Hz, C[3]-H), 1.44–1.42 (4H, m, 2 × C[9]-H, 2 × C[10]-H) 0.92 (3H, t, J = 7.2 Hz, 3 × C[11]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 139.4 C[5], 128.8 (C[6]), 128.2 (C[7]), 126.9 (C[8]), 58.9 (C[4]), 57.2 (C[1/2]), 56.8 (C[1/2]), 55.5 (C[3]), 33.8 (C[9]), 19.3 (C[10]), 13.9 (C[11]). HRMS (ESI) m/z: [M + H]+ calcd for C20H26NO 296.2009; found 296.2021.
(1R)-N-Benzyl-1-phenyl-N-((3-propyloxiran-2-yl)methyl)ethan-1-amine (2g)
It was prepared according to general procedure B using (R)-N-benzyl-1-phenylethan-1-amine (3.89 mmol, 0.822 g) and (3-propyloxiran-2-yl)methyl 4-methylbenzenesulfonate (1.85 mmol, 500 mg). Purification by column chromatography on silica gel (0–10% EtOAc in hexane) afforded 2g as a yellowish oil (0.430 g, 1.39 mmol, 75%). 1H NMR (CDCl3, 300 MHz) δH: 7.45–7.18 (10H, m, C[6-8, 12-14]-H), 4.00 (1H, td, J = 17.9 Hz, 67 Hz, C[9]-H NHCH3), 3.79–3.71 (1H, m, C[4]-H), 3.60–3.46 (1H, m, C[4]-H), 2.77–2.49 (4H, m, C[1]-H, C[2]-H, 2 × C[3]-H), 1.43–1.31 (7H, m, 3 × C[9]-H, 2 × C[15]-H, 2 × C[16]-H), 0.90 (3H, m, 3 × C[17]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 143.7, 142.9, 140.5, 140.4, 128.6, 128.2, 128.1, 127.9, 127.8, 126.8, 126.8, 126.7, 59.0, 58.4, 57.9, 57.7, 57.6, 57.1, 55.2, 55.2, 51.9, 51.7, 33.9, 19.3, 19.23, 16.0, 14.1, 13.9. HRMS (ESI) m/z: [M + H]+ calcd for C14H22NO 310.2165; found 310.2159.
tert-Butyl (4-Methoxybenzyl)((3-propyloxiran-2-yl)methyl)carbamate (2h)
It was prepared according to general procedure C using tert-butyl (4-methoxybenzyl)carbamate (1.85 mmol, 440 mg) and (3-propyloxiran-2-yl)methyl 4-methylbenzenesulfonate (1.85 mmol, 500 mg). Purification by column chromatography on Florisil (0–20% EtOAc in hexane) afforded the title compound as a yellowish oil (436 mg, 1.30 mmol, 70%). 1H NMR (CDCl3, 300 MHz) δH: 7.17 (2H, d, J = 7.2 Hz, 2 × C[9]-H), 6.85 (2H, d, J = 7.2 Hz, 2 × C[10]-H), 4.53 (1H, AB d, J = 15.0 Hz, C[7]-H), 4.37 (1H, d, J = 15.0 Hz, C[7]-H), 3.78 (3H, s, 3 × C[12]-H), 3.63–2.65 (4H, m, C[1]-H, C[2]-H, 2 × C[3]-H), 1.60–1.35 (13H, m, 9 × C[6]-H, 2 × C[13]-H, 2 × C[14]-H), 0.94 (3H, t, J = 6.8 Hz, 3 × C[15]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 158.9 (C[11]), 155.6 (C[4]), 130.3 (C[9]), 129.3 (C[8]), 128.8 (C[9]), 113.9 (C[10]), 80.0 (C[5]), 57.4 (C[1]), 56.8 (C[2]), 55.2 (C[12]), 50.8 (C[7]), 50.3 (C[7]), 47.8 (C[3]), 33.7 (C[13]), 28.5 (C[6]), 19.2 (C[14]), 13.9 (C[15]). HRMS (ESI+) for C19H29NNaO4 ([M + Na]+): calcd for 358.1989; found 358.1982. HRMS (ESI) m/z: [M + H – tBu]+ calcd for C15H22NO4 280.1543; found 280.1533.
tert-Butyl (4-Trifluoromethylbenzyl)((3-propyloxiran-2-yl)methyl)carbamate (2i)
It was prepared according to general procedure C using tert-butyl (4-(trifluoromethy)benzyl)carbamate (1.85 mmol, 510 mg) and (3-propyloxiran-2-yl)methyl 4-methylbenzenesulfonate (1.85 mmol, 500 mg). Purification by column chromatography on Florisil (0–10% EtOAc in hexane) afforded the title compound 2i as a yellowish oil (520 mg, 1.39 mmol, 76%). 1H NMR (CDCl3, 300 MHz) δH: 1H-NMR (CDCl3, 300 MHz) δH: 7.58 (2H, d, J = 7.8 Hz, 2 × C[9]-H), 7.41–7.30 (2H, m, 2 × C[10]-H), 4.66 (1H, AB d, J = 15.0 Hz, C[7]-H), 4.49 (1H, AB d, J = 15.0 Hz, C[7]-H), 3.82–2.66 (4H, m, C[1]-H, C[2]-H, 2 × C[3]-H), 1.55–1.40 (13H, m, 9 × C[6]-H, 2 × C[13]-H, 2 × C[14]-H), 0.94 (3H, t, J = 6.8 Hz, 3 × C[15]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 155.5 (C[4]), 142.6 (q, J = 1.8 Hz, C[9]), 130.0 (C[8]), 127.7 (q, J = 31 Hz, C[11]), 127.3, 125.5 (q, J = 3.6 Hz, C[10]), 129.9 (q, J = 273 Hz, C[12]), 80.6 (C[5]), 56.9 (C[1]), 56.7 (C[2]), 51.2 (C[7]), 50.7 (C[7]), 48.5 (C[3]), 33.6 (C[13]), 28.4 (C[6]), 19.2 (C[14]), 13.9 (C[15]). HRMS (ESI) m/z: [M + H – tBu]+ calcd for C15H19F3NO3 318.1312; found 318.1327.
tert-Butyl (4-tert-Butyl Benzyl)((3-propyloxiran-2-yl)methyl)carbamate (2j)
It was prepared according to general procedure C using tert-butyl (4-(tert-butyl)benzyl)carbamate (1.85 mmol, 490 mg) and (3-propyloxiran-2-yl)methyl 4-methylbenzenesulfonate (1.85 mmol, 500 mg). Purification by column chromatography on Florisil (0–20% EtOAc in hexane) afforded the title compound as a yellowish oil (540 mg, 1.49 mmol, 81%). 1H NMR (CDCl3, 300 MHz) δH: 1H-NMR (CDCl3, 300 MHz) δH: 7.33 (2H, d, J = 7.8 Hz, 2 × C[10]-H), 7.17 (2H, d, J = 7.5 Hz, 2 × C[9]-H), 4.59 (1H, AB d, J = 15.3 Hz, C[7]-H), 4.38 (1H, AB d, J = 15.3 Hz, C[7]-H), 3.56–2.63 (4H, m, C[1]-H, C[2]-H, 2 × C[3]-H), 1.53–1.38 (13H, m, 9 × C[13]-H, 2 × C[14]-H, 2 × C[15]-H), 1.31 (9H, s, 9 × C[6]-H), 0.93 (3H, t, J = 6.3 Hz, C[16]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 155.7 (C[4]), 150.2 (C[11]), 135.2 (C[8]), 127.6 (C[9]), 127.3 (C[9]), 125.4 (C[10]), 80.1 (C[5]), 57.6 (C[2]), 56.8 (C[1]), 57.0 (C[2]), 51.0 (C[7]), 50.6 (C[7]), 48.0 (C[3]), 34.5 (C[12]), 33.7 (C[14]), 31.4 (C[6]), 28.5 (C[13]), 19.2 (C[15]), 13.9 (C[16]). HRMS (ESI) m/z: [M + H – tBu]+ calcd for C18H28NO3 306.2064; found 306.2077.
N-Benzyl-N-methyl-1-(3-((trityloxy)methyl)oxiran-2-yl)methanamine (2k)
It was prepared according to general procedure B using N-benzylmethylamine (4.2 mmol, 0.509 g, 0.542 mL) and (3-((trityloxy)methyl)oxiran-2-yl)methyl 4-methylbenzenesulfonate (2.0 mmol, 1.00 g). Purification by column chromatography on silica gel (0–30% EtOAc in hexane) afforded the title compound as a yellowish oil (0.867 g, 1.93 mmol, 96%). 1H NMR (CDCl3, 300 MHz) δH: 7.44 (6H, d, J = 7.8 Hz, 6 × C[13]-H), 7.30–7.21 (14H, m, Ar-H), 3.55 (1H, AB d, J = 13.0 Hz, C[5]-H), 3.41 (1H, d, J = 13.0 Hz, C[5]-H), 3.32 (1H, dd, J = 10.5 Hz, 6.0 Hz, C[10]-H), 3.18 (2H, m, C[1]-H, C[2]-H), 3.10 (1H, dd, J = 10.5, 4.5 Hz, C[10]-H), 2.57 (1H, dd, J = 13.5, 3.5 Hz, C[3]-H), 2.21 (3H, s, 3 × C[4]-H), 2.18 (1H, dd, J = 13.5, 6.0 Hz, C[3]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 143.7 (C[12]), 138.5 (C[6]), 129.0 (C[13]), 128.6 (C[14]), 128.2 (C[7]), 127.9 (C[8]), 127.1 (C[15]), 127.1 (C[9]), 86.9 (C[11]), 62.4 (C[10]), 62.2 (C[5]), 55.4 (C[3]), 55.1 (C[1]), 54.2 (C[2]), 42.7 (C[4]). HRMS (ESI) m/z: [M + H]+ calcd for C31H32NO2 450.2433; found 450.2443.
N-Benzyl-N-ethyl-1-(3-((trityloxy)methyl)oxiran-2-yl)methanamine (2l)
It was prepared according to general procedure B using N-ethylbenzylamine (4.2 mmol, 0.568 g, 0.625 mL) and (3-((trityloxy)methyl)oxiran-2-yl)methyl 4-methylbenzenesulfonate (2.0 mmol, 1.00 g). Purification by column chromatography on silica gel (0–20% EtOAc in hexane) afforded the title compound as a yellowish oil (0.713 g, 1.54 mmol, 77%). 1H NMR (CDCl3, 300 MHz) δH: 7.44 (6H, d, J = 7.8 Hz, 6 × C[14]-H), 7.30–7.20 (14H, m, Ar-H), 3.67 (1H, AB d, J = 13.0 Hz, C[6]-H), 3.49 (1H, d, J = 13.0 Hz, C[6]-H), 3.29 (1H, dd, J = 10.5 Hz, 6.0 Hz, C[11]-H), 3.18–3.10 (2H, m, C[1]-H, C[2]-H), 3.08 (1H, dd, J = 10.5, 4.5 Hz, C[11]-H), 2.61–2.54 (2H, m, C[3]-H, C[4]-H), 2.52–2.45 (1H, m, C[4]-H), 2.30 (1H, dd, J = 13.5, 6.0 Hz, C[3]-H), 0.98 (3H, t, J = 7.2 Hz, 3 × C[5]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 143.8 (C[13]), 139.4 (C[7]), 128.8 (C[9]), 128.7 (C[14]), 128.2 (C[8]), 127.9 (C[15]), 127.1 (C[16]), 126.9 (C[10]), 86.9 (C[12]), 62.3 (C[11]), 58.3 (C[6]), 55.3 (C[3]), 54.3 (C[1]), 51.5 (C[2]), 47.9 (C[4]), 11.8 (C[5]). HRMS (ESI) m/z: [M + H]+ calcd for C32H34NO2 464.2590; found 464.2596.
N,N-Dibenzyl-1-(3-((trityloxy)methyl)oxiran-2-yl)methanamine (2m)
It was prepared according to general procedure B using dibenzylamine (4.2 mmol, 0.828 g, 0.808 mL) and (3-((trityloxy)methyl)oxiran-2-yl)methyl 4-methylbenzenesulfonate (2.0 mmol, 1.00 g). Purification by column chromatography on silica gel (0–10% EtOAc in hexane) afforded the title compound as a yellowish oil (894 mg, 1.70 mmol, 85%). 1H NMR (CDCl3, 300 MHz) δH: 1H-NMR (CDCl3, 300 MHz) δH: 7.41 (6H, d, J = 7.5 Hz, 6 × C[12]-H), 7.33–7.20 (19H, m, Ar-H), 3.71 (2H, AB d, J = 13.7 Hz, 2 × C[4]-H), 3.47 (2H, d, J = 13.7 Hz, 2 × C[4]-H), 3.24 (1H, dd, J = 9.9 Hz, 5.5 Hz, C[9]-H), 3.20–3.11 (2H, m, C[1]-H, C[2]-H), 3.05 (1H, dd, J = 9.9 Hz, 3.9 Hz, C[9]-H), 2.59 (1H, dd, J = 13.7, 3.1 Hz, C[3]-H), 2.18 (1H, dd, J = 13.7 Hz, 6.2 Hz, C[3]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 143.7 (C[11]), 139.1 (C[5]), 128.8 (C[6]), 128.6 (C[13]), 128.2 (C[7]), 127.8 (C[12]), 127.1 (C[14]), 126.9 (C[8]), 86.9 (C[10]), 62.2 (C[9]), 58.6 (C[4]), 55.3 (C[1]), 54.2 (C[1]), 51.6 (C[2]). HRMS (ESI) m/z: [M + H]+ calcd for C37H36NO2 526.2746; found 526.2733.
tert-Butyl Benzyl((3-((trityloxy)methyl)oxiran-2-yl)methyl)carbamate (2n)
It was prepared according to general procedure C using tert-butyl (benzyl)carbamate (1.2 mmol, 316 mg) and (3-propyloxiran-2-yl)methyl (3-((trityloxy)methyl)oxiran-2-yl)methyl 4-methylbenzenesulfonate (1.2 mmol, 601 mg). Purification by column chromatography on Florisil (0–20% EtOAc in hexane) afforded the title compound as a yellowish oil (372 mg, 0.70 mmol, 58%). 1H NMR (CDCl3, 300 MHz) δH: 7.41 (6H, d, J = 7.8 Hz, 6 × C[15]-H), 7.30–7.18 (12H, m, Ar-H), 7.11–7.05 (2H, m, Ar-H), 4.59 (1H, AB d, J = 15.5 Hz, C[7]-H), 4.32–4.26 (1H, m, C[7]-H), 3.68–2.78 (6H, m, C[1]-H, C[2]-H, 2 × C[3]-H, 2 × C[12]-H), 1.44 (9H, s, 2 × C[6]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 155.6 (C[4]), 143.6 (C[14]), 138.9 (C[8]), 138.1 (C[8]), 128.6 (C[15]), 128.4 (C[10]), 127.9 (C[9]), 127.9 (C[16]), 127.3 (C[11]), 127.1 (C[17]), 87.0 (C[13]), 80.2 (C[5]), 62.3 (C[12]), 54.9 (C[1]), 54.3 (C[2]), 50.9 (C[7]), 45.0 (C[3]), 28.4 (C[6]). HRMS (ESI) m/z: [M + H]+ calcd for C35H38NO4 536.2801; found 536.2816.
N-(4-Methoxybenzyl)-N-methyl-1-(3-((trityloxy)methyl)oxiran-2-yl)methanamine (2o)
It was prepared according to general procedure B using 1-(4-methoxyphenyl)-N-methylmethanamine (2.1 mmol, 0.318 g) and (3-((trityloxy)methyl)oxiran-2-yl)methyl 4-methylbenzenesulfonate (1.0 mmol, 0.50 g). Purification by column chromatography on silica gel (10–40% EtOAc in hexane) afforded the title compound as a yellowish oil (0.363 g, 0.76 mmol, 76%). 1H NMR (CDCl3, 500 MHz) δH: 7.44 (6H, d, J = 7.4 Hz, 6 × C[14]-H), 7.32–7.19 (9H, m, Ar-H), 7.16 (2H, d, J = 5.1 Hz, 2 × C[7]-H), 6.81 (2H, d, J = 5.1 Hz, 2 × C[8]-H), 3.79 (1H, s, 3 × C[10]-H). 3.49 (1H, AB d, J = 12.9 Hz, C[5]-H), 3.38–3.26 (2H, m, C[5]-H, C[11]-H), 3.21–3.14 (2H, m, C[1]-H, C[2]-H), 3.10 (1H, dd, J = 6.3 Hz, 2.7 Hz, C[11]-H), 2.55 (1H, dd, J = 13.2 Hz, 3.3 Hz, C[3]-H), 2.20 (3H, s, 3 × C[4]-H), 2.20–2.10 (1H, m, C[3]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 158.7 (C[9]), 143.7 (C[13]), 130.5 (C[6]), 130.2 (C[7]), 128.6 (C[14]), 127.9 (C[15]), 127.1 (C[16]), 113.6 (C[8]), 86.9 (C[12]), 62.2 (C[11]), 61.7 (C[5]), 55.2 (C[3]), 55.2 (C[10]), 55.1 (C[1]), 54.2 (C[2]), 42.6 (C[4]). HRMS (ESI) m/z: [M + H]+ calcd for C32H34NO3 480.2533; found 480.2523.
N-(3-Methoxybenzyl)-N-methyl-1-(3-((trityloxy)methyl)oxiran-2-yl)methanamine (2p)
It was prepared according to general procedure B using 1-(3-methoxyphenyl)-N-methylmethanamine (2.1 mmol, 0.318 g) and (3-((trityloxy)methyl)oxiran-2-yl)methyl 4-methylbenzenesulfonate (1.0 mmol, 0.50 g). Purification by column chromatography on silica gel (10–40% EtOAc in hexane) afforded the title compound as a yellowish oil (0.424 g, 0.88 mmol, 88%). 1H NMR (CDCl3, 300 MHz) δH: 7.46 (6H, d, J = 7.4 Hz, 6 × C[16]-H), 7.31–7.15 (10H, m, Ar-H), 6.86–6.75 (3H, m, Ar-H), 3.78 (3H, s, 3 × C[12]-H) 3.54 (1H, AB d, J = 13.2 Hz, C[5]-H), 3.38 (1H, AB d, J = 13.2 Hz, C[5]-H), 3.32 (1H, dd, J = 10.1 Hz, 5.6 Hz, C[13]-H), 3.23–3.07 (3H, m, C[1]-H, C[2]-H, C[13]-H), 2.60 (1H, dd, J = 13.5 Hz, 3.3 Hz, C[3]-H), 2.22 (3H, s, 3 × C[4]-H), 2.16 (1H, dd, J = 13.5 Hz, 6.4 Hz, C[3]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 159.6 (C[8]), 143.7 (C[15]), 140.3 (C[6]), 129.2 (C[10]), 128.6 (C[16]), 127.9 (C[17]), 127.1 (C[18]), 121.3 (C[11]), 114.3 (C[7]), 112.6 (C[9]), 86.9 (C[14]), 62.4 (C[5]), 62.1 (C[13]), 55.4 (C[3]), 55.2 (C[12]), 55.2 (C[1]), 54.1 (C[2]), 42.8 (C[4]). HRMS (ESI) m/z: [M + H]+ calcd for C32H34NO3 480.2533; found 480.2535.
N-(4-Fluorobenzyl)-N-methyl-1-(3-((trityloxy)methyl)oxiran-2-yl)methanamine (2q)
It was prepared according to general procedure B using 1-(4-fluorophenyl)-N-methylmethanamine (2.1 mmol, 0.292 g) and (3-((trityloxy)methyl)oxiran-2-yl)methyl 4-methylbenzenesulfonate (1.0 mmol, 0.50 g). Purification by column chromatography on silica gel (10–45% EtOAc in hexane) afforded the title compound as a yellowish oil (0.365 g, 0.78 mmol, 78%). 1H NMR (CDCl3, 300 MHz) δH: 7.44 (6H, d, J = 7.4 Hz, 6 × C[13]-H), 7.32–7.19 (11H, m, Ar-H), 6.95 (2H, t, J = 8.5 Hz, 2 × C[8]-H), 3.51 (1H, AB d, J = 13.0 Hz, C[5]-H), 3.38 (1H, d, J = 13.0 Hz, C[5]-H), 3.32 (1H, dd, J = 10.3 Hz, 6.0 Hz, C[10]-H), 3.15–3.12 (2H, m, C[1]-H, C[2]-H), 3.08 (1H, dd, J = 10.0 Hz, 4.7 Hz, C[10]-H), 2.55 (1H, dd, J = 13.5, 3.4 Hz, C[3]-H), 2.20 (3H, s, 3 × C[4]-H), 2.20–2.10 (1H, m, C[3]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 161.9 (d, J = 243 Hz, C[9]), 143.7 (C[12]), 134.3 (d, J = 2.8 Hz, C[6]) 130.4 (d, J = 7.9 Hz, C[7]), 128.6 (C[13]), 127.9 (C[14]), 127.1 (C[15]), 115.0 (d, J = 21.1 Hz, C[8]), 86.9 (C[11]), 62.2 (C[10]), 61.5 (C[5]), 55.3 (C[3]), 55.0 (C[1]), 54.1 (C[2]), 42.6 (C[4]). HRMS (ESI) m/z: [M + H]+ calcd for C31H31FNO2 468.2333; found 468.2325.
N-Methyl-1-(naphthalen-2-yl)-N-((3-((trityloxy)methyl)oxiran-2-yl)methyl)methanamine (2r)
It was prepared according to general procedure B using N-methyl-1-(naphthalen-2-yl)methanamine (2.1 mmol, 0.360 g) and (3-((trityloxy)methyl)oxiran-2-yl)methyl 4-methylbenzenesulfonate (1.0 mmol, 0.500 g). Purification by column chromatography on silica gel (0–5% MeOH in DCM) afforded the title compound as a yellowish oil (0.332 g, 0.67 mmol, 67%). 1H NMR (CDCl3, 300 MHz) δH: 8.25–8.22 (1H, m, C[9]-H), 7.84–7.79 (1H, m, C[12]-H), 7.79–7.72 (1H, m, C[14]-H), 7.49–7.42 (8H, m, Ar-H), 7.37–7.33 (2H, m, Ar-H), 7.32–7.18 (9H, m, Ar-H), 3.98 (1H, dd, J = 13.2 Hz, C[5]-H), 3.78 (1H, dd, J = 13.2 Hz, C[5]-H), 3.33 (1H, dd, J = 9.9 Hz, 4.8 Hz C[16]-H), 3.25–3.15 (2H, m, C[1]-H, C[2]-H), 3.33 (1H, dd, J = 9.9 Hz, 3.5 Hz C[16]-H), 2.71–2.59 (1H, m, C[3]-H), 2.29–2.25 (4H, C[3]-H, 3 × C[4]-H). 13C{1H} NMR (CDCl3, 300 MHz) δC: 143.7 (C[18]), 134.4 (C[6]), 133.8 (C[8]), 132.4 (C[13]), 128.6 (C[19]), 128.4 (C[20]), 128.0 (C[15]), 127.9 (C[12]), 127.4 (C[9]), 127.1 (C[21]), 125.8 (C[14]), 125.6 (C[7]), 125.1 (C[10]), 124.6 (C[11]), 86.9 (C[17]), 62.1 (C[16]), 60.7 (C[5]), 55.7 (C[3]), 55.2 (C[1]), 55.1 (C[2]), 42.9 (C[4]). HRMS (ESI) m/z: [M + H]+ calcd for C35H34NO2 500.2584; found 500.2576.
N-Benzyl-N-methyl-1-(3-phenyloxiran-2-yl)methanamine (2s)
It was prepared according to general procedure B using N-methylbenzylamine (2.76 mmol, 0.335 g, 0.356 mL) and (3-phenyloxiran-2-yl)methyl 4-methylbenzenesulfonate (1.31 mmol, 0.40 g). Purification by column chromatography on silica gel (10–30% EtOAc in hexane) afforded the title compound as a yellowish oil (0.205 g, 0.81 mmol, 62%). 1H NMR (CDCl3, 300 MHz) δH: 7.37–7.21 (10H, m, C[7-9, 11-13]-H), 3.66 (1H, AB d, J = 13.0, Hz, C[5]-H), 3.63–3.60 (1H, m, C[1]-H), 3.55 (1H, AB d, J = 13.0, Hz, C[5]-H), 3.20–3.12 (1H, m, C[2]-H), 2.80 (1H, dd, J = 13.3 Hz, 3.9 Hz, C[3]-H), 2.58 (1H, dd, J = 13.3 Hz, 6.2 Hz, C[3]-H), 2.35 (3H, s, 3 × C[4]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 138.6 (C[6]), 137.3 (C[10]), 129.0 (C[7]), 128.5 (C[11]), 128.3 (C[8]), 128.1 (C[13]), 127.1 (C[9]), 125.6 (C[12]), 62.6 (C[5]), 61.4 (C[1]), 59.0 (C[2]), 56.9 (C[3]), 43.0 (C[4]). HRMS (ESI) m/z: [M + H]+ calcd for C17H20NO 254.1539; found 254.1536.
N-Benzyl-N-ethyl-1-(3-phenyloxiran-2-yl)methanamine (2t)
It was prepared according to general procedure B using N-benzylethylamine (2.76 mmol, 0.373 g, 0.411 mL) and (3-phenyloxiran-2-yl)methyl 4-methylbenzenesulfonate (1.31 mmol, 0.40 g). Purification by column chromatography on silica gel (15–30% EtOAc in hexane) afforded the title compound 2t as a yellowish oil (0.260 g, 0.97 mmol, 74%). 1H NMR (CDCl3, 300 MHz) δH: 7.38–7.20 (10H, m, C[7-9, 12-14]-H), 3.75 (1H, AB d, J = 13.8, Hz, C[5]-H), 3.67–3.57 (2H, m, C[1]-H C[5]-H), 3.12 (1H, m, C[2]-H) 2.43 (1H, dd, J = 13.8, Hz, 3.9 Hz, C[3]-H), 2.70–2.59 (3H, m, C[2]-H, 2 × C[4]-H), 1.10 (3H, t, J = 6.9, Hz, 3 × C[5]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 139.4 (C[7]), 137.4 (C[11]), 128.8 (C[8]), 128.4 (C[12]), 128.2 (C[9]), 128.1 (C[14]), 126.9 (C[10]), 125.6 (C[13]), 61.6 (C[1]), 58.6 (C[2]), 57.0 (C[6]), 55.1 (C[3]), 48.2 (C[4]), 11.9 (C[5]). HRMS (ESI) m/z: [M + H]+ calcd for C18H22NO 268.1696; found 268.1703.
N,N-Dibenzyl-1-(3-phenyloxiran-2-yl)methanamine (2u)
It was prepared according to general procedure B using dibenzylamine (2.76 mmol, 0.544 g, 0.531 mL) and (3-phenyloxiran-2-yl)methyl 4-methylbenzenesulfonate (1.31 mmol, 0.400 g). Purification by column chromatography on silica gel (0–20% EtOAc in hexane) afforded 2u as a yellowish oil (0.313 g, 95 mmol, 72%). 1H NMR (CDCl3, 300 MHz) δH: 7.44–7.18 (15H, m, C[6-8, 10-12]-H), 3.81 (2H, AB d, J = 13.7, Hz, 2 × C[4]-H), 3.63 (2H, AB d, J = 13.7, 2 × C[4]-H), 3.58–3.53 (1H, m, C[1]-H), 3.20–3.10 (1H, m, C[2]-H), 2.87 (1H, dd, J = 13.8 Hz, 3.7 Hz, C[3]-H) 2.63 (1H, dd, J = 5.8 Hz, 14.1 Hz C[3]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 139.2 (C[5]), 137.3 (C[9]), 128.8 (C[6]), 128.4 (C[10]), 128.3 (C[7]), 128.1 (C[12]), 127.0 (C[8]), 125.6 (C[11]), 61.6 (C[1]), 58.9 (C[4]), 56.7 (C[2]), 55.2 (C[3]). HRMS (ESI) m/z: [M + H]+ calcd for C23H24NO 330.1852; found 330.1852.
N-Methyl-1-(3-propyloxiran-2-yl)-N-(2-(trifluoromethyl)benzyl)methanamine (2v)
It was prepared according to general procedure B using N-methyl-1-(2-(trifluoromethyl)phenyl)methanamine (4.27 mmol, 0.845 g) and (3-propyloxiran-2-yl)methyl 4-methylbenzenesulfonate (2.03 mmol, 0.550 g). Purification by column chromatography on silica gel (0–30% EtOAc in hexane) afforded the title compound as a yellowish oil (0.517 g, 1.80 mmol, 89%). 1H NMR (CDCl3, 300 MHz) δH: 7.83 (1H, J = 7.8 Hz, C[8]-H), 7.61 (1H, d, J = 7.8 Hz, C[11]-H), 7.52 (1H, t, J = 7.6 Hz, C[10]-H), 7.32 (1H, t, J = 7.6 Hz, C[11]-H), 3.76 (1H, AB d, J = 14.7 Hz, C[5]-H), 3.68 (1H, d, J = 14.7 Hz, C[5]-H), 2.95–2.81 (1H, m, C[2]-H), 2.76–2.62 (2H, m C[1]-H, C[3]-H), 2.47 (1H, AB dd, J = 13.2 Hz, 6.2 Hz, C[3]-H), 2.32 (3H, s, 3 × C[4]-H), 1.56–1.39 (4H, m, 2 × C[13]-H, 2 × C[14]-H), 0.96 (3H, t, J = 6.9 Hz, 3 × C[15]-H). 13C{1H} NMR (75 MHz, CDCl3) δC: 138.4 (q, J = 1.5 Hz, C[6]), 131.8 (C[9]), 130.4 (C[11]), 128.5 (q, J = 30 Hz, C[7]), 126.8 (C[10]), 125.7 (q, J = 6 Hz, C[8]), 124.5 (q, J = 273 Hz, C[12]), 59.7 (C[3]), 58.0 (q, J = 1.7 Hz, C[5]), 57.9, 57.1 (C[2]), 56.8 (C[1]), 43.1 (C[4]), 33.9 (C[13]), 19.3 (C[14]), 13.9 (C[15]). HRMS (ESI) m/z: [M + H]+ calcd for C14H22NO 288.1564; found 288.1572.
2-((3-((Trityloxy)methyl)oxiran-2-yl)methyl)isoindoline (2w)
It was prepared according to general procedure B using isoindoline (4.2 mmol, 0.500 g) and (3-((trityloxy)methyl)oxiran-2-yl)methyl 4-methylbenzenesulfonate (2.0 mmol, 1.00 g). Purification by column chromatography on Florisil (0–40% EtOAc in hexane) afforded 2w as a yellowish oil (0.780 g, 1.74 mmol, 87%). 1H NMR (CDCl3, 300 MHz) δH: 7.49–7.46 (6H, d, J = 7.8 Hz, 6 × C[11]-H), 7.33–7.17 (13H, m, Ar-H), 3.98–3.90 (4H, m, 4 × C[4]-H), 3.43 (1H, dd, J = 10.2 Hz, 5.7 Hz, C[8]-H) 3.30–3.21 (2H, m, C[1]-H, C[2]-H) 3.15 (1H, dd, J = 10.2 Hz, 4.5 Hz, C[8]-H) 2.91 (1H, dd, J = 12.9 Hz, 3.6 Hz, C[3]-H), 2.51 (1H, dd, J = 12.9 Hz, 6.6 Hz, C[3]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 143.7 (C[10]), 139.9 (C[5]), 128.6 (C[12]), 127.9 (C[11]), 127.2 (C[6]), 126.8 (C[7]), 122.3 (C[13]), 87.0 (C[9]), 62.1 (C[4]), 59.3 (C[8]), 55.4 (C[1]), 54.4 (C[2]), 53.8 (C[3]). HRMS (ESI) m/z: [M + H]+ calcd for C31H30NO2 448.2271; found 448.2293.
Synthesis of 2-Phenylazetidines
(1-Methyl-2-phenylazetidin-3-yl)methanol (1a)
It was prepared according to general procedure D using 2a (1.39 mmol, 300 mg). Purification by column chromatography on silica gel (0–20% MeOH in DCM) afforded the title compound (1a) as a yellowish oil (261 mg, 1.47 mmol, 87%). 1H NMR (CDCl3, 500 MHz) δH: 7.38 (2H, d, J = 7.8 Hz, Ar-H), 7.32 (2H, t, J = 7.5 Hz, Ar-H), 7.24 (1H, t, J = 7.3 Hz, Ar-H), 3.74 (1H, d, J = 7.8 Hz, C[1]-H), 3.70 (2H, t, J = 4.5 Hz, 2 × C[9]-H), 3.57 (1H, t, J = 6.8 Hz, C[3]-H), 2.75 (1H, t, J = 7.5 Hz, C[3]-H), 2.59 (1H, td, J = 7.0 Hz, 5.9 Hz, C[2]-H), 2.34 (3H, s, 3 × C[8]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 141.9 (C[4]), 128.4 (C[5]), 127.4 (C[7]), 126.7 (C[6]), 73.3 (C[1]), 63.2 (C[9]), 55.6 (C[3]), 44.4 (C[8]), 42.5 (C[2]). HRMS (ESI) m/z: [M + H]+ calcd for C11H16NO 178.1264; found 178.1239.
(1-Ethyl-2-phenylazetidin-3-yl)methanol (1b)
It was prepared according to general procedure D using 2b (1.70 mmol, 325 mg). Purification by column chromatography on silica gel (0–10% MeOH in DCM) afforded the title compound (1b) as a yellowish oil (302 mg, 1.58 mmol, 93%). 1H NMR (CDCl3, 300 MHz) δH: 7.38 (2H, d, J = 7.1 Hz, 2 × C[5]-H), 7.28 (2H, t, J = 7.1 Hz, 2 × C[6]-H), 7.23–7.19 (1H, m, C[7]-H), 3.78–3.70 (2H, d + bs, J = 7.8 Hz, C[1]-H, OH), 3.62–3.52 (2H, m, 2 × C[10]-H), 3.50 (1H, t, J = 6.9 Hz, C[3]-H) 2.69–2.59 (2H, m, C[3]-H, C[8]-H) 2.54–2.40 (2H, m, C[2]-H, C[8]-H) 0.90 (3H, t, J = 7.8 Hz, C[9]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 142.5 (C[4]), 128.3 (C[5]), 127.2 (C[7]), 126.7 (C[6]), 71.7 (C[1]), 62.8 (C[10]), 53.5 (C[3]), 52.8 (C[8]), 42.2 (C[2]), 12.5 (C[9]). HRMS (ESI) m/z: [M + H]+ calcd for C12H17NO 192.1383; found 192.1389.
(1-Benzyl-2-phenylazetidin-3-yl)methanol (1c)
It was prepared according to general procedure D using 2c (1.70 mmol, 431 mg). Purification by column chromatography on silica gel (40–80% EtOAc in hexane) afforded the title compound (1c) as a yellowish oil (105 mg, 0.29 mmol, 25%). Pure TFA salt of 1c can be isolated by preparative HPLC (using 0.1% TFA in 1 L of water and acetonitrile). 1H NMR (DMSO-d6, 500 MHz) δH: 10.27 (1H, bs, NH), 7.47–7.41 (4H, m, Ar-H), 7.41–7.35 (6H, m, Ar-H), 5.29 (1H, bs, C[1]-H), 5.17 (1H, bs, OH), 4.46 (2H, bs, 2 × C[8]-H), 3.92 (2H, bs, 2 × C[3]-H), 3.60–3.55 (2H, m, 2 × C[13]-H), 3.25 (1H, bs, C[2]-H). 13C{1H} NMR (DMSO-d6, 126 MHz) δC: 142.4 (C[4]), 138.2 (C[9]), 128.8 (C[10]), 128.3 (C[5]), 128.2 (C[11]), 127.2 (C[7]), 126.9 (C[12]), 126.7 (C[6]), 71.0 (C[1]), 63.4 (C[13]), 61.9 (C[8]), 53.5 (C[3]), 43.2 (C[2]). HRMS (ESI) m/z: [M + H]+ calcd for C17H19NO 254.1539; found 254.1530.
(1-Methyl-2-phenylazetidin-3-yl)butan-1-ol (1d)
It was prepared according to general procedure D using 2d (0.91 mmol, 200 mg). Purification by column chromatography on silica gel (40–100% EtOAc in hexane) afforded the title compound (1d) as a yellowish oil (172 mg, 0.78 mmol, 86%). 1H NMR (CDCl3, 300 MHz) δH: 7.40 (2H, d, J = 7.2 Hz, 2 × C[5]-H), 7.33 (2H, t, J = 7.3 Hz, 2 × C[6]-H), 7.29–7.22 (1H, m, C[7]-H), 3.75 (1H, d, J = 8.1 Hz, C[1]-H), 3.70–3.62 (1H, m, C[9]-H), 3.55 (1H, t, J = 6.6 Hz, C[3]-H), 2.83 (1H, dd, J = 8.7 Hz, 6.8 Hz, C[3]-H), 2.55–2.44 (2H, m, C[2]-H, OH), 2.33 (3H, s, 3 × C[8]-H), 1.40–1.20 (4H, m, 2 × C[10]-H, 2 × C[11]-H), 0.84 (3H, t, J = 6.6 Hz, 3 × C[12]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 141.8 (C[4]), 128.4 (C[5]), 127.5 (C[7]), 127.2 (C[6]), 72.8 (C[1]), 71.3 (C[9]), 54.4 (C[3]), 46.0 (C[2]), 44.2 (C[8]), 37.4 (C[10]), 18.6 (C[11]), 14.0 (C[12]). HRMS (ESI) m/z: [M + H]+ calcd for C14H22NO 220.1701; found 220.1696.
(1-Ethyl-2-phenylazetidin-3-yl)butan-1-ol (1e)
It was prepared according to general procedure D using 2e (0.89 mmol, 200 mg). Purification by column chromatography on silica gel (40–100% EtOAc in hexane) afforded the title compound (1e) as a yellowish oil (178 mg, 0.76 mmol, 89%). 1H NMR (CDCl3, 300 MHz) δH: 7.42 (2H, d, J = 7.3 Hz, 2 × C[5]-H), 7.33 (2H, t, J = 7.3 Hz, 2 × C[6]-H), 7.29–7.22 (1H, m, C[7]-H), 3.80 (1H, d, J = 8.1 Hz, C[1]-H), 3.75–3.65 (1H, m, C[10]-H), 3.53 (1H, t, J = 6.9 Hz, C[3]-H), 2.76 (1H, t, J = 6.9 Hz, C[3]-H), 2.67–2.54 (1H, m, C[2]-H), 2.54–2.37 (2H, m, 2 × C[8]-H), 1.63 (1H, bs, OH), 1.35–1.26 (4H, m, 2 × C[11]-H, 2 × C[12]-H), 0.93–0.83 (6H, m, 3 × C[9]-H, 3 × C[13]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 142.9 (C[4]), 128.3 (C[5]), 127.2 (C[7]), 127.1 (C[6]), 71.4 (C[1]), 71.2 (C[10]), 52.7 (C[3]), 52.1 (C[8]), 46.1 (C[2]), 37.4 (C[11]), 18.6 (C[12]), 14.0 (C[9]), 12.7 (C[13]). HRMS (ESI) m/z: [M + H]+ calcd for C15H24NO 234.1852; found 234.1862.
(1-Benzyl-2-phenylazetidin-3-yl)butan-1-ol (1f)
It was prepared according to general procedure D using 2f (0.97 mmol, 286 mg). Purification by column chromatography on silica gel (0–10% MeOH in DCM) afforded the title compound (1f) as a yellowish oil (203 mg, 0.71 mmol, 71%). 1H NMR (CDCl3, 300 MHz) δH: 7.45 (2H, d, J = 6.9 Hz, 2 × C[5]-H), 7.38–7.15 (8H, m, Ar-H), 4.00 (1H, d, J = 7.9 Hz, C[1]-H), 3.81 (1H, AB d, J = 12.9 Hz, C[8]-H), 3.74–3.64 (1H, m, C[14]-H), 3.44 (1H, AB d, J = 12.9 Hz, C[8]-H), 3.42–3.35 (1H, m, C[3]-H), 2.85 (1H, t, J = 7.8 Hz, C[3]-H), 2.50–2.35 (1H, m, C[2]-H), 1.61 (1H, bs, OH), 1.38–1.20 (4H, m, 2 × C[15]-H, 2 × C[16]-H), 0.84 (3H, t, J = 6.9 Hz, 3 × C[17]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 142.4 (C[4]), 138.3 (C[9]), 128.8 (C[10]), 128.3 (C[5]), 128.1 (C[11]), 127.3 (C[7]), 127.1 (C[12]), 126.9 (C[6]), 71.2 (C[1]), 70.3 (C[13]), 61.7 (C[8]), 52.2 (C[3]), 46.8 (C[2]), 37.4 (C[14]), 18.6 (C[15]), 14.0 (C[16]). HRMS (ESI) m/z: [M + H]+ calcd for C20H26NO 296.2009; found 296.2008.
1-(2-Phenyl-1-(1-phenylethyl)azetidin-3-yl)butan-1-ol (1g)
It was prepared according to general procedure D using 2g (0.97 mmol, 300 mg). Purification by column chromatography on silica gel (10–100% EtOAc in hexane) afforded the title compound (1g_diastereomer I; 139 mg, 0.45 mmol, 46%) and (1g_diastereomer II; 85 mg, 0.28 mmol%) as a yellowish oil (in overall 70% yield). 1g_diastereomer I:1H NMR (CDCl3, 300 MHz) δH: 7.58 (2H, d, J = 7.4 Hz, C[5]-H), 7.42–7.20 (8H, m, Ar-H), 4.03 (1H, d, J = 7.7 Hz, C[1]-H), 3.72–3.60 (1H, m, C[15]-H), 3.46 (1H, q, J = 6.5 Hz, C[8]-H), 3.19 (1H, t, J = 7.5 Hz, C[3]-H), 2.66 (1H, t, J = 7.5 Hz, C[3]-H), 2.35–2.20 (1H, m, C[2]-H), 1.44 (1H, bs, OH), 1.32–1.15 (4H, m, 2 × C[16]-H, 2 × C[17]-H), 0.94 (3H, d, J = 6.5 Hz, 3 × C[14]-H), 0.82 (3H, t, J = 6.4 Hz, 3 × C[18]-H). 13C{1H} NMR (CDCl3, 300 MHz) δC: 144.7 (C[9]), 144.3 (C[4]), 128.3 (C[11]), 128.2 (C[6]), 127.4 (C[12]), 127.0 (C[13]), 127.0 (C[5]), 126.9 (C[7]), 71.0 (C[15]), 69.8 (C[1]), 68.4 (C[8]), 51.0 (C[3]), 46.4 (C[2]), 37.3 (C[16]), 22.1 (C[12]), 18.6 (C[17]), 14.0 (C[18]). HRMS (ESI) m/z: [M + H]+ calcd for C21H27NO 310.2165; found 310.2174.
1g_diastereomer II
1H NMR (CDCl3, 300 MHz) δH: 7.12–6.98 (7H, m, Ar-H), 6.98–6.90 (3H, m, Ar-H), 3.74 (1H, d, J = 7.8 Hz, C[1]-H), 3.70–3.55 (2H, m, C[3]-H, C[15]-H), 3.41 (1H, q, J = 6.5 Hz C[8]-H), 2.93 (1H, t, J = 7.2 Hz, C[3]-H), 2.54–2. 38 (1H, m, C[2]-H), 1.55 (1H, bs, OH), 1.35–1.15 (7H, m, 3 × C[14]-H, 2 × C[16]-H, 2 × C[17]-H), 0.81 (1H, t, J = 6.8 Hz 3 × C[18]-H). 13C{1H} NMR (CDCl3, 300 MHz) δC: 142.6 (C[4/9]), 142.4 (C[4/9]), 128.0 (C[11]), 127.5 (C[6/C12/C13]), 127.5 (C[6/C12/C13]), 126.7 (C[5]), 126.5 (C[7]), 71.9 (C[15]), 70.4 (C[1]), 68.3 (C[8]), 52.0 (C[3]), 45.4 (C[2]), 37.3 (C[16]), 20.4 (C[12]), 18.5 (C[17]), 13.9 (C[18]). HRMS (ESI) m/z: [M + H]+ calcd for C14H22NO 310.2165; found 310.2169.
tert-Butyl 3-(1-Hydroxybutyl)-2-(4-methoxyphenyl)azetidine-1-carboxylate (1h)
It was prepared according to general procedure D using 2h (0.60 mmol, 200 mg). Purification by preparative HPLC (using 0.4 g of NH4HCO3 in 1 L of water and acetonitrile) afforded the title compound (1h, 84 mg, 0.25 mmol, 42%) as a yellowish oil and 9h (72 mg, 0.21 mmol, 36%) as a yellowish oil.
tert-Butyl 3-(1-Hydroxybutyl)-2-(4-methoxyphenyl)azetidine-1-carboxylate (1h)
1H NMR (CDCl3, 300 MHz) δH(ppm): 1H NMR (300 MHz, CDCl3) δH: 7.28 (2H, d, J = 8.2 Hz, 2 × C[6]-H), 6.88 (2H, d, J = 8.2 Hz, 2 × C[5]-H), 4.87 (1H, d, J = 5.7 Hz, C[1]-H), 3.99 (1H, t, J = 8.4 Hz, C[3]-H), 3.88 (t, J = 7.4 Hz, C[3]-H), 3.81 (4H, s, 3 × C[8]-H, C[12]-H), 2.48–2.35 (1H, m, C[2]-H), 1.81 (1H, bs, OH), 1.55–1.16 (13H, m, 9 × C[11]-H, 2 × C[13]-H, 2 × C[14]-H), 0.90 (t, J = 6.0 Hz, 3 × C[15]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 159.0 (C[7]), 156.7 (C[9]), 133.9 (C[4]), 127.5 (C[5]), 113.9 (C[6]), 79.5 (C[10]), 71.8 (C[12]), 66.0 (C[1]), 55.3 (C[8]), 48.0 (C[3]), 44.9 (C[2]), 37.0 (C[13]), 28.3 (C[11]), 18.7 (C[14]), 14.0 (C[15]). HRMS (ESI) m/z: [M + H]+ calcd for C19H30NO4 336.2169; found 336.2175.
tert-Butyl (E)-(3-Hydroxyhex-1-en-1-yl)(4-methoxybenzyl)carbamate (9h)
1H NMR (CDCl3, 300 MHz) δH: 1H NMR (300 MHz, CDCl3) δH: 7.30–7.02 (1H, bs, C[6]-H), 7.12 (2H, d, J = 8.1 Hz, 2 × C[12]-H), 6.83 (2H, d, J = 8.4 Hz, 2 × C[13]-H), 4.82 (1H, bs, C[5]-H), 4.64 (2H, bs, 2 × C[10]-H), 4.05 (1H, bs, C[4]-H), 3.79 (3H, s, 3 × C[15]-H), 1.61–1.17 (13H, m, 2 × C[2]-H, 2 × C[3]-H, 9 × C[9]-H), 0.87 (3H, t, J = 7.2 Hz, 3 × C[1]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 158.6 (C[14]), 129.3 (C[6]), 128.9 (C[11]), 127.8 (C[12]), 113.9 (C[13]), 112.2 (C[5]), 81.6 (C[8]), 72.1 (C[4]), 55.3 (C[15]), 47.0 (C[10]) 39.9 (C[3]), 28.2 (C[9]), 18.8 (C[2]), 13.9 (C[1]). HRMS (ESI) m/z: [M + H]+ calcd for C19H30NO4 336.2169; found 336.2178.
tert-Butyl 3-(1-Hydroxybutyl)-2-(4-tert-butylphenyl)azetidine-1-carboxylate (1j)
It was prepared according to general procedure D using 2j (1.25 mmol, 450 mg). Purification by preparative HPLC (using 0.4 g of NH4HCO3 in 1 L of water and acetonitrile) afforded the title compound (1j, 198 mg, 0.55 mmol, 47%) as a yellowish oil and 9j (143 mg, 0.43 mmol, 34%) as a yellowish oil.
tert-Butyl 3-(1-Hydroxybutyl)-2-(4-tert-butylphenyl)azetidine-1-carboxylate (1j)
1H NMR (CDCl3, 500 MHz) δH: 7.35 (2H, d, J = 8.3 Hz, 2 × C[6]-H), 7.27 (2H, d, J = 6.9 Hz, 2 × C[5]-H), 4.92 (1H, bs, C[1]-H), 3.99 (1H, t, J = 8.3 Hz, C[3]-H), 3.92 (1H, dd, J = 8.3 Hz, 6.4 Hz, C[3]-H), 3.79 (1H, s, C[13]-H), 2.43 (1H, bs, C[2]-H), 2.38–2.22 (1H, bs, OH), 1.53–1.08 (22H, m, 9 × C[9]-H, 9 × C[12]-H, 2 × C[14]-H, 2 × C[15]-H), 0.92 (3H, t, J = 6.9 Hz, 3 × C[16]-H). 13C{1H} NMR (CDCl3, 126 MHz) δC: 156.8 (C[10]), 150.3 (C[7]), 138.7 (C[4]), 125.8 (C[5]), 125.3 (C[6]), 79.5 (C[11]), 71.5 (C[13]), 66.1 (C[1]), 48.9 (C[3]), 44.6 (C[2]), 36.9 (C[14]), 34.5 (C[8]), 31.4 (C[9]), 28.3 (C[12]), 18.8 (C[15]), 14.0 (C[16]). HRMS (ESI) m/z: [M + H]+ calcd for C22H32NO4 362.2690; found 362.2687.
tert-Butyl (E)-(3-Hydroxyhex-1-en-1-yl)(4- tert-butyl)carbamate (9j)
1H NMR (CDCl3, 300 MHz) δH: 7.32 (2H, d, J = 8.5 Hz, 2 × C[13]-H), 7.11 (2H, d, J = 8.5 Hz, 2 × C[12]-H), 7.18+7.03 (0.5 + 0.5 H, bs. C[6]-H), 4.81 (1H, bs, C[5]-H), 4.66 (2H, bs, 2 × C[10]-H), 4.05 (1H, bs, C[4]-H), 1.67 (1H, bs, OH), 1.52–1.22 (22H, m, 2 × C[2]-H, 2 × C[3]-H, 9 × C[9]-H, 9 × C[19]-H), 0.84 (3H, t, J = 7.5 Hz, 3 × C[1]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 149.9 (C[9]), 138.6 (C[11]), 134.2 (C[14]), 129.3 (C[6]), 126.3 (C[14]), 125.4 (C[13]), 112.3 (C[5]), 81.6 (C[8]), 72.1 (C[4]), 47.4 (C[10]), 39.8 (C[3]), 34.4 (C[15]), 31.4 (C[9]), 28.2 (C[16]), 18.7 (C[2]), 14.0 (C[1]). HRMS (ESI) m/z: [M + H]+ calcd for C22H32NO4 362.2690; found 362.2683.
1-(1-Methyl-2-phenylazetidin-3-yl)-2-(trityloxy)ethan-1-ol (1k)
1.0 mmol Scale Synthesis
It was prepared according to general procedure D using 2k (1.0 mmol, 449 mg). Purification by column chromatography on silica gel (40–100% EtOAc in hexane) afforded the title compound (1k) as a yellowish solid (341 mg, 0.76 mmol, 76%).
Scale-up Synthesis
Potassium tert-butoxide (40 mmol, 4.49 g, 2.0 equiv) was dissolved in dry THF (50 mL) and cooled to −78 °C, using dry ice in acetone, in a Schlenk flask (200 mL) under a nitrogen atmosphere. Diisopropylamine (40 mmol, 4.05 g, 5.65 mL, 2.0 equiv) and a 1.59 M hexane solution of BuLi (46 mmol, 29 mL, 2.3 equiv) were added dropwise into the solution (yellowish). The reaction mixture was stirred for 20 min at −78 °C. Oxirane (2k, 8.98 g, 20 mmol, 1.0 equiv) in dry THF (100 mL) was added dropwise, the dark red mixture was stirred at −78 °C for 2 h. The cold reaction mixture was poured carefully into the mixture of diethyl ether (100 mL) and water (50.0 mL), and then it was allowed to warm up to room temperature. The phases were separated, and the aqueous phase was extracted with diethyl ether (3 × 100 mL). The combined organic layers were washed with brine (1 × 200 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude mixture (8.4 g) was purified by flash chromatography on silica gel (50–100% EtOAc in hexane), affording 1k as an off-white solid (7.01 g, 15.7 mmol, 78%). 1H NMR (CDCl3, 300 MHz) δH: 7.34 (8H, t, J = 9.2 Hz, Ar-H), 7.34 (8H, t, J = 7.8 Hz, Ar-H), 7.24–7.19 (4H, m, Ar-H), 3.92 (1H, td, J = 7.2 Hz, 3.8 Hz, C[9]-H), 3.84 (1H, d, J = 7.8 Hz, C[1]-H), 3.42 (1H, t, J = 7.0 Hz, C[3]-H), 3.00 (1H, dd, J = 9.2 Hz, 3.6 Hz C[10]-H), 2.90 (1H, dd, J = 9.2 Hz, 8.5 Hz, C[10]-H), 2.71 (1H, t, J = 7.4 Hz, C[3]-H), 2.53–2.47 (1H, m, C[2]-H), 2.29 (3H, s, 3 × C[8]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 143.7 (C[12]), 142.3 (C[4]), 128.6 (C[13]), 128.3 (C[5]), 127.9 (C[14]), 127.2 (C[7]), 127.1 (C[15]), 127.0 (C[6]), 86.8 (C[11]), 73.5 (C[9]), 72.6 (C[1]), 65.7 (C[10]), 54.5 (C[3]), 44.2 (C[8]), 43.1 (C[2]). HRMS (ESI) m/z: [M + H]+ calcd for C31H32NO2 450.2433; found 450.2424.
1-(1-Ethyl-2-phenylazetidin-3-yl)-2-(trityloxy)ethan-1-ol (1l)
It was prepared according to general procedure D using 2l (1.0 mmol, 463 mg). Purification by column chromatography on silica gel (20–60% EtOAc in hexane) afforded the title compound (1l) as a yellowish solid (371 mg, 0.80 mmol, 80%). 1H NMR (CDCl3, 300 MHz) δH: 7.39–7.32 (8H, m, Ar-H), 7.32–7.18 (12H, m, Ar-H), 3.93 (1H, td, J = 6.9 Hz, 3.2 Hz, C[10]-H), 3.85 (1H, d, J = 7.8 Hz, C[1]-H), 3.39 (1H, d, J = 7.0 Hz, C[3]-H), 3.02 (1H, dd, J = 9.2 Hz, 3.5 Hz C[11]-H), 2.90 (1H, dd, J = 9.2 Hz, 7.7 Hz, C[11]-H), 2.64–2.51 (2H, m, C[3]-H, C[8]-H), 2.48–2.35 (2H, m, C[2]-H, C[8]-H),2.17 (1H, bs, OH) 0.87 (3H, t, J = 7.2 Hz, 3 × C[9]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 143.7 (C[13]), 143.1 (C[4]), 128.6 (C[14]), 128.2 (C[5]), 127.9 (C[15]), 127.9 (C[7]), 127.1 (C[16]), 127.0 (C[6]), 86.8 (C[12]), 72.7 (C[10]), 71.9 (C[1]), 65.6 (C[11]), 52.7 (C[3]), 52.3 (C[8]), 42.9 (C[2]), 12.7 (C[9]). HRMS (ESI) m/z: [M + H]+ calcd for C32H34NO2 464.2590; found 464.2586.
1-(1-Benzyl-2-phenylazetidin-3-yl)-2-(trityloxy)ethan-1-ol (1m)
It was prepared according to general procedure D using 2m (1.0 mmol, 525 mg). Purification by column chromatography on silica gel (0–25% EtOAc in hexane) afforded the title compound (1m) as a yellowish solid (252 mg, 0.80 mmol, 48%). 1H NMR (CDCl3, 300 MHz) δH: 7.41–7.32 (8H, m, Ar-H), 7.32–7.18 (17H, m, Ar-H), 4.03 (1H, d, J = 7.6 Hz, C[1]-H), 3.99–3.89 (1H, m, C[13]-H), 3.79 (1H, AB d, J = 13.0 Hz, C[8]-H), 3.38 (2H, d, J = 13.0 Hz, C[8]-H), 3.26 (1H, t, J = 7.0 Hz, C[3]-H), 3.01 (1H, dd, J = 9.4 Hz, 7.0 Hz C[14]-H), 2.90 (1H, dd, J = 9.4 Hz, 7.5 Hz C[14]-H), 2.68 (1H, dd, J = 8.3 Hz, 7.0 Hz, C[3]-H), 2.50–2.38 (1H, m, C[2]-H), 2.27 (1H, bs, OH). 13C{1H} NMR (CDCl3, 75 MHz) δC: 143.7 (C[16]), 142.6 (C[4]), 138.3 (C[9]), 128.7 (C[10]), 128.6 (C[17]), 128.2 (C[6]), 128.2 (C[11]), 127.9 (C[18]), 127.9 (C[7]), 127.1 (C[19]), 127.1 (C[5]), 126.9 (C[12]), 86.8 (C[15]), 72.8 (C[13]), 71.2 (C[1]), 65.6 (C[14]), 61.7 (C[8]), 52.6 (C[3]), 43.6 (C[2]). HRMS (ESI) m/z: [M + H]+ calcd for C37H36NO2 526.2746; found 526.2744.
tert-Butyl 3-(1-Hydroxy-2-(trityloxy)ethyl)-2-phenylazetidine-1-carboxylate (1n)
It was prepared according to general procedure D using 2n (1.0 mmol, 535 mg). Purification by column chromatography on silica gel (0–20% EtOAc in hexane) afforded the title compound (1n) as a yellowish solid (215 mg, 0.40 mmol, 40%). 1H NMR (CDCl3, 300 MHz) δH: 7.41–7.35 (6H, m, Ar-H), 7.32–7.20 (14H, m, Ar-H), 5.06 (1H, m, C[1]-H), 4.05–3.95 (1H, m, C[11]-H), 3.89 (1H, J = 8.4 Hz, C[3]-H), 3.68 (1H, dd, J = 8.4 Hz, 6.2 Hz C[3]-H), 3.12 (1H, dd, J = 9.4 Hz, 3.6 Hz C[12]-H), 3.00 (1H, dd, J = 9.4 Hz, 7.4 Hz C[12]-H), 2.55–2.25 (1H, m, C[2]-H), 1.30 (9H, bs, 9 × C[10]-H). 13C{1H} NMR (CDCl3, 126 MHz), δC: 156.5 (C[8]), 143.5 (C[14]), 141.8 (C[4]), 128.5 (C[15]), 128.3 (C[5]), 127.9 (C[16]), 127.2 (C[17]), 127.1 (C[7]), 125.9 (C[6]), 87.0 (C[13]), 79.5 (C[9]), 72.0 (C[11]), 65.3 (C[12]), 60.4 (C[1]), 53.8 (C[3]), 41.6 (C[2]), 28.3 (C[10]). HRMS (ESI) m/z: [M + H]+ calcd for C35H38NO4 536.2801; found 536.2805.
1-(2-(4-Methoxyphenyl)-1-methylazetidin-3-yl)-2-(trityloxy)ethan-1-ol (1o)
It was prepared according to general procedure D using 2o (0.63 mmol, 300 mg). Purification by column chromatography on silica gel (0–20% MeOH in DCM) afforded the title compound (1o) as a yellowish solid (260 mg, 0.54 mmol, 76%). 1H NMR (CDCl3, 300 MHz) δH: 7.45–7.36 (6H, m, 6 × C[14]-H), 7.33–7.24 (11H, m, Ar-H), 6.80 (2H, d, J = 8.6 Hz, 2 × C[6]-H), 3.96–3.88 (1H, m, C[10]-H), 3.77 (3H, s, 3 × C[8]-H), 3.72 (1H, d, J = 7.5, Hz, C[1]-H), 3.37 (1H, t, J = 6.9 Hz, C[3]-H), 3.00 (1H, dd, J = 9.6 Hz, 3.6 Hz, C[11]-H), 2.88 (1H, dd, J = 9.6 Hz, 7.2 Hz, C[11]-H), 2.63 (1H, t, J = 6.9 Hz, C[3]-H), 2.50–2.38 (1H, m, C[2]-H), 2.26 (3H, s, 3 × C[9]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 158.9 (C[7]), 143.7 (C[13]), 134.4 (C[4]), 128.6 (C[14]), 128.2 (C[5]), 127.9 (C[15]), 127.1 (C[16]), 113.7 (C[6]), 86.8 (C[12]), 73.1 (C[1]), 72.5 (C[10]), 65.7 (C[11]), 55.3 (C[8]), 54.4 (C[3]), 44.0 (C[9]), 43.3 (C[2]). HRMS (ESI) m/z: [M + H]+ calcd for C32H34NO3 480.2533; found 480.2538.
1-(2-(3-Methoxyphenyl)-1-methylazetidin-3-yl)-2-(trityloxy)ethan-1-ol (1p)
It was prepared according to general procedure D using 2p (0.83 mmol, 400 mg). Purification by column chromatography on silica gel (0–20% MeOH in DCM) afforded the title compound (1p) as a yellowish solid (290 mg, 0.60 mmol, 72%). 1H NMR (CDCl3, 300 MHz) δH: 7.40–7.32 (6H, m, 6 × C[14]-H), 7.31–7.12 (11H, m, Ar-H), 6.98 (1H, s, C[5]-H), 6.88 (1H, d, J = 7.5 Hz, C[8]-H), 6.76 (1H, dd, J = 7.5 Hz, 1.8 Hz C[7]-H), 3.92 (1H, td, J = 6.6 Hz, 3.6 Hz, C[12]-H), 3.80–3.75 (4H, m, C[1]-H, 3 × C[10]-H), 3.38 (1H, t, J = 6.9 Hz, C[3]-H), 3.02 (1H, dd, J = 9.3 Hz, 3.6 Hz, C[13]-H), 2.92 (1H, dd, J = 9.0 Hz, 7.2 Hz, C[13]-H), 2.64 (1H, t, J = 6.9 Hz, C[3]-H), 2.51–2.42 (1H, m, C[2]-H), 2.29 (3H, s, 3 × C[11]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 159.7 (C[6]), 143.9 (C[4]), 143.7 (C[15]), 129.2 (C[8]), 128.6 (C[16]), 127.8 (C[17]), 127.1 (C[18]), 119.4 (C[9]), 113.1 (C[7]), 112.1 (C[5]), 86.8 (C[14]), 73.5 (C[1]), 72.7 (C[12]), 65.7 (C[13]), 55.2 (C[10]), 54.5 (C[3]), 44.2 (C[11]), 43.1 (C[2]). HRMS (ESI) m/z: [M + H]+ calcd for C32H34NO3 480.2533; found 480.2521.
1-(2-(4-Fluorophenyl)-1-methylazetidin-3-yl)-2-(trityloxy)ethan-1-ol (1q)
It was prepared according to general procedure D using 2q (0.43 mmol, 200 mg). Purification by column chromatography on silica gel (40–100% EtOAc in hexane) afforded the title compound (1q) as a yellowish solid (160 mg, 0.35 mmol, 80%). 1H NMR (CDCl3, 300 MHz) δH: 7.38–7.21 (17H, m, aromatic H), 6.92 (2H, t, J = 8.8 Hz, 2 × C[6]-H), 3.97–3.85 (1H, m, C[9]-H), 3.77 (1H, d, J = 7.8, Hz, C[1]-H), 3.37 (1H, t, J = 6.9 Hz, C[3]-H), 2.98 (1H, dd, J = 9.6 Hz, 3.9 Hz, C[10]-H), 2.88 (1H, dd, J = 9.3 Hz, 7.5 Hz, C[10]-H), 2.66 (1H, t, J = 6.9 Hz, C[3]-H), 2.46–2.36 (1H, m, C[2]-H), 2.26 (3H, s, 3 × C[8]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 162.1 (d, J = 243.3 Hz, C[7]), 143.7 (C[12]), 138.1 (d, J = 2.9 Hz, C[4]), 128.8 (C[5]), 128.6 (C[13]), 127.9 (C[14]), 127.2 (C[15]), 115.1 (d, J = 21.0 Hz, C[6]), 86.9 (C[10]), 72.7 (C[1]), 72.4 (C[9]), 65.8 (C[10]), 54.4 (C[3]), 44.1 (C[8]), 43.3 (C[2]). HRMS (ESI) m/z: [M + H]+ calcd for C31H31FNO2 468.2333; found 468.2322.
1-(1-Methyl-2-(naphthalen-2-yl)azetidin-3-yl)-2-(trityloxy)ethan-1-ol (1r)
It was prepared according to general procedure D using 2r (0.4 mmol, 200 mg). Purification by column chromatography on silica gel (40–100% EtOAc in hexane) afforded the title compound (1r) as a yellowish solid (104 mg, 0.21 mmol, 52%). 1H NMR (CDCl3, 300 MHz) δH: 8.28 (1H, d, J = 8.1 Hz, C[7]-H), 7.84 (2H, t, J = 6.9 Hz, C[10]-H, C[12]-H), 7.73 (1H, d, J = 8.1 Hz, C[13]-H), 7.46–7.34 (18H, m, aromatic H), 4.59 (1H, d, J = 7.5 Hz, C[1]-H), 4.00 (1H, q, J = 5.1 Hz, C[15]-H), 3.46 (1H, t, J = 6.6 Hz, C[3]-H), 2.98–2.84 (2H, m, C[3]-H, C[16]-H), 2.80–2.72 (1H, m, C[16]-H), 2.66–2.54 (1H, m, C[2]-H), 2.29 (3H, s, 3 × C[14]-H). 13C{1H} NMR (CDCl3, 300 MHz) δC: 143.6 (C[18]), 137.8 (C[4]), 133.8 (C[6]), 131.2 (C[11]), 128.6 (C[13]), 128.5 (C[19]), 127.8 (C[20]), 127.7 (C[10]), 127.1 (C[21]), 125.7 (C[7]), 125.6 (C[12]), 125.4 (C[85]), 125.2 (C[9]), 123.4 (C[5]), 86.7 (C[17]), 72.3 (C[15]), 69.7 (C[1]), 66.8 (C[16]), 54.4 (C[3]), 44.6 (C[14]), 44.3 (C[2]). HRMS (ESI) m/z: [M + H]+ calcd for C35H34NO2 500.2584; found 500.2560.
1-Methyl-2-phenylazetidin-3-yl(phenyl)methanol (1s_trans)
It was prepared according to general procedure D using 2s (0.87 mmol, 220 mg). Purification by preparative HPLC (using 0.4 g of NH4HCO3 in 1 L of water and acetonitrile) afforded the title compounds 1s_trans (51 mg, 0.20 mmol, 23%), 1s_cis (15 mg, 0.06 mmol, 7%), 10_trans (48 mg, 0.23 mmol, 19%), and 10_cis (30 mg, 0.23 mmol, 12%) as yellowish oils. Crude product: LC–MS area%: 1s_trans: 31%, 1s_cis: 11%, 10_trans: 33%, 10_cis: 25%
trans-1-Methyl-2-phenylazetidin-3-yl(phenyl)methanol (1s_trans)
1H NMR (DMSO-d6, 500 MHz) δH: 7.25–7.09 (10H, m, aromatic H), 5.44 (1H, bs, OH), 4.60 (1H, d, J = 5.9 Hz, C[9]-H), 3.82 (1H, d, J = 7.8 Hz, C[1]-H), 3.29 (1H, t, J = 6.8 Hz, C[3]-H), 2.91 (1H, dd, J = 8.4 Hz, 6.4 Hz, C[3]-H), 2.45 – (1H, m, C[2]-H), 2.22 (3H, s, 3 × C[8]-H). 13C{1H} NMR (DMSO-d6, 126 MHz) δC: 143.9 (C[10]), 142.5 (C[4]), 127.9 (C[11/C5]), 127.9 (C[11/C5]), 126.9 (C[13]), 126.7 (C[7]), 126.5 (C[12]), 126.2 (C[6]), 72.6 (C[9]), 71.2 (C[1]), 54.4 (C[3]), 47.8 (C[2]), 44.0 (C[8]). HRMS (ESI) m/z: [M + H]+ calcd for C17H20NO 254.1539; found 254.1544.
cis-1-Methyl-2-phenylazetidin-3-yl(phenyl)methanol (1s_cis)
1H NMR (CDCl3, 500 MHz) δH: 7.28–7.18 (8H, m, aromatic H), 7.08–7.04 (2H, m, aromatic H), 4.36 (1H, dd, J = 5.1 Hz, 3.5 Hz, C[9]-H), 3.34 (1H, d, J = 10.7 Hz, C[3]-H), 3.13 (1H, d, J = 9.2 Hz, C[2]-H), 3.09 (1H, dd, J = 9.2 Hz, 3.2 Hz, C[1]-H), 2.81 (1H, dd, J = 10.7 Hz, 5.9 Hz, C[3]-H), 2.50 (1H, bs, OH), 2.20 (3H, s, 3 × C[8]-H). 13C{1H} NMR (CDCl3, 126 MHz) δC: 141.1, 140.4, 128.6, 128.5, 128.1, 127.7, 127.6, 126.7, 79.9 (C[1]), 77.9 (C[9]), 66.1 (C[2]), 65.7 (C[3]), 40.4 (C[8]). HRMS (ESI) m/z: [M + H]+ calcd for C17H20NO 254.1539; found 254.1529.
trans-1-Methyl-4,5-diphenylpyrrolidin-3-ol (10s_trans)
1H NMR (DMSO-d6, 500 MHz) δH: 7.30–7.15 (8H, m, aromatic H), 7.09–7.06 (2H, m, aromatic H), 5.15 (1H, d, J = 5.5 Hz, OH), 4.23–4.17 (1H, m, C[3]-H), 3.84 (1H, d, J = 7.8 Hz, C[1]-H), 3.15 (1H, dd, J = 12.0 Hz, 10.1 Hz, C[4]-H), 2.94 (1H, dd, J = 10.1 Hz, 5.3 Hz, C[2]-H), 2.72 (1H, dd, J = 10.1 Hz, 7.4 Hz, C[4]-H), 2.03 (3H, s, 3 × C[9]-H). 13C{1H} NMR (DMSO-d6, 126 MHz) δC: 141.2, 141.2, 128.2, 128.2, 128.1, 127.6, 127.2, 126.3, 78.1 (C[1]), 76.3 (C[3]), 64.9 (C[2]), 64.9 (C[4]), 40.1 (C[9]). HRMS (ESI) m/z: [M + H]+ calcd for C17H20NO 254.1539; found 254.1549.
cis-1-Methyl-4,5-diphenylpyrrolidin-3-ol (10s_cis)
1H NMR (DMSO-d6, 500 MHz) δH: 7.02–6.98 (4H, m, aromatic H), 6.97–6.90 (4H, m, aromatic H), 6.87–6.84 (2H, m, aromatic H), 5.22 (1H, d, J = 4.5 Hz, OH), 4.36–4.31 (1H, m, C[3]-H), 3.84 (1H, d, J = 7.8 Hz, C[1]-H), 3.66 (1H, dd, J = 9.8 Hz, 6.4 Hz, C[4]-H), 3.22 (1H, dd, J = 7.8 Hz, 3.0 Hz, C[2]-H), 2.35 (1H, dd, J = 9.8 Hz, 5.6 Hz, C[4]-H), 2.15 (3H, s, 3 × C[9]-H). 13C{1H} NMR (DMSO-d6, 125 MHz) δC: 141.0, 139.3, 129.1, 127.9, 127.3, 127.2, 126.1, 125.5, 75.2 (C[3]), 72.9 (C[1]), 64.7 (C[4]), 60.6 (C[2]), 40.7 (C[9]). HRMS (ESI) m/z: [M + H]+ calcd for C17H20NO 254.1539; found 254.1542.
(1-Ethyl-2-phenylazetidin-3-yl)(phenyl)methanol (1t)
It was prepared according to general procedure D using 2t (0.75 mmol, 200 mg). Purification by column chromatography on silica gel (0–20% MeOH in DCM) afforded the title compound (1t) as a white solid (187 mg, 0.70 mmol, 93%). 1H NMR (CDCl3, 300 MHz) δH: 7.31–7.13 (10H, m, Ar-H), 4.67 (1H, d, J = 5.4 Hz, C[10]-H) 3.90 (1H, d, J = 8.1 Hz, C[1]-H), 3.45 (1H, t, J = 6.9 Hz, C[3]-H) 2.94 (1H, t, J = 6.9 Hz, C[3]-H) 2.68–2.40 (3H, m, C[2]-H, 2 × C[8]-H), 0.91 (3H, t, J = 7.2 Hz, 3 × C[9]-H). 13C{1H} NMR (CDCl3, 75 MHz) δC: 142.6 (C[11]), 142.5 (C[4]), 128.4 (C[5]), 128.0 (C[12]), 127.7 (C[14]), 126.9 (C[7]), 126.8 (C[6]), 126.1 (C[13]), 74.0 (C[10]), 70.6 (C[1]), 52.7 (C[3]), 52.3 (C[8]), 47.0 (C[2]), 12.6 (C[9]). HRMS (ESI) m/z: [M + H]+ calcd for C18H22NO 268.1696; found 268.1702.
(1-Benzyl-2-phenylazetidin-3-yl)(phenyl)methanol (1u)
It was prepared according to general procedure D using 2u (0.68 mmol, 225 mg). Purification by column chromatography on silica gel (0–10% MeOH in DCM) afforded the title compound (1u) as a white solid (146 mg, 0.44 mmol, 65%). 1H NMR (CDCl3, 300 MHz) δH: 7.31–7.19 (15H, m, Ar-H), 4.72 (1H, d, J = 7.8 Hz, C[14]-H), 4.10 (1H, d, J = 7.8 Hz, C[1]-H), 3.83 (1H, AB d, J = 12.9 Hz, C[8]-H), 3.45 (1H, AB d, J = 12.9 Hz, C[8]-H), 3.36 (1H, t, J = 6.9, Hz, C[3]-H), 3.03 (1H, t, J = 6.9 Hz, C[3]-H), 2.70–2.59 (1H, m, C[2]-H), 2.09 (1H, bs, OH). 13C{1H} NMR (CDCl3, 75 MHz) δC: 142.7 (C[14]), 142.3 (C[4]), 138.6 (C[9]), 128.9 (C[10]), 128.6 (C[15]), 128.4 (C[5]), 128.3 (C[11]), 128.0 (C[17]), 127.2 (C[7]), 127.1 (C[12]), 127.0 (C[6]), 126.3 (C[17]), 74.2 (C[13]), 70.0 (C[1]), 61.9 (C[8]), 52.7 (C[3]), 48.0 (C[2]). HRMS (ESI) m/z: [M + H]+ calcd for C23H24NO 330.1852; found 330.1866.
Acknowledgments
This work was funded by grants provided by the Hungarian Scientific Research Fund (OTKA K 104528). E.K. is grateful to the National Research, Development and Innovation Office (NKFIH) for Postdoctoral Excellence Award (PD 128612). We thank Miklós Nyerges, Tamás Gáti, and Barbara Balázs (Servier Research Institute of Medicinal Chemistry, Budapest) for spectroscopic assistance. We thank Gábor Turczel for help with experiments and technical support, as well as Imre G. Csizmadia for critical evaluation of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c01310.
NMR spectra of compounds 1, 2, and 9 and data of quantum chemical calculations (PDF)
Author Present Address
⊥ Institute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Magyar tudósok körútja 2, H-1117 Budapest, Hungary and Femtonics Ltd., Tűzoltó utca 59, H-1094 Budapest, Hungary.
Author Present Address
# Department of Organic Chemistry and Technology Budapest University of Technology and Economics, Budafoki út 8, H-1111 Budapest, Hungary.
Author Contributions
E.K. performed experiments and spectroscopic characterization and provided technical assistance. Theoretical calculations were carried out by Z.M. This manuscript was written by E.K., F.F., and Z.M. with comments from all authors.
The authors declare no competing financial interest.
Dedication
† This research paper is dedicated to the memory of Professor Sir Jack Edward Baldwin, who pioneered the selectivity of the ring-closure reactions of alicyclic compounds.
Supplementary Material
References
- Becker M. R.; Richardson A. D.; Schindler C. S. Functionalized Azetidines via Visible Light-Enabled Aza Paternò-Büchi Reactions. Nat. Commun. 2019, 10, 5095 10.1038/s41467-019-13072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandi A.; Cicchi S.; Cordero F. M. Novel Syntheses of Azetidines and Azetidinones. Chem. Rev. 2008, 108, 3988–4035. 10.1021/cr800325e. [DOI] [PubMed] [Google Scholar]
- Singh G. S.; D’hooghe M.; De Kimpe N.. Azetidines, Azetines and Azetes: Monocyclic; Katritzky A. R.; Ramsden C. A.; Scriven E. F. V.; Taylor R., Eds.; Elsevier: Oxford, 2008; pp 1–110. [Google Scholar]
- Clayden J.; Greeves N.; Warren S.. Organic Chemistry, 2nd ed.; Oxford University Press: Oxford, U.K., 2012. [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Shu Y.-Z.; Johnson B. M.; Yang T. J. Role of Biotransformation Studies in Minimizing Metabolism-Related Liabilities in Drug Discovery. AAPS J. 2008, 10, 178–192. 10.1208/s12248-008-9016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Jean D. J.; Fotsch C. Mitigating Heterocycle Metabolism in Drug Discovery. J. Med. Chem. 2012, 55, 6002–6020. 10.1021/jm300343m. [DOI] [PubMed] [Google Scholar]
- Kerns E. H.; Di L.. Drug-Like Properties: Concepts, Structure Design and Methods, 1st ed.; Academic Press, 2008. [Google Scholar]
- Hosny Y.; Abutaleb N. S.; Omara M.; Alhashimi M.; Elsebaei M. M.; Elzahabi H. S.; Seleem M. N.; Mayhoub A. S. Modifying the Lipophilic Part of Phenylthiazole Antibiotics to Control Their Drug-Likeness. Eur. J. Med. Chem. 2020, 185, 111830 10.1016/j.ejmech.2019.111830. [DOI] [PubMed] [Google Scholar]
- Takhi M.; Munikumar M.; Praveena K.; Takhi M.; Rao B. N. V. M.; Ramakanth G.; Sivaranjani J.; Mulik S.; Reddy Y. R.; Narasimha K.; Pallavi R.; Lakshminarasimhan A.; Panigrahi S. K.; Antony T.; Abdullah I.; Lee Y. K.; Ramachandra M.; Yusof R.; Rahman N. A.; Subramanya H.; et al. Discovery of Azetidine Based Ene-Amides as Potent Bacterial Enoyl ACP. Eur. J. Med. Chem. 2014, 84, 382–394. 10.1016/j.ejmech.2014.07.036. [DOI] [PubMed] [Google Scholar]
- Chavan P.; Salve A.; Jadhav S.; Pansare D.; Rai M. Ultrasound Assisted, Synthesis of N-(7-(R)-2-Oxa-8-Azabicyclo[4.2.0]Octan-8-Yl)Isonicotinamide Derivatives and Their Biological Evaluation. J. Heterocycl. Chem. 2020, 57, 1228–1235. 10.1002/jhet.3860. [DOI] [Google Scholar]
- Michalska K.; Chang C.; Maltseva N. I.; Jedrzejczak R.; Robertson G. T.; Gusovsky F.; McCarren P.; Schreiber S. L.; Nag P. P.; Joachimiak A. Allosteric Inhibitors of Mycobacterium Tuberculosis Tryptophan Synthase. Protein Sci. 2020, 29, 779–788. 10.1002/pro.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson P.; Griffin I.; Tucker C.; Smith D.; Oechsle O.; Phelan A.; Stebbing J.; et al. Baricitinib as Potential Treatment for 2019-NCoV Acute Respiratory Disease. Lancet 2020, 395, e30–e31. 10.1016/S0140-6736(20)30304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahraman M.; Govek S. P.; Nagasawa J. Y.; Lai A.; Bonnefous C.; Douglas K.; Sensinta J.; Liu N.; Lee K.; Aparicio A.; J. Qian J.; Shao G.; Prudente R.; Joseph J. D.; Darimont B.; Brigham D.; Heyman R.; Rix P. J.; Hager H.; Smith N. D.; et al. Maximizing ER - α Degradation Maximizes Activity in a Tamoxifen- Resistant Breast Cancer Model: Identification of GDC-0927. ACS Med. Chem. Lett. 2019, 10, 50–55. 10.1021/acsmedchemlett.8b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M.; Demartino M. P.; Kalita B.; Kristam R.. 2-(4-Chlorophenoxy)-N-((1 -(2-(4-Chlorophenoxy)Ethynazetidin-3-Yl)Methyl)Acetamide Derivatives and Related Compounds as ATF4 Inhibitors for Treating Cancer and Other Diseases. WO2019008507A1, 2019.
- Kværnø L.; Werder M.; Hauser H.; Carreira E. M. Synthesis and in Vitro Evaluation of Inhibitors of Intestinal Cholesterol Absorption. J. Med. Chem. 2005, 48, 6035–6053. 10.1021/jm050422p. [DOI] [PubMed] [Google Scholar]
- Feskov I. O.; Kondratov I. S.; Grygorenko O. O.; et al. 3-((Hetera)Cyclobutyl)Azetidines, “Stretched” Analogues of Piperidine, Piperazine, and Morpholine: Advanced Building Blocks for Drug Discovery. J. Org. Chem. 2019, 84, 1363–1371. 10.1021/acs.joc.8b02822. [DOI] [PubMed] [Google Scholar]
- Sonesson C.; Pettersson F.. Novel Azetidine Derivatives Useful as Modulators of Cortical Catecholaminergic Catecholaminergic Neurotransmission. CN109963834A, 2017.
- Samardzic K.; Rodgers K. J. Cell Death and Mitochondrial Dysfunction Induced by the Dietary Non - Proteinogenic Amino Acid l - Azetidine - 2 - Carboxylic Acid (Aze). Amino Acids 2019, 51, 1221–1232. 10.1007/s00726-019-02763-w. [DOI] [PubMed] [Google Scholar]
- Maetani M.; Zoller J.; Melillo B.; Verho O.; Kato N.; Pu J.; et al. Synthesis of a Bicyclic Azetidine with In Vivo Antimalarial Activity Enabled by Stereospeci Fi c, Directed C(Sp3) – H Arylation. J. Am. Chem. Soc. 2017, 139, 11300–11306. 10.1021/jacs.7b06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maianti J. P.; Tan G. A.; Vetere A.; Welsh A. J.; Wagner B. K.; Seeliger M. A.; Liu D. R. Activity of Insulin-Degrading Enzyme. Nat. Chem. Biol. 2019, 15, 565–574. 10.1038/s41589-019-0271-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Žukauskaitė A.; Moretto A.; Peggion C.; De Zotti M.; Šačkus A.; Formaggio F.; De Kimpe N.; Mangelinckx S. Synthesis and Conformational Study of Model Peptides Containing N-Substituted 3-Aminoazetidine-3-Carboxylic Acids. Eur. J. Org. Chem. 2014, 2014, 2312–2321. 10.1002/ejoc.201301741. [DOI] [Google Scholar]
- Žukauskaitė A.; Mangelinckx S.; Buinauskaitė V.; Šačkus A.; De Kimpe N. Synthesis of New Functionalized Aziridine-2- and Azetidine-3-Carboxylic Acid Derivatives of Potential Interest for Biological and Foldameric Applications. Amino Acids 2011, 41, 541–558. 10.1007/s00726-011-0879-1. [DOI] [PubMed] [Google Scholar]
- Feskov I. O.; Chernykh A. V.; Kuchkovska Y. O.; Daniliuc C. G.; Kondratov I. S.; Grygorenko O. O. 3-((Hetera)Cyclobutyl)Azetidines – “Stretched” Analogues of Piperidine, Piperazine and Morpholine: Advanced Building Blocks for Drug Discovery. J. Org. Chem. 2019, 84, 1363–1371. 10.1021/acs.joc.8b02822. [DOI] [PubMed] [Google Scholar]
- Drouillat B.; Dorogan I. V.; Kletskii M.; Burov O. N.; Couty F. Competitive Ring Expansion of Azetidines into Pyrrolidines and/or Azepanes. J. Org. Chem. 2016, 81, 6677–6685. 10.1021/acs.joc.6b01325. [DOI] [PubMed] [Google Scholar]
- Cai W.; Wu J.; Zhang H.; Jalani H. B.; Li G.; Lu H. Rh-Catalyzed Chemoselective [4 + 1] Cycloaddition Reaction toward Diverse 4-Methyleneprolines. J. Org. Chem. 2019, 84, 10877–10891. 10.1021/acs.joc.9b01466. [DOI] [PubMed] [Google Scholar]
- Dolfen J.; Boydas E. B.; Van Speybroeck V.; Catak S.; Van Hecke K.; D’hooghe M. Asymmetric Synthesis of 3,4-Disubstituted 2-(Trifluoromethyl)Pyrrolidines through Rearrangement of Chiral 2-(2,2,2-Trifluoro-1-Hydroxyethyl)Azetidines. J. Org. Chem. 2017, 82, 10092–10109. 10.1021/acs.joc.7b01241. [DOI] [PubMed] [Google Scholar]
- Kovács E.; Faigl F.; Mucsi Z.; Nyerges M.; Hegedűs L. Hydrogenolysis of N- and O-Protected Hydroxyazetidines over Palladium: Efficient and Selective Methods for Ring Opening and Deprotecting Reactions. J. Mol. Catal. A: Chem. 2014, 395, 217–224. 10.1016/j.molcata.2014.08.027. [DOI] [Google Scholar]
- Gleede T.; Rupar P. A.; Wurm F. R.; et al. Aziridines and Azetidines: Building Blocks for Polyamines by Anionic and Cationic Ring-Opening Polymerization. Polym. Chem. 2019, 10, 3257–3283. 10.1039/C9PY00278B. [DOI] [Google Scholar]
- Grimm J. B.; Muthusamy A. K.; Liang Y.; Brown T. A.; Lemon W. C.; Patel R.; Lu R.; Macklin J. J.; Keller P. J.; Ji N.; Lavis L. D. A General Method to Fine-Tune Fluorophores for Live-Cell and in Vivo Imaging. Nat. Methods 2017, 14, 987–994. 10.1038/nmeth.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; English B. P.; Chen J.; Slaughter J. P.; Zhang Z.; Revyakin A.; Patel R.; Macklin J. J.; Normanno D.; Singer R. H.; Lionnet T.; Lavis L. D. A General Method to Improve Fluorophores for Live-Cell and Single-Molecule Microscopy. Nat. Methods 2015, 12, 244–253. 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandioso A.; Palau M.; Bresolí-Obach R.; Galindo A.; Rovira A.; Bosch M.; Nonell S.; Marchán V. High Photostability in Nonconventional Coumarins with Far-Red/NIR Emission through Azetidinyl Substitution. J. Org. Chem. 2018, 83, 11519–11531. 10.1021/acs.joc.8b01422. [DOI] [PubMed] [Google Scholar]
- Bassolino G.; Nançoz C.; Thiel Z.; Bois E.; Vauthey E.; Rivera-Fuentes P. Photolabile Coumarins with Improved Efficiency through Azetidinyl Substitution. Chem. Sci. 2018, 9, 387–391. 10.1039/C7SC03627B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.-C.; Zhang Q.-J.; Zhao W.-X.; Wang X.-D.; Ding X.; Jing T.-T.; Song M.-P. Evaluation of Enantiopure N-(Ferrocenylmethyl)Azetidin-2-Yl(Diphenyl)Methanol for Catalytic Asymmetric Addition of Organozinc Reagents to Aldehydes. J. Org. Chem. 2008, 73, 168–176. 10.1021/jo701943x. [DOI] [PubMed] [Google Scholar]
- Yang X.-C.; Liu M.-M.; Mathey F.; Yang H.; Hua Y.-Z.; Wang M.-C. Access to Chiral 2,5-Pyrrolidinyl Dispirooxindoles via Dinuclear Zinc-Catalyzed Asymmetric Cascade Reactions. J. Org. Chem. 2019, 84, 7762–7775. 10.1021/acs.joc.9b00645. [DOI] [PubMed] [Google Scholar]
- De Kimpe N.3- and 4-Membered Rings, with All Fused Systems Containing 3- and 4-Membered Rings. In Comprehensive Heterocyclic Chemistry II; Padwa A., Ed.; Elsevier: Oxford, U.K., 1996; p 507. [Google Scholar]
- Cromwell N. H.; Phillips B. The Azetidines. Recent Synthetic Developments. Chem. Rev. 1979, 79, 331–358. 10.1021/cr60320a003. [DOI] [Google Scholar]
- Singh G. S.Advances in Synthesis and Chemistry of Azetidines. In Advances in Heterocyclic Chemistry; Scriven E. F. V.; Ramsden C. A., Eds.; Academic Press: 2020; Vol. 130, pp 1–74. [Google Scholar]
- Couty F.; Evano G.; Prim D. Synthesis of Chiral Non Racemic Azetidines. Mini-Rev. Org. Chem. 2004, 1, 133–148. 10.2174/1570193043488845. [DOI] [Google Scholar]
- Zenzola M.; Degennaro L.; Trinchera P.; Carroccia L.; Giovine A.; Romanazzi G.; Mastrorilli P.; Rizzi R.; Pisano L.; Luisi R. Harnessing the Ortho-Directing Ability of the Azetidine Ring for the Regioselective and Exhaustive Functionalization of Arenes. Chem. – Eur. J. 2014, 20, 12190–12200. 10.1002/chem.201403141. [DOI] [PubMed] [Google Scholar]
- Degennaro L.; Zenzola M.; Trinchera P.; Carroccia L.; Giovine A.; Romanazzi G.; Falcicchio A.; Luisi R. Regioselective Functionalization of 2-Arylazetidines: Evaluating the Ortho-Directing Ability of the Azetidinyl Ring and the α-Directing Ability of the N-Substituent. Chem. Commun. 2014, 50, 1698–1700. 10.1039/c3cc48555b. [DOI] [PubMed] [Google Scholar]
- Antermite D.; Degennaro L.; Luisi R. Recent Advances in the Chemistry of Metallated Azetidines. Org. Biomol. Chem. 2016, 15, 34–50. 10.1039/C6OB01665K. [DOI] [PubMed] [Google Scholar]
- Feula A.; Male L.; Fossey J. S. Diastereoselective Preparation of Azetidines and Pyrrolidines. Org. Lett. 2010, 12, 5044–5047. 10.1021/ol102215e. [DOI] [PubMed] [Google Scholar]
- Music A.; Baumann A. N.; Eisold M.; Didier D. Regiodivergent Stereoselective Access to Fused Alkylideneazetidines. J. Org. Chem. 2018, 83, 783–792. 10.1021/acs.joc.7b02786. [DOI] [PubMed] [Google Scholar]
- Gianatassio R.; Kadish D. Direct Alkylation of 1-Azabicyclo[1.1.0]Butanes. Org. Lett. 2019, 21, 2060–2063. 10.1021/acs.orglett.9b00321. [DOI] [PubMed] [Google Scholar]
- Delany P. K.; Hodgson D. M. Synthesis and Homologation of an Azetidin-2-Yl Boronic Ester with α-Lithioalkyl Triisopropylbenzoates. Org. Lett. 2019, 21, 9981–9984. 10.1021/acs.orglett.9b03901. [DOI] [PubMed] [Google Scholar]
- Clader J. W. The Discovery of Ezetimibe: A View from Outside the Receptor. J. Med. Chem. 2004, 47, 1–9. 10.1021/jm030283g. [DOI] [PubMed] [Google Scholar]
- Werder M.; Hauser H.; Carreira E. M. Synthesis and in Vitro Evaluation of Inhibitors of Intestinal Cholesterol Absorption. J. Med. Chem. 2005, 48, 6035–6053. 10.1021/jm050422p. [DOI] [PubMed] [Google Scholar]
- Werder M.; Hauser H.; Carreira E. M. Carbohydrate Sulfonyl Chlorides for Simple, Convenient Access to Glycoconjugates. Org. Lett. 2005, 7, 1145–1148. 10.1021/ol0502127. [DOI] [PubMed] [Google Scholar]
- Smith E. M.; Sorota S.; Kim H. M.; Mckittrick B. A.; Nechuta T. L.; Bennett C.; Knutson C.; Burnett D. A.; Kieselgof J.; Tan Z.; Rindgen D.; Bridal T.; Zhou X.; Jia Y.; Dong Z.; Mullins D.; Zhang X.; Priestley T.; Correll C. C.; Tulshian D.; Czarniecki M.; Greenlee W. Bioorganic & Medicinal Chemistry Letters T-Type Calcium Channel Blockers: Spiro-Piperidine Azetidines and Azetidinones — Optimization, Design and Synthesis. Bioorganic Med. Chem. Lett. 2010, 20, 4602–4606. 10.1016/j.bmcl.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Andresini M.; De Angelis S.; Uricchio A.; Visaggio A.; Romanazzi G.; Ciriaco F.; Corriero N.; Degennaro L.; Luisi R. Azetidine-Borane Complexes: Synthesis, Reactivity, and Stereoselective Functionalization. J. Org. Chem. 2018, 83, 10221–10230. 10.1021/acs.joc.8b01441. [DOI] [PubMed] [Google Scholar]
- Ma X.; Zhao H.; Binayeva M.; Ralph G.; Diane M.; Zhao S.; Wang C.-Y.; Biscoe M. R. A General Approach to Stereospecific Cross-Coupling Reactions of Nitrogen-Containing Stereocenters. Chem 2020, 6, 781–791. 10.1016/j.chempr.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz K. N.; Chiappini N. D.; Du Bois J. Intermolecular Sp3-C–H Amination for the Synthesis of Saturated Azacycles. Org. Lett. 2020, 22, 1687–1691. 10.1021/acs.orglett.9b04096. [DOI] [PubMed] [Google Scholar]
- Quinodoz P.; Drouillat B.; Wright K.; Marrot J.; Couty F. N-Arylazetidines: Preparation through Anionic Ring Closure. J. Org. Chem. 2016, 81, 2899–2910. 10.1021/acs.joc.6b00169. [DOI] [PubMed] [Google Scholar]
- Faigl F.; Kovács E.; Turczel G.; Szöllősy Á.; Mordini A.; Balázs L.; Holczbauer T.; Czugler M. Novel Stereoselective Synthesis of 1,2,3-Trisubstituted Azetidines. Tetrahedron: Asymmetry 2012, 23, 1607–1614. 10.1016/j.tetasy.2012.10.014. [DOI] [Google Scholar]
- Baldwin J. E. Rules for Ring Closure. J. Chem. Soc. Chem. Commun. 1976, 18, 734–736. 10.1039/c39760000734. [DOI] [Google Scholar]
- Gilmore K.; Mohamed R. K.; Alabugin I. V. The Baldwin Rules: Revised and Extended. WIREs Comput. Mol. Sci. 2016, 6, 487–514. 10.1002/wcms.1261. [DOI] [Google Scholar]
- Fiser B.; Cuerva J. M.; Gómez-Bengoa E. Baldwin-Type Rules for Metal-Controlled Intramolecular Migratory Insertions. A Computational Study of Ni, Pd, and Pt Case. Organometallics 2018, 37, 390–395. 10.1021/acs.organomet.7b00812. [DOI] [Google Scholar]
- Gilmore K.; Alabugin I. V. Cyclizations of Alkynes: Revisiting Baldwin’ s Rules for Ring Closure. Chem. Rev. 2011, 111, 6513–6556. 10.1021/cr200164y. [DOI] [PubMed] [Google Scholar]
- Thurner A.; Faigl F.; Tőke L.; Mordini A.; Valacchi M.; Reginato G.; Czira G. Useful Base Promoted Elaborations of Oxiranyl Ethers. Tetrahedron 2001, 57, 8173–8180. 10.1016/S0040-4020(01)00790-6. [DOI] [Google Scholar]
- Dittmer D. C.; Discordia R. P.; Zhang Y.; Murphy C. K.; Kumar A.; Pepito A. S.; Wang Y. A Tellurium Transposition Route to Allylic Alcohols: Overcoming Some Limitations of the Sharpless-Katsuki Asymmetric Epoxidation. J. Org. Chem. 1993, 58, 718–731. 10.1021/jo00055a029. [DOI] [Google Scholar]
- Unkelbach C.; Rosenbaum H. S.; Strohmann C. Direct Benzylic Metalation of a Phenethylamine Derivative: Potassium as the Key to Both Generation and Stabilization of a “Labile Anion”. Chem. Commun. 2012, 48, 10612–10614. 10.1039/c2cc35888c. [DOI] [PubMed] [Google Scholar]
- Kovács E.; Huszka B.; Gáti T.; Nyerges M.; Faigl F.; Mucsi Z. Chemoselective Strategy for the Direct Formation of Tetrahydro-2,5-Methanobenzo[c]Azepines or Azetotetrahydroisoquinolines via Regio- and Stereoselective Reactions. J. Org. Chem. 2019, 84, 7100–7112. 10.1021/acs.joc.9b00798. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Function. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 16, revision C.01; Gaussian Inc.: Wallingford, CT, 2016. [Google Scholar]
- Mucsi Z.; Szabó A.; Hermecz I.; Kucsman Á.; Csizmadia I. G. Modeling Rate-Controlling Solvent Effects. The Pericyclic Meisenheimer Rearrangement of N-Propargylmorpholine N-Oxide. J. Am. Chem. Soc. 2005, 127, 7615–7631. 10.1021/ja042227q. [DOI] [PubMed] [Google Scholar]
- Padwa A.; Dean D. C.; Fairfax D. J.; Xu S. L. Stereo and Electronic Effects in the Rhodium(II)-Mediated Synthesis of Polycyclic Lactones and Lactams from.Alpha.-Diazo Ester and Amide Precursors. J. Org. Chem. 1993, 58, 4646–4655. 10.1021/jo00069a030. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.