

Abstract
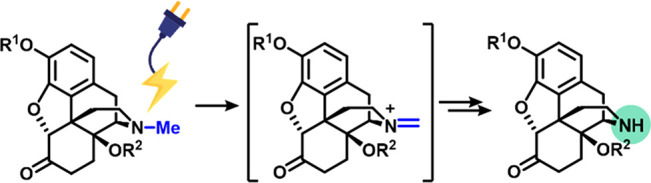
The most challenging step in the preparation of many opioid antagonists is the selective N-demethylation of a 14-hydroxymorphinan precursor. This process is carried out on a large scale using stoichiometric amounts of hazardous chemicals like cyanogen bromide or chloroformates. We have developed a mild reagent- and catalyst-free procedure for the N-demethylation step based on the anodic oxidation of the tertiary amine. The ensuing intermediates can be readily hydrolyzed to the target nor-opioids in very good yields.
The ongoing opioids crisis, which is causing nearly 1000 overdose-related deaths per week in the United States,1 has dramatically increased the demand for opioid antagonists like naloxone or naltrexone.2 The need for these lifesaving antidotes for drug overdose treatment has, unfortunately, also led to a significant rise in their price,3 which reduces their availability to less favored communities.4 Decreasing the cost of production for opioid antagonists via more efficient synthetic routes is therefore highly desired and a very active field of research.5,6 The most challenging step in the preparation of these 14-hydroxy morphinans is the selective removal of the N-methyl group from the relatively complex morphine precursor (Figure 1A). The resulting nor-derivative (i.e., the ensuing secondary amine) is a key intermediate from which a range of essential medicines, including naloxone or naltrexone, can be synthesized by simple realkylation with the corresponding alkyl bromide.5,6 The N-demethylation process is currently carried out using excess amounts of harmful electrophilic reagents like cyanogen bromide (via the von Braun reaction)7 or chloroalkyl formates.8 The combination of stoichiometric amounts of peroxides and acylating agents (classical Polonovski reaction) or metal reductants (nonclassical Polonovsky reaction) has also been applied (Figure 1B).9 Not surprisingly, more benign alternatives have been actively investigated during the past two decades, including palladium-catalyzed10 and photochemical11 aerobic oxidations as well as chemoenzymatic procedures.12 However, these methods have not been adopted by industry.12
Figure 1.

Importance and strategies for the N-demethylation of 14-hydroxy opioids for the preparation of overdose antidotes.
Notably, all N-demethylation reactions previously mentioned entail either an oxidation of the N–CH3 group or withdrawal of the nitrogen electron pair by an electrophilic reagent to initiate the demethylation process. Indeed, iminium cation intermediates have been invoked for the palladium catalyzed10 and photochemical routes11 as well as for the nonclassical Polonovsky reaction.6 We hypothesized that, under suitable electrochemical conditions, the N-methyl group could be anodically oxidized to the corresponding iminium cation in a two-electron process. Trapping of the iminium cation by the 14-hydroxy group or acyl transfer from the same position would generate intermediates that can be readily hydrolyzed to the target nor-derivative (Figure 1C).10 This electrochemical strategy would not require any external oxidant and, ideally, could be carried out in benign solvents under mild conditions, delivering a highly convenient, sustainable,13 and inexpensive N-demethylation methodology.14
At the onset of our investigation, cyclic voltammograms of the 14-hydroxy precursor oxycodone (1a) and its O-acetyl-protected derivative 14-acetyloxycodone (1b) were recorded to assess whether the target tertiary amine could be selectively oxidized (Figure 2A). The presence of a highly activated aromatic ring can cause undesired oxidations, leading to the formation of biaryl dimers.15 Analogous voltammograms were obtained for both compounds. The oxidation of the amine was observed at ca. Ep/2 = 1.1 V vs SCE, following the typical irreversible pattern for tertiary amines.16 The second oxidation peak, corresponding to the oxidation of the aromatic ring, appeared at Ep/2 = 1.6 V vs SCE. In this case, the reversibility of the electron transfer could be observed by increasing the scan rate (Figure S1), indicating a relatively slow degradation (i.e., dimerization) of the oxidized species at the low concentrations utilized for the recording of the voltammograms. Most notably, the difference in oxidation potentials between the two moieties (ca. 0.5 V) pointed to a selective reaction, probably even under galvanostatic conditions.
Figure 2.

(A) Cyclic voltammograms of opioid precursors 1a,b and (B) optimization of the electrolysis conditions using 1a as the model. aGeneral conditions: undivided cell; 0.15 mmol of substrate in 3 mL of solvent; 5 mL of IKA Electrasyn vial; (+)C: graphite anode; (−)Fe: stainless steel cathode. bDetermined by HPLC peak area percent (205 nm). cPercent of product with respect to all peaks except the substrate (HPLC peak area percent, 205 nm).
An initial screening of the reaction conditions was carried out using the electrolysis of oxycodone (1a) as a model, which was expected to provide oxazolidine 2a upon the formation of an iminium cation (Figure 2B).17 All reactions were performed in an undivided cell (IKA Electrasyn) at room temperature. To our delight, the first attempt using graphite as the anode and stainless steel as the cathode material in acetonitrile, using LiClO4 as the supporting electrolyte, provided 29% conversion to 2a and very good selectivity after 2 F/mol of charge (96 min) had been applied (Figure 2B, entry 1). The main side products observed were the expected biaryl dimers.15 (See Figure S3.) Dimerization can take place for both the starting oxycodone (1a) and the oxazolidine electrolysis product (2a), and thus the generation of small amounts of dimers in a late stage of the reaction was expected. A screen of several solvent systems and supporting electrolytes (entries 2–8) revealed that the utilization of quaternary ammonium salts had a significant beneficial influence on the reaction (entries 2 and 3). The poorer performance of the lithium salt could be ascribed to the formation of a complex with the tertiary amine.18 As expected, the addition of protic solvents had a positive effect, providing a source of protons for the concurrent cathodic reduction (entries 6 and 8). The utilization of pure methanol as a solvent resulted in a lower conversion (entry 5), with a 4:1 combination of MeCN and MeOH being the best solvent system (entry 8). Several electrode materials were also evaluated. (See Table S1.) None of the electrode combinations provided significant improvements with respect to graphite/stainless steel. Indeed, the utilization of platinum as an anode material, for example, resulted in lower conversion under otherwise identical conditions (entry 8 vs 9). Excellent results were achieved by applying a 20% excess of electricity (2.4 F/mol, 116 min) under a current of 5 mA in MeCN/MeOH with Et4NBF4 as the supporting electrolyte (entry 10).
With the optimal conditions in hand, several key 14-hydroxy and 14-acetyl opioid precursors were electrolyzed, leading to cyclization to oxazolidines or O,N-acyl transfer, respectively (Figure 3A). The optimal reaction parameters were directly utilized without further reoptimization. The very good conversions and selectivities obtained for all cases enabled a simple workup procedure entailing the evaporation of solvent followed by purification by short-path column chromatography over neutral alumina. In addition to oxycodone (1a) and O-acetyloxycodone (1b), O-acetyl codeinone (1c) was also successfully subjected to the electrochemical oxidation, resulting in a highly selective O,N-acyl transfer (vide infra).10c
Figure 3.

(A) Electrochemical oxazolidination and N-demethylative acyl transfer of several opioid precursors (isolated yields are given). (B) Transfer of the electrochemical reaction to a flow cell and “one-pot” transformation to the nor-derivative.
Opioid antagonists such as naloxone or naltrexone generally feature a 3-hydroxy group (cf. Figure 1), which is generated by O-demethylation of the naturally occurring 3-methoxy opiates, at either an early19 or a late20 stage of the synthetic route. The presence of phenols is particularly problematic during anodic oxidations due to their relatively low oxidation potentials.16 Indeed, the cyclic voltammetry of 3,14-dihydroxy opioids typically shows product degradation starting at ca. 0.6 V vs SCE.21 Gratifyingly, the selective electrochemical N-demethylation of 14-hydroxymorphinone was enabled by first generating its 3,14-O-diacetyl derivative (1d) (Figure 3A). Cyclic voltammograms of the opioid precursor and the diacetyl derivative showed a clear differentiation between the amine and aryl oxidation peaks upon protection, pointing to a selective electrochemical reaction. (See Figure S2.) Indeed, electrochemical N-demethylative acyl transfer under the optimal conditions resulted in 2d in 78% isolated yield. Notably, this compound can be easily transformed to noroxymorphone by acidic workup.10e
To improve the scalability of our electrochemical protocol, the reaction was transferred to a flow electrolysis cell22 using as a model the electrolysis of oxycodone (1a) (Figure 3B). The flow cell consisted of a parallel plate arrangement, with the two electrodes separated by a 0.3 mm chemically resistant Mylar film incorporating a reaction channel.23 (The flow electrolysis cell is described in the Supporting Information.) The reaction mixture was pumped through the cell and recirculated at a flow rate of 2 mL/min until the desired amount of charge had been passed. Using an identical reaction mixture as in batch mode and a current of 10 mA, the outcome of the reaction was analogous. Nearly identical conversion and selectivity to the oxazolidine intermediate as that in batch was obtained. It is worth noting that direct treatment of the crude electrolysis reaction mixture with HCl delivered the target nor-derivative 3a in 75% overall isolated yield. Furthermore, it should be mentioned that no inert atmosphere or anhydrous solvents were required to perform this transformation. This N-demethylation, which generally is executed using rather hazardous reagents in stoichiometric quantities (cf. Figure 1B), here is driven simply by electricity via inexpensive electrode materials, producing hydrogen as a byproduct.
The proposed mechanism for the reaction (Figure 4A) starts with a two-electron oxidation of the tertiary amine with the release of one proton, generating the key iminium cation intermediate. In the case of the 14-hydroxy opioids, rapid intramolecular 1,5-cyclization, with the release of a second proton, generates the oxazolidine intermediate. The two protons released during the process are reduced at the cathode, producing hydrogen gas. In the case of the O-acetyl-protected derivatives, with no hydroxy group available for an intramolecular cyclization, the iminium cation intermediate is trapped by the methanol present as a cosolvent. The resulting N,O-acetal intermediate reacts with a second molecule of methanol, releasing the N-methyl carbon as dimethoxymethane via a cyclic intermediate. Alternatively, the release of dimethoxymethane may directly provide the free secondary amine, which then undergoes rapid O,N-acyl transfer. This mechanism is analogous to the pathway proposed for palladium-promoted acyl-transfer reactions.10c
Figure 4.

(A) Proposed mechanism for the electrochemical oxazolidination and demethylative O,N-acyl transfer of opioids and (B–D) experiments carried out to gain mechanistic insights.
To gain further insights into the reaction mechanism, several experiments were performed. The kinetic isotope effect (KIE) was evaluated using oxycodone-d3 (1a-d3) in a parallel single-component experiment (Figure 4B). A moderate KIE (kH/kD = 1.5) was observed, suggesting that the second oxidation event, with the release of a proton, is the rate-determining step of the reaction. The KIE value is in agreement with a proton-coupled electron transfer (PCET) in which the proton donor is close to the acceptor (the solvent in this case).24 The intermediacy of the iminium cation could be confirmed by its trapping with cyanide and diphenylamine (Figure 4C). This was achieved by generating the iminium ion in a “cation pool”25 at −45 °C using a divided cell (lower temperatures could not be reached in acetonitrile) and adding the nucleophile after the electricity had been turned off. (See the Supporting Information for details.) The trapping products were obtained in low amounts, indicating that the temperature was not sufficiently low to permit accumulation of the cation. To ensure that the observed products were the result of trapping of the iminium cation and not of ring opening of the oxazolidine, the latter was treated with excess amounts of the nucleophilic reagents. No reaction was observed. Finally, direct observation of the iminium ion by infrared spectroscopy was also attempted, again using the “cation pool” methodology (Figure 4D). In this case, an FTIR probe was immersed in the anodic chamber of the divided cell.26 Oxycodone derivative 6-oxyodol (1e), with the ketone group reduced to an alcohol, was used as the substrate to eliminate interference of the carbonyl signal from the IR. Gratifyingly, under electrolysis, a weak peak appeared at ca. 1657 cm–1 that could be ascribed to the C=N stretch of the intermediate.27 The weak signal observed supported the hypothesis that the iminium cation is not sufficiently stable at −45 °C.
In summary, we have designed a catalyst- and reagent-free electrochemical methodology for the N-demethylation of 14-hydroxy opioids, the crucial step in the synthesis of important opioid antagonists such as naloxone or naltrexone. The synthetic strategy is based on the two-electron anodic oxidation of the tertiary amine, generating an iminium ion that rapidly undergoes intramolecular oxazolidination or demethylative O,N-acyl transfer. The procedure has been evaluated for several important opioid API (active pharmaceutical ingredient) precursors including oxycodone and 3,14-diacetylmorphinone. The protocol has been transferred to a flow electrolysis cell, enabling its scale-up. Notably, the key nor-derivatives could be prepared in one pot by simply adding hydrochloric acid to the crude electrolysis reaction mixture. This strategy avoids the use of stoichiometric amounts of hazardous electrophilic reagents and provides the target compounds in good yields.
Acknowledgments
The CC FLOW Project (Austrian Research Promotion Agency FFG No. 862766) is funded through the Austrian COMET Program by the Austrian Federal Ministry of Transport, Innovation and Technology (BMVIT), the Austrian Federal Ministry of Science, Research and Economy (BMWFW), and the State of Styria (Styrian Funding Agency (SFG)).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.0c02424.
Experimental details, HPLC derivatization procedure, cyclic voltammetry, kinetic isotope effect and chemical trapping experiments, FTIR detection of the iminium ion, details of the flow electrolysis cell and setup, and copies of NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hedegaard H.; Miniño A. M.; Warner M.. Drug Overdose Deaths in the United States, 1999–2018; NCHS Data Brief, no. 356; National Center for Health Statistics: Hyattsville, MD, 2020.
- Freeman P. R.; Hankosky E. R.; Lofwall M. R.; Talbert J. C. The Changing Landscape of Naloxone Availability in the United States, 2011 – 2017. Drug Alcohol Depend. 2018, 191, 361–364. 10.1016/j.drugalcdep.2018.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Shah N. D.; Ross J. S. The Rising Price of Naloxone — Risks to Efforts to Stem Overdose Deaths. N. Engl. J. Med. 2016, 375, 2213–2215. 10.1056/NEJMp1609578. [DOI] [PubMed] [Google Scholar]
- a Lozo K. W.; Nelson L. S.; Ramdin C.; Calello D. P. Naloxone Deserts in NJ Cities: Sociodemographic Factors Which May Impact Retail Pharmacy Naloxone Availability. J. Med. Toxicol. 2019, 15, 108–111. 10.1007/s13181-019-00700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lagisetty P.; Bohnert A.; Fendrick A. M.. Meeting The Opioid Challenge: Getting Naloxone to Those Who Need it Most. Health Affairs Blog 2018. [Google Scholar]
- Rinner U.; Hudlicky T. Synthesis of Morphine Alkaloids and Derivatives. Top. Curr. Chem. 2011, 309, 33–66. 10.1007/128_2011_133. [DOI] [PubMed] [Google Scholar]
- Thavaneswaran S.; McCamley K.; Scammells P. J. N-Demethylation of Alkaloids. Nat. Prod. Commun. 2006, 1, 885–897. 10.1177/1934578X0600101008. [DOI] [Google Scholar]
- a Hosztafi S.; Simon C.; Makleit S. Synthesis of N-Demethyl-N-Substituted Dihydroisomorphine and Dihydroisocodeine Derivatives. Synth. Commun. 1992, 22, 1673–1682. 10.1080/00397919208020486. [DOI] [Google Scholar]; b Yu H.; Prisinzano T.; Dersch C. M.; Marcus J.; Rothman R. B.; Jacobson A. E.; Rice K. C. Synthesis and biological activity of 8β-substituted hydrocodone indole and hydromorphone indole derivatives. Bioorg. Med. Chem. Lett. 2002, 12, 165–168. 10.1016/S0960-894X(01)00689-8. [DOI] [PubMed] [Google Scholar]; c Selfridge B. R.; Wang X.; Zhang Y.; Yin H.; Grace P. M.; Watkins L. R.; Jacobson A. E.; Rice K. C. Structure–Activity Relationships of (+)-Naltrexone-Inspired Toll-like Receptor 4 (TLR4) Antagonists. J. Med. Chem. 2015, 58, 5038–5052. 10.1021/acs.jmedchem.5b00426. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Marton J.; Miklòs S.; Hosztafi S.; Makleit S. Synthesis of N-Substituted 7β-Diprenorphine Derivatives. Synth. Commun. 1995, 25, 829–848. 10.1080/00397919508013419. [DOI] [Google Scholar]; e Park H. S.; Lee H. Y.; Kim Y. H.; Park J. K.; Zvartau E. E.; Lee H. A highly selective κ-opioid receptor agonist with low addictive potential and dependence liability. Bioorg. Med. Chem. Lett. 2006, 16, 3609–3613. 10.1016/j.bmcl.2006.02.017. [DOI] [PubMed] [Google Scholar]
- a Wang P. X.; Jiang T.; Cantrell G. L.; Berberich D. W.; Trawick B. N.; Osiek T.; Liao S.; Moser F. W.; McClurg J. P.; Mallinckrodt, Inc. . Process and Compounds for the Production of (+)Opiates. U.S. Patent 20090156818A1, 2009.; b Wang P. X.; Jiang T.; Cantrell G. L.; Berberich D. W.; Trawick B. N.; Liao S.; Mallinckrodt, Inc. . Processes for the Production of (+)-“NAL” Morphinan Compounds. U.S. Patent 20090156820A1, 2009.; c Hosztafi S.; Makleit S. Synthesis of New Morphine Derivatives Containing Halogen in the Aromatic Ring. Synth. Commun. 1994, 24, 3031–3045. 10.1080/00397919408011316. [DOI] [Google Scholar]; d Ninan A.; Sainsbury M. An improved synthesis of noroxymorphone. Tetrahedron 1992, 48, 6709–6716. 10.1016/S0040-4020(01)80016-8. [DOI] [Google Scholar]
- a Endoma-Arias M. A. A.; Cox D. P.; Hudlicky T. General Method of Synthesis for Naloxone, Naltrexone, Nalbuphone, and Nalbuphine by the Reaction of Grignard Reagents with an Oxazolidine Derived from Oxymorphone. Adv. Synth. Catal. 2013, 355, 1869–1873. 10.1002/adsc.201300284. [DOI] [Google Scholar]; b Kok G.; Ashton T. D.; Scammells P. J. An Improved Process for the N-Demethylation of Opiate Alkaloids using an Iron(II) Catalyst in Acetate Buffer. Adv. Synth. Catal. 2009, 351, 283–286. 10.1002/adsc.200800632. [DOI] [Google Scholar]; c Dong Z.; Scammells P. J. New Methodology for the N-Demethylation of Opiate Alkaloids. J. Org. Chem. 2007, 72, 9881–9885. 10.1021/jo071171q. [DOI] [PubMed] [Google Scholar]; d Rosenau T.; Hofinger A.; Potthast A.; Kosma P. A General, Selective, High-Yield N-Demethylation Procedure for Tertiary Amines by Solid Reagents in a Convenient Column Chromatography-like Setup. Org. Lett. 2004, 6, 541–544. 10.1021/ol036319g. [DOI] [PubMed] [Google Scholar]; e Do Pham D. D.; Kelso G. F.; Yang Y.; Hearn M. T. W. One-pot oxidative N-demethylation of tropane alkaloids with hydrogen peroxide and a FeIII-TAML catalyst. Green Chem. 2012, 14, 1189–1195. 10.1039/c2gc16207e. [DOI] [Google Scholar]; f Do Pham D. D.; Kelso G. F.; Yang Y.; Hearn M. T. W. Studies on the oxidative N-demethylation of atropine, thebaine and oxycodone using a FeIII-TAML catalyst. Green Chem. 2014, 16, 1399–1409. 10.1039/C3GC41972J. [DOI] [Google Scholar]; g Li Y.; Ma L.; Jia F.; Li Z. Amide Bond Formation through Iron-Catalyzed Oxidative Amidation of Tertiary Amines with Anhydrides. J. Org. Chem. 2013, 78, 5638–5646. 10.1021/jo400804p. [DOI] [PubMed] [Google Scholar]
- a Carroll R. J.; Leisch H.; Scocchera E.; Hudlicky T.; Cox D. P. Palladium-Catalyzed N-Demethylation/N-Acylation of Some Morphine and Tropane Alkaloids. Adv. Synth. Catal. 2008, 350, 2984–2992. 10.1002/adsc.200800667. [DOI] [Google Scholar]; b Machara A.; Werner L.; Endoma-Arias M. A.; Cox D. P.; Hudlicky T. Improved Synthesis of Buprenorphine from Thebaine and/or Oripavine via Palladium-Catalyzed N-Demethylation/Acylation and/or Concomitant O-Demethylation. Adv. Synth. Catal. 2012, 354, 613–626. 10.1002/adsc.201100807. [DOI] [Google Scholar]; c Machara A.; Cox D. P.; Hudlicky T. Direct Synthesis of Naltrexone by Palladium-Catalyzed N-Demethylation/Acylation of Oxymorphone: The Benefit of C·H Activation and the Intramolecular Acyl Transfer from C-14 Hydroxy. Adv. Synth. Catal. 2012, 354, 2713–2718. 10.1002/adsc.201200677. [DOI] [Google Scholar]; d Gutmann B.; Weigl U.; Cox D. P.; Kappe C. O. Batch and Continuous Flow Aerobic Oxidation of 14-Hydroxy Opioids to 1,3-Oxazolidines – A Concise Synthesis of Noroxymorphone. Chem. - Eur. J. 2016, 22, 10393–10398. 10.1002/chem.201601902. [DOI] [PubMed] [Google Scholar]; e Gutmann B.; Elsner P.; Cox D. P.; Weigl U.; Roberge D. M.; Kappe C. O. Towards the Synthesis of Noroxymorphone via Aerobic Palladium-Catalyzed Continuous Flow N-Demethylation Strategies. ACS Sustainable Chem. Eng. 2016, 4, 6048–6061. 10.1021/acssuschemeng.6b01371. [DOI] [Google Scholar]; f Gutmann B.; Cantillo D.; Weigl U.; Cox D. P.; Kappe C. O. Design and Development of Pd-catalyzed Aerobic N-Demethylation Strategies for the Synthesis of Noroxymorphone in Continuous Flow Mode. Eur. J. Org. Chem. 2017, 2017, 914–927. 10.1002/ejoc.201601453. [DOI] [Google Scholar]; g Mata A.; Cantillo D.; Kappe C. O. An Integrated Continuous Flow Synthesis of a Key Oxazolidine Intermediate to Noroxymorphone from Naturally Occurring Opioids. Eur. J. Org. Chem. 2017, 2017, 6505–6510. 10.1002/ejoc.201700811. [DOI] [Google Scholar]
- a Ripper J. A.; Tiekink E. R.; Scammells P. J. Photochemical N-demethylation of alkaloids. Bioorg. Med. Chem. Lett. 2001, 11, 443–445. 10.1016/S0960-894X(00)00690-9. [DOI] [PubMed] [Google Scholar]; b Chen Y.; Glotz G.; Cantillo D.; Kappe C. O. Organophotocatalytic N-Demethylation of Oxycodone using Molecular Oxygen. Chem. - Eur. J. 2020, 26, 2973–2979. 10.1002/chem.201905505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin M. M.; Augustin J. M.; Brock J. R.; Kutchan T. M. Enzyme morphinan N-demethylase for more sustainable opiate processing. Nat. Sustain. 2019, 2, 465–474. 10.1038/s41893-019-0302-6. [DOI] [Google Scholar]
- a Schäfer H. J. Contributions of organic electrosynthesis to green chemistry. C. R. Chim. 2011, 14, 745–765. 10.1016/j.crci.2011.01.002. [DOI] [Google Scholar]; b Horn E. J.; Rosen B. R.; Baran P. S. Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method. ACS Cent. Sci. 2016, 2, 302–308. 10.1021/acscentsci.6b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wiebe A.; Gieshoff T.; Möhle S.; Rodrigo E.; Zirbes M.; Waldvogel S. R. Electrifying Organic Synthesis. Angew. Chem., Int. Ed. 2018, 57, 5594–5619. 10.1002/anie.201711060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A previous version of this manuscript was deposited in ChemRxiv:; Glotz G.; Kappe C. O.; Cantillo D. ChemRxiv 2020, 10.26434/chemrxiv.12458573.v1. [DOI] [Google Scholar]
- Röckl J. L.; Pollok D.; Franke R.; Waldvogel S. R. A Decade of Electrochemical Dehydrogenative C,C-Coupling of Aryls. Acc. Chem. Res. 2020, 53, 45–61. 10.1021/acs.accounts.9b00511. [DOI] [PubMed] [Google Scholar]
- Roth H. G.; Romero N. A.; Nicewicz D. A. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. 10.1055/s-0035-1561297. [DOI] [Google Scholar]
- The synthesis of oxazolidine 2a and analogues via palladium-catalyzed or photocatalytic oxidation has been suggested to proceed via iminium ion intermediates. (See ref (10).)
- Kawamata Y.; Yan M.; Liu Z.; Bao D.-H.; Chen J.; Starr J. T.; Baran P. S. Scalable, Electrochemical Oxidation of Unactivated C–H Bonds. J. Am. Chem. Soc. 2017, 139, 7448–7451. 10.1021/jacs.7b03539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B.; Šnajdr I.; Machara A.; Endoma-Arias M. A. A.; Stamatatos T. C.; Cox D. P.; Hudlický T. Conversion of Thebaine to Oripavine and Other Useful Intermediates for the Semisynthesis of Opiate-Derived Agents: Synthesis of Hydromorphone. Adv. Synth. Catal. 2014, 356, 2679–2687. 10.1002/adsc.201400445. [DOI] [Google Scholar]
- Andre J.-D.; Dormoy J.-R.; Heymes A. O-Demethylation of Opioid Derivatives with Methane Sulfonic Acid/Methionine: Application to the Synthesis of Naloxone and Analogues. Synth. Commun. 1992, 22, 2313–2327. 10.1080/00397919208019086. [DOI] [Google Scholar]
- Foroughi M. M.; Jahani S.; Rajaei M. Facile Fabrication of 3D Dandelion-Like Cobalt Oxide Nanoflowers and Its Functionalization in the First Electrochemical Sensing of Oxymorphone: Evaluation of Kinetic Parameters at the Surface Electrode. J. Electrochem. Soc. 2019, 166, B1300–B1311. 10.1149/2.0511914jes. [DOI] [Google Scholar]
- a Noël T.; Cao Y.; Laudadio G. The Fundamentals Behind the Use of Flow Reactors in Electrochemistry. Acc. Chem. Res. 2019, 52, 2858–2869. 10.1021/acs.accounts.9b00412. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pletcher D.; Green R. A.; Brown R. C. D. Flow Electrolysis Cells for the Synthetic Organic Chemistry Laboratory. Chem. Rev. 2018, 118, 4573–4591. 10.1021/acs.chemrev.7b00360. [DOI] [PubMed] [Google Scholar]
- Folgueiras-Amador A.; Philipps K.; Guilbaud S.; Poelakker J.; Wirth T. An Easy-to-Machine Electrochemical Flow Microreactor: Efficient Synthesis of Isoindolinone and Flow Functionalization. Angew. Chem., Int. Ed. 2017, 56, 15446–15450. 10.1002/anie.201709717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S. J.; Soudackov A. V.; Hammes-Schiffer S. Analysis of Kinetic Isotope Effects for Proton-Coupled Electron Transfer Reactions. J. Phys. Chem. A 2009, 113, 2117–2126. 10.1021/jp809122y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida J.-i.; Shimizu A.; Hayashi R. Electrogenerated Cationic Reactive Intermediates: The Pool Method and Further Advances. Chem. Rev. 2018, 118, 4702–4730. 10.1021/acs.chemrev.7b00475. [DOI] [PubMed] [Google Scholar]
- Monitoring of the C=O signal of N-acyl iminium cations was accomplished by Yoshida and co-workers:Suga S.; Okajima M.; Fujiwara K.; Yoshida J.-i. “Cation Flow” Method: A New Approach to Conventional and Combinatorial Organic Syntheses Using Electrochemical Microflow Systems. J. Am. Chem. Soc. 2001, 123, 7941–7942. 10.1021/ja015823i. [DOI] [PubMed] [Google Scholar]
- Socrates G.Infrared and Raman Characteristic Group Frequencies Tables and Charts, 3rd ed.; Wiley: New York, 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.