


Abstract
Extracellular vesicles (EVs) hold great promise for transporting CRISPR–Cas9 RNA-guided endonucleases (RNP) throughout the body. However, the cell-selective delivery of EVs is still a challenge. Here, we designed valency-controlled tetrahedral DNA nanostructures (TDNs) conjugated with DNA aptamer, and loaded the valency-controlled TDNs on EV surface via cholesterol anchoring for specific cell targeting. The targeting efficacy of different ratios of aptamer/cholesterol from 1:3 to 3:1 in TDNs on decorating EVs was investigated. TDNs with one aptamer and three cholesterol anchors (TDN1) efficiently facilitated the tumor-specific accumulation of the EVs in cultured HepG2 cells and human primary liver cancer-derived organoids, as well as xenograft tumor models. The intracellular delivery of RNP by TDN1-EVs successfully realized its subsequent genome editing, leading to the downregulation of GFP or WNT10B in specific cells. This system was ultimately applied to reduce the protein expression of WNT10B, which presented remarkable tumor growth inhibition in vitro, ex vivo and in vivo, and could be extended to other therapeutic targets. The present study provides a platform for the directional display of aptamer on surface labeling and the EVs-based Cas9 delivery, which provides a meaningful idea for future cell-selective gene editing.
INTRODUCTION
Gene therapy based on CRISPR/Cas9 system has gained widespread attention for the potentials of precise genome editing to cure genetic diseases, such as Alzheimer's disease, immune deficiency diseases, and cancer (1). With the guidance of single guide RNA (sgRNA), Cas9 can efficiently regulate target gene expression of cancer cells (2,3), and therefore become a powerful tool for curing cancer. Nowadays, viral vectors or liposome-based carriers are widely used in CRISPR/Cas9 studies (4,5). Adeno-associated viruses (AAVs) based Cas9 delivery has the potential to establish gene therapy in a wide variety of target tissue, however, the risk of immunogenicity limits the practical application of virus-based gene therapy (6). Although liposomes via surface modification may provide relative cell-type specificity, the liver toxicity and rapid clearance of liposomal Cas9 systems bring big challenges to clinical application (7). Recently, extracellular vesicles (EVs) have displayed great potential as endogenous nanovehicles for efficient drug delivery.
EVs are cell-derived membrane vesicles, which serve as carriers for cell-to-cell communication. Meanwhile, since EVs are immune-privileged, they are hardly cleared by the immune defense system and avoid the hypersensitivity reactions (8–10). It seems that EVs can specifically transport their contents from one cell to another even over long distances (11). Moreover, our data demonstrated that EVs presented less cytotoxicity and enabled more efficient cellular interactions when compared with liposome (Supplementary Figures S1 and S2). The human embryonic kidney cell line HEK293T is widely used to produce EVs for the delivery of Cas9/sgRNA complex due to its capacity for high yield of EVs, minimal intrinsic biological cargos and ease of culture. Nevertheless, major challenges for applications of natural EVs are its poor targeting ability and the off-target effects (12–14). Noteworthy, EVs’ potential cell-type specific interactions were determined by their surface ligands (15,16). Thus surface modifications of EVs could be used to specifically direct EVs to tumor cells by receptor-ligand binding, and different approaches have been proposed for this aim (17–21). Among distinct targeting approaches, DNA aptamers offer several unique advantages over Abs or other probes, because they are cheap, readily available and non-immunogenic. Furthermore, DNA molecules can add a net negative charge to EVs, and thereby reducing the non-specific binding of EVs to non-target cells (22,23). It's well known that the recognition of DNA aptamer greatly relies on the orientation and entanglement between the surface which it stands and the target receptor. Current reports indicated that single-strand DNA aptamers still require a series of ‘backfillers’ including mercaptohexanol (MCH), oligo (ethylene glycol)-terminated thiols and so on, to minimize lateral interactions between single-strand DNA aptamers (24–26). Previous study has demonstrated that 3D tetrahedral DNA nanostructures (TDNs) can offer specific orientation of DNA scaffold without steric hindrances (27–29). Thus, in the present study, TDNs were applied to precisely anchor DNA aptamer on the EVs’ membrane, to give better control of the DNA recognition units. Typically, a TDN is a DNA nanostructure with a spatial conformation composed of four vertex corners, which indicates the potential of chemical modification at these four vertices. For specific targeting of cancer cells, TLS11a aptamers that can bind to HepG2 cells are integrated at the apex of TDNs (30). In our design, TDNs conjugated with DNA aptamers/cholesterol molecules at these four vertices could be divided into three nanostructures according to the distribution ratio of DNA aptamers and cholesterol molecules (1:3, 2:2 and 3:1), namely TDN1, TDN2 and TDN3. We demonstrated that the DNA aptamer/cholesterol ratio-controlled modification of the TDNs could regulate the display of DNA aptamers on the EVs’ surface and thereby present enhancing the cellular uptake of EVs in varying degrees. Based on the highly programmable and predictable nature of DNA nanotechnology, we could achieve heterogeneous assemblies of these TDNs to construct a cell-specific targeted delivery system of EVs with different efficiency. Here, the TDNs-based targeting strategy for exosomal delivering CRISPR/Cas9 to liver cancer cells was presented in Scheme 1.
Scheme 1.
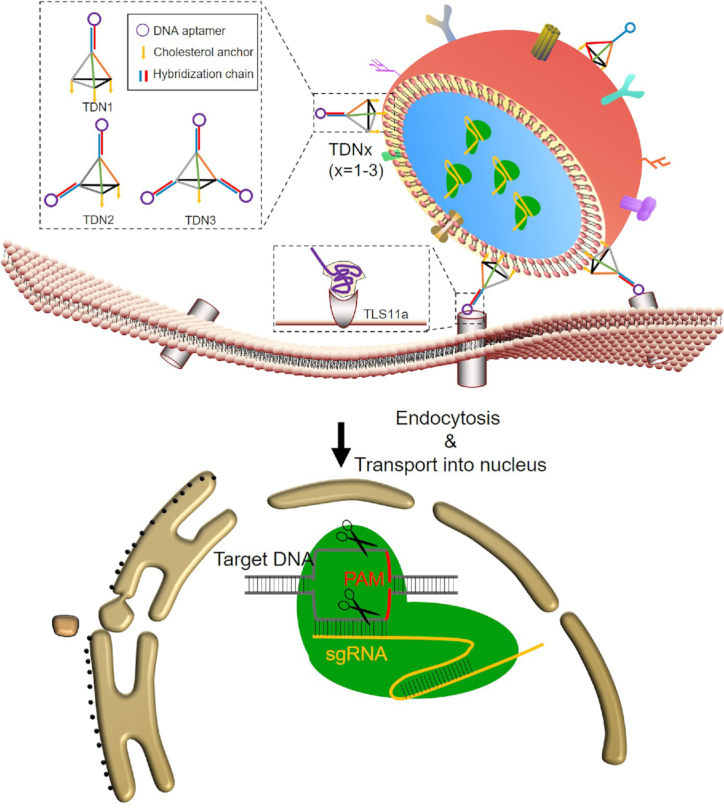
Design and the targeting delivery of TDNs-EVs.
MATERIALS AND METHODS
Materials
All Chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified and were used as received. DNA oligonucleotides were purchased from SBS tenetech Co., Ltd. (Beijing, China). HiScribe T7 High Yield RNA Synthesis Kit, Endonuclease III, T7 Endonuclease I was purchased from New England Biolabs (USA). BCA Protein Assay Kit, OptiPrep, Triton X-100, GeneJET Genomic DNA Purification Kit were purchased from Thermo Fisher Scientific (USA). S. pyogenes Cas9 protein was purchased from novoprotein (Shanghai, China). PrimeSTAR® Max DNA Polymerase was purchased from takara (Japan).
Cell culture
HEK293T, HepG2, MCF-7 and HELA cell lines were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). These cells were grown in DMEM (Gibico), which was supplemented with 10% (vol/vol) FBS (Gibico), 1% penicillin–streptomycin, and 2 μg/ml blasticidin at 37°C in a humidified atmosphere of 5% CO2. Cells were harvested by treatment with 0.25% trypsin–DTA (Gibico).
Extracellular vesicles (EVs) purification
HEK293T cells were incubated with DMEM with 10% exosome-free FBS (Gibico) for 2 days. The cell medium was first spun at 300g for 10 min to remove dead cells. Then the supernatant was spun at 10 000g for 30 min at 4°C. An aliquot of 10 ml of supernatant was transferred to an ultracentrifuge tube which there are 500 μl of Optiprep solution containing 60% iodixanol (Sigma-Aldrich) at the bottom and spun at 100 000g for 70 min at 4°C in a Beckman SW40 rotor. The EVs-enriched cushion was carefully aspirated between the iodixanol and supernatant. Then the EVs-enriched cushion was washed by PBS and spun at 100 000g for 70 min at 4°C. The EVs samples were quantified by BCA assay and stored at 4°C for further study.
Transcription and purification of single-guide RNA (gRNA)
The sgRNA was transcribed in vitro with HiScribe T7 High Yield RNA Synthesis Kit according to protocol. Transcription templates encoding a T7 promoter followed by the sgRNA were synthesized with the sgRNA containing a GFP targeting sequence (sgGFP) and a WNT10B targeting sequence (sgWNT10B) (Supplementary Table S1). The transcribed RNA was extracted by Spin Column RNA Cleanup & Concentration Kit (Sangon Biotech) to purify RNA samples. Purified RNA was analyzed by agarose gel electrophoresis and quantified with Nanodrop 2000c (Thermo Scientific).
Preparation of ribonucleoproteins (RNPs)
Cas9 protein (162 kDa ≈ 162 000 g /mol) was diluted to the desired concentration (10 mM) with PBS and incubated with sgRNA (molecular weight ≈ (132 × 320.5) + 159.0 = 42465 g/mol) at the molar ratio (1:1) at 37°C for 10 min. The resulting RNP was cooled to room temperature (RT) and was either used immediately or frozen at –80°C for later use.
Loading of EVs
For sonication, the RNP solutions or Alexa Fluor 488 (AF488)-labeled sDNAs with EVs were sonicated (2 kHz, 5% power, 20 cycles by 2 s pulse/1 s pause) in the ice-bath.
For freeze–thaw cycles (F/T), the RNP solution or AF488-labeled sDNA were added to EVs, incubated at RT for 30 min. And then the resulting samples were immediately frozen by liquid nitrogen, and then thawed at RT. The above F/T process was repeated three times.
After the loading of RNP or AF488-labeled sDNA, the loaded EVs were purified by ultracentrifugation at 100 000g for 70 min at 4°C. All the pellets and the supernatants were collected and subjected to determine the loading efficiency by western blot and BCA assay. The loading efficiency of RNP for different TDNs-EVs formulations was determined via western blot. The protein content in the particles was quantified via densitometry analysis on the respective gel bands. After separation of the unloaded Cas9 RNP from EVs, the loading percent of Cas9 RNP was determined by comparing the amount of Cas9 present in purified Cas9 RNP loaded EVs versus the amount of Cas9 present in the unpurified Cas9 RNP loaded EVs and the loading efficiency was calculated through the equation below:
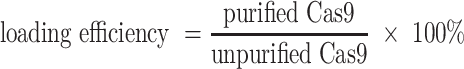 |
All the RNP loading experiments were performed in duplicate.
Preparation of tetrahedrons (TDNs)
Equimolar quantities of four strands (Supplementary Table S2; sequence a, b or b-CS, c or c-CS and d-CS) for the formations of TDNs with different number of cohesive ends were mixed in buffer (20 mM Tris, 16 mM MgCl2, pH 8.0) at 95°C for 10 min and then cooled to 4°C for 910 min (0.1°C/min) (Supplementary Figure S3). The construction of TDNs with different ratio of aptamers/cholesterol was assembled by mixing TDN-xCS (x = 1–3) with apt-s, and incubating under 37°C for 120 min in buffer (20 mM Tris, 16 mM MgCl2, pH 8.0) respectively. For the TDNs with fluorescent labels, sequence apt-s was labeled by AF488. After the synthesis of TDNs, the DNA nanoparticles solutions were stored at 4°C for further study. At last, the TDNs were characterized by agarose gel electrophoresis (AGE) in TAE (40 mM Tris-acetate, 1 mM EDTA, 16 mM MgCl2) buffer.
TDNs decoration of RNP-loaded EVs
A modified heat-shock protocol was used to anchor the TDNs on the surface of EVs. The RNP-loaded EVs were incubated with TDNs at the weight ratio of 1:3 (EVs: TDNs, the EVs were measured by protein amount) at 37°C for 1h, then rapidly left on ice for 1 h under gentle shaking condition. The TDNs decorated EVs were purified by ultracentrifugation at 100 000g for 70 min at 4°C. All the pellets were collected and stored at 4°C for further study. An AGE test in TAE (40 mM Tris-acetate, 1 mM EDTA) buffer was used to demonstrate the success of TDNs decoration. The decoration efficiency was measured after a modified heat-shock process for TDNs decoration. In brief, the resulting TDNs-EVs were precipitated down with Exo-Fect™ (SBI System Biosciences) and the undecorated DNA nanostructures were collected from the supernatant after ultracentrifugation. The concentration of free DNA and total input DNA were determined and the decoration efficiency was calculated through the equation below:
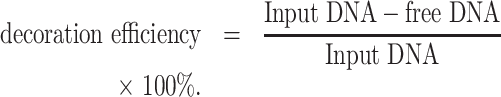 |
Characterization of different TDNs-EVs formulations by dynamic light scattering (DLS) and transmission electron microscope (TEM)
The effective hydrodynamic diameters and the zeta potentials of native EVs, or RNP-loaded EVs and TDNs-EVs were measured by DLS using the ZetaPlus' Zeta Potential Analyzer (Brookhaven Instruments, Santa Barbara, CA, USA) equipped with a 35 mW solid state laser (658 nm laser).
The morphologies of native EVs and TDNs-EVs were investigated by TEM. The samples were drop-cast on glow discharged copper grids with pure carbon support film and incubated for 10−15 min and then washed with ultrapure water. Finally, the sample grids were negatively stained with 1% uranyl acetate solution. Then the TEM images of these nanocarriers were captured by Jeol 2100F at an acceleration voltage of 200 kV.
Fluorescence resonance energy transfer (FRET) decrease assay
The AF488-labeled TDNs –Evs (the aptamers of TDNs were labeled by AF488) and AF488-labeled TDNs were incubated with 20 μM DiI for 1 h at 37°C. The free dye was removed by ultracentrifugation at 110 000 g for 70 min. The phenomena of FRET can be used to detect the interaction of DNA aptamers in TDNs and the surface of EVs as the emission peak of AF488 (Donor) overlaps with the excitation peak of DiI (Acceptor) thus allowing FRET to occur. The recovery of DiI fluorescence was detected using the LS-55 luminescence spectrometer (PerkinElmer) with excitation at 488 nm, emission at 500–600 nm. The FRET efficiency was calculated based on the donor emission spectrum, which was used a classical equation as following: 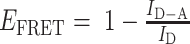 . And the interaction effect (IE) between AF488-lablled aptamers and Dil-lablled membrane, were calculated by:
. And the interaction effect (IE) between AF488-lablled aptamers and Dil-lablled membrane, were calculated by: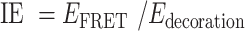 , where Edecoration is the decoration efficiency of the TDNs.
, where Edecoration is the decoration efficiency of the TDNs.
Preparation of liposomes
Liposomes were prepared using the extrusion method. The AF488-labeled sDNAs and resulting RNP were encapsulated in 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) liposomes. Briefly, the lip mixture were dried under vacuum to remove all the organic solvent. Then the dried DOPC were hydrated by adding cargo (in PBS) at a 500:1 molar ratio (DOPC/cargo (RNP were counted for Cas9 protein). Then the liposome was homogenized by extrusion through a 0.1 μm polycarbonate membrane using the Avanti Mini-Extruder; at least 17 passages were performed.
AF488-labeled sDNA loading efficiency analysis
To determine the loading efficiency of AF488-labeled sDNA loaded inside the liposome or EVs, a lysis buffer consisting of 200 μl 10% Triton X-100 solution, and 1800 μl Tris–EDTA Buffer (pH 8.0) was added to the prepared liposome or EVs solution, resulting in the destabilization of the lipid structure of the nanoparticles and sDNA release. Then the fluorescences of the released sDNA after centrifugation and the total loading sDNA during production were measured by LS-55 luminescence spectrometer (PerkinElmer). At last, the sDNA loading efficiency was calculated using the equation below:
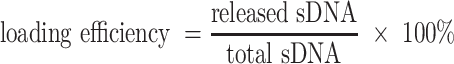 |
Internalization assay
The HepG2 and MCF-7 cells were seeded in 12-well plate before the assay. After the adherence to the plate, the cells were incubated with different AF488-labeled sDNAs-loaded nanoplatforms (1 μg sDNA/ well, by weight of sDNA) for 8 h. After washing with PBS, the samples were observed by Olympus IX81 in FITC and bright field channels. For each sample, fluorescence was quantified using the mean value of n > 3 pictures.
Flow cytometry analysis
The HepG2, MCF-7, HEK293T and Hela cells were seeded in 12-well plate before the assay. After the adherence to the plate, the cells were incubated with sDNA and TDN1-EVs-sDNA (1 μg/well, by weight of sDNA) for 8 h. The nanocarriers-treated cells were rinsed with PBS twice, trypsinized, permeabilized, stained with PI and collected. The cells were re-suspended with PBS and determined by a BD FACSVerse flow cytometer (BD). The results were analyzed with Flowjo.
Human liver organoids (PLOs) culture
The experiment was reviewed and approved by the Clinical Research and Experimental Animal Ethics Committee of the First Affiliated Hospital of Sun Yat-sen University (2018 [43]). Human tissue and specimens were obtained by surgical resection from patients of primary liver cancer. Written informed consents were all obtained from patients. The tumor surgical resections were mechanically disintegrated into small pieces (0.5–1 mm3) and incubated with the digestion solution, which contained 2.0 mg/ml collagenase IV (Sigma-Aldrich), 0.1 mg/ml DNase (Sigma) and 10 μM Y-27632 (Sigma) for 30 min. Then the digested cell suspention was filtered through 100 μm nylon net and centrifuged for 5 min at 300 G. Cells were seeded in the reduced growth factor BME2 (Basement Membrane Extract, Type 2; R&D) with the cell density at 5000–10 000/100 ul per well in a 24-multi well plate. Incubate the plate at 37°C for 20min until the BME2 wsa solidified. Added 100 ul per well organoid expansion medium. The composition is Advanced DMEM/F12 (GIBCO) supplemented with 1% penicillin/streptomycin (GIBCO), 1% Glutamax (Sigma), 10 mM HEPES (Sigma), 1:50 B27 supplement (GIBCO), 1:100 N2 supplement (GIBCO), 1.25 mM n-acetyl-l-cysteine (Sigma), 10% Rspo-1 conditioned medium(homemade), 30% Wnt3a conditioned medium (homemade), 10 mM nicotinamide (Sigma), 10 nM recombinant human [Leu15]-Gastrin I (Sigma), 50 ng/ml recombinant human EGF (PeproTech), 100 ng/ml recombinant human FGF10 (PeproTech), 25 ng/ml recombinant human HGF (PeproTech), 5 μM A8301 (Tocris), 25 ng/ml Noggin (PeproTech) and 10 μM Y27632 (Tocris). The organoid expansion medium was changed every 3 days and the organoids were passaged for every two weeks using 2.0 mg/ml collagenase IV. Organoids were cryopreserved in 10% DMSO solution prior for freezing and the Organoids’ passage <30 were used in experiments. All the Organoids were regularly checked for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza).
Tracking of the uptake of PLOs
The PLOs samples were first stained with CellTrace Blue (C34574, Thermo Scientific), followed by incubating with complete medium for 5 min, and then washed with twice in DMEM/F12. Cells were then resuspended in PBS and incubated with a mixture of anti-CD45-PE (BioLegend, #368509) for 30 min on ice in the dark. Then the PLOs were seeded on confocal culture dishes and incubated with EVs-AF488-labeled sDNA, Apt-EVs-AF488-labeled sDNA and Apt-TDN1-EVs-AF488-labeled sDNA at the dose of 5 μg sDNA per well for 24 h followed by washing with PBS. At last, the samples were observed by lionheartFX (Biotek). The colocalization signals of the captured images between AF488-labeled sDNA and CD45 were analyzed by imageJ.
In vivo biodistribution study of TDN1-EVs
The HepG2 subcutaneous hepatocellular cancer models were established by injecting 0.1 ml of HepG2 cell suspension (1 × 107/mouse) into the back of female BALB/c nude mice using 50% (v/v) Matrigel. The animals were purchased from Shanghai Model Organisms Center, Inc. All experimental protocols were conducted within Sun Yat-sen University guidelines for animal research and were approved by the Sun Yat-sen University Institutional Animal Care and Use Committee (IACUC): Approval Number IACUC-DD-2017–181.
After 2 weeks, when the tumors grew to 80−120 mm3 size, the animals were randomly assigned into four groups (3 mice/group). The mice were intravenous injected with 100 μl of DiR solution (0.3 mg/ml) and several groups of DiR stained nanoparticles including liposome, EVs and TDN1-EVs (0.1 mg/kg), respectively. Then the mice were anesthetized for imaging by IVIS spectrum (Xenogen, USA) at 0.5 h and 4 h postinjection. After 12 h treatment, all animals were sacrificed and the organs (liver, spleen, kidney, heart, lungs and brain) and tumors were collected for ex vivo imaging. All the captured images and the quantitative analysis of the biodistribution in major organs were processed by Aura Imaging Software (https://spectralinvivo.com/software/).
GFP gene disruption assay
Lentiviral vector carrying GFP gene has been successfully constructed and maintains high expression in HepG2 cells. HepG2/GFP cells were first seeded in 12-well plates. Then the cells were treated with PBS, Lipo-RNP (sgGFP), EVs-RNP and TDN1-EVs-RNP (1 μg/well, by weight of Cas9 protein). After incubation for 4h, the medium was replaced with fresh full serum medium. Two days after the delivery, the cells were analyzed by Olympus IX81. The fluorescent intensity of GFP protein was determined according to the captured images and analyzed by ImageJ. For the flow cytometry analysis, the cells were washed, trypsinized and collected. At last, the cells were analyzed by a BD FACSVerse flow cytometer.
Sequencing and T7EI assay to detect genomic modifications
Genomic DNA of HepG2/GFP cells was harvested 2 das after the treatment. Genomic DNA of cells were purified by GeneJET Genomic DNA Purification Kit (Thermo Scientific). The sgGFP targeted genomic locus was amplified using primers 5′-ATGGTGAGCAAGGGCGAGGAGCTGTTC-3′ and 5′- TTACTTGTACAGCTCGTCCATGCCGAG-3′ and purified by gel extraction. The DNA samples were cloned into pEasy-Blunt plasmids and sequenced.
The DNA samples were also incubated with T7EI (10 U/μl, NEB) at 37°C for 15 min. An AGE assay was used to determine the indel formation.
WNT10B gene disruption assay
HepG2 cells were first seeded in six-well and 96-well plates. Then the cells were treated with PBS, Lipo-RNP (sgWNT10B), EVs-RNP and TDN1-EVs-RNP (2 μg (six-well) or 200 ng (96-well)/well, by weight of Cas9 protein). After incubation for 4h, the medium was replaced with fresh full serum medium. Two days after the delivery, the cells were subjected to CCK8 assay or harvested for purification of total protein and genomic DNA. For CCK8 assay, the cells in 96-well plates were added with CCK8 solution and incubated for another 1 h at 37°C. Then the absorbance measurements were immediately read at a wavelength of 450 nm while cell viability was calculated accordingly. The protein samples were subjected to western blot analysis. Total protein extracts were subjected to SDS-polyacrylamide gel electrophoresis (PAGE). After electrophoresis, the proteins were transferred to poly (vinylidene fluoride) (PVDF) membranes (Millipore). To block non-specific binding sites, the membranes were treated for 1 h with TBST (mixture of Tris-buffered saline and Tween-20) containing 5% milk. Subsequently the membranes were incubated with the primary antibody against WNT10B (ab70816, abcam) or GAPDH (14C10, CST) for 1 h overnight at 4°C. After washing, signals were detected by HRP-conjugated secondary anti-rabbit antibody and were visualized using proteinsimple software.
Deep sequencing
Deep sequencing was used to confirm the presence of indels surroding surrounding the predicted Cas9 cleavage site around WNT10B for PLO models and xenograft models. The sample were collected at the end point of the study and the genomic DNA of tumor cells was extracted by a Universal Genomic DNA Extraction Kit (D2100, Solarbio) The genomic DNA was used to amplify the targeted human WNT10B locus with primers 5′-TCTTGGTTCCCAGGGCTCTA-3′ and 5′- GCCTCCGCTCAGCTTAATCT-3′ and purified by gel extraction. The targeted deep sequencing library was constructed based on the protocol of the Nextera XT DNA Library Prep Kit. Then the Illumina MiSeq was used to perform the high-throughput sequencing and the FASTQ files were generated for data analysis. FASTQ files were merged using FLASH version 1.2.11. Cas-Analyzer tool (http://www.rgenome.net/cas-analyzer/!) was used to determine the indel rates. The input parameter was full human WNT10B WT sequence: TCTTGGTTCCCAGGGCTCTAAGCAATGAGATTCTGGGCCTGAAGTTGCCTGGCGAGCCGCCGCTGACGGCCAACACCGTGTGCTTGACGCTGTCCGGCCTGAGCAAGCGGCAGCTAGGCCTGTGCCTGCGCAACCCCGACGTGACGGCGTCCGCGCTTCAGGGTCTGCACATCGCGGTCCACGAGTGTCAGCACCAGCTGCGCGACCAGCGCTGGAACTGCTCCGCGCTTGAGGGCGGCGGCCGCCTGCCGCACCACAGCGCCATCCTCAAGCGCGGTGAGCCTGGCTGGGCGGGTCGGCGCTGCCGCGGAGGCTAGGGGGTGGGCGGCAGGGCGAGGCGCGCGGAGGCGGGAAGGGATGGTTTCGCGGCTCCGGGAGGGGCCTCTCCTGGAATTCCCCGCCCCCCCCGGAGCCGCTCTGAGCCAGGTCTGGCCACTCGTCTGGGCCCCTGCGCGCCTAACTTCCACCTGCGTGCTGCCAGCGAGGCAGCTGGGTCCCTGCTCTCCTACCCTAGGGCCCCCCTTTCCTGCCTCCGCTCAGCTTAATCT. Other settings were followed with the default values.
Ex vivo tumor inhibition assays with TDN1-EVs
PLOs were treated with PBS, Lipo-RNP(sgWNT10B), EVs-RNP and TDN1-EVs-RNP (5 μg/well, by weight of Cas9 protein). After incubation for 4 h, the medium was replaced with organoid expansion medium. A green-fluorescent caspase 3/7 probe (C10723, Thermo Scientific) was used to determine apoptotic rate of the PLOs while the Celltrace far red (C34572, Thermo Scientific) was used to visualize the organoids. The apoptosis process of the PLOs was visualized by lionheartFX (Biotek) and the captured images were used to quantified the organoid killing upon nanocarrier treatments after 48 h.
In Vivo antitumor activity study with TDN1-EVs
The animals were purchased from Shanghai Model Organisms Center, Inc. All experimental protocols were conducted within Sun Yat-sen University guidelines for animal research and were approved by the Sun Yat-sen University Institutional Animal Care and Use Committee (IACUC): Approval Number IACUC-DD-2017–181. First HepG2 tumor-bearing female BALB/c nude mice were established as described above. When the tumor volume was 80−120 mm3 at 14 days after cell implantation, the mice were randomly divided into 5 groups (5 mice / group) and intravenously injected with 100 μl of saline, Lipo-RNP (0.1 mg/kg), EVs-RNP(0.1 mg/kg) and TDN1-EVs-RNP (0.1 mg/kg (low) and 1.0 mg/kg (high)). The growths of tumor volume were measured every four days. The estimated volume was calculated according to the formula: tumor volume (mm3) = 0.52 × length × width2. For the evaluation of the in vivo safety of TDN1-EVs-RNP treatment, the levels of serological markers were tested after all mice were sacrificed. Then the major organs (liver, spleen, kidney, heart, lungs and tumors) were collected and fixed in formalin. The tumors were weighed and all the samples were subjected to H&E staining.
Evaluation of cytokines induction by TDN1-EVs in mice
Systemic inflammation was assessed by measuring the immune-related cytokines in the BALB/c mice that had been injected with Saline, Lipo-RNP and TDN1-EVs-RNP at the dose of 0.1 mg/kg. Mice were sacrificed either 6 or 24 h after the injection and their plasma was collected and analyzed by using Mouse ELISA MAX Deluxe sets (BioLegend, USA) following the protocol provided by the manufacturer to measure the stimulated cytokine levels of interleukin-6 (IL-6), IL-10, tumor necrosis factor-α (TNF-α) and Interferon-γ (IFN-γ). Experiment results were repeated for at least three independent measurements.
Statistical analysis
All results were presented as mean ± SD. Statistical analysis was performed based on Student's t-test (one-tailed) using GraphPad Prism 6. The difference between experimental groups and control groups were considered statistically significant when P < 0.05 or highly significant when P < 0.01.
RESULTS
Construction of RNP-loaded EVs with valency-controlled TDNs
This delivery system consists of three parts, the outer TDNs (targeting units), the extracted exosomes (loading vehicles), and the inner layer of RNP complex (functional ingredients). The TDNs were successfully self-assembled by four-component strands via a programmable ‘soft lithography’ method owing to their highly specific Watson–Crick base pairing (31). A triethylene glycol (TEG) cholesterol was conjugated to the 5′ end of the sense strand of DNAs used in the present study, which can enhance anchorage of the TDNs onto the EVs surface (32). As an elegant example, the defined-valent TDNs with 1 DNA aptamer and 3 cholesterol anchors (namely TDN1) were synthesized by 3 cholesterol-modified strands, 1 DNA strand with the cohesive end and 1 aptamer strands with a complementary overhang to another cohesive end (Figure 1A and Supplementary Figure S3). With an increasing number (1–3) of DNA aptamer display strands that were used to form TDNs, we constructed a series of TDNs with increasing valency of DNA aptamers (from 1 to 3) including TDN1, TDN2 and TDN3 (Figure 1B). The assembly efficiencies of TDN2 and TDN3 were observed to be different from that of TDN1, which indicated the impact of cholesterol modified DNA strands on the DNA self-assembly of TDNs. Then a standard ultracentrifugation procedure was used to purify the EVs from HEK293T. Dynamic light scattering (DLS) revealed that the size distribution of isolated native EVs was ranged from 50 to 250 nm, with a peak of around 130 nm. The western blotting data confirmed the positive staining for EV marker proteins TSG101 and CD63 (Supplementary Figure S4). Additionally, no notable physical disruption of EVs was observed by ultracentrifugation pelleting according to electron microscopy imaging. Cas9 protein and the sgRNA targeting GFP or WNT10B were characterized and mixed to form an RNP complex before loading of EVs (Supplementary Figures S5 and S6). Sonication and repeated freeze-thaw cycles (F/T) were applied for RNP incorporation into EVs and evaluated. Both of these two methods could partly reform and reshape EVs, which can enable RNP diffusion across lipid bilayers of EVs. At last we achieved more than 30% of loading efficiency by F/T (Figure 1C). Moreover, significant size increases were found after sonication or F/T of EVs with RNP. The obtained RNP loaded EVs were in the range from 100 to 150 nm (Supplementary Table S3). After self-assembly of TDNs, these DNA nanostructures were decorated on the surface of loaded EVs by a modified heat-shock process. The increase in the membrane fluidity induced by the temperature change could be well organized during the decoration of TDNs, leading to the insertion of cholesterol anchors. The TEM images revealed that the engineered EVs appeared as flattened spheres whereas the native EVs were full spheres (Supplementary Figure S7). After loading of RNPs in EVs, we decorated EVs surface with different TDNs via a modified heat-shock process. Although there are different numbers of cholesterol anchors in these DNA nanostructures, which might theoretically affect the DNA decoration, the results demonstrated that all of them showed more than 50% decoration efficiency, and thus has the complexation efficiency needed for developing aptamers display (Figure 1D and Supplementary Figure S8). Moreover, the stability of the TDNs on the EVs was accessed and found that TDN1 on EVs were more stable when compared with other TDNs in DNA degradation assay (Supplementary Figure S9). After decorating EVs with TDNs, no significant change in EVs size was observed while the zeta-potential was decreased since DNA nanostructures were negatively charged (Figure 1E). These results together suggested that our loading process did not significantly alter the morphology of EVs, which could exhibit similar characteristics as native EVs did.
Figure 1.
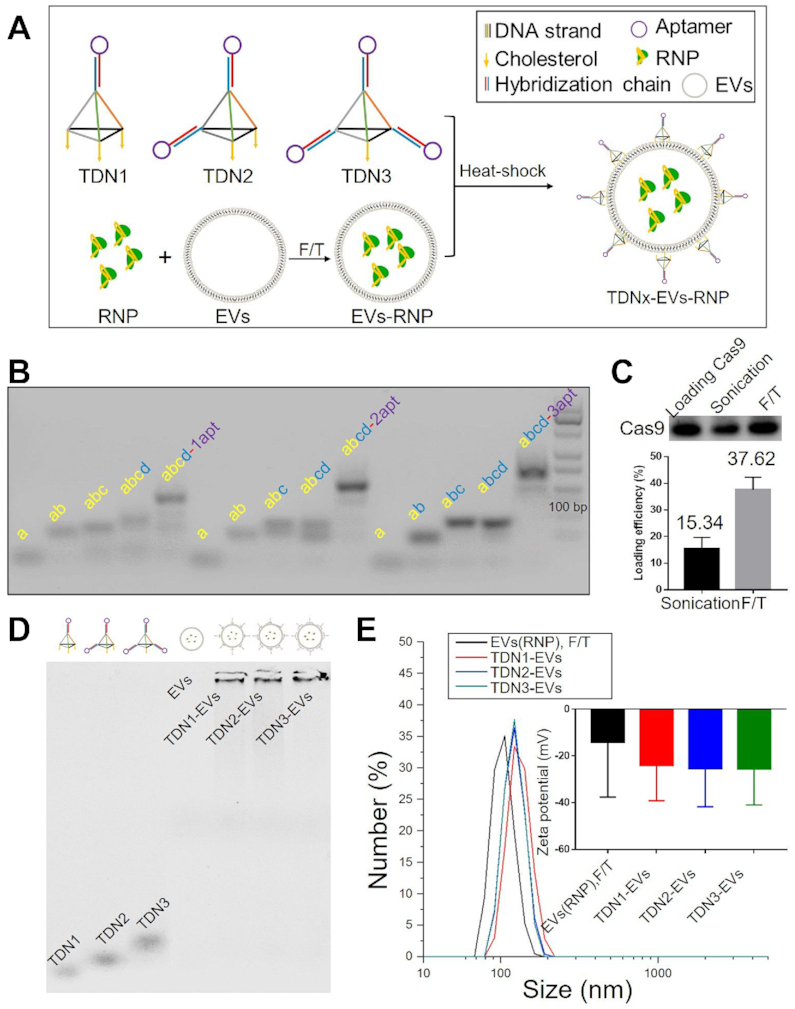
Characterization of TDNx-EVs (x = 1–3). (A) Loading RNPs into EVs and decoration of TDNs. (B) Self-assemblies of DNA nanostructures. (C) Loading efficiency of RNPs by different techniques. (D) AGE-characterization of TDNs decoration. (E) DLS for size analysis and zeta potential measurements for TDNs-EVs, scale bar, 100 nm.
The valency of TDNs control the orientation of DNA aptamer on EVs
In this study, the distribution ratios of DNA aptamers and cholesterol molecules (1:3, 2:2 and 3:1) for TDNs were used to control the surface display of aptamers. To illustrate the valence-controlled capability of surface display of TDNs on EVs, we first used fluorescence resonance energy transfer (FRET) to investigate the orientations of the TDNs and the interactions between aptamers and the EVs membrane. Alexa Fluor 488 (AF488) was labeled the aptamer strands of TDNs and Dil was labeled the lipid bilayer of EVs, respectively. Figure 2A shows a schematic diagram for a FRET experiment to demonstrate the display of aptamer ligands. As we know, FRET has been widely used to identify the interactions between two molecules. Specifically, FRET signals could arise from the interaction of the AF488-labelled aptamers (donor) and the Dil-labelled membrane (acceptor), which indicate the change in the distance or orientation of the donor and acceptor. The presence of the FRET signal first confirmed the successful anchor of TDNs onto EVs membrane (Figure 2B). An intense emission band around 540–580 nm was observed when compared to the FRET donor band of AF488 around 520 nm, which suggested that the binding of the TDNs on the EVs membrane resulted in the conformational alteration of the TDNs (Figure 2C). Next, the FRET efficiencies of FRET between aptamers of different TDNs and membranes were evaluated and found that the FRET signal was sensitive to the addition of the cholesterol anchors in the DNA nanostructures. Indeed, TDNs with three cholesterol anchors exhibited remarkably high FRET efficiency, which implies efficient energy transfer from aptamer to the membrane at its surface. Consistently, a decrease from 3 to 1 in the numbers of membrane anchors of TDNs (from TDN1 to TDN3) leads to an increase in the emission peak of the AF488 donor (500–520 nm) and a subsequent reduction in the emission peak of the DiI acceptor (555–582 nm). Meanwhile, the measured loading efficiencies of different TDNs were used to correct acquired the FRET efficiencies, which could quantity the interaction effects between aptamers and EVs membrane. As expected, theTDN3 might follow a free-swaying mode of its aptamer strands and exert preferential encounters with the DiI-labelled membrane, thus the interaction effect was estimated to be the largest one among three TDNs. Similarly, the aptamers of TDN2 seemed to entangle with EVs membrane in two directions and fall on the EVs surface, where the interaction effect slightly decreased. In contrast, TDN1 was built to orthogonally stand on the lipid membrane with three legs and maintain an exposed display of aptamers, which results in the lowest value of interaction effect. It seems as if the three arms that can act as a hook to lock the aptamer in place. If so, such conformation would prevent the energy transfer between aptamers and EVs membrane for TDN1-EVs. These findings indicated that the DNA aptamer assembly on EVs membrane could be controlled via hybridizing different ratios of aptamer/cholesterol among these TDNs.
Figure 2.

Characterization of valency-controlled display of DNA aptamers in nano-scale. (A) FRET analysis of the formation of TDNx-EVs. (B) Schematic illustration of different displays of valence-controlled TDNx (x = 1–3) on EVs surface. (C) Characterization of the interaction effect between aptamers and lipid membrane for different TDNx-EVs
The surface display of TDN1 on EVs enhance the cell-specific delivery
The surface display of DNA aptamers was further confirmed by cellular internalization assay. AF488-labeled sDNAs were first encapsulated within the TDNs engineered EVs and liposome. Then cells were incubated with these fluorescent nanoparticles and detected by a confocal laser scanning microscope. We first evaluated the targeting capacity of TLS11a aptamer and observed an obvious binding of TLS11a when the aptamer was used to treat with HepG2 cells (Supplementary Figure S10). Then the AF488-labeled sDNA was encapsulated in order to characterize the cellular uptake of the developed nanoparticles (Supplementary Figure S11). Our cell imaging results of HepG2 (TLS11a specific) and MCF-7 (TLS11a non-specific) cells revealed that the intracellular accumulations of HepG2 were significantly benefited from the EVs-based delivery and DNA nanostructure decoration when compared with MCF-7 (Figure 3A). The quantitative cellular uptake increased monotonically with decreasing valence of cholesterol anchors in TDNs (Figure 3B), suggesting that the display of aptamer on EVs surface could change the binding between EVs and cells. However, the TDNs could also act as a double-edged sword in enhancing cellular uptake due to its negative charge. For example, the surface charge effect of DNA nanostructures was greater than the binding force of the aptamer of DNA for TDN3-EVs, which resulted in the reduction of cellular uptake of TDN3-EVs when compared with that of EVs. Together, these results indicated that the TDNs, especially TDN1, allowing for better display of aptamers on the surface of EVs, increased EVs’ targeting efficiency to the recipient cancer cells. Since TDN1-EVs showed the highest delivery efficacy in HepG2 cells, we choose TDN1-EVs as our final delivery nanovehicle in the following study. The cellular uptake efficacy of TDN1-EVs was much lower in all the studied non-liver cell lines when compared with that in HepG2 cells (Supplementary Figure S12). Besides, human primary liver cancer-derived organoid (PLO) model was established to verify the targeting efficacy of TDN1-EVs. Unlike cell lines, organoids are three-dimensional micro-organs which are highly similar to the original tissues and organs (33). Tumor organoids can be maintained for months and grow to a diameter of about 1 mm, conserving the molecular and cellular composition, as well as genetic features of the original tumors. The results showed that the TDN1-EVs exhibited larger fluorescent cargo accumulation in the organoids when compared with liposome and EV groups, indicating that the DNA nanostructure is vital for improving the targeted delivery (Figure 3C). Since the TDN1-EVs possess a rigid display of aptamers on the surface with TDNs, the affinity between nanoparticles and tumor organoids was significantly enhanced (Figure 3D). Notably, human PLO models contained a few CD45+ leukocytes, which could be used to differentiate targeted delivery from non-targeted delivery (34). The fluorescence of sDNA by liposome delivery was mostly co-localized in CD45+ leukocytes. By contrast, <10% of CD45+ leukocytes were double-stained in TDN1-EVs group (Supplementary Figure S13). Moreover, the flow cytometry also revealed that only a few CD45+ cells from THP-1 cells could interact with TDN1-EVs (Supplementary Figure S14).
Figure 3.

Targeting effects of TDNx-EVs (x = 1–3) in vitro, ex vivo and in vivo. (A) Cellular uptake of indicated AF488-labelled sDNA loaded vehicles in HepG2 and MCF-7 cells, scale bar, 10 μm. (B) Quantification of intracellular fluorescence accumulation in HepG2 and MCF-7 cells over 8 h (n = 5). (C) Quantification of intracellular fluorescence accumulation in PLO models over 24 h. (D) Bright field, Celltrace blue, CD45-PE, sDNA-AF488 and merged pictures of the organoids exposing different nanoparticles (n = 3), scale bar, 50 μm. (E) Fluorescence images of HepG2 tumor-bearing mice treated with different foumlations at 0.5 and 4 h postinjection. (F) Biodistribution of fluorescent nanoparticles in major organs collected after 12 h of intravenous injection.
To further investigate the in vivo targeting effects of TDN1-EV biomaterials, a xenograft tumor model was established by subcutaneous injection of HepG2 into BALB/c nude mice, and then after 28 days, the fluorescent dye DiR-stained EVs or liposomes (Lipo) were intravenously injected into HepG2 tumor-bearing mice, and analysis by in vivo fluorescence imaging of living mice. Given the long duration of EVs, EVs-based nanoplatform exhibited a higher accumulation than that of Lipo group (Figure 3E). However, nanoparticles without targeting molecule, especially for EVs, failed to reach the tumor, resulting in a rapid accumulation in liver, which was similar to free DiR (Figure 3F and Supplementary Figure S15), which suggested that EVs lacks tumor targeting ability. As expected, TDN1-EVs achieved successful delivery into tumor, as indicated by a significantly higher level of fluorescence in tumors than major organs when compared with EVs group and Lipo group. Taking together, we demonstrated that TDN1-EVs was capable of hepatocellular carcinoma cell-specific delivery.
Exploring the feasibility of gene-editing with TDN1-EVs-RNP
We next evaluated the gene editing capability of the TDN1-EVs. The GFP-expressing HepG2 cells (HepG2/GFP) were used to evaluate the feasibility of gene-editing by RNP-loaded nanoparticles. Carriers containing RNP (targeting GFP gene) could cut the target DNA fragments into two bands with a size of 383 and 338 bp, respectively, as confirmed by the T7E1 assay (Figure 4A). The results revealed that the TDN1-EVs reduced fluorescence in about 43% of the cells when compared with the PBS group, whereas the Lipo-RNP had a weak effect on GFP expression (Figure 4B). The PCR-based sequencing analysis revealed that there were indel mutations at the GFP-specific gRNAs editing sites in PCR products (Figure 4C and Supplementary Figure S16). TDN1-EVs performed better for GFP knockdown than their counterparts, including Liposome and Apt-EVs, which resulted in a significant rise of GFP negative cells (Figure 4D, E). To be noted, the newly developed TDNs-based exosomal carriers showed a potent capability of loading and delivering CRISPR/Cas9 RNP complexes for gene editing, which may present great potential in gene therapy of various diseases.
Figure 4.

Gene editing by TDN1-EVs-deliveryed Cas9/sgGFP system. (A) T7EI assay of HepG2/GFP cells treated with PBS, Lipo-RNP, EVs-RNP and TDN1-EVs-RNP (B) Flow-cytometry analysis revealed that the population of GFP negative cells were significantly increased by TDN1-EVs-RNP when compared with other treatments. (C) The gene edting of GFP targeting site based on the clone sequencing analysis. (D) Fluorescence images of HepG2/GFP cells showing the GFP knockdown of PBS, Lipo-RNP, EVs-RNP and TDN1-EVs-RNP. (E) Quantification of green fluorescence in HepG2 cells over 72 h (n = 3), scale bar, 10 μm.
CRISPR/Cas9 mediated WNT10B inhibition produced anti-tumor effects on hepatocellular cancer with TDN1-EVs-RNP
Recent studies have shown an aberrant overexpression of WNT10B gene in tissues and cultured cells from hepatocellular cancer (35,36). The current report has revealed that siRNA-induced gene silencing WNT10B could reduce the viability and inhibit the migration of HepG2 (37). However, the working mechanism of siRNA is largely limited to its transient transfection, which cannot maintain a prolonged effect of tumor inhibition. In this study, RNP contained a sgRNA that targets the exon of WNT10B, was constructed as cargo for gene editing. CRISPR knockdown of WNT10B expression with TDN1-EVs in HepG2 cells and PLOs were investigated. The protein expression levels of WNT10B were reduced significantly in the TDN1-EVs-treated cells, whereas no differences in the WNT10B expression were observed in other groups compared to PBS-treated cells (Figure 5A). The result indicated the capability of the novel developed RNP nano-delivery system to successful CRISPR inhibition of WNT10B in HepG2 cells, which also suggested the potential of TDN1-EVs in repressing tumor growth via the transcriptional down-regulation of endogenous WNT10B. The antitumor effect of TDN1-EVs in HepG2 cells was evaluated. The cells in TDN1-EVs group exhibited significantly lower viability (59%) than any other comparative groups, suggesting the viability of HepG2 cells was decreased as a result of RNP delivery (Figure 5B). Similarly, high-throughput sequencing indicated that negligible editing was observed upon PBS incubation ex vivo, while TDN1-EVs induced effective editing in WNT10B gene (Figure 5C and Supplementary Tables S4 and S5). In addition to determining the suppressing effects by TDN1-EVs in cultured cancer cell lines, PLO model was also applied to assess the efficiency of tumor destruction by these nanoparticles. A green fluorescent caspase-3/7 probe was used to detect cell apoptosis of tumor organoids. The inhibition of ex vivo cancer growth was more pronounced than that in in vitro hepatocellular cancer studies (Figure 5D; Supplementary Figure S17). For all samples tested, exposure of tumor organoids to EVs and TDN1-EVs led to widespread apoptosis in organoids, whereas Liposome exhibited a little anti-tumor effect on PLOs. By inhibiting the essential proteins for WNT/β-catenin signaling, TDN1-EVs showed strong RNP-mediated apoptosis of the tumor cells in PLOs. Based on the successful tumor inhibition in vitro and ex vivo, additional studies were performed to investigate the potential of TDN1-EVs-RNP in cancer gene therapy. In comparison with the saline group, treated with Lipo-RNP and EVs-RNP did not significantly suppress tumor development (Figure 5E, F). However, tumor development was terminated by TDN1-EVs-RNP treatment. Tumor inhibition was found in the mice injected with two different doses of TDN1-EVs-RNP (Figure 5G). The results also suggested that high dose of TDN1-EVs-RNP triggered the cell apoptosis in xenograft tumor tissues (Supplementary Figure S18). Encouraged by these results, the gene editing of TDN1-EVs-RNP was also investigated. Of importance, the gene editing observed in the high dose of TDN1-EVs-RNP was close to 5 fold greater than that performed by the EVs-RNP (Supplementary Tables S6 and S7). No obvious change in body weights was observed while the histopathological analysis of H&E-stained major organ sections including heart, liver, spleen, lung and kidney found no significantly toxic pathological changes after administration (Supplementary Figures S19 and S20), indicating limited toxicity of our developed nanoparticles. Considering a high concentration of EVs was accumulated in the liver, another concern about safety is the liver and kidney toxicity of TDN1-EVs-RNP. Although the increased level of ALT in a high dose of TDN1-EVs-RNP suggested the induced liver toxicity, the decreased value of AST/ALT hints this toxicity is possibly mild and reversible (Supplementary Figure S21). Meanwhile, the BUN and CR levels in the mouse blood did not show a significant increase among all samples. Moreover, negligible variations of inflammatory cytokine were detected in plasma derived from TDN1-EVs-RNP-treated mice (Supplementary Figure S22). Overall, the inhibition of tumor growth confirmed the feasibility of applying the CRISPR/Cas9 system for cancer treatment with the aid of a TDNs-based targeted delivery system, which indicated that TDN1-EVs presented its potential in the development of novel CRISPR/Cas9-based cancer gene therapies.
Figure 5.

Anti-tumor effects of TDN1-EVs delivered Cas9/sgWNT10B gene editing system in vitro, ex vivo and in vivo. (A) Western blot analysis of WNT10B protein expression in HepG2 cells treated by different Cas9/sgWNT10B nanocarriers. (B) Cell growth inhibition was observed after WNT10B knockdown (n = 5). (C) The percentage indel rates in PLOs based on sequencing. (D) Quantification of organoid killing upon different Cas9/sgWNT10B nanocarriers (n = 3). (E) Representative images of the BALB/c nude mice bearing HepG2 xenograft tumors after intravenous injection with different Cas9/sgWNT10B nanocarriers and the xenograft tumors harvested from the mice. (F) The volume growth curves of xenograft tumors from HepG2 tumor-bearing mice during treatment (n = 5). (G) Comparison of the weight of the harvested tumors from the mice after treatment.
DISCUSSION
CRISPR/Cas9 hold great promise as therapeutic uses, given its role in facilitating effective regulation of gene expression both in vitro and in vivo. Various approaches have been developed to efficiently deliver CRISPR/Cas9 systems in recent studies (38–40). Here we developed a novel delivery vehicle for CRISPR/Cas9 based on EVs, which can naturally transfer biomolecules in the human body. However, one major limitation of the therapeutic use of EVs is the lack of selectivity. This study aimed to optimize a 3D DNA nanostructure surface modification for specific cell-targeting EVs and evaluate the application of CRISPR/cas9 technology to knockdown gene expression of interest. Approaches have been developed to over-express protein ligands on the surface of EVs, which could be used to enhance the specificity of therapeutic EVs (41–43), generating protein ligands on EVs is only limited to the donner cells. Therefore, it would be acceptable to use our surface engineered strategy to precisely assemble DNA aptamers on EVs in the laboratory.
To summarize, our study demonstrated the modification of EVs surface can regulate the affinity of EVs for certain cells. We used the 3D DNA nanotechnology to reprogram natural EVs’ surface for cell-specific delivery of Cas9 in in vitro cultured cancer cells, ex vivo PLO models and in vivo xenograft models. We were able to tune the display of aptamers on the EVs surface by regulating the cholesterol/aptamer ratio in TDNs. The cellular accumulation of these RNP-loaded nanoparticles was found to be dependent on their surface properties, demonstrating the impacts of the aptamer/cholesterol-based, valency-controlled decoration of TDNs. With 3 cholesterol anchors of TDNs, the DNA aptamer on the EVs showed an oriented surface display of targeting modules for cancer-specific binding. Thus an efficient targeting capacity of TDN1-EVs was observed, which could be used to deliver RNPs to specific cells in current work. The cargo of vehicles could be also extended to siRNA, DNA plasmids, and other enzymes. Overall, we established a potential strategy for cell-specific exosomal delivery of RNPs with DNA nanotechnology, and we believe that such an approach may eventually allow in vivo delivery of genome-editing enzymes to targeted cell types.
Supplementary Material
Contributor Information
Jialang Zhuang, Guangdong Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, Guangdong 510006, China.
Jizhou Tan, Department of Interventional Oncology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, P. R. China.
Chenglin Wu, Department of Interventional Oncology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, P. R. China; Organ Transplantation Center, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, P. R. China.
Jie Zhang, Guangdong Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, Guangdong 510006, China.
Ting Liu, Department of Interventional Oncology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, P. R. China.
Chunhai Fan, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Dongchuan Rd 800, Shanghai 200240, P. R. China.
Jiaping Li, Department of Interventional Oncology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, P. R. China.
Yuanqing Zhang, Guangdong Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, Guangdong 510006, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [81601571, 81771932, 81671797, 81971719, 2019YFA0111500, 81601391, 81171441]; Guangdong Natural Science Fund for Distinguished Young Scholars [2017A030306016, 2016A030306004]; Major scientific and technological project of Guangdong Province [2017B030308006, 2017A030313825]; Major program for tackling key problems of Guangzhou City, China [201704020144]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Savic N., Schwank G.. Advances in therapeutic CRISPR/Cas9 genome editing. Transl Res. 2016; 168:15–21. [DOI] [PubMed] [Google Scholar]
- 2. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yi L., Li J.. CRISPR–Cas9 therapeutics in cancer: promising strategies and present challenges. Biochim. Biophys. Acta. 2016; 1866:197–207. [DOI] [PubMed] [Google Scholar]
- 4. Wang H.X., Li M., Lee C.M., Chakraborty S., Kim H.W., Bao G., Leong K.W.. CRISPR/Cas9-based genome editing for disease modeling and therapy: challenges and opportunities for nonviral delivery. Chem. Rev. 2017; 117:9874–9906. [DOI] [PubMed] [Google Scholar]
- 5. Liu C., Zhang L., Liu H., Cheng K.. Delivery strategies of the CRISPR–Cas9 gene-editing system for therapeutic applications. J. Control. Release. 2017; 266:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nault J.C., Datta S., Imbeaud S., Franconi A., Mallet M., Couchy G., Letouze E., Pilati C., Verret B., Blanc J.F. et al.. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015; 47:1187–1193. [DOI] [PubMed] [Google Scholar]
- 7. Yin H., Kanasty R.L., Eltoukhy A.A., Vegas A.J., Dorkin J.R., Anderson D.G.. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014; 15:541–555. [DOI] [PubMed] [Google Scholar]
- 8. Al-Nedawi K., Meehan B., Micallef J., Lhotak V., May L., Guha A., Rak J.. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008; 10:619–624. [DOI] [PubMed] [Google Scholar]
- 9. Thery C., Ostrowski M., Segura E.. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009; 9:581–593. [DOI] [PubMed] [Google Scholar]
- 10. Zhang B., Yin Y., Lai R.C., Tan S.S., Choo A.B., Lim S.K.. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev. 2014; 23:1233–1244. [DOI] [PubMed] [Google Scholar]
- 11. Salido-Guadarrama I., Romero-Cordoba S., Peralta-Zaragoza O., Hidalgo-Miranda A., Rodriguez-Dorantes M.. MicroRNAs transported by exosomes in body fluids as mediators of intercellular communication in cancer. Onco Targets Ther. 2014; 7:1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yanez-Mo M., Siljander P.R., Andreu Z., Zavec A.B., Borras F.E., Buzas E.I., Buzas K., Casal E., Cappello F., Carvalho J. et al.. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles. 2015; 4:27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tao S.C., Guo S.C., Zhang C.Q.. Modularized extracellular Vesicles: The dawn of prospective personalized and precision medicine. Adv. Sci. (Weinh.). 2018; 5:1700449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gudbergsson J.M., Jønsson K., Simonsen J.B.k., Johnsen K.B.. Systematic review of targeted extracellular vesicles for drug delivery - Considerations on methodological and biological heterogeneity. J. Control. Release. 2019; 306:108–120. [DOI] [PubMed] [Google Scholar]
- 15. Colao I.L., Corteling R., Bracewell D., Wall I.. Manufacturing Exosomes: A promising therapeutic platform. Trends Mol. Med. 2018; 24:242–256. [DOI] [PubMed] [Google Scholar]
- 16. Mathieu M., Martin-Jaular L., Lavieu G., Thery C.. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019; 21:9–17. [DOI] [PubMed] [Google Scholar]
- 17. O Loughlin A.J., Mäger I., de Jong O.G., Varela M.A., Schiffelers R.M., El Andaloussi S., Wood M.J.A., Vader P.. Functional delivery of lipid-conjugated siRNA by extracellular vesicles. Mol. Ther. 2017; 25:1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ye Z., Zhang T., He W., Jin H., Liu C., Yang Z., Ren J.. Methotrexate-Loaded extracellular vesicles functionalized with therapeutic and targeted peptides for the treatment of glioblastoma multiforme. ACS Appl. Mater. Interfaces. 2017; 10:12341–12350. [DOI] [PubMed] [Google Scholar]
- 19. Piffoux M., Silva A.K.A., Wilhelm C., Gazeau F., Tareste D.. Modification of extracellular vesicles by fusion with liposomes for the design of personalized biogenic drug delivery systems. ACS Nano. 2018; 12:6830–6842. [DOI] [PubMed] [Google Scholar]
- 20. Pi F., Binzel D.W., Lee T.J., Li Z., Sun M., Rychahou P., Li H., Haque F., Wang S., Croce C.M. et al.. Nanoparticle orientation to control RNA-loading and ligand display on extracellular vesicles for cancer regression. Nat. Nanotechnol. 2018; 13:82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jc Bose R., Uday Kumar S., Zeng Y., Afjei R., Robinson E., Lau K., Bermudez A., Habte F., Pitteri S.J., Sinclair R. et al.. Tumor cell-derived extracellular Vesicle-coated nanocarriers: an efficient theranostic platform for the cancer-specific delivery of anti-miR-21 and imaging agents. ACS Nano. 2018; 12:10817–10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun H., Zu Y.. Aptamers and their applications in nanomedicine. Small. 2015; 11:2352–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li J., Fan C., Pei H., Shi J., Huang Q.. Smart drug delivery nanocarriers with self-assembled DNA nanostructures. Adv. Mater. 2013; 25:4386–4396. [DOI] [PubMed] [Google Scholar]
- 24. Pei H., Zuo X., Zhu D., Huang Q., Fan C.. Functional DNA nanostructures for theranostic applications. Acc. Chem. Res. 2013; 47:550–559. [DOI] [PubMed] [Google Scholar]
- 25. Bujold K.E., Lacroix A.l., Sleiman H.F.. DNA nanostructures at the interface with biology. Chem. 2018; 4:495–521. [Google Scholar]
- 26. Wang Z., Qin W., Zhuang J., Wu M., Li Q., Fan C., Zhang Y.. Virus-mimicking cell capture using heterovalency magnetic DNA nanoclaws. ACS Appl. Mater. Interfaces. 2019; 11:12244–12252. [DOI] [PubMed] [Google Scholar]
- 27. Li Q., Zhao D., Shao X., Lin S., Xie X., Liu M., Ma W., Shi S., Lin Y.. Aptamer-modified tetrahedral DNA nanostructure for tumor-targeted drug delivery. ACS Appl. Mater. Interfaces. 2017; 9:36695–36701. [DOI] [PubMed] [Google Scholar]
- 28. Jiang D., Sun Y., Li J., Li Q., Lv M., Zhu B., Tian T., Cheng D., Xia J., Zhang L.. Multiple-armed tetrahedral DNA nanostructures for tumor-targeting, dual-modality in vivo imaging. ACS Appl. Mater. Interfaces. 2016; 8:4378–4384. [DOI] [PubMed] [Google Scholar]
- 29. Liu Q., Ge Z., Mao X., Zhou G., Zuo X., Shen J., Shi J., Li J., Wang L., Chen X. et al.. Valency-controlled framework nucleic acid signal amplifiers. Angew. Chem. Int. Ed. 2018; 57:7131–7135. [DOI] [PubMed] [Google Scholar]
- 30. Shangguan D., Meng L., Cao Z.C., Xiao Z., Fang X., Li Y., Cardona D., Witek R.P., Liu C., Tan W.. Identification of liver cancer-specific aptamers using whole live cells. Anal. Chem. 2008; 80:721–728. [DOI] [PubMed] [Google Scholar]
- 31. Lin M., Wang J., Zhou G., Wang J., Wu N., Lu J., Gao J., Chen X., Shi J., Zuo X. et al.. Programmable engineering of a biosensing interface with tetrahedral DNA nanostructures for ultrasensitive DNA detection. Angew. Chem. Int. Ed. 2015; 54:2151–2155. [DOI] [PubMed] [Google Scholar]
- 32. Bunge A., Loew M., Pescador P., Arbuzova A., Brodersen N., Kang J., Dahne L., Liebscher J., Herrmann A., Stengel G. et al.. Lipid membranes carrying lipophilic cholesterol-based oligonucleotides–characterization and application on layer-by-layer coated particles. J. Phys. Chem. B. 2009; 113:16425–16434. [DOI] [PubMed] [Google Scholar]
- 33. Gao D., Vela I., Sboner A., Iaquinta P.J., Karthaus W.R., Gopalan A., Dowling C., Wanjala J.N., Undvall E.A., Arora V.K.. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014; 159:176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dijkstra K.K., Cattaneo C.M., Weeber F., Chalabi M., van de Haar J., Fanchi L.F., Slagter M., van der Velden D.L., Kaing S., Kelderman S. et al.. Generation of Tumor-Reactive T cells by Co-culture of peripheral blood lymphocytes and tumor organoids. Cell. 2018; 174:1586–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shibata T., Aburatani H.. Exploration of liver cancer genomes. Nat. Rev. Gastroenterol. Hepatol. 2014; 11:340. [DOI] [PubMed] [Google Scholar]
- 36. Ji J., Yamashita T., Wang X.W.. Wnt/beta-catenin signaling activates microRNA-181 expression in hepatocellular carcinoma. Cell & Bioscience. 2011; 1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu G., Fan X., Sun L.. Silencing of Wnt10B reduces viability of heptocellular carcinoma HepG2 cells. Am. J. Cancer Res. 2015; 5:1911. [PMC free article] [PubMed] [Google Scholar]
- 38. Li Y., Bolinger J., Yu Y., Glass Z., Shi N., Yang L., Wang M., Xu Q.. Intracellular delivery and biodistribution study of CRISPR/Cas9 ribonucleoprotein loaded bioreducible lipidoid nanoparticles. Biomater. Sci. 2019; 7:596–606. [DOI] [PubMed] [Google Scholar]
- 39. Liu Q., Zhao K., Wang C., Zhang Z., Zheng C., Zhao Y., Zheng Y., Liu C., An Y., Shi L. et al.. Multistage delivery nanoparticle facilitates efficient CRISPR/dCas9 activation and tumor growth suppression in vivo. Adv. Sci. 2019; 6:1801423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rouet R., Thuma B.A., Roy M.D., Lintner N.G., Rubitski D.M., Finley J.E., Wisniewska H.M., Mendonsa R., Hirsh A., de Oñate L et al.. Receptor-mediated delivery of CRISPR–Cas9 endonuclease for cell-type-specific gene editing. J. Am. Chem. Soc. 2018; 140:6596–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ohno S.-i., Takanashi M., Sudo K., Ueda S., Ishikawa A., Matsuyama N., Fujita K., Mizutani T., Ohgi T., Ochiya T.. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013; 21:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang J.-H., Forterre A.V., Zhao J., Frimannsson D.O., Delcayre A., Antes T.J., Efron B., Jeffrey S.S., Pegram M.D., Matin A.C.. Anti-HER2 scFv-directed extracellular vesicle-mediated mRNA-based gene delivery inhibits growth of HER2-positive human breast tumor xenografts by prodrug activation. Mol. Cancer Ther. 2018; 17:1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kooijmans S.A.A., Aleza C.G.m., Roffler S.R., van Solinge W.W., Vader P., Schiffelers R.M.. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J. Extracell. Vesicles. 2016; 5:31053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.