


Abstract
The protein kinase Gcn2 is a central transducer of nutritional stress signaling important for stress adaptation by normal cells and the survival of cancer cells. In response to nutrient deprivation, Gcn2 phosphorylates eIF2α, thereby repressing general translation while enhancing translation of specific mRNAs with upstream ORFs (uORFs) situated in their 5′-leader regions. Here we performed genome-wide measurements of mRNA translation during histidine starvation in fission yeast Schizosaccharomyces pombe. Polysome analyses were combined with microarray measurements to identify gene transcripts whose translation was up-regulated in response to the stress in a Gcn2-dependent manner. We determined that translation is reprogrammed to enhance RNA metabolism and chromatin regulation and repress ribosome synthesis. Interestingly, translation of intron-containing mRNAs was up-regulated. The products of the regulated genes include additional eIF2α kinase Hri2 amplifying the stress signaling and Gcn5 histone acetyl transferase and transcription factors, together altering genome-wide transcription. Unique dipeptide-coding uORFs and nucleotide motifs, such as ‘5′-UGA(C/G)GG-3′, are found in 5′ leader regions of regulated genes and shown to be responsible for translational control.
INTRODUCTION
In response to diverse cellular stresses, ribosomes reprogram global protein synthesis to optimize utilization of nutrients and energy and reconfigure the proteome to mitigate stress damage (1,2). For example, during limitation for amino acids, Gcn2 (EIFKA4) phosphorylates the α subunit of eIF2, thereby reducing delivery of initiator tRNAs to ribosomes which culminates in lowered global protein synthesis. Concurrently, phosphorylation of eIF2α (eIF2α-P) enhances translation of select mRNAs, such as Saccharomyces cerevisiae Gcn4 and mammalian Atf4, which direct gene expression for stress adaptation (3,4) in both normal cells and cancer (5,6).
The underlying process by which ribosomes delineate which mRNAs are translationally repressed, not affected, or preferentially translated involves cis-acting elements embedded in target mRNAs. A critical cis-acting regulator is upstream ORFs (uORFs) that can be present in the 5′-leaders of mRNAs (7). uORFs are suggested to be present in over 50% of mammalian and 10% of yeast mRNAs. Ribosomes associated with eIF2 and its bound initiator tRNA engage the 5′-cap of mRNAs and then processively scan 5′- to 3′- in search of an initiation codon. Often uORFs are bypassed due to the poor context of its AUG start codon or, when translated, lower downstream initiation of the main coding sequence (CDS). In some cases, uORFs allow ribosomes to reinitiate downstream if the ribosome remains linked to the mRNA after translation termination and resumes scanning for subsequent translation initiation (8). Under special arrangements, the inhibition caused by uORFs can be alleviated when Gcn2 or one of the three other eIF2α kinases present in mammals, Hri (EIF2AK1) Pkr (EIF2AK2), and Perk (EIFAK3), is activated by different stress stimuli (9). Yeast Gcn4 (3) and mammalian Atf4 (4) mRNAs possess such inhibitory uORFs, along with a 5′-proximal uORF allowing for re-initiation downstream. Upon stress and induced eIF2α-P, delayed translation re-initiation allows for ribosomes to scan through one or more downstream inhibitory uORFs and instead initiate translation at the CDS. In this way, the Gcn4 and Atf4 are well translated only during cellular stress and induced eIF2α-P.
Given that multiple mammalian protein kinases direct translational control of key stress response genes via phosphorylation of eIF2α, this pathway in mammals has been referred to as the integrated stress response (10). By comparison, S. cerevisiae features the single eIF2α kinase Gcn2, which is present in virtually all eukaryotes, and this translation control process is referred to as general amino acid control (GAAC) (3). It is noteworthy that other eukaryotes, including insects, plants and fungi, express different combinations of these eIF2α kinases or variants thereof. For example, fission yeast Schizosaccharomyces pombe expresses gcn2 and two orthologs of Hri, designated hri1 and hri2 (11). The expression of distinct collections of eIF2α kinases among different eukaryotic organisms indicates that the conserved translation control scheme is well positioned to respond to their distinct developmental, physiological and environmental stresses.
While recent studies revealed various mechanisms of uORF-dependent regulation (9), there is less known about the participation of other types of cis-regulatory elements in the differential translation in response to eIF2α-P. Furthermore, there are still important gaps in our knowledge about the extent of preferential translation induced by eIF2α-P and the physiological consequences of the translational control during stress. To gain a quantitative genome-wide overview of translational control by Gcn2 and pursue new mechanisms of translational regulation, we conducted translational profiling using polysome profiling of the S. pombe model system challenged with histidine depletion. Combined with previous ribosome profiling data from S. pombe treated under similar conditions (12), our results reveal that unique uORF and nucleotide motifs found in mRNA 5′ leader regions coordinately regulate translation genome-wide by functional groups.
MATERIALS AND METHODS
Yeast strains and plasmids
Schizosaccharomyces pombe strains used in this study are described in Supplementary File. Plasmids used are listed in Supplementary Table S1 and generated using the oligodeoxyribonucleotides listed in Supplementary Table S2.
mRNA translation analysis by polysome profiling
To assess the translation status of mRNAs, cell extracts containing poly-ribosomes (polysomes) were prepared after 5-min cycloheximide (CHX) treatment (50 μg/ml) of the cells, and resolved by sucrose gradient-velocity sedimentation (13), all as described in Supplementary Materials and Methods. Amounts of specific mRNAs in the gradients or total lysate were assessed by microarray hybridization or quantitative PCR (qPCR) analysis and normalized by external RNA standards (see Supplementary Materials and Methods). Microarray data was deposited to GEO as GSE143299.
Bioinformatics analysis
For each (i) of 6657 poly(A) RNAs in S. pombe, we obtained relative molar abundance, ai, from transcriptional analysis and its distribution in seven polysomal fractions, f i,n (n = 1–7), under four conditions, WT with or without 3AT (average of three independent experiments) and gcn2Δ with or without 3AT (1 experiment). We then calculated ribosome density (δi, the average number of ribosomes associated with mRNA per 1-kb coding region) for mRNA species i as the measurement of its translation efficiency, using the obtained f i,n, as follows:
 |
where Nn is the number of ribosomes associated with mRNA in each fraction that was determined by simulation (see Supplementary Materials and Methods for details).
To evaluate distribution of a specific group (m) of mRNAs in polysomes, the abundance of ribosomes associated with this group of mRNAs was computed as follows:
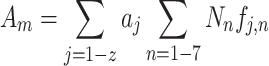 |
where j is a member of mRNA in this group and z is the number of mRNAs belonging to this group.
To generate a list of translationally controlled genes, we ranked genes and listed groups TA1, TA2 and TA3 by 3AT-induced fold change in δ greater in WT than 1.025 (TA1), 1.05 (TA2) or 1.06 (group TA3), gcn2+ dependency and no transcriptional regulation (see Supplementary Materials and Methods for details). Alternatively, we used a spike-free method (ppm scaling) to rank similarly translationally controlled genes and made the list TA4 (see Supplementary Materials and Methods for details). For gene ontology (GO) enrichment analysis, we used PANTHER (14). The consensus motifs found in mRNAs of translationally controlled groups were analyzed by REFINE (15,16), which runs MEME (17) after short sequence enrichment.
Luciferase reporter assays
S. pombe transformants carrying the dual luciferase plasmids were grown in EMMC-His-Leu and treated with 10 mM 3AT. Fixed A600 units of culture were collected and subjected for Dual GloR Luciferase Assay System, as described in Supplementary Materials and Methods.
Re-analysis of ribosome profiling data
To determine ribosome occupancy of uORFs in 3AT-treated wild-type and gcn2Δ fission yeasts, we used the ribosome profiling data from (12). Details of uORF analysis are described in Supplementary Materials and Methods.
RESULTS
Translational profiling of S. pombe by polysome fractionation
3-Aminotrizole (3AT) is a potent inducer of starvation stress by inhibiting the imidazole glycerol phosphate dehydratase (encoded by S. pombe His5), effectively reducing the level of histidine in an otherwise prototrophic strain (18). As a result, eIF2α∼P by Gcn2 and the accompanying reduction in eIF2-GTP lead to an overall reduction in translation initiation. This decrease of global translation initiation during the starvation stress results in a reduction in large polysomes, accompanied by increased monosomes, as observed with 3AT-treated yeast (Figure 1A). Of note, histidine starvation altered polysome-to-monosome (P/M) ratio from 4.2 to 3.3, without altering the overall ribosome abundance measured directly from the A254 profile. However, Gcn2 can also mediate transcriptional response termed GAAC through affecting translation of key transcription factors or chromatin modifiers (12,18,19) and that GAAC involves transcriptional repression. In fact, ribosomal protein mRNAs, which make up ∼50% in mole of total mRNA in yeast (20), are less abundant under the 3AT-induced starvation (see below). Thus, the transcriptional (or posttranscriptional) regulation might account, in part, for the observed reduction in polysomes. We therefore attempted to carefully compare the effect of translational versus transcriptional control on total or specific groups of mRNAs, as described in Table 1.
Figure 1.

Polysome profiling analysis of S. pombe histidine starvation response. (A) A wild-type yeast strain was grown in EMM-Complete (EMMC) (left) or EMM-C-His till A600 = 0.3–0.5 and 10 mM 3AT (inhibitor of histidine synthesis) was added to the latter medium for 30 min (right). After cycloheximide treatment, cell extracts were prepared and resolved by sucrose gradient velocity sedimentation as described (53), as A254 profile was being monitored (shown on Top panels). (B) Representative microarray data of gradient fractions. Fixed amounts of RNA isolated from each fraction were processed for DNA microarray hybridization, after the addition of external RNA standards (spike RNAs). The mRNA abundance across the gradient was normalized by spike RNA hybridization signals and presented as percentage (%) distribution as fi, n after correction by the fraction of RNA used for hybridization reactions. Values for indicated genes under four conditions, WT or gcn2Δ with or without 3AT, are presented. The diagram to the right of the gene name depicts its genomic structure; brown boxes, exons; horizontal lines, introns; and blue boxes, UTR.
Table 1.
Translational control of groups of mRNAs during histidine starvation. We used polysomal microarray data to compute the total molar amount of ribosomes bound to a specified group of mRNA in WT and gcn2Δ cells (columns D, E, H, I; odd numbered rows), relative to total amount of ribosomes bound to all 6657 poly(A) RNA species in WT without 3AT. Likewise, we used conventional microarray data from total RNA to compute the total amount of the specified group of mRNAs (columns D, E, H, I; even numbered rows). 3AT Up, the percentage of the value in 3AT compared to that in control. ΔUp, (3AT Up Ribo/3AT Up mRNA) – 1.
| A | B | C | D | E | F | G | H | I | J | K | row # |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | gcn2- | ||||||||||
| N | Control | 3AT | 3AT Up | ΔUp | Control | 3AT | 3AT Up | ΔUp | |||
| Total poly(A) RNA | 6657 | Ribo # | 100.00% | 81.69% | 81.69% | 1.58% | 85.76% | 83.23% | 97.05% | −1.22% | 1 |
| mRNA | 100.00% | 80.42% | 80.42% | 90.26% | 88.68% | 98.25% | 2 | ||||
| Total mRNA | 5154 | Ribo # | 99.67% | 80.49% | 80.76% | 1.69% | 85.49% | 82.82% | 96.88% | −1.25% | 3 |
| mRNA | 99.36% | 78.90% | 79.41% | 89.63% | 87.94% | 98.11% | 4 | ||||
| Ribosome | 142 | Ribo # | 38.73% | 26.00% | 67.12% | −1.09% | 29.29% | 27.68% | 94.50% | −2.44% | 5 |
| mRNA | 47.01% | 31.90% | 67.86% | 39.66% | 38.42% | 96.87% | 6 | ||||
| Chromatin | 418 | Ribo # | 2.11% | 2.16% | 102.47% | 3.46% | 2.02% | 2.10% | 103.72% | −0.38% | 7 |
| mRNA | 2.02% | 2.00% | 99.05% | 1.98% | 2.06% | 104.12% | 8 | ||||
| Chromatin in TA1 | 262 | Ribo # | 1.01% | 0.98% | 97.01% | 8.01% | 0.96% | 0.94% | 97.28% | −0.22% | 9 |
| mRNA | 1.03% | 0.92% | 89.82% | 0.98% | 0.95% | 97.49% | 10 | ||||
| Chromosome organization | 442 | Ribo # | 2.57% | 2.15% | 83.51% | 0.29% | 2.44% | 2.86% | 117.36% | 2.25% | 11 |
| mRNA | 2.35% | 1.96% | 83.27% | 2.31% | 2.65% | 114.78% | 12 | ||||
| Chromosome organization in TA1 | 264 | Ribo # | 1.02% | 1.00% | 97.62% | 7.98% | 0.96% | 0.95% | 99.30% | 0.26% | 13 |
| mRNA | 1.03% | 0.93% | 90.40% | 0.98% | 0.97% | 99.04% | 14 | ||||
| RNA metabolism | 744 | Ribo # | 10.13% | 7.76% | 76.59% | 4.01% | 8.15% | 8.39% | 102.90% | 0.71% | 15 |
| mRNA | 10.71% | 7.89% | 73.64% | 9.22% | 9.42% | 102.18% | 16 | ||||
| RNA processing | 508 | Ribo # | 8.09% | 5.98% | 74.00% | 4.05% | 6.30% | 6.63% | 105.19% | 1.52% | 17 |
| mRNA | 8.84% | 6.28% | 71.12% | 7.50% | 7.77% | 103.61% | 18 | ||||
To evaluate the genome-wide contribution of translational control in mRNA distribution across polysomes, we performed microarray hybridization experiments, using seven gradient fractions collected from wild type (WT, three experiments) and gcn2Δ strain (1 experiment) grown in the presence and absence of 10 mMT 3AT for 30 nim, and thereby measured relative molar amount of mRNA in each fraction (translational control analysis). We also isolated mRNA from total cell lysates to measure the total relative molar abundance of mRNAs (transcriptional control analysis). We isolated RNA from each fraction, as described (13) (also see Materials and Methods and SI), and examined the quality of RNA samples by testing the integrity of rRNA found in each fraction prior to every hybridization experiment (Supplementary Figure S1).
The transcriptional control analysis indicated that 3AT decreases the total molar abundance of 6657 poly(A) RNA species to 80.42% (Table 1, row 2, columns D and E). The majority (99.36%) of poly(A) RNA is protein-coding mRNA (while the rest, 0.64%, is non-coding RNA); thus, 3AT likewise decreases the total mRNA abundance to 78.90% (Table 1, row 4, columns D and E). Ribosomal protein mRNA accounts for 47% in mole of the total RNA, which is decrease to 32% by 3AT (Table 1, row 6, columns D and E; also see Supplementary Figure S2A). This decrease accounts for ∼75% of decrease in mRNA molar amount caused by 3AT. Importantly, all of these changes (transcriptional repression) are largely mediated by Gcn2, as they are alleviated by gcn2Δ (Table 1, rows 2, 4, 6, columns H and I). Fission yeast mating and the attendant meiosis require nitrogen starvation (21). To examine if amino acid starvation is a part of the signals induced by this stimulus, we compared genes transcriptionally regulated by 3AT with those regulated by nitrogen starvation at an early stage (22). As shown in Supplementary Figure S2B, regulated genes significantly overlap regardless of whether their transcript abundance is increased (P = 1.9e−56) or decreased (P = 1.1e−18). Importantly, more than half of the overlapping genes alter their expression upon 3AT treatment in Gcn2-dependent manner. Thus, Gcn2-dependent amino acid starvation response is an early component of nitrogen starvation response that leads to mating and meiosis, in agreement with the previous finding that gcn2 is required for mating (23).
To evaluate translational control, we obtained translational profiling data (fi,1∼ fi,7 representing distribution of mRNA i, across the seven sucrose gradients) for all 6657 poly(A) RNA species in S. pombe by normalizing the hybridization signals with external RNA standards (spike-dependent scaling, see Materials and Methods and Supplementary File). Figure 1B shows examples of microarray data for mRNA species showing dramatic gcn2-dependent 3AT-induced translation. We computed the number of ribosomes associated with every mRNA species i by the equation Σ Nn f i,n for n = 1–7 (Nn is the number of ribosomes associated with mRNA found in each fraction and determined after computer simulation; see Figure 2A and Materials and Methods) and the relative molar amount of ribosomes bound to the mRNA by ai Σ Nnfi,n for n = 1–7, where ai is the relative molar amount of the mRNA. We used this equation to compare the amounts of ribosome bound to the distinct groups of mRNAs between 3AT-treated and untreated yeasts. As shown in Table 1, row 1, columns D and E, 3AT treatment decreased the amount of ribosome bound to total poly(A) RNA to 81.69%, slightly higher than the decrease in the total poly(A) RNA abundance (row 2). Because the total ribosome abundance was not altered after 3AT treatment (Figure 1A), this suggests that, per mRNA amount, there are 1.58% more ribosomes loaded to the total RNA during 3AT stress (ΔUp; see Table 1, row 1, column G, and Figure 2B, blue bar; note that, in general, mRNAs were depleted of ribosomes upon stress due to decrease in poly(A) RNA transcription). This net increase in ribosome binding to total poly(A) RNA was not observed in gcn2Δ (Table 1, row 1, column K, and Figure 2B, red bar), indicating that the increase observed in WT is due to Gcn2 function. As described below, we propose that the net increase in ribosome loading observed in WT is due to active, Gcn2-mediated ribosome redistribution (i.e. translational control) between distinct mRNA subsets.
Figure 2.

Determination of ribosome density (δi) by polysome profiling and analysis of genes translationally controlled by Gcn2. (A), the ribosome number indicated was assigned to fractions 3–7, based on simulation of ribosome mass in each fraction (blue bars) in comparison to A254 profile. δi was computed for every mRNA using these values. (B) Graphs indicated ribosome density fold increase by 3AT plotted against fold increase in mRNA amount (transcription rate) by 3AT. This is shown for WT and gcn2Δ cells. Note a smaller variation in y axis in gcn2Δ, as Gcn2 is responsible for transcriptional induction. Both WT and gcn2Δ show a positive correlation between more mRNA translation and higher δi. (C) The rate of 3AT-induced ribosome loading against different groups of mRNAs. ΔUp values in WT (blue bars) and gcn2Δ (red bars) yeasts, as defined in Table 1, are shown for the groups of poly(A) RNAs designated to the left of the graph. Left column, the number of RNA species in each group. (D) Analysis of mRNAs with one or more introns. Table shows the P value for enrichment of these mRNAs in the indicated list of genes/mRNAs. Graph, the total abundance (blue) and ribosome load (red) of mRNAs of this type was computed and compared to the values for total RNA in the cell. (E), the UGA(C/G)GG-like motif. Four motifs obtained in our motif analysis of the group TA4 under the following conditions are presented to the left, K = 6, 0th, K = 7, 0th, K = 6, 5th and K = 7, 5th (see Supplementary Methods for details) from top. Numbers to the right of each motif are MEME e value for enrichment in TA4 and the frequency of mRNA with each motif found in TA4. Venn diagram indicates the number of genes with this motif or more strict UGACGG sequence (in parenthesis) inside or outside of the group TA4. For mRNAs with these motifs, see Supplementary Table S6.
Importantly, this method allowed us to directly compute the ‘ribosome density’, δi, of every mRNA by ΣNnfi,n/(CDS length in kb) as the measurement of translational efficiency. On average, δi is 4.53 ± 0.92 in WT and 3.94 ± 1.3 in gcn2Δ (n = 6657, with untreated yeasts). 3AT increases δi by 1.04 ± 0.09 fold in WT. There is no correlation between 3AT-induced change in δi and the number of His codons found in mRNAs (Supplementary Figure S2C). Thus, the former is not due to differential effect of histidine starvation on mRNAs with His codons through, for example, ribosome stalling at these codons (24). Instead, the data agrees with the notion that this parameter largely reflects the change in regulation of individual mRNA at the level of translation initiation, rather than elongation. Intriguingly, there is a significant correlation between fold-increase in δi (∼ translation efficiency) and fold-increase in transcript abundance, both in WT and gcn2Δ cells (Figure 2C, P = 8.2e–66 and 6.7e–13, respectively). This is in agreement with the idea that newly synthesized transcripts are translated more frequently than existing mRNAs (25).
Translational control of groups of mRNAs encoding related functions
In order to examine which functional groups of mRNA are preferentially translated after 3AT treatment dependent on Gcn2, we listed genes whose δi is increased 1.06-, 1.05- and 1.025-fold by 3AT in a manner dependent on Gcn2 as groups TA1, TA2 and TA3 (N = 1664, 1846 and 2303, respectively; see Supplementary Table S3 for gene lists and Supplementary File for details of ranking method). Important, rpc34, nse4 and tfa2 listed in Figure 1B are among top 10 displaying the highest fold increase in δi by 3AT. We also ran an alternative ranking method (ppm scaling) through lane-by-lane comparison of gradient distribution signals (Supplementary Figure S3) and made the additional list of Gcn2-dependent translationally controlled genes termed TA4 (N = 1779, see Supplementary Table S3 for gene list). As shown in Supplementary Table S4, analysis by PANTHER database (14) indicated that all these groups are enriched for genes involved in chromatin formation and RNA metabolism. In addition, the groups TA1–3 are enriched for genes involved in chromosome organization and RNA processing. Importantly, all the four groups are enriched against genes involved in making ribosomes, in agreement with general translational inhibition by gcn2 during histidine starvation, which would decrease the need for ribosome synthesis.
To verify these trends through translational control analysis, we computed ribosome loading differences within the defined subsets of mRNA in response to 3AT treatment. As shown in Table 1, 39% of the ribosomes are bound to 142 mRNA species encoding ribosomal proteins (row 5, column D) - less ribosome per mRNA (row 6) due to shorter coding regions. As shown in row 5, column G, ΔUp is negative for WT (Figure 2B), indicating that the ribosomes dissociate from these mRNAs despite the general trend of ribosome association. Thus, this group of mRNA is negatively controlled at the translational level, consistent with their negative enrichment in the groups TA1–4 (Supplementary Table S4).
In contrast, 418 mRNA species encoding chromatin components (e.g. RNA polymerase subunits and chromatin remodelers and modifiers) are translationally up-regulated dependent upon Gcn2 (Table 1, rows 7 and, ΔUp = 3.46% in WT and −0.38% in gcn2Δ; also see Figure 2B). The trend is even stronger with 262 selected mRNA species included in TA1 (ΔUp = 8.01% in WT; Table 1 rows 9 and 10, and Figure 2B). Likewise, 774 and 508 mRNA mRNAs involved in RNA metabolism and RNA processing also displayed Gcn2-dependent translational up-regulation (Table 1, rows 15 and 17, and Figure 2B). Although the group of 442 mRNA involved in chromosome organization did not display Gcn2-dependent translational control as a whole (minor ΔUp in WT and a higher value in gcn2Δ, row 11), a strong translational control (ΔUp) was observed for 264 selected such mRNAs included in group TA1 (row 13) (Figure 2B). In conclusion, the translational control analysis of the microarray experiments supports the idea that translational control operates against functionally related groups of mRNA. Furthermore, we conclude that the net increase in ribosome binding by total RNA (ΔUp = 1.58%) is due to active redistribution of mRNA-bound ribosomes by Gcn2, in favor of chromatin and RNA regulation and in disfavor of ribosome synthesis.
Other characteristics of mRNA enriched in the groups TA1∼4
We noted that the genes with high δi, including rpc34 and nse4 (Figure 1B), tended to have introns at a higher frequency than random distribution. Indeed, our statistical analyses showed that the groups TA1∼4 are enriched for mRNAs with introns (Figure 2D, left). As shown in Figure 2D, the abundance of intron-containing mRNA is not altered by 3AT treatment or the gcn2Δ mutation (right graph, WT, blue bars). However, ∼25% more ribosomes are loaded onto this class of mRNA in response to 3AT (WT, red bars), in a manner dependent on gcn2 (gcn2, red bars). These trends were consistently observed with mRNAs containing various numbers of introns (Supplementary Table S5). As mentioned above, the lists TA1–3 are enriched for genes involved in RNA processing, which include mRNA splicing functions. Furthermore, mRNA with more introns are expressed more strongly under 3AT-induced starvation in a manner dependent on Gcn2 (Supplementary Table S5, even numbered rows). It is likely that more active splicing process during the stress arrangement allows for a tighter coupling between mRNA processing, export and translation, in agreement with the idea that newly synthesized mRNAs tend to get translated better in S. pombe (25) (also see Figure 2C).
Next, we searched for the consensus motifs found in 5′-, 3′-UTR and the entire mRNAs of the translationally controlled groups (H. Kato and K. Asano, unpublished results). Here, we report an interesting motif enriched in the 5' UTR of the mRNAs of the list TA4 (Figure 2E, left; see Materials and Methods). This motif comprises four overlapping motifs, together represented as ‘UGA(C/G)GG-like’ motif. 98 genes contain this motif in their 5-’UTR of which 51 are found in the list TA4 (Figure 2E and Supplementary Figure S4A, and Table S6). Interestingly, this motif is similar to ‘TGACGT’, the binding site for Atf1/Pcr1 heterodimer (26,27). However, we propose that this motif works at the RNA level, because the complimentary motif, CCGTCA, is not enriched in the list TA4. Moreover, none of the degenerate UGA(C/G)GG-like motif contained UGACGU. Furthermore, of UGACGN where N is G, A, C or U, only UGACGG is enriched significantly in the list TA4 (Figure 2D). Conversely, G at the sixth position is disfavored in degenerate TGACGT motifs found in the leader region of agl1 as a transcriptional control signal (26). As shown in Supplementary Figure S4B, the UGA(C/G)GG-like motifs found in TA4 display distinct distribution within the mRNA leader regions (red line), compared to those found outside TA4 (blue line). The motifs in TA4 occur most frequently at two locations, 20–30% and 80–90%, toward the start codon, suggesting that they might regulate ribosomal access to either 5′-cap or the start codon through local mRNA structures (Supplementary Figure S4B). Below we verify that the UGA(C/G)GG-like motif, along with the 5′-terminal sequence of its mRNA, is responsible for translational control of hrd1 encoding ubiquitin protein ligase E3 subunit listed in all TA1∼4 (also see Supplementary Figure S5).
Translational control by uORFs depends on Gcn2
uORFs are considered as a major type of cis regulatory elements that are used to up-regulate translation of myriads of genes (9). Indeed, the lists TA1∼4 contained gcn5 (histone acetyl transferase) (19), hri2 (heme-binding eIF2α kinase) (18) and fil1 (GATA transcription factor) (12), which are known to possess uORFs in their leader regions and encode a protein involved in the starvation response (see Supplementary Figure S5 for microarray data). Translational control of fil1 has been recently characterized (12) although the role of individual uORFs in its regulation remains unclear (see Supplementary text and Figure S6 for role of uORFs in fil1 control).
To characterize the role of uORFs in translational control of hri2 and gcn5, we generated a dual luciferase plasmid vector expressing a Renilla luciferase (Rluc) gene from a fixed AUG context and firefly luciferase (Fluc) from DNA inserted at an MCS (see Materials and Methods). As a positive control, we first cloned S. cerevisiae GCN4 leader region with 4 uORFs (28) into the MCS of the dual reporter plasmid (Figure 3A, top). The transformant of WT yeast carrying the GCN4-Fluc Rluc reporter was grown in EMMC-LH and subjected to dual luciferase assay after 0, 30 min and 60 min of 3AT treatment. The Fluc/Rluc expression ratio was normalized to the time-0 value from the original dual luciferase vector in the same strain. As shown in Figure 3A, row 2, we verified 3AT-induced reporter expression from the GCN4-Fluc reporter: Before 3AT treatment, the presence of the 4 GCN4 uORFs repressed Fluc expression compared to the expression from the vector, which was then partially derepressed by 3-fold after 1 h of 3AT treatment.
Figure 3.

uORF-dependent regulation of hri2. (A) Transformants of S. pombe wild type (WT) or isogenic gcn2 Δ strain carrying dual luciferase plasmids with 5′ leader from indicated genes were grown in a minimal medium (EMMC-Leu-His) and assayed for dual luciferase after 3AT addition. Schematics to the left indicates the arrangement of uORFs (shaded boxes – orange, positive element; blue, negative element), drawn to scale, preceding the Fluc coding region (large empty box). The luciferase mRNA is transcribed from a weakened nmt1 promoter (gray box) with its transcription start site (arrow) located 72-bp upstream of the MCS. Graphs indicate normalized luciferase expression compared to untreated vector control with bars indicating standard error of the mean (SE). Blue, expression at time 0. Red and green, 10 mM 3AT treatment for 30 and 60 min, respectively. (B) Luciferase assay was done with indicated mutant constructs altering hri2 leader region in WT and gcn2Δ strains. Horizontal bar indicates the region left in the construct.
Likewise, as shown in Figure 3A, left, rows 3–4, the constructs bearing uORFs from S. pombe hri2 and gcn5 also displayed a significant 2–3-fold increase in reporter expression in response to 1 hr-long 3AT treatment. In all three cases, the 3AT-triggered induction was lost when the activity was measured in gcn2Δ cells, verifying Gcn2-dependent control (Figure 3A, right graph).
Translational control of hri2 involves a dipeptide uORF (MI) as positive element
To investigate contribution of uORFs in its translational control, we generated a series of 5′ deletion in the Hri2 leader region and their start codon mutants (Figure 3B, schematics to the left). Then, their expression was measured in WT and gcn2Δ cells (Figure 3B, graphs to the right). As shown by blue bars in Figure 3B, row 2, in both WT and gcn2Δ, the deletion of the first uORF (uORF1) increased reporter expression by 3-fold compared to intact hri2 reporter (Figure 3A, row 4) in the absence of the stress, but not to the vector level (Figure 3A, row 1; 100% in the scale). Thus, uORF1 partially inhibits hri2 translation. Further deletion of uORF2 appeared to increase reporter expression in WT and did so clearly in gcn2Δ (blue bars, row 4), suggesting that uORF2 is inhibitory as well. Finally, the deletion of uORFs1∼3 leaving uORF4 intact dropped reporter expression to ∼20% compared to the vector expression (blue bars, row 8). This last result indicates that uORF4 also works to inhibit hri2 translation.
In GCN4 translational control model (3), two uORFs with opposing roles must be present in the 5′ leader region of the controlled gene. The positive uORF element is located 5′ of the other, allowing re-initiation at a start codon located downstream. In the absence of stress, ribosomes re-initiate at the second negative uORF element, inhibiting translation from the main CDS. With stress, re-initiating ribosomes bypass the negative element, allowing translation of the main CDS. Our results suggest that uORF3 serves as the positive element, while uORF4 serves as the negative element, because two deletion constructs with uORFs 3 and 4 intact allows reporter expression inducible by 3AT (Figure 3B, rows 2 and 4). In contrast, if uORFs 1 or 2 and uORF4 are left intact, reporter expression is no longer 3AT-inducible (rows 1 and 3). Together, we propose that (i) uORFs 3 and 4 work analogously to GCN4 uORFs 1 and 4, respectively, allowing hri2 3AT-inducible. However, in hri2, uORF4 seems to be more frequently bypassed without a stress than GCN4 uORF4, allowing basal reporter expression higher in the d2 construct (Figure 3B, row 4). Additionally, (ii) uORFs 1 and 2 partially inhibit overall hri2 expression, making its 3AT-induced level somewhat modest (Figure 3A, row 4, red bar).
Our identification of uORF3 (AUG-AUC) encoding a dipeptide MI as positive element supports the idea that shorter uORFs tend to allow re-initiation at downstream start codons at a higher frequency (7). In addition, dipeptide-coding uORFs, MC (AUG-UGU) or MM (AUG-AUG), are suggested to be the positive and negative elements for fil1 control, respectively (see Supplementary text and Figure S6). In agreement with these assessments of positive elements, the codons AUC and UGU are listed among 26 sense codons displaying stronger re-initiation potentials than wild-type UGC codon, when placed at the last codon of GCN4 uORF1 (8). In contrast, the codon AUG introduced to the same position of GCN4 uORF1 is expected to inhibit re-initiation. Thus, we decided to test the effect of replacing the codon AUC of hri2 uORF3 with codons UGU (MC) or AUG (MM). We also changed it to a stop codon UGA (M-stop; AUG-UGA), since M-stop uORFs are frequently observed in S. pombe mRNA and yet allows downstream initiation of the main CDS at varying frequencies, in agreement with previous study replacing the last codon of GCN4 uORF1 with a stop codon (8). Interestingly, none of these changes allowed 3AT-inducible reporter expression (Figure 3B, WT, rows 5–7), indicating that the hri2 uORF3 sequence is optimized for its function under the specific context of the mRNA, similar to GCN4 uORF1 (8). However, the motif MM reduced expression from the d2 construct in both WT and gcn2Δ (Figure 3B, row 6), in agreement with the idea that the uORF with this motif is more refractory to downstream re-initiation and therefore used as a negative element in the fil1 uORF system. The motifs MC and M-stop allowed reporter expression equivalent to the original d2 construct (Figure 3B, WT, rows 5 and 7), as expected for a motif allowing downstream re-initiation. In gcn2Δ cells where Gcn2 signaling is completely absent, the expression from WT d2 construct is significantly higher than that from the MC d2 construct (Figure 3B, gcn2Δ, rows 4 and 5), suggesting that the inability of the latter to induce reporter expression in gcn2+ cells is due to a lower frequency of the MC-coding uORF3 to allow downstream re-initiation. Thus, the nucleotide composition of dipeptide-coding uORFs plays important roles in translational control by affecting their re-initiation potentials. Below, we provide evidence that dipeptide-coding uORFs are enriched in the leader regions of translationally controlled genes and that the sequence composition of their second codon can influence their re-initiation potentials (see below Figure 5 and Supplementary Figure S8).
Figure 5.

Analysis of uORF codon composition suited for re-initiation. (A) 680 uORFs showing RPKM > 6 in the ribosome profiling data (12) were selected (see Supplementary Methods for detail), and relationship between the nucleotide compositions of their codons and their uORF/CDS ratio were analyzed and plotted. The box below indicates the definition of codon numbers relative to the stop codon of the uORFs analyzed. Significant P values were highlighted in red. Beyond pyrimidine-rich (more pyrimidine or MPyr) versus purine-rich (more purine or MPur), the following comparisons were made but not shown, because significant differences were not observed: only purine versus only pyrimidine; only CG versus only AT; of only AT, more A versus more T; of only CG, more G versus more C; more C or G versus more A or T. (B) Using the uORF/CDS dataset for 680 uORFs, comparisons were conducted at the position 1 defined in panel A (or the last codon prior to stop codon) by limiting the uORF size to encompassing two amino acid-long to n amino acid-long (where n is 3∼19). Shown are the comparisons yielding significant differences among those tested for the six categories designated in panel A. OCG, only C or G. OAT, only A or T.
Translational control of gcn5 by uORFs
To study uORFs regulating gcn5, we started by examining ribosome profiling data from 3AT-treated yeasts (12) (Figure 4A and Supplementary Figure S7). As shown in Figure 4B, columns 1–2, gcn5 uORF3 is strongly translated in WT before the 3AT treatment, and its level of translation, as measured by ribosome density in RPKM, is decreased following the stress (red bars). In contrast, 3AT increases the translation of the main gcn5 ORF (blue bars). This inverse relation between the uORF and main ORF translation was not observed in gcn2Δ (Figure 4A, right graph in purple). This Gcn2-dependent response is entirely as expected for the GCN4 translational control model, which is also observed with fil1 uORF4 (Supplementary Figure S6B). Thus, we propose that uORF3 works as the negative element for gcn5 regulation.
Figure 4.

Translational control of gcn5. (A) The graphs on top present read counts from the ribosome profiling data in the wild-type control experiments (12) plotted against the nucleotide positions within the 5′ half of gcn5 mRNA. Plots are color-coded by three reading frames presented on bottom. Schematics in the middle represent uORF structures and the main CDS also color-coded similarly. Trips-vis was used for the graphical presentation (54). Ribosome density at each ORF is presented italicized as RPKM. (B) Left graph, ribosome densities in RPKM for gcn5 CDS (blue) and uORF3 (red) are presented in RPKM for four experiments with WT and gcn2Δ strains (12). Right graph, the ribosome density ratio of uORF3 against gcn5 CDS. (C) The dual luciferase reporter with Gcn5::Fluc missing the start codon of uORF1 (row 2) and changing that of uORF2 (row 3) were generated and used for dual luciferase assay in the indicated strain grown in the presence of 3AT for 60 min. Schematics to the left show the uORF structures of wild type and mutant versions of the reporter constructs, as in Figure 3. (D) 680 uORFs displaying reasonable ribosome protection were chosen to study Gcn2- and uORF-dependent regulation (see SI Methods for details). 3AT-induced fold-change in uORF/CDS values in WT (ratioWT) is plotted against 3AT-induced fold-change in these values in gcn2Δ (ratio gcn2), Red lines indicate the threshold of ratio gcn2 = 1 and ratio WT = 0.5. Red dots, 28 uORFs suggested to display Gcn2-dependent induction of CDS translation (see Supplementary Table S7 for their list).
Since uORFs 1 and 2 did not display ribosome protection in the available dataset (Figure 4A), we performed the mutagenesis of the gcn5::Fluc Rluc reporter construct. As shown in Figure 4C, the elimination of uORF1 substantially increased reporter expression, making it unresponsive to 3AT (row 2), while that of uORF2 displayed 3AT-inducible reporter expression (row 3) comparable to the original reporter (row 1). These results suggest that uORF1 works mainly as the positive element, but uORF2, when uORF1 is leaky scanned, can also contribute to gcn5 control.
Genome-wide analysis of uORF-mediated translational control in S. pombe
Because the comparison of uORF/CDS translation ratio between WT and gcn2Δ is a good indicator of uORF-mediated translational control (Figure 4B and Supplementary Figure S6B), we selected 680 uORFs with reasonable translation signatures from the ribosome profiling data (out of 17 986 possible uORFs) and examined the uORF/CDS ratio under the four different conditions—WT or gcn2Δ and with or without 3AT. As shown in Figure 4D, the comparison of 3AT-induced changes in this ratio between WT and gcn2Δ (ratioWT and ratiogcn2, respectively) identified 28 uORFs potentially involved in translational control (red dots), as they displayed the fold change in uORF/CDS ratio of ≤0.5 in WT and ≥1 in gcn2Δ. The 28 uORFs included gcn5-uORF3 and fil1-uORF4 (MM) as expected, as well as fil1-uORF1 (MC) and uORFs found in three other mRNAs encoding putative transcription factors, Prt1, Prz1 and SPCC777.02 (Supplementary Table S7). These factors are listed in at least one of the TA groups (Supplementary Table S3). Five of the 28 listed uORFs, including that of prt1, are the sole uORF of the mRNA (Supplementary Table S7), suggestive of a mechanism distinct from the paired uORF system as found in gcn5 or fil1 (29).
On average, gcn2 represses 3AT-induced change in uORF/CDS ratio (Supplementary Figure S8A). Conversely, 3AT increases the fold change in uORF/CDS ratio caused by gcn2Δ (Supplementary Figure S8B). Thus, gcn2 appears to regulate 3AT-induced changes in uORF/CDS ratio genome-wide. To re-evaluate the type of uORFs involved in 3AT-induced CDS translation, we divided the uORFs into four groups based on change in uORF/CDS ratio by gcn2Δ in the presence of 3AT (Supplementary Figure S8C, panel 1). As shown in Supplementary Figure S8C, panels 3 and 4, the top group (Q1, n = 170) displayed the lowest ratioWT and the highest ratiogcn2, indicative of typical Gcn2-dependent 3AT-induced translational control similar to gcn5 or fil1. The dipeptide-encoding uORF is significantly enriched in this group, while the frequency of longer uORF is progressively lower (Supplementary Figure S8C, panel 5). Thus, the dipeptide-coding uORFs seems to be the most favored uORF element in this type of control. In contrast, the top group (Q1) displaying the largest change in uORF/CDS ratio by gcn2Δ in the absence of 3AT shows the highest ratio WT and the lowest ratiogcn2 (Supplementary Figure S8D, panels 1–4). Since 3AT increases uORF/CDS ratio for most uORFs in this group (panel 3), this group of uORFs enhances the repression of CDS translation in response to 3AT and independent of gcn2 (panel 4). Monopeptide-coding uORF or M-stop is significantly enriched in this group (Supplementary Figure S8D, panel 5), suggesting that this motif is at least in part responsible for 3AT-induced but Gcn2-independent depletion of the ribosome from mRNA (Table 1).
Lastly, we analyzed uORF/CDS ratio in WT without 3AT as proxy for re-initiation frequency. Our analysis of uORF base composition indicated that pyrimidine-rich codon is more favored at position 1 or 2 from the stop codon to promote re-initiation than purine-rich codon (Figure 5A). If we limit the analysis to uORFs encoding 2–5 to 2–7 amino acids, codons with only A or U are additionally favored at the position 1 than codons with only G or C (Figure 5B). These results agree with the previous work delineating the requirement for uORF1’s ability to re-initiate in the control of GCN4 translation (8).
Translational control by the 5′-UGA(C/G)GG-3′ motif
We chose hrd1 encoding ubiquitin ligase with a 53-nt long 5′-UTR containing the UGA(C/G)GG motif, but without any uORF. As shown in Figure 6A, row 2, the reporter translation from the WT hrd1 construct was slightly increased at 30 min after 3AT treatment and stayed until 1hr of the treatment. This is in contrast to other constructs with similar Kozak contexts (rpl702 and rsm23), showing a higher translation rate before the treatment but a strong inhibition after 3AT-induced amino acid starvation (rows 6 and 7). In gcn2Δ cells, hrd1 translation was derepressed before the stress and inhibited after it, similar to control constructs (rpl702 and rsm23) in WT (Figure 6B, row 2). These results indicate that hrd1 mRNA is normally repressed in a Gcn2-dependent manner, which is alleviated after starvation.
Figure 6.

Motif-dependent regulation. (A) S. pombe WT or (B) gcn2Δ transformants carrying dual luciferase plasmids with indicated 5′-UTR sequences before Fluc are grown and assayed as in Figure 3. hrd1 d6, hrd1 (motif) and hrd1 d22 are deletion derivatives of wild-type hrd1 shown on top, with TGA(C/A)GG-like motifs highlighted in red. In parenthesis a part of the BamHI site used for cloning is shown with its distance in bp from the adenine residue at the transcription start point (TSP). Italicized, sequence from the vector portion. (C) S. pombe WT, atf1Δ, pcr1Δ, int6Δ or gcn2Δ transformants carrying indicated dual luciferase plasmids with WT or mutant hrd1 leader sequences were grown in EMMC-LH and assayed for Fluc/Rluc activity. The normalized Fluc activities relative to WT control were presented. Data for WT and gcn2Δ were taken from panels A and B. (D) Predicted secondary structure of hrd1 5′ UTR with the location of UGACGG highlighted.
As expected, deletion of the 6-nt motif termed d6 disrupted Gcn2-dependent repression, allowing the reporter to express at a higher level than with the motif and decrease with starvation (Figure 6A, B, rows 3). However, the shorter segment with just the motif in hrd1(motif) did not restore Gcn2-dependent repression and stress-resistant translation (Figure 6A, B, row 4), indicating that the motif is necessary but not sufficient for hrd1 translational control. Deletion of the 5′-terminal 22 nt residues in d22 also disrupted Gcn2-dependent repression (Figure 6A, B, row 5), indicating that the regulation requires both the 6-nt motif and the 5′-terminal residues.
Next, we investigated other genes than gcn2 involved in hrd1 translational control. We tested int6 encoding eIF3e subunit (30), and atf1 and pcr1 encoding stress-responsive transcription factors (31,32). int6 was suggested to be involved in specific translational control of atf1 mRNA (19), and shown here by qPCR analysis of polysome fractionation samples that Int6/eIF3e promotes polysomal incorporation of atf1 mRNA in response to histidine starvation (Supplementary Figure S9). atf1 and pcr1 were chosen because Atf1/Pcr1 heterodimer not only binds ‘5′-TGACGT-3′’ motif at the DNA level (26,27), but also is suggested to bind the same motif found in RNA to stabilize mRNA (33). Here, we took advantage of the fact that the motif- or 5′-terminal deletion of hrd1 derepress hrd1 translation, in a manner dependent on gcn2 (Figure 6A-B and 6C, column 5), and tested the effect of these mutations on the hrd1-reporter expression in int6Δ, atf1Δ and pcr1Δ mutants. As shown in Figure 6C, the 2–3-fold increase in hrd1 expression caused by d6 and d22 in WT yeast was eliminated or compromised by any of these mutations. These results provide genetic evidence that hrd1 translational control involves Atf1, Pcr1 and eIF3e/Int6, besides Gcn2.
DISCUSSION
Quantitative overview of translational control during histidine starvation
Hereby, our translational profiling through polysome fractionation presented a quantitative overview of Gcn2-dependent translational control during histidine starvation in S. pombe. In response to the starvation, total mRNA molar abundance decreases by ∼20%, of which three quarters is due to decrease in ribosomal protein mRNA abundance (Table 1 and Supplementary Figure S2A). However, 1.6% more ribosomes (per mRNA molecule) are loaded to the total mRNAs after the stress. Given that a yeast cell contains 200 000 ribosomes (20) and that ∼80% of the ribosomes are bound to mRNA in our polysome fractions, the net increase of 1.6% bound to mRNA accounts for ∼2500 ribosomes. As shown in Table 1, this ribosome binding is the consequence of differential binding rates to specific subsets of mRNAs. We found that, during the stress, mRNAs encoding chromatin components and RNA regulation are preferentially translated, and yet, that those encoding ribosomal proteins are modestly depleted of ribosomes. Importantly, these functional groups are not the well-known GCN4 or Fil1 targets (12,34). Moreover, all these translational control changes, as well as ribosomal mRNA abundance changes, are due to the Gcn2 function (Table 1), and in agreement with the cellular needs of transcriptional regulation (general amino acid control response) and the slower rate of protein biosynthesis by ribosomes, as evidenced by the polysome profile changes (Figure 1A).
Our translational control analyses also indicated that mRNA with introns are better translated during the starvation (Figure 2D and Supplementary Table S5). Introns are known to prevent genotoxic DNA:RNA hybrids (also known as R-loops) generated during DNA transcription (35). In S. pombe, 710 mRNAs with three or more introns are expressed better (by 8%) during the stress arrangement (Supplementary Table S5), which would likely contribute to preventing R-loop formation in the time of insult. However, the effect of the stress on translation of intron-containing mRNA is even more dramatic: 2510 mRNAs with any intron are loaded with 25% more ribosomes after the stress. Again, Gcn2 regulates both the stress-induced translation and transcription of the genes with introns (Supplementary Table S5). Given the recent discovery that introns protect budding yeast from starvation (36), it would be intriguing to find out the physiological benefits of translation of intron-containing mRNAs during the histidine starvation response.
Translational control by uORFs in S. pombe
How are ∼2000 genes regulated coordinately at the level of translation? It is well established that, in S. cerevisiae and mammals, a combination of two uORFs in GCN4 and ATF4 mRNA, respectively, each playing the positive and negative role, mediates eIF2∼P-dependent translational induction of the main CDS by a delayed re-initiation mechanism (3,4). Similarly, we found that an analogous combination of uORFs regulates Gcn2-dependent translational control of hri2 (Figure 3) and gcn5 (Figure 4). Moreover, uORF-mediated control was reported for fil1 encoding a GATA transcription factor governing ∼10% of the S. pombe amino acid control response (12). Interestingly, the positive uORF elements in hri2 and fil1 leaders and the negative uORF element in fil1 are dipeptide-coding uORFs, AUG-AUC-UGA (MI), AUG-UGU-UAA (MC) and AUG-AUG-UAA (MM), respectively. Replacement of the MI-coding sequence with MC-coding sequence in hri2 mRNA leader disrupted 3AT-inducible reporter expression (Figure 3B), indicating that the dipeptide motifs are unexchangeable. It has been long known that an uORF carrying an AU-rich codon immediately preceding its stop codon allows re-initiation (8). In agreement with this, our genome-wide analysis of the ribosome profiling data (12) indicated that uORFs carrying AU-only or pyrimidine-rich codon before its stop codon tend to allow a high level of downstream CDS translation (Figure 5). In the case of hri2 and fil1, the positive uORF elements indeed carry a pyrimidine-rich codon (AUC and UGU) before the stop codon. However, the large deviation in uORF/CDS ratio observed with pyrimidine-rich or purine-rich codons (Figure 5) suggests that an RNA element outside of this codon, such as a sequence immediately following the stop codon (8), additionally governs the propensity of these uORFs to allow downstream re-initiation. Our bioinformatics analysis also identified ∼170 mRNAs whose uORF/CDS translation ratio is decreased by starvation in Gcn2-dependent manner (Figure 4D and Supplementary Figure S8C), similar to gcn5 uORF3 (Figure 4B) and fil1 uORF4 (Supplementary Figure S6B). The uORFs in these mRNA may work as a part of paired uORF system as found in S. cerevisiae GCN4, or work alone to normally inhibit CDS translation while the stress alleviates this inhibition by various previously defined mechanisms (9,29) (Supplementary Table S7). Together, these results indicate that uORF is a common regulatory element in eIF2∼P-induced translation in S. pombe.
As mentioned above, our translational control analysis also indicated that a subset of mRNAs such as those encoding ribosomal proteins is depleted of ribosomes upon 3AT-induced starvation stress (Table 1). We found that mRNAs with the shortest uORF, M-stop, tends to inhibit downstream CDS translation more strongly after the stress (Supplementary Figure S8D). In contrast to the regulation mediated by the dipeptide uORFs, this regulation is independent of Gcn2. Thus, we propose that M-stop at least in part provides the mechanism for negative regulation of mRNA translation in response to 3AT-induced starvation.
Conservation of uORF regulation in diverse fungi
We previously showed that gcn2Δ allows delayed and diminished eIF2α phosphorylation during histidine starvation in the presence of Hri1/Hri2 (19). The uORF-dependent translational control of Hri2, as shown in Figure 3, would contribute to enhancing eIF2α phosphorylation likely through oxidative stress associated with metabolic perturbation that activates Hri1/Hri2 (18). Thus, it appears that fission yeast is able to modulate eIF2α signaling through dissecting stress inputs into amino acid availability and oxidative stress intensity. Fission yeast is not alone among the members of the phylum Ascomycota that contain this additional eIF2α kinase, Hri (Figure 7A and Supplementary Figure S10A). Interestingly, Hri is present in Aspergillus species belonging to distantly related filamentous fungi (the subphylum Pezizomycotina) (37,38). As shown in Figure 7B, a similar arrangement of uORFs is found in Hri mRNAs from Aspergillus species. Moreover, A. nidulans HriA mRNA has uORF encoding the dipeptide MI (AUG AUU UGA) as possible positive element. Therefore, the coupling of the dual eIF2α kinases, Gcn2 and Hri, through uORF-dependent regulation of the latter appears to be a conserved strategy found across diverse fungi.
Figure 7.

Conservation and diversity of translational control in fungi. (A) Left diagram depicts a simplified fungal tree of life. Columns 1–3 indicate the presence of eIF2α kinases Gcn2 and Hri, and the translational regulator 5MP, a competitive inhibitor mimicking a part of translation factor eIF5 (55) (for phylogenetic trees of Hri and 5MP, see Supplementary Figure S10). Columns 4 and 5 list the Gcn4/CpcA/Cpc1 ortholog and whether its translational control is of the type found for S. cerevisiae GCN4. The schematics to the right depict the uORF arrangement, drawn to scale, of mRNA coding for the relevant Gcn4 homolog (56,57), where boxes indicate uORFs (colored yellow for positive, and blue for negative element) or the main CDS (shaded). The conserved proteins listed in columns 1–4 are as follows: from Aspergillus representing the class Eurotiomycetes, A. fumigatus CpcC (37) and A. nidulans AN2246 (38) (Gcn2 homologs), A. nidulans HriA (AN7321, XP_680590) (38), and A. fumigatus CpcA (58) and A. nidulans CpcA (56); from Neurospora representing the class Sordariomycetes, Cpc3 (59) and Cpc1 (57); from Saccharomyces representing the class Saccharomycetes, Gcn2 and Gcn4 (3); from Neolecta representing Neolectales, OLL26911 (Gcn2) and OLL24616 (CpcA); from Taphrinomycotina incertae sedis, Saitoella, XP_019023598 (Gcn2), XP_019024465 (Hri), XP_019027573 (5MP) and XP_019021532 (CpcA); and from Coprinopsis representing the subphylum Agaricomycotina class Agaricomycetes, XP_001828226 (Gcn2), XP_001830176 (5MP) and Cpc1 (57). (B-D) uORFs found in Hri (B), Gcn5 (C) and Fil1 (D) mRNAs in Schizosaccharomyces (Taphrinomycotina) and Aspergillus (Pezizomycotina) species. Their arrangement was depicted in the schematics as defined for Gcn4/CpcA mRNAs as in (A). Bars within the boxes indicate additional AUG start codons that initiate uORF in the same reading frame. For di- or tripeptide-coding uORFs, the coded peptides are listed by arrows. Accession numbers of the depicted mRNAs are; for Hri, XM_013160615 (Hri2), AN7321 in AspGD (HriA), XM_025542713, and XM_025617590; for Gcn5, XM_002173929; and for Fil1, NM_001022957, XM_013163848 and XM_013169711. In (D), the nucleotide sequences of di- or tripeptide coding uORFs found in fil1 mRNA are listed with start and stop codons underlined and pyrimidines in other codons boldfaced.
In contrast, uORF-dependent regulation for Gcn5 (Figure 4) is limited to Schizosaccharomyces species (Figure 7C), because, in Aspergillus, mRNAs of the gcn5 ortholog gcnE typically have a short 5′-UTR of ∼40–80 bases devoid of upstream AUG. In the case of uORF-dependent Fil1 regulation, similar uORF arrangement including the dipeptide MC or tripeptide motifs, containing pyrimidine-rich codons, is conserved across closely related Schizosaccharomyces species (Figure 7D). However, Fil1, a GATA zinc-finger transcription factor, is found only in the subphylum Taphrinomycotina to which Schizosaccharomyces belongs (39). We propose that some of the translational control modules found in S. pombe evolved in response to the opportunity brought about by the lack of the Gcn4/CpcA-dependent system governing the expression of the majority of the genome during nutrient stress signaling (see below).
Translational control by nucleotide motifs in S. pombe
Our study also revealed a unique nucleotide-dependent regulation involving Gcn2. The UGA(C/G)GG-like motif is potentially found in 98 genes (∼2%) of the fission yeast genome and is similar to but distinct from TGACGT motif defined as Atf1/Pcr1 transcription factor heterodimer binding site (Figure 6 and Supplementary Figure S4). Analogous to this finding, the TISU (Translation Initiator of Short 5′ UTR) element was proposed to work at both the transcriptional and translational levels (40). It is defined by 5′-SAASAUGGCGGC-3′ (where start codon is underlined and S is G or C), and shown to direct translation from start codons with very short 5′ UTRs (41,42). It also overlaps with binding sites for transcription factors (e.g. YY1) regulating ∼4.5% of the human genome. Moreover, at the DNA level, the 5′TOP element that mediates mTOR-induced mRNA translation (43) is long known to be a part of the transcription initiator signal as well (44,45). Is the evolution of these bifunctional nucleotide motifs coincidental? In eukaryotes, the DNA binding sites for transcription factors are often transcribed in 5′-UTR. So, they might be able to evolve these elements into a translational regulatory sequence at the RNA level. In the case of TISU, it appears that the mammalian ribosome evolved to react differently at this site, allowing translation from very short 5′UTR that is otherwise prevented (46).
Because Gcn2 is not fully activated prior to starvation stress, we hypothesize that the UGA(C/G)GG-like motif is bound by an RNA binding protein that is regulated by low level of Gcn2 phosphorylation in unstressed cells (47). Based on our genetic studies, this RNA-binding protein may be Atf1 and/or Pcr1 that otherwise binds DNA in the nucleus (Figure 6C). Similar to the UGACGU element in mRNAs that is suggested to be bound by Atf1 (33), the UGA(C/G)GG motif in hrd1 mRNA is found in a stem-loop structure (Figure 6D). Together with the precise sequence requirement for translational control of hrd1, the novel mode of regulation warrants further studies involving cutting-edge genomic, genetic and biochemical approaches.
CONCLUSIONS AND PERSPECTIVES
While our studies highlight a common mechanism of translational control across fungi and higher eukaryotes, it is apparent that S. pombe lacks the Gcn4/CpcA/Cpc1 homolog and its uORF-dependent regulation, which play a major role in nutritional stress response across diverse fungi (Figure 7). Clearly, its ancestor rewired regulatory networks to mitigate this loss. We believe this was possible because of its presumed ‘antiquity’ (21) disallowing this transcription factor to predominate the major stress response. In fact, only two other members of the related yeasts belonging to Taphrinomycotina are known to contain CpcA (39) (Figure 7A). In agreement with their close relationship to the fungal common ancestor, Saitoella complicata, a member of Taphiromycotina, contains not only Hri but also the ancient translational regulator 5MP (48) (Figure 7A), which is conserved in most eukaryotes but otherwise entirely absent in the phylum Ascomycota (49) (Supplementary Figure S10B). To rewire regulatory networks at the translational level, the ancestor of S. pombe must have taken advantage of all the available toolkits, such as uORFs, either Gcn4-type (this study) or attenuator-type leader peptide (29), and possibly RNA switches (Figure 6), most of which are found also in prokaryotes (50–52). We believe that the common ancestor of the animals (the phylum Metazoa) has taken similar steps to begin to evolve their appreciated complexity.
DATA AVAILABILITY
GEO accession number, GSE143299.
In-house scripts are available at https://github.com/hkatomed/Chikashigeetal.
Supplementary Material
ACKNOWLEDGEMENTS
We are strongly indebted to Ron Wek and Tom Baird, University of Indiana School of Medicine, for their advice and assistance during the initial phase of this study, Richard Todd, Kansas State University, for assistance in phylogenetic analyses of fungal proteins and proofreading, and Akira Yamashita, Genetics Institute, Japan, for timely gifts of yeast strains.
Contributor Information
Yuji Chikashige, Advanced ICT Research Institute, National Institute of Information and Communications Technology, Kobe, Hyogo 651-2492, Japan.
Hiroaki Kato, Department of Biochemistry, Shimane University School of Medicine, Izumo, Shimane 693-8501, Japan.
Mackenzie Thornton, Molecular Cellular and Developmental Biology Program, Division of Biology, Kansas State University, Manhattan, KS 66506, USA.
Whitney Pepper, Molecular Cellular and Developmental Biology Program, Division of Biology, Kansas State University, Manhattan, KS 66506, USA.
Madelyn Hilgers, Molecular Cellular and Developmental Biology Program, Division of Biology, Kansas State University, Manhattan, KS 66506, USA.
Ariana Cecil, Molecular Cellular and Developmental Biology Program, Division of Biology, Kansas State University, Manhattan, KS 66506, USA.
Izumi Asano, Molecular Cellular and Developmental Biology Program, Division of Biology, Kansas State University, Manhattan, KS 66506, USA.
Haana Yamada, Molecular Cellular and Developmental Biology Program, Division of Biology, Kansas State University, Manhattan, KS 66506, USA; Department of Advanced Transdisciplinary Sciences, Faculty of Advanced Life Science, Hokkaido University, Sapporo, Hokkaido 060-0810, Japan.
Chie Mori, Advanced ICT Research Institute, National Institute of Information and Communications Technology, Kobe, Hyogo 651-2492, Japan.
Cheyenne Brunkow, Molecular Cellular and Developmental Biology Program, Division of Biology, Kansas State University, Manhattan, KS 66506, USA.
Carter Moravek, Molecular Cellular and Developmental Biology Program, Division of Biology, Kansas State University, Manhattan, KS 66506, USA.
Takeshi Urano, Department of Biochemistry, Shimane University School of Medicine, Izumo, Shimane 693-8501, Japan.
Chingakham Ranjit Singh, Molecular Cellular and Developmental Biology Program, Division of Biology, Kansas State University, Manhattan, KS 66506, USA.
Katsura Asano, Molecular Cellular and Developmental Biology Program, Division of Biology, Kansas State University, Manhattan, KS 66506, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
JSPS KAKENHI [JP18K05556, JP19H05264 to H.K.]; K-INBRE program Pilot Grant [P20 GM103418], National Institutes of Health (to K. A.); Kansas State University (KSU) Terry Johnson Cancer Center (to K. A.); National Institutes of Health [GM125671 to K.A.]; KSU Arts and Science Research Scholarship (to W.P., M.H., A.C., C.B., C.M.); K-INBRE Semester Scholarship [P20 GM103418 to M.H., M.T.]; K-INBRE Star Trainee program [P20 GM103418]; Goldwater Scholarship (to M.T.). Funding for open access charge: NIH [GM125671].
Conflict of interest statement. None declared.
REFERENCES
- 1. Dever T.E. Gene-specific regulation by general translation factors. Cell. 2002; 108:545–556. [DOI] [PubMed] [Google Scholar]
- 2. Asano K. Dubitzky W., Wolkenhauser O., Cho K-.H., Yokota H.. Encyclopedia of Systems Biology. 2013; NY: Springer; 2278–2282. [Google Scholar]
- 3. Hinnebusch A.G., Dever T.E., Asano K.. Mathews M.B., Sonenberg N., Hershey J.W.B.. Translational Control in Biology and Medicine. 2007; NY: Cold Spring Harbor Lab Press; 225–268. [Google Scholar]
- 4. Vattem K.M., Wek R.C.. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ye J., Kumanova M., Hart L.S., Sloane K., Zhang H., De Panis D., Bobrovnikova-Marjon E., Alan Diehl J, Ron D., Koumenis C.. The GCN2-ATF4 pathway is critical for tumor cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010; 29:2082–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wek R.C., Staschke K.A.. How do tumors adapt to nutrient stress. EMBO J. 2010; 29:1946–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Asano K. Why is start codon selection so precise in eukaryotes. Translation. 2014; 2:e28387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grant C.M., Hinnebusch A.G.. Effect of sequence context at stop codons on efficiency of reinitiation in GCN4 translational control. Mol. Cell. Biol. 1994; 14:606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Young S.K., Wek R.C.. Upstream open reading frames differentially regulate gene-specific translation in the integrated stress response. J. Biol. Chem. 2016; 291:16927–16935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ron D., Harding H.P.. Mathews M.B., Sonenberg N., Hershey J.W.B.. Translational Control in Biology and Medicine. 2007; NY: Cold Spring Harbor Lab Press; 345–386. [Google Scholar]
- 11. Zhan K., Vattem K.M., Bauer B.N., Dever T.E., Chen J.-J., Wek R.C.. Phosphorylation of eukaryotic initiation factor 2 by heme-regulated inhibitor kinase-related protein kinases in Schizosaccharomyces pombe is important for resistance to environmental stress. Mol. Cell. Biol. 2002; 22:7134–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Duncan C.D.S., Rodríguez-López M., Ruis P., Bähler J., Mata J.. General amino acid control in fission yeast is regulated by a nonconserved transcription factor, with functions analogous to Gcn4/Atf4. Proc. Natl Acad. Sci. U.S.A. 2018; 115:E1829–E1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Teske B.F., Baird T.D., Wek R.C.. Methods for analyzing eIF2 kinases and translational control in the unfolded protein response. Methods Enzymol. 2011; 493:333–356. [DOI] [PubMed] [Google Scholar]
- 14. Thomas P.D., Campbell M.J., Kejariwa A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., Narechania A.. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003; 13:2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hogan D.J., Riordan D.P., Gerber A.P., Herschlag D., Brown P.O.. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008; 6:e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Riordan D.P., Herschlag D., Brown P.O.. Identification of RNA recognition elements in the Saccharomyces cerevisiae transcriptome. Nucleic Acids Res. 2011; 39:1501–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bailey T.L., Bodén M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nemoto N., Udagawa T., Ohira T., Jiang L., Hirota K., Wilkinson C.R.M., Bähler J., Jones N., Ohta K., Wek R.C. et al.. The roles of stress-activated Sty1 and Gcn2 kinases and protooncoprotein homologue Int6/eIF3e in responses to endogenous oxidative stress during histidine starvation. J. Mol. Biol. 2010; 404:183–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Udagawa T., Nemoto N., Wilkinson C., Narashimhan J., Watt S., Jiang L., Zook A., Jones N., Wek R.C., Bähler J. et al.. Int6/eIF3e promotes general translation and Atf1 abundance to modulate Sty1 MAP kinase-dependent stree response in fission yeast. J. Biol. Chem. 2008; 283:22063–22075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Warner J.R. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 1999; 24:437–440. [DOI] [PubMed] [Google Scholar]
- 21. Hoffman C.S., Wood V., Fantes P.A.. An ancient yeast for young geneticists: a primer on the schizosaccharomyces pombe model system. Genetics. 2015; 201:403–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chikashige Y., Tsutsumi C., Yamane M., Okamasa K., Haraguchi T., Hiraoka Y.. Meiotic proteins Bqt1 and Bqt2 tether telomeres to form the bouquet arrangement of chromosomes. Cell. 2006; 125:59–69. [DOI] [PubMed] [Google Scholar]
- 23. Martín R., Berlanga J.J., de Haro C.. New roles of the fission yeast eIF2α kinases Hri1 and Gcn2 in response to nutritional stress. J. Cell Sci. 2013; 126:3010–3020. [DOI] [PubMed] [Google Scholar]
- 24. Guydosh N.R., Green R.. Dom34 rescues ribosomes in 3. untranslated regions. Cell. 2014; 156:950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lackner D.H., Beilharz T.H., Marguerat S., Mata Juan, Watt Stephen, Schubert F., Preiss T., Bähler J.. A network of multiple regulatory layers shapes gene expression in fission yeast. Mol. Cell. 2007; 26:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kato H., Kira S., Kawamukai M.. The transcription factors Atf1 and Pcr1 are essential for transcriptional induction of the extracellular maltase Agl1 in fission yeast. PLoS One. 2013; 8:e80572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Steiner W.W., Smith G.R.. Optimizing the nucleotide sequence of a meiotic recombination hotspot in Schizosaccharomyces pombe. Genetics. 2005; 169:1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mueller P.P., Hinnebusch A.G.. Multiple upstream AUG codons mediate translational control of GCN4. Cell. 1986; 45:201–207. [DOI] [PubMed] [Google Scholar]
- 29. Hood H.M., Neafsey D.E., Galagan J., Sachs M.S.. Evolutionary roles of upstream open reading frames in mediating gene regulation in fungi. Annu. Rev. Microbiol. 2009; 63:385–409. [DOI] [PubMed] [Google Scholar]
- 30. Akiyoshi Y., Clayton J., Phan L., Yamamoto M., Hinnebusch A.G., Watanabe Y., Asano K.. Fission yeast homolog of murine Int-6 protein, encoded by mouse mammary tumor virus integration site, is associated with the conserved core subunits of eukaryotic translation initiation factor 3. J. Biol. Chem. 2001; 276:10056–10062. [DOI] [PubMed] [Google Scholar]
- 31. Kanoh J., Watanabe Y., Ohsugi M., Iino Y., Yamamoto M.. Schizosaccharomyces pombe gad7+ encodes a phosphorpotein with a bZIP domain, which is required for proper G1 arreset and gene expression under nitrogen starvation. Genes Cells. 1996; 1:391–408. [DOI] [PubMed] [Google Scholar]
- 32. Watanabe Y., Yamamoto M.. Schizosaccharomyces pombe pcr1+ encodes a CREB/ATF protein involved in regulation of gene expression for sexual development. Mol. Cell. Biol. 1996; 16:704–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gao J., Wagnon J.L., Protacio R.M., Glazko G.V., Beggs M., Raj V., Davidson M.K., Wahls W.P.. A stress-activated, p38 mitogen-activated protein kinase-ATF/CREB pathway regulates posttranscriptional, sequence-dependent decay of target RNAs. Mol. Cell. Biol. 2013; 33:3026–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Natarajan K., Meyer M.R., Jackson B.M., Slade D., Roberts C., Hinnebusch A.G., Marton M.J.. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol. Cell. Biol. 2001; 21:4347–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bonnet A., Grosso A.R., Elkaoutari A., Coleno E., Presle A., Sridhara S.C., Janbon G., Géli V., de Almeida S.F., Palancade B.. Introns protect eukaryotic genomes from transcription-associated genetic instability. Mol. Cell. 2017; 67:608–621. [DOI] [PubMed] [Google Scholar]
- 36. Parenteau J., Maignon L., Berthoumieux M., Catala M., Gagnon V., Abou Elela S.. Introns are mediators of cell response to starvation. Nature. 2019; 565:612–617. [DOI] [PubMed] [Google Scholar]
- 37. Sasse C., Bignell E.M., Hasenberg M., Haynes K., Gunzer M., Braus G.H., Krappmann S.. Basal expression of the Aspergillus fumigatus transcriptional activator CpcA is sufficient to support pulmonary aspergillosis. Fungal Genet. Biol. 2008; 45:693–704. [DOI] [PubMed] [Google Scholar]
- 38. De Souza C.P., Hashmi S.B., Osmani A.H., Andrews P., Ringelberg C.S., Dunlap J.C., Osmani S.A.. Functional analysis of the Aspergillus nidulans kinome. PLoS One. 2013; 8:e58008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Todd R.B., Zhou M., Ohm R.A., Leeggangers H.A., Visser L., de Vries R.P.. Prevalence of transcription factors in ascomycete and basidiomycete fungi. BMC Genomics. 2014; 15:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Elfakess R., Sinvani H., Haimov O., Svitkin Y., Sonenberg N., Dikstein R.. Unique translation initiation of mRNAs-containing TISU element. Nucleic Acids Res. 2011; 39:7598–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sinvani H., Haimov O., Svitkin Y., Sonenberg N., Tamarkin-Ben-Harush A., Viollet B., Dikstein R.. Translational tolerance of mitochondrial genes to metabolic energy stress involves TISU and eIF1-eIF4GI cooperation in start codon selection. Cell Metab. 2015; 21:479–492. [DOI] [PubMed] [Google Scholar]
- 42. Haimov O., Sehrawat U., Ben-Harush A.T., Bahat A., Uzonyi A., Will A., Hiraishi H., Asano K., Dikstein R.. Dynamic and competitive interaction of eIF4G1 with eIF4E and eIF1 underlie scanning dependent and independent translation. Mol. Cell. Biol. 2018; 38:e00139–00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Levy S., Avni D., Hariharan N., Perry R.P., Meyuhas O.. Oligopyrimidine tract at the 5′ end of mammalian ribosomal protein mRNAs is required for their translational control. Proc. Natl Acad. Sci. U.S.A. 1991; 88:3319–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hariharan N., Perry R.P.. Functional dissection of a mouse ribosomal protein promoter: significance of the polypyrimidine initiator and an element in the TATA-box region. Proc. Natl Acad. Sci. U.S.A. 1990; 87:1526–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shibui-Nihei A., Ohmori Y., Yoshida K., Imai J., Oosuga I., Iidaka M., Suzuki Y., Mizushima-Sugano J., Yoshitomo-Nakagawa K., Sugano S.. Functional dissection of a mouse ribosomal protein promoter: significance of the polypyrimidine initiator and an element in the TATA-box region. Gene. 2003; 87:1526–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haimov O., Sinvani H., Martin F., Ulitsky I., Emmanuel R., Tamarkin-Ben-Harush A., Vardy A., Dikstein R.. Efficient and accurate translation initiation directed by TISU involves RPS3 and RPS10e binding and differential eukaryotic initiation factor 1A regulation. Mol. Cell. Biol. 2017; 37:e00150-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Singh C.R., Udagawa T., Lee B., Wassink S., He H., Yamamoto Y., Anderson J.T., Pavitt G.D., Asano K.. Change in nutritional status modulates the abundance of critical pre-initiation intermediate complexes during translation initiation in vivo. J. Mol. Biol. 2007; 370:315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Riley R., Haridas S., Wolfe K.H., Lopes M.R., Hittinger C.T., Göker M., Salamov A.A., Wisecaver J.H., Long T.M., Calvey C.H. et al.. Comparative genomics of biotechnologically important yeasts. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:9882–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hiraishi H., Oatman J., Haller S., Blunk L., McGivern B., Morris J., Papadopoulos E., Guttierrez W., Gordon M., Bokhari W. et al.. Essential role of eIF5-mimic protein in animal development is linked to control of ATF4 expression. Nucleic Acids Res. 2014; 42:10321–10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Asano K., Mizobuchi K.. An RNA Pseudoknot as the molecular switch for translation of the repZ gene encoding the replication initiator of IncIa plasmid ColIb-P9. J. Biol. Chem. 1998; 273:11815–11825. [DOI] [PubMed] [Google Scholar]
- 51. Yanofsky C. Transcription attenuation: once viewed as a novel regulatory strategy. J. Bacteriol. 2000; 182:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Breaker R.R. Riboswitches and the RNA world. Cold Spring Harb. Perspect. Biol. 2012; 4:a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee B., Udagawa T., Singh C.S., Asano K.. Yeast phenotypic assays on translational control. Methods Enzymol. 2007; 429:139–161. [DOI] [PubMed] [Google Scholar]
- 54. Kiniry S.J., O’Connor P.B.F., Michel A.M., Baranov P.V.. Trips-Viz: a transcriptome browser for exploring Ribo-Seq data. Nucleic Acids Res. 2019; 47:D847–D852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singh C.R., Watanabe R., Zhou D., Jennings M.D., Fukao A., Lee B.-J., Ikeda Y., Chiorini J.A., Fujiwara T., Pavitt G.D. et al.. Mechanisms of translational regulation by a human eIF5-mimic protein. Nucleic Acids Res. 2011; 39:8314–8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hoffmann B., Valerius O., Andermann M., Braus G.H.. Transcriptional autoregulation and inhibition of mRNA translation of amino acid regulator gene cpcA of filamentous fungus Aspergillus nidulans. Mol. Biol. Cell. 2001; 12:2846–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ivanov I.P., Wei J., Caster S.Z., Smith K.M., Michel A.M., Zhang Y., Firth A.E., Freitag M., Dunlap J.C., Bell-Pedersen D. et al.. Translation initiation from conserved Non-AUG codons provides additional layers of regulation and coding capacity. mBio. 2017; 8:e00844-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krappmann S., Bignell E.M., Reichard U., Rogers T., Haynes K., Braus G.H.. The Aspergillus fumigatus transcriptional activator CpcA contributes significantly to the virulence of this fungal pathogen. Mol. Microbiol. 2004; 52:785–799. [DOI] [PubMed] [Google Scholar]
- 59. Sattlegger E., Hinnebusch A.G., Barthelmess I.B.. cpc-3, the Neurospora crassa homologue of yeast GCN2, encodes a polypeptide with juxtaposed eIF2a kinase and histidyl-tRNA synthetase-related domains required for general amino acid control. J. Biol. Chem. 1998; 273:20404–20416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GEO accession number, GSE143299.
In-house scripts are available at https://github.com/hkatomed/Chikashigeetal.