


Abstract
Heterogeneous nuclear ribonuclear protein K (hnRNPK) is an abundant RNA-binding protein crucial for a wide variety of biological processes. While its binding preference for multi-cytosine-patch (C-patch) containing RNA is well documented, examination of binding to known cellular targets that contain C-patches reveals an unexpected breadth of binding affinities. Analysis of in-cell crosslinking data reinforces the notion that simple C-patch preference is not fully predictive of hnRNPK localization within transcripts. The individual RNA-binding domains of hnRNPK work together to interact with RNA tightly, with the KH3 domain being neither necessary nor sufficient for binding. Rather, the RG/RGG domain is implicated in providing essential contributions to RNA-binding, but not DNA-binding, affinity. hnRNPK is essential for X chromosome inactivation, where it interacts with Xist RNA specifically through the Xist B-repeat region. We use this interaction with an RNA motif derived from this B-repeat region to determine the RNA-structure dependence of C-patch recognition. While the location preferences of hnRNPK for C-patches are conformationally restricted within the hairpin, these structural constraints are relieved in the absence of RNA secondary structure. Together, these results illustrate how this multi-domain protein's ability to accommodate and yet discriminate between diverse cellular RNAs allows for its broad cellular functions.
INTRODUCTION
Heterogeneous nuclear ribonucleoprotein K (hnRNPK) is an abundant, single-stranded nucleic acid binding protein whose RNA binding activity regulates gene expression at many levels, including transcription, RNA splicing and stability, and translation (1–3). In addition to binding RNA, hnRNPK also interacts with many proteins, such as p53 and RNA helicase DDX3X, to affect a variety of signaling pathways including the DNA damage and oxidative stress responses (4,5), and its haploinsufficiency and homozygous lethality demonstrate its importance for survival (6). hnRNPK’s association with poly-cytosine DNA (7) can additionally affect gene expression through promoter recognition (8). Resulting from its ability to impact many aspects of gene expression, hnRNPK overexpression is linked to cancer progression and poor prognosis (9).
hnRNPK is a complex multi-domain protein with four putative RNA binding domains: three KH domains and an arginine glycine (RG/RGG) rich linker domain (Figure 1). As a multidomain protein with nucleic acid-binding activity paramount to its function, there has been interest in both defining which of hnRNPK’s four protein domains contribute to nucleic acid interactions as well as refining what nucleic acid features lead to a productive interaction. Protein truncation studies followed by various in vitro analyses with cytosine-rich RNA and DNA ligands have been reported in an effort to define the minimal nucleic acid binding motif of the protein (10–15). While available biochemical and structural data emphasize hnRNPK’s utilization of its KH3 domain to achieve cytosine specific recognition of both DNA and RNA (16,17), contributions to nucleic acid interaction from the rest of the protein, previously evaluated through qualitative pull-down or gel shift experiments with various protein constructs and cytosine-rich oligonucleotides, remain unresolved (10,13,15,18). Similarly, studies attempting to define the breadth of nucleic acids sequences recognized by hnRNPK in an unbiased manner through SELEX, yeast three-hybrid, and genome-wide pull-down techniques (18–23) commonly find enrichment of at least one simple core sequence of 4–6 cytosines in a row bound to hnRNPK (19,21,22). However, a quantitative understanding of longer RNA sequence preferences of full-length hnRNPK, particularly in the context of RNA structure, which is prevalent in cells (24), is not available.
Figure 1.
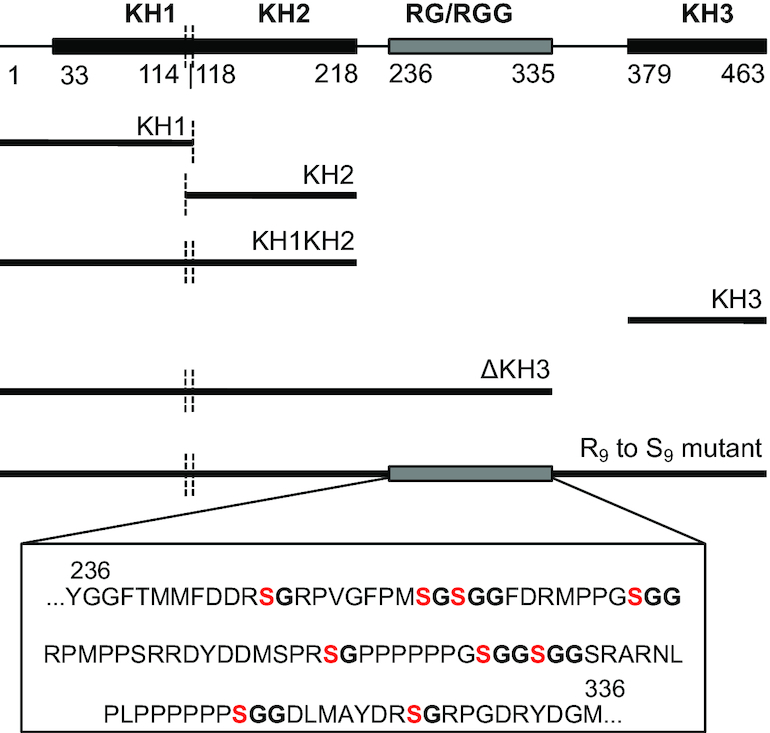
Domain map of full-length hnRNPK with various domain deletion mutants diagramed below. R to S mutations are specifically noted within the sequence of the RG/RGG domain.
Recent studies have revealed that hnRNPK also interacts with a number of long non-coding RNAs (lncRNAs). Some of these hnRNPK-lncRNA interactions have been shown to play roles in neural cell differentiation (25) and mediation of transcription factor response pathways (26), while others have been found to promote oncogenesis (10,27–29). hnRNPK’s interaction with Xist lncRNA is one of the most well-defined in this lncRNA class, where hnRNPK-Xist binding has been shown to be essential for X-chromosome dosage compensation in Eutherian mammals (30,31). Xist RNA is one of the first lncRNAs discovered (32), and, in normal development, its expression marks the onset of X chromosome inactivation (XCI). During early stages of female embryonic development, expression of Xist is randomly initiated from one of the two X chromosomes (33). Xist RNA subsequently coats its parent chromosome in cis (34) and results in chromosomal compaction, transcriptional silencing (33), and the deposition of heterochromatic epigenetic marks (35) to maintain the silenced state of the same chromosome across further cell divisions. Xist is a large RNA, 17.9 kb in the mouse and 19.3 kb in humans, and is both spliced and poly-adenylated (36). Xist displays an overall low sequence conservation across species with the exception of conserved repeat domains A-F (32,36). Mechanistically, Xist exerts its activity through many direct protein partners, one of which is hnRNPK (30,37). hnRNPK links the activity of polycomb complexes 1 and 2 (PRC1 and PRC2) to XCI and is essential for the deposition of inactive X-associated H2AK119ub and H3K27me3 marks, allowing for inherited epigenetic silencing across cell generations (31). Furthermore, deletion analysis in murine Xist reveal that within the tens of kilobases that make up the transcript, hnRNPK interacts predominantly with the cytosine-rich B repeat region (31,38).
Here, we sought to quantitatively define the protein and RNA features that drive hnRNPK-RNA recognition to biologically relevant targets. A suite of biologically relevant RNAs were studied, and hnRNPK sequence preferences refined through analysis of publicly available cellular hnRNPK–RNA interactions obtained from the ENCODE project. We find that hnRNPK’s RG/RGG domain is responsible for much of the protein's affinity for RNA, while the C-terminal KH3 domain, which had previously been ascribed specificity for C-patches, is largely dispensable. The impact of presenting C-patches at specific locations within a structured RNA was investigated using Xist-derived RNAs as a platform, and these experiments define the RNA features that allow hnRNPK to bind to structured RNAs. We found that hnRNPK retains its preference for multi cytosine-patch-containing RNA in the context of structured RNA, with a more precise preference for the disposition and orientation of these patches within a structural context that is relieved upon loss of that secondary structure.
MATERIALS AND METHODS
Cloning, expression, and purification of wild type and mutant hnRNPK proteins
Mouse and human hnRNPK are 100% identical. Human/mouse hnRNPK cDNA was obtained from Addgene in a pcDNA3.1 vector. R9 to S9 mutant cDNA (R247S, R256S, R258S, R268S, R287S, R296S, R299S, R316S, R326S) was ordered as a gBlock from Integrated DNA Technologies (IDT). Primers containing SalI and XhoI restriction enzyme cut sites on the 5′-end and 3′-end respectively of the cDNA corresponding to the protein construct of interest (full-length, R9 to S9, KH1, KH2, KH3, KH1KH2 or ΔKH3) were used to PCR amplify cDNA inserts for ligation into a pET28b vector containing an N-terminal 10xHis-SUMO tag (39) using the Quick Ligation kit (NEB). All plasmids were verified by Sanger sequencing (Quintara Biosciences).
Recombinant proteins were expressed in Escherichia coli and purified for use in RNA binding assays. BL21(DE3) or Rosetta (DE3) expression cells (Novagen) were transformed with plasmids described above (NEB protocol). Briefly, frozen competent cells were thawed and added to 1 μl of plasmid. Cells are left on ice for 20 min, heat shocked at 37°C for 90 s, then placed back on ice for 5 min. 500–600 μl of LB media is added to the cells and they are left to recover at 37°C for at least 1 hour. Then cells are plated on LB plates with antibiotic. One liter cultures of 2xYT media were grown at 37°C for 3 h to an OD600 of 1.0. Cells were flash-cooled on ice for 10 min, then 1 mM isopropyl β-d-thiogalactopyranoside was added to induce protein expression overnight at 20°C. Cells were harvested by centrifugation (5000 RCF) and pellets were stored at −20°C until purification.
All proteins were subjected to a 3-column purification protocol. Cells pellets were thawed and resuspended in 100 ml lysis buffer (750 mM KCl, 750 mM NaCl, 1 M urea, 50 mM Tris (pH 8.3 at 25°C), 10% glycerol, 10 mM imidazole, 1% Triton X-100) with one cOmplete EDTA-free EASYpack protease inhibitor tablet (Roche). Cells were lysed via sonication on ice with a Misonix Sonicator 3000 for 4–6 min in pulses of 15 s followed by 35 s of rest. Lysate was cleared by centrifugation (15 000 RCF, 30 min, 4°C) and incubated with 10 ml of precleared Ni-NTA beads for 10 min at 4°C after which unbound proteins were cleared by gravity flow. Beads were washed twice; once with 100 ml of lysis buffer, and once with 100 ml of modified lysis buffer containing 20–30 mM imidazole. Protein was eluted with 50–100 ml of lysis buffer containing 300–350 mM imidazole. The elution was transferred to 6–8K MWCO dialysis tubing (Spectra/Por – Spectrum Labs) and dialyzed in 4 l of size exclusion column buffer (500 mM urea, 270 mM KCl, 30 mM NaCl, 50 mM Tris (pH 8.3 at 25°C), 5% glycerol, 1 mM DTT) for 2 h at 4°C. One mg of in-house His-tagged ULP1 SUMO-protease (39) was added to the partially dialyzed eluate, and His-SUMO tag cleavage and further dialysis was allowed to proceed overnight at 4°C. Dialyzed eluate was cleared of protein precipitate, when present, by centrifugation or filtration prior to flowing over the second Ni-NTA column (11 ml Ni-NTA beads precleared in size column buffer). Flow through containing cleaved hnRNPK was concentrated to 1–2 ml for injection onto either the HiLoad 16/600 Superdex 200 (constructs ≥35 kDa) or the HiLoad 16/600 Superdex 75 size exclusion columns (constructs <35 kDa) (GE Healthcare) using spin concentrators (Vivaspin Turbo). Size exclusion fractions containing purified protein were combined, concentrated to between 90–500 μM, aliquoted, flash frozen, and stored at −70°C. One liter of growth typically yielded 2 mg of purified protein. (ϵ in M−1cm−1 for protein constructs: Full-length = 41,830; R9 to S9 mutant = 41,830; KH1 = 1490; KH2 = 2980; KH3 = 4,470; KH1KH2 = 4470; ΔKH3 = 14 900) (40).
Oligonucleotide preparation
All oligonucleotide sequences used in binding assays are listed in Supplemental Table S1. RNAs were prepared by run-off in vitro transcription using T7 RNA polymerase and dsDNA templates according to published protocols (41). dsDNA templates were generated as follows. For transcripts larger than 43 nts, cDNAs of RNAs of interest with a 5′ T7 promoter flanked by EcoRI and BamHI restriction sites on the 5′- and 3′-ends, respectively, were ordered as gBlocks and cloned into pUC19. Plasmids were verified by Sanger sequencing (Quintara Biosciences) and standard PCR was performed to generate dsDNA template for run-off transcription. For transcripts less than or equal to 43 nts, sense and antisense DNA oligonucleotides corresponding to the cDNA of the RNA construct preceded by the T7 promoter were ordered (IDT) and annealed (2 μM, 5 min 95°C, slow cool to 25°C in 10 mM Tris (pH 7.5), 1 mM EDTA, 50 mM NaCl) to create dsDNA template for run-off transcription. RNAs generated by run-off transcription were purified by denaturing PAGE (1× TBE/8 M urea) (42). Purified RNA concentrations and quality were assessed by the A260 and the A260/A280 ratio. Concentrations were calculated using the extinction coefficient provided for each RNA sequence by the Scripps extinction coefficient calculator (adapted and utilized in: http://www.fechem.uzh.ch/MT/links/ext.html). Typically, a 200 μl transcription reaction yielded 2.5 nmol of RNA and had a A260/A280 ratio of 2.0.
Purified RNA was 3′-end-labeled with fluorescein-5-thiosemicarbazide (FTSC) to perform fluorescence anisotropy binding assays using a previously published protocol (43). At typical labeling efficiencies we obtain good signal to noise for the use of RNA in binding assays at concentrations between 1 and 3 nM. FTSC-labeled RNA was stored in dark amber tubes at −20°C. M5′ ssDNA was ordered synthesized with a conjugated fluorescein at the 5′-end for use in fluorescence anisotropy binding assays (IDT).
Purified RNA was 5′-end-labeled with 32P for use in protein activity assays and RNase structure probing assays. 50 pmol of RNA was dephosphorylated with calf intestinal phosphatase (NEB) for 1 h at 37°C. Phenol–chloroform extraction followed by ethanol precipitation was performed to purify the RNA. 5′-End-labeling with [γ-32P]ATP was carried out as described previously (44). RNA used in structure probing was subject to further denaturing PAGE purification (42). 32P-labeled RNA was stored at −20°C and used within 2 weeks.
Fluorescence anisotropy (FA) binding assays
To perform binding studies, RNA ligand concentrations were held constant and as low as possible while still maintaining good signal/noise (1–3 nM). Purified labeled RNA was snap cooled in 1× binding buffer (135 mM KCl, 15 mM NaCl, 50 mM Tris (pH 8.3 at 25°C), 10% glycerol) at 2× final concentration and the carrier molecule, tRNALeu, was added at 2× final concentration (3.6 μM). Protein dilutions were performed separately in 1× binding buffer at 2× final concentration. Protein and RNA were then mixed in a 1:1 volume ratio in a 20 μl reaction and allowed to come to equilibrium at room temperature in the dark for 40–60 min. Flat-bottom low-flange 384-well black polystyrene plates (Corning) were used. Perpendicular and parallel fluorescence intensities were measured using a ClarioStar Plus FP plate reader (BMG Labtech) and anisotropy values were calculated for each protein titration point where anisotropy = (I‖ – I⊥)/(I‖ + 2*I⊥) and correlates directly with fraction bound. Associated anisotropy was plotted as a function of the log of activity corrected protein concentration. To determine the KD the data were fit to the simplified binding isotherm, anisotropy = O + (S*P)/(KD + P) with KaleidaGraph where S and O are saturation and offset respectively, and P is the protein concentration. All binding reactions were performed in triplicate or more using different protein dilutions on separate days. Standard errors of the mean were calculated and reported. ΔG is calculated from the fitted KD, ΔG = RT*ln(KD). Statistical significance is determined for differences between averages of apparent dissociation constants using the two-tailed paired t-test. KD determination for the interaction of full-length hnRNPK to the B motif RNA by FA was verified by EMSA (Supplemental Figure S1).
Activity binding assays by EMSA
Activity binding assays were performed to ensure accurate comparison of binding constants between various protein preparations and protein mutants. Full-length hnRNPK and ΔKH3 were assayed for activity by performing EMSA binding assays with the B motif RNA (45). Binding was performed as described above except B motif RNA was held constant at a final concentration of 1–2 μM (1 nM of radiolabeled RNA was supplemented with unlabeled RNA). Samples were loaded onto a pre-electrophoresed 5% acrylamide 0.25× TBE native gel and were electrophoresed for 15 min at 200 V. Gels were dried on Whatman paper using a BioRad Model 583 Gel Dryer, and dried gels were exposed to a phosphor screen overnight (GE Healthcare). Screens were imaged on a Typhoon imager (GE Healthcare) and quantified with ImageQuant software (GE Healthcare). Molar ratio of protein to RNA was plotted against fraction bound and activity was calculated by multiplying the slope of the best-fit line in the linear range by the stoichiometry of binding observed in the gel (2:1 for full-length; 3:1 for ΔKH3). Corrections between 0.75 and 1.2 were used (Supplemental Figure S2). Other protein constructs exhibited KDs too weak to assess for activity; 100% activity coefficients were assumed and the lower-limit KD was reported.
RNA structure probing assays
RNase structure probing assays were performed in 10 μl reactions; RNase I cleaves non-specifically at single-stranded regions of RNA and was used at 0.0083 U/μl (ThermoScientific), and RNase T1 cleaves at single-stranded guanines and was used at 1 U/μl (ThermoScientific). Gel purified 32P-labeled RNA was supplemented with unlabeled RNA to a final concentration of 1 μM. RNA was snap cooled in 9 μl of 50 mM Tris (pH 8.3 at 25°C), 135 mM KCl, 15 mM NaCl. 1 μl of 10× RNase I or RNase T1 was added and allowed to react with folded RNA at room temperature for 20 s and 5 min, respectively. The reaction was stopped and quenched with 40 μl of 8 M urea. Alkaline hydrolysis reactions for each RNA were performed simultaneously to create a nucleotide-resolution ladder (46). Depending on the size of the RNA, 6% or 8% sequencing gels were pre-electrophoresed at 55 W for 1–4 h, sample was loaded while running at 10 W, and loaded gels were allowed to electrophorese at 55 W for 45–55 min. Gels were dried and exposed to a phosphor screen overnight. Screens were imaged on a Typhoon imager (GE Healthcare) at non-saturating intensity at 100 μm resolution.
Fluorescence-based melting assays were performed in 25 μl reactions to probe for the presence of RNA secondary structure in a high throughput manner. 2–3 μM RNA was snap cooled in 24 μl 1× melting buffer (100 mM NaCl, 10 mM PIPES pH 7.0 at 25°C). SYBR Green I dye (Sigma Aldrich) was added to a final concentration of 2× the manufacturer's specifications. Samples were transferred to Ultra-Fast PCR/R1 plates (Life Science Products) and fluorescence was monitored while samples were slowly heated from 25°C to 95°C at a rate of 1.2°C/min (Applied Biosystems StepOnePlus). Melting temperatures were determined by first derivative analysis of fluorescence; a single peak and the x-axis value at its maximum correspond to a homogenously folded RNA and its apparent melting temperature.
Determination of RNA motifs bound by hnRNPK in K562 and HepG2 cells
Human hnRNPK eCLIP data were acquired from the Yeo lab through the ENCODE Consortium (47,48) for K562 and HepG2 cell types (experiment ENCSR268ETU, file ENCFF918XJQ and experiment ENCSR828ZID, file ENCFF855CPQ, respectively). Downloaded data was compared to human genome assembly GRCh38 (hg38) using the bedtools getfasta function to obtain RNA sequences for motif analysis (49), and sequences shorter than 20 nucleotides were filtered out. Background Markov models (first order) were generated from input reads for each cell type (experiment ENCSR669DKA for K562 and experiment ENCSR354KAS for HepG2) using the fasta-get-markov tool in the MEME suite. K562 or HepG2 hnRNPK eCLIP peak-derived sequences and corresponding background models were used as input for MEME (multiple EM for motif elicitation) (50), and the motif length was specified to be between 15 and 35 nucleotides long. We ran MEME on the two sequence datasets from the different cell types separately and found the top 5 motifs enriched in each cell type. HOMER was also used to generate motifs for comparison to MEME (51). Specifically, we used HOMER’s findMotifsGenome.pl function with the K562 peak file (ENCFF918XJQ) and the hg38 genome assembly to build motifs 21 nucleotides in length (Supplemental Figure S3).
C-richness, C patch abundance and hnRNPK eCLIP signature sliding window analysis
A cytosine patch (C-patch) is defined as three cytosines or more in a row, and cytosine content or C-richness is defined as the percent representation of cytosines within a given window of sequence. For 26 transcripts, sliding window analysis was performed to calculate how C-patch abundance and C-richness varied across the entire length of the transcript. Specifically, windows of a defined nucleotide length shift over one nucleotide at a time from the 5′-end to the 3′-end until the end of the transcript is reached. Each window was associated with the position of its first nucleotide within the transcript. For each window, the number of C-patches and C-richness was determined, and both metrics were plotted relative to window position across transcript length. These metrics were then compared to the transcript's hnRNPK eCLIP reads signature from replicate 1 of the K562 sample. To overlay either C-richness or C-patch abundance with eCLIP signature to make patterns more easily comparable, z-scores for the eCLIP reads and the C-richness or C-patch abundance were calculated with the built-in z-score python script from SciPy and plotted together (52). eCLIP reads were converted to reads per window before calculating z-scores to match the other analyses.
Alignment of Xist from various mammals
Alignments were generated using Clustal Omega multiple sequence alignment (53). All options used were default. Accession numbers for the sequences aligned are as follows: vole (M. transcaspicus), AJ310127.1; mouse (M. musculus), NR_001463.3; mole (S. orarius), DQ845733.1; human (H. sapiens), NR_001564.2; cow (B. taurus), NC_037357.1:77161577–77198299.
RESULTS
hnRNPK interacts productively and differentially with a set of diverse biologically relevant ligands in vitro
hnRNPK participates in a broad range of biological processes, and this wide-spread participation is believed to be achieved through its direct interaction with a range of cellular RNAs that share the common characteristic of being cytosine rich (20,29,54). To determine if these interactions are indeed direct, as well as to characterize the range of affinities that define these interactions, we curated and created a set of hnRNPK-relevant RNAs as well as two unrelated control RNAs to test in vitro (43,55). These hnRNPK-relevant RNAs include elements from mouse Xist and other lncRNAs, several mRNAs (19,29,31), a nuclear enriched RNA motif (54), and two variations of a consensus motif generated in a yeast three-hybrid study that contain differential cytosine content (19) (Table 1). The RNAs are between 25 and 43 nucleotides in length and all contain varying representation of cytosines.
Table 1.
hnRNPK binds biologically relevant RNA in vitro across a range of affinities. Cytosines predicted to be single-stranded within RNA sequences used for in vitro binding assays are highlighted in red. Average apparent binding constants and associated standard errors to full-length hnRNPK are reported; experiments are performed in triplicate or more. (*) indicate in vitro tested RNA that do not associate with hnRNPK in vivo. Representative binding curves can be found in Supplemental Figure S5.

|
To understand how the intrinsic affinity of hnRNPK varies for its known targets, we expressed and purified hnRNPK to high homogeneity (Supplemental Figure S4) and used an equilibrium fluorescence anisotropy (FA) binding assay to measure binding affinities between recombinant full-length hnRNPK and in vitro transcribed RNAs from our curated set (Supplemental Figure S5). We find that the eight biologically relevant RNAs bind to hnRNPK with submicromolar affinities while the two unrelated RNAs fail to bind productively (Table 1). Within the eight biologically relevant RNAs, six are able to bind full-length hnRNPK tightly (KDs between 44 ± 3 nM and 120 ± 10 nM) while the RNA derived from the 3′ UTR of Cdk6 and the nuclear enriched Sirloin motif bound more weakly at 450 ± 60 nM and 690 ± 20 nM, respectively (Table 1). All of these RNAs are cytosine-rich and contain at least one tri-cytosine patch (‘CCC’), so we expected they would bind equivalently. The observed 15-fold range of affinities to biologically validated RNAs was unexpected, suggesting that there could be a more complex motif that supports high affinity hnRNPK binding.
hnRNPK’s KH3 domain is neither necessary nor sufficient for interaction with RNAs
To investigate the reason for the variability in RNA-binding activity exhibited by hnRNPK, we first looked to the KH3 domain of hnRNPK. The KH3 domain of hnRNPK is the most well studied and has been implicated as largely responsible for the nucleic acid binding activity observed by the full-length protein (11,13,15). Furthermore, structural characterization of this domain in complex with short cytosine-rich DNA illustrates how cytosines are accommodated base-specifically (16,17), providing experimental context that could explain the strong cytosine preferences displayed by the full-length protein.
First we asked if the KH3 domain alone is sufficient for nucleic acid binding, as KH3 binds short DNA and RNA oligonucleotides in the low micromolar range and is proposed to be specific for cytosine (17). We expressed and purified the KH3 domain alone and assessed how well it could bind to DNA and RNA ligands. Consistent with prior reports, we find that KH3 alone interacts with a short ssDNA (a 10mer, ATATTCCCTC, M5′) as expected (observed KD = 8.7 ± 1.3 μM, reported as 3.0 ± 1.5 μM (16), Supplemental Figure S6). To assess whether KH3 alone is sufficient for the interaction, we compared this to how tightly full-length hnRNPK interacts with M5′ ssDNA. Full-length hnRNPK recognizes M5′ ssDNA with low micromolar affinity (KD = 1.4 ± 0.2 μM, Supplemental Figure S6), indicating that KH3, while it binds weaker than full-length, is sufficient to interact with M5′ ssDNA in the same low micromolar affinity regime as the full-length protein. Likewise, we find that the KH3 domain alone binds several of the biologically derived RNA ligands quite weakly (Table 2, Supplemental Figure S7), with a conservative lower-limit estimate of the KD >10 μM. Thus, while the KH3 domain alone is sufficient for full-length hnRNPK’s interaction with a 10mer ssDNA, it is not sufficient for the full-length protein's interaction with the longer biologically derived RNAs.
Table 2.
KH3 generally contributes marginally to hnRNPK binding affinity. Table depicts average dissociation constants with standard errors for full-length hnRNPK and the KH3 and ΔKH3 constructs to several biologically derived RNAs. Fold changes are only listed between ΔKH3 and full-length hnRNPK. The P values from two-tailed t-tests determine binding constant averages that differ from each other as indicated with statistical significance (*P< 0.05; **P< 0.01). Representative binding curves can be found in Supplemental Figures S5, S7A and S9.
| RNA | Full-length hnRNPK KDApp (nM) | ΔKH3 KDApp (nM) | Fold change (ΔKH3 to full-length) | KH3KDApp (nM) |
|---|---|---|---|---|
| B motif RNA | 61 ± 6 | 140 ± 30 | **2.3 | >15000 |
| Sirloin motif | 690 ± 20 | 760 ± 40 | 1.1 | >15000 |
| Ucp2 | 44 ± 3 | 87 ± 9 | *2.0 | 11100 ± 600 |
| Rab7 | 110 ± 20 | 360 ± 80 | **3.2 | >15000 |
| CDK6 3′UTR | 450 ± 60 | 710 ± 50 | *1.6 | 12900 ± 700 |
| MYU lncRNA | 70 ± 7 | 120 ± 10 | *1.7 | >15000 |
| Env8 | 1000 ± 200 | 2300 ± 400 | **2.3 | >15000 |
Next, to determine the necessity of the KH3 domain in RNA binding, we performed FA binding assays with a truncated version of hnRNPK that lacks the KH3 domain (ΔKH3). The impact of the loss of this domain on RNA binding was modest. Of the seven RNAs tested, four show a statistically significant 2- to 3-fold loss in affinity relative to full-length hnRNPK (Table 2, Supplemental Figures S8 and S9). The weaker binding RNAs, such as the CDK6 3′UTR and Sirloin motif, were insensitive to the loss of the KH3 domain, showing modest (<2-fold) changes in affinity for ΔKH3 relative to full-length hnRNPK binding. Thus, while the KH3 domain may play a part in cytosine recognition, as supported by structural characterization (16,17), this domain is not necessary for interaction with biologically derived cytosine-rich RNAs and contributes only marginally to affinity. The fact that KH3 is neither necessary nor sufficient for interaction with RNA indicates that domains outside of KH3 may play larger roles in hnRNPK-RNA recognition than previously appreciated.
hnRNPK binds to RNAs at regions containing multiple cytosine patches in vivo
Given the lack of an obvious sequence motif within the curated set of biologically relevant hnRNPK binding RNAs, we next sought to determine which specific features in an RNA allow for tight association with hnRNPK. A short simple (A)GCCC(A) recognition motif has been identified for hnRNPK through recent CLIP and RNA Bind-n-Seq studies (22,38), consistent with earlier yeast three-hybrid screen studies that support recognition of C-rich consensus sequences (19). Furthermore, previous structural studies inform how a short C-rich sequence can interact with the KH3 domain alone (16,17). This interaction is, however, much weaker than what we observed with longer RNAs and the full-length protein. Moreover, even though the curated set of biologically relevant ligands all contained a tri-cytosine patch of ‘CCC’, the wide range of binding affinities achieved with the full-length protein suggests that there is more to hnRNPK–RNA recognition outside of this simple RNA sequence motif contacting the KH3 domain.
To better understand the RNA sequence features that allow for tight binding by the full-length protein in cells, we first analyzed available deep sequencing data of RNA bound by full-length hnRNPK to specifically define a longer (>6 nt) RNA motif. Publicly available enhanced crosslinking immunoprecipitation (eCLIP) data for hnRNPK in two different cell lines, HepG2 and K562, was analyzed using the MEME tool (50) to identify enriched motifs between 15 and 35 nucleotides in length. Since eCLIP data commonly have low signal to noise (56), we conservatively filtered the raw available data to only analyze eCLIP peak-derived RNA sequences strictly reproduced in two replicates for each cell type. Specifically, 2970 and 5661 unique RNA sequences for K562 and HepG2 cells, respectively, were reproduced in two replicates and thus were utilized in our motif analysis. MEME was used to generate five enriched motifs for each cell type. We found the motifs from both cell types contained multiple cytosine patches (as defined by three or more cytosines in a row) interspersed with one or two other nucleotides (Figure 2A, Supplemental Figures S10 and S11). Notably, these motifs are also substantially deficient in purines (Supplemental Figures S10 and S11). These findings are consistent with a recent report which found a very similar motif in a different cell line, MCF10A (57). That motif is 20 nucleotides in length, composed of mainly three-cytosine patches, and is also purine-depleted. Thus, longer motif analysis of deep-sequenced immunoprecipitated RNA performed by us and others reproducibly indicate that hnRNPK binds to RNA containing tandem cytosine patches in multiple cell types in vivo.
Figure 2.

hnRNPK binds transcripts at locations with patches of cytosines in cells. (A) The top RNA motif generated from RNA sequences bound to hnRNPK in HepG2 cells (above) and K562 cells (below). (B) eCLIP read coverage and cytosine patch sliding window analysis of human XIST from K562 cells; windows are 75 nt in length; eCLIP reads are from ENCODE experiment ENCFF894NKS; eCLIP read coverage and the number of cytosine patches found are plotted separately (top and middle); z-scores are calculated for each and overlaid on the same graph for easy visualization (bottom).
We then used the FIMO (Find Individual Motif Occurrences) tool (58) to ask how closely each of the biologically relevant RNA sequences we performed binding with matched the top motif found for the K562 cell line. We find that almost all of the biologically relevant RNAs match this motif well (P < 0.01) (Supplemental Table S2) while the unrelated control RNAs, as well as one of the two yeast three-hybrid RNAs, do not statistically significantly match the motif (P > 0.01). Notably, both the 3′ UTR of the Cdk6 element and the Sirloin motif element matched the eCLIP-derived motif with statistical significance, but bound weakly, while the yeast three-hybrid 1 patch motif did not match the motif but bound tightly, suggesting that similarity to eCLIP-derived motifs is largely, but not completely, predictive of high affinity hnRNPK-RNA interactions.
Cytosine patch abundance and percentage of cytosines correlate with but do not guarantee interaction with hnRNPK in cells
Given the discovery of robust binding motifs for hnRNPK in in-cell associated RNAs, we next asked whether the number of cytosine patches, and more broadly, if general cytosine percentage was predictive of the location where hnRNPK interaction occurs within a transcript. To investigate this, we analyzed 26 unique RNA transcripts chosen from the list of eCLIP-derived sequences used in our motif analysis (Supplemental Table S3). We performed a sliding window analysis down the entire length of each transcript measuring C-richness, C-patch abundance, and hnRNPK eCLIP read coverage. Visual comparison of these positional cytosine enrichment and eCLIP read coverage analyses for the 26 transcripts finds that wherever we see a strong hnRNPK eCLIP signal, there exists a high percentage of cytosines, and in particular, cytosine patches (Supplemental Figures S12 and S13). Notably, in some cases, like for human XIST, the eCLIP signal tracks exceptionally well with C-patch abundance, and patch abundance is strikingly predictive (Figure 2B). There are cytosine patch-containing regions of other transcripts, however, where an eCLIP signal is not found, which could be due to the limited accessibility of those regions upon folding or occlusion by the binding other proteins. Thus, while the presence of multiple cytosine patches appears to be necessary for interaction with hnRNPK in vivo, it is not always sufficient and does not guarantee an interaction with hnRNPK.
The combination of in vitro tested biologically relevant RNAs and findings from the eCLIP analysis support a consistent recognition motif comprised of multiple cytosine patches. However, hnRNPK appears to discriminate between elements containing this simple motif, as evidenced by the differential affinities observed in vitro, where biologically validated RNAs bind in a range over an order of magnitude (44–690 nM). Moreover, the simple presence of the consensus motif in an RNA in vivo is not strictly predictive nor is it exclusively descriptive of all ligands that interact tightly with hnRNPK. The observations that hnRNPK is able to tightly associate with ligands that fall outside of this description and that regions of RNAs that contain the motif do not always show an association in vivo suggest that hnRNPK recognition depends on more than primary sequence alone.
Recent advances in in vivo RNA secondary structure probing indicate that many RNAs are structured in cells (24), leading us to evaluate the structural context of the C-patches in the RNAs studied. The RNAs tested exhibit diverse predicted secondary structures (Supplemental Figure S14), suggesting that the presentation of their cytosine patches in the context of these diverse structures might explain the differences in affinities observed. To test the structural context of the C-patches explicitly, we developed an RNA platform that would allow us to systematically probe structure-dependent RNA sequence preferences of hnRNPK using the B motif RNA derived from mouse Xist. We chose this motif because, unlike many of the other biological motifs, it has a stable secondary structure while having single-stranded regions comprised primarily of six cytosine patches.
The B repeat region of Xist is represented by a model 41 nt RNA
Xist RNA contains several conserved repeat regions, designated A–F, and murine eCLIP data reveals that hnRNPK strongly and preferentially associates with the B-repeat region of Xist in vivo (38) (Figure 3A). This association is necessary for function, as deletion of either hnRNPK or the B repeat region results in a failure of Xist to elicit polycomb mark deposition across its parent chromosome leading to a subsequent defect in gene silencing (31). The B repeat region in mouse Xist spans nucleotides 2839–3024 (∼200 nucleotides long) and contains over 30 repeats of the cytosine-rich sequence of (A/G)CCCC. Alignment of murine Xist with other mammalian Xist sequences highlights the widespread conservation of these cytosine-rich repeats, with some variability in the number of these repeats in other species (Supplemental Figure S15).
Figure 3.
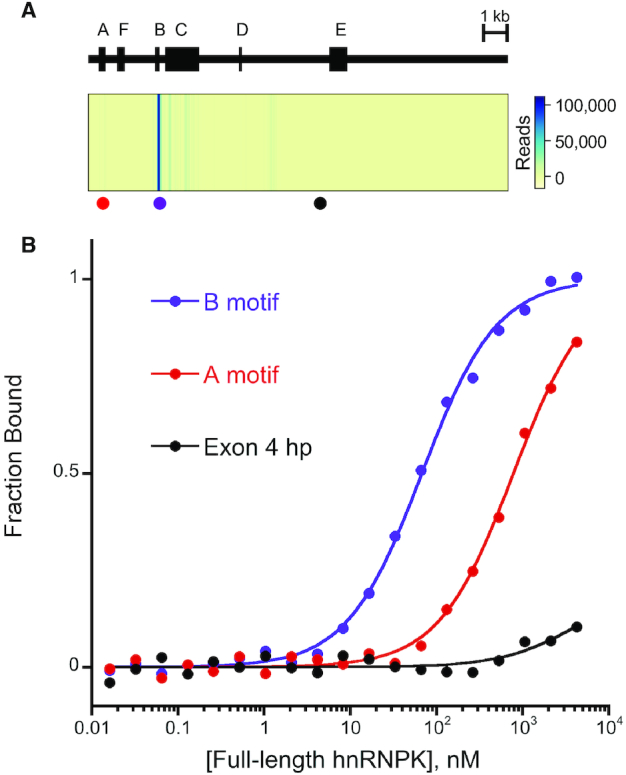
hnRNPK’s binding specificity for Xist's B repeat region in vivo is recapitulated in vitro. (A) Domain map of murine Xist (NR_001463) showing repeat regions A–F (above); heatmap of eCLIP reads (GEO code: GSM2325771) with locations of associated RNA sequences tested in vitro depicted by colored dots (below). (B) Representative normalized FA binding curves for RNA sequences from Xist to full-length hnRNPK.
Consistent with the propensity for many longer RNAs to display conformational heterogeneity in vivo (59,60), computational structure prediction of this B repeat region using mFold (61) yields multiple RNA conformations with similar energies (Supplemental Figure S16). These predicted structures, however, all contain similar features. Specifically, short double-stranded hairpin regions interspersed with regularly spaced internal loops composed mainly of cytosines are predicted widely throughout the B repeat region (Supplemental Figure S16). Moreover, similar predictions of the B repeat regions from cow, mole, and vole Xist also show hairpins with single-stranded cytosines (Supplemental Figure S17). To develop a model of this region, we screened several murine Xist-derived constructs for folding homogeneity and identified a 41 nt sequence within the B repeat region that is exclusively predicted to form a single secondary structure representative of the hairpin features predicted of the full-length B repeat region. This model RNA contains six patches of 3–4 consecutive single-stranded cytosines interspaced evenly between short duplex regions of Watson-Crick base pairs. Strikingly, the exclusivity of this structure prediction is experimentally supported by fluorescence-based melting temperature measurements that indicate the formation of a single RNA structure (Supplemental Figure S18). Additionally, treatment with RNase I provide compelling experimental evidence of the formation of this structure in solution, and results in a cleavage pattern that strongly reflects the single-stranded regions of the structure prediction (Supplemental Figure S18). We conclude that this B repeat region motif RNA (hereafter referred to as the ‘B motif RNA’) represents a homogeneously folded, tractable structural unit of the B repeat region of mouse Xist and pursued investigation of its interaction with hnRNPK.
Full-length hnRNPK binds preferentially to the B motif RNA in vitro
To determine if the specific in vivo interaction between hnRNPK and the B repeat is replicated in vitro, we used FA binding assays to measure binding affinities between full-length hnRNPK and in vitro transcribed RNAs derived from various regions of Xist. hnRNPK binds tightly to the B motif RNA with a KDApp of 61 ± 6 nM, binds weakly to an 85 nucleotide A repeat region motif RNA from Xist at 620 ± 90 nM, and exhibits no detectable binding to a 77 nucleotide conserved hairpin (62) from Xist's exon 4 (Figure 3A, B). All three RNA elements are predicted to form secondary structure by mFold (Supplemental Figures S14 and S16), however, neither the A repeat nor exon 4 are predicted to present a significant accessible patch of cytosines. Thus, full-length hnRNPK displays the same preference for the B repeat region of Xist in vitro as suggested by murine eCLIP data in vivo.
hnRNPK’s RG/RGG domain is necessary to achieve the high affinity interaction with the B motif RNA
Using the hnRNPK-B motif RNA interaction as a model for how hnRNPK binds structured RNAs, we continued experiments to narrow down a necessary and sufficient domain of hnRNPK for RNA binding. hnRNPK contains four putative RNA binding domains; two predicted KH domains reside on the N terminal half of the protein and an RG/RGG rich region precedes a structurally characterized third KH domain to make up the C terminal half (Figure 1). Since KH3 is neither sufficient nor necessary for binding RNA, we questioned if either of the less well-characterized KH1 or KH2 domains were solely responsible for hnRNPK’s interaction with its biological ligand. To test this, we expressed and purified KH1 and KH2 domains alone (Supplemental Figure S19) and performed FA binding assays with the B motif RNA. Similar to KH3, we find that neither KH1 nor KH2 alone are sufficient for interacting with the B motif (Table 3). Furthermore, while KH2 alone shows some evidence of binding at mid-micromolar protein concentrations similar to KH3, KH1 alone shows no detectable binding at all up to 50 μM protein (Supplemental Figure S7).
Table 3.
hnRNPK protein domain analysis with the B motif RNA. Average apparent binding constants and associated standard errors observed for the full-length protein and protein mutants with the B motif RNA are listed; n = number of replicates done for each experiment; relative KDs are to that of full-length hnRNPK. Representative binding curves can be found in Supplemental Figures S7B and S20.
| Protein | K D A pp, nM | n | K rel |
|---|---|---|---|
| Full-length | 61 ± 6 | 40 | 1.0 |
| KH1 | DNB | 3 | >160 |
| KH2 | >10 000 | 3 | >160 |
| KH3 | >10 000 | 6 | >160 |
| KH1KH2 | >10,000 | 5 | >160 |
| ΔKH3 | 140 ± 30 | 17 | 2.3 |
| R9 to S9 mutant | 430 ± 40 | 6 | 7.0 |
While KH1 and KH2 as individual domains could not productively interact with the B motif RNA, they are very close together in sequence spanned by a negligible linker (Figure 1). Since these two domains are the only predicted folded domains of the ΔKH3 mutant, which accommodates the B motif reasonably tightly, we posited they might form an extended binding module such that both domains together are required for RNA interaction. To test this, we created a subsequent truncation mutant from ΔKH3, mutant KH1KH2, that represents the N-terminal half of the protein (Figure 1). Strikingly, binding data for KH1KH2 resembled the binding data for KH2 and KH3; KH1KH2 was unable to bind the B motif RNA tightly and exhibits partial binding at mid-micromolar protein concentrations (Supplemental Figure S7). As a result, we could only provide a conservative estimated KD >10 μM (Table 3). This result, in combination with the tight binding achieved by the ΔKH3 variant, highlights the importance of the RG/RGG rich linker between KH2 and KH3 in hnRNPK’s ability to achieve a high affinity interaction with the B motif RNA.
To collectively assess the importance of the positively charged arginines within these small, unstructured RG/RGG motifs in the hnRNPK-B motif RNA interaction, we made an R9 to S9 protein mutant of full-length hnRNPK where nine arginines in an RG or RGG motif within amino acids 236–336 were simultaneously changed to serines (Figure 1). Rather than deleting all nine RG or RGG motifs, this R9 to S9 strategy preserves the total length of the linker between KH2 and KH3, thereby allowing us to maintain any synergy that might exist between the KH domains due to relative spacing. When we perform binding with this R9 to S9 mutant and the B motif RNA, we find the R9 to S9 mutant binds the B motif with a 7-fold weaker affinity than full-length wild type hnRNPK (430 ± 40 nM, Table 3, Supplemental Figure S20). These data suggest that the high affinity between hnRNPK and the B motif RNA is not exclusively due to arginines within the RG/RGG motifs, although it remains possible that the remaining 11 arginines within the linker could still be contributing to binding. Furthermore, replacement of these arginines to serines did not impact DNA binding, which remained weak at 3.0 ± 0.1 μM (Supplemental Figure S20). Together, our domain analyses suggest that hnRNPK’s RNA binding activity, unlike its DNA binding activity, requires more than a single region of hnRNPK. The domains of hnRNPK appear to be purposed to different binding functions, with the RG/RGG linker playing an essential role in hnRNPK’s ability to interact with long RNAs.
The C-patches within the B motif RNA are required for hnRNPK binding
To determine if the single-stranded cytosine patches within the B motif RNA are required for high affinity binding in the context of this scaffold, we created a cytosine-deplete variant of the B motif RNA predicted to take on the same singular predicted secondary structure, where all single stranded cytosines are replaced with uridines (C to U variant) and tested its binding to hnRNPK (Figure 4A). The predicted conservation of secondary structure for this ‘C to U’ variant RNA is supported by melting temperature and RNase I accessibility data (Supplemental Figure S21). We find that substituting these cytosine patches en masse to uridine completely abolishes hnRNPK’s ability to bind (Figure 4B). Thus, wild type (WT) B motif RNA and its C to U variant RNA provides a modular RNA platform to determine sequence and structural requirements for hnRNPK recognition.
Figure 4.
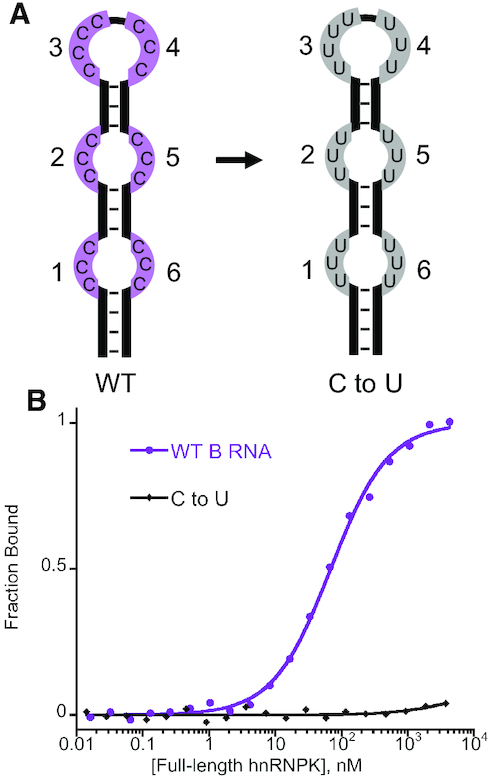
hnRNPK recognizes single-stranded cytosines in the B motif RNA. (A) Stylized cartoon depicting the six patches of single-stranded cytosines in the wild-type (WT) B motif RNA (purple) that are changed to uridines in the C to U RNA variant (gray); patches are numbered from the 5′- to the 3′-end of the RNA hairpin. (B) Representative normalized binding curves for the WT B motif RNA and the C to U RNA variant to full-length hnRNPK.
hnRNPK requires a minimum of three C-patches for high affinity binding within the B motif RNA
To test the role of individual C-patches in achieving high affinity binding with hnRNPK, we made a series of RNA constructs utilizing this B motif RNA scaffold where individual sets of C-patches were replaced with U-patches. As with the C to U variant, we found that replacing any one patch or more of cytosines with uridines in any combination results in RNAs that all, except for three variants, exclusively retain the same predicted RNA secondary structure as the WT B motif RNA. Three variants are predicted to form the same bulged hairpin structure as WT B motif RNA in addition one other less stable structure (Supplemental Figure S22). Fluorescence-based melting temperature assays on these, as well as on all other structured variants, supports the presence of a single hairpin structure, indicating that WT B motif RNA and all of its mutants consistently form the same secondary structure in solution (Supplemental Figure S22).
Using this validated structural scaffold, we next sought to determine whether high affinity hnRNPK binding requires all six C-patches or if a smaller number of C-patches would suffice. We replaced C-patches with U-patches one at a time from the 3′-to 5′-end and measured binding affinities with these RNAs and full-length hnRNPK. Binding performed with these RNAs results in a range of observed affinities ranging from WT affinity to no observable binding at all (Figure 5A, B, Supplemental Figures S23–S25). We find that RNAs containing five, four, and three C-patches all bind hnRNPK with WT affinity (variants (1,2,3,4,5), (1,2,3,4) and (1,2,3) respectively, Figure 5A, B, C). Binding decreases 5.9-fold when only two C-patches remain (variant (1,2)), and binding is completely lost if only one C-patch is present (variant (1)) (Figure 5A, B). Thus, in the context of the structured B motif, three C-patches are necessary and sufficient for a full binding interaction. hnRNPK is able to accommodate two C-patches to achieve a moderate interaction and is unable to bind just one C-patch.
Figure 5.
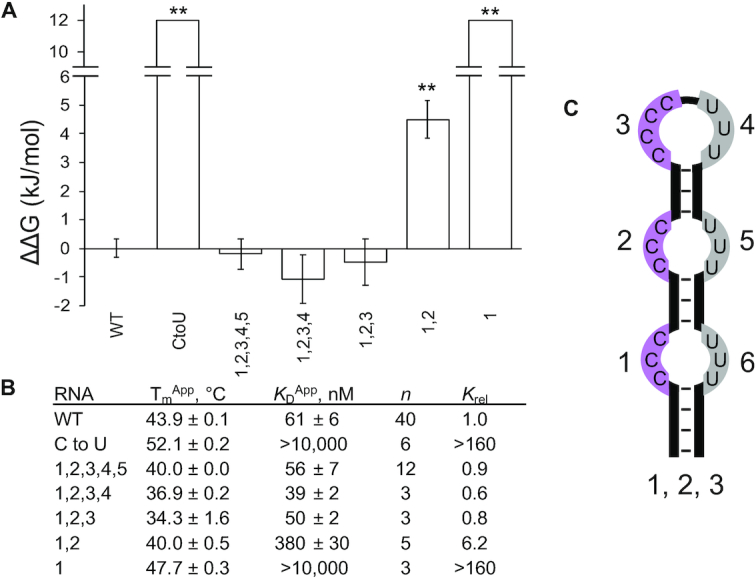
Cytosine-patch minimization of the B motif RNA and binding to full-length hnRNPK. (A) Average ΔΔGs of binding for B motif RNA variants to full-length hnRNPK with associated standard errors; WT is the wild-type B motif RNA, CtoU is the C to U variant, and the names of other variants describe which positions along the RNA hairpin contain patches of cytosines. (B) Apparent melting temperatures of RNA variants with associated standard errors (n = 3); apparent dissociation constants of RNA variants to full-length hnRNPK with associated standard errors, n is the number of binding replicates, relative KDs are to that of the wild-type B motif RNA. The P values from two-tailed t-tests determine binding constant averages that differ from that of WT as indicated with statistical significance (**P < 0.01). Representative binding curves can be found in Supplemental Figures S23–S25. (C) Stylized cartoon illustrating the cytosine patch numbering scheme using variant (1, 2, 3) as an example.
Since there was a such a dramatic loss of binding when transitioning from two C-patches to one C-patch, we asked how the relative positioning of two C-patches affected binding. First, we performed binding with variants (1,3) and (1,6) to ask if the two C-patches need to be adjacent in primary sequence or if they could be adjacent in structural space and find that both (1,3) and (1,6) fail to bind appreciably to hnRNPK (Figure 6A, B, Supplemental Figure S24). Thus, in the structured context, two C-patches need to be adjacent in primary sequence in order to achieve the optimal interaction with hnRNPK. Next, we asked if the position of these two adjacent C-patches within the hairpin influences hnRNPK recognition by moving these two adjacent C-patches sequentially from the 5′-end to the 3′-end of the hairpin and performed binding with these and the full-length protein. Variant (1,2) binds the tightest at 380 nM, variants (2,3) and (5,6) show a loss in affinity to 710 and 730 nM respectively, and variants (3,4) and (4,5) bind weakly in the micromolar range (Figure 6A, B; Supplemental Figure S24). These results demonstrate that hnRNPK recognition of C-patches depends on positioning within the RNA structure, and more specifically, suggests that hnRNPK strongly disfavors interacting with C-patches in the terminal loop found in B motif RNA. Moreover, the ability of variant (2,3) to bind with submicromolar affinity compared with the inability of variant (4,5) to bind well emphasizes the importance of the 5′-most C-patch residing in an internal loop rather than a terminal loop.
Figure 6.
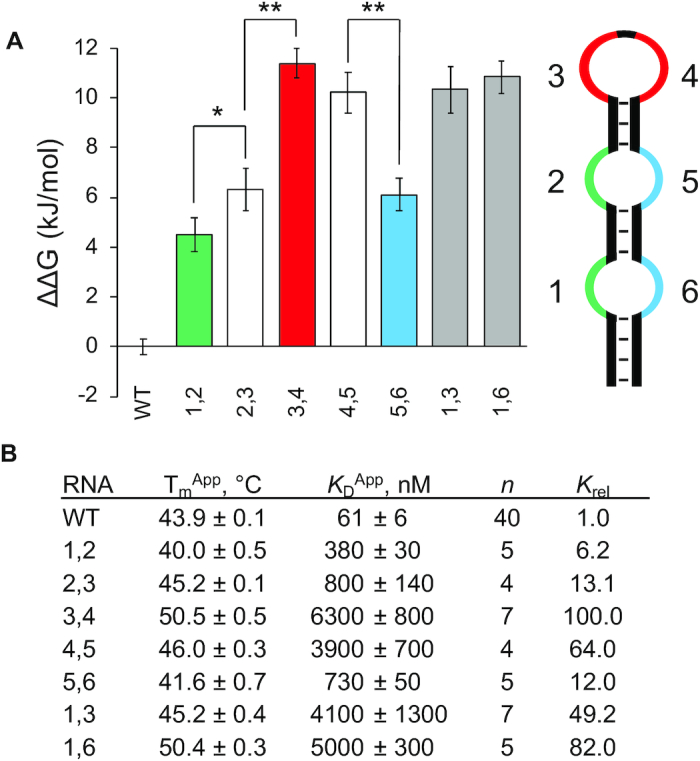
Two-cytosine-patch variants of the B motif RNA and binding to full-length hnRNPK. (A) Average ΔΔGs of binding for two-patch B motif RNA variants to full-length hnRNPK with associated standard errors; WT is the wild-type B motif RNA, names of other variants describe which positions along the RNA hairpin contain patches of cytosines (left); stylized cartoon of the B motif RNA structure (right). Colors correspond to adjacent internal loop cytosine patches (green and blue) and adjacent terminal loop cytosine patches (red). (B) Apparent melting temperatures of RNA variants with associated standard errors (n = 3); apparent dissociation constants of RNA variants to full-length hnRNPK with associated standard errors, n is the number of binding replicates, relative KDs are to that of the wild-type B motif RNA. The P values from two-tailed t-tests determine binding constant averages that differ from each other as indicated with statistical significance (*P < 0.05, **P < 0.01). Representative binding curves can be found in Supplemental Figure S24.
hnRNPK interacts with all three-C-patch variants of the B motif RNA productively, but achieves 10-fold differentiation in affinity through their relative placement
Since many in vivo hnRNPK-bound RNAs contain more than two patches of cytosines, we asked how the relative positioning of a higher number of C-patches within the RNA structure would influence hnRNPK recognition. Because the three-C-patch RNA from the minimization experiments was sufficient to achieve WT affinity, we created all 20 possible RNA variants that contained unique combinations of three C-patches and measured their binding affinities to hnRNPK (Supplemental Figures S25 and S26). We find that hnRNPK is able to bind this entire set of variants with submicromolar affinity, but that the variation in affinity suggested discrimination between ligands, with affinities ranging from 0.8 to 8.0-fold that of WT B motif RNA (Figure 7A, B).
Figure 7.
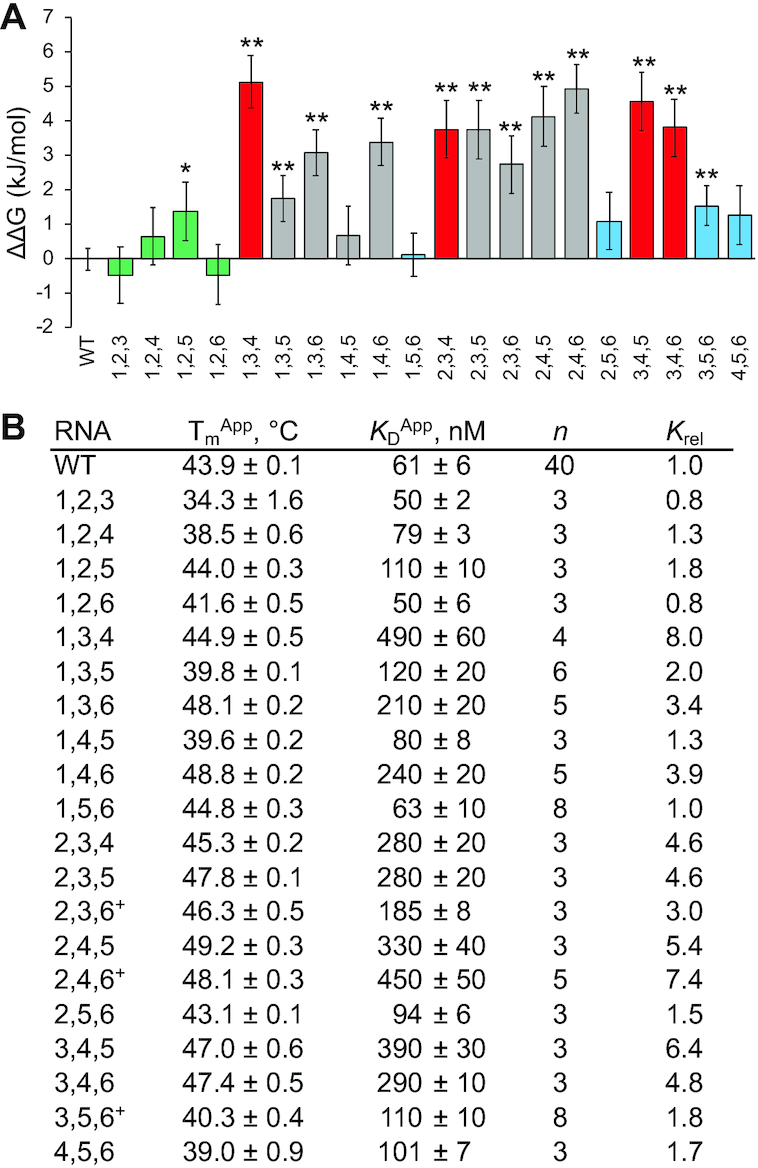
Three-cytosine-patch variants of the B motif RNA and binding to full-length hnRNPK. (A) Average ΔΔGs of binding for three-patch B motif RNA variants to full-length hnRNPK with associated standard errors; WT is the wild-type B motif RNA, names of other variants describe which positions along the RNA hairpin contain patches of cytosines, and colors correspond to the stylized cartoon in Figure 6 and indicate 1,2,X (green), X,3,4 or 3,4,X; (red) and X,5,6 (blue) variants. Gray indicates RNA variants that don’t fit into the above three categories. (B) Apparent melting temperatures of RNA variants with associated standard errors (n = 3); apparent dissociation constants of RNA variants to full-length hnRNPK with associated standard errors, n is the number of binding replicates, relative KDs are to that of the wild-type B motif RNA, (+) indicate variants that have one other predicted secondary structure in addition to the B motif hairpin (Supplemental Figure S22). The P values from two-tailed t-tests determine binding constant averages that differ from that of WT as indicated with statistical significance (*P < 0.05, **P < 0.01). Representative binding curves can be found in Supplemental Figures S25 and S26.
Implications of these results are two-fold. First, the data show hnRNPK is able to recognize the ligand with appreciable apparent affinity when presented with a high enough number of small binding sites within a set length of RNA. Second, because hnRNPK binds some three-C-patch ligands an order of magnitude better than others (KD’s range between 50 and 490 nM), hnRNPK is still able to discriminate between and prefers certain spatial arrangements of C-patches. Moreover, analysis of the differences in affinities between three-C-patch variants and the position of these C-patches yield insights that are consistent with findings from the two-C-patch experiment. Specifically, hnRNPK binds the tightest when there are at least two adjacent C-patches in a row, and when these two adjacent patches are not part of the terminal loop (C-patches at positions 3 and 4 in the hairpin). In fact, all ligands that contain C-patches at positions 1,2,X or X,5,6 are able to bind within 2-fold of WT B motif RNA while ligands that contain C-patches at positions X,3,4 or 3,4,X bind 4-fold or more weaker than WT B motif RNA (Figure 7A, B). These results show that high affinity binding between hnRNPK and the B motif RNA requires three C-patches at a minimum and is also highly dependent on the position of those patches relative to RNA structure.
Ablation of B-motif secondary structure rescues high-affinity binding by hnRNPK
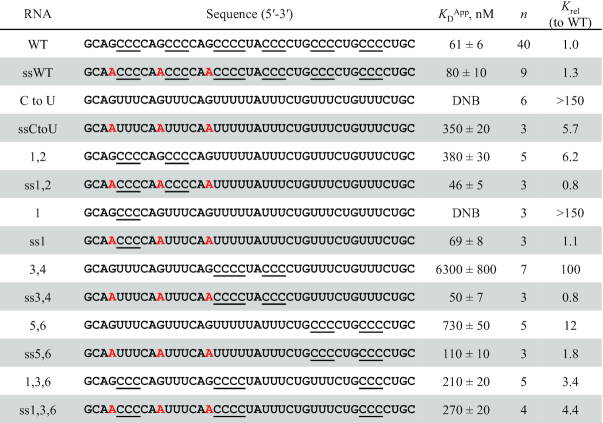
The above analysis suggests that hnRNPK binding is highly sensitive to the disposition of C-patches within structured RNAs. This observation raises the question of whether this selectivity is relaxed in unstructured RNAs. To better understand the role of the B motif RNA’s structure in binding affinity, we altered the sequence of the WT B motif RNA and six of the C-patch substituted RNA variants to disrupt the stability of the hairpin. In each single-stranded variant (denoted with ss), three guanines were mutated to adenines to eliminate GC pairing and destabilize the hairpin (Table 4). Secondary structure prediction on these ssRNAs using mFold did not return any stable folds, consistent with melting assays that confirmed that the folding of each ss variant is disrupted compared to its more stable counterpart (Supplemental Figures S27 and S28). We then performed FA binding assays with hnRNPK and compared the KDApp of each RNA to its ss variant (Table 4, Supplemental Figure S29). The ssWT B motif RNA maintains a similar affinity to hnRNPK relative to its structured counterpart, the WT B motif RNA. However, while the full C to U variant showed no observable binding in the hairpin structure, the ssCtoU variant binds only 5.7-fold weaker than the WT B motif RNA. This rescue effect can be seen with the C-patch RNAs as well. Constructs that bind with greater than 10-fold weaker affinity, like RNA variants (3,4) and (5,6), or that show no measurable binding like variant (1) maintain affinity within 2-fold of WT when forced to be single-stranded (Table 4, Supplemental Figure S29). Taken together, these results suggest that hnRNPK–RNA interactions that are sensitive to structural constraints can recover high affinity binding when these constraints are lifted. This effect is likely due to the flexibility of the ligand to adopt more binding competent conformations when the hairpin structure is no longer enforced. These results are consistent with the observation that a stringent binding motif did not emerge from the analysis of biological ligands. While hnRNPK is able to bind both single-stranded and structured RNAs with high affinity, it is more tolerant of the relative disposition of C-patches in single-stranded RNA and more discriminating of their location in constrained RNA structures.
Table 4.
hnRNPK tolerates C to U B motif RNA variants in the absence of RNA structure. Single-stranded cytosine patches are underlined and guanine to adenine mutations used to disrupt RNA folding are highlighted in red. Average apparent binding constants and associated standard errors to full-length hnRNPK are reported as well as binding constants relative to the WT B motif RNA (Krel); experiments are performed in triplicate or more. DNB = does not bind/no observable binding. Representative binding curves can be found in Supplemental Figure S29.
|
DISCUSSION
hnRNPK is one of several ubiquitous, high abundance RNA-binding proteins that are characterized by the presence of multiple tandem interaction domains and their ability to interact with a large range of targets (63). How these proteins use their multiple domains to engage with and discriminate between a range of biological targets has been addressed using in vitro biochemical preferences and, more recently, comprehensive analysis of their in vivo bound RNAs (21,22,57,64). While many protein binding preferences have been distilled down to simple motifs (22,64), how well these motifs recapitulate biological behavior is an outstanding question.
The binding preference of hnRNPK has been expressed as containing patches of clusters of cytosine nucleotides (15,19,22,57). We observe a similar pattern in available eCLIP data, with the consensus defined by patches of three to four Cs interspersed by 2–3 weakly predicted nucleotides (Figure 2A). While the set of known hnRNPK-interacting RNAs consistently contain C-patches, we report here that the binding affinity for these RNAs spans more than a 10-fold range of values and does not necessarily follow the trend anticipated from the consensus. For example, Rab7 contains only one stretch of consecutive cytosines but binds well, while the Sirloin motif and Cdk6 3′UTR contain several stretches of cytosines yet bind weakly (Table 1).
These data clearly show that the binding preferences of hnRNPK cannot be exclusively represented by a single consensus sequence. In addition to containing C-patches, the hnRNPK binding profile is notably depleted of purines, suggesting that the binding site may disfavor secondary structure. The structural context dependence of hnRNPK interaction is particularly evident when analyzing how hnRNPK binds the B repeat of Xist as part of its essential role in mediating XCI. Using a consensus ligand derived from this B repeat region, we found that hnRNPK requires at least two adjacent cytosine patches, and interacts tightest when these two adjacent cytosine patches reside within internal loops. Our data suggest that simple motif predictions for complex binding events may be overly simplistic and provide an inaccurate view of binding events that occur through alternate modes of accommodation. Rather, biochemical analysis of bound RNAs analyzed in light of the consensus motifs suggests a far more complex set of recognition rules that need to take into account, in the case of hnRNPK, the interplay of C-patch spacing and secondary structure formation.
The complexity inherent in hnRNPK RNA recognition could be due to a differential utilization of the individual RNA-binding domains contained within full-length hnRNPK. Prior analyses of hnRNPK suggested that individual KH domains could recapitulate high affinity and specificity of the full-length protein (13,15). In contrast, the data presented here reveals that multiple domains, at minimum two KH domains and the RG/RGG domain, are needed. Surprisingly, KH3, the focus of prior structural analysis (16,17), contributes modestly to binding of the biologically targeted RNAs investigated here. Dissection of the individual domains of hnRNPK that contribute to binding further highlights the RG/RGG domain's importance in mediating tight affinity for the B motif RNA.
This central role of hnRNPK’s RG/RGG domain is consistent with emerging themes regarding the involvement of intrinsically disordered RG/RGG motifs in RNA biology (65–67). These RG/RGG domains are highly represented in the RNA-binding proteome (67–69), and are frequently present in conjunction with other types of RNA-binding domains such as KH domains or RRMs within individual proteins (70). In some proteins such as FMRP, FUS, and hnRNPU, the roles of these domains have been explored and are essential for interaction with nucleic acids, although specificity imparted by these domains can be degenerate (65,66). Additionally, the crystal structure of an RGG peptide from FMRP with its in vitro selected SC1 RNA shows how important contacts made by arginines facilitate nucleic acid recognition by these short motifs (71). Previous studies investigating the role of hnRNPK’s RG/RGG region in nucleic acid binding assessed a subset of the RG or RGG repeats and concluded the region was unimportant (14,18,23), but our findings here indicate the opposite is true. In these RG/RGG-containing multidomain proteins, RG/RGG domains are thought to serve as affinity anchors while more specific binding properties are provided by neighboring domains, such as in the case of the fused in sarcoma/translocated in sarcoma (FUS/TLS) protein (72). In addition to contributing to high affinity RNA-binding, hnRNPK’s RG/RGG domain also interacts with other proteins (73,74). Thus hnRNPK, like many other multidomain RNA-binding proteins, utilizes its multiple RNA-binding domains to achieve broad specificity, likely enabling hnRNPK to fulfill its diverse biological roles by mediating both protein and RNA interactions (75).
Regarding the question of how predictive the consensus motif is of cellular localization, we found that hnRNPK binding affinities for individual regions of its binding partner Xist mirror its observed binding specificity in vivo (38). This correlation suggests that hnRNPK-RNA binding in cells, and therefore hnRNPK function, is dictated and driven by intrinsic binding preferences. This correlation between in vivo site specificity and in vitro affinities, however, is not universal for all RNA-binding proteins, suggesting that other factors, such as protein binders and accessibility, can modulate these intrinsic binding preferences. Thus, strategies that rely on using in vivo association may miss the mark. Finally, the observation that biologically associated RNAs bind with a range of affinity may serve to provide functional discrimination, as tight versus weak binding might serve a specific function for several of the tested ‘biologically relevant RNAs’ that are bound by hnRNPK.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank lncRNA members of the Wuttke and Batey labs for helpful discussions. We would also like to thank Leslie Glustrom and Alexandra Barbour for comments and edits on the manuscript as well as thank our reviewers for thoughtful points raised during the review of the manuscript.
Notes
Present address: Deborah Wuttke, Department of Biochemistry, University of Colorado, Boulder, CO 80309, USA.
Present address: Robert Batey, Department of Biochemistry, University of Colorado, Boulder, CO 80309, USA.
Contributor Information
Meagan Y Nakamoto, Department of Biochemistry, University of Colorado, Boulder, CO 80309-0596, USA.
Nickolaus C Lammer, Department of Biochemistry, University of Colorado, Boulder, CO 80309-0596, USA.
Robert T Batey, Department of Biochemistry, University of Colorado, Boulder, CO 80309-0596, USA.
Deborah S Wuttke, Department of Biochemistry, University of Colorado, Boulder, CO 80309-0596, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [T32GM065103 to M.Y.N. and N.C.L., R01GM120347 to R.T.B. and D.S.W.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Ostareck D.H., Ostareck-Lederer A., Wilm M., Thiele B.J., Mann M., Hentze M.W.. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-Lipoxygenase translation from the 3′ end. Cell. 1997; 89:597–606. [DOI] [PubMed] [Google Scholar]
- 2. Moumen A., Magill C., Dry K.L., Jackson S.P.. ATM-dependent phosphorylation of heterogeneous nuclear ribonucleoprotein K promotes p53 transcriptional activation in response to DNA damage. Cell Cycle. 2013; 12:698–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nazarov I.B., Bakhmet E.I., Tomilin A.N.. KH-domain poly(C)-binding proteins as versatile regulators of multiple biological processes. Biochem. Mosc. 2019; 84:205–219. [DOI] [PubMed] [Google Scholar]
- 4. Moumen A., Masterson P., O’Connor M.J., Jackson S.P.. hnRNP K: An HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell. 2005; 123:1065–1078. [DOI] [PubMed] [Google Scholar]
- 5. Good A.L., Haemmerle M.W., Oguh A.U., Doliba N.M., Stoffers D.A.. Metabolic stress activates an ERK/hnRNPK/DDX3X pathway in pancreatic β cells. Mol. Metab. 2019; 26:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gallardo M., Lee H.J., Zhang X., Bueso-Ramos C., Pageon L.R., McArthur M., Multani A., Nazha A., Manshouri T., Parker-Thornburg J. et al.. hnRNP K is a haploinsufficient tumor suppressor that regulates proliferation and differentiation programs in hematologic malignancies. Cancer Cell. 2015; 28:486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matunis M.J., Michael W.M., Dreyfuss G.. Characterization and primary structure of the poly(C)-binding heterogeneous nuclear ribonucleoprotein complex K protein. Mol. Cell. Biol. 1992; 12:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takimoto M., Tomonaga T., Matunis M., Avigan M., Krutzsch H., Dreyfuss G., Levens D.. Specific binding of heterogeneous ribonucleoprotein particle protein K to the human c-myc promoter, in vitro. J. Biol. Chem. 1993; 268:18249–18258. [PubMed] [Google Scholar]
- 9. Gallardo M., Hornbaker M.J., Zhang X., Hu P., Bueso-Ramos C., Post S.M.. Aberrant hnRNP K expression: All roads lead to cancer. Cell Cycle Georget. Tex. 2016; 15:1552–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li D., Wang X., Mei H., Fang E., Ye L., Song H., Yang F., Li H., Huang K., Zheng L. et al.. Long noncoding RNA pancEts-1 promotes neuroblastoma progression through hnRNPK-Mediated β-catenin stabilization. Cancer Res. 2018; 78:1169–1183. [DOI] [PubMed] [Google Scholar]
- 11. Ito K., Sato K., Endo H.. Cloning and characterization of a single-stranded DNA binding protein that specifically recognizes deoxycytidine stretch. Nucleic Acids Res. 1994; 22:53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tomonaga T., Levens D.. Heterogeneous nuclear ribonucleoprotein K is a DNA-binding transactivator. J. Biol. Chem. 1995; 270:4875–4881. [DOI] [PubMed] [Google Scholar]
- 13. Dejgaard K., Leffers H.. Characterisation of the nucleic-acid-binding activity of KH domains different properties of different domains. Eur. J. Biochem. 1996; 241:425–431. [DOI] [PubMed] [Google Scholar]
- 14. Siomi H., Chol M., Sloml M.C., Nussbaum R.L., Dreyfuss G.. Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMRl that causes fragile X syndrome. Cell. 1994; 77:33–39. [DOI] [PubMed] [Google Scholar]
- 15. Vries I.S.N., Brendle A., Bähr-Ivacevic T., Benes V., Ostareck D.H., Ostareck-Lederer A.. Translational control mediated by hnRNP K links NMHC IIA to erythroid enucleation. J. Cell Sci. 2016; 129:1141–1154. [DOI] [PubMed] [Google Scholar]
- 16. Braddock D.T., Baber J.L., Levens D., Clore G.M.. Molecular basis of sequence-specific single-stranded DNA recognition by KH domains: solution structure of a complex between hnRNP K KH3 and single-stranded DNA. EMBO J. 2002; 21:3476–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Backe P.H., Messias A.C., Ravelli R.B.G., Sattler M., Cusack S.. X-ray crystallographic and NMR studies of the third KH domain of hnRNP K in complex with single-stranded nucleic acids. Struct. Lond. Engl. 2005; 13:1055–1067. [DOI] [PubMed] [Google Scholar]
- 18. Paziewska A., Wyrwicz L.S., Bujnicki J.M., Bomsztyk K., Ostrowski J.. Cooperative binding of the hnRNP K three KH domains to mRNA targets. FEBS Lett. 2004; 577:134–140. [DOI] [PubMed] [Google Scholar]
- 19. Klimek-Tomczak K., Wyrwicz L.S., Jain S., Bomsztyk K., Ostrowski J.. Characterization of hnRNP K Protein–RNA interactions. J. Mol. Biol. 2004; 342:1131–1141. [DOI] [PubMed] [Google Scholar]
- 20. Ostrowski J., Wyrwicz L., Rychlewski L., Bomsztyk K.. Heterogeneous nuclear ribonucleoprotein K protein associates with multiple mitochondrial transcripts within the organelle. J. Biol. Chem. 2002; 277:6303–6310. [DOI] [PubMed] [Google Scholar]
- 21. Thisted T., Lyakhov D.L., Liebhaber S.A.. Optimized RNA targets of two closely related triple KH domain proteins, heterogeneous nuclear ribonucleoprotein K and αCP-2KL, suggest distinct modes of RNA recognition. J. Biol. Chem. 2001; 276:17484–17496. [DOI] [PubMed] [Google Scholar]
- 22. Dominguez D., Freese P., Alexis M.S., Su A., Hochman M., Palden T., Bazile C., Lambert N.J., Van Nostrand E.L., Pratt G.A. et al.. Sequence, structure, and context preferences of human RNA binding proteins. Mol. Cell. 2018; 70:854–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moritz B., Lilie H., Naarmann-de V.I.S., Urlaub H., Wahle E., Ostareck-Lederer A., Ostareck D.H.. Biophysical and biochemical analysis of hnRNP K: arginine methylation, reversible aggregation and combinatorial binding to nucleic acids. Biol. Chem. 2014; 395:837–853. [DOI] [PubMed] [Google Scholar]
- 24. Sun L., Fazal F.M., Li P., Broughton J.P., Lee B., Tang L., Huang W., Kool E.T., Chang H.Y., Zhang Q.C.. RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol. 2019; 26:322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin N., Chang K.-Y., Li Z., Gates K., Rana Z.A., Dang J., Zhang D., Han T., Yang C.-S., Cunningham T.J. et al.. An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol. Cell. 2014; 53:1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huarte M., Guttman M., Feldser D., Garber M., Koziol M.J., Kenzelmann-Broz D., Khalil A.M., Zuk O., Amit I., Rabani M. et al.. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010; 142:409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Z., Zhou C., Chang Y., Zhang Z., Hu Y., Zhang F., Lu Y., Zheng L., Zhang W., Li X. et al.. Long non-coding RNA CASC11 interacts with hnRNP-K and activates the WNT/β-catenin pathway to promote growth and metastasis in colorectal cancer. Cancer Lett. 2016; 376:62–73. [DOI] [PubMed] [Google Scholar]
- 28. Gallardo M., Malaney P., Aitken M.J.L., Zhang X., Link T.M., Shah V., Alybayev S., Wu M.-H., Pageon L.R., Ma H. et al.. Uncovering the role of hnRNP K, an RNA-binding protein, in B-cell lymphomas. J. Natl. Cancer Inst. 2019; 112:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kawasaki Y., Komiya M., Matsumura K., Negishi L., Suda S., Okuno M., Yokota N., Osada T., Nagashima T., Hiyoshi M. et al.. MYU, a target lncRNA for Wnt/c-Myc signaling, mediates induction of CDK6 to promote cell cycle progression. Cell Rep. 2016; 16:2554–2564. [DOI] [PubMed] [Google Scholar]
- 30. Chu C., Zhang Q.C., da Rocha S.T., Flynn R.A., Bharadwaj M., Calabrese J.M., Magnuson T., Heard E., Chang H.Y.. Systematic discovery of Xist RNA binding proteins. Cell. 2015; 161:404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pintacuda G., Wei G., Roustan C., Kirmizitas B.A., Solcan N., Cerase A., Castello A., Mohammed S., Moindrot B., Nesterova T.B. et al.. hnRNPK recruits PCGF3/5-PRC1 to the Xist RNA B-repeat to establish polycomb-mediated chromosomal silencing. Mol. Cell. 2017; 68:955–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brown C.J., Hendrich B.D., Rupert J.L., Lafrenière R.G., Xing Y., Lawrence J., Willard H.F.. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992; 71:527–542. [DOI] [PubMed] [Google Scholar]
- 33. Penny G.D., Kay G.F., Sheardown S.A., Rastan S., Brockdorff N.. Requirement for Xist in X chromosome inactivation. Nature. 1996; 379:131–137. [DOI] [PubMed] [Google Scholar]
- 34. Clemson C.M., McNeil J.A., Willard H.F., Lawrence J.B.. XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J. Cell Biol. 1996; 132:259–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Żylicz J.J., Bousard A., Žumer K., Dossin F., Mohammad E., da Rocha S.T., Schwalb B., Syx L., Dingli F., Loew D. et al.. The implication of early chromatin changes in X chromosome inactivation. Cell. 2019; 176:182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brockdorff N., Ashworth A., Kay G.F., McCabe V.M., Norris D.P., Cooper P.J., Swift S., Rastan S.. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell. 1992; 71:515–526. [DOI] [PubMed] [Google Scholar]
- 37. McHugh C.A., Chen C.-K., Chow A., Surka C.F., Tran C., McDonel P., Pandya-Jones A., Blanco M., Burghard C., Moradian A. et al.. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015; 521:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cirillo D., Blanco M., Armaos A., Buness A., Avner P., Guttman M., Cerase A., Tartaglia G.G.. Quantitative predictions of protein interactions with long noncoding RNAs. Nat. Methods. 2017; 14:5–6. [DOI] [PubMed] [Google Scholar]
- 39. Mossessova E., Lima C.D.. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell. 2000; 5:865–876. [DOI] [PubMed] [Google Scholar]
- 40. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A.. Walker J.M. Protein identification and analysis tools on the ExPASy server. The Proteomics Protocols Handbook. 2005; Totowa: Humana Press; 571–607. [Google Scholar]
- 41. Milligan J.F., Groebe D.R., Witherell G.W., Uhlenbeck O.C.. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987; 15:8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nilsen T.W. Gel Purification of RNA. Cold Spring Harb. Protoc. 2013; 2013:doi:10.1101/pdb.prot072942. [DOI] [PubMed] [Google Scholar]
- 43. Parsonnet N.V., Lammer N.C., Holmes Z.E., Batey R.T., Wuttke D.S.. The glucocorticoid receptor DNA-binding domain recognizes RNA hairpin structures with high affinity. Nucleic Acids Res. 2019; 47:8180–8192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hom R.A., Wuttke D.S.. Human CST prefers G-Rich but not necessarily telomeric sequences. Biochemistry. 2017; 56:4210–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hellman L.M., Fried M.G.. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007; 2:1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nilsen T.W. Preparing size markers for gel electrophoresis. Cold Spring Harb. Protoc. 2013; 2013:1186–1189. [DOI] [PubMed] [Google Scholar]
- 47. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K. et al.. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinforma. Oxf. Engl. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-Regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J. et al.. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nature Methods. 2020; 17:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Madeira F., Park Y. mi, Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A.R.N., Potter S.C., Finn R.D. et al.. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019; 47:W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lubelsky Y., Ulitsky I.. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature. 2018; 555:107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Polaski J.T., Holmstrom E.D., Nesbitt D.J., Batey R.T.. Mechanistic insights into Cofactor-Dependent coupling of RNA folding and mRNA Transcription/Translation by a cobalamin riboswitch. Cell Rep. 2016; 15:1100–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wheeler E.C., Nostrand E.L.V., Yeo G.W.. Advances and challenges in the detection of transcriptome-wide protein–RNA interactions. WIREs RNA. 2018; 9:e1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Malik N., Yan H., Moshkovich N., Palangat M., Yang H., Sanchez V., Cai Z., Peat T.J., Jiang S., Liu C. et al.. The transcription factor CBFB suppresses breast cancer through orchestrating translation and transcription. Nat. Commun. 2019; 10:2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grant C.E., Bailey T.L., Noble W.S.. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu Z., Zhang Q.C., Lee B., Flynn R.A., Smith M.A., Robinson J.T., Davidovich C., Gooding A.R., Goodrich K.J., Mattick J.S. et al.. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell. 2016; 165:1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Smola M.J., Christy T.W., Inoue K., Nicholson C.O., Friedersdorf M., Keene J.D., Lee D.M., Calabrese J.M., Weeks K.M.. SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fang R., Moss W.N., Rutenberg-Schoenberg M., Simon M.D.. Probing Xist RNA structure in cells using targeted Structure-Seq. PLoS Genet. 2015; 11:e1005668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Geuens T., Bouhy D., Timmerman V.. The hnRNP family: insights into their role in health and disease. Hum. Genet. 2016; 135:851–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nostrand E.L.V., Freese P., Pratt G.A., Wang X., Wei X., Xiao R., Blue S.M., Chen J.-Y., Cody N.A.L., Dominguez D. et al.. A large-scale binding and functional map of human RNA binding proteins. Nature. 2020; 583:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Calabretta S., Richard S.. Emerging roles of disordered sequences in RNA-binding proteins. Trends Biochem. Sci. 2015; 40:662–672. [DOI] [PubMed] [Google Scholar]
- 66. Ozdilek B.A., Thompson V.F., Ahmed N.S., White C.I., Batey R.T., Schwartz J.C.. Intrinsically disordered RGG/RG domains mediate degenerate specificity in RNA binding. Nucleic Acids Res. 2017; 45:7984–7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Basu S., Bahadur R.P.. A structural perspective of RNA recognition by intrinsically disordered proteins. Cell. Mol. Life Sci. 2016; 73:4075–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hentze M.W., Castello A., Schwarzl T., Preiss T.. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018; 19:327–341. [DOI] [PubMed] [Google Scholar]
- 69. Zagrovic B., Bartonek L., Polyansky A.A.. RNA-protein interactions in an unstructured context. FEBS Lett. 2018; 592:2901–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gerstberger S., Hafner M., Tuschl T.. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014; 15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vasilyev N., Polonskaia A., Darnell J.C., Darnell R.B., Patel D.J., Serganov A.. Crystal structure reveals specific recognition of a G-quadruplex RNA by a β-turn in the RGG motif of FMRP. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E5391–E5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Loughlin F.E., Lukavsky P.J., Kazeeva T., Reber S., Hock E.-M., Colombo M., Von Schroetter C., Pauli P., Cléry A., Mühlemann O. et al.. The solution structure of FUS bound to RNA reveals a bipartite mode of RNA recognition with both sequence and shape specificity. Mol. Cell. 2019; 73:490–504. [DOI] [PubMed] [Google Scholar]
- 73. Mikula M., Dzwonek A., Karczmarski J., Rubel T., Dadlez M., Wyrwicz L.S., Bomsztyk K., Ostrowski J.. Landscape of the hnRNP K protein–protein interactome. PROTEOMICS. 2006; 6:2395–2406. [DOI] [PubMed] [Google Scholar]
- 74. Bomsztyk K., Denisenko O., Ostrowski J.. hnRNP K: one protein multiple processes. BioEssays. 2004; 26:629–638. [DOI] [PubMed] [Google Scholar]
- 75. Mackereth C.D., Sattler M.. Dynamics in multi-domain protein recognition of RNA. Curr. Opin. Struct. Biol. 2012; 22:287–296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.