


Abstract
Eukaryotic DNA is organized in nucleosomes, which package DNA and regulate its accessibility to transcription, replication, recombination and repair. Here, we show that in living cells nucleosomes protect DNA from high-energy radiation and reactive oxygen species. We combined sequence-based methods (ATAC-seq and BLISS) to determine the position of both nucleosomes and double strand breaks (DSBs) in the genome of nucleosome-rich malignant mesothelioma cells, and of the same cells partially depleted of nucleosomes. The results were replicated in the human MCF-7 breast carcinoma cell line. We found that, for each genomic sequence, the probability of DSB formation is directly proportional to the fraction of time it is nucleosome-free; DSBs accumulate distal from the nucleosome dyad axis. Nucleosome free regions and promoters of actively transcribed genes are more sensitive to DSB formation, and consequently to mutation. We argue that this may be true for a variety of chemical and physical DNA damaging agents.
INTRODUCTION
Eukaryotic DNA is organized in nucleosomes, which both help package the DNA and regulate its accessibility to transcription, replication and recombination. The accessibility of damaged DNA for its repair is also controlled by nucleosomes, and chromatin needs to be disassembled prior to repair (1). However, even prior to their repair, the occurrence of damages to DNA might be modulated by nucleosomes. The correlation between nucleosome distribution and the occurrence of DNA damage has been inferred indirectly from the distribution of mutations (2,3), which at least in part arise from the error prone repair of damaged DNA. However, this correlation is subject to a number of limitations, the most significant of which is that the mechanics of DNA repair is expected to determine whether a DNA damage is repaired correctly (with no mutation arising) or incorrectly (causing a mutation). The need for DNA repair for the production of mutations thus obfuscates the relation between the distribution of mutations and the distribution of DNA damage. Moreover, mutations also arise from incorrect replication, further complicating the relationship between mutations and DNA damage. To overcome these limitations, here we investigated experimentally the genomic distribution and occupancy of nucleosomes and the distribution of double strand breaks (DSBs) after ionizing irradiation.
DNA damage is caused either by spontaneous depurination and deamination of DNA bases, or by chemical or physical agents, among which high-energy photons (ionizing radiation, IR). IR can break DNA strands either by colliding directly with the phosphodiester backbone, or by splitting water molecules into hydrogen and hydroxyl radicals (a form of reactive oxygen species, ROS) that can react with DNA and produce several types of DNA damage. Notably, ROS are continuously produced in the cell by mitochondrial metabolism and by several biochemical reactions, and IR-induced ROS actually promote the formation of mitochondrially derived ROS (4). Among ROS-induced DNA damages, single strand breaks (SSBs) and double strand breaks (DSBs) are prominent, with a much larger amount of IR-induced SSBs relative to DSBs (5); in fact, most DSBs are the outcome of two close SSBs on opposite strands of DNA (6). Whether and to what extent nucleosomes protect DNA from IR-induced DSBs has been studied only in vitro (7,8). These studies have broadly established that nucleosomes reduce the average incidence of DSBs on bulk chromatin. However, the distribution of DSBs in the genome is arguably as important as their total number, and has not been addressed.
Nucleosomes are assembled fairly regularly over DNA: about 150 bp are wrapped around the histone octamer and linker DNA (∼40 bp in humans and 20 bp in yeast) separates consecutive nucleosomes (9). The positioning of nucleosomes is indirectly dictated by the DNA sequence, with nucleosomes assembling on the more flexible sequences (10,11). Nucleosomes are not present all of the time on all nucleosomal sites, however; the fraction of cells where a specific nucleosomal site is covered (or alternatively, the fraction of time a specific site is covered by a nucleosome in a cell) is called occupancy. Occupancy depends on how favorable the sequence is for nucleosome assembly, and on changes in chromatin organization brought about by chromatin remodeling complexes, transcription, replication, binding of transcription factors and histone post-translational modifications (12–15).
We and others have described several instances in which nucleosomal occupancy is altered at genomewide level: cell senescence (16,17), embryonic stem cell identity (18,19), activation of macrophages (20,21), and mutation of the DNA chaperone HMGB1 in mammalian cells, or Nhp6A/B in yeast (22). At least in the case of the lack of HMGB1 or Nhp6A/B, histones are reduced by 20%. The positioning of nucleosomes in Nhp6A/B mutants is maintained, but occupancy is decreased substantially and not uniformly (22). We thus took advantage of nucleosome modulation in mammalian cells after HMGB1 depletion to correlate nucleosome occupancy with DSB formation.
Our laboratory is interested in malignant mesothelioma (MM), an almost incurable tumor arising from asbestos exposure (23). While characterizing mouse models of MM, we realized that the mouse cell line AB22, which reproduces relevant features of human epithelioid MM (24), is histone-rich relative to primary mesothelial cells (PMC) derived from the peritoneal cavity of BALB/c mice. We partially reverted the high genomewide nucleosomal occupancy of MM cells by knocking down HMGB1. We then compared nucleosome-rich unmodified MM cells and nucleosome-depleted MM cells with respect to their sensitivity to radiation-induced DNA damage, and integrated information about genomewide nucleosome positioning and occupancy (obtained with the Assay for Transposase Accessible Chromatin and sequencing, ATAC-seq (25)) with information about the location of IR-induced DSBs (obtained by Breaks Labelling In-Situ and Sequencing, BLISS (26)). To confirm our results, we repeated this analysis by integrating ATAC-seq and BLISS datasets obtained in human MCF-7 breast carcinoma cells.
Overall, our results obtained in living cells confirm that nucleosomes shield DNA from damage, and further indicate at nucleotide resolution that nucleosomal occupancy, or the probability of a nucleosome occupying a specific site, is by far the dominant factor in determining whether a sequence will be damaged or not.
MATERIALS AND METHODS
HMGB1 down-modulation by RNA interference
In mesothelioma AB22 cells
The pTRIPZ plasmids (Dharmacon) are described in Supplementary Figure S1B. The shRNA sequence was designed to target the second exon of the murine Hmgb1 transcript and adapted to the miR-30 sequence for optimal expression in mammalian cells (27). The control shRNA sequence (shCTRL) does not target any mammalian transcript (Dharmacon). The designed template oligos were synthesized and then amplified by PCR to introduce cloning restriction enzyme sites using the primers below. shHMGB1-cloning template: 5′_TGCTGTTGACAGTGAGCGATGACAAGGCTCGTTATGAAAGTAGTGAAGCCACAGATGTACTTTCATAACGAGCCTTGTCACTGCCTACTGCCTCGGA_3′, the annealing sequence for PCR primers is underlined and the sequence complementary to the HMGB1 mRNA sequence at position 550–570 in NCBI Refseq NM_001313894 is in bold type.
miR30-XhoI FW: 5′_CAGAAGGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCG_3′
miR30-MluI REV: 5′_CTAAAGTAGCCCCTTACGCGTCCGAGGCAGTAGGCA_3′
The PCR fragment and recipient plasmid pTRIPZ were digested with XhoI and MluI and ligated to obtain pTRIPZ-shHMGB1 (shHMGB1).
For lentiviral particle production, HEK 293T cells were cultured in DMEM plus 10% fetal bovine serum, 100 U/ml penicillin, 100 U/ml streptomycin, glutamine and sodium pyruvate (all from Gibco). Two hours before transfection, the medium was changed to IMDM (Sigma) containing 10% FBS, 100 U/ml penicillin, 100 U/ml streptomycin, 2 mM glutamine. Nine million low-passage HEK 293T cells were transfected with the packaging plasmids pDM2-VSVG and pCVM-ΔR8.91 and the vectors pTRIPZ-shHMGB1 or pTRIPZ-shCTRL using the calcium phosphate method. A plasmid mix solution (7 μg pDM2-VSVG, 28 μg pCMV-ΔR8.91, 32 μg pTRIPZ-shHMGB1) was added to 125 mM CaCl2 to a final volume of 1250 μl and incubated at room temperature for 5 minutes; DNA-calcium phosphate precipitate was formed by dropwise addition of 1250 μl of 2× HBS (to a final concentration of 140.5 mM NaCl; 50 mM HEPES; 0.75 mM Na2HPO4 pH 7.12); the precipitated complexes were immediately added dropwise to the cell media. Virus-containing media was collected 24 and 48 h after transfection, centrifuged 5 min at 200 g and filtered through 0.22 μm membranes. The virus preparation was then concentrated by ultracentrifugation (20 000g for 2 h) and resuspended in PBS. Titration was performed by transducing HEK293T cells with serial 1:10 dilutions of the viral preparation. One day after transduction, expression of the Red Fluorescence Protein (RFP) was induced by incubating cells with 1 μg/ml doxycycline (Sigma) for three days. At the end of induction, cells were collected, fixed in 1% paraformaldehyde and the percentage of RFP+ cells was measured using a BD Accuri C6 cytometer. The concentration of Transducing Units (TU) was determined using the following formula and considering the dilution of virus preparation that resulted in <20% of RFP+ cells:
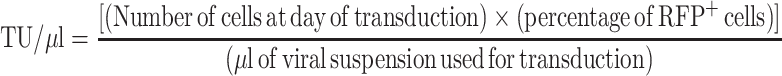 |
In MCF-7 breast cancer cells
Briefly, MCF-7 were transfected with plasmid HMGB1shRNA-pSuperior.puro (28) or, as a mock control, with the empty vector pSuperior.puro (Invitrogen) and selected with puromycin. (Sequence: 5′-GGATATTGCTGCATATCGA, position 1412–1430 in NCBI Refseq NM_001313893.1). Single resistant clones were picked, amplified, and analyzed for HMGB1 expression by western blot. Clone shB1/MCF-7, with <10% HMGB1 remaining, was used.
Mice, primary mesothelial cells and cell lines
Animal experiments had been reviewed and approved by the Animal Care and Use Committees (IACUC) of Ospedale San Raffaele, which include ‘ad hoc’ members for ethical issues. All experiments were performed in accordance with the approved guidelines. Animals were housed in the Institute's Animal Care Facilities, which meet international standards. Certified veterinarians who are responsible for health monitoring, animal welfare supervision, experimental protocols and procedures revision regularly checked them.
Murine primary mesothelial cells (PMC) were isolated and cultured as described (29). Briefly, 6–10 weeks old BALB/c male mice were euthanized by CO2 inhalation and their skin was removed from the abdomen. The peritoneal cavity was flushed twice with PBS to remove blood cells. We injected about 10 ml of warm 0.25% Trypsin/0.002% EDTA (Sigma); fluid with mesothelial cells was then collected after 10 min. A further lavage of peritoneal cavity with warm cell culture medium was also collected and pooled. Cells from each mouse were centrifuged, resuspended in fresh RPMI 1640 medium supplemented with 2 mM glutamine, 20% (v/v) fetal bovine serum (FBS), 10 μM 2-mercaptoethanol, 20 mM HEPES (Gibco), 400 μg/l hydrocortisone (Sigma), 100 U/ml penicillin 100 U/ml streptomycin, and seeded in 60 mm culture dishes, then cultured for 8–9 days.
Murine malignant mesothelioma (MM) AB22 cells were obtained from Cell Bank Australia and cultured in RPMI 1640 supplemented with 10% (v/v) FBS, 2 mM l-glutamine and 100 U/ml penicillin/streptomycin. shCTRL-MM and shHMGB1-MM were obtained by infection with pTRIPZ-shCTRL or pTRIPZ-shHMGB1 viral particles at a multiplicity of infection equal to 20; transduced cells were then selected with 4 μg/ml puromycin. Cells were cultured in RPMI 1640, as above, using tetracycline-free FBS (Euroclone). shRNA expression was induced by adding 1 μM doxycycline (Sigma) to the culture medium. All the experiments were performed after 7–10 days from doxycycline addition. Both primary and MM cells were cultured in humidified incubator at 37°C with 5% oxygen.
Cell synchronization in G1 phase was achieved by starving sub-confluent cells in medium containing 0.1% FBS overnight and releasing them from starvation in regular medium for 5 h. Cell cycle distribution was assessed by cytofluorimetry analysis of DNA content.
Human mesothelioma REN and the mesothelial LP9 cell lines were a kind gift of Haining Yang and Michele Carbone (Cancer Center, University of Hawai‘i) and cultured in DMEM/F12 supplemented with 10% (v/v) FBS, 2 mM l-glutamine and 100 U/ml penicillin/streptomycin.
Human breast cancer MCF-7 and normal mammary epithelial MCF10A cell lines were purchased from ATCC. MCF-7 cells were cultured in DMEM (Dulbecco's modified Eagle's medium, Gibco) supplemented with 10% (v/v) FBS, 2 mM l-glutamine and 100 U/ml penicillin/streptomycin. MCF10A were cultured in DMEM/F12 1:1 supplemented with 2 mM l-glutamine and 100 U/ml penicillin/streptomycin, 5% horse serum (Gibco), 0.5 μg/ml hydrocortisone (Sigma), 100 ng/ml cholera toxin (Sigma), 10 μg/ml human insulin (Sigma), 20 ng/ml hEGF (Preprotech). Cells were maintained sub-confluent in humidified incubator at 37°C with 5% CO2.
Western blotting
Whole-cell extracts were prepared by direct lysis of cells in SDS-PAGE sample buffer (aka Laemmli buffer: 62.5 mM Tris–HCl pH 6.8, 25% (v/v) glycerol, 2% (w/v) sodium dodecyl sulphate, 5% (v/v) 2-mercaptoethanol). Samples were denatured for 5 min at 95°C and loaded onto SDS polyacrylamide protein gels (10% or 14% acrylamide/bisacrylamide, 29:1). Proteins separated by SDS-PAGE were transferred onto 0.2 μm nitrocellulose membranes using the Trans-Blot® Turbo™ Transfer System (Bio-Rad). Membranes were blocked for 1 hour at room temperature in 5% nonfat milk/TBST (20 mM Tris, pH 7.5, 137 mM NaCl, 0.1% Tween 20), washed in TBST and incubated overnight with primary antibody at 4°C. After three washes in TBST, filters were incubated with a secondary antibody conjugated to HRP for 1 h at RT. Proteins were visualized by chemiluminescence (Western Blot Luminol Reagent, SantaCruz) and acquired using Bio-Rad ChemiDoc Imaging system. Quantification of band intensities was performed with ImageLab software (Bio-Rad) on 16-bit images. The following primary antibodies were used for western blotting: rabbit α-H3 (1:1000; ab1791 Abcam), rabbit α-HMGB1 (1:1000; ab18256 Abcam), rabbit α-GAPDH (1:10 000; G9545 Sigma).
SDS-PAGE gels are loaded with lysate samples containing an equal number of cells, with the exception of the blots for quantification of histone H3 where DNA concentration in whole cell extracts was quantified in order to load equal amount of corresponding DNA in lysates. DNA quantification was performed with Quant-iT™ PicoGreen® dsDNA Kit (Invitrogen) following manufacturer instructions and fluorescence measured using a Victor3 plate reader (Perkin-Elmer, excitation and emission filters: 485 nm/535 nm, exposure 1.0 s). DNA concentration of the samples was determined by interpolation of the fluorescence intensity values of the samples with the standard curve.
For the quantification of histone H3 by western blotting, Coomassie staining was used to normalize for minimal sample concentration variability. Briefly, SDS-PAGE gels were stained using InstantBlue™ Coomassie Protein Stain (Expedeon), transferred to nitrocellulose membrane. Coomassie stained membranes are further hybridized as described above.
SDS-PAGE, western blots and H3 quantifications for MCF-7, MC10A, LP9 and REN were performed as described (20).
Immunofluorescence and image analysis
Cells grown on coverslips were fixed in 4% PFA in PHEM buffer (120 mM PIPES, 50 mM HEPES, 20 mM EGTA, 8 mM MgSO4, buffered to pH 7.0 with KOH) for 10 min at room temperature. After fixation, cells were washed with PBS and incubated 5 min in permeabilization buffer (0.2% Triton X-100, 20 mM HEPES, 300 mM sucrose, 50 mM NaCl, 3 mM MgCl2) then blocked in 4% BSA/PBS. Coverslips were incubated over night at 4°C with gentle agitation with primary antibodies diluted in 0.2% BSA/PBS to the final concentration. After three rinses with 0.2% BSA/PBS, cells were incubated with a secondary fluorescent antibody in 0.2% BSA/PBS at RT for 1 h, washed three times, and then mounted with mounting medium containing Hoechst 33342 for DNA counterstaining. The rabbit polyclonal α-HMGB1 antibody (1:1000; ab18256 Abcam) and an AlexaFluor488 conjugated secondary antibody (Molecular Probes) were used. A Leica TCS SP5 confocal microscope with a 20×/NA0.75 objective and Airy3 pinhole was used to acquire 16-bit images that were processed with a semi-automatic image quantification pipeline based on nuclear pixel identification in the UV channel. Total HMGB1 intensity was expressed in arbitrary unit (AU) (20).
Cell cycle analysis
Cells were collected, fixed in cold 70% ethanol overnight at −20°C, washed once in 5% FBS/PBS and then resuspended in PBS containing 10 μg/ml RNase A (Sigma) and 10 μg/ml propidium iodide (Sigma) for 1 h at room temperature. Samples were acquired with a BD Canto II cytometer. Cells were gated to exclude doublets and cell cycle distributions were estimated by deconvolution of the DNA content profiles using FCS Express (De Novo Software) and expressed as percentage of cells in each cell cycle phase.
Alkaline comet assay
The alkaline comet assay was performed as previously described (30). Cells were irradiated and placed on ice to minimize the processing of DNA breaks, immediately resuspended in 0.5% low-melting-point agarose in PBS and deposited on a microscope slide pre-coated with a thin layer of 1% agarose in PBS. The slides were incubated in lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris pH 10, 1% Triton X-100) for 1 h at 4°C and then in alkaline electrophoresis buffer (1 mM EDTA, 300 mM NaOH) for 20 min to allow DNA unwinding. Electrophoresis was performed at 4°C for 30 min at a constant voltage (0.8 V/cm). After electrophoresis, slide pH was neutralized by 5 min incubation in 0.4 M Tris, pH 7.5. DNA staining was performed by overlaying 60 μl of ethidium bromide (20 μg/ml) on each slide. Comets were imaged using a Zeiss AxioVision microscope and analysed with CaspLab software (31) to calculate the tail moment, which is the product of the percentage of tail DNA and the tail length. More than 150 comets were scored per sample.
RNA sequencing
Total RNA from G1-synchronized MM cells was isolated using the NucleoSpin RNA II kit (Macherey-Nagel). RNA integrity was tested using Agilent RNA 6000 Nano Kit (RIN>9) and Ribosomal RNA was removed using RiboMinus Gold™ Technology (ThermoFisher). Libraries of cDNA were prepared with TruSeq RNA Library Prep Kit (Illumina) and sequenced using NextSeq500 (Illumina) 75 bp pair-end up to 30 million reads. After trimming the adapter sequences from reads (Trimmomatic), sequences were aligned to the mouse mm10 genome using the STAR (v2.5.3a) (https://github.com/alexdobin/STAR/) and counted using featureCounts (http://bioconductor.org/packages/release/bioc/html/Rsubread.html) with the gene annotation from Gencode (version M13). The R/Bioconductor package edgeR was used to obtain normalized RPKM expression values (http://bioconductor.org/packages/release/bioc/html/edgeR.html).
ATAC-seq (Assay for Transposase Accessible Chromatin sequencing)
ATAC-seq was performed on permeabilized nuclei of shCTRL-MM and shHMGB1-MM cells synchronized in G1 phase following a detailed protocol (25), using 75 000 cells/sample. Libraries were sequenced using NextSeq-500 Illumina sequencing system and up to 400 million 150 bp pair-end reads were produced. After trimming low-quality bases and adapters (Trimmomatic), reads were aligned to the mouse genome (GRCm38/mm10) using bwa mem (v0.7.15-r1140). Properly paired primary alignments with MAPQ >15 were saved while reads mapping to the mitochondrial chromosome were removed. The deconvolution of the fragment size distribution (Figure 2B) originated three populations: the first one was described by an exponential curve and contained fragments with size <100 bp representing the nucleosome free regions (NFRs). The second and third populations were described by Gaussian curves and contained the mononucleosomal (186–282 bp) and dinucleosomal (>282 bp) fragment populations. Such cutoffs minimized the overlap between the populations.
Figure 2.

HMGB1 knock-down MM cells have fewer nucleosomes and lower nucleosome occupancy genomewide. (A) Summary of ATAC-seq protocol. Open Chromatin regions (OCRs) correspond to regions with a high frequency of transposition events. (B) Fragment size distribution of ATAC-seq libraries obtained from shCTRL and shHMGB1 MM cells. Shaded areas (0–100 and 186–282 bp) indicate the sizes of the ATAC-seq fragments corresponding to NFRs and mononucleosomes. (C) NUCs and NFRs called by NucleoATAC, normalized for the number of base pairs belonging to OCRs in both cell lines. (D) Distribution of mononucleosome reads normalized by library size on a section of chromosome 10 (bins of 1 kb, scalebar) in shCTRL (blue) and shHMGB1 (yellow) MM cells. The gray track represents the difference between yellow and blue tracks. (E) Positioning of nucleosomes on 3 kb around the promoter of the Fbxo30 gene in shCTRL (blue) and shHMGB1 (yellow) MM cells. Rectangles mark the positions of the called nucleosomes. (F) Comparison of genomewide nucleosome positioning. Frequency distribution of the distance (in either direction) between the dyad axes of paired nucleosomes in shCTRL and shHMGB1 MM cells. The mode of the distribution is 16 bp. (G) Nucleosome positioning around TSS of all genes; the mean cross-correlation signal is centred at the TSS. (H) Scatter density plot of nucleosome occupancy score in shCTRL (x axis) and shHMGB1 (y axis) cells, as calculated by NucleoATAC tool. Each point is colored by its estimated local density (scalebar). A dashed line for perfect linear relationship (x = y) is indicated. Dashed numbered boxes are drawn to help visualization of differences between shCTRL and shHMGB1 nucleosome occupancy.
Open chromatin regions (OCRs) were called using the MACS2 software, with the following parameters: -q 0.05 –broad -f BAMPE (OCR bed intervals). The regions were also extended 100 bp up- and downstream, and merged when in overlap. We used the NucleoATAC software (32) to call nucleosomes and NFRs. NFRs are listed in the nfrpos.bed.gz results file. Nucleosomal occupancy and Nucleosomal positioning were calculated with occ and nuc tools of NucleoATAC.
BLISS (Breaks Labeling In Situ and Sequencing)
To map DNA breaks in native chromatin, 5 × 104 shCTRL or shHMGB1 cells were seeded on coverslips, synchronized in G1 as described before and irradiated with 0, 3, 5 and 10 Gy of gamma rays from a cesium-137 source. MCF-7 cells were seeded on coverslips at the same density, pre-treated 1 hour with 10 mM N-acetyl-l-cysteine (Sigma, A9165) or vehicle (PBS) in culture medium, then irradiated with 0 or 10 Gy of gamma rays. Immediately after irradiation, cells were fixed with 4% paraformaldehyde in PBS and subsequently subjected to the described protocol (33), with only minor changes: genomic DNA was isolated from cells using the phenol:chloroform method; libraries were prepared from purified RNA using TruSeq Small RNA library preparation kit (Illumina) and sequenced (2 × 150 bp paired-end, NextSeq500, Illumina) at 20–30 M reads per sample. We applied the bioinformatic pipeline described by Yan et al. (26) to identify and quantify DSBs, with minor modifications regarding how to manage the unique molecular identifier (UMI) present in reads. While Yan et al. (26) used custom scripts, we took advantage of the umi tools to first extract and then align reads with bwa software. Umi tools was also applied to deduplicate and aggregate the UMIs (34).
The number of DSBs in each sample was estimated using an in silico saturation experiment (Figures 3D, 5A and Supplementary Figure S3A). Briefly, a random downsampling of the original FASTQ produced four read populations representing the 25, 50, 75 and 100% of the total. DSBs at saturation were estimated using the hyperbolic formula DSB = r * DSBmax/r + k where DSBmax is the DSB number at saturation, r the number of sequenced reads and k is a constant (hyperbolic fitting by nls function in R).
Figure 3.

Irradiation of nucleosome-poor MM cells produces more DNA breaks. Quantification of DNA breaks by alkaline comet assay in shCTRL and shHMGB1 cells irradiated with increasing doses of γ rays. (A) Representative images of DNA comets. (B) Distributions of comet tail moment ( = % DNA in Tail × Tail Length) in N >120 cells per group; boxes and whiskers correspond to 25–75 and 5–95 percentiles, respectively; horizontal line is the median. Statistical analysis was performed with two-way ANOVA: the differences between cell types, between IR doses, and the interaction between cells types and doses are all highly significant, P< 0.0001. The significance of the differences between cell types at each dose was estimated applying Bonferroni's post-test; NS, not significant; *** P< 0.001. This experiment was repeated twice with comparable results. (C) Summary of the BLISS protocol: living cells were exposed or not to radiation, immediately fixed, and DSB free ends were labelled in situ by adapter ligation. Sequencing of BLISS libraries generated reads that accumulate bidirectionally from the original break (piles of black lines). Arrows: endogenous breaks; strikes: radiation induced breaks). (D) Estimated number of DSBs in shCTRL (blue) and shHMGB1 (yellow) cells irradiated with 0, 3, 5 and 10 Gy; dots and lines represent biological replicates and means, respectively. The fitting for DSB estimation is shown in Supplementary Figure S3A. Statistics: two-way ANOVA. The effects of irradiation dose, genotype and their interaction are all significant (P = 0.018, P = 0.002, P = 0.0016, respectively). (E) DSB distribution in the 10 kb around the transcription start site (TSS) of the 10% most transcribed (magenta line) or the 10% least transcribed genes (black line). The frequency of DSBs in each 0.5 kb window is the percentage of total DSBs in the 10 kb around the TSS; in a uniform distribution each 0.5 kb would contain 5% of the total DSBs. The DSB distributions in shCTRL and shHMGB1 MM cells irradiated with 10 Gy shown here are similar to those in all the other samples (Supplementary Figure S3D).
Figure 5.

DSBs anti-correlates with nucleosome occupancy in human MCF-7 cells. (A) Interrogation of public datasets: The probability of harboring endogenous DSBs anticorrelates with nucleosome occupancy. This hypothesis was tested on BLISS and ATAC datasets produced and published by other groups: ATAC-seq GSE101736, GSM2714245; BLISS GSE136365, GSM4047452 (left) and GSM4047456 (right). Each dot represents one percentile in nucleosome occupancy. The x-axis, log scale, is the nucleosome occupancy value; the y-axis, log scale, is the probability to harbor one or more DSBs. Statistics: Pearson correlation on the log-transformed values. (B) Estimated number of DSBs in MCF-7 cells, expressed as fold change over the paired control sample (not treated not irradiated). MCF-7 cells were pre-treated (magenta) or not (green) with 10 mM N-acetyl-l-cysteine (NAC) for 1 h and irradiated or not with 10 Gy; dots and lines represent biological replicates and averages, respectively. Statistics: two-way ANOVA test (paired experiments). Irradiation dose (P = 0.0063) and interaction of irradiation and NAC treatment (P = 0.043) are statistically significant; NAC treatment (P = 0.28) is not. (C) The probability of harboring DSBs anticorrelates with nucleosome occupancy. Each dot represents one percentile in nucleosome occupancy (ATAC-seq raw data from GSE101736). The x-axis, log scale, is the nucleosome occupancy value; the y-axis, log scale, is the probability to harbor one or more DSBs. Statistics: Pearson correlation on the log-transformed values.
To evaluate experimental reproducibility in biological replicates (Supplementary Figure S3C), we used deepTools to compare the number of deduplicated DSBs (BAM files) in 100 kb windows after normalization by library and genome size. Zero values deriving from windows with no DSB coverage were removed using the –skipZeros option. Distances between datasets were obtained by Pearson correlation, clustered with the ‘complete’ algorithm and plotted in a heatmap using the R package pheatmap.
Integration of nucleosome positioning and occupancy with DSB events
Nucleosome positioning and occupancy
Coverage values (bigwig files) for chromosomes 10 visualized in Figure 2D, E and Supplementary Figure S2 were generated with the IGV Genome Browser (DOI: 10.1038/nbt.1754) using a 1 kb binning to visualize nucleosome positioning.
To assess differences in nucleosome positioning between the two genotypes, we calculated the absolute value of the distance between dyad axes of paired nucleosomes detected in shCTRL and shHMGB1; the frequency distribution was plotted using R (Figure 2F).
Occupancy was calculated by NucleoATAC on nucleosomes called either in shCTRL or shHMGB1 cells. The scatterplot in Figure 2H compares the density distribution of nucleosomes with similar occupancy values in shCTRL and shHMGB1. Briefly, the density for each data point (x,y) is estimated by counting the number of points in the neighborhood (kernel density), including the query point. The neighborhood is an area defined by a fixed radius value provided as an argument to the function. In order to get colors, density values are scaled between 1 to 256, which is the number of colors in the scalebar.
Relative enrichment of DSBs around the TSS of expressed genes
RNA-Seq data were used to calculate the expression levels of all the genes in three biological replicates from shCTRL and shHMGB1 cells. A ranking based on mean RPKM values was produced and the 10% more and less expressed genes in the two cell types were extracted.
From the BLISS bed file obtained after UMI deduplication and aggregation, we extracted the number of DSBs present in windows of 500 bp around the TSS (–5 to +5 kb) using the giggle tool (https://github.com/ryanlayer/giggle), which performs at faster speed than bedtools. The frequency of DSBs in each 500 bp window was calculated as the sum of DSBs in that region in all genes divided by the total detected DSBs in the 10 kb around the TSS (Figure 3E).
Fold enrichment of DSBs in OCRs, nucleosomes and NFRs
For each shCTRL and shHMGB1 sample, OCR intervals (MACS2, OCR bed files) were intersected with DSB positions using bedtools (https://academic.oup.com/bioinformatics/article/26/6/841/244688).
DSBs falling inside OCRs or not were counted using the -v option of the bedtools intersect function. The number of DSBs inside or outside OCRs were normalized by the number of the base pairs inside or outside OCRs, respectively. The fold enrichment (FE) of DSB events in OCRs relative to whole genome was then calculated as FE = (DSBs in OCRs/bp in OCRs) / (total DSBs/genome size in bp). The same procedure was applied for nucleosomes and NFRs called by NucleoATAC.
To disprove the null hypothesis that DSBs are homogeneously distributed in the genome (fold enrichment = 1), we first generated a collection of random regions equivalent in number and size to OCRs from ATAC-seq experiments. To do so, we shuffled one thousand times the genomic locations of the OCRs using shuffleBed from the BEDTools Suite. We used the same procedure to test the statistical significance of the fold enrichment of nucleosomes and NFRs.
The nucleosome positioning cross-correlation bedgraph track (smoothed version) produced by NucleoATAC was used to produce the metadata profiles around nucleosome dyad positions and around TSS of genes using deepTools (Figures 2F and 4D, respectively).
Figure 4.

Nucleosomes shield DNA from DSBs. (A) DSBs are preferentially associated to OCRs. The fold enrichment of DSBs falling in OCRs relative to the entire genome is plotted here as mean and standard deviation of two replicates per experimental condition. The difference between the observed enrichment and the enrichment in a null distribution of randomly generated genomic sequences equal in size and number to the observed OCRs is statistically significant (P = 0.001; statistics described in Materials and Methods and shown in Supplementary Figure S4A). Not irradiated shCTRL and shHMGB1 MM cells are shown as representative result; the same results hold true for all BLISS samples (Supplementary Figure S4A). (B) DSBs are enriched in NFRs. The fold enrichment relative to the entire genome is calculated as in (A) considering separately NUCs or NFRs in OCRs. Statistics: P = 0.001, calculated as in panel A and shown in Supplementary Figure S4B,C. (C) Nucleosomes damaged by a DSB have lower occupancy. The distribution of occupancy values is shown for intact nucleosomes and DSB-containing nucleosomes in two representative samples (shCTRL-MM cells irradiated or not with 10 Gy). Boxes and whiskers correspond to 25–75 and 5–95 percentiles, respectively; horizontal line is the median. Statistics, Mann-Whitney test. (D) DSBs accumulate far from the nucleosome dyad axis. DSB relative coverage (left y-axis, reads per genome coverage) on metadata profiles centred on nucleosome dyads (black line; right y-axis: cross-correlation normalized signal from NucleoATAC). Blue lines represent DSB coverage averaged on the two replicates of unirradiated (fill) and 10 Gy irradiated (dashed) shCTRL cells. (E) The probability of harboring DSBs decreases as nucleosome occupancy increases. Each dot represents one percentile in nucleosome occupancy. The x-axis, log scale, is the nucleosome occupancy value; the y-axis, log scale, is the probability to harbor one or more DSBs. The linearity in the log-log plot indicates a power distribution. Statistics: Pearson correlation on the log-transformed values. This result holds true for all the other BLISS samples (Supplementary Figure S4D).
Correlation between nucleosomal occupancy and DSBs
The nucleosomes called by NucleoATAC in shCTRL and shHMGB1 ATAC-Seq experiments were intersected with DSB positions of the corresponding samples using bedtools. The number of DSBs found in every called nucleosome was recorded, including the zero DSB events. The distribution of DSBs/nuc in the 1–10 range showed a linear relationship (log scale) with nucleosome frequencies (Supplementary Figures S3B and S5A). For higher DSB/nuc values, nucleosome frequency is negligible and such outliers would increase the noise in the fitting procedure. Therefore, we considered for the analyses only nucleosomes bearing 1–10 DSBs. Afterwards, nucleosomes were divided in one hundred percentiles of occupancy, as calculated by NucleoATAC. For each percentile, the number of DSB events were used to estimate the parameters of negative binomial distribution (using fitdist function of the fitdistrplus R package). The estimates were used to calculate the probability of the events using the dnbinom function in R. The log of probabilities of DSB occurrence was Pearson correlated to the log of the mean value of occupancy (Figure 4E).
RESULTS
Mesothelioma cells have a high histone content
To test the histone content in the mouse MM cell line AB22 relative to primary mesothelial cells (PMC) derived from the peritoneal cavity of BALB/c mice, we synchronized PMC and MM cells in G1 to minimize fluctuations in DNA and histone content occurring during the cell cycle (Supplementary Figure S1A). Due to the stoichiometry of histones in nucleosomes, histone H3 quantification is a reliable proxy for the abundance of H4, H2A and H2B histones (22). MM cells contain a significantly higher amount of histone H3 compared to PMC (Figure 1A and Supplementary Figure S1E). Notably, the human mesothelioma cell line REN also contains a higher amount of histone H3 relative to the non-transformed mesothelial cell line L9 (Figure 1B). We thus used AB22 MM cells (henceforth called MM cells) as our experimental model.
Figure 1.
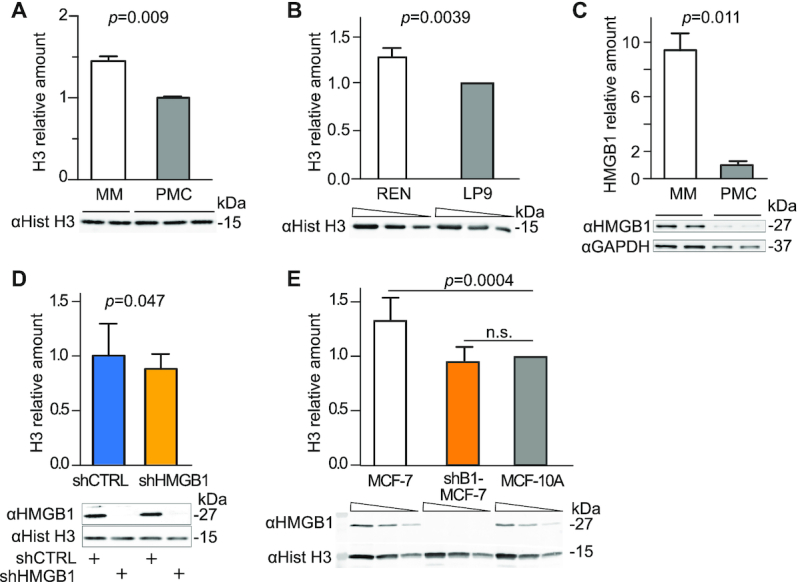
Malignant Mesothelioma cells have a high histone content that can be partially reverted by HMGB1 downmodulation. (A) Histone H3 levels in whole cell lysates of malignant mesothelioma (MM) cells and Primary Mesothelial Cells (PMC) were assessed by quantitative Western blotting. The intensity of Coomassie staining was used as loading control and normalization (Supplementary Figure S1E). Bars and error bars represent average and standard deviation, respectively (biological replicates are shown). Statistics: unpaired t-test. This experiment was repeated twice with comparable results. (B) Histone H3 levels in REN human mesothelioma cell line and its non-tumoral counterpart, human mesothelial cell line LP9. The relative amount of H3 histone in REN cell relative to LP9 cells (normalized to 1) was tested in two different experiments. Below the plot, a representative western blot is shown in which three sequential 1:2 dilutions for each sample are loaded (wedges). Bars and error bars represent average fold-change and standard deviation. Statistics: one-sample t-test. (C) HMGB1 is highly expressed in MM cells as compared to PMC. Total cell lysates were analysed by quantitative Western blot; HMGB1 level after normalization to GAPDH levels is shown. Bars and error bars represent average and standard deviation, respectively (n = 2; biological replicates). Statistics: unpaired t-test. This experiment was repeated twice with comparable results. (D) Histone H3 levels are lower in MM cells when HMGB1 is downregulated by a targeted shRNA (shHMGB1). shHMGB1 and shCTRL cell extracts are compared by quantitative western blot. H3 bands are quantified and normalized with the Coomassie staining (Supplementary Figure S1F). Mean and standard deviation of H3 levels are shown (n = 7 biological replicates in independent experiments). Statistics: one-sample t test with a hypothetical mean = 1. (E) Histone H3 levels in whole cell lysates of human breast cancer MCF-7, MCF-7 knocked-down for HMGB1 (shB1/MCF-7) and mammary epithelium MCF-10A cells (set to 1). Quantification as in panel B. Statistics: one-sample t test (n = 4 independent experiments). MCF-7 cells and shB1-MCF-7 cells are statistically different (P = 0.0022 unpaired t test); n.s. = not significant.
We had proven that HMGB1 downmodulation decreases the histone content in primary mouse embryonic fibroblasts (22), as HMGB1 acts as a DNA chaperone in nucleosome formation. We first confirmed that MM cells express HMGB1 at high levels (Figure 1C), and then tested whether depleting HMGB1 in MM cells would reduce their histone content. We transduced MM cells with lentiviral vectors expressing either a specific shRNA directed against HMGB1 (shHMGB1) or a scrambled sequence (shCTRL) under the control of a tetracycline-responsive promoter (Supplementary Figure S1B). We obtained cell populations with stably integrated vectors but not expressing shRNAs. Doxycycline efficiently induced HMGB1 depletion in 3 days; the depletion was maintained for up to 14 days (Supplementary Figure S1C) without affecting the viability of MM cells. After 7 days of doxycycline treatment, the depletion of HMGB1 in shHMGB1 MM cells was remarkably homogeneous within the cell population (Supplementary Figure S1D). After synchronization in G1, we observed by Western blotting a small but significant reduction of 10–15% in histone H3 content in shHMGB1 cells compared to shCTRL cells (Figure 1D and Supplementary Figure S1F).
We also compared the histone content in the human cell line MCF-7, representative of invasive breast cancer carcinoma, relative to the MCF-10A cell line, representative of non-malignant breast epithelial cells. MCF-7 cells contained a higher level of H3 histone relative to MCF-10A cells, and this level was significantly reduced after HMGB1 knockdown (Figure 1E).
Overall, our results show that human and murine mesothelioma cell lines and a human breast cancer cell line have a high abundance of histone proteins as compared to normal counterparts, while HMGB1 knockdown decreases their nuclear histone content.
Histone-depleted MM cells have fewer nucleosomes and a lower genomewide nucleosomal occupancy
A reduced amount of histone proteins should result in an alteration in nucleosome organization along the genome. In fact, a yeast mutant bearing a deletion of the Nhp6a/b genes, coding for the yeast proteins functionally equivalent to mammalian HMGB1, has low histone protein content, displays increased DNA accessibility and reduced nucleosome occupancy genomewide; notably, nucleosome positioning is scarcely affected (22). We thus explored whether nucleosome positioning and occupancy is affected in shHMGB1 MM cells.
We applied ATAC-seq (25) to identify nucleosome-free regions and nucleosomes within highly accessible chromatin (open chromatin regions, OCRs, Figure 2A) in G1-synchronized shCTRL and shHMGB1 cells. We obtained DNA fragments with a typical size distribution: a high peak of small fragments (<100 bp) deriving mostly from regions of DNA not covered by nucleosomes, followed by two clear peaks corresponding to DNA fragments with the size of one or two nucleosomes (Figure 2B). Reads belonging to the one-nucleosome size distribution and aligned to the reference genome were extracted and normalized per genome coverage.
We used the computational tool NucleoATAC (32) to identify sequences that are nucleosome-free or that are organized in nucleosomes. Overall, NucleoATAC called about 10% fewer nucleosomes and 4% more NFRs, per kb of sequenced DNA, in shHMGB1 than shCTRL cells (Figure 2C). These results are completely in line with the biochemical analyses, and indicate that fewer nucleosomes form in shHMGB1 cells, which contain less histones.
The gross distribution of reads corresponding to nucleosomal DNA (Figure 2D, in bins of 1 kb; Supplementary Figure S2, whole chromosome) is variable along chromosome 10, representative of all 20 mouse chromosomes, but comparable between shCTRL and shHMGB1 samples. The differences between the shHMGB1 track and the shCTRL track (gray track) are small in both directions. Even at base-pair resolution, the positions of nucleosomes were similar in shCTRL and shHMGB1 MM cells (Figure 2E). Genomewide, the location of the dyad axis of corresponding nucleosomes in shCTRL and shHMGB1 MM cells was offset by only about 16 bp on average (Figure 2F). Nucleosome positioning at transcription start sites was nearly identical in shCTRL and shHMGB1 MM cells (Figure 2G). Thus, the genomewide positioning of nucleosomes was hardly changed by reducing the histone content of MM cells.
NucleoATAC also computes the nucleosome occupancy for each sequence; its value is calculated at the base pair level (32). We computed the occupancy of all nucleosomes called in shHMGB1 and in shCTRL datasets: each sequence i has an associated occupancy xi in shHMGB1 cells and yi in shCTRL cells. We then plotted the nucleosome occupancy score (x,y) in a density plot where the y-axis represents occupancy in the shHMGB1 cells, and the x-axis represents occupancy in the shCTRL cells, respectively (Figure 2H).). We found that the fraction of nucleosomes with occupancy between 0.25 and 0.4 in shCTRL cells (dotted area 2 in Figure 2H) is larger than the fraction of nucleosomes with similar occupancy in shHMGB1-MM cells (dotted area 1). These results indicate that sequences that have a high probability of forming nucleosomes in shCTRL cells also a high probability of forming nucleosomes in shHMGB1 cells, whereas sequences that have a low probability of forming nucleosomes in shCTRL cells have an even lower probability of forming nucleosomes in shHMGB1 cells. Thus, nucleosomal occupancy is not uniformly decreased over the genome of shHMGB1 cells but is lower where the occupancy is already low in shCTRL cells. This is comparable with results obtained in Nhp6a/b mutants in yeast (22).
Overall, our results show that HMGB1 downmodulation in MM cells changes chromatin conformation by decreasing the number of nucleosomes and their relative occupancy, but not their position.
Cells with fewer nucleosomes accumulate more DNA damage
We previously showed that IR causes more DNA breaks in nucleosome-poor Hmgb1−/− MEFs than in wt MEFs (22); therefore we expected that nucleosome depletion in MM cells would increase the sensitivity of their DNA to IR.
We exposed shCTRL and shHMGB1 MM cells to increasing doses of gamma rays from a cesium source, and immediately cooled and processed the cells, thus avoiding the repair of DNA breaks by DNA repair mechanisms. Using an alkaline comet assay we evaluated the extent of DNA damage by quantifying the amount of DNA that migrates out of lysed cells in an electric field.
As expected, in both cell types the amount of DNA breaks increased with IR dose (Figure 3A). Of note, at any given IR dose, shHMGB1 cells contained significantly more DNA breaks than shCTRL cells; non-irradiated shHMGB1 and shCTRL cells were not statistically different (Figure 3B).
Thus, even a modest decrease in histone content corresponds to a significant increase in the susceptibility of genomic DNA to breaks induced by IR.
We then investigated the physical relation between decreased nucleosome occupancy and increased susceptibility to IR-induced DNA damage. We G1-synchronized shCTRL and shHMGB1 MM cells and irradiated them with 0, 3, 5 and 10 Grays. The cells were then immediately paraformaldehyde-fixed to block the onset of any DNA repair event. We located DSBs in the genomic sequence using BLISS (26) (Figure 3C). We mapped the positions of DSBs on the reference genome and estimated the maximal number of DSB per library (Supplementary Figure S3A). In both cell types, the number of DSBs increased with the dose of gamma radiation and was significantly higher at each dose in shHGMB1 compared to shCTRL cells (Figure 3D).
A strong correlation between DSB location and transcribed sequences has long been observed (26,35). To identify transcribed sequences in MM cells, we profiled gene expression by RNAseq in G1-synchronized shCTRL and shHMGB1 cells. We then calculated the relative enrichment of DSBs within 5 kb of Transcription Start Sites (TSS) in the top 10% or bottom 10% expressed genes. We found, as expected, that DSBs are enriched at the TSS of the most transcribed genes, with no clear difference between the two genotypes (Figure 3E, Supplementary Figure S3D).
Overall, these results show that cells with fewer nucleosomes are more sensitive to radiation-induced SSBs and DSBs. We find the expected enrichment of DSBs over the TSS of expressed genes in both shHMGB1 and shCTRL MM cells.
Nucleosomes protect DNA from damage
We next integrated the information on nucleosome positioning and occupancy (derived by ATAC-seq) with the location of DSBs (derived by BLISS). We first located DSBs and asked whether they are enriched in OCRs. Indeed, DSBs are strongly enriched within OCRs relative to the whole genome, both for shCTRL and shHMGB1 cells and regardless of the dose of radiation used (Figure 4A and Supplementary Figure S4A).
We then considered separately sequences in OCRs occupied by nucleosomes (NUCs) and nucleosome-free regions (NFRs), as identified by ATAC-seq. In both shCTRL and shHMGB1 cells, DSBs are enriched in NFRs as compared to nucleosome-occupied regions, suggesting that the presence of a nucleosome might interfere with ROS accessibility to DNA. We then sorted all the nucleosomes identified by NucleoATAC into two groups: nucleosomes on sequences where at least one DSB was mapped (DSB present), and nucleosomes on sequences with no identified DSB (DSB absent). In shCTRL cells, the occupancy of nucleosomes where DSBs were present was significantly lower than the occupancy of those where DSBs were absent, both in the case of endogenous DSBs (0 Gy radiation) or IR-induced DSBs (Figure 4C). Thus, DNA protected by nucleosomes with higher occupancy has a lower probability of suffering a DSB.
To visualize the relative locations of DSBs and nucleosomes genomewide, we averaged the positioning signal of all nucleosomes identified by NucleoATAC in shCTRL cells, and superimposed the probability of finding a DSB at each nucleotide starting from the dyad axis (Figure 4D). This procedure is equivalent to mapping all DSBs relative to the dyad axis of a meta-nucleosome. An inverse correlation between the probability of being occupied by a nucleosome and the presence of DSBs is clearly visible. These data indicate that DSBs occur with higher probability on sequences that are distant from the nucleosome dyad.
We then calculated the correlation between nucleosome occupancy and the probability of harboring a DSB. We produced one hundred sets of nucleosomes based on their relative occupancy; each set represents a one-percentile increase in occupancy. We then calculated the probability density function of being damaged by a DSB, using the actual number of DSBs found on the sequences contained in each set of increasingly occupied nucleosomes. As seen in Figure 4E, the probability of harboring one or more DSBs strongly anti-correlated with the occupancy, for both endogenous and IR-induced DSBs, and in both shCTRL and shHMGB1 cells. The correlation was extremely significant and replicated with each dose of irradiation (Supplementary Figure S4D). On average, the probability of being damaged by a DSB is ∼4-fold lower (0.02 versus 0.09) for a nucleosome with 99–100% occupancy, relative to a nucleosome with 20% occupancy (which can be modeled as a nucleosome that insists on a specific sequence 20% of the time). Notably, we could interpolate a straight line through the log-log transformation of data points, indicating that the probability of harboring a DSB follows a power distribution. The slope was −0.7 to −0.8; a slope close to −1 means the probability of DSB formation is directly proportional to the probability of the sequence being nucleosome-free at the time of irradiation.
Overall, our analysis shows that DSBs occur mainly in regions of accessible chromatin (OCRs) and with much higher probability in nucleosome-free regions and in sequences with lower nucleosomal occupancy. Thus, DSBs mainly occur on sequences stably or temporarily not occupied by a nucleosome.
Anti-correlation between DSB probability and nucleosomal occupancy in the human cell line MCF-7
To extend our data to other cell lines, we looked for public datasets where cells had been analyzed both by BLISS and ATAC-seq. Datasets in GSE136365 describing the DSBs distribution in non-irradiated human MCF-7 breast cancer cells (36) were intersected with ATAC-seq data from control-treated MCF-7 cells, GSE101736 (37). Indeed, a highly significant negative correlation between nucleosome occupancy and probability of harboring a DSB was clearly present in MCF-7 cells either transfected with a control scrambled siRNA, or exposed to DMSO as control for a pharmacological treatment (Figure 5A).
We could not find any example of BLISS analysis of irradiated cells, and we therefore proceeded to generate our own BLISS data on MCF-7 cells irradiated or not with 10 Gy. We also pre-exposed cells to the anti-oxidant N-acetyl-l-cysteine (NAC) for 1 h, in order to confirm that ROS are involved in the generation of DSBs, as already reported (38). Indeed, irradiation significantly increased the number of DSBs, whereas NAC pre-exposure largely suppressed this increase (Figure 5B). We calculated with NucleoATAC the nucleosome occupancy in MCF-7 cells, based on the raw data available in GSE101736. We then divided nucleosomes in 100 sets of increasing occupancy and calculated the probability of each set of nucleosomes of harboring a DSB (based on our own BLISS data) (Figure 5C and Supplementary Figure S5B). A higher proportion of highly occupied nucleosomes were identified by NucleoATAC on MCF-7 cells, relative to our data on MM cells; this was possibly due to size selection of the DNA to be sequenced, partially excluding small fragments (37, and Materials and Methods). Despite these potential confounding factors, the inverse correlation between nucleosome occupancy and the probability of harboring a DSB is extremely significant in MCF-7 cells, irradiated and non-irradiated, and pre-exposed or not to NAC. The slopes of the interpolations in log-log plots for all MCF-7 cell samples (−0.7 to −1.0) is comparable to the slope in MM cells (−0.7 to −0.8).
Thus, we find a convincing inverse correlation between nucleosome occupancy and the probability of harboring a DSB in a human cell line, in addition to mouse MM cells.
DISCUSSION
Nucleosomes shield DNA from radiation-induced DNA damage
By intersecting ATAC-seq and BLISS data, which allow to map the distribution of nucleosomes and DSBs over the genome, respectively, we show here that in living cells nucleosomes effectively shield DNA from DSBs produced by ionizing radiation.
To derive our conclusions, we took advantage of murine malignant mesothelioma (MM) cells, which contain ∼1.5-fold more histones than their normal mesothelial cell counterpart, and reduced their histone and nucleosome content by knocking down HMGB1, a DNA chaperone that facilitates nucleosome formation. Previously, we had shown that HMGB1 depletion, either genetic or by silencing, causes histone and nucleosome depletion in MEFs and HeLa cells (22). Of note, the histone reduction obtained by HMGB1 silencing in MM cells is small—about one nucleosome in ten is missing, overall (Figures 1 and 2)—but is sufficient to significantly increase the sensitivity to DNA damage, as assessed by both comet assays and NGS-based assays (Figures 3 and 4). Even more notably, the positioning of nucleosomes is hardly affected when nucleosomes are depleted, in accordance with the view that the position of nucleosomes is dictated (albeit indirectly) by the DNA sequence itself (10,11). The invariant location of nucleosomes enables us to use the data obtained in the two MM cell lines as replicate of each other, since a specific sequence is interrogated twice, once in the control cells and once in the nucleosome-depleted cells. Indeed, the relation between nucleosome occupancy and DSB occurrence holds true in both cell lines, and is also true in human MCF-7 cells that we used for data replication.
An inverse correlation between the packaging of DNA into chromatin and the incidence of radiation-induced DSBs had already been noticed in vitro (7,8), but we extend those early results to living cells and to nucleosome occupancy at base pair resolution. The anti-correlation between the probability of DSB occurrence and nucleosome occupancy is both high and extremely significant, and follows a power law, since it can be interpolated by a straight line after a log-log transformation. Notably, the slope of the line interpolating the log-log transformed data is −0.7 to −0.8 in MM cells, where a slope of −1 would indicate an inverse linear relationship between the two variables. Since occupancy can be interpreted as the amount of time a given sequence spends packaged into a nucleosome, we infer that DSB formation is almost inversely proportional to the amount of time the sequence is occupied by a nucleosome. The probability of DSB formation on sequences not packaged in nucleosomes, e.g. on NFRs, is about five times as big as the probability of DSBs forming on sequences of highest occupancy, which are almost always packaged into a nucleosome (Figure 4E). We observed a comparable anti-correlation between nucleosome occupancy and DSB formation in the human MCF-7 line.
The genomic distribution of DSBs
A notable conclusion from our data is that DSB distribution is very similar in irradiated and non-irradiated cells. This is in agreement with the consensus that the DNA-reactive species in both irradiated and non-irradiated cells are the same –reactive oxygen species (ROS). N-Acetyl-l-cysteine (NAC), a ROS scavenger, reduces the amount of DNA damage upon irradiation (38, and Figure 5).
It is tempting to speculate that the probability of DSB formation within a nucleosome is proportional to the solvent accessible surface of DNA, which is estimated to range between one-quarter to one-third relative to free DNA, due to the occlusion of the surface of contact with the histones. However, this would imply that NFRs are not protein-bound, which is clearly not the case because NFRs are preferentially bound by transcription factors. Possibly, the solvent accessible surface of DNA within nucleosomes in living cells is even lower than that estimated in dilute solutions, reflecting a tight packing of neighboring nucleosomes in the nucleus. This would readily explain the different accessibility of DNA bound by transcription factors and that of DNA in nucleosomes.
We note that if solvent accessibility of nucleosome-packaged DNA is the main route of DNA damage caused by ROS, nucleosomes might also provide a barrier to alkylating agents and other DNA reactants. As for UV, the other widespread physical mutagen, it has already been noticed that nucleosomes in vitro protect from the formation of cyclobutane pyrimidine dimers (CPDs) because of the distortion of the planarity between consecutive base pairs (39). In fact, the DNA in contact with the nucleosome is protected from CPDs induced by UV irradiation in melanoma cells and the probability of a CPD to occur does anticorrelate with the nucleosome stability (40). Notably, the depletion of nucleosomes in yeast increases the susceptibility of DNA to UV irradiation (41).
Beside ROS, there is another proposed source of DSBs: stalled replication forks, which are processed to DSBs to allow the continuation of replication after the repair of the DNA template. Stalled forks are not expected to correlate to prior nucleosomal occupancy, as nucleosomes are removed to allow replication. In our experiments on MM cells, cells were synchronized in G1 at the time of irradiation, and therefore very few were replicating their DNA and might have contained stalled replication forks. The experiments on MCF-7 were performed on cycling cells; the results were broadly similar to the results obtained in G1-synchronized MM cells.
Our analyses (Figure 3E) are concordant with old (35) and new findings (26) showing that DSBs are more frequently associated with gene promoters of highly transcribed genes. Yan et al. (26) measured DNA damage in cells exposed to etoposide, which traps topoisomerases on torsionally stressed DNA. We suggest that our result and those by Yan et al. are similar because of two different mechanistic reasons: promoter sequences are more accessible to ROS because of the low occupancy by nucleosomes, and are more prone to etoposide-induced damage because promoters accumulate toposoisomerases.
Nucleosomal occupancy and radiation sensitivity
In our work, we used MM cells because they contain a high amount of histones and nucleosomes, especially in relation to their normal counterpart, primary mesothelial cells (PMCs). The comparison of MM and PMCs shows that PMCs have ample space for additional nucleosomes and, conversely, that a lot of the genome is not covered by nucleosomes at any one time in PMCs. Our analysis also suggests that MM cancer cells may be less prone to DNA damage than PMCs, which may have relevance for radiotherapy. It also suggests that cell types with very low nucleosomal occupancy, such as embryonic stem (ES) cells (18,19) or senescent cells (16,17), may be exquisitely sensitive to DNA damage. Notably, ES cells minimize ROS damage by relying for energy production on glycolysis rather than ROS-producing oxidative phosphorylation, and by expressing antioxidant defences, yet they have a constantly activated DNA damage response (DDR) (42); this is possibly a consequence of higher DNA vulnerability to damage.
CONCLUSION AND PERSPECTIVE
Overall, we have shown that nucleosomes effectively shield DNA from ionizing radiation and ROS, and we argue that this may be true for a variety of chemical and physical DNA damaging agents. The consequence is that genome regions depleted from nucleosomes, such as promoters of actively transcribed genes, are more sensitive to DNA damage, and consequently to mutation deriving from incorrect repair. This creates an evolutionary dilemma: DNA protected by nucleosomes is more resistant to damage (has a thicker shield), but nucleosomes restrict the accessibility of critical gene control regions such as promoters and enhancers (the shield is heavy and inconvenient). We suggest that different cell types might pack their DNA somewhat differently to respond to this dilemma; conversely, organisms might select for high nucleosome occupancy to reduce the rate of mutation of specific sequences.
DATA AVAILABILITY
Metrics and quality checks for NGS data are reported in Supplementary Table S1. ATAC-seq, BLISS and RNA-seq data have been deposited in the Gene Expression Omnibus database. Data are registered as GSE134798 (Nucleosomes protect DNA from irradiation-induced double strand breaks in living cells, 22 July 2020).
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge Francesco De Marchis for technical assistance, and Luca Michetti, Dr Samuel Zambrano and Dr Massimo P. Crippa for useful discussions. We are grateful to the Center for Omics Sciences @OSR (COSR) for guidance on libraries preparation and for sequencing.
Author contribution: F.B. generated cell lines, performed most wet experiments, analyzed the data and contributed to the manuscript; J.G.M. and E.M. were responsible for computational analysis; L.H. and A.Z. performed comet assays and other unreported experiments; A.A. and M.E.B. conceived and directed the study, and were involved in all aspects of the experimental design, data analysis and manuscript preparation. All authors critically reviewed the text and figures.
Contributor Information
Francesca Brambilla, Università Vita-Salute San Raffaele, via Olgettina 58, 20132 Milan, Italy.
Jose Manuel Garcia-Manteiga, IRCCS San Raffaele Scientific Institute, via Olgettina 58, 20132 Milan, Italy; Center for Omics Sciences, IRCCS San Raffaele Scientific Institute, Milan, Italy.
Emanuele Monteleone, Università Vita-Salute San Raffaele, via Olgettina 58, 20132 Milan, Italy.
Lena Hoelzen, Università Vita-Salute San Raffaele, via Olgettina 58, 20132 Milan, Italy; Faculty of Biology, Albert-Ludwigs-University Freiburg, D79104 Freiburg, Germany.
Angelica Zocchi, Università Vita-Salute San Raffaele, via Olgettina 58, 20132 Milan, Italy.
Alessandra Agresti, IRCCS San Raffaele Scientific Institute, via Olgettina 58, 20132 Milan, Italy.
Marco E Bianchi, Università Vita-Salute San Raffaele, via Olgettina 58, 20132 Milan, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
AIRC [IG-18623]; CNR Epigen (to M.E.B.); AIRC [IG-18687]; ‘Sovem s.r.l. [CF00631140126, donazione liberale’ to A.A.]; Italian Ministry of Health with ‘5 × 1000 funds’, 2019. Funding for open access charge: AIRC [2016 IG18687 to A.A.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Hauer M.H., Gasser S.M.. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017; 31:2204–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tolstorukov M.Y., Volfovsky N., Stephens R.M., Park P.J.. Impact of chromatin structure on sequence variability in the human genome. Nat. Struct. Mol. Biol. 2011; 18:510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sasaki S., Mello C.C., Shimada A., Nakatani Y., Hashimoto S.-i., Ogawa M., Matsushima K., Gu S.G., Kasahara M., Ahsan B. et al.. Chromatin-associated periodicity in genetic variation downstream of transcriptional start sites. Science. 2009; 323:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamamori T., Yasui H., Yamazumi M., Wada Y., Nakamura Y., Nakamura H., Inanami O.. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012; 53:260–270. [DOI] [PubMed] [Google Scholar]
- 5. Cannan W.J., Pederson D.S.. Mechanisms and consequences of double-strand DNA break formation in chromatin. J. Cell. Physiol. 2016; 231:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Milligan J.R., Ng J.Y., Wu C.C., Aguilera J.A., Fahey R.C., Ward J.F.. DNA repair by thiols in air shows two radicals make a double-strand break. Radiat. Res. 1995; 143:273–280. [PubMed] [Google Scholar]
- 7. Takata H., Hanafusa T., Mori T., Shimura M., Iida Y., Ishikawa K., Yoshikawa K., Yoshikawa Y., Maeshima K.. Chromatin compaction protects genomic DNA from radiation damage. PLoS One. 2013; 8:e75622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elia M.C., Bradley M.O.. Influence of chromatin structure on the induction of DNA double strand breaks by ionizing radiation. Cancer Res. 1992; 52:1580–1586. [PubMed] [Google Scholar]
- 9. Bednar J., Horowitz R.A., Grigoryev S.A., Carruthers L.M., Hansen J.C., Koster A.J., Woodcock C.L.. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:14173–14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Segal E., Fondufe-Mittendorf Y., Chen L., Thåström A., Field Y., Moore I.K., Wang J.-P.Z.P., Widom J., Thastrom A., Field Y. et al.. A genomic code for nucleosome positioning. Nature. 2006; 442:772–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ortiz V., de Pablo J.J.. Molecular origins of DNA flexibility: sequence effects on conformational and mechanical properties. Phys. Rev. Lett. 2011; 106:238107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rowley M.J., Corces V.G.. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018; 19:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paul M.R., Hochwagen A., Ercan S.. Condensin action and compaction. Curr. Genet. 2019; 65:407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Talbert P.B., Meers M.P., Henikoff S.. Old cogs, new tricks: the evolution of gene expression in a chromatin context. Nat. Rev. Genet. 2019; 20:283. [DOI] [PubMed] [Google Scholar]
- 15. Klemm S.L., Shipony Z., Greenleaf W.J.. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019; 20:207–220. [DOI] [PubMed] [Google Scholar]
- 16. O’Sullivan R.J., Kubicek S., Schreiber S.L., Karlseder J.. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010; 17:1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feser J., Truong D., Das C., Carson J.J., Kieft J., Harkness T., Tyler J.K.. Elevated histone expression promotes life span extension. Mol. Cell. 2010; 39:724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karnavas T., Pintonello L., Agresti A., Bianchi M.E.. Histone content increases in differentiating embryonic stem cells. Front. Physiol. 2014; 5:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ricci M.A., Manzo C., García-Parajo M.F., Lakadamyali M., Cosma M.P.. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015; 160:1145–1158. [DOI] [PubMed] [Google Scholar]
- 20. De Toma I., Rossetti G., Zambrano S., Bianchi M.E.E., Agresti A.. Nucleosome loss facilitates the chemotactic response of macrophages. J. Intern. Med. 2014; 276:454–469. [DOI] [PubMed] [Google Scholar]
- 21. Nair R.R., Mazza D., Brambilla F., Gorzanelli A., Agresti A., Bianchi M.E.. LPS-challenged macrophages release microvesicles coated with histones. Front. Immunol. 2018; 9:1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Celona B., Weiner A., Di Felice F., Mancuso F.M., Cesarini E., Rossi R.L., Gregory L., Baban D., Rossetti G., Grianti P. et al.. Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol. 2011; 9:e1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carbone M., Ly B.H., Dodson R.F., Pagano I., Morris P.T., Dogan U.A., Gazdar A.F., Pass H.I., Yang H.. Malignant mesothelioma: facts, myths, and hypotheses. J. Cell. Physiol. 2012; 227:44–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mezzapelle R., Rrapaj E., Gatti E., Ceriotti C., Marchis F. De, Preti A., Spinelli A.E., Perani L., Venturini M., Valtorta S. et al.. Human malignant mesothelioma is recapitulated in immunocompetent BALB/c mice injected with murine AB cells. Sci. Rep. 2016; 6:22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J.. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015; 2015:21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yan W.X., Mirzazadeh R., Garnerone S., Scott D., Schneider M.W., Kallas T., Custodio J., Wernersson E., Li Y., Gao L. et al.. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017; 8:15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paddison P.J., Cleary M., Silva J.M., Chang K., Sheth N., Sachidanandam R., Hannon G.J.. Cloning of short hairpin RNAs for gene knockdown in mammalian cells. Nat. Methods. 2004; 1:163–167. [DOI] [PubMed] [Google Scholar]
- 28. Trisciuoglio L., Bianchi M.E.. Several nuclear events during apoptosis depend on caspase-3 activation but do not constitute a common pathway. PLoS One. 2009; 4:e6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bot J., Whitaker D., Vivian J., Lake R., Yao V., McCauley R.. Culturing mouse peritoneal mesothelial cells. Pathol. Res. Pract. 2003; 199:341–344. [DOI] [PubMed] [Google Scholar]
- 30. Hartmann A., Agurell E., Beevers C., Brendler-Schwaab S., Burlinson B., Clay P., Collins A., Smith A., Speit G., Thybaud V. et al.. Recommendations for conducting the in vivo alkaline Comet assay. Mutagenesis. 2003; 18:45–51. [DOI] [PubMed] [Google Scholar]
- 31. Końca K., Lankoff A., Banasik A., Lisowska H., Kuszewski T., Góźdź S., Koza Z., Wojcik A.. A cross-platform public domain PC image-analysis program for the comet assay. Mutat. Res. - Genet. Toxicol. Environ. Mutagen. 2003; 534:15–20. [DOI] [PubMed] [Google Scholar]
- 32. Schep A.N., Buenrostro J.D., Denny S.K., Schwartz K., Sherlock G., Greenleaf W.J.. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 2015; 25:1757–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang F., Bienko M., Crosetto N., Yan W.X., Mirzazadeh R., Garnerone S., Scott D., Schneider M.W., Kallas T., Custodio J. et al.. Breaks labeling in situ and sequencing (BLISS). Protoc. Exch. 2017; doi:10.21203/rs.2.1448/v2. [Google Scholar]
- 34. Smith T., Heger A., Sudbery I.. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 2017; 27:491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yunis J.J., Soreng A.L., Bowe A.E.. Fragile sites are targets of diverse mutagens and carcinogens. Oncogene. 1987; 1:59–69. [PubMed] [Google Scholar]
- 36. Hazan I., Monin J., Bouwman B.A.M., Crosetto N., Aqeilan R.I.. Activation of oncogenic super-enhancers is coupled with DNA repair by RAD51. Cell Rep. 2019; 29:560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Porter J.R., Fisher B.E., Baranello L., Liu J.C., Kambach D.M., Nie Z., Koh W.S., Luo J., Stommel J.M., Levens D. et al.. Global inhibition with specific activation: How p53 and MYC redistribute the transcriptome in the DNA double-strand break response. Mol. Cell. 2017; 67:1013–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kurashige T., Shimamura M., Nagayama Y.. Differences in quantification of DNA double-strand breaks assessed by 53BP1/γH2AX focus formation assays and the comet assay in mammalian cells treated with irradiation and N-acetyl-L-cysteine. J. Radiat. Res. 2016; 57:312–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schieferstein U., Thoma F.. Modulation of cyclobutane pyrimidine dimer formation in a positioned nucleosome containing poly(dA·dT) tracts. Biochemistry. 1996; 35:7705–7714. [DOI] [PubMed] [Google Scholar]
- 40. Brown A.J., Mao P., Smerdon M.J., Wyrick J.J., Roberts S.A.. Nucleosome positions establish an extended mutation signature in melanoma. PLoS Genet. 2018; 14:e1007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Giavara S., Kosmidou E., Hande M.P., Bianchi M.E., Morgan A., D’Adda Di Fagagna F., Jackson S.P.. Yeast Nhp6A/B and mammalian Hmgb1 facilitate the maintenance of genome stability. Curr. Biol. 2005; 15:68–72. [DOI] [PubMed] [Google Scholar]
- 42. Vitale I., Manic G., De Maria R., Kroemer G., Galluzzi L.. DNA damage in stem cells. Mol. Cell. 2017; 66:306–319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Metrics and quality checks for NGS data are reported in Supplementary Table S1. ATAC-seq, BLISS and RNA-seq data have been deposited in the Gene Expression Omnibus database. Data are registered as GSE134798 (Nucleosomes protect DNA from irradiation-induced double strand breaks in living cells, 22 July 2020).