


Abstract
Gene losses provide an insightful route for studying the morphological and physiological adaptations of species, but their discovery is challenging. Existing genome annotation tools focus on annotating intact genes and do not attempt to distinguish nonfunctional genes from genes missing annotation due to sequencing and assembly artifacts. Previous attempts to annotate gene losses have required significant manual curation, which hampers their scalability for the ever-increasing deluge of newly sequenced genomes. Using extreme sequence erosion (amino acid deletions and substitutions) and sister species support as an unambiguous signature of loss, we developed an automated approach for detecting high-confidence gene loss events across a species tree. Our approach relies solely on gene annotation in a single reference genome, raw assemblies for the remaining species to analyze, and the associated phylogenetic tree for all organisms involved. Using human as reference, we discovered over 400 unique human ortholog erosion events across 58 mammals. This includes dozens of clade-specific losses of genes that result in early mouse lethality or are associated with severe human congenital diseases. Our discoveries yield intriguing potential for translational medical genetics and evolutionary biology, and our approach is readily applicable to large-scale genome sequencing efforts across the tree of life.
INTRODUCTION
The placental mammal radiation exhibits tremendous phenotypic diversity (1). This myriad form and function arose from gain, loss, and modification of inherited traits from one generation to the next. Genomic sequence comparisons reveal that, similarly, genes arose, were modified, and died through evolutionary time across different lineages. While it is nearly impossible to be certain that a gene locus no longer emits any kind of coding or non-coding transcript, ancestral ‘gene loss’ by virtue of losing or dramatically altering coding potential is well-established (2) and could contribute to the sequence basis of phenotypic evolution.
Previous studies have identified a few intriguing examples of gene losses that are associated with phenotypic traits. For example, the enzyme gene GULO is inactivated in independent mammalian lineages that likely consequently lost the ability to synthesize Vitamin C, which must instead be constantly supplied by their diet (3,4); multiple visual system genes are no longer functional in subterranean mammals which live primarily in darkness (5,6); taste receptor and other genes are lost in fully aquatic cetaceans (7–9); renal transporter genes URAT1, GLUT9 and OAT1 are dead in fruit-eating bats, which could have facilitated their frugivorous diet (10); the immune genes MX1 and MX2 are eroded in toothed whales, possibly making them more susceptible to certain viral pathogens (11); and the loss of PON1 in several marine mammals may increase their vulnerability to agricultural pesticide pollution (12). Systematic annotation of gene losses across species could, therefore, not only reveal fascinating evolutionary events and genotype-phenotype relationships, but could also highlight ‘natural knockout’ models for human pathologies that point to compensating molecular pathways in species missing otherwise-indispensable genes (2,3,13).
Manual annotation, such as ENCODE-HAVANA (14), is the gold standard for labeling pseudogenized genes but has only been applied to a handful of species (human, mouse, rat, and zebrafish) and is not scalable for hundreds to thousands of newly sequenced species (15). Automating the discovery of gene losses is scalable but challenging. For example, Sharma et al. (10) recently attempted an automated method using whole-genome multiple-sequence alignment to identify genes with inactivating mutations. However, their overly inclusive approach reportedly predicted hundreds of gene losses per species across 62 mammals, of which only 21 losses in total are discussed in the paper, presumably because manual inspection deemed many loss predictions inconclusive at best. A similar technique was used to further semi-manually annotate 85 gene losses in cetaceans (16). Other computational approaches for gene annotation, like the Ensembl pipeline (17), combine ab initio predictions with protein sequence evidence (18) and with gene models inferred from RNA-seq data. However, they focus only on annotating intact genes and do not attempt to distinguish decaying genes from those that only confoundingly appear so in regions of low sequence coverage, poor sequence quality, or dubious alignment calls. A clear need exists for a method that automatically calls gene losses with high confidence.
We developed a novel, conservative approach for identifying gene loss events and applied it to 58 mammalian genomes, using human assembly hg38 as the reference. We hypothesized that the most effective strategy for declaring loss of an ancestral gene function is to demonstrate that it is highly sequence-eroded compared to other genes in the same genome. While it is impossible to guarantee that no gene transcript is expressed from the orthologous locus (especially if a known promoter remains intact), the erosion of gene sequence implies that the gene does not maintain its original function.
To that end, we used pairwise whole-genome alignments to locate the orthologs of human protein-coding genes in each of the 58 query species by applying a novel synteny-aware mapping procedure. We employed a Mahalanobis distance-based model to identify the most highly mutated orthologs in each genome in a way that normalizes for the baseline extent of sequence divergence between this species and the reference (human). We considered these per-genome predictions to be likely eroded and functionally altered orthologs in the sequenced individual, and thus possibly the species represented by the assembly. Lastly, we applied a stringent phylogenetic filter, requiring that all final candidate erosions were supported by observations from at least two closely related species and affected ancestral genes—in order to minimize confounders due to assembly artifacts, private (individual-specific) mutations, or reference genome biases. Though very conservative, our approach uncovered hundreds of high-confidence ortholog erosion (hcoErosion) events, affecting over 50 mammals, including some very surprising gene losses with intriguing evolutionary and biomedical implications. Importantly, our method was designed to allow easy addition of genomes to the analysis, such as the those from countless newly sequenced mammals, birds (19), vertebrates (20), insects (21), etc.
MATERIALS AND METHODS
Genome assemblies, reference genome and protein-coding gene annotations
We used genome assemblies of 59 mammalian species (listed in Supplementary Table S1) in this study, with human genome assembly GRCh38/hg38 as reference. From Ensembl biomart release 86, we downloaded lists of gene identifiers and counted how many unique ‘protein_coding’ gene identifiers are found. Of the 59 aligned mammalian species, 34 species had gene annotation in Ensembl. For our reference gene set, we started with 22 072 unique human protein-coding genes annotated in Ensembl and excluded gene annotations from un-placed and un-localized scaffolds, leaving 19 729 protein-coding genes. We then obtained coordinates for segmental duplications (genomic fragments larger than 1 kb with >90% sequence identity to other genomic fragments) from the UCSC genome browser and excluded from our analysis genes with 10% or more overlap with any segmentally duplicated fragment. We also restricted the reference gene set to genes having ‘complete’ Ensembl transcripts. These filters generated our reference gene set of 17 860 unique human protein-coding genes for mapping across mammalian genomes. We collected assembly N50 measures for each genome from NCBI: https://www.ncbi.nlm.nih.gov/assembly.
Mapping orthologs across mammalian genomes
We used Jim Kent’s BLASTZ-based, whole-genome pairwise alignment chains (22) to map—when possible with high confidence—each reference gene to a single orthologous position in each of the 58 query species (Figure 1A). First, we assigned every exonic (CDS+UTR) base in the canonical transcript of the reference gene to the highest-scoring chain (in terms of UCSC chain alignment scores) that contains the base in its alignment (Figure 1B1– B2). The chain to which most number of exonic bases get assigned was picked as the best chain (Cb) for that gene (Figure 1B3). Since chains are preferentially ordered based on the alignment score before assigning exonic bases (Figure 1B1), the best chain was also the chain with highest conservation of synteny for those aligning bases, and was treated as the most likely ortholog for that gene. Next, this procedure was repeated after removing the best chain (Cb) from consideration to pick another chain as the second-best chain (Csb) for the same gene (Figure 1B4). This chain Csb was treated as containing a likely paralog of the gene. To ensure that the orthologous chain was easily differentiable from paralogs, we required the second-best chain to have an alignment score at least 20 times lower than that of the best chain. To also ensure high conservation of synteny , we required the number of bases in the aligning blocks of the best chain be at least 20 times greater than the number of bases in the gene itself—i.e. gene-in-synteny ≥ 20, where gene-in-synteny = length of Cb/length of gene. We also required unique mapping of coordinates between reference and query genomes, such that if two or more reference genes were mapped to the same query location, all overlapping mappings were discarded.
Figure 1.

Chain-picking method to map genes from a reference genome to an orthologous locus in query species genome. (A) We designed a conservative gene-by-gene scanning approach that maps genes from a reference genome (here human, GRCh38) to their orthologous locations in target mammalian genomes based on pairwise genome chain alignments. For each reference gene, our approach first picks two chains: the best chain (Cb) and the second-best chain, Csb (panel B). The chain Cb is then assigned to a gene g if (i) it has a significantly better alignment score relative to the other chain Csb so that the orthologous region can be unambiguously distinguished from remaining paralogs, (ii) it is much larger than the coding size of g such that it ensures sufficient conservation of synteny for the flanking genomic context, and (iii) it maps g to a locus in the target genome that no other gene g′ is mapped to, so as to ensure 1-to-1 mapping of genes to query locus. (B) Detailed illustration of the procedure to pick the best-chain (Cb) and the second-best chain (Csb) for gene g. In the case of multiple chains at a locus (gene, g), our procedure first assigns each exonic (CDS+UTR) base of g to the highest-scoring chain overlapping with the base-pair, and then identifies the chain (Cb) to which the most exonic bases of g were assigned. The procedure is repeated to identify another chain (Csb) after Cb is hidden. (C) SERPINA10 is a known loss of function gene in dogs (34), and hence lacks any annotations or orthology mapping from human to dog. This pairwise, chain-based approach enables precise mapping of an intact gene from a reference assembly to sequence-eroded loci in another species.
In each species, for every gene to which an orthologous chain was assigned, we used the alignments derived from the chain to determine the orthologous amino acid sequence for every transcript of that gene. Because exon boundaries sometimes shift for evolutionary or alignment reasons, we excluded the first and last two amino acid positions in each exon for all downstream analyses (23). Gaps in chain alignments found in regions containing an assembly gap in the query species were masked out to avoid confusing them with true deletions in the query genome. Note that, while UCSC genome browser views of multiple sequence alignments are shown for compact visualization in the Results section, only pairwise alignments were used for the underlying computation described here.
Outlier detection model
To identify the genes orthologs that have a surprisingly high number of amino acid substitutions and deletions in each of the 58 query species, we used an outlier detection approach. We first used the derived amino acid sequence of each transcript for every gene in a query species, as described above, to compute a feature vector 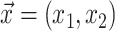 , where x1 is the fraction of the transcript amino acids deleted in the query species and x2 is the fraction of non-synonymous amino acid substituted in the remaining portion of the gene transcript compared to the orthologous gene transcript sequence in the reference species (human).
, where x1 is the fraction of the transcript amino acids deleted in the query species and x2 is the fraction of non-synonymous amino acid substituted in the remaining portion of the gene transcript compared to the orthologous gene transcript sequence in the reference species (human).
We then used 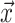 across all genes in the query species to compute the mean vector,
across all genes in the query species to compute the mean vector, 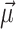 , and a covariance matrix, S. Mahalanobis distance (MD) (24,25) measures how ‘far’ a given gene with feature vector,
, and a covariance matrix, S. Mahalanobis distance (MD) (24,25) measures how ‘far’ a given gene with feature vector, 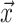 , is from the distribution of all genes in the query species, as follows:
, is from the distribution of all genes in the query species, as follows:
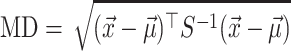 |
(1) |
Since most genes exhibit relatively few non-synonymous substitutions and deletions (as placental mammals are relatively closely related, see Figure 2B), a large value of MD implies extensive erosion of the gene. Conversely, a small value of MD implies that the gene is well-conserved. Since the coding sequences are expected to diverge more with increasing phylogenetic distance between reference and query species, we computed a different distribution (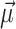 and S) for each query species. If all transcripts of an ortholog had MD greater than an absolute threshold (MD > 15), we marked the gene an outlier (✗) since it is likely sequence-eroded and nonfunctional in the species represented by the query genome. If all transcripts of an ortholog exhibited MD ≤ 5, we regarded the gene sequence-conserved and functional (✓) in the species represented by the query genome. All other orthologs that were not categorized into either of the former two groups were marked with undetermined (?, see Figure 2C).
and S) for each query species. If all transcripts of an ortholog had MD greater than an absolute threshold (MD > 15), we marked the gene an outlier (✗) since it is likely sequence-eroded and nonfunctional in the species represented by the query genome. If all transcripts of an ortholog exhibited MD ≤ 5, we regarded the gene sequence-conserved and functional (✓) in the species represented by the query genome. All other orthologs that were not categorized into either of the former two groups were marked with undetermined (?, see Figure 2C).
Figure 2.

A pipeline to call hcoErosions. (A) Using pairwise chained alignments, we determined an orthologous location for human coding genes in each genome (Figure 1). Then we computed the amount of amino acid deletion and substitution to find, for each each genome, the outlier genes among all the genes that were mapped (i.e., likely eroding genes). (B) Scatter plot showing the distribution of the mouse (mm10) genes along the two features (% amino acids substituted and % amino acids deleted) for calculating Mahalanobis Distance (MD). The histogram along each axis shows the distribution of points when projected on the respective axis; for example, the peak in the histogram parallel to the y-axis illustrates a high density of overlapping points abutting the x-axis. Per-species likely eroded genes (in red, such as known losses of Cetp and Eys) have much higher values of % amino acids substituted and % amino acids deleted compared to intact (in green) and undetermined genes (in black). (C) Histogram of the MD values for mouse genes along with the threshold values (dashed vertical lines) for conserved (✓, MD ≤ 5), undetermined (?), and likely eroded (†, MD > 15) genes. (D) An example illustration of applying phylognetic filtering. We call hcoErosion (×, e.g. in mouse and squirrel) if a gene is marked likely eroded (MD > 15) in more than one species in a clade that is flanked by at least one group in which the gene is conserved (✓). An undetermined gene in an individual species (?, e.g. in rat) is inferred as eroded if it resides in a clade with hcoErosion. The likely eroded (†) gene in cow, however, is not considered a hcoErosion since the finding is not supported by another adjacent species.
Phylogenetic filtering
We used the phylogenetic tree of the 59 mammalian species (including human; Supplementary Figure S1) to identify which of the outlier orthologs would be considered to have undergone hcoErosion (Figure 2D illustrates this procedure). For each gene, we recorded which species exhibit a sequence-conserved ortholog (based on the results of the outlier detection model described above; also see Figure 2B, C). Since the gene under consideration is already annotated to be functionally present in hg38 by Ensembl, we inferred by parsimony that the last common ancestor of human and of all species containing the conserved gene likely had a functional copy of this gene with a sequence similar to that of its extant functional orthologs. Finally, we search all subtrees starting from the earliest ancestral species marked in this fashion with gene conservation to identify specific groups where hcoErosion has occurred. A group in which hcoErosion has occurred (Figure 2D) is one in which there are at least two species with the gene marked as likely eroded (all transcripts with MD > 15) and no leaf-node species with the gene marked as sequence-conserved (MD ≤ 5).
Curation of UniProt data and details of FPR (False Positive Rate) estimation in mouse and rat
To measure the rate of false positives in hcoErosion predictions for mouse and rat, we obtained experimentally curated coding gene models downloaded as protein sequences with ‘protein level’ or ‘transcript level’ evidence from UniProt (18) (release-17_01; ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete). We found 16 385 and 7591 gene models in mouse and rat, respectively. We then mapped those amino acid sequences to their respective genomes (mouse sequences to mm10; rat sequences to rn6) using protein BLAT (26) with default settings (minimum mapping score 30; minimum sequence identity 25; maximum intron size 750 000). After mapping these protein sequences, we computed what proportion of orthologs predicted to be affected by hcoErosion is contradicted by empirical evidence. Specifically, for a given species and its genome, any predicted hcoErosion-affected orthologs that deviate by >20% in sequence length, share <90% identity, or disagree with the locus of the relevant UniProt sequence were considered false predictions. Lastly, since we had 17 860 tested proteins in the reference gene set, we conservatively scaled up the number of conflicts in mouse and rat by a factor of 1.09 (=17 860/16 385) and 2.35 (=17 860/7591), respectively, to estimate our false positive rate (FPR) while accounting for potentially missing UniProt entries.
Curation of lethal loss-of-function genes in mouse models
We used gene function annotations from the MGI Phenotype Ontology (Mouse Genome Informatics, http://www.informatics.jax.org/) to derive a subset of genes whose disruption leads to severe developmental perturbations and lethal consequences (27). The MGI Phenotype Ontology catalogs spontaneous, induced, and genetically-engineered mouse mutations and their associated phenotypes. Ontology data (containing 8949 phenotypic terms; v6.04) were lifted over from mouse to human, resulting in 609 253 (canonicalized) gene-phenotype associations. We selected all 50 unique phenotypic terms (Supplementary Table S2) with the string ‘lethal’ in their description (e.g., MP:0008762 embryonic lethality; MP:0008569 lethality at weaning; MP:0006206 embryonic lethality between somite formation and embryo turning), identifying a total of 3617 genes associated with causing early lethality when disrupted.
Analysis of hcoErosion genes implicated in HGMD
The professional version (16.2) of the Human Gene Mutation Database (28) contains 165 939 entries of known disease-causing gene mutations (referenced to GRCh38 assembly) and their respective phenotypes. We obtained the list of 4014 monogenic disease genes (i.e. genes with at least one ‘DM’ mutation) from this database, of which 3711 genes were also present in our reference gene set.
Analysis of genic variation intolerance with RVIS and pLI scores
Residual Variation Intolerance Score (RVIS) is a unified genome-wide metric that quantifies the extent to which genes tolerate coding mutation load (i.e. the higher the score, the more a gene can tolerate common genetic variation without negative consequences) (29). We used the pre-computed RVIS percentiles based on Exome Aggregation Consortium variants with Minor Allele Frequency 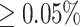 (http://genic-intolerance.org/data/RVIS_Unpublished_ExACv2_March2017.txt) to identify hcoErosion genes with scores in the bottom quintile likely to be intolerant to functional variation in humans. We performed a two-tailed Mann–Whitney U test to assess whether there is a significant difference between the RVIS percentile distribution of hcoErosions compared to the reference gene set. We also screened for any additional hcoErosion genes that have a high probability of being loss-of-function intolerant (pLI) score of ≥0.9 using publicly available data (https://storage.googleapis.com/gnomad-public/release/2.1.1/constraint/gnomad.v2.1.1.lof_metrics.by_gene.txt.bgz); we used the threshold of ≥0.9 per the convention established by pLI score developers Lek et al. for flagging genes that are very likely intolerant to functional variation (30).
(http://genic-intolerance.org/data/RVIS_Unpublished_ExACv2_March2017.txt) to identify hcoErosion genes with scores in the bottom quintile likely to be intolerant to functional variation in humans. We performed a two-tailed Mann–Whitney U test to assess whether there is a significant difference between the RVIS percentile distribution of hcoErosions compared to the reference gene set. We also screened for any additional hcoErosion genes that have a high probability of being loss-of-function intolerant (pLI) score of ≥0.9 using publicly available data (https://storage.googleapis.com/gnomad-public/release/2.1.1/constraint/gnomad.v2.1.1.lof_metrics.by_gene.txt.bgz); we used the threshold of ≥0.9 per the convention established by pLI score developers Lek et al. for flagging genes that are very likely intolerant to functional variation (30).
Statistical significance of depletion
To estimate statistical significance of depletion (31) for the hcoErosion group of genes relative to categories such as mouse lethal genes or HGMD disease genes or genes with pLI ≥0.9, we used the Fisher exact test to compute a one-sided P-value:
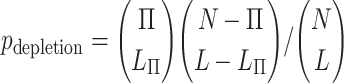 |
(2) |
In the above equation, N and L denote the total number of genes and the hcoErosion subset of genes in the analysis, respectively. Similarly, Π and LΠ denotes the subset of N and L associated with a gene category such as HGMD disease genes or mouse lethal genes.
We also compute a ‘fold change’ for depletion as 1/Foldenrichment:
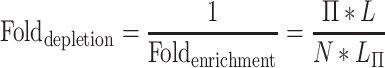 |
(3) |
Tissue and pathway enrichment of hcoErosion genes
To determine if expression of the 412 unique hcoErosion genes might be enriched in particular tissues, we used the TissueEnrich tool (32), providing the hcoErosion gene names as input. To determine if the 412 unique hcoErosions are enriched in biological functions and/or compartments, we used the GREAT v4.0.4 tool (33), providing the Ensembl 86 human canonical transcription start sites as input, considering terms annotating ≥30 and ≤1000 genes, and using the ’basal plus extension’ association rule. We report all tested ontology terms with a hypergeometric FDR Q-value <0.05 and fold enrichment ≥2.
RESULTS
Pre-existing gene annotation methods fail to identify and determine the functional state for thousands of orthologs
Of the 59 mammalian genomes (including the human reference) that we used in our study, Ensembl gene predictions (release 86) were available for 34 genomes. The number of protein-coding gene annotations varied widely (Figure 3A), from 11 765 annotations in alpaca to 22 285 annotations in human. When plotting the number of Ensembl predicted genes against the assembly N50, it is clear that the annotation completeness is artifactually dependent on the level of assembly contiguity (Figure 3A). For instance, absence of the critical chordate development gene PAX6 in the dog annotation set is likely due to a sequencing gap in the genome assembly (Figure 3B). Similarly, while TP53 and multiple adjacent genes are absent from the alpaca annotation set, its genome includes a region with high sequence similarity to the human TP53 ortholog (Figure 3C), implying a potentially missed prediction. Labour-intensive, manual pseudogene annotation pipelines are similarly incomplete, tackling a handful of species and still working on key species pseudogene sets (https://www.gencodegenes.org/). Based on these observations, we devised an improved approach for identifying a high-confidence, conservative subset of gene loss events across the 58 placental mammals, using human as the reference.
Figure 3.

Incomplete coding gene annotations in genomic databases. (A) Of the 59 mammalian genomes used in this study, 34 have Ensembl (release 86) gene annotations. The number of gene annotations per genome correlates with assembly quality (as measured by N50 contig length), suggesting that gene loss should not be inferred from a missing ENSEMBL ortholog in a lower N50 genome. (B) Top: pax6 Ensembl gene model artefactual absence (gray highlight) in the dog genome assembly due to a sequencing gap. The genome browser view shows the minus strand. Bottom: The orthologous position of PAX6 (and flanking genes) in the human genome. (C) Top: TP53 Ensembl gene model absence (gray highlight) in the alpaca genome within a conserved region.The genome browser view shows the minus strand. Bottom: The orthologous position of TP53 (and flanking genes) in the human genome.
Mapping gene orthologs across mammalian genomes
To identify ab initio orthologs of human genes across the 58 query species, we used whole-genome pairwise alignments (i.e. Jim Kent’s BLASTZ-based chains (22)) to map genes from human coordinate space to each of the other genomes. Chains identify conserved sequences not only in coding regions (covering 2% of the human genome) but also in non-coding, gene-regulatory regions (covering 5–10% of the human genome) in which the genes are interspersed. As such, chains facilitate accurately determining genomic orthology between species. Using these 58 pairwise alignment chains anchored to the human reference, we determined the single locus in the aligning (query) genome that exhibited the greatest sequence similarity and conservation of synteny with each human protein-coding gene. Specifically, each successfully mapped gene was (i) positioned within a chain that had alignment and gene-in-synteny scores sufficiently higher than the next-highest-scoring (likely paralogous) chain and (ii) did not overlap in genomic space with any other gene mappings proposed in the previous step (see Materials and Methods, Figure 1A, B). In total, our mapping procedure identified 11 155–17 690 orthologs per species (median: 16 782; Figure 2A) across the 58 non-human mammals considered.
Importantly, this chain-based mapping approach worked correctly for previously known gene losses, accurately identifying pseudogenized orthologs with extensive sequence erosion compared to the ancestral gene. For example, much of the exonic portions of SERPINA10 are absent in the dog genome and do not yield a functional gene product (34). As such, Ensembl does not offer a corresponding gene annotation in the dog genome assembly (canFam3), which would leave ambiguity about the existence and function this ortholog in dogs if not for experimental evidence. Our approach, however, precisely located the remnants of SERPINA10 in the canFam3 assembly and allowed for assessing the semantic status (reading frame integrity and occurrence of substitution, insertion, and deletion) of the remaining chain-mapped exons (Figure 1C).
Per-species identification of likely eroded orthologs using a Mahalanobis distance outlier model
Because functional gene inactivation will be accompanied by relaxation of purifying selection on all genomic regions encoding the gene, we used the extent of coding sequence erosion to declare the loss of gene function. In particular, to account for mutation rate heterogeneity across different branches of the mammalian tree, we restricted our results to orthologs that appear extremely eroded (using outlier detection) in sequence relative to all other mapped genes in the same given species. Unlike Sharma et al. (10), who used single-event features, such as in-frame stop codons and frameshift indels, we considered two different aggregate features of sequence erosion—i.e. the fraction of amino acids affected by substitutions and the fraction of exonic amino acids out-right deleted with respect to the sequence of the human ortholog. This way, we avoided making spurious outlier calls caused by some mutations, such as stop codons near the end of a gene or multiple frameshift indels that restore the original frame, which do not necessarily lead to loss of the ortholog’s function. Additionally, before declaring outliers, we masked out (i.e. did not consider) alignment gaps that could have resulted from sequencing gaps in the target species (see Materials and Methods).
For outlier detection, we used a Mahalanobis distance (MD) (24,25) based model (see Materials and Methods) to mark a small fraction of the orthologs (typically 1–2% or 120–284 genes per species, median of 192; Figure 2A) as outliers for their extreme fraction both of amino acid substitutions and deletions. We considered these outlier orthologs to be likely eroded in sequence and function in the species represented by the given genome assembly. For instance, by performing these methods on the 17 836 genes confidently mapped to the mm10 mouse assembly (Figure 2B), we correctly identified previously discovered gene losses for Cetp (35) and Eys (36) (plus other novel predictions) as outliers (MD > 15), compared to uneroded, relatively sequence-conserved orthologs (MD ≤ 5), including genes Tp53 (37), Notch1 (38) and Sox11 (39) (Figure 2B, C) with indispensable function in mice.
Phylogenetic filtering to identify high-confidence ortholog erosions in multiple related species
Two caveats must be considered for the per-species gene erosion predictions: Firstly, not all genes in the human reference set were necessarily present in functional form in the common ancestor shared by human and the species under consideration. Sequences in other species that have some resemblance (such as paralogs) to a functional human gene, and that appear eroded, but were never present/functional in their common ancestor with human, could be confounded for gene erosion. Secondly, the erosion could be real but private to the single sequenced individual (i.e. not fixed in the species at-large).
To address these caveats, we applied strict phylogenetic filters to restrict the final candidate list to the highest-confidence predictions (Figure 2D). First, we required a final candidate to have an uneroded, sequence-conserved ortholog (MD ≤ 5) in at least one other mammal in addition to human such that the eroding species is internal to the subtree spanned by human and the other sequence conserving species. With the assumption of parsimony, this first step ensures that the candidate ortholog was present, of functional importance, and under purifying selection in the common ancestor of human and the eroding species. Second, even more importantly, we also required that each final candidate was considered likely eroded in two or more phylogenetically neighboring species (such as rat/mouse or manatee/elephant/cape elephant shrew) sharing a common ancestor with human in which the gene was declared conserved. Doing so minimized false positives from gene losses that are real but not fixed in a species, as well as those resulting from assembly, alignment, and mapping artifacts. We designated the final candidates as high-confidence ortholog erosions or hcoErosions.
In all, we identified 412 genes that were eroded in one or more multi-species groups , yielding 2192 and 539 unique gene-species and gene-clade pairs, respectively (Figure 2A, Supplementary Table S3). Our hcoErosion predictions include many previously known functional losses such as Cetp (35), Eys (36), and Tcn1 (40) in mouse, ZP1 in bovine genomes (41), CRYGB in subterranean mammals (5), GSDMA in whales (10), and RHBG in fruit bats (10). The number of hcoErosions per species ranged from 5 in aardvark to 88 in brush-tailed rat and chinchilla (median of 41, Figure 4A).
Figure 4.

Distribution of hcoErosions across species and estimation of False Positive Rate (FPR). (A) Number of hcoErosions predicted in different species. (B) If hcoErosions are directly inferred by applying our phylogenetic filtering (Figure 2D) to missing gene annotations in Ensembl (instead of eroded genes from Figure 2C), the estimated FPR (red bars) in mouse and rat is significantly higher than in our set of predicted hcoErosions.
To estimate the false positive rate (FPR) for these results, we tabulated how many predicted hcoErosions for mouse and rat were contradicted by proteomic or even transcriptomic evidence from UniProt (18) (see Methods). We found 6/68 (8.82%) and 2/68 (2.94%) hcoErosion predictions in mouse and rat conflicting with UniProt evidence, respectively (Supplementary Table S4). We conservatively re-scaled the conflicts to account for potentially missing UniProt entries but still found the FPR estimate to be <10% in both species (Figure 4B, see Materials and Methods). In contrast, if we applied our phylogenetic filtering (Figure 2D) directly to the orthologs lacking Ensembl annotations for mouse and rat, the FPR for both species would be >30% (Figure 4B), highlighting the importance of our orthologous chain mapping and outlier detection approach.
Selection and variation in mammalian genes exhibiting multi-species ortholog erosion
We found that our final hcoErosion predictions (Supplementary Table S3), containing 412 unique human orthologs are depleted of genes causing lethality when inactivated in developing mice (P-value < 2.2e–16; fold = 8.43; Figure 5A), of known human HGMD disease-causing genes (P-value < 1.3e–11, fold = 3.17; Figure 5B), and of those with RVIS and pLI scores predicted to be intolerant to functional variation in humans (Figure 5C, P-value < 4.1e–24, fold = 19.94 and Figure 5D, P-value < 2.7e–35). Despite this depletion, 10 hcoErosions do involve genes whose knockout mouse models do not survive beyond early development (Table 1, see Methods). Likewise, 27 hcoErosions involve HGMD (28) genes implicated in severe human congenital disorders (Table 2, see Materials and Methods). Furthermore, three hcoErosion genes are pLI-predicted to be deleterious in human patients when mutated (Table 3, see Materials and Methods) (30). Using the Residual Variation Intolerance Score (RVIS) (29), which is a gene-based measure of intolerance to common genetic variation (with minor allele frequencies greater than 0.05%), we also found six genes predicted to be affected by hcoErosion that have an RVIS in the bottom quintile of all scored human genes (patients when mutated, see Materials and Methods).
Figure 5.

Selection and variation in mammalian genes affected by hcoErosion. hcoErosions are depleted of (A) genes whose inactivation in mouse models leads to lethality in early development; (B) genes with known Mendelian disease mutations (HGMD genes); and (C) genes with pLI scores ≥0.9. (D) We compare the genome-wide distribution of Residual Variation Intolerance Score (RVIS) percentiles to those of genes affected by hcoErosion. By and large, hcoErosion genes are more tolerant of common functional variation in humans, suggesting that selection over coding genes is similar across mammalians. Several genes at the lower tail of the RVIS percentiles are highlighted (see Table 3 for details).
Table 1.
Genes implicated in mammalian hcoErosions (high confidence ortholog erosions) that result in lethality when inactivated in mouse models
| Gene | Mouse knockout phenotype | Species with gene hcoErosion |
|---|---|---|
| CA5B | Preweaning lethality, incomplete penetrance | Killer whale, dolphin |
| CCDC94 | Embryonic lethality | Black flying-fox, megabat, microbat, big brown bat |
| David‘s myotis bat | ||
| DMBT1 | Embryonic lethality | (I) Naked mole-rat, brush-tailed rat, guinea pig, chinchilla |
| (II) Goat, sheep, Tibetan antelope, cow | ||
| HAP1 | Lethality during fetal growth through weaning | Naked mole-rat, brush-tailed rat, guinea pig, chinchilla |
| MESP1 | Embryonic lethality | Naked mole-rat, brush-tailed rat, guinea pig, chinchilla |
| MLKL | Embryonic lethality | Cat, dog, ferret, panda, pacific walrus, Weddell seal, horse, white rhinoceros |
| PKD1L1 | Lethality throughout fetal growth and development | Alpaca, Bactrian camel, killer whale, dolphin, goat, sheep, Tibetan antelope, cow |
| RESP18 | Prenatal lethality | Microbat, David's myotis bat |
| SERPINA10 | Lethality, incomplete penetrance | (I) Cat, dog, ferret, panda, pacific walrus, Weddell seal |
| (II) Rabbit, pika | ||
| ZNF565 | Embryonic lethality | Pacific walrus, Weddell seal |
(I), (II) Independent hcoErosion loss events: (I) species belonging to group I (II) species belonging to group II.
Table 2.
Genes implicated in mammalian hcoErosions that harbor monogenic disease variants in human patients (disease mutations in HGMD (28))
| Gene | Associated human monogenic disease | Species with gene hcoErosion |
|---|---|---|
| A2ML1 | • Noonan syndrome | Naked mole-rat, brush-tailed rat, guinea pig, chinchilla |
| • Otitis media (susceptibility to) | ||
| CETP | • Higher/Lower HDL cholesterol level | (I)Bactrian camel, alpaca |
| • Hyperalphalipoproteinaemia | (II)Goat, sheep, Tibetan antelope, cow | |
| • Cholesterol ester transfer protein deficiency | ||
| CRYBB3 | • Cataract | Microbat, big brown bat, David‘s myotis bat |
| CRYGB | • Congenital cataract | Cape golden mole, tenrec |
| EYS | • Retinitis pigmentosa | Mouse, rat, Chinese hamster, golden hamster, prairie vole |
| • Retinal dystrophy | ||
| • Peripheral dystrophy | ||
| • Usher syndrome | ||
| FCN2 | • FCN2 deficiency | Goat, sheep, Tibetan antelope, cow |
| GCKR | • Elevated HDL-cholesterol | Microbat, big brown bat, David‘s myotis bat |
| • Hypertriglyceridaemia | ||
| HMGCS2 | • Mitochondrial HMG-CoA synthase deficiency | Black flying-fox, megabat |
| HSD3B2 | • 3 beta-hydroxysteroid dehydrogenase deficiency | Goat, sheep, Tibetan antelope, cow |
| • Adrenal hyperplasia | ||
| • Hypospadias, non-syndromic | ||
| • Idiopathic hypospadias | ||
| • Pseudohermaphroditism | ||
| KLKB1 | • Prekallikrein deficiency | Dolphin, killer whale |
| KRT3 | • Corneal dystrophy, Meesmann | Mouse, rat |
| MESP1 | • Tetralogy of Fallot | Naked mole-rat, brush-tailed rat, guinea pig, chinchilla |
| • Ventricular septal defect | ||
| MMP21 | • Heterotaxy | Pig, dolphin, killer whale, Bactrian camel, alpaca, goat, sheep, Tibetan antelope, cow |
| MYD88 | • MYD88 deficiency | Brush-tailed rat, chinchilla |
| OPTC | • Glaucoma, primary open angle | Cape golden mole, tenrec, microbat, big brown bat, David‘s myotis bat |
| PCSK9 | • Hypercholesterolaemia | (I)Cat, dog, ferret, panda, pacific walrus, Weddell seal |
| • Low/High LDL cholesterol | (II)Goat, sheep, Tibetan antelope, cow | |
| PLA2G2A | • Colorectal cancer | Dolphin, killer whale |
| RIPPLY1 | • Intellectual disability | Black flying-fox, megabat |
| SFXN4 | • Colorectal cancer | (I)Chinese hamster, golden hamster, prairie vole |
| • Mitochondriopathy and macrocytic anaemia | (II)Naked mole-rat, brush-tailed rat, guinea pig, chinchilla | |
| STAP1 | • Hypercholesterolaemia | Dolphin, killer whale |
| TCN1 | • Transcobalamin I deficiency | Mouse, rat |
| TNFRSF6B | • Systemic lupus erythematosus | Mouse, rat, Chinese hamster, golden hamster, prairie vole |
| UMOD | • Chronic kidney disease | Microbat, big brown bat, David‘s myotis bat, black flying-fox, megabat |
| • Congenital anomalies of the kidney and urinary tract | ||
| • FJHN/MCKD syndrome | ||
| • Glomerulocystic kidney disease | ||
| • Uromodulin-associated kidney disease | ||
| • Hyperuricaemic nephropathy | ||
| XAF1 | • Multiple sessile serrated adenoma | Cat, dog, ferret, panda, pacific walrus, Weddell seal |
| ZNF543 | • IgA nephropathy | Cat, dog, ferret, panda, pacific walrus, Weddell seal |
| ZNF589 | • Intellectual disability | Ferret, panda, pacific walrus, Weddell seal |
| ZP1 | • Infertility | Pig, dolphin, killer whale, Bactrian camel, alpaca, goat, sheep, Tibetan antelope, cow |
(I), (II) Independent hcoErosion loss events: (I) species belonging to group I (II) species belonging to group II.
Table 3.
Mammalian hcoErosions that either have an RVIS (Residual Variation Intolerance Score) in the bottom quintile of all human genes analyzed and/or have a pLI (probability of being loss-of-function intolerant) score ≥ 0.9 (✓)
| Gene | RVIS percentile | gnomAD pLI score ≥ 0.9 | Species with hcoErosion |
|---|---|---|---|
| UBC | 4.19% | (I) Hedgehog, Star-nosed mole, Shrew | |
| (II)Naked mole-rat, Guinea Pig, Chinchilla, Brush-tailed rat | |||
| (III)Ferret, Panda, Pacific walrus, Weddell seal | |||
| (IV)Alpaca, Bactrian camel | |||
| DDX60 | 6.02% | Tibetan antelope, Sheep, Domestic goat, Cow | |
| PCDHA7 | 10.27% | Panda, Weddell seal, Ferret, Pacific walrus | |
| ADAM30 | 10.78% | N/A | Guinea Pig, Brush-tailed rat, Naked mole-rat, Chinchilla |
| ZYG11B | 14.34% | ✓ | Rhesus macaque, Crab-eating macaque |
| PPP2R2D | 18.43% | ✓ | Tibetan antelope, Sheep, Domestic goat, Cow |
| DCAF12L1 | 22.73% | ✓ | Naked mole-rat, Guinea Pig, Chinchilla, Brush-tailed rat |
(I), (II), (III), (IV ) Independent hcoErosion loss events:
(I) species belonging to group I (II) species belonging to group II (III) species belonging to group III (IV) species belonging to group IV.
Erosion of GCKR in microbats may facilitate their high-fat insectivorous diet
Microbats and megabats, the two primary suborders of bats, have vastly different diets. Microbats have extremely fat-rich, insect-based diets (42), while megabats have fruit-based diets depleted of fat (43). We found that GCKR (Glucokinase regulatory protein; OMIM: 600842), variants of which are associated with elevated high-density lipoprotein (HDL) cholestrol in human (44), is specifically eroded in microbats (see Figure 6 and Table 2). This potentially provides a genotypic basis for an observation by Widmaier et al. (45) who found that HDL-cholestrol levels in a lactating microbat species (Tadarida brasiliensis mexicana) were 10-fold higher in comparison to three species of megabats. Specifically, they proposed that these extraordinarily high HDL levels, possibly facilitated by GCKR erosion, could explain why microbats are protected from atherosclerosis and other common diseases associated with high-fat intake.
Figure 6.

GCKR hcoErosion in microbats. A genome browser view of GCKR (Glucokinase regulatory protein) on the human genome (GRCh38) and a mammalian multiple alignment highlight coding lesions in microbat. Positions of deleted genomic fragments (black) and non-synonymous substitutions (green) are highlighted for David’s myotis and big brown bat (both of microchiroptera). GCKR loss could explain why microbats are protected from common diseases associated with high-fat intake.
Erosion of PKD1L1 and MMP21 observed in artiodactyla (even-toed ungulates) but causes prenatal lethality in mice and congenital heart defects in humans
Pkd1l1 (polycystin kidney disease protein 1-like 1) null mutations in mice result in a high rate of embryonic lethality (46) (Table 1) characterized by misregulated nodal signaling (47) and anomalous Left–Right (L–R) symmetry patterning (48,49) (Figure 7A). Indeed, humans with homozygous loss of function in PKD1L1 (OMIM: 609721) present with laterality defects ranging from Situs Inversus Totalis to heterotaxy and with congenital heart disorders, all of which can be deleterious early in life (50). As such, it was remarkable to find PKD1L1 hcoErosion, with deletion of as much as 98% of coding bases in some species, in a clade of nine even-toed ungulates (including cetaceans, see Figure 7B). Congruent with this finding, an Ensembl annotation for PKD1L1 does not exist for the cow, sheep, dolphin, alpaca and pig genomes, and we detected no orthology between human PKD1L1 to any other gene in these genomes. In contrast, we found high sequence homology for the human protein sequence of PKD1L1 in the genomes of outgroup lineages such as horse and elephant, suggesting that the gene is intact in those species, and lost in artiodactyla (even-toed ungulates).
Figure 7.

PKD1L1 and MMP21 hcoErosions in even-toed ungulates. (A) PKD1L1 (Polycyctic kidney disease protein 1-like 1) and MMP21 (Matrix Metalloproteinase 21) are both related to the Nodal signaling pathway, and null mutations in either gene results in severe laterality disorders with congenital heart defects. (B) A genome browser view of PKD1L1 (on the human genome, GRCh38) and a mammalian multiple alignment highlight a clade-specific lesion of the gene in 9 even-toed ungulate species. Positions of deleted fragments (black) and non-synonymous substitutions (green) are highlighted for cow and killer whale. (C) MMP21 has undergone hcoErosion in the same ancestor. Positions of deleted fragments (black) and non-synonymous substitutions (green) are highlighted for cow and dolphin. The morphological defect Double Outlet Right Ventricle has significantly higher incidence in cattle with congenital heart disorder relative to human and other animals with this condition (55) (perhaps because hcoErosion of both PKD1L1 and MMP21 naturally occurs in artiodactyla but not in other mammals).
Interestingly, we also discovered a related hcoErosion in the same group of even-toed ungulates (including cetaceans) affecting the gene MMP21 (Matrix Metalloproteinase 21; OMIM: 608416; Figure 7C) that encodes a component of Nodal signaling (50) and is also implicated in heterotaxy (51) and in congenital heart disorders (52,53) (Figure 7 A). Interestingly, Double Outlet Right Ventricle—a cardiac disorder associated with mutation in mouse Mmp21 (54) and with mutation in human PKD1L1 (50)—has significantly higher incidence in cattle with congenital heart disorder relative to human and other animals with this condition (55) (presumably because hcoErosion of bothPKD1L1 and MMP21 naturally occurs in artiodactyla but not in other mammals).
Erosion of endocrine regulator GPRC6A evolved in odontoceti (toothed whales) but is associated with testicular insufficiency in humans and mice
GPRC6A (OMIM:613572) is a G protein-coupled receptor that plays an important role in metabolic and endocrine regulation (56). Highly expressed in Leydig cells of the testis (in addition to many other tissues), GPRC6A is activated by testosterone, osteocalcin, basic amino acids, and various cations (57,58). In mouse models, Gprc6a ablation results in dramatic decrease of testosterone levels, smaller testis size, and reduced sperm count (59). Similarly in humans, loss-of-function mutations in GPRC6A are associated with testicular insufficiency (subfertility, altered sperm parameters, low circulating testosterone levels, and high circulating luteinizing hormone levels) (60). A number of studies also found that GPRC6A is associated with prostate cancer progression and could serve as a potential drug target for this malignancy (58,61).
GPRC6A is a highly conserved gene across mammals and even has orthologs in species as distant as zebrafish (Figure 8), suggesting it may have arisen early in vertebrate evolution. It is highly remarkable then, that we observed hcoErosion of GPRC6A in dolphin and killer whale—where over 50% of amino acids are deleted in both species (including the entirety of exons 5 and 6), and the remaining regions contain several frameshift indels and nonsense mutations (Figure 8). The gene is well-conserved in outgroup bovidae (domestic goat, sheep, Tibetan antelope, and cow), suggesting it was functional in the cetartiodactyla ancestor (superorder of odontoceti and bovidae) but was inactivated and then allowed to erode in the evolutionary past of toothed whales. GPRC6A erosion in toothed whales is especially striking when considering that toothed whales exhibit 7–25 times greater testis-to-body mass ratio relative to other mammals (62). Alternatively, it is notable that Gprc6a-null mice also exhibit overall higher body fat percentage (59), which could offer a hypothesis for how blubber evolved in cetaceans.
Figure 8.

GPRC6A hcoErosion in toothed whales. A genome browser view of GPRC6A (on the human genome, GRCh38) and a mammalian multiple alignment highlight coding lesions in dolphin and killer whale. Positions of deleted fragments (black) and non-synonymous substitutions (green) are shown for both species. GPRC6A is a key hormonal and metabolic regulation protein, and the knockout of GPRC6A in male mice results in over 6-fold decrease of testosterone levels and reduced size and weight of the testis (59). Notably, toothed whales, like dolphin and killer whale, exhibit 7–25 times greater testis-to-body mass ratio relative to other mammals despite hcoErosion of GPRC6A (62).
DISCUSSION
In this work, we devised a novel approach to automate the survey of genomes for instances of coding gene sequence erosion. Knowledge of gene losses has far-reaching applications in studying the morphological and physiological adaptations of different species (10), in discovering alternative molecular pathways for critical genes (2), in medical genetics (13) and even in animal conservation programs (12). Our approach is both fully automated and includes steps to greatly minimize false positive predictions, which should appeal to downstream experimental predictions.
In particular, our conservative approach addresses a number of challenges that hindered previous attempts to automate screening. The two biggest potential confounders are mistaking paralogs for orthologs, and confusing sequencing assembly or alignment artifacts for real genomic changes. Our mapping procedure using whole-genome pairwise alignments is conservation of synteny-aware, is robust to sequencing gaps and other artifacts, and produces strictly one-to-one ortholog assignments that are not confounded by gene duplication events (paralogs) across species. Pairwise alignments for orthology mapping are more reliable than multi-sequence alignments (MSAs)—such as those generated by TBA/MULTIZ (63)—because, unlike MSAs, pairwise alignments help guarantee that the entire gene alignment maintains synteny and collinearity in both species and also allow us to ignore alignments where orthology cannot be determined with high confidence. To enrich for high-confidence gene erosion predictions, we used a Mahalanobis distance-based metric to refine the candidates to just those orthologs exhibiting extreme amounts of amino acid deletion and substitution relative to other genes in that species. To further minimize erroneous predictions, we distilled out just those candidates that are considered likely eroded in at least two closely related species but sequence-conserved in at least one other outgroup besides human.
Our method for detecting gene losses is, by design, necessarily conservative in order to maintain high accuracy and may not capture all gene loss events. For example, because our orthology mapping procedure (Figure 1) requires some portion of the reference gene to overlap at the query genome locus where the gene loss has occurred, loss events that involved (i) complete deletion of the gene region; (ii) massive rearrangement disrupting conservation of synteny or (iii) sequence decay beyond what can be detected in the whole-genome alignment will not be identified as hcoErosions. Additionally, the phylogenetic filter discards all gene losses occurring only in a single species, some of which may well be fixed in this one species. Despite this conservative strategy, our method discovered 2192 hcoErosion events across 52 species (Supplementary Table S3).
Although the false positive rate (FPR) of our hcoErosion predictions is low (<10%, see Figure 4B), various factors can still contribute to its false positives. For instance, poor sensitivity of whole genome aligners can make entire gene exons appear to be missing (deleted) in a query species (64,65), particularly for genes with a high mutation rate, such as positively selected genes. Sensitive approaches to whole genome alignments have been shown to recover missing exons (66) and may help further reduce the already low FPR for hcoErosion predictions. We provide details on hcoErosion candidates which contradicted UniProt evidence (Supplementary Table S4) in order to facilitate future work investigating and minimizing the few remaining false positive results of our pipeline.
The evolutionary mechanisms contributing to these functionally significant gene losses were likely diverse. Some may have resulted from regressive evolution (i.e. degeneration of formerly useful anatomical and physiological functions), such as the degraded OPTC vision gene (67) in nocturnal microbats or the CRYGB erosion in subterranean cape golden moles (5). Other losses may have contributed to species adaptation, such as erosion of the epithelium gene GSDMA (68,69) in cetaceans which have skin adaptations to life in the water (10). Interestingly, we also found that skin-related terms were the most over-represented when we performed tissue and pathway enrichment analysis of our 412 unique hcoErosion genes (Supplementary Table S5 and Figure S2).
Species with hcoErosion that tolerate functional changes known to cause disease in humans provide intriguing potential for medical genetics (3,13). Some of these ‘knockout experiments of nature’ may tolerate and even derive selective advantage from gene loss(es) because of factors specific to their habitat niche. Others, however, may have evolved compensatory suppressor mutations to genetically negate otherwise deleterious consequences. It is these latter cases that could be harnessed for treating human congenital disorders (3,13). For instance, despite the erosion of GPRC6A in toothed whales, these magnificent creatures have relatively massive testes for their size—the categorically opposite phenotype for this gene when inactivated in mice and humans. We posit that further examination of odontoceti could reveal complementary testis development pathways that might inform therapy for male hypogonadism.
Extension of this gene loss survey to additional or newly sequenced species across the metazoan tree of life will likely reveal more such evolutionary curiosities with great clinical potential. To that end, we emphasize the ease with which genomes can be added to analyses using our approach. Each additional genome would independently be aligned to the reference assembly—without the high computational cost of realigning all genomes, as is necessary for methods requiring multiple-sequence alignments. Thus, our approach is easily scalable for the ever-increasing number of new species being sequenced (15,70).
DATA AVAILABILITY
The source code for the automated hcoErosions pipeline is available at http://bitbucket.org/bejerano/hcoerosions.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Bejerano Laboratory, particularly M.J. Berger, K. Jagadeesh, J. Birgmeier and B. Yoo, for technical advice and helpful discussions; Hiram Clawson and the UCSC Genome Browser team for providing the mammalian alignment chains, multiple sequence alignments and technical advice; Robin Jia for his contribution to the code for extracting coding sequences from alignment chains; D. Cooper and P. Stenson for Human Gene Mutation Database (HGMD) access; and Leigh Darryl Quarles for his insights into GPRC6A function.
Notes
Present address: Yatish Turakhia, Genomics Institute, University of California, Santa Cruz, CA 95064, USA.
Contributor Information
Yatish Turakhia, Department of Electrical Engineering, Stanford University, Stanford, CA 94305, USA.
Heidi I Chen, Department of Developmental Biology, Stanford University, Stanford, CA 94305, USA.
Amir Marcovitz, Department of Developmental Biology, Stanford University, Stanford, CA 94305, USA.
Gill Bejerano, Department of Developmental Biology, Stanford University, Stanford, CA 94305, USA; Department of Computer Science, Stanford University, Stanford, CA 94305, USA; Department of Biomedical Data Science, Stanford University, Stanford, CA 94305, USA; Department of Pediatrics, Stanford University, Stanford, CA 94305, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
H. I. Chen was funded in part by a National Science Foundation Graduate Research Fellowship Program [1656518] and by a National Institutes of Health Genetics and Developmental Biology Training Program [5T32GM007790]. G. Bejerano is funded by the National Human Genome Research Institute [R01HG008742]. Funding for open access charge: NIH [R01HG008742].
Conflict of interest statement. None declared.
REFERENCES
- 1. O’Leary M.A., Kaufman S.. MorphoBank: phylophenomics in the ‘cloud’. Cladistics. 2011; 27:529–537. [DOI] [PubMed] [Google Scholar]
- 2. Albalat R., Cañestro C.. Evolution by gene loss. Nat. Rev. Genet. 2016; 17:379–391. [DOI] [PubMed] [Google Scholar]
- 3. Hiller M., Schaar B.T., Indjeian V.B., Kingsley D.M., Hagey L.R., Bejerano G.. A ‘forward genomics’ approach links genotype to phenotype using independent phenotypic losses among related species. Cell Rep. 2012; 2:817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marcovitz A., Jia R., Bejerano G.. ‘Reverse Genomics’ Predicts Function of Human Conserved Noncoding Elements. Mol. Biol. Evol. 2016; 33:1358–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Partha R., Chauhan B.K., Ferreira Z., Robinson J.D., Lathrop K., Nischal K.K., Chikina M., Clark N.L.. Subterranean mammals show convergent regression in ocular genes and enhancers, along with adaptation to tunneling. eLife. 2017; 6:e25884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Emerling C.A., Springer M.S.. Eyes underground: regression of visual protein networks in subterranean mammals. Mol. Phylogenet. Evol. 2014; 78:260–270. [DOI] [PubMed] [Google Scholar]
- 7. Jiang P., Josue J., Li X., Glaser D., Li W., Brand J.G., Margolskee R.F., Reed D.R., Beauchamp G.K.. Major taste loss in carnivorous mammals. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:4956–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McGowen M.R., Gatesy J., Wildman D.E.. Molecular evolution tracks macroevolutionary transitions in Cetacea. Trends. Ecol. Evol. 2014; 29:336–346. [DOI] [PubMed] [Google Scholar]
- 9. Strasser B., Mlitz V., Fischer H., Tschachler E., Eckhart L.. Comparative genomics reveals conservation of filaggrin and loss of caspase-14 in dolphins. Exp. Dermatol. 2015; 24:365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharma V., Hecker N., Roscito J.G., Foerster L., Langer B.E., Hiller M.. A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nat. Commun. 2018; 9:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Braun B.A., Marcovitz A., Camp J.G., Jia R., Bejerano G.. Mx1 and Mx2 key antiviral proteins are surprisingly lost in toothed whales. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:8036–8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meyer W.K., Jamison J., Richter R., Woods S.E., Partha R., Kowalczyk A., Kronk C., Chikina M., Bonde R.K., Crocker D.E. et al.. Ancient convergent losses of Paraoxonase 1 yield potential risks for modern marine mammals. Science. 2018; 361:591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Emerling C.A., Widjaja A.D., Nguyen N.N., Springer M.S.. Their loss is our gain: regressive evolution in vertebrates provides genomic models for uncovering human disease loci. J. Med. Genet. 2017; 54:787–794. [DOI] [PubMed] [Google Scholar]
- 14. Harrow J., Denoeud F., Frankish A., Reymond A., Chen C.-K., Chrast J., Lagarde J., Gilbert J.G., Storey R., Swarbreck D. et al.. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006; 7:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. of Scientists G. K.C. Genome 10K: a proposal to obtain whole-genome sequence for 10 000 vertebrate species. J. Hered. 2009; 100:659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huelsmann M., Hecker N., Springer M.S., Gatesy J., Sharma V., Hiller M.. Genes lost during the transition from land to water in cetaceans highlight genomic changes associated with aquatic adaptations. Sci. Adv. 2019; 5:eaaw6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R. et al.. Ensembl 2020. Nucleic Acids Res. 2019; 48:D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bateman A., Martin M.J., O’Donovan C., Magrane M., Alpi E., Antunes R., Bely B., Bingley M., Bonilla C., Britto R. et al.. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017; 45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang G., Li C., Li Q., Li B., Larkin D.M., Lee C., Storz J.F., Antunes A., Greenwold M.J., Meredith R.W. et al.. Comparative genomics reveals insights into avian genome evolution and adaptation. Science. 2014; 346:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Venkatesh B. Evolution and diversity of fish genomes. Curr. Opin. Genet. Dev. 2003; 13:588–592. [DOI] [PubMed] [Google Scholar]
- 21. Clark A.G., Eisen M.B., Smith D.R., Bergman C.M., Oliver B., Markow T.A., Kaufman T.C., Kellis M., Gelbart W., Iyer V.N. et al.. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007; 450:203–218. [DOI] [PubMed] [Google Scholar]
- 22. Kent W.J., Baertsch R., Hinrichs A., Miller W., Haussler D.. Evolution’s cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:11484–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marcovitz A., Turakhia Y., Chen H.I., Gloudemans M., Braun B.A., Wang H., Bejerano G.. A functional enrichment test for molecular convergent evolution finds a clear protein-coding signal in echolocating bats and whales. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:21094–21103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mahalanobis P.C. On the Generalized Distance in Statistics. 1936; National Institute of Science of India. [Google Scholar]
- 25. De Maesschalck R., Jouan-Rimbaud D., Massart D.L.. The mahalanobis distance. Chemometr. Intell. Lab. 2000; 50:1–18. [Google Scholar]
- 26. Kent W.J. BLAT the BLAST-like alignment tool. Genome Res. 2002; 12:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dickinson M.E., Flenniken A.M., Ji X., Teboul L., Wong M.D., White J.K., Meehan T.F., Weninger W.J., Westerberg H., Adissu H. et al.. High-throughput discovery of novel developmental phenotypes. Nature. 2016; 537:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stenson P.D., Mort M., Ball E.V., Evans K., Hayden M., Heywood S., Hussain M., Phillips A.D., Cooper D.N.. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017; 136:665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petrovski S., Gussow A.B., Wang Q., Halvorsen M., Han Y., Weir W.H., Allen A.S., Goldstein D.B.. The intolerance of regulatory sequence to genetic variation predicts gene dosage sensitivity. PLOS Genet. 2015; 11:e1005492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rivals I., Personnaz L., Taing L., Potier M.-C.. Enrichment or depletion of a GO category within a class of genes: which test?. Bioinformatics. 2006; 23:401–407. [DOI] [PubMed] [Google Scholar]
- 32. Jain A., Tuteja G.. TissueEnrich: tissue-specific gene enrichment analysis. Bioinformatics. 2019; 35:1966–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010; 28:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Derrien T., Thézé J., Vaysse A., André C., Ostrander E.A., Galibert F., Hitte C.. Revisiting the missing protein-coding gene catalog of the domestic dog. BMC genomics. 2009; 10:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hogarth C.A., Roy A., Ebert D.L.. Genomic evidence for the absence of a functional cholesteryl ester transfer protein gene in mice and rats. Comp. Biochem. Phys. B. 2003; 135:219–229. [DOI] [PubMed] [Google Scholar]
- 36. Abd El-Aziz M.M., Barragan I., O’Driscoll C.A., Goodstadt L., Prigmore E., Borrego S., Mena M., Pieras J.I., El-Ashry M.F., Safieh L.A. et al.. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat. Genet. 2008; 40:1285–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bowling S., Di Gregorio A., Sancho M., Pozzi S., Aarts M., Signore M., D Schneider M., Barbera J.P.M., Gil J., Rodríguez T.A.. P53 and mTOR signalling determine fitness selection through cell competition during early mouse embryonic development. Nat. Commun. 2018; 9:1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Su R.-W., Strug M.R., Jeong J.-W., Miele L., Fazleabas A.T.. Aberrant activation of canonical Notch1 signaling in the mouse uterus decreases progesterone receptor by hypermethylation and leads to infertility. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:2300–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hargrave M., Wright E., Kun J., Emery J., Cooper L., Koopman P.. Expression of the Sox11 gene in mouse embryos suggests roles in neuronal maturation and epithelio-mesenchymal induction. Dev. Dyn. 1997; 210:79–86. [DOI] [PubMed] [Google Scholar]
- 40. Greibe E., Fedosov S., Nexo E.. The cobalamin-binding protein in zebrafish is an intermediate between the three cobalamin-binding proteins in human. PLoS One. 2012; 7:e35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goudet G., Mugnier S., Callebaut I., Monget P.. Phylogenetic analysis and identification of pseudogenes reveal a progressive loss of zona pellucida genes during evolution of vertebrates. Biol. Reprod. 2008; 78:796–806. [DOI] [PubMed] [Google Scholar]
- 42. Phillips C.J., Phillips C.D., Goecks J., Lessa E.P., Sotero-Caio C.G., Tandler B., Gannon M.R., Baker R.J.. Dietary and flight energetic adaptations in a salivary gland transcriptome of an insectivorous bat. PLoS One. 2014; 9:e83512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Voigt C.C., Zubaid A., Kunz T.H., Kingston T.. Sources of assimilated proteins in Old and New World phytophagous bats. Biotropica. 2011; 43:108–113. [Google Scholar]
- 44. Singaraja R.R., Tietjen I., Hovingh G.K., Franchini P.L., Radomski C., Wong K., Stylianou I.M., Lin L., Wang L., Mitnaul L. et al.. Identification of four novel genes contributing to familial elevated plasma HDL cholesterol in humans. J. lipid Res. 2014; 55:1693–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Widmaier E., Gornstein E., Hennessey J., Bloss J., Greenberg J., Kunz T.. High plasma cholesterol, but low triglycerides and plaque-free arteries, in Mexican free-tailed bats. Am. J. Physiol.-Reg. I. 1996; 271:R1101–R1106. [DOI] [PubMed] [Google Scholar]
- 46. Vogel P., Read R., Hansen G.M., Freay L.C., Zambrowicz B.P., Sands A.T.. Situs inversus in Dpcd/Poll-/-, Nme7-/- , and Pkd1l1-/- mice. Vet. Pathol. 2010; 47:120–131. [DOI] [PubMed] [Google Scholar]
- 47. Field S., Riley K.-L., Grimes D.T., Hilton H., Simon M., Powles-Glover N., Siggers P., Bogani D., Greenfield A., Norris D.P.. Pkd1l1 establishes left-right asymmetry and physically interacts with Pkd2. Development. 2011; 138:1131–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grimes D.T., Keynton J.L., Buenavista M.T., Jin X., Patel S.H., Kyosuke S., Vibert J., Williams D.J., Hamada H., Hussain R. et al.. Genetic analysis reveals a hierarchy of interactions between polycystin-encoding genes and genes controlling cilia function during left-right determination. PLoS Genet. 2016; 12:e1006070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kamura K., Kobayashi D., Uehara Y., Koshida S., Iijima N., Kudo A., Yokoyama T., Takeda H.. Pkd1l1 complexes with Pkd2 on motile cilia and functions to establish the left-right axis. Development. 2011; 138:1121–1129. [DOI] [PubMed] [Google Scholar]
- 50. Vetrini F., DâĂŹAlessandro L.C.A., Akdemir Z.C., Braxton A., Azamian M.S., Eldomery M.K., Miller K., Kois C., Sack V., Shur N. et al.. Bi-allelic mutations in PKD1L1 are associated with laterality defects in humans. Am. J. Hum. Genet. 2016; 99:886–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perles Z., Moon S., Ta-Shma A., Yaacov B., Francescatto L., Edvardson S., Rein A. J. J.T., Elpeleg O., Katsanis N.. A human laterality disorder caused by a homozygous deleterious mutation in MMP21. J. Med. Genet. 2015; 52:840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Akawi N., McRae J., Ansari M., Balasubramanian M., Blyth M., Brady A.F., Clayton S., Cole T., Deshpande C., Fitzgerald T.W. et al.. Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4,125 families. Nat. Genet. 2015; 47:1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li Y., Klena N.T., Gabriel G.C., Liu X., Kim A.J., Lemke K., Chen Y., Chatterjee B., Devine W., Damerla R.R. et al.. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015; 521:520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eppig J.T., Smith C.L., Blake J.A., Ringwald M., Kadin J.A., Richardson J.E., Bult C.J.. Mouse genome informatics (MGI): resources for mining mouse genetic, genomic, and biological data in support of primary and translational research. Methods Mol. Biol. 2017; 1488:47–73. [DOI] [PubMed] [Google Scholar]
- 55. Michaëlsson M., Ho S.Y.. Congenital Heart Malformations in Mammals: An Illustrated Text. 2000; London, UK: Imperial Collage Press. [Google Scholar]
- 56. Clemmensen C., Smajilovic S., Wellendorph P., Bräuner-Osborne H.. The GPCR, class C, group 6, subtype A (GPRC6A) receptor: from cloning to physiological function. Brit. J. Pharmacol. 2014; 171:1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pi M., Quarles L.D.. Multiligand specificity and wide tissue expression of GPRC6A reveals new endocrine networks. Endocrinology. 2012; 153:2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pi M., Quarles L.D.. GPRC6A regulates prostate cancer progression. Prostate. 2012; 72:399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pi M., Chen L., Huang M.-Z., Zhu W., Ringhofer B., Luo J., Christenson L., Li B., Zhang J., Jackson P.D. et al.. GPRC6A null mice exhibit osteopenia, feminization and metabolic syndrome. PLoS ONE. 2008; 3:e3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. De Toni L., Di Nisio A., Speltra E., Rocca M.S., Ghezzi M., Zuccarello D., Turiaco N., Ferlin A., Foresta C.. Polymorphism rs2274911 of GPRC6A as a Novel Risk Factor for Testis Failure. J. Clin. Endocr. Metab. 2016; 101:953–961. [DOI] [PubMed] [Google Scholar]
- 61. Ye R., Pi M., Cox J.V., Nishimoto S.K., Quarles L.D.. CRISPR/Cas9 targeting of GPRC6A suppresses prostate cancer tumorigenesis in a human xenograft model. J. Exp. Clin. Canc. Res.: CR. 2017; 36:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kenagy G.J., Trombulak S.C.. Size and function of mammalian testes in relation to body size. J. Mammal. 1986; 67:1–22. [Google Scholar]
- 63. Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D. et al.. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004; 14:708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McLean C.Y., Reno P.L., Pollen A.A., Bassan A.I., Capellini T.D., Guenther C., Indjeian V.B., Lim X., Menke D.B., Schaar B.T. et al.. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature. 2011; 471:216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sharma V., Hiller M.. Increased alignment sensitivity improves the usage of genome alignments for comparative gene annotation. Nucleic Acids Res. 2017; 45:8369–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Turakhia Y., Goenka S.D., Bejerano G., Dally W.J.. Darwin-WGA: A co-processor provides increased sensitivity in whole genome alignments with high speedup. 2019 IEEE International Symposium on High Performance Computer Architecture (HPCA). 2019; IEEE; 359–372. [Google Scholar]
- 67. Acharya M., Mookherjee S., Bhattacharjee A., Thakur S. K.D., Bandyopadhyay A.K., Sen A., Chakrabarti S., Ray K.. Evaluation of the OPTC gene in primary open angle glaucoma: functional significance of a silent change. BMC Mol. Biol. 2007; 8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Saeki N., Kuwahara Y., Sasaki H., Satoh H., Shiroishi T.. Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm. Genome. 2000; 11:718–724. [DOI] [PubMed] [Google Scholar]
- 69. Saeki N., Kim D.H., Usui T., Aoyagi K., Tatsuta T., Aoki K., Yanagihara K., Tamura M., Mizushima H., Sakamoto H. et al.. GASDERMIN, suppressed frequently in gastric cancer, is a target of LMO1 in TGF-beta-dependent apoptotic signalling. Oncogene. 2007; 26:6488–6498. [DOI] [PubMed] [Google Scholar]
- 70. Lewin H.A., Robinson G.E., Kress W.J., Baker W.J., Coddington J., Crandall K.A., Durbin R., Edwards S.V., Forest F., Gilbert M.T.P. et al.. Earth BioGenome project: sequencing life for the future of life. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:4325–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code for the automated hcoErosions pipeline is available at http://bitbucket.org/bejerano/hcoerosions.