

Abstract
Hepatitis C virus (HCV) replication requires annealing of a liver specific small-RNA, miR-122 to 2 sites on 5′ untranslated region (UTR). Annealing has been reported to (a) stabilize the genome, (b) stimulate translation and (c) promote the formation of translationally active Internal Ribosome Entry Site (IRES) RNA structure. In this report, we map the RNA element to which small RNA annealing promotes HCV to nucleotides 1–44 and identify the relative impact of small RNA annealing on virus translation promotion and genome stabilization. We mapped the optimal region on the HCV genome to which small RNA annealing promotes virus replication to nucleotides 19–37 and found the efficiency of viral RNA accumulation decreased as annealing moved away from this region. Then, by using a panel of small RNAs that promote replication with varying efficiencies we link the efficiency of lifecycle promotion with translation stimulation. By contrast small RNA annealing stabilized the viral genome even if they did not promote virus replication. Thus, we propose that miR-122 annealing promotes HCV replication by annealing to an RNA element that activates the HCV IRES and stimulates translation, and that miR-122 induced HCV genome stabilization is insufficient alone but enhances virus replication.
INTRODUCTION
Hepatitis C Virus (HCV) is a flavivirus that causes chronic infections of the liver and can lead to liver cirrhosis and hepatocellular carcinoma (1,2). The genome of HCV is a 9.6 kb long positive sense RNA that consists of a 5′ untranslated region (UTR), a polyprotein coding region, and a 3′UTR region. The 5′ and 3′ UTRs are highly structured and required for genome translation and replication (3,4).
The 5′UTR is a structured RNA that forms 4 stem loops (SL), SLI, SLII, SLIII and SLIV. SLII, SLIII and SLIV comprise the internal ribosomal entry site (IRES) that drives cap-independent HCV translation (Figure 1A) (5,6). SLII is divided into two parts, SLIIa which induces SLII to form a bent structure that directs SLIIb, to the ribosomal E-site in the head region of the 40S subunit, facilitating 80S ribosome assembly (7–9). The first 42 nucleotides on the 5′UTR are not considered part of the IRES and forms SLI and an RNA structured element created by annealing of two copies of microRNA-122 (miR-122), a host microRNA found in human liver cells (10–13). This tri-molecular structure is required for virus replication (11–14).
Figure 1.
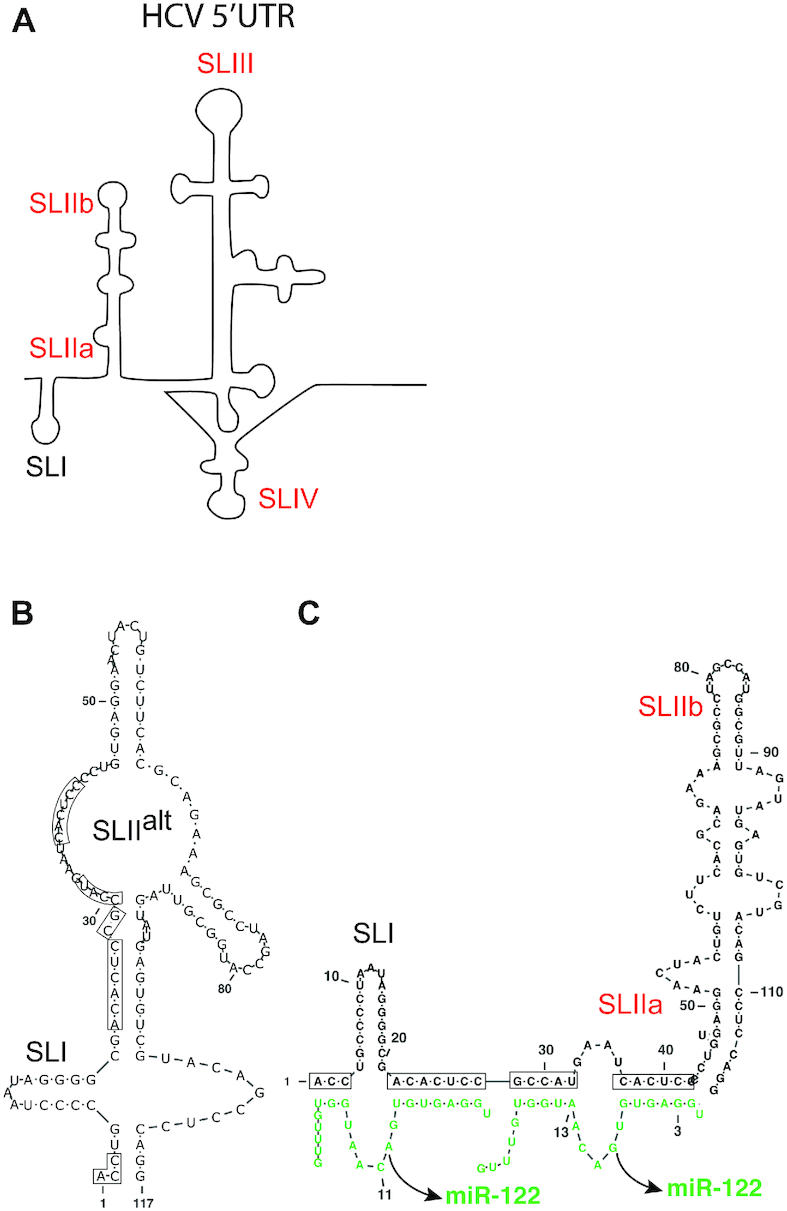
HCV 5′ UTR RNA structures. (A) Schematic representation of HCV 5′UTR stem loops; SLI, SLIIa, SLIIb, SLIII and SLIV are indicated. The 5′ 117 nucleotide RNA fragment alone is predicted to form SLIIalt (B), and annealing of 2 copies of miR-122 is hypothesized to favour formation of the canonical SLII and the active IRES structure but this has yet to be experimentally validated. (C) The miR-122 binding nucleotides are shown within black boxes. Stem loops indicated in red are parts of the IRES.
MicroRNAs (miRNAs) are small RNAs about 21–23 nucleotides long and are central to mRNA regulation by miRNA gene silencing (15,16). miRNAs silence genes in association with a host Argonaute (Ago) protein within an RNA induced silencing complex (RISC) and direct the protein complex to the 3′ UTR of an mRNA by annealing with imperfect sequence complementarity (17). miRNAs target an mRNA by annealing to an ∼6–7 nucleotide seed site binding at the 5′ end of the miRNA and an accessory site binding on the 3′ end (18). This leaves a loop of mismatched nucleotides between the seed and accessory sites and overhangs on the 5′ and 3′ ends of the miRNA that do not bind to the target mRNA. Such complex miRNA:target RNA interactions regulates the expression of targeted mRNAs by inducing their translation suppression and degradation (19). Two copies of miR-122 anneal in conjunction with Ago to the HCV genome in a similar manner, including seed and accessory binding sites (20) but in this case activate instead of silence the viral genome.
HCV replication is undetectable in the absence of miR-122 and the mechanism behind miR-122’s stimulatory effect is not fully understood. miR-122 stabilizes the viral RNA by protecting it from degradation by host exonuclease Xrn1, and phosphatases DOM3Z and DUSP11 (21–23). However, simultaneous knock-down of these three enzymes cannot completely rescue HCV replication in the absence of miR-122, suggesting that other roles exist (23). miR-122 also promotes HCV translation and recent reports hypothesize that miR-122 and Ago modify the HCV RNA genome to induce the translationally active 5′UTR IRES structure (Figure 1B and C) (12,13,24). Finally, miR-122 has been reported to directly induce genome amplification (25,26). However, the relative impact of these functions on miR-122 directed HCV replication promotion is unknown (27).
We previously showed that annealing of small interfering RNAs (siRNA) to the HCV 5′ UTR can mimic the pro-viral activity of miR-122 (13). Like miRNAs, siRNAs are also 21–23 nucleotides in length and associate with Ago proteins, but based on associating with Ago2 and perfect sequence match with their targets, induce mRNA cleavage and gene knockdown (28,29). However, when siRNA cleavage activity was blocked by using Ago2 knockout cells, siRNAs that anneal to the miR-122 binding region on the HCV genome promoted virus replication, some as efficiently as miR-122. That siRNA annealing promoted HCV, suggested that the complex annealing pattern formed by miR-122 on the HCV genome was not required for the pro-viral activity and also provide a method to assess the impact of small RNA annealing to other locations on the genome on HCV replication (13). Using a panel of HCV genome-targeting siRNAs, we found that annealing between nucleotides 1 and 44 in HCV 5′UTR, promoted HCV replication, and annealing within the IRES, NS5B and 3′UTR regions did not. We thus define a regulatory 5′ UTR RNA element to which small RNA annealing regulates the HCV lifecycle. Small RNAs that annealed to different locations on the 5′ UTR promoted virus replication with different efficiencies, and we found that replication promotion correlated with translation stimulation. Finally, like miR-122, annealing of the siRNAs to the 5′ UTR also stabilized the viral genome, but the siRNAs did so regardless of whether they could promote replication or not, suggesting that genome stabilization alone is insufficient for HCV replication promotion. Thus, our current model for the pro-viral mechanism of miR-122 replication posits that its role is to stimulate translation by shifting the 5′ UTR RNA folding equilibrium toward the translationally active IRES conformation and that genome stabilization is insufficient alone but enhances replication induced by translation stimulation.
MATERIALS AND METHODS
Plasmids
Full-length HCV Renilla Luciferase (Rluc) reporter genome constructs pJ6/JFH-1 RLuc (p7-RLuc2A) and the non-replicative version pJ6/JFH-1 RLuc (p7-RLuc2A) GNN were provided by Dr C.M. Rice (30). For translation suppression assays the miR-122 suppression firefly luciferase (Fluc) reporter plasmid, pFLuc JFH-1 5′UTR × 2 (11) was modified to contain a single copy of the complete 5′UTR (pFluc JFH-1 5′UTR) or NS5B-3′UTR (pFluc JFH-1 NS5B-3′UTR) region of the genome by replacing the fragment between restriction sites SpeI and SacII. To generate the complete 5′ UTR fragment we used PCR and the forward primer 5′GCCACTAGTACGACGGCCAGTGAATTC3′; and reverse primer 5′CAGCCGCGGATCGATGACCTTACCCACG3′, to generate the NS5B-3′UTR region we used forward primer 5′GCCACTAGTAATGTGTCTGTGGCGTTGG3′; reverse primer 5′CAGCCGCGGAAACAGCTATGACCATGA3′ and the pJ6/JFH-1 RLuc (p7-RLuc2A) plasmid as a template. Each Forward primer has sequence for restriction site, SpeI, and each Reverse primer has sequence for SacII. The control plasmid pRL-TK was obtained from Promega (Madison, USA). A pT7 Fluc containing plasmid, herein called pT7 Fluc (Promega, Madison, USA), was used to generate control mRNA for translation assays.
In vitro RNA transcription
To generate full length viral RNA the plasmid pJ6/JFH-1 RLuc (p7-RLuc2A) or related mutants were linearized by digestion with XbaI and RNA was made by using the MEGA Script T7 High Yield Transcription Kit (Life Technologies, Burlington, Canada). The transcription process was performed using the suggested manufacturer's protocol. Fluc mRNA transcript was prepared by digesting the plasmid, pT7 Fluc mRNA, with XmnI and mRNA was prepared using the mMessage mMachine mRNA synthesis kit (Life Technologies, Burlington, Canada) using manufacturer's protocol.
Small interfering RNA (siRNA) design and sequence
HCV targeting siRNAs that anneal to regions in IRES, NS5B coding region and 3′UTR were designed using online software, i-score, https://www.med.nagoya-u.ac.jp/neurogenetics/i_Score/i_score.html (31). The sequence of siRNA JFH-1 6367 (si6367) was adapted from the siRNA described previously to inhibit the HCV con1 genotype, by modifying the sequence to match the same region in JFH-1 GACCCACAAACACCAAUUCCC (32). The control siRNA (siControl) target sequence is GAGAGUCAGUCAGCUAAUCA and does not anneal to the virus genome. All siRNAs that anneal to the HCV genome were designed to have 21 nucleotides; 19 nucleotides complementary to target site and 2 UU overhangs on the 3′ end for incorporating into RISC complex, unless stated otherwise. These siRNAs were synthesized by GE Lifesciences Dharmacon. Anti-miR-122, miRIDIAN microRNA Human hsa-miR-122-5p-Hairpin Inhibitor (IH-300591-06-0050), were purchased from Dharmacon Horizon Discoveries (Chicago, USA).
Cell culture
miR-122 knockout (miR-122 KO) Huh-7.5 (33), Ago2 knockout (Ago2 KO) Huh-7.5 cells (13) and DROSHA/Ago2 KO cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 0.1 nM non-essential amino acids (Wisent, Montreal, Canada) and 100 ug/ml Pen/Strep (Invitrogen, Burlingtion, Canada). miR-122 knockout Huh-7.5 cells were a kind gift from Dr Matthew Evans. DROSHA/Ago2 KO Huh 7.5 cells were generated from DROSHA KO Huh 7.5 cells (34) (a gift from Dr Charlie Rice) by using the CRISPR–Cas9 genome editing techniques (35).
siRNA suppression assay
To assess the ability of an siRNA to suppress mRNA translation and assess whether the siRNAs are functional in the RISC, we assessed their impact in a transient suppression assay. For this assay we used reporter plasmids (pFluc JFH-1 5′UTR/pFuc JFH-1 NS5B-3′UTR) that express Fluc mRNAs containing the siRNA target sequence from HCV in their 3′UTRs. We also used an Rluc expressing control plasmid, pRL-TK to normalize transfection efficiency (Promega, Madison, USA). The day before transfection 8 × 104 miR-122 KO cells/well were plated in a 24-well dish and incubated overnight. The next day, the cells were transfected with 100 ng of each of pRL-TK and pFluc JFH-1 (5′UTR or NS5B-3′UTR) and 0.1 pmol of a particular test siRNA. The transfection mixture was prepared using 1 μl lipofectamine 2000 according to the suggested manufacture's protocol (Life Technologies, Burlington, Canada). The cells were incubated at 37°C, 5% CO2 after transfections and after 48 h were lysed using passive lysis buffer and assayed for Fluc and Rluc activity using a dual luciferase assay kit (Promega, Madison, USA) (13).
HCV replication assay
Ago2 KO Huh-7.5 cells were co-electroporated as described previously (36) with 5 ug J6/JFH-1(p7-Rluc2a) RNA or related point mutant viral RNAs, 60 pmol of test or control siRNAs and 60 pmol of anti-miR-122. In all samples in an experiment the amount of small RNAs added per sample was equivalent, and if necessary siControl was added to balance the amount of small RNA (13). Cells were harvested 2, 24, 48 and 72 h post-electroporations and assayed for Rluc expression. HCV replication was assessed based on Rluc expression and was normalized to a positive control sample in which replication of J6/JFH-1(p7-Rluc2a) RNA was supported by endogenous cellular miR-122 (Endo miR-122).
Generation of Ago2/ DROSHA knockout Huh-7.5 cells
To generate the double knockout Huh 7.5 cells, we used CRISPR–Cas9 gene editing system to knockout Ago2 in DROSHA knockout Huh 7.5 cells provided to us by Dr. Charlie Rice (34). Three synthetic guide RNAs (AAUACCUGUUAACUCUCCUC-140585131; UAAUUUGAUUGUUCUCCCGG-140585231, GGCGCAGGAGGUGCAAGUGC-140585310) were designed using the Synthego knockout design tool (https://design.synthego.com/#/) in such a way that the guide RNAs would result in a frameshift deletion in the early region of the exon 2 in the Ago2 gene. DROSHA knockout cells were transfected with the TrueCut™ Cas9 Protein v2 (Invitrogen, Thermo Fisher Scientific, Vilnius, Lithuania) and the guide RNAs (Synthego CRISPRevolution EZkit, Redwood City, USA) using the Lipofectamine™ CRISPRMAX™ Cas9 Transfection Reagent (Invitrogen, Thermo Fisher Scientific, Carlsbad, USA) according to the manufacturer's protocol (Quick Reference: Invitrogen, Thermo Fisher Scientific, Lipofectamine CRISPRMAX Transfection Reagent Pub. No.: MAN0014545). Forty eight hours post-transfection, the cells were passaged, and a portion of the cells collected to test the knockout efficiency. Knockout efficiency was assessed by sanger sequencing of a PCR product that amplified the Crispr targeted region of the Ago2 gene, generated using primers- forward: 5′ATTCATGCTGCCTCATCTCTCC3′ and reverse: 5′CGGAAGAAGGTATGAGGCAA3′. PCR was performed using the PfuUltra II Fusion High-fidelity DNA polymerase (Agilent, California, USA) using genomic template DNA extracted using QuickExtract DNA extraction solution (Epicentre/ Lucigen, Wisconsin, USA). DNA was prepared by harvesting the cells in the QuickExtract DNA extraction solution and heating the samples at 65°C for 15 min, 68°C for 15 min and 98°C for 10 min. The ABI file obtained after Sanger sequencing was examined for indels on the Synthego ICE tool (https://ice.synthego.com/#/) and the knockout efficiency was found to be 90%. An array dilution method was used to isolate single cell colonies and condition media was used to encourage the growth of the single clones. Condition medium was obtained by collecting the medium used for growing the knockout pool of cells, filtered using a 0.22 μm filter and stored at −20°C prior to use. Successful knockout of both Ago2 alleles in the DROSHA knockout cells was identified based on analysis for the loss of siRNA knockdown phenotype based on HCV replication promotion by 5′ UTR annealing siRNAs and confirmed by western blot and genome sequencing.
Phenotypic analysis of DROSHA/Ago2 knockout cells
To confirm knockout of Ago2 in the DROSHA/Ago2 KO cells we assessed the ability of the cells to support HCV replication promotion by si18-36, as compared to DROSHA KO wild type cells in which the siRNA will knock-down and thus fail to promote HCV. Isolated clones of putative DROSHA/Ago2 double knockout cells were seeded in 24-well plate 24 h prior to transfection with 1 ug of J6/ JFH-1(p7-Rluc2a) RNA and 12 pmol small RNAs. miR-122 was used as positive control and siControl was used as negative control. Transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, USA) as per manufacturer's protocol. Cells were harvested 48 h post-transfection and Rluc expression was measured.
Western blot
Knockout of Ago2 was confirmed by western blot analysis for the expression of Ago2. Putative DROSHA/Ago2 knockout cells were treated with 1x SDS lysis buffer (with 1% 1M DTT) and heated at 95°C for 5 min. The proteins were then separated using a 7.5% SDS-PAGE gel and transferred to a nitrocellulose membrane (GE healthcare Lifesciences, Amersham Protran 0.45 NC membranes, Freiburg, Germany). The membrane was blocked with 5% skimmed milk (BD Difco) and probed with 1:1000 diluted primary anti-Ago2 rat monoclonal antibody clone 11A9 (Millipore Sigma/ Merck KGaA MABE 253, Darmstadt, Germany), 1:25 000 diluted primary mouse monoclonal anti-beta actin antibody (AC-15) (Abcam ab6276, Cambridge, USA) and subsequently with 1:40 000 diluted secondary peroxidase conjugated AffiniPure Goat Anti-Rat IgG (H+L) (Jackson Immunoresearch 112-035-003, West Grove, USA) and 1:25 000 diluted secondary HRP conjugated Goat anti-mouse IgG (H+L) (BioRad, Mississauga, Canada). The blot was developed using Clarity Western ECL substrate (BioRad, Mississauga, Canada) and imaged with BioRad ChemiDoc MP Imaging system.
HCV translation assay
To assess siRNA promotion of HCV translation, DROSHA/Ago2 KO Huh-7.5 cells were co-electroporated with 5ug of non-replicative mutant HCV genomic RNA, J6/JFH-1(p7-Rluc2a) GNN, 1ug of control T7 Fluc mRNA, and 60 pmol of test siRNA. As positive control J6/JFH-1(p7-Rluc2a) GNN was electroporated with miR-122, since the DROSHA/Ago2 KO Huh-7.5 cells lack miR-122 expression. As a negative control, J6/JFH-1(p7-Rluc2a) GNN was electroporated with siControl. Cells were harvested 4 h post-electroporations and assayed for Rluc and Fluc expression.
Luciferase assay
Luciferase expression was measured by using Firefly, Renilla, or Dual luciferase kits (Promega, Madison, USA) as suggested by the manufacturer's protocols. Cells were washed once in Dulbecco's phosphate-buffered saline then lysed with 100 ul of passive lysis buffer. 10 ul of the cell extract was mixed with the appropriate luciferase assay substrate and light emission was measured by using a Glomax 20/20 Luminometer (Promega, Madison, USA).
RNA purification
Cells were harvested into 1 ml of Trizol and total cellular RNA was isolated using the manufacturer's provided protocol (Life Technologies, Burlington, Canada).
HCV genome stabilization assay and northern blots
To assess the impact of miR-122 and the siRNAs on HCV genome stability we assessed the amount of non-replicative HCV RNA present in cells at various times post-electroporation using northern blot analysis. For each assay, 32 × 106 DROSHA/Ago2 knockout Huh-7.5 cells were electroporated (in 4 cuvettes) with 40 μg of HCV J6/JFH-1(p7-Rluc2a) GNN RNA and 240 pmol of one of the small RNAs (si15-33, si19-37, si26-44 or si27-45) or miR-122 (as a positive control) or siControl (as a negative control). Cells from the four electroporation cuvettes were pooled and plated onto four 10 cm plates. Cells for the 0 min time point were harvested immediately after electroporation, and the others were incubated at 37°C and harvested at 30, 60 and 120 min post-electroporation. Total cellular RNA was harvested using Trizol as recommended by the manufacturer (Life Technologies, Burlington, Canada), and 10 ug were separated on an 0.8% agarose gel and transferred to a Nylon membrane (GE Healthcare Limited, Buckinghamshire, England) as described previously (36). The transferred RNAs were crosslinked to the membrane using a UV crosslinker (Spectrolinker XL-1000) at X100 μJ/cm2 for 12 s and cut in half to probe for HCV RNA and GAPDH separately. The radioactive DNA probes used were prepared using Prime-a-Gene Labeling System kit (U1100, Promega, Madison, WI, USA), and radiolabeled dCTP (PerkinElmer, Boston, USA). The probes were generated from a 3 kb BamHI-to-EcoRV DNA fragment of the pJ6/JFH-1 RLuc (p7-RLuc2A) plasmid or a 1.3 kb cDNA fragment of human GAPDH. Radioactive bands were detected by exposing the membranes overnight on a phosphorscreen and scanned using a Phosphoimager (Typhoon, GE Healthcare Life Sciences, Mississauga, Canada). Band signal intensities were quantified using Image Studio Lite version 5.2.5.
RNA structure prediction analysis
RNA structure predictions were done using the RNA prediction software ‘RNA structure’ available from the website by the Matthews lab at https://rna.urmc.rochester.edu/index.html. (37). Single RNA structure predictions were performed using algorithm ‘fold’ and structure predictions of two interacting RNA molecule were predicted using algorithm ‘bifold’. Dot-bracket files for the six lowest free energy structures were generated using the RNA fold command in ‘RNAstructure’ and RNA images were generated from them using VARNA (VARNA GUI applet) (38).
Statistical analysis
All data are displayed as the mean of three or more independent experiments, and error bars indicate standard deviation of the mean. Where appropriate, one-way ANOVA was performed using Graph Pad Prism version 8.3 for MacOS (San Diego, USA, www.graphpad.com). In graphs, statistical significance is indicated as follows: *P < 0.0332; **P < 0.0021; ***P < 0.0002; ****P < 0.0001.
RESULTS
siRNA annealing to nucleotides between 15 and 44 on 5′UTR promotes virus replication
Annealing of miR-122 to two complementary sequences on 5′UTR of the HCV genome is required for detectible HCV replication in cell culture (27,39). In our previous work, we showed that HCV replication was promoted efficiently by 5′ UTR targeting siRNAs when siRNA-directed cleavage activity was abolished by using Ago2 knockout cells (Ago2 KO) (13). Thus, replication promotion does not require the annealing pattern exhibited by miR-122, but we hypothesized that it may be impacted by the annealing location.
To test this hypothesis, we determined the range of genome locations to which small RNA annealing can promote HCV replication. First, we designed and tested an array of siRNAs with target sequences that walk the 5′UTR between nucleotides 10–47 at single nucleotide resolution (Figure 2A). Each siRNA was 19 nucleotides long and contained two 3′ UU (uracil) overhangs to ensure RISC loading (Supplementary Table S1). The siRNAs were named based on the 19 nucleotide positions on the HCV genome to which they bind, from si10-28 to si29-47 (Figure 2A).
Figure 2.

Mapping the region on the HCV 5′ UTR to which siRNAs binding promotes the HCV lifecycle. (A) Diagrammatic representation of HCV genome showing 5′UTR polyprotein coding region and 3′ UTR. The first 55 nucleotides of HCV 5′ UTR are shown interacting with two copies of miR-122 (green). siRNAs designed to walk the 5′UTR with single nucleotide resolution are also shown. Black lines represent siRNAs that do not promote replication and red lines represent ones that do. (B) siRNA suppression assay results with siControl (grey bar), bars are colour coded based on their suppression activities. siRNAs that do not suppress translation (black bars) and that suppress translation (red bars). (C) Six lowest delta free energy structure of HCV 5′ 117 nucleotide RNA fragment alone, with small RNAs that promote virus replication (miR-122 and si19-37) and with small RNA that does not promote replication (si27-45). Structure predictions were performed using an online software, ‘RNAstructure’ and have not been experimentally validated. (D) The pro-viral activity of the siRNAs was assessed using HCV replication assays in which the activity of miR-122 is antagonized using anti-miR-122. Ago2 knockout cells were co-electroporated with HCV J6/ JFH-1(p7-Rluc2a) RNA, anti-miR-122 and the indicated siRNA and harvested at 2 h (gray bars), 24 h (light gray bars), 48 h (blue bars) and 72 h (red bars) post-electroporation. HCV replication was measured based on Rluc expression and is presented as % relative to Rluc expression from HCV RNA supported by endogenous miR-122 at 72 h post-electroporation (Endo miR-122). The data are the average of at least three independent experiments and error bars represent the standard deviation. Statistical significance was determined using one-way ANOVA on the relative 72-h values where, *P < 0.0332; **P < 0.0021; ***P < 0.0002; ****P < 0.0001. (E) Diagram depicting the siRNA suppression assay and (F) replication assay respectively.
To confirm RISC loading, the siRNAs were tested for their ability to knockdown gene expression in a suppression assay (Figure 2E). For suppression assays, we transfected cells with a plasmid, pFluc JFH-1 5′UTR, that expresses an mRNA encoding Fluc having the HCV miR-122 binding region/siRNA target sites in its 3′ UTR. This plasmid was co-transfected in miR-122 KO Huh 7.5 cells (cells that express Ago2), with a test siRNA and knockdown was measured based on Fluc expression compared to cells transfected with a control siRNA (siControl). Fluc expression levels were assessed relative to Rluc expression from a transfection control plasmid, pRL-TK. We observed that siRNAs binding from nucleotides 14 onwards were significantly (P value < 0.0001) able to suppress Fluc expression at different levels compared to siControl, implying that they were actively incorporated into the RISC complex (Figure 2B). Most siRNAs that bound within SLI did not knockdown Fluc and thus did not enter RISC, likely due to hairpin structure formation. Inactive siRNAs were omitted from further analyses.
After confirming RISC incorporation, we determined the ability of the siRNAs to promote HCV replication in replication assays. For these assays we electroporated Ago2 KO Huh 7.5 cells with J6/JFH-1(p7-Rluc2a) RNA, anti-miR-122, an antagonist of endogenous miR-122, and a test siRNA. Anti-miR-122 is a locked nucleic acid that anneals to and inactivates miR-122. The limited complementarity between the test siRNAs and anti-miR-122 was insufficient to inhibit siRNA activity (personal communication with Dharmacon). HCV replication was assessed 2, 24, 48 and 72 h post-electroporation based on luciferase expression as a proxy for HCV replication (Figure 2F). We electroporated J6/JFH-1(p7-Rluc2a) RNA without anti-miR-122 as a positive control to measure HCV RNA replication induced by endogenous miR-122 (Endo miR-122). RLuc expression in this sample at 72 h post-electroporation was deemed 100% and used to calculate relative luciferase levels in the rest of the samples. The negative control (siControl) (Figure 2D) containing viral RNA, anti-miR-122 and siControl (an HCV non-targeting siRNA) confirmed abolition of HCV replication by the miR-122 antagonist. In other samples the addition of the indicated siRNA reinstated HCV replication to the relative levels shown (Figure 2D). An siRNA that anneals within the NS5B coding sequence (si6367) did not promote HCV replication but siRNAs binding between nucleotides 15 and 44 did. HCV replication was promoted most efficiently by si19-37 and was similar to replication induced by endogenous miR-122 (Endo miR-122), and less efficient replication induction was observed using siRNAs that annealed to locations moving away from nucleotides 19–37 in either direction. si27-45 and any siRNAs binding beyond si27-45 did not promote virus replication (Figure 2D) (Supplementary Figure S1A).
In support of the hypothesis that miR-122 annealing reshapes the 5′ UTR to form the translationally active HCV IRES structure (12,13,24) there was a general correlation between the predicted ability of an siRNA to induce the translationally active structure and its ability to promote virus replication (Supplementary Figure S2). For example, si19-37, an siRNA that promoted efficient virus replication, was predicted to stimulate the translationally favorable structure and si27-45, an siRNA that did not promote virus replication did not (Figure 2C).
Defining the 3′ boundary of the domain to which siRNA annealing promotes HCV replication
We identified the 3′ boundary of the region to which siRNA annealing promoted HCV RNA as nucleotide 44. In our siRNA walk, si26-44 was the last siRNA to promote virus replication (P value < 0.0001) and si27-45 and those that bound further in the 3′ direction did not. Based on this, we hypothesized that either annealing to nucleotide 26 was essential for promotion, or siRNA annealing to nucleotide 45 and onwards was detrimental. To distinguish between these possibilities, we designed two additional siRNAs, si26-45 and si27-44 (Figure 3A). Both siRNAs were active in suppression assays, however only si27-44 promoted HCV replication (Figure 3B and C). This indicated that annealing to nucleotide 45, inhibited promotion activity and annealing to nucleotide 26 was not essential (Figure 3B). Thus, we have defined the 3′ boundary of the region to which small RNA annealing can promote HCV replication as nucleotide 44.
Figure 3.
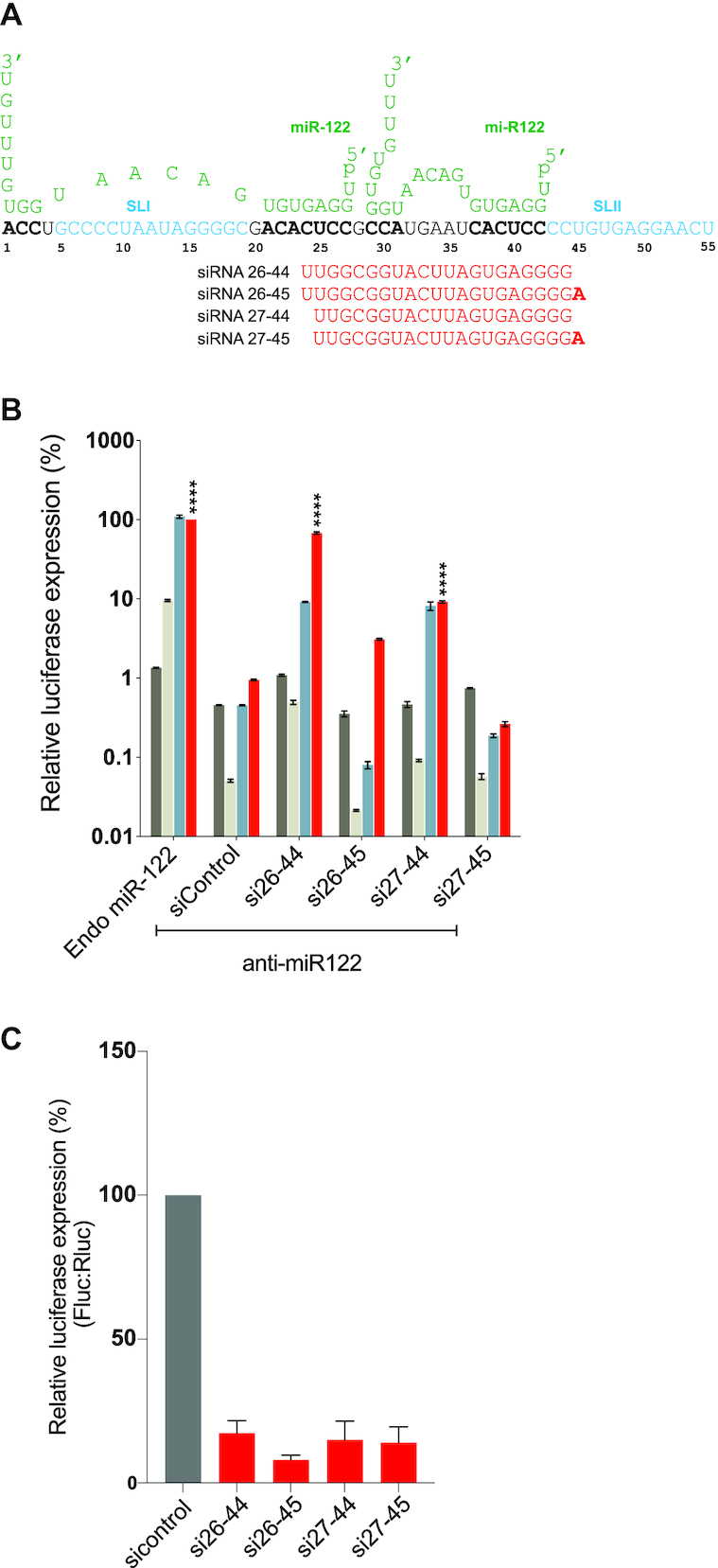
Mapping the 3′boundary of small RNA annealing induced replication promotion. (A) Diagrammatic representation of the first 55 nucleotides of HCV 5′ UTR are shown interacting with two copies of miR-122 (green). siRNA binding to terminal nucleotide 44 and 45 are shown and nucleotide 45 is shown in bold. (B) The pro-viral activity of the siRNAs was assessed using HCV replication assays in which the activity of miR-122 is antagonized using anti-miR-122. Ago2 knockout cells were co-electroporated with HCV J6/ JFH-1(p7-Rluc2a) RNA, anti-miR-122 and the indicated siRNA and harvested at 2 h (gray bars), 24 h (light gray bars), 48 h (blue bars) and 72 h (red bars) post-electroporation. HCV replication was measured based on Rluc expression and is presented as % relative to Rluc expression from HCV RNA supported by endogenous miR-122 at 72 h post-electroporation (Endo miR-122). The data are the average of at least three independent experiments and error bars represent the standard deviation. Statistical significance was determined using one-way ANOVA on the relative 72-h values where, *P < 0.0332; **P < 0.0021; ***P < 0.0002; ****P < 0.0001. (C) siRNA suppression assay results with siControl (gray bar) and that suppress translation (red bars).
Defining the 5′ boundary of siRNAs binding and the role of annealing to the 5′ terminus (nucleotides 1–3) and generation of an RNA overhang
The 5′ boundary to which siRNA annealing promoted HCV replication was nucleotide 15 (Figure 2D). Si14-32 did not promote detectable HCV replication and siRNAs that anneal nearer to the 5′ UTR did not enter RISC and thus could not be tested (Figure 2B). Previous reports showed that replication promotion by miR-122 was enhanced by annealing of miR-122 to nucleotides on the 5′ terminus of HCV and by the generation of a 3′ overhang (20). To assess the impact of siRNA binding to the 5′ terminus (nucleotides 1–3) we compared HCV replication promotion by a small RNA that binds to the 5′ terminus with one that does not (Figure 4). We designed an siRNA, si1-3-21-36, that binds to 21–36 and to the 5′ terminal 3 nucleotides and found that it promoted replication ∼4-fold more efficiently than si1-3mm–21-36, that also binds to nucleotides 21–36 but not the 5′ terminus (Figure 4A and B). Both siRNAs were active in our suppression assays (Supplementary Figure S3E). Indeed, HCV replication promoted by si1-3-21-36 was around 2-fold higher than the most efficient siRNA, si19-37, identified in Figure 2D. Thus, end annealing is not required for pro-viral impact of small RNA annealing to the 5′ UTR but has a positive effect.
Figure 4.
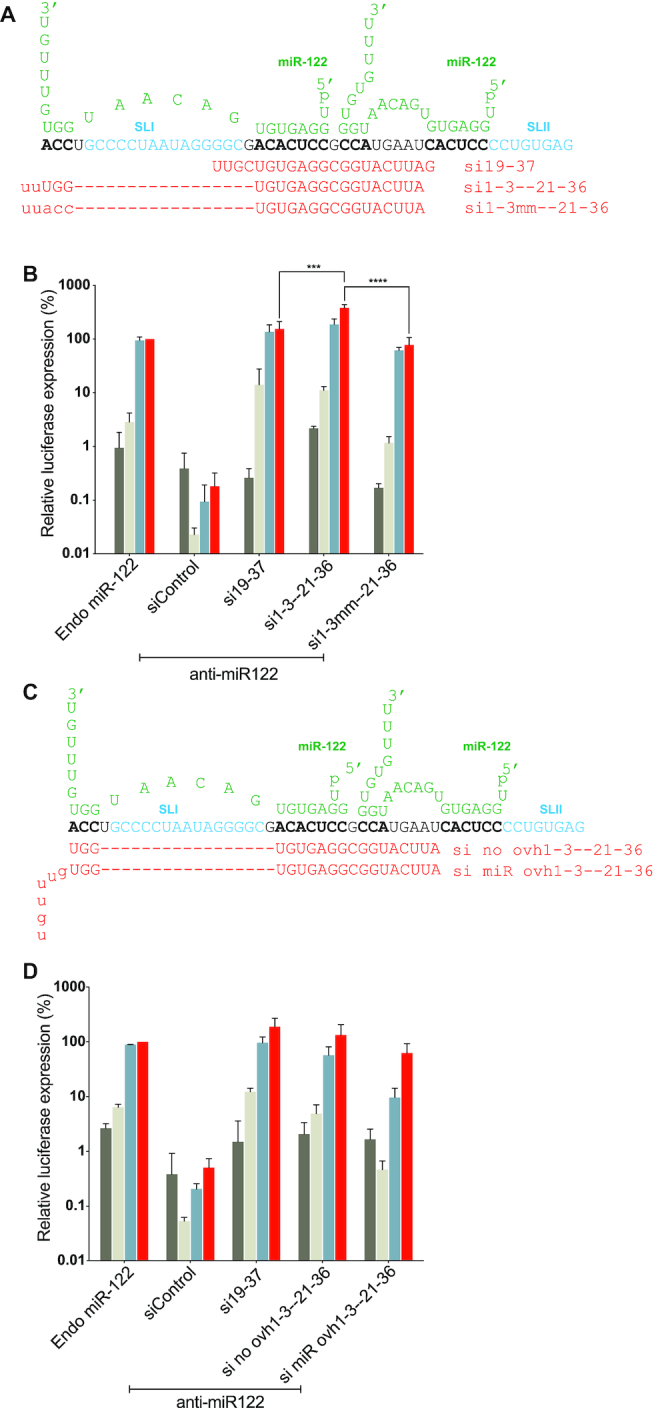
HCV replication promotion by siRNA annealing to the 5′ terminus by an siRNA generated 3′ overhang. (A and C) Diagram of the first 50 nucleotides of HCV 5′ UTR interacting with two copies of miR-122 (green). miR-122 binding sites on HCV genome are shown in bold characters. Uppercase letters represent nucleotides in the siRNAs that anneal to the HCV RNA and lowercase letters represent nucleotides that do not. (A) Sequences of si19-37, si1-3-21-36 and si1-3mm-21-36 are shown. (C) Schematic representation of si no ovh1-21-36 and si miR ovh1-3mm-21-36 binding on HCV genome. (B and D) Graphs showing HCV replication promotion by siRNAs that bind or do not bind to the 5′ terminus and with siRNAs that do or do not generate a 3′ overhang. Ago2 knockout cells were electroporated with HCV J6/JFH-1(p7-Rluc2a) RNA, anti-miR-122 and the indicated siRNA and harvested at 2 h (gray bars), 24 h (light gray bars), 48 h (blue bars) and 72 h (red bars) post-electroporation. HCV replication was measured based on Rluc expression and is presented as % relative to Rluc expression from HCV RNA supported by endogenous miR-122 at 72 h post-electroporation (Endo miR-122). Data represents the average three independent experiments and error bars represent the standard deviation. Statistical significance was determined using one-way ANOVA on 72-h values where, *P < 0.0332; **P < 0.0021; ***P < 0.0002; ****P < 0.0001.
The annealing of miR-122 to binding site 1 on the HCV genome generates a 7-nucleotide overhang of the HCV 5′ terminus that was reported to contribute to the efficiency of HCV replication promotion by miR-122 (20). Since the siRNAs used in this study only consisted of 2 UU overhangs, we knew that a long miR-122-like overhang was not essential for replication promotion, but we wanted to test if having such an overhang contributes to replication. To test this, we assessed replication promotion by an siRNA that has a miR-122-like overhang, ‘si mir ovh1-3-21-36’, with one that does not, ‘si no ovh1-3-21-36’ (Figure 4C). Both siRNAs were active in our suppression assays (Supplementary Figure S3E) and predicted to induce the translationally active IRES structure (Supplementary Figure S4C) but we found that the miR-122-like overhang did not enhance replication promotion, and in fact, decreased HCV replication efficiency (Figure 4D). This experiment confirmed that generation of a 5′ overhang is not required for and may hinder HCV replication.
siRNA annealing to the HCV IRES do not promote nor inhibit HCV replication
To determine whether siRNAs binding on other regions of HCV genome can promote virus replication, we designed siRNAs using the online software, i-score, to determine best target sites within the HCV IRES region (31). We designed six siRNAs that bind to various sites on the IRES regions namely si38-56, si42-60, si73-91, si88-106, si317-338 and si339-357 (Figure 5A). We validated the knockdown activity of these siRNAs to confirm their incorporation into RISC, and, five out of six showed suppression activity (Supplementary Figure S3A). However, none of the active siRNAs promoted replication in HCV replication assays (Figure 5B).
Figure 5.
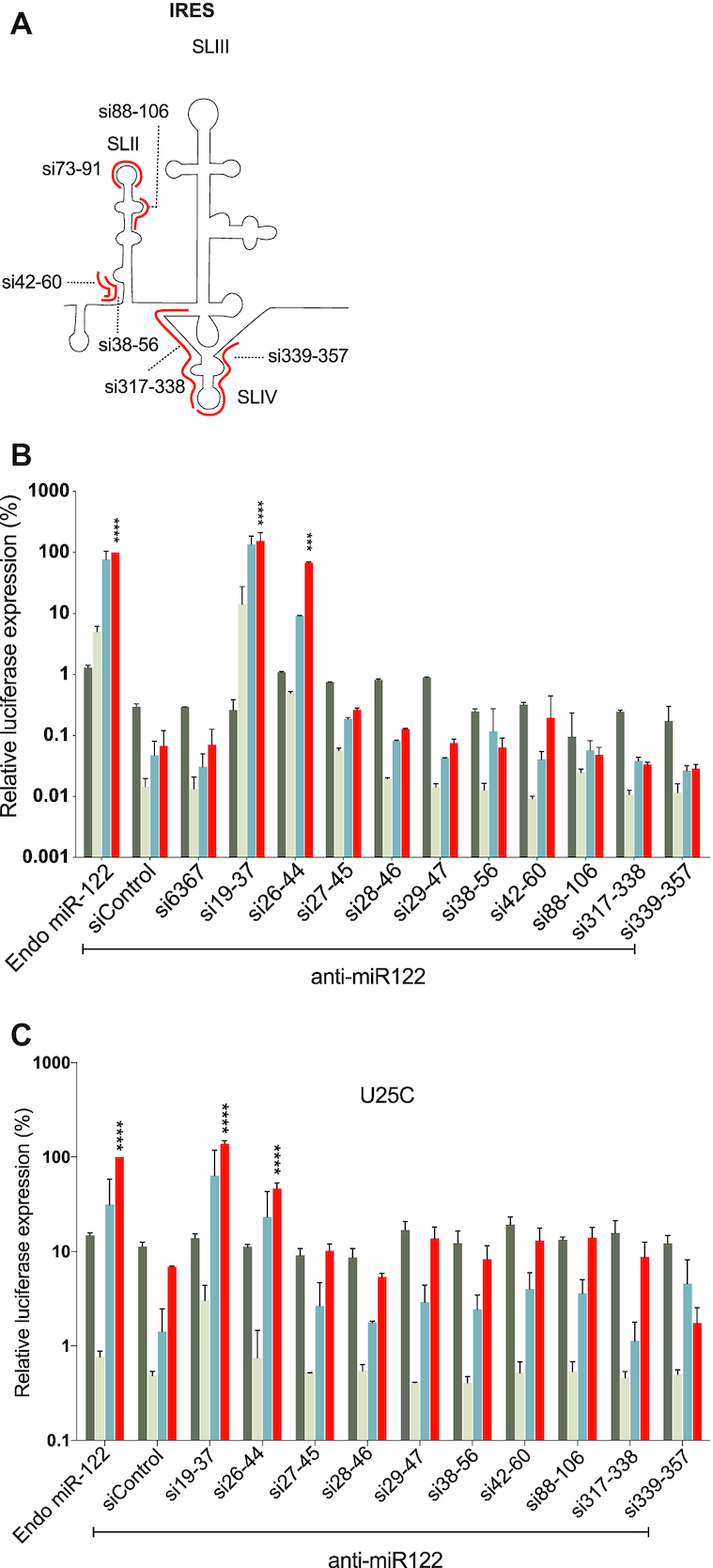
Assessing the pro-or antiviral activity of siRNA binding within the HCV IRES. (A) A cartoon diagram of the HCV IRES including stem loops II, III and IV and the locations of annealing of six siRNAs that target the IRES are shown in red. (B) The pro-viral activity of the siRNAs was assessed using replication assays in which the activity of miR-122 is antagonized by anti-miR-122. Ago2 knockout cells were electroporated with HCV J6/JFH-1(p7-Rluc2a) RNA, anti-miR-122 and the indicated siRNA and harvested at 2 h (gray bars), 24 h (light gray bars), 48 h (blue bars) and 72 h (red bars) post-electroporation. (C) The pro- or anti-viral effects of the indicated siRNAs was assessed based on their influence on miR-122-independent replication of a U25C mutant J6/JFH-1(p7-Rluc2a) similar to the experiments described in B. For B and C, HCV replication was measured based on Rluc expression and is presented as % relative to Rluc expression from HCV RNA supported by endogenous miR-122 at 72 h post-electroporation (Endo miR-122). The data are the average of at least three independent experiments and error bars represent the standard deviation. Statistical significance was determined using one-way ANOVA on the relative 72 h values where, *P < 0.0332; **P < 0.0021; ***P < 0.0002; ****P < 0.0001.
Small RNAs that anneal within the IRES and downstream of nucleotides 45, and thus within SLII of the IRES did not promote detectible HCV replication. We hypothesized that these siRNAs may have failed to promote HCV because they anneal to and interfere with the activity of the IRES and thus inhibit HCV replication. To test this hypothesis, we assessed inhibition of HCV by the IRES targeting siRNAs. For these assays we used a mutant HCV RNA, U25C, that can replicate independent from miR-122, and thus replicated in Ago2 knockout cells even when miR-122 is antagonized. This assay was used instead of miR-122-promoted replication to eliminate the possible influence of annealing competition between miR-122 and the siRNAs tested. Using this assay, we observed no HCV replication promotion or inhibition by the IRES-binding siRNAs, while replication was promoted by the positive control siRNA, si19-37 (Figure 5C). Thus, our data suggests that IRES-binding siRNAs do not promote nor inhibit HCV replication.
siRNA annealing to the alternative predicted miR-122 binding sites do not promote HCV replication
Other potential miR-122 binding sites have been reported in the NS5B coding region and the 3′UTR region of HCV genome (40–43). We therefore also assessed whether siRNAs binding on any of these potential miR-122 binding sites affected HCV replication. We started by designing siRNAs binding to seven predicted miR-122 binding sites in NS5B and 3′UTR region. For convenience we named these siRNAs according to the region they bind and in serial order, namely siNS5B1, siNS5B2, siNS5B3, siNS5B4, si3′UTR1, si3′UTR2 and si3′UTR3 (Supplementary Figure S1B and C). In order to test these in our suppression assay, we replaced the 5′UTR with NS5B-3′UTR regions in suppression plasmid to have target sites for our siRNAs. We observed that each of seven siRNAs were able to suppress Fluc expression suggesting they all entered RISC (Supplementary Figure S3B). To assess their role in HCV replication, we performed HCV replication assays and observed that Rluc expression after addition of the NS5B and 3′ UTR targeting siRNAs was comparable to the negative controls (siControl and si6367) (Supplementary Figure S1D). Thus, siRNA binding to other predicted miR-122 binding sites did not promote virus replication. Overall our data show that small RNAs that bind between nucleotides 15–44 on the HCV 5′ UTR promote HCV replication.
Defining the minimum annealing requirements required for HCV life-cycle promotion
Next we wanted to define the minimum annealing requirements and optimum location for the pro-viral activity of small RNA annealing (Figure 6). To determine this, we designed siRNA analogues of the most efficient siRNA, si19-37, but having sequence matches ranging from 7 to 17 nucleotides on the 3′ or 5′ ends (Figure 6A). For example: si19(21-37) is 19 nucleotides long with 17 nucleotides (21–37) matching the HCV 5′ UTR (Figure 6A). Similarly, si19(23-37), si19(25-37), si19(27-37), si19(27-37), si19(29-37) and si19(31-37) have between 15 and 7 matching nucleotides. si19(21-37), si19(23-37) promoted HCV replication as efficiently as si19-37 and the others did not promote at all (Figure 6C). From the 3′ end we generated siRNAs with sequence matches of 16, si19(19-35), 14, si19(19-33) and 12, si19(19-31) matches (Figure 6A) and only si19(19–35) promoted HCV replication (Figure 6D). These data highlighted annealing to nucleotides 23 to 35 as being required for replication promotion and an siRNA that annealed only to these 13 nucleotides also stimulated HCV replication, albeit poorly (Figure 6A–C). Suppression assay of all si19-37 analogs showed that siRNA knockdown activity decreased with fewer annealing nucleotides; however, those that promoted replication were all active (Supplementary Figure S3C and D). Thus, an siRNA must anneal to at least 13 nucleotides at location 23–35 to promote replication but annealing to 15 nucleotides that span this region is required for efficient replication promotion. This region is located within single and double stranded regions of the SLIIalt RNA structure (Figure 6B) and thus might be optimum location for access, and 15 nucleotides the minimum annealing strength required to modify the 5′ UTR RNA structure. Thus, in total we have defined a regulatory RNA element between nucleotides 1 and 44 of the HCV 5′ UTR to which small RNA annealing promotes the HCV lifecycle.
Figure 6.

Determining the minimum annealing requirement of si19-37 analogues. (A) Diagrammatic representation of first 50 nucleotides of HCV 5′ UTR are shown interacting with two copies of miR-122 (green). Sequences of si19-37 analogues at their binding positions are shown and the number of annealing nucleotides for each analogue siRNA is indicated. siRNAs in red promote HCV replication and siRNAs in black do not promote HCV replication. Small lettered characters in siRNAs show mismatched nucleotides that do not bind on HCV RNA (gray) while capital lettered characters are nucleotides that bind on the RNA. The green box shown around nucleotides 23–35 which were common in case of all siRNAs that promote HCV replication. (B) Predicted secondary structures of 117 nucleotides of HCV 5′UTR alone and with miR-122 showing the 13 nucleotide optimum annealing location required for small RNA dependent HCV replication. Structure predictions were performed using an online software, ‘RNAstructure’ and have not been experimentally validated. (C and D) The activity of the si19-37 analogues was assessed using replication assays in which the activity of miR-122 is antagonized by anti-miR-122. Ago2 knockout cells were electroporated with HCV J6/JFH-1(p7-Rluc2a) RNA, anti-miR-122 and the indicated siRNA and harvested at 2 h (gray bars), 24 h (light gray bars), 48 h (blue bars) and 72 h (red bars) post-electroporation. HCV replication was measured based on Rluc expression and is presented as % relative to Rluc expression from HCV RNA supported by endogenous miR-122 at 72 h post-electroporation (Endo miR-122). Data represent the average of three independent experiments and error bars represent the standard deviation. Statistical significance was determined using one-way ANOVA on 72-h values where, *P < 0.0332; **P < 0.0021; ***P < 0.0002; ****P < 0.0001.
siRNA promotion of virus replication correlates with promotion of virus translation
Two confirmed functions of miR-122 are promotion of HCV translation and stabilization of the viral genome, however, the relative contributions of each to HCV life-cycle promotion are unknown (27,39,44). We hypothesized that if stimulation of translation or genome stabilization is a key mechanism by which miR-122 promotes virus replication then the ability of an siRNA to promote replication will correlate with its ability to stimulate translation or stabilize the viral genome. To test this hypothesis, we assessed siRNA stimulation of translation and genome stabilization by panels of siRNAs that promote HCV with varying efficiencies.
To assess siRNA translation promotion, we measured the ability of miR-122 and an array of siRNAs to promote translation of a non-replicative HCV J6/JFH-1(Rluc2a) GNN RNA in DROSHA/Ago2 double KO cells. DROSHA/Ago2 double KO cells were generated to provide a background that lacked both miR-122 expression and Ago2 associated siRNA cleavage activity and allowed us to remove the miR-122 antagonist from our assays. DROSHA/Ago2 double KO cells were generated from DROSHA knockout cells using Crispr/Cas9 and the knockout of Ago2 was confirmed based on the ability of the cells to use si18-36 to promote instead of knockdown HCV replication (Figure 7A) and by western blot analysis showing abolished Ago2 expression (Figure 7B), (Supplementary Figure S1E). To assess HCV translation promotion by the siRNAs, cells were electroporated with viral RNA and siRNAs and translation efficiency was measured based on Rluc expression vs a co-electroporated Fluc mRNA control (Figure 7C). By using an array of siRNAs that promote replication with different efficiencies we found that the levels of translation stimulation correlated with their ability to promote replication (Figure 7D and E). siRNAs that promoted efficient HCV replication, (Figure 7D, red bars) also efficiently stimulated translation (Figure 7E, red bars), and siRNAs that promoted HCV replication to a moderate level also promoted translation less efficiently (Figure 7D and E, orange bars). Finally, siRNAs that promoted HCV replication poorly or not at all, displayed little or no translation stimulation (Figure 7D and E, yellow and black bars). Thus, our data suggests that small RNA dependent promotion of HCV replication depends on its ability to promote virus translation. However, within the group of siRNAs that promoted virus replication efficiently (Figure 7D and E, red bars) all of the siRNAs promoted virus replication with similar efficiencies but varied in their translation stimulation ability. This suggests there is a relatively low threshold amount of translation stimulation required for efficient HCV replication.
Figure 7.
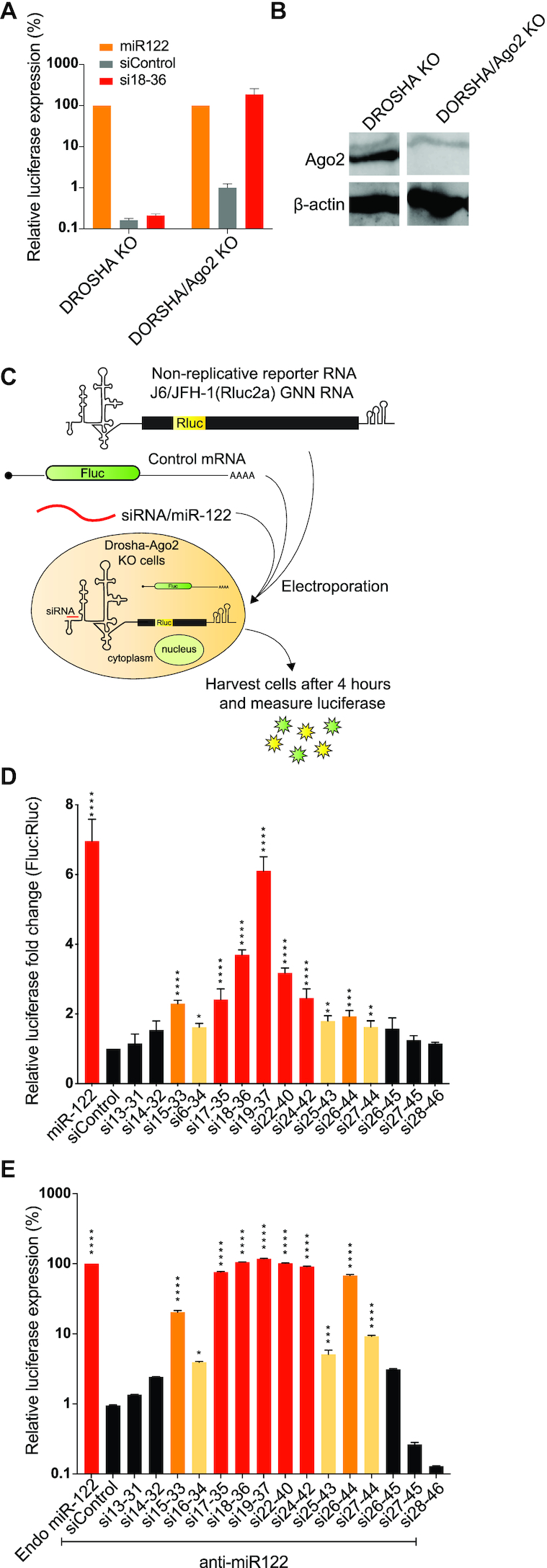
small RNAs annealing promotes translation of HCV. (A) Phenotypic analysis of DROSHA/Ago2 double KO cells. Graph showing HCV replication induction by siRNAs in DROSHA KO cells vs DROSHA/Ago2 double KO cells. (B) Western blot images showing the absence of Ago2 protein in DROSHA/Ago2 double KO cells vs the presence of Ago2 protein in DROSHA KO cells. The uncropped blot is shown in Supplementary Figure S1E. (C) Schematic diagram showing HCV translation assay. (D) Transient translation assays performed in DROSHA/Ago2 double KO cells using non-replicative viral RNA, J6/ JFH-1(p7-Rluc2a) GNN, and the indicated siRNAs and an mRNA expressing Fluc as an electroporation control. Samples were harvested at 4 h and translation was accessed based on Rluc expression vs the co-electroporated Fluc mRNA control. Data represent the average of at least five independent experiments and error bars represent the standard deviation. Statistically significant differences between siRNAs and siControl was assessed by one-way ANOVA, *P < 0.0332; **P < 0.0021; ***P < 0.0002; ****P < 0.0001. (E) Graph showing replication promotion at 72 h post-infection by siRNAs used in the translation assays performed in Ago2 KO cells. (D and E) siRNAs that promoted HCV translation and replication efficiently are coloured red, moderately efficient ones are coloured orange and low efficient are coloured yellow. siRNAs that do not promote replication are coloured black.
siRNA annealing-induced HCV genome stabilization is not sufficient to promote virus replication
In addition to stimulating HCV translation, miR-122 annealing also stabilizes the HCV genomic RNA by protecting it from cellular pyrophosphatases, DOM3Z and DUSP11, and the exonuclease Xrn1 (23). To test for a linkage between siRNA induced replication and virus genome stabilization we investigated HCV RNA genome stability in presence of miR-122, a control siRNA, siControl and two test siRNAs (Figure 8) (Supplementary Figure S5). We chose si19-37 because it promotes efficient replication, efficient translation, is predicted to induce the translationally active IRES, and was shown previously to stabilize the viral genome (13) (Figure 7D, E and 2C). si27-45 was chosen since it does not promote replication, or translation and is not predicted to form translationally active structure (Figure 7D, E and 2C). miR-122 was used as a positive control and siControl was used as a negative control. For the stability assays HCV J6/JFH-1(p7-Rluc2a) GNN RNA, a non-replicative HCV RNA and an siRNA (or miR-122, or control) were electroporated in DROSHA/Ago2 double KO cells and total RNA was harvested at 0, 30, 60 and 120 min post-electroporation. To determine the half-life of HCV GNN RNA northern blots were performed (Figure 8A). As expected, the half-life of the viral RNAs was extended by miR-122 annealing (Figure 8B–D) and si19-37. However, contrary to our expectations si27-45 stabilized the HCV genome, even though it did not promote HCV replication (Figure 8B–D and Supplementary Figure S5). This suggests that small RNA annealing stabilizes HCV genomic RNA but stabilization alone is not sufficient to promote HCV genome replication. Thus, stimulation of translation appears to be linked with miR-122 promotion of HCV replication, and genome stabilization, while stimulatory, is not sufficient alone.
Figure 8.
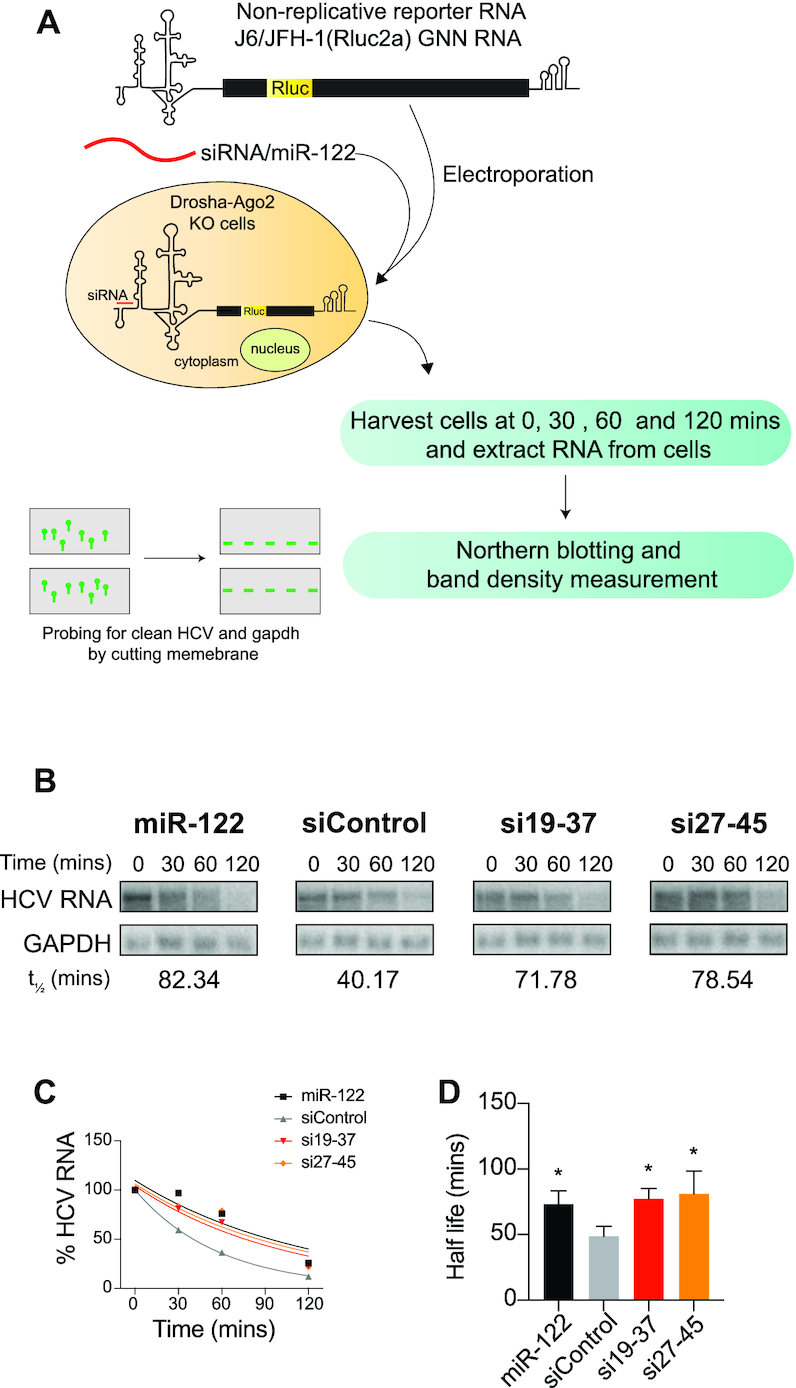
Genome stabilization by small RNAs that do and do not promote HCV replication. (A) Schematic diagram showing HCV stabilization assay. (B) Northern blot analyses of HCV genomic RNA quantities during stability assays. Assays are shown for HCV RNA with annealing of miR-122, two different siRNAs, or siControl. Bands were quantified using ImageStudio Lite and were plotted as a one phase decay curve. These data are representative of three independent experiments performed with each of the two siRNAs along with control small RNAs (miR122 and sicontrol). (C) Decay curves for each sample (miR-122/siControl/19-37/si27-45) were generated and half-lives obtained from these decay curves are noted in part B. The uncropped blots are shown in Supplement Figure S4. (D) The average half-lives and standard deviations obtained from three independent experiments are plotted. Blots, decay curves, and half-lives for the other two experiments are shown in Supplementary Figure S5. Decay curves and half-lives were calculated by Graphpad Prism.
DISCUSSION
miR-122 binding to two sites on HCV 5′UTR is required for efficient HCV replication (Figure 2A). We and others previously reported a hypothesis that the pro-viral activity of miR-122 was mediated by annealing induced RNA structural changes to the HCV 5′ UTR to induce the translationally active 5′ UTR IRES structure (12,13,24). We also showed that HCV replication was stimulated by siRNAs as efficiently as by miR-122 if their siRNA-directed cleavage activity was abolished by using Ago2 KO cells (13). In this study, we have defined an RNA element located between nucleotides 1 and 44 on the HCV 5′ terminus to which small RNA annealing induces the HCV lifecycle and identified distinct impacts of small RNA annealing on translation and genome stabilization, further clarifying the underlying mechanism of replication promotion by miR-122.
We identified that small RNA annealing to nucleotides 1–3 and 15–44 promote HCV replication and that annealing to nucleotide 45 (SLIIa) and beyond do not (Figures 2-4). RNA structure prediction algorithms show that the ability of siRNAs to promote replication is related to its predicted ability to induce the translationally active 5′ UTR RNA structure, including formation of SLI, SLIIa, and SLIIb (Figure 2C) (Supplementary Figure S2) and support the hypothesis that, like miR-122, small RNA annealing promotes virus replication by favouring the formation of the translationally active IRES RNA structures. However, this hypothesis has not been confirmed by us or by others using biochemical and biophysical methods (12,13,24).
We also mapped a region between nucleotides 23–35 as the optimum region of annealing (Figure 6B and C). Annealing to these 13 nucleotides was the minimum annealing requirement to promote replication and annealing to 15 nucleotides, 23–37, was the minimum annealing required to promote efficient replication. This annealing location comprises most of miR-122 binding seed site 1 (nucleotides 21–27) and the accessory miR-122 binding site 2 (nucleotides 29–32) and suggests that binding to miR-122 binding seed site 1 and accessory site 2 may be the minimum requirement for efficient small RNA dependent HCV replication, but also shows that binding beyond the accessory site to nucleotides 33–36 that are not bound by miR-122, can also contribute to replication promotion. This data also supports a previous finding that miR-122 site 1 behaves similar to a conventional miRNA:target interaction where binding to a seed site is important (45), and that miR-122 binding site 2 has higher affinity owing to extended base pairing to the accessory site. This data may also explain why annealing of two copies of miR-122 is required for efficient HCV replication since annealing of one copy does not fulfill the requirement of annealing to 15 nucleotides.
We also speculate that the location of nucleotides 23–35 may be optimal for small RNA annealing to activate the HCV IRES. This region resides within both based-paired and non-base paired regions of SLIIalt in the non-canonical 5′ UTR structure (Figure 6B) and the non-base paired region may allow efficient small RNA annealing, and 15 base pairs of annealing is sufficient strength to induce the translationally active IRES structure. In addition, a previous report proposed that miR-122 annealing to binding site 2 positioned Ago2 such that it contacts SLIIs and potentially modulates the function of the HCV IRES (24). Perhaps annealing at position 23–35 is the optimal location to position Ago for this interaction. Biophysical analyses of RNA-Ago complexes induced by annealing of siRNAs that promote the HCV lifecycle with varying efficiencies could clarify the molecular details of pro-viral nucleoprotein structures.
A previous report showed that miR-122 binding to the extreme 5′ terminus of the HCV genome was required for efficient HCV replication (20). By contrast, our data indicates that small RNA annealing to the 5′ terminal region is not essential but enhances HCV replication promotion (Figure 4A and B). The previous report also showed that the seven nucleotide 3′miR-122 overhang contributes to virus replication (20). However, our study indicates that a 2UU overhang was sufficient to promote HCV replication, and that generation of a miR-122-like 3′ overhang was not necessary and, in fact, was detrimental to HCV replication (Figure 4C and D).
We also determined that siRNA binding to other regions on HCV genome, including the HCV IRES does not stimulate virus replication. In addition, IRES annealing siRNAs neither promote nor inhibit HCV replication (Figure 5B and C) and thus do not appear to disrupt IRES structure and function. Therefore, small RNA annealing induced RNA structure changes appear to be specific to the HCV 5′ terminal region and the active HCV IRES structure may be too stable to be disrupted by small RNA annealing.
At least seven more miR-122 binding sites were predicted in the HCV genome and were speculated to also affect the virus life cycle, (11,40,41,43). However, none of the siRNAs that bound to the predicted miR-122 binding sites promoted replication (Supplementary Figure S1B). Our data therefore suggests that the two miR-122 binding sites on HCV 5′UTR are the only active binding sites that promote HCV replication. This data is in agreement with a recently published report in which mutation of the other miR-122 binding sites had no influence on HCV replication (40).
That miR-122 stimulates HCV translation was first reported in 2008 and has been confirmed by several groups (4,12,46). We hypothesized that if translation stimulation was a key mechanism by which miR-122 is promoting HCV replication then the ability of an siRNA to stimulate translation will correlate with its ability to promote virus replication. Our data support this hypothesis and showed a correlation between siRNAs that promoted replication efficiently and their ability to stimulate translation (Figure 7D and E). However, siRNAs that promoted replication as efficiently as miR-122 (si17-35, si18-36, si19-37, si22-40, si22-42) stimulated translation with a range of abilities, from 2- to 7-fold, and only si19-37 stimulated translation as efficiently as miR-122. Thus, small changes in annealing location have a dramatic effect on translation stimulation and differences in efficiency may be due to differential annealing site access or altered Ago positioning. However, in spite of different abilities to stimulate translation all of these small RNAs promoted replication similarly and suggest that a minimum threshold of translation stimulation is required for efficient HCV replication, at least in cell culture. However, the annealing pattern of miR-122 appears to be optimized for efficient translation stimulation, and thus optimal translation stimulation by miR-122 may be important for HCV infections in humans.
Annealing of miR-122 to the HCV 5′ UTR stabilizes the viral genome, and this has been proposed as a mechanism by which miR-122 promotes HCV replication (13,25). Further, it was speculated that the mechanism of protection is the generation of a double stranded 5′ terminus by miR-122 binding that protect it from cellular pyrophosphatases and exonuclease (Figure 2A) (20). We showed previously that si19-37 annealing also stabilizes the HCV genome, and that annealing to the extreme 5′ terminus was not required (13). In this report we show that annealing of si27-45 to the 5′ UTR stabilizes the HCV genome even though it does not promote replication (Figure 8). This confirms that 5′ end annealing is not required for genome stabilization and also that small RNA induced genome stabilization alone is not sufficient to promote virus replication. We propose that stabilization likely functions to enhance replication that is induced by translation stimulation.
Finally, based on our findings we present a model in which annealing of miR-122 or position specific small RNAs to a 5′ terminal regulatory RNA element induces the formation of the active viral IRES and stimulates virus translation. We propose that the minimal IRES may intrinsically fold into a translationally active structure and that the 5′ terminal RNA element and miR-122 annealing modulates this intrinsic structure in some way. In addition, miR-122 or position-independent small RNA annealing stabilizes the viral genome, but stabilization alone is insufficient to promote the virus life cycle (Figure 9). However, whether miR-122 or small RNA annealing directly affect HCV RNA replication remains unclear, as does the dynamics of miR-122 annealing during specific events in the virus lifecycle such as replication and virion assembly. In addition, the roles of miRNA associated proteins like Ago and Ago complexes in HCV promotion by miR-122 remain to be clarified.
Figure 9.

Summary model figure. We have developed a model for the mechanism of miR-122 promotion of HCV. In our model, the (A) HCV 5′ UTR RNA forms SLIIalt a non-canonical 5′ UTR structure in the absence of small RNA annealing but (B) the canonical SLII structure of the active HCV IRES when bound with 2 copies of miR-122 or with siRNAs. Structure predictions in (A) and (B) were performed using an online software, ‘RNAstructure’ and have not been experimentally validated. (A and B) Boxed nucleotides represent miR-122 binding sites. and the colours of nucleotides represent RNA replication efficiency when siRNAs are bound to those nucleotides. Small RNA induction or stabilization of the canonical IRES structure promotes virus translation leading to enhanced virus replication. Small RNA annealing also stabilizes the viral genome, but genome stabilization alone is not sufficient to promote the HCV lifecycle. We propose that small RNAs annealing in association with host Argonaute proteins are responsible for RNA structure changes and viral genome stabilization. It is unknown if genome amplification is affected by miR-122 or whether replication initiation is regulated by the canonical or non-canonical structures (dotted arrow lines). All together we propose that a key role of miR-122 is to induce the canonical 5′UTR IRES structure and promote virus translation and that genome stabilization has a secondary enhancing role but is insufficient alone.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge Charlie Rice (The Rockefeller University) for providing the pJ6/JFH-1(p7Rluc2A), DROSHA knockout, and Huh-7.5 cells. We also thank Matthew Evans for miR-122 knockout cells.
Authors’ contributions: R.K., S.G., J.Q.K. and J.A.W. designed and performed the experiments, and analyzed the data; R.K. and J.A.W. wrote the manuscript.
Contributor Information
Rasika D Kunden, Department of Biochemistry, Microbiology and Immunology, University of Saskatchewan, Saskatoon, Saskatchewan S7N 5E5, Canada.
Sarah Ghezelbash, Department of Biochemistry, Microbiology and Immunology, University of Saskatchewan, Saskatoon, Saskatchewan S7N 5E5, Canada.
Juveriya Q Khan, Department of Biochemistry, Microbiology and Immunology, University of Saskatchewan, Saskatoon, Saskatchewan S7N 5E5, Canada.
Joyce A Wilson, Department of Biochemistry, Microbiology and Immunology, University of Saskatchewan, Saskatoon, Saskatchewan S7N 5E5, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research [MOP-133458]; Canadian Foundation for Innovation [18622 to J.A.W.]; Canadian Network on Hepatitis C (CanHepC) Training Program, Doctoral Research Fellowships (to R.K.); Natural Sciences and Engineering Research Council of Canada, Undergraduate Summer Research Awards (NSERC-USRA) (to S.G.). Funding for open access charge: University of Saskatchewan-Bridge Funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Rijnbrand R., Bredenbeek P.J., Haasnoot P.C., Kieft J.S., Spaan W.J., Lemon S.M.. The influence of downstream protein-coding sequence on internal ribosome entry on Hepatitis C Virus and other flavivirus RNAs. RNA. 2001; 7:585–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perz J.F., Armstrong G.L., Farrington L.A., Hutin Y.J.F., Bell B.P.. The contributions of hepatitis B virus and Hepatitis C Virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006; 45:529–538. [DOI] [PubMed] [Google Scholar]
- 3. Dustin L.B. Innate and adaptive immune responses in chronic HCV infection. Curr. Drug Targets. 2017; 18:826–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sagan S.M., Chahal J., Sarnow P.. cis-Acting RNA elements in the Hepatitis C Virus RNA genome. Virus Res. 2015; 206:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson A.G., Grosely R., Petrov A.N., Puglisi J.D.. Dynamics of IRES-mediated translation. Philos. Trans. R. Soc. B Biol. Sci. 2017; 372:20160177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fraser C.S., Doudna J.A.. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat. Rev. Microbiol. 2007; 5:29–38. [DOI] [PubMed] [Google Scholar]
- 7. Spahn C.M.T., Kieft J.S., Grassucci R.A., Penczek P.A., Zhou K., Doudna J.A., Frank J.. Hepatitis C Virus IRES RNA-Induced changes in the conformation of the 40S ribosomal subunit. Science. 2001; 291:1959–1962. [DOI] [PubMed] [Google Scholar]
- 8. Lukavsky P.J., Kim I., Otto G.A., Puglisi J.D.. Structure of HCV IRES domain II determined by NMR. Nat. Struct. Mol. Biol. 2003; 10:1033–1038. [DOI] [PubMed] [Google Scholar]
- 9. Paulsen R.B., Seth P.P., Swayze E.E., Griffey R.H., Skalicky J.J., Cheatham T.E., Davis D.R.. Inhibitor-induced structural change in the HCV IRES domain IIa RNA. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:7263–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jopling C.L., Yi M., Lancaster A.M., Lemon S.M., Sarnow P.. Modulation of Hepatitis C Virus RNA abundance by a Liver-Specific MicroRNA. Science. 2005; 309:1577–1581. [DOI] [PubMed] [Google Scholar]
- 11. Jopling C.L., Schütz S., Sarnow P.. Position-dependent function for a tandem MicroRNA miR-122-binding site located in the Hepatitis C Virus RNA genome. Cell Host Microbe. 2008; 4:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schult P., Roth H., Adams R.L., Mas C., Imbert L., Orlik C., Ruggieri A., Pyle A.M., Lohmann V.. microRNA-122 amplifies Hepatitis C Virus translation by shaping the structure of the internal ribosomal entry site. Nat. Commun. 2018; 9:2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Amador-Cañizares Y., Panigrahi M., Huys A., Kunden R.D., Adams H.M., Schinold M.J., Wilson J.A.. miR-122, small RNA annealing and sequence mutations alter the predicted structure of the Hepatitis C Virus 5′ UTR RNA to stabilize and promote viral RNA accumulation. Nucleic Acids Res. 2018; 46:9776–9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Israelow B., Mullokandov G., Agudo J., Sourisseau M., Bashir A., Maldonado A.Y., Dar A.C., Brown B.D., Evans M.J.. Hepatitis C Virus genetics affects miR-122 requirements and response to miR-122 inhibitors. Nat. Commun. 2014; 5:5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee Y., Jeon K., Lee J.-T., Kim S., Kim V.N.. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002; 21:4663–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lam J.K.W., Chow M.Y.T., Zhang Y., Leung S.W.S.. siRNA versus miRNA as therapeutics for gene silencing. Mol. Ther. Nucleic Acids. 2015; 4:e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hutvágner G., Zamore P.D.. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002; 297:2056–2060. [DOI] [PubMed] [Google Scholar]
- 18. Brennecke J., Stark A., Russell R.B., Cohen S.M.. Principles of MicroRNA–target recognition. PLOS Biol. 2005; 3:e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O’Brien J., Hayder H., Zayed Y., Peng C.. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018; 9:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Machlin E.S., Sarnow P., Sagan S.M.. Masking the 5′ terminal nucleotides of the Hepatitis C Virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:3193–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Y., Masaki T., Yamane D., McGivern D.R., Lemon S.M.. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of Hepatitis C Virus replication. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:1881–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shimakami T., Yamane D., Jangra R.K., Kempf B.J., Spaniel C., Barton D.J., Lemon S.M.. Stabilization of Hepatitis C Virus RNA by an Ago2–miR-122 complex. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amador-Cañizares Y., Bernier A., Wilson J.A., Sagan S.M.. miR-122 does not impact recognition of the HCV genome by innate sensors of RNA but rather protects the 5′ end from the cellular pyrophosphatases, DOM3Z and DUSP11. Nucleic Acids Res. 2018; 46:5139–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chahal J., Gebert L.F.R., Gan H.H., Camacho E., Gunsalus K.C., MacRae I.J., Sagan S.M.. miR-122 and Ago interactions with the HCV genome alter the structure of the viral 5′ terminus. Nucleic Acids Res. 2019; 47:5307–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Masaki T., Arend K.C., Li Y., Yamane D., McGivern D.R., Kato T., Wakita T., Moorman N.J., Lemon S.M.. miR-122 stimulates Hepatitis C Virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe. 2015; 17:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fukuhara T., Kambara H., Shiokawa M., Ono C., Katoh H., Morita E., Okuzaki D., Maehara Y., Koike K., Matsuura Y.. Expression of MicroRNA miR-122 facilitates an efficient replication in nonhepatic cells upon infection with Hepatitis C Virus. J. Virol. 2012; 86:7918–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilson J.A., Huys A.. miR-122 promotion of the Hepatitis C Virus life cycle: sound in the silence. WIREs RNA. 2013; 4:665–676. [DOI] [PubMed] [Google Scholar]
- 28. Meister G., Landthaler M., Patkaniowska A., Dorsett Y., Teng G., Tuschl T.. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell. 2004; 15:185–197. [DOI] [PubMed] [Google Scholar]
- 29. Carthew R.W., Sontheimer E.J.. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009; 136:642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jones C.T., Murray C.L., Eastman D.K., Tassello J., Rice C.M.. Hepatitis C Virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007; 81:8374–8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ichihara M., Murakumo Y., Masuda A., Matsuura T., Asai N., Jijiwa M., Ishida M., Shinmi J., Yatsuya H., Qiao S. et al.. Thermodynamic instability of siRNA duplex is a prerequisite for dependable prediction of siRNA activities. Nucleic Acids Res. 2007; 35:e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilson J.A., Richardson C.D.. Hepatitis C Virus replicons escape RNA interference induced by a short interfering RNA directed against the NS5b coding region. J. Virol. 2005; 79:7050–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hopcraft S.E., Azarm K.D., Israelow B., Lévêque N., Schwarz M.C., Hsu T.-H., Chambers M.T., Sourisseau M., Semler B.L., Evans M.J.. Viral determinants of miR-122-independent Hepatitis C Virus replication. mSphere. 2015; 1:e00009-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luna J.M., Scheel T.K.H., Danino T., Shaw K.S., Mele A., Fak J.J., Nishiuchi E., Takacs C.N., Catanese M.T., de Jong Y.P. et al.. Hepatitis C Virus RNA functionally sequesters miR-122. Cell. 2015; 160:1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Doudna J.A., Charpentier E.. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 36. Wilson J.A., Zhang C., Huys A., Richardson C.D.. Human Ago2 is required for efficient MicroRNA 122 regulation of Hepatitis C Virus RNA accumulation and translation. J. Virol. 2011; 85:2342–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reuter J.S., Mathews D.H.. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010; 11:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Darty K., Denise A., Ponty Y.. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009; 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thibault P.A., Huys A., Amador-Cañizares Y., Gailius J.E., Pinel D.E., Wilson J.A.. Regulation of Hepatitis C Virus genome replication by Xrn1 and MicroRNA-122 binding to individual sites in the 5′ untranslated region. J. Virol. 2015; 89:6294–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bernier A., Sagan S.M.. Beyond sites 1 and 2, miR-122 target sites in the HCV genome have negligible contributions to HCV RNA accumulation in cell culture. J. Gen. Virol. 2019; 100:217–226. [DOI] [PubMed] [Google Scholar]
- 41. Nasheri N., Singaravelu R., Goodmurphy M., Lyn R.K., Pezacki J.P.. Competing roles of microRNA-122 recognition elements in Hepatitis C Virus RNA. Virology. 2011; 410:336–344. [DOI] [PubMed] [Google Scholar]
- 42. Jopling C.L. Regulation of Hepatitis C Virus by microRNA-122. Biochem. Soc. Trans. 2008; 36:1220–1223. [DOI] [PubMed] [Google Scholar]
- 43. Schult P. Functional dissection of the Hepatitis C Virus non-structural proteins and miR-122 in viral replication and translation. 2017; PhD Thesis.
- 44. Sarnow P., Sagan S.M.. Unraveling the mysterious interactions between Hepatitis C Virus RNA and liver-specific MicroRNA-122. Annu. Rev. Virol. 2016; 3:309–332. [DOI] [PubMed] [Google Scholar]
- 45. Mortimer S.A., Doudna J.A.. Unconventional miR-122 binding stabilizes the HCV genome by forming a trimolecular RNA structure. Nucleic Acids Res. 2013; 41:4230–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Henke J.I., Goergen D., Zheng J., Song Y., Schüttler C.G., Fehr C., Jünemann C., Niepmann M.. microRNA-122 stimulates translation of Hepatitis C Virus RNA. EMBO J. 2008; 27:3300–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.