


Abstract
Epigenetic regulation is important for establishing lineage-specific gene expression during early development. Although signaling pathways have been well-studied for regulation of trophectoderm reprogramming, epigenetic regulation of trophectodermal genes with histone modification dynamics have been poorly understood. Here, we identify that plant homeodomain finger protein 6 (PHF6) is a key epigenetic regulator for activation of trophectodermal genes using RNA-sequencing and ChIP assays. PHF6 acts as an E3 ubiquitin ligase for ubiquitination of H2BK120 (H2BK120ub) via its extended plant homeodomain 1 (PHD1), while the extended PHD2 of PHF6 recognizes acetylation of H2BK12 (H2BK12Ac). Intriguingly, the recognition of H2BK12Ac by PHF6 is important for exerting its E3 ubiquitin ligase activity for H2BK120ub. Together, our data provide evidence that PHF6 is crucial for epigenetic regulation of trophectodermal gene expression by linking H2BK12Ac to H2BK120ub modification.
INTRODUCTION
Epigenetic regulators including writers, readers and erasers function to determine cell fate as critical players for the crosstalk of histone modifications and regulation of transcriptional memory (1–6). The gene expression program in cell fate determination is tightly regulated, and each cell exhibits a unique gene expression signatures achieved by activating or silencing lineage-specific genes (7–10). When embryonic stem cells (ESCs) are differentiated into three germ layer lineages, the lineage-specific gene expression is marked by various histone modifications. In particular, the role of H3K4me3 as a transcriptional activation mark is implicated in cell fate decision and stem cell maintenance by specifically exhibited on the promoter of lineage-specific genes (11,12). Further, crosstalk between several histone modifications elaborates on the dynamics of transcription regulation for stem cell maintenance and differentiation. For example, increased H2BK120ub levels lead to open and accessible chromatin conformations during stem cell differentiation (13,14), and trigger subsequent H3K4me3 modification for transcriptional activation (15). Ash2l, one of the core subunits of mixed-lineage leukemia (MLL) complex, is known to act as a reader for H2BK120ub, thereby linking H2BK120ub to H3K4me3 on the active promoters during ESC differentiation (16). Therefore, precise H2BK120ub regulation is essential for ESC differentiation, although the underlying mechanisms on how to determine specific lineages remain largely unknown. Notably, levels of histone acetylation are almost undetectable in pluripotent ESCs but significantly increase when stem cells leave the undifferentiated states (17–19). However, the precise mechanism whereby stem cells proliferate and differentiate into specific lineage by linking histone acetylation to H2BK120ub and/or H3K4me3, or even what epigenetic regulators determine the fate of these stem cells, is not yet understood.
Epigenetic readers are key players of histone modification crosstalk by recognizing various posttranslational modifications (PTMs) and serving as platforms for loading various epigenetic regulators. There are several domains for reading specific PTMs, including bromodomain for acetylation and chromodomain for methylation. Besides, the PHD finger mainly recognizes methylated lysine on histones such as H3K4me2/3, which is associated with the transcription start site of active genes (20). PHD fingers read combinatorial histone PTMs, thereby playing a critical role in histone modifications crosstalk. PHD fingers belong to the Zinc-binding superfamily and the tandem PHD domain detects two zinc ions by conserved cysteine and histidine residues. Unlike other epigenetic reader domains, PHD fingers are known to be relatively diverse in functions or structures. PHD fingers of some proteins recognize non-methylation of H3 lysine 4 (21,22). Some kinds of PHD fingers exist as double form or extended form, which captures four zinc ions or three zinc ions, respectively (23). Moreover, histone PTM-reading capability of PHD fingers is further improved by tandem combination of other enzymatic motifs (24,25). These cooperative abilities enable PHD fingers to have a functional diversity, thereby playing a critical role in crosstalk of histone modifications.
Of the PHD finger-containing protein (PHF) families (26), PHF6, which has two extended PHDs, is known as a key factor in leukemia with well-known leukemia disease-driving mutations (27,28). PHF6 functions as a transcriptional regulator by interacting with histones in leukemia (29), implicating the role of PHF6 as a reader of histone modification. However, the extended PHD2 domain of PHF6 has been shown to read double-stranded DNA but not histones in vitro (30). Furthermore, genetic mutations in PHF6 have been found to induce Börjeson–Forssman–Lehmann (BFL) syndrome (31–33). Patients with this syndrome show symptoms like irregular teeth, intellectual disability, and hypogonadism, which are typical features of early developmental defects. It has been shown that homozygote Phf6-knockout (KO) mice exhibit perinatal lethality, indicating that PHF6 plays an important role during developmental process (34).
Although there is a substantial understanding on the functional consequences of H2B ubiquitination in ESC differentiation, relatively little is known about the preceding inputs of PTM that modulate the H2BK120ub mark. The established enzymatic complex for introducing H2BK120ub in mammals consists of the E2 enzyme UBCH6 and the heterodimeric RING-type E3 ligase RNF20/40 (35). The enzymatic machinery that initiates H2BK120ub operates on chromatin that is compositionally varied by several histone PTMs (36). Which pre-existing histone modifications influence the levels of H2BK120ub in ESC differentiation and the players connecting the prior PTM to H2BK120ub are mostly unidentified thus far. Here, we identify PHF6 as an E3 ubiquitin ligase for mono-ubiquitination of H2BK120 through extended PHD2 domain-mediated interaction with H2BK12Ac. Our results provide evidence that PHF6 functions as a pivotal regulator of histone crosstalk during the differentiation of ESCs into a trophectodermal lineage.
MATERIALS AND METHODS
Cell culture and reagents
A conditional Oct4-depleted (ZHBTc4) mouse ESC line was described previously (37). Briefly, in the first 1–2 passages, ZHBTc4 ESCs were maintained on mouse primary embryonic fibroblast feeders. After stabilization, ESCs were cultured under feeder-free conditions, using 0.1% gelatin-coated culture dishes. ESCs were maintained in Dulbecco's modified Eagle's medium (DMEM; Welgene), supplemented with 15% fetal bovine serum (FBS; Hyclone), 0.055 mM β-mercaptoethanol, 2 mM l-glutamine, 0.1 mM non-essential amino acids, 5,000 units/ml of penicillin/streptomycin (GIBCO) and 1000 units/ml of leukemia inhibitory factor (LIF) (Chemicon) in a humidified incubator at 37°C with 5% CO2. All cell lines used in the study were regularly tested for mycoplasma contamination. For trophectoderm reprogramming of ZHBTc4, 0.5 μg/ml doxycycline (DOX) (#D3072, SIGMA) was treated for 2 days. For neural ectoderm differentiation of ZHBTc4, 10 μg/ml all-trans-retinoic acid (RA) (#R2625, SIGMA) was treated in culture condition without LIF for 4 days. Transfection experiment in mouse ESCs was performed using Lipofectamine 3000 (Invitrogen) according to the manufacturer's protocol. Small interfering RNA (siRNA) sequences for knockdown of CBP and p300 were designed from http://gesteland.genetics.utah.edu/siRNA_scales. siRNA sequence for knockdown of RNF20 was used as previously reported (38). The following antibodies were used: anti-PHF6 (#68262) from Novus; anti-Nanog (ab21624), anti-H3 (ab1791), anti-H2B (ab1790), anti-H4(ab10158), anti-H3K4me3 (ab8580), anti-H3K4me1 (ab8895), anti-H3K27me3 (ab6002), anti-H3K27Ac (ab4729), anti-H3K9me3 (ab8898), anti-H2BK12Ac (ab195494), anti-RNF40 (ab191309) and anti-Cdx2 (ab157524) from Abcam; anti-USP44 (sc-377203), anti-p300 (sc-584), anti-CBP (sc-1211), anti-Oct4 (sc-5279), anti-GAPDH (sc-25778) and anti-GST (sc-459) from Santa Cruz biotechnology; anti-RNF20 (A300-714A) from Bethly laboratories; anti-H2BK120ub (#39623) from Active Motif; anti-Flag (#F3165) and anti-β-actin (#A1978) from Sigma; anti-His (#G020) from Abm.
Short-hairpin RNAs (shRNAs)
To generate shRNA knockdown cells, lentiviral shRNA constructs were transfected along with viral packaging plasmids (psPAX2 and pMD2.G) into HEK293T cells. 1 day after transfection at 60–70% confluency of cells, transfected cells were changed new fresh media and maintained 24 h for virus collecting. After virus collecting, the media were filtered and mixed with 4× Lenti-X collector (#631232, TAKARA), and incubated at 4°C overnight. Cells were infected by concentrated virus with polybrene. shRNA sequences were as follows:
shPhf6
fwd 5′-CCGGGTTCAGCTCACAACAACATCACTCGAGTGATGTTGTTGTGAGCTGAACTTTTTG-3′ rev 5′-AATTCAAAAAGTTCAGCTCACAACAACATCACTCGAGTGATGTTGTTGTGAGCTGAAC-3′.
shRnf20
fwd 5′- CCGGCGCATCATCCTTAAACGTTATCTCGAGATAACGTTTAAGGATGATGCGTTTTTG-3′ rev 5′- AATTCAAAAACGCATCATCCTTAAACGTTATCTCGAGATAACGTTTAAGGATGATGCG -3′.
shRnf40
fwd 5′- CCGGGACCACTCTAATCGAACCCATCTCGAGATGGGTTCGATTAGAGTGGTCTTTTTG-3′ rev 5′- AATTCAAAAAGACCACTCTAATCGAACCCATCTCGAGATGGGTTCGATTAGAGTGGTC-3′.
Generation of Phf6 KO mouse ESCs
We used CRISPR-Cas9 system for Phf6 KO mouse ESCs. Single guide RNA (sgRNA) designs for Phf6 targeting were performed from GPP sgRNA Designer (CRISPRko) (39). Selected sgRNAs were cloned into the pRGEN-U6 vector, and transfected into ZHBTc4. After single colony selection with puromycin treatment, we obtained Phf6 KO colonies, checked by immunoblotting, and confirmed frame-shift mutation by Sanger-sequencing.
Embryoid bodies (EB) formation
From ZHBTc4 ESCs, EBs were created by the hanging-drop method, starting with 1000 cells/drop. After 2 days in the drop state, each drop begins to grow on a non-coated sterile cover glass for immunocytochemistry, and on a round-shaped 96-well plate for live-cell imaging. In all culture situations, media were fed through addition per day and plate movement was also fixed to a minimum for minimizing the physical impact on the EBs. All EBs were cultured using ESC media without LIF.
Alkaline phosphatase (AP) staining
For AP staining, wild-type (WT) and Phf6 KO ZHBTc4 were cultured on the 0.1% gelatin-coated cover glass in 12-well plates. 104 cells were seeded per well. After 2 days, AP staining was performed using the Alkaline Phosphatase Detection Kit (#SCR004, Millipore). Staining was performed according to the provided protocol. Briefly, each cell was fixed by 4% paraformaldehyde for 1–2 min, and react with the solution (Naphthol/Fast Red Violet mix) for 15–20 min at room temperature. After the reaction, washout solutions and make slides for imaging.
Live cell imaging
For tracking growth of ZHBTc4 ESCs and EBs, JuLI Stage real-time cell history recorder (NanoEnTeK) was used. For tracking ESC growth, 104 cells were seeded into 0.1% gelatin-coated 12-well culture dishes, and cell confluency was recorded every 12 h. After the recording is complete, cell growth curves were analyzed by software provided by JuLI. In the case of EB formation, each drop was moved into each well of 96-well round-shaped dishes and cell morphology was recorded every 12 h. To calculate the out-layer cell area, images taken at 7 days were calculated using the ImageJ program.
Immunocytochemistry
EB spheroids for immunofluorescence microscopy were grown on coverslips prior to the experiment (1 EB spheroid per coverslip). EBs were fixed with 2% paraformaldehyde in PBS for 10 min and then were washed two times with DPBS at room temperature. Fixed cells were permeabilized with 0.5% Triton X-100 in PBS (PBS-T) for 5 min at room temperature. Blocking was performed with 10% FBS in 0.1% PBS-T for 30 min. For staining, cells were incubated with primary antibodies for 4 h at room temperature, washed four times with 0.1% PBS-T, and incubated for 1 h with fluorescent labeled secondary antibodies (Invitrogen). Cells were washed and mounted by VECTASHIELD (H-1200, Vector Laboratories) with DAPI (Sigma). Fluorescence was visualized on a Zeiss LSM700 confocal microscope (Carl Zeiss).
Whole-cell lysate preparation
All cells were briefly rinsed with ice-cold PBS before collection. For whole-cell lysates, the cells were resuspended in the lysis buffer (50 mM Tris–HCl pH 8.0, 200 mM NaCl and 0.5% NP-40) supplemented with protease inhibitors and sonicated using a Branson Sonifier 450 at output 4 and a duty cycle of 20 for 10–12 pulses. All lysates were quantified by the Bradford method and analysed by SDS-PAGE.
Far-Western blot analysis
GST-PHF6 proteins were purified using GST beads in Escherichia coli. Histone extracts from ZHBTc4 ESCs were separated by SDS-PAGE. After separating histone octamers in order of H3, H2B, H2A and H4 according to their sizes, 0.1 μg/μl of purified GST-PHF6 proteins were added to binding buffer (100 mM NaCl, 20 mM Tris–HCl [pH 7.6], 10% glycerol, 0.1% Tween-20, 50 μM ZnCl2, 2% skim milk powder and 1 mM DTT), and allowed to conjugate with histone extracts on membrane. After binding of GST-PHF6 on histones, immunoblot analysis was performed using anti-GST antibody.
In vitro histone peptide pulldown assay
0.5 μg of biotinylated histone peptides mixed with 0.3 μg of GST-eluted proteins in assay buffer (250 mM NaCl, 50 mM Tris–HCl [pH 7.5], 0.05% NP-40 and 50 μM ZnCl2) were incubated overnight. Then, 50% slurry of streptavidin beads were added and further incubated for 1 h. After removing non-specific interactions by washing, beads were boiled in sampling buffer and peptide–protein interaction was detected by immunoblotting. Biotinylated histone peptides of modified histone H3 were purchased from Boston BioChems, and those of modified histone H2B were from JPT peptide technologies.
In vitro histone peptide binding array
Histone peptide binding array kit was purchased from ActiveMotif (#13005). The following assays were progressed according to the provided protocol. Briefly, blocking a kit by 5% milk in TTBS (10 mM Tris–HCl [pH 7.5], 150 mM NaCl, and 0.05% Tween-20) at 4°C overnight. After blocking, the kit was incubated with 0.3 μg of eluted GST-PHF6 in binding buffer (100 mM KCl, 20 mM HEPES [pH 7.9], 1 mM EDTA, 10% glycerol and 0.1 mM DTT) at room temperature for 1 h. Then, GST primary antibody and secondary antibody were incubated on the kit sequentially at room temperature for 1 h. Detection was by ECL develop. Washed three times with TTBS between each step.
Protein expression and purification
PHF6 WT and E223S mutant (MT) were expressed as GST fusion proteins in E. coli Rosetta (DE3) cells. Cells were harvested after growing at 20°C overnight following induction with 0.25 mM isopropyl 1-thio-β-d-galactopyranoside (IPTG), and lysed using Emulsiflex C3 (Avestin) in phosphate buffered saline (PBS) supplemented with 500 mM NaCl and 1 mM phenylmethanesulfonyl fluoride (PMSF). After centrifugation at 14 000 rpm for 15 min, the supernatant was loaded onto a glutathione agarose column (Thermo Scientific). After column washing with equilibrium buffer (20 mM HEPES–NaOH [pH 7.5] and 300 mM NaCl), each protein was eluted with elution buffer (100 mM HEPES–NaOH [pH 7.5], 300 mM NaCl and 20 mM reduced glutathione). Each GST-PHF6 (WT and E223S) was further purified with a HiTrap SP cation exchange column followed by a Superdex 200 size exclusion column that was pre-equilibrated with gel filtration buffer (20 mM HEPES–NaOH [pH 7.5] and 150 mM NaCl). For the control experiment, GST was expressed in E. coli Rosetta (DE3) cells and purified similarly as described above, except for using a HiTrap Q anion exchange column instead of the HiTrap SP column.
MicroScale thermophoresis (MST) measurement
Binding affinity measurement was performed with Nanotemper Monolith NT.115pico. H2B peptides (1:20) WT and those with K12Ac modification were purchased from Genscript and each GST-PHF6 (WT and E223S MT) and GST were labeled with the dye NT-647 (Cy5) (Lumiprobe). Labeled GST and GST-PHF6 were used at a concentration of ∼90 nM. Each H2B peptide was diluted from ∼880 μM to 26 nM in MST buffer (20 mM HEPES–NaOH [pH 7.5], 150 mM NaCl, 0.05% Tween 20 and 0.5 mg/ml BSA) and incubated with labeled protein at room temperature for 10 min. MST measurements were performed at 22°C with 40% MST power and 25% LED power (GST-PHF6 WT) or 12% LED power (GST-PHF6 E223S and GST). Each data set was analyzed using the MO. Affinity Analysis Software (Nanotemper technologies).
Micrococcal nuclease (MNase) digestion
For making nucleosomes without H2BK12Ac or H2BK120ub, H2B-Flag WT/K12R/K120R were transfected into cells. Then, cells were lysed by MNase lysis buffer (50 mM Tris–HCl [pH 7.5], 1 mM CaCl2, 0.2% NP-40, Inhibitor Complex) and treated 50 U MNase (#M0247S, NEB) for making mono-nucleosomes. After MNase digestion, H2B-Flag containing mono-nucleosomes were pull down by Flag-M2 bead overnight, then these mono-nucleosomes were eluted by 3× Flag peptides (#F4799, Sigma).
In vitro ubiquitination assay
For substrates, H2B-Flag WT/K12R were transfected into Phf6 KO ZHBTc4 and H2B-Flag containing mono-nucleosomes were purified by MNase digestion. After purification, these nucleosomes were mixed with E1, E2, E3 enzyme, ubiquitin, 50 μM ZnCl2, and 10× buffer (500 mM Tris–HCl [pH 7.5], 20 mM ATP, 10 mM MgCl2, 2 mM DTT) in 50 μl volume, then incubated at 37°C for 1hr. To stop the reaction, sampling buffer was added and samples were boiled at 100°C for 10 min. E1, E2 (UBCH3, UBCH6), and ubiquitin were purchased from Boston Biochems. Purified GST-PHF6 WT/ MTs were considered as E3 ligases in reaction.
In vitro GST-pulldown assay
To check the interaction between PHF6 and UCBH3 as E2 and E3 relations, we ubiquitinated His-UBCH3 by in vitro ubiquitination assay. For UBCH3-ubiquitin conjugation, 50 ng of E1, 0.5 μg of His-UBCH3 and ubiquitin were incubated with the ubiquitination assay buffer in 30 μl volume at 37°C for 1 h. Next, 1 μg of ubiquitin-conjugated His-UBCH3 were incubated with bead-bound GST-only or GST-PHF6 WT or MTs in 1 ml volume of the pulldown-assay buffer (20 mM Tris–HCl [pH 7.8], 125 mM NaCl, 10% glycerol, 0.1% NP-40, and protease inhibitors) at 4°C, overnight. After reactions, washout the beads with the same buffer four times and boiled beads with sampling buffer.
Chromatin-Immunoprecipitation (ChIP) and MNase-ChIP assays
The ChIP assays were basically conducted as previously described (40). For ChIP assay, cells were cross-linked by 1% formaldehyde at RT for 15 min. Nuclear pellets were sonicated in RIPA buffer. For MNase-ChIP, nuclear pellets were lysed in MNase lysis buffer, and reacted with MNase at 37°C for 10 min instead of sonication. Reverse-crosslinking was performed at 65°C, overnight. For reverse-crosslinking and immunoblotting of MNase-ChIP samples, immunoprecipitated beads were mixed with 2× sampling buffer and boiled at 100°C, 45 min. DNAs were isolated by purification columns (#28105, QIAGEN). Eluted DNAs were detected by quantitative RT-PCR (qRT-PCR). All reactions were performed as triplicates. ChIP-qRT-PCR primers used in this study were annotated in Supplementary Table S1.
Quantitative RT-PCR
Total RNAs were extracted using Trizol (Invitrogen) and reverse transcription was performed from 1 to 2 μg of total RNAs using the M-MLV cDNA Synthesis kit (Enzynomics). The abundance of mRNA was detected by an ABI prism 7500 system or BioRad CFX384 with SYBR TOPreal qPCR 2X PreMix (Enzynomics). The quantity of mRNA was calculated using ddCt method and Gapdh and β-actin were used as controls. All reactions were performed as triplicates. The qRT-PCR primers used in this study were annotated in Supplementary Table S1.
RNA-sequencing analysis
Total RNAs were extracted from WT and Phf6 KO cells, respectively, in the presence or absence of DOX. Then, the stranded mRNA-seq library was prepared following Illumina's TruSeq protocol. After the production of raw data by HiSeq platform, the reads were pre-processed to remove adaptors and bases with low quality by Trimmomatic (v0.36) (41). Next, STAR (v2.5.3) was used to align the reads (42), and Transcripts Per Million (TPM) was calculated with RSEM (v1.3.0) (43). Using read counts per gene, differentially expressed genes (DEG) were identified with DESeq2 (v1.18.1) for all six possible pair-wise comparisons between WT and Phf6 KO under normal condition, DOX treatment and RA treatment (44). k-means clustering was performed for the union of four sets of DEGs in R (v 3.4.3). For Gene Set Enrichment Analysis (GSEA), a phenotype label was defined as 2:2:50:10 = WT-DOX:KO-DOX:WT + DOX:KO + DOX, and Pearson correlation coefficient per gene was used for ranking. Enrichment score was then computed for the gene sets in molecular signature database (MSigDB) v6.2. (45,46). To analyze the difference in DOX reactivity between WT and Phf6 KO cells for each cluster, we first obtained the z-score mean of the genes in each cluster for each sample. Thereafter, the difference in the mean values according to the presence or absence of DOX in each of the WT and Phf6 KO cells was determined, which was regarded as the DOX reactivity of WT and Phf6 KO cells. Finally, the differences in DOX reactivity between WT and Phf6 KO cells by clusters were calculated and considered to be DOX reactivity according to the presence or absence of PHF6 per cluster.
Response difference = |(∑(+DOX z-scores) – ∑(–DOX z-scores) in WT)/n – (∑(+DOX z-scores) – ∑(–DOX z-scores) in KO)/n |
Statistical analysis
All experiments were performed independently at least three times. Values are expressed as mean ± SEM. Significance was analyzed using ANOVA test. A P-value of <0.05 was considered statistically significant.
RESULTS
Phf6 deficiency impairs trophectoderm reprogramming
To examine the potential role of PHF6 during early development, we generated Phf6 KO ESCs and explored whether deletion of Phf6 affects ESC pluripotency and differentiation (Figure 1A). We used ZHBTc4 mouse ESC line, which is engineered to allow inducible depletion of Oct4 by tetracycline (Tc) or DOX treatment. It has been established that ESCs are reprogrammed into early trophectoderm lineage after suppression of Oct4 (47,48). Thus, this inducible system of ZHBTc4 has an advantage of simultaneously mimicking the whole lineages of early-stage embryos in vitro. Immunoblot analysis revealed that PHF6 expression was completely depleted in Phf6 KO ZHBTc4 ESCs (hereafter Phf6 KO ESCs) (Figure 1B). Knockdown of Phf6 by shRNA also showed significantly decreased PHF6 expression in ZHBTc4 ESCs (Figure 1B).
Figure 1.

Phf6 deficiency caused defects in trophectoderm differentiation from ESCs. (A) Schematics of the strategy for Phf6 gene knockout (KO) using guide RNA and the CRISPR-Cas9 nuclease system. The guide RNA sequence and altered sequences of Phf6 KO ESCs are indicated. (B) Immunoblot analysis showing PHF6 depletion in Phf6 KO ESCs and Phf6 knockdown by shRNA in ESCs. Nanog as a positive control, and β-actin as a loading control. (C) Growth curves of Phf6 KO ESCs compared to those of WT ESCs and growth rate curves of shPhf6 in ESCs compared to those of shNS in ESCs, respectively. (D) Comparison of pluripotency measured by AP staining in WT, Phf6 KO, shNS, and shPhf6 ESCs. Images were photographed at 10× magnification. (E) Schematics of DEG analysis between WT and Phf6 KO ZHBTc4 during DOX-induced trophectoderm reprogramming and RA-induced neural ectoderm differentiation. (F) Comparison expressions of Top 50% genes (n = 7389) between WT and Phf6 KO ZHBTc4. Cutoff (|fold change of (Phf6 KO versus WT)| = 3) was shown in red dotted line. Significant down-regulated genes were shown in red dot (n = 15), up-regulated genes shown in blue dot (n = 1). (G) Gene Ontology (GO) analysis of chemical-dependent induced genes between DOX treatment and RA treatment. Genes with fold-change >3 were selected as significantly up-regulated genes by chemical treatment. Cutoff of significant biological process = log10(adjusted P-value = 0.1). (H) GO analysis of Phf6-dependently expressed genes in chemical-induced genes. Cutoff of significant biological process = log10(adjusted P-value = 0.05).
We compared WT and Phf6 KO ESCs to determine whether PHF6 affects key features of ESCs, including self-renewal and pluripotency. The cellular growth rate of WT and Phf6 KO ESCs did not differ significantly from each other (Figure 1C). Furthermore, alkaline phosphatase (AP) activity between WT and Phf6 KO ESCs to examine the effect of PHF6 on pluripotency of ESCs revealed comparable AP staining without loss of ESC morphological features (Figure 1D), indicating that PHF6 deficiency did not affect ESC pluripotency. Consistent with the results of Phf6 KO ESCs, the pluripotency and growth properties of Phf6 shRNA-knockdown ESCs did not significantly differ from those of control shRNA-knockdown ESCs (Figure 1C and D). Next, we compared the transcriptome of WT and Phf6 KO ESCs by performing mRNA-sequencing and differentially expressed gene (DEG) analysis (Figure 1E). We found that expressions of only a few genes were affected by PHF6, but not overall expressions of top 50% highly expressed genes, in ESCs (Figure 1F). Further, the expressions of several ESC marker genes, including Oct4, Nanog, Sox2 and Klf4, were not affected by Phf6 depletion (Figure 1F). Together, these data indicate that PHF6 is not responsible for maintaining ESC pluripotency.
Next, in order to investigate whether PHF6 has lineage-specific roles during differentiation, we compared gene expression profiles between WT and Phf6 KO ESCs treated with DOX or retinoic acids (RA), which reprograms ESCs into early trophectoderm or differentiates ESCs into neural ectoderm, respectively (Figure 1E). We performed Gene Ontology (GO) analysis for significantly up-regulated genes by DOX or RA treatment. The GO terms indicated the reprogramming of ESCs to trophectoderm lineage, which is placenta development by DOX treatment, and differentiation of ESCs to neural ectoderm lineage, which is nervous system development by RA treatment (Figure 1G). We then identified DEGs by comparing WT and Phf6 KO under DOX or RA treatment. As expected, only a few genes were the shared DEGs between the different differentiation states. Interestingly, GO analysis revealed that the DEGs of Phf6 KO under DOX treatment were significantly enriched in placenta development, whereas the DEGs of Phf6 KO under RA treatment showed no significant functional enrichment (Figure 1H). Taken together, these results indicate that PHF6 functions in a lineage-specific manner and has an important role in trophectoderm reprogramming.
PHF6 is a transcriptional activator for trophectodermal genes
To further identify PHF6-dependent gene clusters during trophectoderm reprogramming, we performed unsupervised k-means clustering (k = 6) for DEGs between WT and Phf6 KO ESCs with or without DOX treatment (Figure 2A). In each cluster, we measured the differences in DOX response between WT and Phf6 KO ESCs. Cluster 1 showed the strongest differential DOX response between WT and Phf6 KO ESCs (Figure 2B), suggesting that regulation of gene expression within cluster 1 is highly dependent on PHF6. Gene expression within cluster 1 exhibited little or no difference between WT and Phf6 KO in the ESC states (-DOX) but showed considerable decrease in Phf6 KO ESCs after induction during reprogramming states (+DOX) (Figure 2C). In addition, we also found that the differential expression between WT and Phf6 KO for DOX-repressed clusters (clusters 2 and 4) was not as high as that of DOX-activated clusters (Figure 2B). The most significant term of GO for cluster 1 was placenta development that is biologically associated with trophectoderm reprogramming (Figure 2D). These results indicate that PHF6 functions as a transcriptional activator during trophectoderm reprogramming.
Figure 2.

RNA-seq analysis reveals PHF6 as a transcriptional activator for trophectodermal genes. (A) Heatmap of k-means clustering of variably expressed genes in WT and Pfh6 KO ESCs with or without DOX treatment (n = 1775, k = 6). Genes were grouped into six clusters on the basis of expression similarity. (B) Differences in DOX response for WT and Phf6 KO ESCs. DOX response is calculated using average of z-score differences of genes for DOX treatment in ESCs. (C) z-score centroids of Cluster 1 genes. The center of the cluster is highlighted as red. (D) Functional Gene Ontology (GO) analysis of Cluster 1. Placenta development was the most enriched terms in Cluster 1. (E) Representative gene set enrichment analysis (GSEA) of DEGs. The gene sets involved in embryonic placenta development and trophoblast giant cell differentiation were significantly enriched according to phenotypic labels. The pattern of media gene expression from centroids (D) was used for standards of significance. (F) Trophectodermal genes that were down-regulated in Phf6 KO ESCs in comparison with WT ESCs. The color bar represents the gradient of log2-fold-changes in each comparison. Positively correlated genes from GSEA were selected. (G) qRT-PCR analysis of trophectodermal genes in WT and Phf6 KO ESCs with or without DOX treatment. mRNA levels of each gene were determined as relative values for Gapdh and relatively compared based on WT - DOX. Statistical significance was calculated by ANOVA test (*P < 0.05, **P < 0.01, ***P < 0.001).
To identify the subset of genes whose expression depends on PHF6 during trophectoderm reprogramming, Gene Set Enrichment Analysis (GSEA) was performed by correlating with PHF6 dependency in cluster 1. GSEA revealed that the gene sets for embryonic placenta development and trophoblast giant cell differentiation were significantly enriched in cluster 1 (Figure 2E). From the gene sets, PHF6-dependent genes included Cdx2, Gata2, Esx1, Plac1, Ascl2, and Wnt7b that are crucial for placenta development (Figure 2F). qRT-PCR analysis confirmed that the trophectodermal genes are regulated in PHF6-dependent manner (Figure 2G). Together, the transcriptome profiles indicate that PHF6 directly activates the expression of genes involved in trophectoderm lineage determination.
PHF6 activates trophectodermal genes for trophectoderm lineage determination
Since PHF6 is responsible for transcriptional activation of genes for trophectoderm lineage determination, we tested the possibility that PHF6 plays an important role in trophectoderm lineage determination process. For this, we utilized embryoid body (EB) formation method and examined the role of PHF6 in differentiation process by comparing EBs derived from Phf6 KO and WT ESCs. It has been shown by previous studies that differentiating EBs by adherent culture methods could generate not only the EB core that is differentiated into three germ layers but also outer cell layer that is differentiated into trophectoderm lineages (49–51). By employing this method, we found that Phf6 KO EBs failed to form the outer cell layer in contrast to WT EBs (Figure 3A). CDX2 has been known as a master transcription factor of trophectoderm lineage specification and differentiation by counteracting function of Oct4 (52). Immunostaining of WT EBs with an antibody against CDX2, a trophectoderm marker, showed that CDX2 is highly expressed in the outer cell layer of the EBs (Figure 3B). Intriguingly, PHF6 coexpressed with CDX2 in the outer cell layers, including the migratory zone (Figure 3C). Furthermore, the mRNA levels of trophectodermal genes including Cdx2, Plac1, Ascl2 and Gata2 during EB differentiation in Phf6 KO EBs were not significantly as high as those in WT EBs (Figure 3D). These data indicate that PHF6 is crucial for transcriptional activation of trophectodermal genes for differentiation into trophectoderm lineages.
Figure 3.
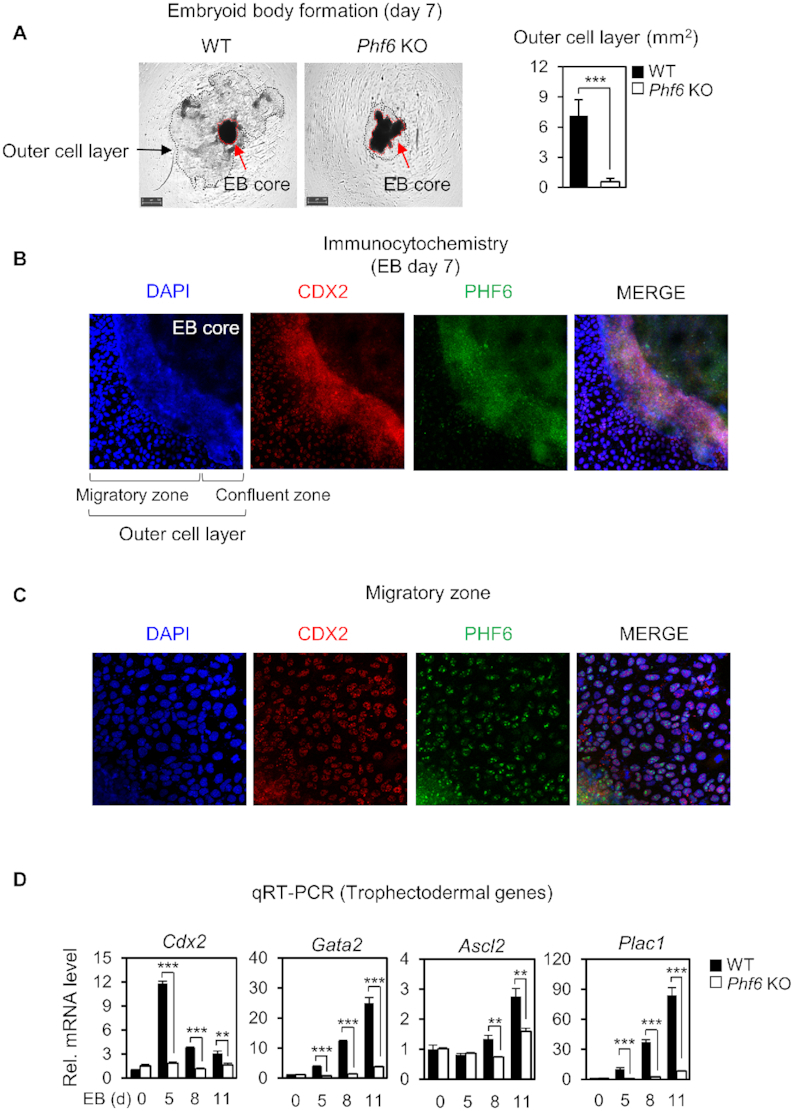
PHF6 activates trophectodermal genes for trophectoderm lineage determination. (A) Morphological features of EBs from WT and Phf6 KO ESCs on Day 7 (left). Each EBs were grown and observed separately. Magnification ×10. The average areas (mm2) of the outer cell layer were compared between WT (n = 11) and Phf6 KO (n = 9) EBs (right). Statistical significance was calculated by ANOVA test (*P < 0.05, **P < 0.01, ***P < 0.001). (B, C) Immunostaining of CDX2 and PHF6 in the outer cell layer in WT EBs. Magnification ×100 (B) and ×200 (C). (D) qRT-PCR analysis of the trophectodermal genes during EB differentiation in WT and Phf6 KO ESCs. WT and Phf6 KO EBs were maintained without LIF and harvested at the indicated days. mRNA levels of each gene were determined as relative values for Gapdh or β-actin and relatively compared based on WT day 0. Statistical significance was calculated by ANOVA test (*P < 0.05, **P < 0.01, ***P < 0.001).
PHF6 regulates the H2BK120ub levels via preceding H2BK12Ac recognition
Next, we examined whether PHF6 plays a role in transcriptional regulation of trophectodermal genes by regulating histone recognition and modification. For this, we performed far-western blot analysis on histone extracts obtained from ZHBTc4 ESCs along with recombinant proteins including GST-PHF6, to check the interaction between PHF6 and histones, and potential specific histone modifications that can be recognized by PHF6. As a result, we found that histones H2B and H3 selectively interacted with recombinant GST-PHF6 proteins (Figure 4A). Next, we applied an in vitro peptide binding array to identify specific modifications of H2B and H3 recognized by PHF6. Consistent with previous far-western blot data, the in vitro peptide binding array confirmed the specific binding of PHF6 to peptides of histones H2B and H3. The top five enriched histone modifications were H2BK15Ac, H3K27Ac, H2BK12Ac, H3R26me2a and H3K27me2 (Figure 4B). Further, we conducted an in vitro peptide pulldown assay for each H2B or H3 modification to identify specific histone modification recognized by PHF6, and confirmed that PHF6 specifically binds H2BK12Ac in vitro (Figure 4C).
Figure 4.

PHF6 reads H2BK12Ac and writes mono-ubiquitination of H2BK120. (A) Far-western blotting analysis of histone proteins with either GST or GST-PHF6 proteins. Assays were performed on histone extracts obtained from ZHBTc4 ESCs. Histone extracts were separated by H3, H2B, H2A and H4 according to size on the SDS-PAGE gel. (B) Top five ranked histone modifications that show the highest affinity with GST-PHF6. The screening was performed by using a histone peptide array kit. (C) Histone peptide pulldown analysis was performed with GST-PHF6 for binding of the top five modified histone peptide candidates and nearby modifications. (D) Amino acid sequences of the extended PHD1 (ePHD1) and extended PHD2 domains (ePHD2) of PHF6 orthologues in diverse species are aligned. The amino acids with red characters indicate the negatively charged region in the ePHD2. The amino acid alignment was performed the ClustalX. (E) In vitro peptide binding assay was performed with GST-PHF6 WT or E223S MT. (F) MST binding curves of H2B peptides (1:20) (non-modified and K12Ac) with GST-PHF6 WT and E223S mutant (left axis) and GST (right axis) as a negative control. Error bars represent the standard deviation of three independent experiments. The measured KD value is shown for each binding curve. * N.B. represents no apparent binding. (G) Immunoblot analysis conducted using the indicated antibodies in WT and Phf6 KO ESCs with DOX treatment. (H) Immunoblot analysis performed using the indicated antibodies in ESCs in the absence or presence of siRNA against p300 or CBP after DOX treatment. (I) Comparison of correlation between K12Ac and K120ub using H2B K12R / K120R MTs. MNase digestion was performed to elute mono-nucleosomes containing these H2B WT and MTs, and the modification states of these ectopic-H2B containing mono-nucleosomes were confirmed by immunoblot analysis. (J) RNF20 and RNF40 protein levels by immunoblot between WT and Phf6 KO ESCs with DOX treatment. (K) Co-immunoprecipitation assay was performed to detect the interaction between the endogenous PHF6 with USF44, RNF20, or RNF40 in ESCs with or without DOX treatment. (L) Immunoblot of H2BK120ub levels between knockdown of Phf6, Rnf20, and Rnf40 with or without DOX treatment. (M) qRT-PCR analysis of Cdx2, and Gata2 in Phf6, Rnf20 and Rnf40 knockdown ESCs with DOX treatment. mRNA levels of each gene were determined as relative values for Gapdh and relatively compared based on shNS + DOX. Statistical significance was calculated by ANOVA test (*P < 0.05, **P < 0.01, ***P < 0.001). (N) ChIP assays were performed on the promoters of Cdx2 and Gata2 using anti-H2BK120ub antibody in Phf6, Rnf20, and Rnf40 knockdown ESCs with DOX treatment. Statistical significance was calculated by ANOVA test (*P < 0.05, **P < 0.01, ***P < 0.001).
To further clarify whether the ability of PHF6 to read H2BK12Ac is conferred by its specific domains exemplified by extended PHDs, we compared the amino acid sequences of the two extended PHDs of PHF6. Intriguingly, the extended PHD2, but not the extended PHD1, was found to contain a portion of the negatively charged amino acids (Figure 4D). In the case of the acetylation-recognition by PHD, it was reported that the carbonyl oxygen of the negatively charged amino acid is important to interact with the acetyl amide of acetylation (53). The crystal structure of the extended PHD2 indicates that four glutamic acids (E219, E220, E221 and E223) with negative charges are key residues forming an acetylated substrate recognition motif. Among the E to S MTs, which impair the charge without inducing structural perturbations of the protein, the E223S MT caused significant loss of H2BK12 acetyl-lysine recognition in vitro (Figure 4E). In addition, binding affinity measurements using MST analysis showed that GST-PHF6 WT has ∼5-fold higher affinity for H2BK12Ac peptide than H2B WT peptide, whereas GST-PHF6 E223S bind both WT and K12Ac peptides with similarly low affinities (Figure 4F). These data indicate that the glutamic acid-rich motif in the extended PHD2 of PHF6 is crucial for H2BK12Ac recognition.
Since H2BK12Ac is a transcriptional activation marker (54), we hypothesized that acetyl reading activity of PHF6 is crucial for regulating epigenetic changes of lineage-specific genes. Therefore, we examined various histone modifications including H2BK120ub, H3K4me3, H3K27me3, H3K9me3 and H2BK12Ac in WT and Phf6 KO ESCs. Among them, the H2BK120ub levels were considerably reduced in Phf6 KO ESCs compared to those in WT ESCs upon DOX treatment (Figure 4G). Significant reduction of H2BK120ub in Phf6 KO ESCs allowed us to check for potential crosstalk between H2BK120ub and H2BK12Ac. Given that CBP/p300 functions as an acetyltransferase of several lysine residues of histone H2B including K12 (55), we knocked down p300 or CBP by siRNA in WT and Phf6 KO ESCs with DOX treatment and checked whether knockdown of p300 or CBP affects H2BK120ub levels. Intriguingly, knockdown of p300 or CBP showed reduction of both H2BK120ub and H2BK12Ac levels with DOX treatment, indicating crosstalk between H2BK120ub and H2BK12Ac (Figure 4H). More directly, H2BK12R acetylation deficient mutation led to marked reduction in H2BK120ub level compared to that noted for H2B WT, whereas H2BK120R ubiquitination deficient mutation did not affect H2BK12Ac levels (Figure 4I). These data indicate that PHF6 functions to regulate the H2BK120ub levels via preceding H2BK12Ac recognition.
The proteins possessing PHDs typically work as linker proteins to recruit other modifying enzymes for alteration of modification status. Therefore, we checked the possibility of whether PHF6 recruits other E3 ubiquitin ligases or deubiquitinases to regulate the H2BK120ub levels. RNF20/40 has been shown to function as an E3 ubiquitin ligase of H2BK120ub and USP44 as a deubiquitinase during ESC differentiation (56). We examined the RNF20/40 and USP44 expression levels to verify whether the expressions of these enzymes are regulated by PHF6, but neither of them showed altered expression levels in Phf6 KO ESCs compared to those in WT ESCs (Figure 4J). Moreover, the co-immunoprecipitation assay revealed that PHF6 binds neither RNF20/40 nor USP44 (Figure 4K). These data exclude the possibility that PHF6 regulates the H2BK120ub levels via RNF20/40 and USP44. To further examine whether the function of PHF6 for the regulation of H2BK120 levels is independent of E3 ub ligase function of RNF20/40, we compared H2BK120ub levels upon knockdown of Phf6, Rnf20 and Rnf40 with or without DOX treatment. Knockdown of Rnf20/40, but not Phf6, led to the reduction of H2BK120ub level in the absence of DOX treatment (Figure 4L), indicating that RNF20/40 is responsible for activation of target genes involved in ESC differentiation as previously reported (56). However, knockdown of Phf6 resulted in reduction of H2BK120ub level only upon DOX treatment (Figure 4L). More importantly, knockdown of Phf6 specifically reduced mRNA levels of trophectoderm marker genes including Cdx2 and Gata2, whereas knockdown of Rnf20 and Rnf40 failed to downregulate Cdx2 and Gata2 mRNA levels upon DOX treatment (Figure 4M). Further, ChIP assay revealed that knockdown of Phf6, but neither Cdx2 nor Gata2, reduced H2BK120ub levels on the promoters of Cdx2 and Gata2 (Figure 4N). These data indicate that PHF6 specifically regulates the H2BK120ub levels for transcriptional activation of trophectodermal genes.
PHF6 has an E3 ubiquitin ligase activity for histone H2BK120
Previous studies have reported that PHD domain itself is enough for exerting an E3 ligase activity (57–60). Since PHF6 recognizes H2BK12Ac through an extended PHD2 and that there exists a PHF6 dependency between K12Ac and K120ub in H2B, we examined whether PHF6 has an E3 ubiquitin ligase activity for H2BK120ub via PHDs. First, we eluted mono-nucleosome that contains Flag-tagged H2BK12R acetylation deficient MT and H2B-WT from Phf6 KO ESCs using MNase digestion. Then, we performed in vitro ubiquitination assay by mixing eluted mono-nucleosomes as a substrate with purified GST-PHF6 (Figure 5A). We found that PHF6 functions as an E3 ubiquitin ligase when UBCH3, but not UBCH6, works together as its E2 partner (Figure 5B). Moreover, PHF6 failed to ubiquitinate H2BK120 with introduction of the H2BK12R MT, further confirming that PHF6 functions as an E3 ubiquitin ligase for H2BK120ub via preceding H2BK12Ac recognition. Next, we determined the extended PHD1 is responsible for exerting the E3 ubiquitin ligase activity of PHF6. In vitro ubiquitination assay revealed that PHF6 WT is able to ubiquitinate H2BK120; however, neither C82A (functional activity dead MT of extended PHD1 by disrupting Zinc ion capturing) nor E223S (H2BK12Ac unrecognizable MT of extended PHD2) were able to ubiquitinate H2BK120 (Figure 5C). Together, these in vitro results indicate that PHF6 recognizes H2BK12Ac through extended PHD2 and subsequently ubiquitinates H2BK120 residue through extended PHD1.
Figure 5.

PHF6 is an acetyl-dependent E3 ubiquitin ligase for histone H2B on K120. (A) Flowchart of in vitro ubiquitination assay. (B) In vitro ubiquitination assay using H2B WT or K12R MT-containing mono-nucleosomes. Mono-nucleosomes were purified from Phf6 KO ZHBTc4 with DOX treatment. (C) In vitro ubiquitination assay using H2B-Flag WT nucleosomes with GST-PHF6 WT, C82A or E223S MTs. (D) Expression levels of ectopic PHF6 WT, C82A or E223S MTs in Phf6 KO ZHBTc4 with DOX treatment. (E) Schematics of extended PHD1 of PHF6. Black characters are PHD core residues for Zinc capturing; red characters are new candidates of E3 ligase activity core residues which are highly conserved only in extended PHD1. (F) In vitro ubiquitination assay with several PHF6 MTs. (G) In vitro GST-pulldown assay of His-UBCH3 with GST, GST-PHF6 WT or MTs. (H) Schematics of functions of each domain in PHF6.
Next, we rescued PHF6 WT, C82A or E223S MT in Phf6 KO ESCs. Unexpectedly, we observed that the expression levels of C82A MT were significantly decreased in ESCs (Figure 5D). The previous study has shown that the BFL syndrome mutations in the core PHD1 of PHF6 (C45Y and C99F) resulted in the reduced protein stability of PHF6. Therefore, we searched for other MTs in extend PHD1 which are responsible for exerting E3 ligase activity of PHF6 without affecting protein stability. Previous reports have stated that PHD domain shows structural resemblance to the RING domain that is known to possess E3 ligase activity (61,62). Since hydrophobic residues nearby Zinc capturing core residues are crucial for RING domains in E3 ligase (63–65), we examined the possibility whether these nearby hydrophobic residues within PHD1 are important for exerting its intrinsic E3 ligase activity of PHF6 (Figure 5E). Prediction of specific conserved residues in extended PHD1 and generation of various hydrophobic residue MTs by site-direct mutagenesis allowed us to perform the in vitro ubiquitination assay with PHF6 WT and several MTs. Intriguingly, M125 and R129 residues turned out to be indispensable for E3 ligase activity of PHF6 (Figure 5F). In addition, both PHF6 WT and E223S MT bound to UBCH3 E2 enzyme, whereas M125A and R129A MTs failed to bind UBCH3 in vitro, suggesting that PHD1 of PHF6 is crucial for exerting E3 ligase activity (Figure 5G). Based on these findings, we propose a previously unrecognized regulatory molecular basis by which PHF6 serves as an E3 ubiquitin ligase that specifically links preceding H2BK12Ac to H2BK120ub (Figure 5H).
PHF6 is crucial for activation of trophectodermal genes via H2BK120 ubiquitination
Since extended PHD2 domain of PHF6 is responsible for recognition of H2BK12Ac and extended PHD1 domain of PHF6 is for exerting E3 ligase activity, we further checked these activities of PHF6 in ESCs with DOX treatment. First, we performed MNase-ChIP analysis to validate that E223 residue is important for H2BK12Ac recognition of PHF6. Indeed, the binding of the PHF6 E223S MT to H2BK12Ac-mono-nucleosome was significantly reduced, whereas PHF6 WT and E3 ligase MT M125A retained H2BK12Ac-mono-nucleosome binding ability (Figure 6A). We also found that PHF6 WT failed to bind H3K4me1-mono-nucleosome, which is an enhancer marker. These results support that PHF6 is recruited on the promoter regions of target genes by recognizing H2BK12Ac. Next, we checked the H2BK120ub levels after restoring expression of PHF6 WT or MTs. Although PHF6 WT reconstitution almost completely restored H2BK120ub levels, introduction of neither H2BK12Ac unrecognizable MT E223S nor E3 ligase activity dead MTs M125A/129A restored H2BK120ub levels in Phf6 KO ESCs with DOX treatment (Figure 6B). Further, we performed ChIP assays on the promoters of Cdx2 and Gata2 in WT and Phf6 KO ESCs with or without DOX treatment. DOX treatment led to the increased recruitment of PHF6 along with increased H2BK120ub levels on the promoters of these genes in WT ESCs, but not in Phf6 KO ESCs (Figure 6C). However, ChIP assays on the promoter of Phf6-independent gene Msx2 showed no significant change in recruitment of PHF6 and the level of H2BK120ub between WT and Phf6 KO ESCs (Figure 6D). Since H2BK120ub is prerequisite for H3K4me3 at promoter regions, we also checked H3K4me3 level at these regions. We confirmed that H3K4me3 levels at Cdx2 and Gata2 promoters are increased H2BK120ub and Phf6 dependently upon DOX treatment (Figure 6C). However, ChIP assays at the promoter of PHF6-independent gene Msx2 failed to show significant difference in H3K4me3 levels (Figure 6D). Next, we generated cell lines stably expressing PHF6 WT or MTs in Phf6 KO ZHBTc4 by lenti-viral infection, and compared the mRNA levels of trophectodermal genes in Phf6 KO ESCs reconstituted with PHF6 WT or MTs (Figure 6E). qRT-PCR analysis revealed that reconstitution of PHF6 MTs in Phf6 KO ESCs failed to induce mRNA levels of trophectodermal genes including Cdx2, Ascl2, Wnt7b, Fgfr2 and Plac1, compared to those of target genes in PHF6 WT-reconstituted Phf6 KO ESCs (Figure 6F).
Figure 6.

The acetylation-ubiquitination crosstalk at H2B by PHF6 is critical for trophectodermal gene activation. (A) Immunoblot of MNase-ChIP assay with anti-H2BK12Ac and anti-H3K4me1 antibodies for checking PHF6-binding mono-nucleosomes. Immuno-precipitation of chromatin was performed using anti-Flag antibody. Flag-PHF6 WT, M125A and E223S were over-expressed in Phf6 KO ESC with DOX treatment for 2 days. Blotting of each antibody in IP: IgG and IP: Flag was in the same exposure. 1% input was used. (B) Immunoblot analysis of histone H2BK120ub and PHF6 levels from over-expression of pLVX-PHF6 WT and MTs. (C) ChIP assays were performed on the promoters of Cdx2 and Gata2, which are trophectodermal and Phf6-dependently expressed genes, using anti-IgG, anti-PHF6, anti-H2BK120ub, anti-H3K4me3, and anti-H2BK12Ac antibodies in WT and Phf6 KO ESCs with or without DOX treatment. Statistical significance was calculated by ANOVA test (* P < 0.05, ** P < 0.01, *** P < 0.001). (D) ChIP assays were performed on the promoter of Msx2, which is Phf6-independently induced gene by DOX treatment, using anti-IgG, anti-PHF6, anti-H2BK120ub, anti-H3K4me3 and anti-H2BK12Ac antibodies in WT and Phf6 KO ESCs with or without DOX treatment. Statistical significance was calculated by ANOVA test (* P < 0.05, ** P < 0.01, *** P < 0.001). (E, F) Generation of pLVX-PHF6 WT or MTs stable cells by lenti-viral infection in Phf6 KO ESC. Immunoblot analysis of histone H2BK120ub and PHF6 levels of Phf6 WT or MTs stable-rescued Phf6 KO ESCs with DOX treatment (E). qRT-PCR analysis of the trophectodermal genes in Phf6 WT or MTs stable-rescued Phf6 KO ESCs with DOX treatment. mRNA levels of each gene were determined as relative values for Gapdh and relatively compared based on Phf6 WT stable cells (F). Statistical significance was calculated by ANOVA test (* P < 0.05, ** P < 0.01, *** P < 0.001).
DISCUSSION
We discovered sequential events of histone modification crosstalk for lineage-specific gene expressions regulated by PHF6 during trophectoderm reprogramming. PHF6 functions as a reader by recognizing H2BK12 acetylation via the extended PHD2, then as a writer by triggering H2BK120 ubiquitination through the extended PHD1, leading to the transcriptional activation of trophectodermal gene expressions (Figure 7).
Figure 7.
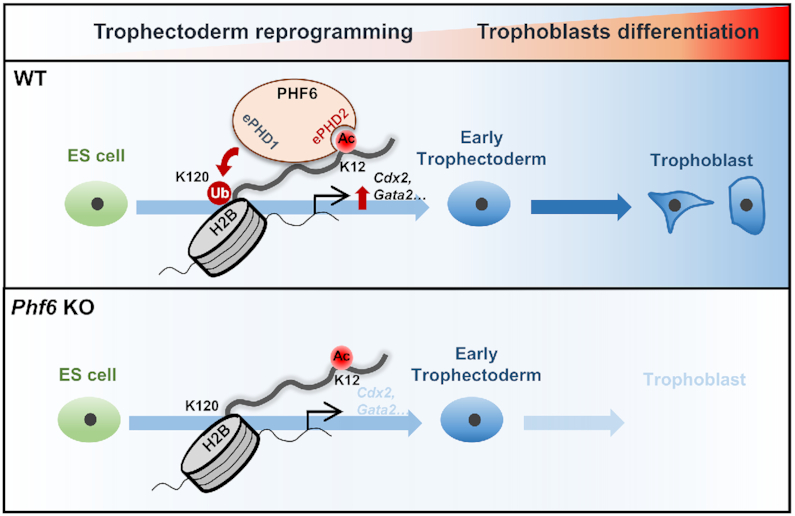
Schematics of PHF6 function as an E3 ubiquitin ligase of H2BK120 via H2BK12Ac recognition for activation of trophectodermal genes Schematic models show PHF6 functions as a transcriptional activator of trophectodermal genes during trophectoderm reprogramming. PHF6 recognizes H2BK12Ac via extended PHD2 domain and ubiquitinates H2BK120 residues via its extended PHD1 domain on the promoter of trophectodermal genes (upper). However, when PHF6 is depleted, H2BK120ub levels on the promoter of trophectodermal genes are not increased, leading to the failure of transcriptional activation of trophectodermal genes (bottom).
The covalent transfer of ubiquitin to substrate proteins involves E1, E2 and E3 enzymes. Among these enzymes, >600 different E3 enzymes provide proper specificity for substrate recognition. The conserved domains of E3 enzyme for substrate recognition read the specific sequence elements on their dedicated substrates. In many cases, the interaction of E3 ligase complex to the substrates often requires PTM of substrates, and several PTM-dependent ubiquitinations conform to these criteria, including phosphorylation, methylation and acetylation. Notably, we identify PHF6 as an E3 ubiquitin ligase for H2BK120 stimulated by recognizing preceding acetylation of H2BK12. We propose that H2BK12 acetylation by p300 acetyltransferase triggers H2BK120 ubiquitination through simultaneous recognition and enzymatic activities within PHF6. H2AX is reported to be a histone substrate controlled by acetylation-dependent ubiquitination via the TIP60–UBC13 complex, however, its acetyl recognition motif has not been identified (66). More recently, the double PHD finger of Dpf3b has been found to associate with acetylated lysine on histone peptides (53). Here, we found that the acetylation recognition motif of PHF6 is the glutamic acid-rich motif in the extended PHD2 of PHF6. Therefore, we speculate that other PHF family members possessing the glutamine acid-rich motif might have function to recognize acetylated substrates as in the case of PHF6.
Compared with other histone-binding modules, the PHD domain has more multifaceted recognizing properties (23,26). Histone modification-reading capability of PHD fingers is further improved by cooperative ability obtained from tandem combination of two or more PHD fingers. We show that the two extended PHD domains of PHF6 work as one functional unit in both recognition and regulation of H2B that is modulated by two functionally distinct modifications in transcription, triggered by H2BK12Ac and further promoted by H2BK120ub. H2BK12Ac is known as a marker of transcription activation, enriched dominantly within the promoter region of highly expressed genes. However, very little is known about the precise mechanism of H2BK12Ac-dependent transcription activation. Here, we found that preceding acetylation on H2BK12 residue by CBP/p300 acetyltransferase provides a platform for recruitment of PHF6 to specific trophectodermal gene locus which might be required for pre-initiation of transcription, followed by consequent ubiquitination of H2BK120 by PHF6 for initiation of transcriptional activation. Therefore, we believe that our study is the first case to reveal recognition of acetylated H2B through the acetyl-lysine binding PHD2 domain of PHF6 and then ubiquitination by E3 ligase PHD1 domain of PHF6.
The first genetically modified mouse model for Phf6 was reported to show that PHF6 is important for BFL syndrome (34). Phf6 C99F knock-in mice showed defects in neurodevelopment which are several symptoms of BFL syndrome. Phf6 KO mice showed a perinatal phenotype, indicating that PHF6 is important for developmental process. Further, function of PHF6 as a regulator for hematopoietic stem cells (HSCs) and leukemia has been reported (67–69). Although development of HSCs is believed to be derived from mesoderm in general, recent reports have shown the possibility of the existence of HSCs originating from placenta, along with the presence of HSCs in placenta (70–72). These in vivo studies support that PHF6 is highly likely to play an important role during trophectoderm lineage determination. Several KO mice of key trophectodermal genes have a perinatal lethality phenotype. From International Mouse Phenotype Consortium (IMPC) (73–75), KO mice of several genes such as Gata2, Notch1, Yap1 and Wnt6 have shown preweaning lethality as in the case of Phf6 KO mice. These genes have important function in cell fate determination during early development (76–80). From mouse genome informatics (MGI), several KO mice of trophectoderm-related genes including Tfap2a, Cited2 and Fgfr2 have shown perinatal lethality similar to the phenotype of Phf6 KO mice. On the basis of these KO mouse phenotype and in vivo results, we suggest the probability that PHF6 plays an important function during trophectoderm lineage development.
Compared to conserved PHD finger capturing two zinc ions, but extended PHD fingers have an additional half domain, called pre-PHD. Intriguingly, PHF6 has two extended PHD fingers. We first provided evidence that specific acidic-rich motif in pre-PHD of extended PHD2 is important for acetylation reading activity of PHF6 in vitro. However, given that far-western blotting and histone peptide array results showed that PHF6 recognizes not only H2BK12Ac but also histone H3 residues, we cannot exclude the possibility that core PHD2 of PHF6 might have an additional ability to recognize other regions of histones than H2BK12Ac. Taken together, we demonstrate that the acetylation-ubiquitination crosstalk by the pivotal regulator PHF6 adds another layer of orchestrated complexity to the epigenetic regulation of trophectodermal genes. Our findings shed light on understanding the molecular basis and potential therapeutic application for congenital disabilities and clinical reproductive disorders that may originate from defects in early embryonic development.
DATA AVAILABILITY
RNA sequencing data for this publication have been deposited in the NCBI Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE144298.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jonghwan Kim in the University of Texas at Austin for critical reading of the manuscript and for helpful comments. ZHBTc4 ESCs were kindly provided by Hitoshi Niwa (Cell Bank-RIKEN, Japan).
Contributor Information
Sungryong Oh, Creative Research Initiatives Center for Epigenetic Code and Diseases, Department of Biological Sciences, Seoul National University, Seoul 08826, South Korea.
Kyungjin Boo, Creative Research Initiatives Center for Epigenetic Code and Diseases, Department of Biological Sciences, Seoul National University, Seoul 08826, South Korea.
Jaebeom Kim, Creative Research Initiatives Center for Epigenetic Code and Diseases, Department of Biological Sciences, Seoul National University, Seoul 08826, South Korea.
Seon Ah Baek, Creative Research Initiatives Center for Epigenetic Code and Diseases, Department of Biological Sciences, Seoul National University, Seoul 08826, South Korea.
Yoon Jeon, Graduate School of Cancer Science and Policy, Research Institute, National Cancer Center, Goyang 10408, South Korea.
Junghyun You, Department of Biological Sciences, Seoul National University, Seoul 08826, South Korea.
Ho Lee, Graduate School of Cancer Science and Policy, Research Institute, National Cancer Center, Goyang 10408, South Korea.
Hee-Jung Choi, Department of Biological Sciences, Seoul National University, Seoul 08826, South Korea.
Daechan Park, Department of Biological Sciences, College of Natural Sciences, Ajou University, Suwon 16499, South Korea.
Ji Min Lee, Department of Molecular Bioscience, College of Biomedical Sciences, Kangwon National University, Chuncheon 24341, South Korea.
Sung Hee Baek, Creative Research Initiatives Center for Epigenetic Code and Diseases, Department of Biological Sciences, Seoul National University, Seoul 08826, South Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Creative Research Initiatives Program (Research Center for Epigenetic Code and Diseases) [2017R1A3B1023387]; Science Research Center program [NRF-2016R1A5A1010764 to S.H.B.]; Basic Science Research Program [NRF-2018R1D1A1A02085592 to J.M.L., NRF-2019R1C1C1008181 to D.P., NRF-2015R1D1A1A01058037 to K.B.] from the National Research Foundation (NRF) grant funded by the Korean government; Foundation for Medical Innovation [FMI, 0543-20200023 to S.O.]; National Cancer Center [NCC-2010271 to H.L.]. Funding for open access charge: Creative Research Initiatives Program [2017R1A3B1023387].
Conflict of interest statement. None declared.
REFERENCES
- 1. Reik W., Dean W., Walter J.. Epigenetic reprogramming in mammalian development. Science. 2001; 293:1089–1093. [DOI] [PubMed] [Google Scholar]
- 2. Lee D.Y., Teyssier C., Strahl B.D., Stallcup M.R.. Role of protein methylation in regulation of transcription. Endocr. Rev. 2005; 26:147–170. [DOI] [PubMed] [Google Scholar]
- 3. Allis C.D., Berger S.L., Cote J., Dent S., Jenuwien T., Kouzarides T., Pillus L., Reinberg D., Shi Y., Shiekhattar R. et al.. New nomenclature for chromatin-modifying enzymes. Cell. 2007; 131:633–636. [DOI] [PubMed] [Google Scholar]
- 4. Boland M.J., Nazor K.L., Loring J.F.. Epigenetic regulation of pluripotency and differentiation. Circ. Res. 2014; 115:311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morey L., Santanach A., Di Croce L.. Pluripotency and epigenetic factors in mouse embryonic stem cell fate regulation. Mol. Cell. Biol. 2015; 35:2716–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guo G., von Meyenn F., Rostovskaya M., Clarke J., Dietmann S., Baker D., Sahakyan A., Myers S., Bertone P., Reik W. et al.. Epigenetic resetting of human pluripotency. Development. 2017; 144:2748–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Braude P., Bolton V., Moore S.. Human gene expression first occurs between the four- and eight-cell stages of preimplantation development. Nature. 1988; 332:459–461. [DOI] [PubMed] [Google Scholar]
- 8. Prioleau M.N., Huet J., Sentenac A., Mechali M.. Competition between chromatin and transcription complex assembly regulates gene expression during early development. Cell. 1994; 77:439–449. [DOI] [PubMed] [Google Scholar]
- 9. Popowski M., Templeton T.D., Lee B.K., Rhee C., Li H., Miner C., Dekker J.D., Orlanski S., Bergman Y., Iyer V.R. et al.. Bright/Arid3A acts as a barrier to somatic cell reprogramming through direct regulation of Oct4, Sox2, and Nanog. Stem Cell Rep. 2014; 2:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rhee C., Lee B.K., Beck S., Anjum A., Cook K.R., Popowski M., Tucker H.O., Kim J.. Arid3a is essential to execution of the first cell fate decision via direct embryonic and extraembryonic transcriptional regulation. Genes Dev. 2014; 28:2219–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X., Wang C., Liu W., Li J., Li C., Kou X., Chen J., Zhao Y., Gao H., Wang H. et al.. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature. 2016; 537:558–562. [DOI] [PubMed] [Google Scholar]
- 12. Pan G., Tian S., Nie J., Yang C., Ruotti V., Wei H., Jonsdottir G.A., Stewart R., Thomson J.A.. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007; 1:299–312. [DOI] [PubMed] [Google Scholar]
- 13. Wright D.E., Wang C.Y., Kao C.F.. Histone ubiquitylation and chromatin dynamics. Front Biosci (Landmark Ed.). 2012; 17:1051–1078. [DOI] [PubMed] [Google Scholar]
- 14. Chen S., Li J., Wang D.L., Sun F.L.. Histone H2B lysine 120 monoubiquitination is required for embryonic stem cell differentiation. Cell Res. 2012; 22:1402–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakanishi S., Lee J.S., Gardner K.E., Gardner J.M., Takahashi Y.H., Chandrasekharan M.B., Sun Z.W., Osley M.A., Strahl B.D., Jaspersen S.L. et al.. Histone H2BK123 monoubiquitination is the critical determinant for H3K4 and H3K79 trimethylation by COMPASS and Dot1. J. Cell Biol. 2009; 186:371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu L., Lee S.Y., Zhou B., Nguyen U.T., Muir T.W., Tan S., Dou Y.. ASH2L regulates ubiquitylation signaling to MLL: trans-regulation of H3 K4 methylation in higher eukaryotes. Mol. Cell. 2013; 49:1108–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Markowetz F., Mulder K.W., Airoldi E.M., Lemischka I.R., Troyanskaya O.G.. Mapping dynamic histone acetylation patterns to gene expression in nanog-depleted murine embryonic stem cells. PLoS Comput. Biol. 2010; 6:e1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saraiva N.Z., Oliveira C.S., Garcia J.M.. Histone acetylation and its role in embryonic stem cell differentiation. World J Stem Cells. 2010; 2:121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Podobinska M., Szablowska-Gadomska I., Augustyniak J., Sandvig I., Sandvig A., Buzanska L.. Epigenetic modulation of stem cells in neurodevelopment: the role of methylation and acetylation. Front. Cell Neurosci. 2017; 11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sanchez R., Zhou M.M.. The PHD finger: a versatile epigenome reader. Trends Biochem. Sci. 2011; 36:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lan F., Collins R.E., De Cegli R., Alpatov R., Horton J.R., Shi X., Gozani O., Cheng X., Shi Y.. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007; 448:718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Org T., Chignola F., Hetenyi C., Gaetani M., Rebane A., Liiv I., Maran U., Mollica L., Bottomley M.J., Musco G. et al.. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep. 2008; 9:370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gatchalian J., Kutateladze T.G.. Zhou M.M. Histone Recognition. 2015; Cham: Springer International Publishing; 27–47. [Google Scholar]
- 24. Fair K., Anderson M., Bulanova E., Mi H., Tropschug M., Diaz M.O.. Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol. Cell. Biol. 2001; 21:3589–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rack J.G.M., Lutter T., Kjaereng Bjerga G.E., Guder C., Ehrhardt C., Varv S., Ziegler M., Aasland R.. The PHD finger of p300 influences its ability to acetylate histone and non-histone targets. J. Mol. Biol. 2014; 426:3960–3972. [DOI] [PubMed] [Google Scholar]
- 26. Musselman C.A., Kutateladze T.G.. Handpicking epigenetic marks with PHD fingers. Nucleic. Acids. Res. 2011; 39:9061–9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Vlierberghe P., Palomero T., Khiabanian H., Van der Meulen J., Castillo M., Van Roy N., De Moerloose B., Philippe J., Gonzalez-Garcia S., Toribio M.L. et al.. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010; 42:338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Vlierberghe P., Patel J., Abdel-Wahab O., Lobry C., Hedvat C.V., Balbin M., Nicolas C., Payer A.R., Fernandez H.F., Tallman M.S. et al.. PHF6 mutations in adult acute myeloid leukemia. Leukemia. 2011; 25:130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meacham C.E., Lawton L.N., Soto-Feliciano Y.M., Pritchard J.R., Joughin B.A., Ehrenberger T., Fenouille N., Zuber J., Williams R.T., Young R.A. et al.. A genome-scale in vivo loss-of-function screen identifies Phf6 as a lineage-specific regulator of leukemia cell growth. Genes Dev. 2015; 29:483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu Z., Li F., Ruan K., Zhang J., Mei Y., Wu J., Shi Y.. Structural and functional insights into the human Borjeson-Forssman-Lehmann syndrome-associated protein PHF6. J. Biol. Chem. 2014; 289:10069–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Crawford J., Lower K.M., Hennekam R.C., Van Esch H., Megarbane A., Lynch S.A., Turner G., Gecz J.. Mutation screening in Borjeson-Forssman-Lehmann syndrome: identification of a novel de novo PHF6 mutation in a female patient. J. Med. Genet. 2006; 43:238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Berland S., Alme K., Brendehaug A., Houge G., Hovland R.. PHF6 deletions may cause Borjeson-Forssman-Lehmann syndrome in females. Mol. Syndromol. 2011; 1:294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zweier C., Kraus C., Brueton L., Cole T., Degenhardt F., Engels H., Gillessen-Kaesbach G., Graul-Neumann L., Horn D., Hoyer J. et al.. A new face of Borjeson-Forssman-Lehmann syndrome? De novo mutations in PHF6 in seven females with a distinct phenotype. J. Med. Genet. 2013; 50:838–847. [DOI] [PubMed] [Google Scholar]
- 34. Cheng C., Deng P.Y., Ikeuchi Y., Yuede C., Li D., Rensing N., Huang J., Baldridge D., Maloney S.E., Dougherty J.D. et al.. Characterization of a mouse model of Borjeson-Forssman-Lehmann syndrome. Cell Rep. 2018; 25:1404–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu B., Zheng Y., Pham A.D., Mandal S.S., Erdjument-Bromage H., Tempst P., Reinberg D.. Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol. Cell. 2005; 20:601–611. [DOI] [PubMed] [Google Scholar]
- 36. Minsky N., Shema E., Field Y., Schuster M., Segal E., Oren M.. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat. Cell Biol. 2008; 10:483–488. [DOI] [PubMed] [Google Scholar]
- 37. Hay D.C., Sutherland L., Clark J., Burdon T.. Oct-4 knockdown induces similar patterns of endoderm and trophoblast differentiation markers in human and mouse embryonic stem cells. Stem Cells. 2004; 22:225–235. [DOI] [PubMed] [Google Scholar]
- 38. Ooga M., Suzuki M.G., Aoki F.. Involvement of histone H2B monoubiquitination in the regulation of mouse preimplantation development. J. Reprod. Dev. 2015; 61:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. et al.. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016; 34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim D., Nam H.J., Lee W., Yim H.Y., Ahn J.Y., Park S.W., Shin H.R., Yu R., Won K.J., Bae J.S. et al.. PKCalpha-LSD1-NF-kappaB-signaling cascade is crucial for epigenetic control of the inflammatory response. Mol. Cell. 2018; 69:398–411. [DOI] [PubMed] [Google Scholar]
- 41. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E. et al.. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003; 34:267–273. [DOI] [PubMed] [Google Scholar]
- 46. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo Y., Mantel C., Hromas R.A., Broxmeyer H.E.. Oct-4 is critical for survival/antiapoptosis of murine embryonic stem cells subjected to stress: effects associated with Stat3/survivin. Stem Cells. 2008; 26:30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Whyte W.A., Bilodeau S., Orlando D.A., Hoke H.A., Frampton G.M., Foster C.T., Cowley S.M., Young R.A.. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012; 482:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gerami-Naini B., Dovzhenko O.V., Durning M., Wegner F.H., Thomson J.A., Golos T.G.. Trophoblast differentiation in embryoid bodies derived from human embryonic stem cells. Endocrinology. 2004; 145:1517–1524. [DOI] [PubMed] [Google Scholar]
- 50. Golos T.G., Giakoumopoulos M., Garthwaite M.A.. Embryonic stem cells as models of trophoblast differentiation: progress, opportunities, and limitations. Reproduction. 2010; 140:3–9. [DOI] [PubMed] [Google Scholar]
- 51. Xu X., Shi D., Liu Y., Yao Y., Dai J., Xu Z., Chen D., Teng H., Jiang Q.. In vivo repair of full-thickness cartilage defect with human iPSC-derived mesenchymal progenitor cells in a rabbit model. Exp. Ther. Med. 2017; 14:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Suwińska A., Czołowska R., Ozdzeński W., Tarkowski A.K.. Blastomeres of the mouse embryo lose totipotency after the fifth cleavage division: expression of Cdx2 and Oct4 and developmental potential of inner and outer blastomeres of 16- and 32-cell embryos. Dev. Biol. 2008; 322:133–144. [DOI] [PubMed] [Google Scholar]
- 53. Zeng L., Zhang Q., Li S., Plotnikov A.N., Walsh M.J., Zhou M.M.. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010; 466:258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang Z., Zang C., Rosenfeld J.A., Schones D.E., Barski A., Cuddapah S., Cui K., Roh T.Y., Peng W., Zhang M.Q. et al.. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008; 40:897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ganai S.A., Banday S., Farooq Z., Altaf M.. Modulating epigenetic HAT activity for reinstating acetylation homeostasis: a promising therapeutic strategy for neurological disorders. Pharmacol. Ther. 2016; 166:106–122. [DOI] [PubMed] [Google Scholar]
- 56. Fuchs G., Shema E., Vesterman R., Kotler E., Wolchinsky Z., Wilder S., Golomb L., Pribluda A., Zhang F., Haj-Yahya M. et al.. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol. Cell. 2012; 46:662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ivanov A.V., Peng H., Yurchenko V., Yap K.L., Negorev D.G., Schultz D.C., Psulkowski E., Fredericks W.J., White D.E., Maul G.G. et al.. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell. 2007; 28:823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu Z., Xu S., Joazeiro C., Cobb M.H., Hunter T.. The PHD domain of MEKK1 acts as an E3 ubiquitin ligase and mediates ubiquitination and degradation of ERK1/2. Mol. Cell. 2002; 9:945–956. [DOI] [PubMed] [Google Scholar]
- 59. Wang J., Muntean A.G., Wu L., Hess J.L.. A subset of mixed lineage leukemia proteins has plant homeodomain (PHD)-mediated E3 ligase activity. J. Biol. Chem. 2012; 287:43410–43416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zucchelli C., Tamburri S., Filosa G., Ghitti M., Quilici G., Bachi A., Musco G.. Sp140 is a multi-SUMO-1 target and its PHD finger promotes SUMOylation of the adjacent bromodomain. Biochim. Biophys. Acta Gen. Subj. 2019; 1863:456–465. [DOI] [PubMed] [Google Scholar]
- 61. Capili A.D., Schultz D.C., Rauscher I.F., Borden K.L.. Solution structure of the PHD domain from the KAP-1 corepressor: structural determinants for PHD, RING and LIM zinc-binding domains. EMBO J. 2001; 20:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Matthews J.M., Bhati M., Lehtomaki E., Mansfield R.E., Cubeddu L., Mackay J.P.. It takes two to tango: the structure and function of LIM, RING, PHD and MYND domains. Curr. Pharm. Des. 2009; 15:3681–3696. [DOI] [PubMed] [Google Scholar]
- 63. Zheng N., Wang P., Jeffrey P.D., Pavletich N.P.. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000; 102:533–539. [DOI] [PubMed] [Google Scholar]
- 64. Deshaies R.J., Joazeiro C.A.. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009; 78:399–434. [DOI] [PubMed] [Google Scholar]
- 65. Plechanovova A., Jaffray E.G., Tatham M.H., Naismith J.H., Hay R.T.. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012; 489:115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ikura T., Tashiro S., Kakino A., Shima H., Jacob N., Amunugama R., Yoder K., Izumi S., Kuraoka I., Tanaka K. et al.. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol. Cell. Biol. 2007; 27:7028–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Soto-Feliciano Y.M., Bartlebaugh J.M.E., Liu Y., Sanchez-Rivera F.J., Bhutkar A., Weintraub A.S., Buenrostro J.D., Cheng C.S., Regev A., Jacks T.E. et al.. PHF6 regulates phenotypic plasticity through chromatin organization within lineage-specific genes. Genes Dev. 2017; 31:973–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wendorff A.A., Quinn S.A., Rashkovan M., Madubata C.J., Ambesi-Impiombato A., Litzow M.R., Tallman M.S., Paietta E., Paganin M., Basso G. et al.. Phf6 loss enhances HSC Self-Renewal driving tumor initiation and leukemia stem cell activity in T-ALL. Cancer Discov. 2019; 9:436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Miyagi S., Sroczynska P., Kato Y., Nakajima-Takagi Y., Oshima M., Rizq O., Takayama N., Saraya A., Mizuno S., Sugiyama F. et al.. The chromatin-binding protein Phf6 restricts the self-renewal of hematopoietic stem cells. Blood. 2019; 133:2495–2506. [DOI] [PubMed] [Google Scholar]
- 70. Alvarez-Silva M., Belo-Diabangouaya P., Salaün J., Dieterlen-Lièvre F.. Mouse placenta is a major hematopoietic organ. Development. 2003; 130:5437–5444. [DOI] [PubMed] [Google Scholar]
- 71. Dzierzak E., Speck N.A.. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat. Immunol. 2008; 9:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gao X., Xu C., Asada N., Frenette P.S.. The hematopoietic stem cell niche: from embryo to adult. Development. 2018; 145:dev139691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Brown S.D., Moore M.W.. The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mamm. Genome. 2012; 23:632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Brown S.D., Moore M.W.. Towards an encyclopaedia of mammalian gene function: the International Mouse Phenotyping Consortium. Dis Model Mech. 2012; 5:289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Koscielny G., Yaikhom G., Iyer V., Meehan T.F., Morgan H., Atienza-Herrero J., Blake A., Chen C.K., Easty R., Di Fenza A. et al.. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2014; 42:D802–D809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rayon T., Menchero S., Nieto A., Xenopoulos P., Crespo M., Cockburn K., Cañon S., Sasaki H., Hadjantonakis A.K., de la Pompa J.L. et al.. Notch and hippo converge on Cdx2 to specify the trophectoderm lineage in the mouse blastocyst. Dev. Cell. 2014; 30:410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Krendl C., Shaposhnikov D., Rishko V., Ori C., Ziegenhain C., Sass S., Simon L., Muller N.S., Straub T., Brooks K.E. et al.. GATA2/3-TFAP2A/C transcription factor network couples human pluripotent stem cell differentiation to trophectoderm with repression of pluripotency. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E9579–E9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Frum T., Murphy T.M., Ralston A.. HIPPO signaling resolves embryonic cell fate conflicts during establishment of pluripotency in vivo. Elife. 2018; 7:e42298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rivron N.C., Frias-Aldeguer J., Vrij E.J., Boisset J.C., Korving J., Vivié J., Truckenmüller R.K., van Oudenaarden A., van Blitterswijk C.A., Geijsen N.. Blastocyst-like structures generated solely from stem cells. Nature. 2018; 557:106–111. [DOI] [PubMed] [Google Scholar]
- 80. Knofler M., Haider S., Saleh L., Pollheimer J., Gamage T., James J.. Human placenta and trophoblast development: key molecular mechanisms and model systems. Cell. Mol. Life Sci. 2019; 76:3479–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data for this publication have been deposited in the NCBI Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE144298.