

Abstract
Zerebrale Anfälle gehören zu den häufigsten neurologischen Erkrankungen des Kindes- und Jugendalters. In der Praxis hat sich die Einteilung in epileptische Anfälle, nichtepileptische Reaktionen (Gelegenheitskrämpfe) und nichtepileptische Anfälle bewährt. Beschrieben werden Pathophysiologie, Ätiologie und Diagnostik sowie Symptomatik der epileptischen Anfälle und Eigenheiten der wichtigsten epileptischen Syndrome, ergänzt durch typische EEG-Bilder. Anhand von Tabellen und Schemata erfolgt die Darstellung der Akut- und Langzeittherapie einschließlich der Vorgehensweise beim Status epilepticus. Differentialdiagnostisch finden sich Ausführungen zur Problematik der Fieberkrämpfe und dem Erscheinungsbild nichtepileptischer Anfälle.
Zerebrale Anfälle
Grundlagen
Unter zerebralen Anfällen versteht man eine plötzliche, vorübergehende, meist kurz (Sekunden bis Minuten) dauernde Funktionsänderung eines Teils oder des gesamten Gehirns mit sensorischen, motorischen, vegetativen oder psychischen Reiz- bzw. Hemmungserscheinungen.
Epidemiologie
Zerebrale Anfälle sind ein häufiges Ereignis. Etwa 4–5% aller Kinder erleiden im Laufe der Kindheit mindestens einen Anfall. In der überwiegenden Zahl handelt es sich um sog. Gelegenheitsanfälle.
Systematik
Folgende Formen werden unterschieden:
epileptische Anfälle,
epileptische Reaktionen (Gelegenheitsanfälle),
nichtepileptische Anfälle.
Epileptische Anfälle
Definition
Als Symptom einer Epilepsie gelten epileptische Anfälle nur dann, wenn sie chronisch-rezidivierend und weitestgehend unabhängig von äußeren Einflussfaktoren auftreten.
Epidemiologie
Mit einer Prävalenz von 5–10‰ und einer Inzidenz von etwa 0,4‰ gehören die Epilepsien zu den häufigsten chronischen Erkrankungen des Kindesalters. Die Hälfte der Epilepsien manifestiert sich vor dem 10. Lebensjahr, etwa 2/3 vor dem 20. Lebensjahr.
Pathophysiologie
Der epileptische Anfall ist Ausdruck einer abnormen und exzessiven elektrischen Entladung größerer Neuronenverbände. Basismechanismus ist dabei eine paroxysmale Depolarisation der Nervenzellmembran.
Dieser pathologische Entladungsvorgang wird u. a. ausgelöst durch:
Imbalancen zwischen inhibitorischen und exzitatorischen Transmittersubstanzen,
Veränderungen der Membranrezeptoren und Ionenkanäle (Natrium- und Kalziumkanäle),
Störungen des neuronalen Energiestoffwechsels.
Ätiologie
Epileptische Anfälle sind multifaktoriell bedingt. Die Manifestation resultiert aus dem Zusammenwirken von genetischer Disposition und exogenen Realisationsfaktoren: prä-, peri- und postnatale Hirnschäden, Phakomatosen, metabolisch-genetische Erkrankungen, chromosomale Aberrationen.
Klassifikation
Nach der gegenwärtig international gültigen Klassifikation werden epileptische Anfälle in fokale (partielle, herdbezogene) Anfälle mit einfacher und komplexer Symptomatik, fokale Anfälle mit sekundärer Generalisation, generalisierte (Absencen; myoklonische, klonische, tonische, atonische, tonisch-klonische Anfälle) und nichtklassifizierbare Anfälle unterteilt.
Grundsätzlich ist zwischen epileptischen Anfällen und epileptischen Syndromen zu unterscheiden.
Diese Differenzierung ist zweckmäßig, da unterschiedliche Anfälle bei gleichen Syndromen, bzw. gleiche Anfälle bei unterschiedlichen Syndromen auftreten können.
Einfache fokale Anfälle
Beim einfach fokalen Anfall bleibt das Bewusstsein erhalten. Es finden sich motorische, sensible, sensorische, autonome und psychische Symptome. Die motorischen (Kloni) oder sensiblen Erscheinungen (Kribbeln, Taubheitsgefühl) treten an umschriebenen Körperstellen (Gesicht, Extremitäten) auf. Eine Ausbreitung auf die gesamte Körperhälfte („march of convulsion“, Jackson-Anfall) oder Übergang in einen tonisch-klonischen Anfall (fokales Grand mal) ist möglich. Bei längerer Anfallsdauer kann es zu einer postparoxysmalen temporären Lähmung einer Extremität bzw. Körperseite kommen (Todd-Lähmung).
Komplexe fokale Anfälle
Kernsymptom ist die Bewusstseinsveränderung. Häufig geht eine Aura (visuell, auditiv, gustatorisch, olfaktorisch, vertiginös) voraus. Nicht selten zeigen sich Automatismen (Schmatz-, Kau-, Schluckbewegungen, Nesteln, Treten und Scharren mit den Füßen), unartikulierte Laute oder szenische Ausgestaltung (Umherlaufen, An- und Auskleiden). Das Anfallsende ist meist durch eine über mehrere Minuten anhaltende Reorientierungsstörung gekennzeichnet.
Absencen
Die Absence ist charakterisiert durch eine unvermittelt einsetzende und ebenso plötzlich endende Bewusstseinspause von 15–30 s. Das Kind unterbricht seine Tätigkeit, das Gesicht wird starr und leer. Die Augen sind halb geöffnet und meist leicht nach oben gerichtet.
Myoklonische Anfälle
Myoklonische Anfälle äußern sich als blitzartige symmetrische Muskelzuckungen im Bereich von Schulter und Armen, gelegentlich auch unter Beteiligung der Beine. Bei heftigen Myoklonien kann es zum Sturz kommen.
Klonische Anfälle
Unter klonischen Anfällen versteht man symmetrische rhythmische Muskelkontraktionen, wobei die Abfolge von längerer Dauer ist als bei Myoklonien. Das Verteilungsmuster entspricht dem der Myoklonien.
Tonische Anfälle
Tonische Anfälle sind gekennzeichnet durch eine wenige Sekunden dauernde symmetrische axorhizomelische Muskelverkrampfung mit Beugung des Kopfs und Anheben der Arme. Beim Auftreten im Stehen ist plötzliches Hinstürzen möglich.
Atonische (astatische) Anfälle
Bei atonischen Anfällen kommt es zu einem plötzlichen Tonusverlust, sodass die Kinder hinstürzen. Der Anfall ist so kurz, dass die Patienten rasch wieder aufstehen. Häufig geht der Atonie eine Myoklonie voraus (myoklonisch-astatischer Anfall).
Tonisch-klonische Anfälle
Meist ohne Vorboten stürzen die Patienten zu Boden. Der Anfallsablauf gliedert sich in eine tonische und eine klonische Phase. Nach Abklingen der Kloni kommt es zur Erschöpfung und meist tiefem Schlaf. Begleitphänomene sind häufig Gesichtszyanose, Mydriasis, Anstieg von Blutdruck und Herzfrequenz, vermehrter Speichelfluss, Einnässen und seitlicher Zungenbiss.
Myoklonien, tonische, tonisch-klonische und atonische Anfälle können sowohl primär generalisierten als auch fokalen Ursprungs sein.
Epileptische Syndrome
Klassifikation
Grundlage der Einteilung epileptischer Syndrome sind 2 unterschiedliche Dichotomien:
Anfallssymptomatik (fokal, generalisiert),
Ätiopathogenese (idiopathisch, symptomatisch).
Die Klassifikation der Epilepsiesyndrome ist aus Tab. 7.1 ersichtlich.
| Fokale Epilepsien | Idiopathisch mit altersgebundenem Beginn |
Benigne Epilepsie im Kindesalter mit zentrotemporalen Spikes und „sharp waves“ Epilepsie im Kindesalter mit okzipitalen „sharp slow waves“ |
| Symptomatisch | ||
| Kryptogen | ||
| Generalisierte Epilepsien | Idiopathisch mit altersgebundenem Beginn |
Benigne familiäre Neugeborenenkrämpfe Benigne Neugeborenenkrämpfe Benigne myoklonische Epilepsie des Kleinkindalters Epilepsie mit pyknoleptischen Absencen (Pyknolepsie) Juvenile Absenceepilepsie Juvenile myklonische Epilepsie Aufwach-Grand-mal-Epilepsie |
| Idiopathisch und/oder symptomatisch altersgebunden |
Blitz-Nick-Salaam-Krämpfe (West-Syndrom) Lennox-Gastaut-Syndrom Epilepsie mit myoklonisch-astatischen Anfällen Epilepsie mit myoklonischen Absencen |
|
| Symptomatisch | ||
| Epilepsien, die nicht als fokal oder generalisiert bestimmbar sind | Mit sowohl generalisierten als auch fokalen Anfällen |
Neugeborenenkrämpfe Schwere myoklonische Epilepsie des Kleinkindalters Epilepsie mit anhaltenden Spike-wave-Entladungen im synchronisierten Schlaf Aphase-Epilepsie-Syndrom (Landau-Kleffner-Syndrom) |
| Ohne eindeutige generalisierte oder fokale Zeichen | ||
| Spezielle Syndrome |
Gelegenheitsanfälle (Fieberkrämpfe, Alkohol, Drogen, Schlafentzug) Oligoepilepsien (einzelne scheinbar unprovozierte Anfälle) Epilepsien mit speziellen Formen der Anfallsauslösung Chronisch progrediente Epilepsia partialis continua des Kindesalters |
Benigne Partialepilepsie mit zentrotemporalen Spitzen (Rolandi-Epilepsie)
Dies ist die häufigste Form kindlicher fokaler Epilepsien. Das Manifestationsalter liegt zwischen dem 2. und 12. Lebensjahr. Die Anfälle treten überwiegend im Schlaf auf. Charakteristisch sind Missempfindungen im Bereich von Mundhöhle, Zunge und Gesicht, gefolgt von hemifazialen Kloni unter Einbeziehung der Kaumuskulatur (typisch vermehrter Speichelfluss). Das Bewusstsein ist in der Regel erhalten, die Kinder können aber nicht sprechen. Das EEG zeigt zentrotemporale „sharp waves“ (Abb. 7.1). Die Anfälle sistieren mit Beginn der Pubertät.

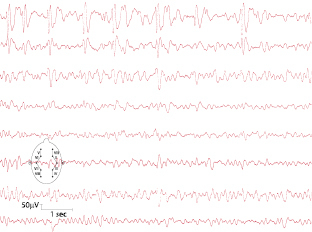
Bei Vorliegen eines bioelektrischen Status im Schlaf ist mit einer ungünstigen Entwicklung zu rechnen.
Seltene Varianten sind das Landau-Kleffner-Syndrom (erworbene epileptische Aphasie) und das Pseudo-Lennox-Syndrom.
Epilepsie mit pyknoleptischen Absencen
Der Erkrankungsgipfel liegt zwischen dem 5. und 9. Lebensjahr. Kennzeichnend ist das täglich gehäufte Auftreten von Absencen. Die Prognose ist günstig. Bei etwa 30% der Patienten können jedoch im Verlauf tonisch-klonische Anfälle hinzutreten. Abzugrenzen ist die frühkindliche und juvenile Absenceepilepsie. Das EEG zeigt während einer Absence generalisierte bilateral-synchrone, regelmäßige 3/s „spike-waves“ (Abb. 7.2).

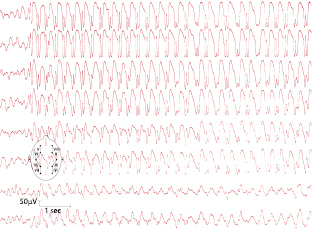
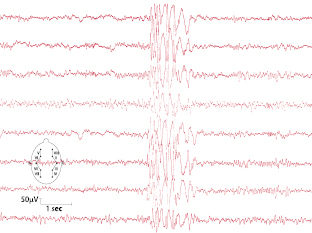
Juvenile myoklonische Epilepsie
Hauptmanifestationsalter ist das 12.–20. Lebensjahr. Bevorzugt nach dem morgendlichen Erwachen zeigen sich meist bilateral-synchron einzelne bzw. repetitive, teilweise sehr heftige (impulsive) Myoklonien im Bereich von Armen und Schultergürtel. Das Bewusstsein ist erhalten. Der Verlauf ist selbst bei Hinzukommen tonisch-klonischer Anfälle günstig. Im EEG finden sich kurze, generalisierte Paroxysmen von unregelmäßigen „spike-“ und „polyspike-waves“ (Abb. 7.3).

Aufwach-Grand-mal-Epilepsie
Betroffen sind Jugendliche ab dem 10. Lebensjahr. Die Anfälle treten typischerweise in den beiden ersten Stunden nach dem Erwachen auf. Begünstigende Faktoren sind Schlafentzug, Stress, Alkoholgenuss, visuelle Reize (Fernsehen, Spielautomaten – fotosensible Epilepsie). Eine Kombination mit Myoklonien und Absencen ist nicht selten. Bei ordnungsgemäßer Therapie und geregelter Lebensführung ist die Prognose günstig.
Epilepsie mit Blick-Nick-Salaam-Krämpfen (BNS-Anfälle, West-Syndrom)
Der Anfallsbeginn liegt zwischen dem 3. und 7. Lebensmonat. Es kommt zu blitzartigen Myoklonien mit Hochreißen der Arme, Anheben des Rumpfs und der Beine. Weitere Anfallsformen sind Nickanfälle und dem orientalischen Salaam-Gruß ähnelnde tonische Anfälle. Charakteristisch ist das serienhafte Auftreten. Häufige Ursachen sind frühkindliche Hirnschäden, neurokutane Syndrome (tuberöse Hirnsklerose), ZNS-Fehlbildungen und metabolische Störungen. Bioelektrisches Korrelat ist die Hypsarrhythmie (diffuses Auftreten von spannungshohen Theta- und Deltawellen mit Einlagerung von „spikes“, „sharp waves“, unregelmäßigen „spike-waves“ sowie „sharp-slow waves“; Abb. 7.4). Die Therapie ist schwierig, in den meisten Fällen zeigen die Patienten komplexe Entwicklungsstörungen.


Lennox-Gastaut-Syndrom
Dieses Syndrom ist gekennzeichnet durch das Auftreten unterschiedlicher Anfallsformen (tonisch, tonisch-klonisch, tonisch-astatisch, myoklonisch-astatisch, Nick- und fokale Anfälle, atypische Absencen) zwischen dem 2. und 7. Lebensjahr. Bei ca. 20% der Fälle geht ein West-Syndrom voraus. Die Prognose ist sehr ungünstig. Neben Therapieresistenz findet sich häufig eine deutliche Retardierung. Das EEG ist charakterisiert durch meist generalisierte, frontal betonte, langsame 2–2,5/s „sharp-“ bzw. „spike-slow waves“: Spike-wave-Variant-Muster (Abb. 7.5).

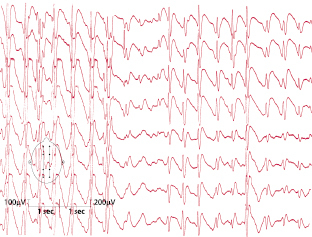
Epilepsie mit myoklonisch-astatischen Anfällen
Leitsymptom sind myoklonische und atonisch-astatische Anfälle. Heftige Anfälle mit plötzlichem Hinstürzen sind nicht selten. Prädilektionsalter ist das 1.–5. Lebensjahr. Die Behandlung gestaltet sich meist problematisch. Im EEG zeigen sich kurze generalisierte Paroxysmen unregelmäßiger 2–3/s „spike-waves“ (Abb. 7.6).


Diagnostik
Wesentliche Voraussetzung für eine exakte Diagnosestellung ist eine akribisch und ggf. wiederholt erhobene Anamnese. Unerlässlich ist die Durchführung eines Elektroenzephalogramms. Bei fokalen Anfällen bzw. bei Verdacht auf eine symptomatische Genese ist der Einsatz bildgebender Verfahren erforderlich.
Differenzialdiagnose
Differenzialdiagnostisch sind alle epileptischen Reaktionen (Abschn. 7.1.4) und nichtepileptischen Anfälle (Abschn. 7.4) zu berücksichtigen.
Akuttherapie
Zur Unterbrechung des Anfalls erfolgt die langsame i.v.-Injektion von Lorazepam, Diazepam oder Clonazepam (Dosierung Abb. 7.7). Ist eine i.v.-Applikation nicht möglich, können alternativ Diazepam-rectal-Tube, Lorazepam, Clonazepam und Midozolam bukkal bzw. Midozolam intranasal verabreicht werden.

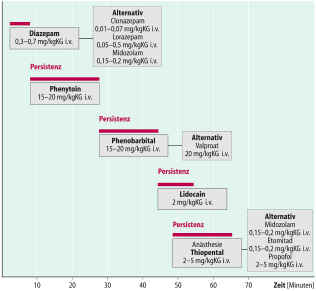
Nach dem ersten Anfall ist die Einleitung einer Langzeittherapie meist nicht indiziert.
Langzeittherapie
Da es nur bei 65% der betroffenen Kinder zu wiederholten epileptischen Anfällen kommt und das individuelle Rezidivrisiko nicht eindeutig voraussagbar ist, kann nach einem ersten Anfall in der Regel mit einer Langzeittherapie zugewartet werden. Sofern die Indikation gegeben ist, muss zur Vermeidung gravierender Nebenwirkungen die Einstellung stufenweise erfolgen (bevorzugter Einsatz von Retardpräparaten). Eine allmähliche Beendigung der Therapie kann nach 3- bis 5-jähriger Anfallsfreiheit vorgenommen werden. Die zum Einsatz kommenden Antiepileptika sind Tab. 7.2 zu entnehmen.
| Anfalls- bzw. Epilepsieform | 1. Wahl | 2. Wahl | 3. Wahl |
|---|---|---|---|
| Frühkindliche tonisch-klonische Anfälle | Valproat |
Brom Phenobarbital |
|
| Benigne Partialepilepsien |
Sultiam Oxcarbazepin |
Valproat Levetirazetam |
Clobazam |
| Einfache und komplexe fokale Anfälle, fokales Grand mal |
Oxcarbazepin Lamotrigin |
Levetirazetam Valproat Topiramat Zonisamid Sultiam Lacosamid |
Phenytoin Primidon Phenobarbital |
| Absencen |
Valproat Ethosuximid |
Lamotrigin | Mesuximid |
| Myoklonien, Aufwach-Grand-mal | Valproat | Lamotrigin | Primidon |
| BNS-Anfälle |
Initialversuch mit Pyridoxin ACTH Kortikoide Vigabatrin |
Sultiam Valproat Topiramat |
Ketogene Diät |
| Dravet-Syndrom | Valproat |
Topiramat + Brom Stiripentol + Clobazam |
Levetirazetam Ethosuximid Mesuximid |
| Myoklonisch-astatische Anfälle | Valproat |
Ethosoximid Lamotrigin |
Mesuximid |
| Lennox-Gastaut-Syndrom | Valproat |
Lamotrigin ACTH Kortikoide Topiramat Rufinamid Felbamat |
Prognose
Anfallsfreiheit ist bei etwa 60%, eine deutliche Reduktion der Anfallsfrequenz bei weiteren 20% der Patienten erreichbar. Eine nicht unbedeutende Zahl mit therapieresistenten Anfällen profitiert von epilepsiechirurgischen Eingriffen oder einer N.-vagus-Stimulation.
Soziale Aspekte
Neben der medikamentösen Therapie ist bei anfallskranken Kindern besonders auf eine ihrem Leistungsvermögen angepasste soziale Eingliederung und Förderung zu achten. Unnötige Restriktionen sind zu vermeiden. Gegen die Teilnahme am Sport bestehen grundsätzlich keine Bedenken (lediglich Sportarten mit Absturzgefahr sind nicht geeignet). Schwimmen sollte nur unter besonderer Aufsicht erfolgen.
Bis auf wenige Ausnahmen (sorgfältige Abwägung bei parenteraler Impfung gegen Typhus, Paratyphus, Gelbfieber und Cholera) können alle Impfungen durchgeführt werden.
Status epilepticus
Definition
Unter einem Status epilepticus versteht man eine länger als 15 min andauernde epileptische Anfallstätigkeit mit oder ohne Beeinträchtigung des Bewusstseins. Zu unterscheiden ist zwischen konvulsivem und nichtkonvulsivem Status. Bewusstseinseinengung, Stupor oder Dämmerzustand können das alleinige Symptom des nichtkonvulsiven Status sein. Eine Erkennung ist dann nur durch das EEG möglich.
Cave
Der Status epilepticus ist eine Notfallsituation und bedarf dringlich stationärer Behandlung.
Therapie
Die grundsätzliche Vorgehensweise bei Status epilepticus besteht in:
Erhaltung vitaler Funktionen,
Unterbrechung des Status (Abb. 7.7),
Behandlung systemischer Komplikationen (Hirndruck, Blutdruck, Blutzucker, Azidose, Elektrolyte),
Abklärung der Ätiologie.
Epileptische Reaktionen
Definition
Als epileptische Reaktionen (Gelegenheits- oder Okkasionskrämpfe) werden Konvulsionen bezeichnet, die nur im Rahmen akuter zerebraler bzw. extrazerebraler Erkrankungen auftreten. Sie sistieren nach Behandlung oder Abklingen der jeweils auslösenden Situation.
Ätiologie
Wichtige Ursachen von Gelegenheitsanfällen sind:
Fieber,
intrakranielle Infektionen (Meningitis, Enzephalitis),
Schädel-Hirn-Trauma,
hypoxisch-ischämische Enzephalopathie,
Elektrolytstörungen (Hypo-/Hypernatriämie, Hypokalzämie),
metabolische Störungen (Hypoglykämie, angeborene Stoffwechseldefekte, Vitamin-B6-Mangel).
Fieberkrämpfe
Als Fieberkrämpfe (Infektkrämpfe) werden Konvulsionen verstanden, die zwischen dem 6. Lebensmonat und dem 5. Lebensjahr im Rahmen hochfieberhafter Infekte auftreten.
Klinik
Fieberkrämpfe sind mit einem Anteil von 50% die häufigste Form der Gelegenheitskrämpfe. Fieber als Realisationsfaktor sowie eine genetische und Altersdisposition sind die wesentlichsten pathogenetischen Faktoren. Die Anfälle treten meist im Fieberanstieg auf und sind phänomenologisch nicht von tonisch-klonischen Anfällen zu unterscheiden. In 10–15% der Fälle zeigen sich fokale Symptome. Prolongierte Halbseitenkrämpfe können von neurologischen Ausfällen und fokaler Epilepsie gefolgt sein: Hemikonvulsion-Hemiplegie-Epilepsie-Syndrom (HHE-Syndrom).
Diagnose
Man unterscheidet einfache und komplizierte Fieberkrämpfe.
Bei jedem Krampfanfall unter Fieber ist stets an eine entzündliche Erkrankung des ZNS zu denken und im Zweifelsfall durch eine Lumbalpunktion auszuschließen.
Kriterien komplizierter Fieberkrämpfe
Dauer >15 min
Fokale Anfallssymptome
Mehrfaches Auftreten innerhalb 24 h
Akuttherapie
Neben der Fiebersenkung erfolgt die Unterbrechung des Anfalls wie oben beschrieben (Abschn. 7.1.2).
Dauertherapie
Eine Langzeittherapie sollte nur nach sorgfältiger Abwägung eingeleitet werden. Als engere Indikationen gelten:
Anfallsdauer von mehr als 15 min,
fokale Anfallssymptome,
Anfallsserien,
Kombination von mindestens 2 der o. g. Risikofaktoren.
Anfallsprophylaxe
Bei Temperaturen über 38°C erfolgt die prophylaktische Antipyrese. Eine Anfallsprophylaxe mit Diazepam-Suppositorien sollte erst nach wiederholten Fieberkrämpfen erwogen werden.
Prognose
Die Kinder nehmen in der Regel eine normale Entwicklung. Das Epilepsierisiko beträgt bei einfachen Fieberkrämpfen 1–2%, bei komplizierten 10–15%.
Neugeborenenkrämpfe
Die Neugeborenenkrämpfe werden in Kap. 4.11 beschrieben.
Nichtepileptische Anfälle
Nichtepileptische paroxysmale Phänomene können in ihrem Erscheinungsbild epileptischen Anfällen sehr ähnlich sein und somit fehlgedeutet werden. Eine Gegenüberstellung ist aus Tab. 7.3 ersichtlich.
| Epileptische Anfälle | Nichtepileptische Anfälle |
|---|---|
| Tonische Anfälle | Paroxysmale kinesiogene Choreoathetose, transitorische Dystonie des Säuglings, benigner paroxysmaler Tortikollis, alternierende Hemiplegie mit Dystonie des Kleinkindalters, Hyperekplexie, Sandifer-Syndrom |
| Atonische Anfälle | Synkopen, Affektanfälle, blasse Reflexsynkopen, benigne paroxysmale Vertigo |
| Myoklonien | Benigne Schlafmyoklonien des Säuglings, Tics, Schauderanfälle, Spasmus nutans, Muskelzittern des Neugeborenen (Jitteriness) |
| Absencen | Tagträumen |
| Einfach fokale Anfälle | Migräne mit Aura |
| Komplex fokale Anfälle | Pavor nocturnus, Somnambulismus, dysphrenische Migräne, Hyperventilationssyndrom, Wutanfälle, Bewegungsstereotypien (Jactatio, Masturbation), Narkolepsie |
Psychogene Anfälle
Kennzeichnend ist reaktives, situationsgebundenes Auftreten, demonstrativ theatralischer Ablauf, Zusammensinken ohne Verletzungen, unkoordiniertes Umherschlagen und partiell erhaltene Reaktion. Im Gegensatz zu epileptischen Anfällen findet sich in der Regel keine Erhöhung des Prolaktinspiegels.
Synkopen
Unter Synkopen (Kollaps, Ohnmacht) versteht man Zustände kurzzeitiger Bewusstlosigkeit infolge kardiovaskulär bedingter zerebraler Ischämie. Bei vagovasalen Synkopen führen komplexe zentrale Reflexmechanismen, die über vagale Efferenzen vermittelt werden, zum Kreislaufversagen. Auslösende Situationen sind langes Stehen (orthostatische Dysregulation: allmähliches Hinsinken nach Vorboten wie Übelkeit, Schwindel, Schwarzwerden vor den Augen) bzw. Angst, Schreck oder Schmerz (z. B. Blutentnahmen). Im Einzelfall ist blitzartiges Hinstürzen begleitet von tonischer Haltung und Myoklonien möglich (konvulsive Synkope). Bei kardialen Synkopen ist u. a. an das Long-QT-Syndrom zu denken (Romano-Ward-Syndrom: autosomal-dominant; Jervell-Lange-Nielsen-Syndrom assoziiert mit Taubheit: autosomal-rezessiv).
Blasse Reflexsynkopen
Bei meist banalen Kopftraumen stürzen die Kinder nach einem Aufschrei plötzlich zu Boden, sind blass und bewusstlos. Ursache ist ein hypersensitiver Vagusreflex, der über eine kurze Asystolie zu einer zerebralen Minderperfusion führt.
Affektanfälle
Hauptmanifestationsalter ist das 2.–4. Lebensjahr. Hervorgerufen durch emotionale Belastungen (Trotz, Wut, Angst) kommt es zu heftigem Schreien und Anhalten der Atmung in Exspiration. Die Folge sind Zyanose, Bewusstseinstrübung und allmähliches Hinsinken. Dramatische Verläufe mit tonischen Streckkrämpfen und einzelnen Kloni sind möglich.
Pavor nocturnus
Betroffen sind meist Knaben im Vorschul- und Schulalter. Etwa 1–2 h nach dem Einschlafen setzen sich die Kinder nach einem initialen lauten Schrei im Bett auf, gestikulieren und zeigen einen angstbesetzten Gesichtsausdruck. Zuspruch und Trost sind wirkungslos. Gewöhnlich schlafen die Kinder nach Abklingen der Symptome ruhig weiter. Als Begleiterscheinungen oder alleinige Manifestation kann es zu ziellosem Umherlaufen kommen (Somnambulismus).