

Abstract
Die rheumatischen Erkrankungen gehören zusammen zu den häufigsten chronischen Erkrankungen bei Kindern und Jugendlichen. Meist ist die Ursache unbekannt, aber wichtige Schritte der Pathogenese sind erforscht und haben oft zu neuen Behandlungsmethoden geführt, sodass die Prognose sich in den letzten Jahren deutlich gebessert hat. Bedeutsam ist die juvenile idiopathische Arthritis (JIA) mit den Subgruppen frühkindliche Oligoarthritis, Polyarthritis, systemische Arthritis (M. Still), Enthesistis assoziierte Arthritis, auch juvenile Spondylarthritis genannt. Die Behandlung besteht aus nichtsteroidalen Antirheumatika, intraartikulären, Low-dose- und Puls-Steroiden, langsam wirkenden Medikamenten wie Methotrexat und Biologika. Zu den wichtigen Differenzialdiagnosen gehören die akuten transienten Arthritiden, besonders die Coxitis fugax. Die meisten nfektionsassoziierten Arthritiden wie reaktive Arthritiden, Arthritiden bei Chlamydien- oder viralen Infektionen, akutes rheumatisches Fieber und die durch Borrelia burgdorferi hervorgerufene Lyme-Arthritis zeigen eine transiente Arthritis. Die septische Arthritis ist ein rheumatologischer Notfall und muss unmittelbar nach Anlage von Erregerkulturen antibiotisch behandelt werden.
Juvenile idiopathische Arthritis
Definition
Unter der Bezeichnung „juvenile idiopathische Arthritis“ (JIA) fasst man eine Gruppe von Gelenkentzündungen zusammen, die vor dem 16. Lebensjahr beginnen, länger als 6 Wochen dauern und für die keine andere Ursache gefunden werden kann.
Epidemiologie
Die Prävalenz beträgt 1 auf 1000 Kinder (bis zum 16. Lebensjahr). Das führende Symptom ist eine Arthritis im Sinne einer chronischen Synovialitis mit möglicher Beteiligung weiterer Gelenkstrukturen (Gelenkknorpel und subchondraler Knochen, Abb. 12.1). Daneben können bindegewebige Strukturen anderer Organe betroffen sein.

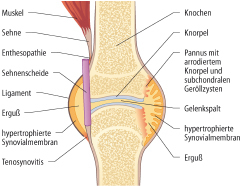
Histopathologie
Histopathologisch finden sich wenige polymorphkernige Zellen sowie ein vornehmlich mononukleäres entzündliches Infiltrat, bestehend aus Lymphozyten, Plasmazellen und Makrophagen. Das Synovialgewebe ist hyperplastisch und stark vaskularisiert (→ Gelenkerguss). Ein Granulationsgewebe (Pannus) führt zu Erosionen des gesunden Knorpels und Knochens (→ Gelenkzerstörung).
Klinik
Schon Tage vor Auftreten der Gelenksymptome können die Kinder abgeschlagen, blass und müde wirken – das gilt auch für Rezidive. Andererseits gibt es auch den Beginn der Arthritis aus völligem Wohlbefinden, wobei von den Eltern häufig Bagatelltraumen als vermeintliche Auslöser der Erkrankung angeführt werden.
Wichtigstes Symptom ist die Arthritis, eine nicht traumatisch bedingte Schwellung, Erguss oder schmerzhafte Bewegungseinschränkung in mindestens einem Gelenk. Die Schmerzhaftigkeit findet man häufig nur bei der endgradigen Bewegungseinschränkung. Häufig findet sich eine Überwärmung der darüber liegenden Haut. Eine Rötung spricht eher für eine Phlegmone oder eine septische Arthritis. Das Symptom „Arthritis“ ist ausschließlich durch die physikalische Untersuchung definiert und so am Patienten zu finden. Die Gelenkuntersuchung mit Inspektion, Palpation, Feststellung der Bewegungseinschränkung nach der Neutral-Null-Methode und Funktionsprüfung muss alle Gelenke umfassen, den unterschiedlichen physiologischen Bewegungsumfang in Abhängigkeit vom Alter berücksichtigen (je jünger desto beweglicher) und bedarf besonders beim Kleinkind der Übung und Erfahrung. Eine Morgensteifigkeit findet sich nicht so regelhaft wie bei der rheumatoiden Arthritis des Erwachsenen.
Subtypen
Eine Unterscheidung von verschiedenen Subtypen der juvenilen idiopathischen Arthritis ist sinnvoll, weil sie sich in Therapie, Komplikationen und Prognose unterscheiden (Tab. 12.1). Nicht selten ist die Zuordnung eines Patienten zu einem Subtypus der juvenilen idiopathischen Arthritis nicht möglich.
| Rheumafaktornegative Polyarthritis | Rheumafaktorpositive Polyarthritis | (Frühkindliche) Oligoarthritis | Undifferenzierte juvenile Spondylarthritis | Still-Syndrom | |
|---|---|---|---|---|---|
| Relative Häufigkeit | 20% | <5% | 40% | 20% | 5% |
| Rheumafaktor | – | + | – | – | – |
| Antinukleäre Antikörper | 25% + | 75% + | Bis zu 50% + | – (außer Psoriasisarthritis) | – |
| Durchschnittsalter bei Beginn [Jahre] | 5 | 12 | 2 | 10 | 4 |
| Geschlechtsverteilung | w > m | w >> m | w >> m | m > w | w = m |
| HLA-Assoziation | DR8 | DR4 | DR5, DR8 | B27 | Keine |
| Uveitis | Selten | Keine | Bis zu 40% | Bis zu 20% | Keine |
| Sakroileitis | Keine | Keine | Keine | Möglich | Keine |
| Gelenkprognose | Im Einzelfall ungewiss | Frühe Knorpelerosion möglich | Gut | Gut | Progression möglich |
| Bemerkungen | Kann sich auch aus der frühkindlichen Oligoarthritis entwickeln (extended Oligoarthritis) | Entspricht adulter rheumatoider Arthritis | Bleibende Augenschäden in bis zu 10% | Übergang in ankylosierende Spondylitis möglich | Fieber, Ausschlag, Organomegalie, Lymphknotenschwellung |
Die Internationale Klassifikation „juvenile idiopathische Arthritis“ umfasst als weitere Subgruppen die Psoriasisarthritis und die Enthesitis-assoziierte Arthritis und überlappt dadurch mit der juvenilen Spondylarthritis (Abschn. 12.2). Zudem wird die aus einer Oligoarthritis hervorgehende Polyarthritis als eigene Subgruppe aufgefasst. Schließlich gibt es die Gruppe der nicht zuzuordnenden oder mehr als einer Gruppe zuzuordnenden Fälle.
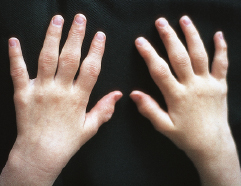
Bei den polyartikulären Verlaufsformen (≥5 Gelenke mit Arthritis) wird eine rheumafaktornegative von der selteneren rheumafaktorpositiven Form unterschieden. Die polyartikuläre Form ohne Rheumafaktor kann prinzipiell alle Gelenke außer der lumbalen Wirbelsäule aber einschließlich der Halswirbelsäule, der Krikoarythenoidgelenke (Heiserkeit) und der Kiefergelenke betreffen. Oft ist das Muster symmetrisch und schließt neben großen Gelenken die kleinen Fingergelenke, besonders die Metakarpophalangealgelenke und die proximalen Interphalangealgelenke ein (Abb. 12.2). Initial kann Fieber auftreten. Da beim Kind und Jugendlichen ein ausgesprochener Bewegungsdrang besteht und der Umgang mit Gleichaltrigen fast immer eine körperliche Betätigung beinhaltet (Spielplatz, Sportverein, Diskothek), führt die Erkrankung unbehandelt oft zu einer schweren Beeinträchtigung der psychosozialen Entwicklung mit Adaptationsphänomenen. Der Befall der Hand mit schmerzhafter ulnarer Fehlstellung im Handgelenk kann zur verminderten Schreibleistung führen.

Unter den Formen mit oligoartikulärem Befall (≤4 betroffene Gelenke) ist die (frühkindliche) Oligoarthritis die häufigste. Meist sind weibliche Kleinkinder betroffen. Die fast immer vorhandene Gonarthritis beeinträchtigt die evtl. gerade erworbene Fähigkeit zu laufen (Abb. 12.3). Bei Befall eines Kiefergelenks kommt es bei der Mundöffnung zum Abweichen des Unterkiefers zur betroffenen Seite und mittelfristig zur Mikrogenie. Wichtig ist das häufige Vorkommen einer chronischen Iridozyklitis. Diese lässt sich vor Auftreten von Komplikationen wie Synechien oder Bandkeratopathie nur in der Spaltlampenuntersuchung erkennen, da das Auge unauffällig erscheint und die Kinder bei einseitigem Befall weiterhin lesen können.


Cave
Bei zu später oder unzureichender Behandlung können die Kinder bleibenden Visusverlust erleiden oder erblinden.
Die mit dem HLA-Klasse-I-Antigen B27 assoziierte Enthesitis-verbundene Arthritis wird auch als undifferenzierte juvenile Spondylarthritis (Abschn. 12.2) bezeichnet. Weiter wird die Psoriasisarthritis als eigene JIA-Subgruppe abgegrenzt.
Die systemische Verlaufsform (M. Still) beginnt mit extraartikulären Manifestationen:
über Wochen intermittierendes, hektisches (rasche Temperatursprünge) Fieber,
stammbetontes, flüchtiges, nur während des Fiebers sichtbares kleinfleckiges, konfluierendes, blassrosa Exanthem (Abb. 12.4),
Hepatosplenomegalie,
generalisierte Lymphadenopathie,
Pleuritis,
Perikarditis.


Nur wenige Kinder haben initial Gelenksymptome. Nach Wochen oder Monaten und allmählichem Verschwinden der systemischen Manifestationen entwickeln die Kinder eine Oligo- oder Polyarthritis. Zunächst handelt es sich um „Fieber ungeklärter Ursache“ und erst nach Auftreten einer Arthritis bestätigt sich die Diagnose JIA mit systemischem Beginn.
Diagnose
Die Diagnose wird klinisch gestellt durch den Nachweis der chronischen Arthritis und den Ausschluss anderer Ursachen.
Labor
Es finden sich häufig Entzündungszeichen bei den Laboruntersuchungen: erhöhtes C-reaktives Protein, erhöhte Blutkörperchensenkungsgeschwindigkeit, beim M. Still auch Leukozytose mit Linksverschiebung im Differenzialblutbild.
Cave
Besonders bei Befall nur eines oder weniger Gelenke können alle Laborwerte unauffällig sein.
Bei länger dauernder Erkrankung kommt es zur Anämie mit hohem Ferritinspiegel. Die Bestimmung des Rheumafaktors ist diagnostisch wenig hilfreich, da er nur bei wenigen Fällen von juveniler idiopathischer Arthritis nachzuweisen ist und meist auf andere Erkrankungen wie Kollagenose oder Infektion hinweist. Niedrigtitrige antinukleäre Antikörper finden sich häufig bei der frühkindlichen Oligoarthritis und sind mit erhöhtem Risiko für eine chronische Iridozyklitis assoziiert.
Gelenkpunktion
Bei Beginn der Erkrankung, insbesondere bei Monarthritis, ist zum Ausschluss einer septischen Arthritis manchmal eine Gelenkpunktion indiziert. Wenn eine Osteomyelitis (mit lokalisiertem metaphysärem Druckschmerz) klinisch nicht ausgeschlossen werden kann, sollte, evtl. unter sonographischer Kontrolle, ein subperiostaler Abszess zur Erregeranzucht in Analgosedierung punktiert werden.
Sonographie
Sonographisch kann ein Erguss im Hüftgelenk oder anderen Gelenken nachgewiesen werden. Daneben können eine Tenosynovitis, Verdickung der Synovialis oder andere Weichteilveränderungen festgestellt werden. Dopplersonographisch kann Entzündung dokumentiert werden.
Röntgen
Röntgenaufnahmen sind in den Hintergrund getreten und werden nicht mehr regelhaft angefertigt, wenn andere bildgebende Methoden die gleiche Information liefern können.
MRT
Mittels Magnetresonanztomographie können alle Gelenkstrukturen dargestellt und das Ausmaß von Entzündung und Pannusbildung in der Gadolinium-Kontrastmittel-Darstellung beurteilt werden. Dadurch ist eine diagnostische Arthroskopie im Kindesalter häufig entbehrlich geworden.
Differenzialdiagnose
Im Vordergrund der differenzialdiagnostischen Überlegungen stehen septische (oder tuberkulöse) Arthritis, Osteomyelitis, akute transiente Arthritiden wie die Coxitis fugax, infektassoziierte Arthritiden wie reaktive Arthritis, Lyme-Arthritis, akutes rheumatisches Fieber und virale Arthritiden (Kap. 14), familiäres Mittelmeerfieber, Sepsis, akute lymphatische Leukämie, Neuroblastom, Kollagenosen und Schmerzverstärkungssyndrome wie die häufigen sog. „Wachstumsschmerzen“.
Therapie
Der „Erfolg“ einer bestimmten Behandlung, insbesondere das Erreichen einer Remission, ist kritisch zu werten, da dies im individuellen Fall auch dem Spontanverlauf entsprechen kann. Nach längerem Verlauf können bleibende Schäden am Bewegungsapparat mit Behinderung auftreten, die heute aber unter entsprechender rechtzeitiger und konsequenter Therapie meist vermeidbar sind.
Ziel der Therapie ist die Erhaltung oder Wiedergewinnung der vollständigen Gelenkfunktion, die Unterdrückung der Entzündung sowie die psychosoziale Reintegration des Kindes und der Familie.
Der Erfolg der Behandlung wird durch Symptomlisten und Fragebögen zum Ausmaß der Beeinträchtigung am besten beschrieben. Kinder mit chronischer Arthritis sollten an einem darin erfahrenen Zentrum mit betreut werden.
Aufklärung
Wichtig ist die ausführliche Aufklärung der Eltern und einsichtsfähiger Jugendlicher, bei denen die Nennung der Diagnose „Rheuma“ zunächst Erstaunen und dann Angst auslöst. Es ist eine wichtige ärztliche Aufgabe darzustellen, dass trotz fehlender kausaler Therapie unter den heute verfügbaren Möglichkeiten der Behandlung die Erkrankung ohne bleibende Schäden ausheilen kann. Die Kinder sollen zu einem normalen Leben ermuntert werden. Psychologische und soziale Beratung der Eltern und/ oder strukturierte Schulungen von Eltern und Schulkindern können sinnvoll sein. Längere Krankenhausaufenthalte oder Maßnahmen, die zur Immobilisierung von Gelenken führen, sind zu vermeiden.
Pharmakotherapie
Leichte Fälle können mit nichtsteroidalen Antirheumatika(Naproxen, Ibuprofen, Diclofenac, Meloxicam, Indometacin) behandelt werden. Nebenwirkungen sind vor der Pubertät selten und betreffen meist den Magen mit Bauchschmerzen, Übelkeit, Bluterbrechen und Teerstühlen. Außerdem sind Nebenwirkungen des Zentralnervensystems (Konzentrationsstörungen) und selten der Niere (Papillennekrose bei ungenügender Hydrierung) möglich.
Bei Oligoarthritiden sind intraartikuläre Steroide, evtl. in Kombination mit langsam wirkenden Medikamenten wie Methotrexat, Sulfasalazin oder Hydroxychloroquin, Mittel der Wahl. Bei Polyarthritiden ist Methotrexat 10–12 mg/m2 Körperoberfläche, oral 1-mal wöchentlich, wirksam; höhere Dosen können besser wirksam sein, müssen aber subkutan verabreicht werden. Die Applikation intraartikulärer Steroide erfordert besondere Erfahrung und erfolgt häufig in Analgosedierung. Während die intraartikulären Steroide rasch fast immer zur Besserung führen, muss man bei den langsam wirkenden Medikamenten bis zu 3 Monate auf den Wirkungseintritt warten, der auch ausbleiben kann. Nebenwirkungen von Methotrexat betreffen den Gastrointestinaltrakt, die Leber und das blutbildende System. Wird bei Hydroxychloroquin eine Tagesdosis von 5 mg/kgKG nicht überschritten, ist auch in der Langzeitanwendung nicht mit Nebenwirkungen wie einer Schädigung der Retina zu rechnen.
Hochdosierte systemische Kortikosteroide (Prednison, 2 mg/kgKG) sind aufgrund ihrer hohen Rate an schweren Nebenwirkungen auf wenige Indikationen beschränkt: Systemischer Verlauf, symptomatische Peri-/Myokarditis, chronische Iridozyklitis, die auf lokale Anwendung von Kortikosteroiden nicht anspricht. In anderen Fällen kommen neben der intraartikulären Gabe Kortikosteroide als hochdosierte i.v.-Pulstherapie mit Methylprednisolon, 20 mg/kgKG/Tag über 3 Tage oder niedrig unterhalb der Cushing-Schwelle dosiert (Prednison 0,25 mg/kgKG p.o.) zum Einsatz. Da Kortikosteroide rasch wirken, kann man durch ihre Gabe die Zeit bis zum Ansprechen auf langsam wirkende Medikamente überbrücken.
Wenn Methotrexat bei Patienten mit Polyarthritis nicht ausreichend wirksam ist, werden Biologicals eingesetzt, wie die subkutan zu applizierenden Tumornekrosefaktor-α-Inhibitoren Etanercept oder Adalimumab, der Interleukin (IL)-6-Antagonist Tocilizumab oder der Kostimulationshemmer Abatacept, worunter es oft zur raschen Besserung kommt. Selten treten schwere Infektionen darunter auf. Vor Beginn der Behandlung müssen eine Tuberkulose und eine Hepatitis B ausgeschlossen werden.
Beim M. Still ist häufig eine Behandlung mit Tocilizumab indiziert. Alternativ können IL-1-Blocker wie Anakinra oder Canakinumab eingesetzt werden.
Vor dem Einsatz immunsuppressiver Medikamente sollten möglichst alle von der STIKO empfohlenen Impfungen gegeben worden sein.
Physikalische Therapie
Die meisten Kinder mit JIA benötigen zu bestimmten Zeiten Krankengymnastik. Ziele der Therapie sind: Erhaltung der Gelenkfunktion, wenn möglich Wiederherstellung des Bewegungsumfangs sowie Vermeidung der Osteoporose und der Inaktivitätsatrophie von Muskulatur und Bandapparat. Wiedererlernen von komplexen motorischen Abläufen wie dem Laufen. Die Kinder sollen zur sportlichen Betätigung zunächst ohne Notenstress und in der Freizeit ermuntert werden.
Orthopädisch-chirurgische Maßnahmen
Eine Immobilisierung von Gelenken ist schädlich. Sehr selten ist bei therapieresistenten monarthritischen Formen eine Synovektomie indiziert. Bei Zerstörung des Hüftgelenks kann auch im Wachstumsalter eine Totalendoprothese notwendig werden. Bei Kindern mit Beteiligung des Kiefergelenks sind kieferorthopädische Maßnahmen mit Tragen einer den Processus condylaris mobilisierenden Schiene indiziert.
Augenuntersuchung
Zu jedem Zeitpunkt der Erkrankung und sogar Jahre nach ausgeheilter Arthritis kann bei Kindern mit frühkindlicher Oligoarthritis eine chronische Iridozyklitis auftreten, sodass diese Kinder mindestens für 5 Jahre alle 3 Monate vom Ophthalmologen an der Spaltlampe untersucht werden müssen.
Die chronische Iridozyklitis wird zuerst mit topischen Steroiden, Mydriatika und nichtsteroidalen Antirheumatika behandelt. Kommt es innerhalb von 2 Wochen zu keiner Besserung, so sind systemische Steroide einzusetzen, bei langfristiger Behandlung auch Methotrexat oder Adalimumab.
HLA-B27-assoziierte juvenile Spondylarthritiden
Definition
Zu den juvenilen Spondylarthritiden gehört eine Gruppe von Erkrankungen (Tab. 12.2), die neben einer peripheren Arthritis Folgendes gemeinsam haben:
möglicher Befall des Achsenskeletts einschließlich der Iliosakralgelenke,
Entzündung periartikulärer Strukturen wie Sehnenansätze, Sehnenscheiden und Bänder,
Assoziation mit dem HLA-B27.
| Undifferenzierte juvenile Spondylarthritis | Nicht den anderen Spondylarthritisden zuzuordnen (Übersicht) |
| Reaktive Arthritis | Arthritis nach intestinaler oder urogenitaler Infektion |
| Psoriasisarthritis | Arthritis bei Psoriasis oder psoriasiformem Ausschlag |
| Arthritis bei chronisch-entzündlicher Darmerkrankung | Arthritis bei Morbus Crohn oder Colitis ulcerosa |
| Juvenile ankylosierende Spondylitis | Radiologische Veränderungen der Iliosakralgelenke |
Da es im Kindes- und Jugendalter seltener zur klinischen Beteiligung des Achsenskeletts kommt, ist der Name der Erkrankung missverständlich.
Undifferenzierte juvenile Spondylarthritis
Bei der undifferenzierten juvenilen Spondylarthritis ist die Erkrankung (noch) nicht so weit fortgeschritten, dass eine der anderen juvenilen Spondylarthritiden diagnostiziert werden kann. Sie ist nach der frühkindlichen Oligoarthritis die zweithäufigste chronisch entzündliche Gelenkerkrankung im Kindes- und Jugendalter. Die Arthritis ist meist asymmetrisch, bevorzugt die untere Extremität und verläuft häufig ohne Knorpelerosion. Zusätzliche Symptome sind Fersenschmerzen (Enthesitis: Entzündung der Ansatzpunkte von Ligamenten, Sehnen und Faszien am Knochen), Tenosynovitis (Abb. 12.1), akute vordere Uveitis mit hochrotem Auge und Lichtscheu sowie eine Karditis mit Aorteninsuffizienz. Die Diagnose wird häufig anhand der Kriterien der European Spondyloarthropathy Study Group (ESSG) gestellt. Zum Teil können diese Patienten auch als JIA bzw. Enthesitis-assoziierte Arthritis klassifiziert werden.
ESSG-Kriterien zur Diagnose einer Spondylarthritis
Entzündliche Rückenschmerzen oder periphere Arthritis (asymmetrisch oder überwiegend untere Extremität) und mindestens einer der folgenden Punkte:
HLA-B27-assoziierte Erkrankung in der Familienanamnese
Psoriasis
Entzündliche Darmerkrankung
Urethritis/Zervizitis oder akuter Durchfall innerhalb eines Monats vor Arthritisbeginn
Wechselseitige Gesäßschmerzen
Enthesitis
Radiologischer Nachweis einer Sakroileitis
Diese Kriterien wurden für Erwachsene erarbeitet. Der Beginn der Arthritis soll vor Vollendung des 16. Lebensjahres liegen
Reaktive Arthritis
Die reaktive Arthritis tritt nach gastrointestinalen oder urogenitalen Infektionen durch Yersinien, Shigellen, Salmonellen, Campylobacter oder Chlamydien auf. Etwa 1–4 Wochen nach der Infektion kommt es zu einer oligoartikulären, gelegentlich auch polyartikulären asymmetrischen Arthritis. Die reaktive Arthritis kann sehr schmerzhaft sein und mit Fieber einhergehen. Bei Vorliegen von Augenveränderungen, wie Konjunktivitis, Keratitis oder Iritis spricht man auch von Reiter-Syndrom. Meist verschwindet die Erkrankung nach einigen Wochen bis Monaten, nur selten kommt es zu einem langfristigen Verlauf, bei dem die Erkrankung in eine ankylosierende Spondylitis übergehen kann.
Psoriasisarthritis
Bei der juvenilen Psoriasisarthritis geht die Gelenkmanifestation nicht selten den Hauterscheinungen voraus, oder aber die kutanen Symptome, wie schuppende Erytheme am Haaransatz und Tüpfelnägel, reichen für die Diagnose einer Psoriasis (noch) nicht aus. Die Psoriasisarthritis zeigt eine familiäre Häufung, beginnt als asymmetrische Oligoarthritis und kann auch als Polyarthritis auftreten, oft mit Befall der distalen Interphalangealgelenke. Charakteristisch ist der Befall eines einzelnen kleinen Fingergelenks einer Hand und eine Daktylitis (Schwellung und evtl. Rötung eines ganzen Strahls mit Entzündung aller Weichteile einschließlich der Gelenke; Abb. 12.5). Die Zuordnung der Psoriasisarthritis zu den Spondylarthritiden ist umstritten, zumal die Assoziation mit dem HLA-B27 nur bei Patienten mit Befall der Wirbelsäule besteht.


Entzündliche Darmerkrankungen
Bei Colitis ulcerosa und beim M. Crohn kommt es in etwa 10% der Fälle zu einer Beteiligung großer peripherer Gelenke. Die Stärke der Arthritis korreliert mit der Aktivität der Darmerkrankung. Mit erfolgreicher Therapie der Grunderkrankung wird auch die Arthritis positiv beeinflusst. Nur bei Kindern, die HLA-B27-positiv sind, kann sich eine juvenile ankylosierende Spondylitis entwickeln.
Juvenile ankylosierende Spondylitis (JAS)
Bei der JAS (M. Bechterew) kommt es zu einer Entzündung der kleinen Wirbelgelenke und der Iliosakralgelenke mit nachfolgender Einsteifung der Wirbelsäule. Die Erkrankung kann in der Kindheit beginnen, gewöhnlich mit Entzündung großer peripherer Gelenke. Oft erst Jahre später kommt es dann zur charakteristischen Beteiligung der Sakroiliakalgelenke und der lumbodorsalen Wirbelsäule, die mit tiefen, oft nächtlichen Rückenschmerzen und einer zunehmenden Versteifung der Wirbelsäule einhergeht.
Diagnose
Die Entzündungszeichen entsprechen denen bei juveniler idiopathischer Arthritis. Der Rheumafaktor ist immer negativ. Nur bei der Psoriasisarthritis können antinukleäre Antikörper vorkommen, einschließlich des Risikos einer chronischen Iridozyklitis. Der Nachweis des HLA-B27 sollte die klinische Diagnose einer juvenilen Spondylarthritis nur bestätigen, sein Fehlen die Diagnose nicht in Zweifel ziehen. Das HLA-B27 ist ein genetischer Marker und kein Krankheitsindikator, der bei 6–10% der deutschen Bevölkerung vorhanden ist, während er bei 60% (reaktive Arthritis) bis 95% (ankylosierende Spondylitis) der Patienten mit juveniler Spondylarthritis gefunden wird.
Eine Sakroiliitis kann mittels Kernspintomographie nachgewiesen werden, die Sicherung einer ankylosierenden Spondylitis erfolgt röntgenologisch.
Therapie
Die Behandlung entspricht im Wesentlichen der der juvenilen idiopathischen Arthritis (Abschn. 12.1). Charakteristischerweise kommt es zu einer raschen Besserung der Schmerzen bei Gabe nichtsteroidaler Antirheumatika. Sulfasalazin beeinflusst häufig die periphere Arthritis großer Gelenke günstig, besonders bei Vorhandensein des HLA-B27. Bei Befall des Achsenskeletts ist intensive krankengymnastische Behandlung der Wirbelsäule indiziert. Schwere Verläufe werden mit Tumornekrosefaktor-α-Inhibitoren behandelt. Die akute vordere Uveitis wird mit topischen Steroiden behandelt und führt selten zu bleibenden Schäden.
Infektassoziierte Arthritiden
Definition
Infektassoziierte Arthritiden haben eine bekannte Ätiologie und führen mit Ausnahme der unbehandelten septischen Arthritis nicht zur raschen Zerstörung des Gelenks.
Pathogenese
Die Pathogenese infektassoziierter Arthritiden ist teilweise bekannt. Stoffwechselaktive Chlamydien lassen sich regelmäßig im Gelenk von Patienten mit Chlamydienarthritis nachweisen. Bei der Lyme-Arthritis ist die PCR im Gelenk oft positiv, bei der reaktiven Arthritis finden sich die Lipopolysaccharide der gramnegativen Bakterien im Gelenk, während beim akuten rheumatischen Fieber molekulares Mimikry wirksam ist. Es gibt also ein pathogenetisches Spektrum mit abnehmender Erregerpräsenz im Gelenk.
Klinik
Oft handelt es sich um eine selbstbegrenzte Arthritis, die unter dem Bild einer akuten transienten Arthritis (Arthritisdauer unter 6 Wochen) verläuft.
Die häufigste akute transiente Arthritis ist die Coxitis fugax (transiente Synovitis), bei der der Erreger allerdings noch unbekannt ist. Akut kommt es zu Humpeln und Schmerzen in Hüfte oder Knie, die nach einigen Tagen spontan verschwinden. Anfangs muss eine septische Koxitis ausgeschlossen werden. Bleibt die Bewegungseinschränkung bestehen, können sich auch eine chronische Arthritis oder ein M. Perthes herausstellen.
Die Rötelnvirusarthritis kann auch nach der Impfung, aber seltener als nach der Wildvirusinfektion, auftreten. Nach der Pubertät nimmt die Häufigkeit der Rötelnvirusarthritis beim weiblichen Geschlecht stark zu. Bei einer Monarthritis im Rahmen von Windpocken sollte man zunächst an eine septische Arthritis durch Staphylococcus aureus denken. Nach Infektion mit gramnegativen Darmkeimen, wie Yersinien, Salmonellen, Campylobacter oder Shigellen und nach einer Infektion mit Chlamydien kann es zur reaktiven Arthritis kommen. Die Chlamydienathritis findet sich bei sexuell aktiven Jugendlichen. Die Lyme-Arthritis ist eine späte Manifestation der Lyme-Borreliose und kann Monate bis Jahre nach der Infektion auftreten, meist als episodische Monarthritis des Kniegelenks.
Diagnose
Oft ergibt sich die Diagnose aus typischen Begleiterscheinungen, z. B. Exanthem bei Parvovirus-B19-Arthritis oder vorangehender Durchfall bei reaktiver Arthritis. Im Zweifelsfall muss eine septische Arthritis ausgeschlossen werden. Wegen der Vielzahl möglicher Erreger (Tab. 12.3) und den entsprechenden Kosten einer kompletten Diagnostik, begnügt man sich bei akuter transienter Arthritis häufig mit der Verdachtsdiagnose einer Infektarthritis. Die entsprechende Diagnostik sollte durchgeführt werden, wenn das Ergebnis eine therapeutische Konsequenz hat, wie die antibiotische Therapie bei der Lyme-Arthritis, oder die Prognose beeinflusst, wie bei einer juvenilen Spondylarthritis mit Nachweis der Yersinien-Infektion.
| Erkrankung | Erreger |
|---|---|
| Chlamydienarthritis | Chlamydien |
| Lyme-Arthritis | Borrelia burgdorferi |
| Reaktive Arthritisa | Yersinien, Salmonellen, Campylobacter, Shigellen |
| Akutes rheumatisches Fieberb | Streptokokken der Gruppe A |
| Virusarthritis | Rötelnvirus, Parvovirus B19, Mumpsvirus, Hepatitis-B-Virus, Varicella-Zoster-Virus, Ross-River-Virus, Hepatitis-C-Virus |
Therapie
Die Behandlung ist symptomatisch mit nichtsteroidalen Antirheumatika. Daneben sind bei einigen Arthritiden spezifische Therapien möglich:
Reaktive Arthritiden durch gramnegative Darmkeime bessern sich unter antibiotischer Therapie nicht, eine intraartikuläre Steroidtherapie kann jedoch sinnvoll sein.
Die Chlamydienarthritis wird ab dem 10. Lebensjahr mit Doxycyclin, 200 mg/Tag behandelt.
Nach einer Behandlung mit Ceftriaxon 50 mg/kgKG/Tag i.v. in einer Dosis für 14 Tage und/oder 200 mg Doxycyclin für 4 Wochen verschwindet die Arthritis bei fast 80% der Patienten mit Lyme-Arthritis.
Prognose
Die Prognose ist meist gut. Einige Fälle reaktiver Arthritis können in eine undifferenzierte juvenile Spondylarthritis übergehen. 10% der Patienten mit Lyme-Arthritis sprechen nicht auf antibiotische Therapieversuche an.
Systemischer Lupus erythematodes (SLE)
Definition
Diese seltene Erkrankung ist charakterisiert durch in Schüben auftretende Entzündungsreaktionen in mehreren Organen und die Bildung von Autoantikörpern. Es kommt zur Ablagerung zirkulierender komplementbindender Immunkomplexe z. B. in den Glomerula. Unbehandelt ist der systemische Lupus erythematodes mit einer hohen Letalität belastet. Anfangssymptome sind Gewichtsverlust, Abgeschlagenheit, Fieber, Hepatosplenomegalie, generalisierte Lymphknotenvergrößerung und verschiedene Hautausschläge. Die häufigsten Organmanifestationen sind in Tab. 12.4 dargestellt.
| Organsystem | Art der Schädigung |
|---|---|
| Haut und Schleimhaut | Schmetterlingsförmiges Erythem* (Abb. 12.6), Photosensitivität*, Ulzera der Mund- oder Nasenschleimhaut*, Raynaud-Phänomen, diskoider Lupus* |
| Gelenke | Nichterosive Arthritis* |
| Serosa | Pleuritis oder Perikarditis*, Peritonitis |
| Herz | Endo-/Myokarditis |
| Niere | Glomerulonephritis* (Proteinurie oder Zylinder), nephrotisches Syndrom, Urämie, arterielle Hypertension |
| Zentralnervensystem | Enzephalopathie* (Anfälle oder organisches Psychosyndrom) |
| Auge | Retinopathie, Papillenödem |
| Blut bildendes System | Zytopenie* (Anämie, Thrombopenie, Leukopenie), Gerinnungsstörungen |
| Allgemeines | Fieber, Gewichtsabnahme, Organomegalie |
| Immunserologie | Hochtitrige antinukleäre Antikörper*, Antikörper gegen Doppelstrang-DNA oder Sm-Kernantigen* |
Der Nachweis von 4 der 11 mit * gekennzeichneten Kriterien erlaubt die Diagnose „systemischer Lupus erythematodes“ mit einer Sensitivität und Spezifität von jeweils 96%


Diagnose
Die Diagnose eines systemischen Lupus erythematodes erfordert ein hohes Maß an Aufmerksamkeit für den wiederkehrenden Befall mehrerer Systeme.
Labor
Die Bestätigung der Diagnose erfolgt durch den Nachweis von hochtitrigen antinukleären Antikörpern (homogenes Muster) und Antikörpern gegen Doppelstrang-DNA, evtl. auch anderen spezifischen Autoantikörpern (Histon, Sm, SS-A, SS-B, Antiphospholipid). Die Erniedrigung des Komplementfaktors C3 ist ein Zeichen aktiver Erkrankung, besonders bei Patienten mit Nephritis. Während Entzündungszeichen wie BSG, CRP und Immunglobuline erhöht sind, zeigt das Blutbild eine Zytopenie.
Weitere Diagnostik
Die weitere Diagnostik richtet sich nach dem Organbefall, z. B. Lumbalpunktion, EEG und Kernspintomographie des Schädels bei Befall des Zentralnervensystems. Bei Verdacht auf Lupus-Nephritis mit Nachweis von Erythrozyten, Eiweiß und Zylindern im Urin kann eine Nierenbiopsie bei der Therapieplanung hilfreich sein.
Therapie
Angesichts des Fehlens einer spezifischen Therapie kann der Entzündungsprozess nur symptomatisch angegangen werden. Sonnenbestrahlung ist zu vermeiden, Vitamin D ist zu substituieren. Die Intensität der pharmakologischen Behandlung richtet sich nach der Stärke der Erkrankung und soll bleibende Schäden, z. B. an Niere oder Zentralnervensystem verhindern. Alle Patienten erhalten Hydroxychloroquin. Der Erfolg der Behandlung wird an einer Normalisierung des Allgemeinzustands und der Listen bewerteter Symptome sowie am Rückgang der Entzündungszeichen erkannt. Später fällt auch der Titer der Doppelstrang-DNA-Antikörper ab. Sein Wiederanstieg kündigt einen drohenden weiteren Schub der Erkrankung an.
Systemische Gaben von Kortikosteroiden mit Prednison oder besser als Puls-Steroide sind häufig initial notwendig. Sobald wie möglich soll die Kortikosteroiddosis reduziert werden. Azathioprin und Mycophenolat-Mofetil werden als steroidsparende Medikamente eingesetzt. Das akute Nierenversagen ist eine lebensbedrohliche Komplikation und erfordert oft die i.v.-Gabe von Cyclophosphamid. In schweren Fällen kann der CD20-Antikörper Rituximab eingesetzt werden.
Verlauf, Prognose
Die Prognose ist durch den Einsatz der antiinflammatorischen und symptomatischen Therapie deutlich gebessert worden. Ein 5-Jahres-Überleben von über 90% ist an größeren Zentren die Regel. Die Langzeitprognose bleibt aber unsicher und wird durch den Befall von Niere, Herz und Zentralnervensystem und Komplikationen der Therapie wie Sepsis infolge Immunsuppression bestimmt.
Lupusähnliche Krankheitsbilder
Nach der Einnahme verschiedener Medikamente wie Sulfonamide, Hydralazin und Antikonvulsiva wird ein medikamenteninduzierter Lupus beobachtet, der milder verläuft und nach Absetzen der Medikamente reversibel ist. Neugeborene von Müttern mit systemischem Lupus erythematodes oder Sjögren-Syndrom können durch transplazentar übertragene Antikörper Symptome eines neonatalen Lupus entwickeln. Die Symptome an der Haut (an einen diskoiden Lupus erinnernd mit erythematösen, leicht schuppenden Effloreszenzen, die zentral abblassen und so einen ringförmigen Charakter haben) und die Zytopenie bilden sich in den ersten Lebensmonaten zurück. Hingegen können die mütterlichen Anti-Ro-Antikörper am Herzen des Feten zum AV-Block führen, der irreversibel ist und eine Therapie mit α-Sympathikomimetika und Herzschrittmacher notwendig machen kann. Gelegentlich führt erst die Erkrankung des Neugeborenen zur Diagnose der mütterlichen Kollagenose, die Mutter kann aber auch asymptomatische Trägerin der Anti-Ro-Autoantikörper sein. Sie sollte mit Hydroxychloroquin in der Schwangerschaft behandelt werden
Juvenile Dermatomyositis
Definition
Die juvenile Dermatomyositis ist eine entzündliche Erkrankung unbekannter Ursache von Muskel und Haut, selten weiterer Organe. Im Gegensatz zur Dermatomyositis des Erwachsenen gibt es keine Assoziation mit Malignomen.
Klinik
Die Symptome beginnen schleichend über viele Wochen oder akut, und umfassen allgemeines Krankheitsgefühl, Fieber und Ermüdbarkeit. Typisch ist die Muskelschwäche mit Bevorzugung der proximalen Muskulatur. An den Oberlidern treten bläulich-livide Verfärbungen und im Gesicht Erytheme und ödematöse Schwellungen auf. Über Ellenbogen, Knien und Malleoli und besonders über den Metakarpophalangeal- und proximalen Interphalangealgelenken kommt es zu erythematösen schuppenden Hautveränderungen (Abb. 12.7). Haut- und Muskelsymptome können mit einer zeitlichen Differenz auftreten, die isolierte Polymyositis ist im Kindesalter aber sehr selten. Subkutane und intermuskuläre Verkalkungen können nach schwerem Verlauf auftreten und prognosebestimmend werden, wenn sie zur Immobilisierung führen. Nekrosen über subkutanen Kalkplatten oder sehr schmerzhafte, Kalk absondernde Hautulzera sind möglich.


Diagnose
Die Muskelkraft muss semiquantitativ mit bewerteten Listen gemessen werden. Als Zeichen der Myositis kommt es zum Anstieg der muskelspezifischen Enzyme (GOT, Kreatinkinase LDH). Muskelantikörper können diagnostische und prognostische Hinweise liefern. Die Myositis kann mittels Kernspintomographie (T2-gewichtetes Bild) und Muskelbiopsie (lymphozytäre Infiltration) gesichert werden. Das EMG erfordert die Mitarbeit des Patienten. Häufig sind antinukleäre Antikörper nachweisbar. Die typischen Hautveränderungen zusammen mit den Muskelsymptomen machen die Diagnose leicht. Differenzialdiagnostisch kommen virale Myositiden oder neuromuskuläre Erkrankungen in Betracht.
Therapie
Hautpflege, Mobilisierung und Krankengymnastik sind besonders bei bereits längerem Verlauf wichtig. Kortikosteroide sind effektiv in der Unterdrückung der Entzündung. Die Steroid-Puls-Therapie in Kombination mit niedrigdosierten oralen Steroiden ist wirksam. Hydroxychloroquin wirkt sich günstig auf die Hautveränderungen aus, intravenöse Immunglobuline können steroidsparend wirken. Oft wird frühzeitig Methotrexat gegeben.
Prognose
Unbehandelt hat die Erkrankung eine schlechte Prognose. Durch Beteiligung der Schlund- und Atemmuskulatur oder gastrointestinale Perforation können lebensgefährliche Komplikationen auftreten, die Intensivpflege und Respiratorbehandlung erfordern. Oft heilt die Erkrankung unter Therapie nach jahrelangem Verlauf aus.
Sklerodermie
Klinik
Die zirkumskripte/lokalisierte Sklerodermie ist eine chronisch-entzündliche Erkrankung der Haut und darunter liegender Strukturen mit nachfolgender Atrophie. Die Hautsymptome können asymmetrisch herdförmig (Morphea) oder in einer bandförmigen Anordnung (lineare Sklerodermie) vorkommen (Abb. 12.8). Bei der systemischen Sklerose finden sich neben symmetrischen Hauterscheinungen systemische Befunde:
Synovitis,
Lungenfibrose,
pulmonale Hypertonie,
ösophageale Dysfunktion,
Perikarditis,
Raynaud-Phänomen mit akraler Nekrosen,
Nephritis mit Hochdruck und eingeschränkter Nierenfunktion.


Die häufigere lokalisierte Form kann zur Zerstörung von Extremitäten und zur Entstellung des Gesichts führen, die systemische Sklerose kann durch renale, pulmonale oder kardiale Komplikationen tödlich enden.
Diagnose
Typische Laborbefunde existieren nicht, entzündliche Zeichen fehlen. Oft sind die antinukleäre Antikörper, evtl. mit nukleolärem Muster, und Scl-70-Antikörper positiv.
Therapie
Eine spezifische Therapie ist nicht bekannt. Methotrexat scheint aber einen positiven Einfluss zu haben. Wichtig sind intensive Hauptpflege und die Vermeidung von einem Vasospasmus auslösenden Faktoren wie Kälte. Nifedipin wird zur Behandlung des Raynaud-Phänomens und ACE-Hemmer bei Nierenbefall eingesetzt. Eine pulmonale Hypertonie kann mit Endothelin-Antagonisten, Prostaglandinen und Phosphatdiesterase-Inhibitoren therapiert werden. Wichtig ist der Einsatz der Physiotherapie, um Kontrakturen durch die narbigen Veränderungen zu vermeiden.
Mischkollagenose
Diese Erkrankung, auch „mixed connective tissue disease“ oder Sharp-Syndrom genannt, vereint Symptome des systemischen Lupus erythematodes, der juvenilen idiopathischen Arthritis, der juvenilen Dermatomyositis und/oder der systemischen Sklerose.
Das klassische Sharp-Syndrom ist definiert durch den Nachweis von Antikörpern gegen extrahierbares ribonukleäres Protein (Anti-RNP-Antikörper) unter den antinukleären Antikörpern. Die Behandlung entspricht der des systemischen Lupus erythematodes.
Vaskulitissyndrome
Eine Vaskulitis, d. h. eine Entzündung in der Wand oder Umgebung der Blutgefäße wird als begleitendes Symptom bei den meisten rheumatischen Erkrankungen gefunden. Eine Gruppe von Erkrankungen zeigt die Vaskulitis als Hauptsymptom.
Nach Ausschluss anderer rheumatischer Erkrankungen kann die Diagnose „Vaskulitis“ vermutet werden, wenn mehrere Organsysteme auf sonst nicht erklärliche Weise im Verlauf einer Erkrankung befallen werden und wenn Myalgien, Arthralgien, Hauteffloreszenzen, Bauchschmerzen, Nephritis, Hochdruck, neurologische Manifestationen, Lungeninfiltrate, nasale Symptome oder ungewöhnliche kardiale Symptome, wie Erkrankung der Herzkranzgefäße oder unerklärtes Herzversagen auftreten. Die klassischen Vaskulitiden sind Periarteriitis nodosa, Granulomatose mit Polyangiitis (Wegener), eosinophile Granulomatose mit Polyangiitis (Churg-Strauss) und Takayasu-Arteriitis, die selten auch im Kindesalter vorkommen können. Im Folgenden werden 2 gut definierte, im Kindesalter häufige Vaskulitiden vorgestellt.
Purpura Schönlein-Henoch
Definition
Diese auch anaphylaktoide Purpura genannte Vaskulitis ist charakterisiert durch Hauterscheinungen mit typischer Lokalisation und einem charakteristischen Muster von Organbeteiligungen. Immunhistologisch lassen sich vorwiegend an Kapillaren, aber auch an Arteriolen und Venolen IgA-haltige Immunkomplexe nachweisen. Per definitionem sind die Hauterscheinungen für die Diagnose unerlässlich.
Klinik
Die Hautläsionen sind an den unteren Extremitäten, dem Glutäalbereich, seltener auch an den Streckseiten der oberen Extremitäten und am Stamm lokalisiert. Die Einzeleffloreszenz kann vielgestaltig sein. Meistens beginnt sie als makulopapulöses Erythem mit einem Durchmesser von 1–3 mm, in die es zur petechialen oder flächenhaften Blutung kommt, sodass die charakteristische palpable Purpura entsteht, die zu großen Purpurabezirken konfluieren kann (Abb. 12.9). Die in der Regel symmetrisch auftretenden Erscheinungen jucken nicht. Angioödeme können im Bereich der Extremitäten, aber auch im Gesicht auftreten. Schmerzhafte transiente Gelenkschwellungen, besonders der Sprunggelenke, und ausgeprägten periartikulären Schwellungen, auch am Fußrücken, entwickeln sich in etwa 2/3 der Fälle.


Eine gastrointestinale Beteiligung in Form kolikartiger Bauchschmerzen, Erbrechen und blutiger Stühle kann auf eine gehäuft vorkommende Invagination hinweisen. Eine renale Beteiligung mit Erythrozyturie kommt in etwa ¼ der Fälle vor. Ein kleiner Teil dieser Patienten entwickelt eine chronische Nierenerkrankung (Kap. 25). Sehr selten ist eine Beteiligung des Zentralnervensystems (Anfälle, Paresen). Gelegentlich tritt die Purpura Schönlein-Henoch rezidivierend auf.
Therapie
Eine Therapie mit Kortikosteroiden ist bei schwerer intestinaler Beteiligung indiziert in der Absicht, eine Invagination zu verhindern. Die Glomerulonephritis mit Halbmondbildung nach Purpura Schönlein-Henoch kann durch Pulssteroide günstig beeinflusst werden. Die Prognose ist bis auf die wenigen Fälle mit schweren gastrointestinalen Komplikationen oder chronischer Glomerulonephritis gut.
Kawasaki-Erkrankung
Definition
Diese auch mukokutanes Lymphknotensyndrom genannte Vaskulitis ist charakterisiert durch hohes, lang anhaltendes Fieber und weitere typische Befunde (Tab. 12.5; Abb. 12.10).
| Symptome | Häufigkeit |
|---|---|
| Fieber (>4 Tage) | 100% |
|
Hautveränderungen der Extremitäten: - Palmar-/Plantarerythem (früh) - Schuppung (spät) |
70% |
| Exanthem (polymorph) | 80% |
|
Orale Veränderungen: - Trockene rote (Lack)lippen mit vertikaler Fissur - Himbeerzunge - Erythem der Mundschleimhaut |
90% |
| Konjunktivitis (bilateral ohne Exsudat) | 85% |
| Lymphknotenvergrößerung (zervikal, evtl. unilateral) | 70% |

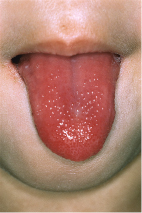
Klinik
Weitere Erscheinungen sind Urethritis, Arthritis, aseptische Meningitis, Durchfall, Gallenblasenhydrops und Verschlussikterus. Während in der Akutphase der Erkrankung Myokarditis, Herzinsuffizienz und Perikarditis vorkommen, sind die im weiteren Verlauf auftretenden Folgen der echokardiographisch zu untersuchenden Koronararteriitis (Infarkt, Stenose, Aneurysma) für die Gesamtprognose der Erkrankung entscheidend. Bei Säuglingen sind die Befunde häufig wenig charakteristisch ausgebildet und manchmal weist erst der Nachweis von Koronaraneurysmen auf die Kawasaki-Erkrankung hin.
Diagnose
Die Diagnose eines Kawasaki-Syndroms sollte immer erwogen werden, wenn therapieresistentes Fieber bei einem jungen Kind auftritt.
Die klinischen Symptome ermöglichen die Diagnose. Laborauffälligkeiten sind eine Leukozytose mit erhöhter Blutsenkungsgeschwindigkeit und erhöhtem CRP, eine Thrombozytose (in der 2. Krankheitswoche beginnend) und eine sterile Pyurie.
Therapie
Durch eine hochdosierte i.v.-Immunglobulingabe (2 g/kgKG, einmalig über 8–16 h) und Acetylsalizylsäure (ASS; 30 mg/kgKG) kann der Entzündungsprozess oft rasch unter Kontrolle gebracht und die Rate von kardialen Komplikationen deutlich gesenkt werden. Bei Nichtansprechen werden erneute Immunglobuline, Steroide, Anakinra oder Infliximab gegeben. In der Abklingphase der Erkrankung ist wegen der Gefahr der Koronarthrombose eine Thrombozytenaggregationshemmung mit ASS 3 mg/kgKG/Tag wichtig.
Alle Kinder mit Kawasaki-Erkrankung sollen klinisch und echokardiographisch nachuntersucht werden (zeitliches Maximum der Koronarveränderungen 4. Krankheitswoche). Späte Todesfälle aufgrund einer Herzbeteiligung kommen vor.
Weitere rheumatische Erkrankungen
Akutes rheumatisches Fieber
Grundlagen
Das akute rheumatische Fieber ist eine entzündliche Erkrankung, die sich an Gelenken, Herz, Zentralnervensystem und Haut manifestiert. Eine Infektion mit Streptokokken der Gruppe A, z. B. als Tonsillitis, geht den Erkrankungserscheinungen 2–5 Wochen voraus.
Epidemiologie, Pathophysiologie
Es findet sich ein erhöhter oder ansteigender Antistreptolysintiter, der Nachweis der Streptokokken gelingt aufgrund der Latenzperiode zwischen Tonsillitis und akutem rheumatischen Fieber nicht immer. In Europa ist diese Erkrankung sehr selten geworden, unter schlechten sozioökonomischen Bedingungen spielt sie eine wichtige Rolle.
Es handelt sich um eine Autoimmunerkrankung mit Kreuzreaktivität zwischen Streptokokkenantigenen und menschlichen Bindegewebsstrukturen, sodass sich durch die Streptokokkeninfektion hervorgerufene Antikörper bei prädisponierten Patienten an Wirtsantigene binden.
Klinik
Die klinischen Erscheinungen sind vielgestaltig und können in verschiedenen Kombinationen auftreten:
Neben Fieber sind Gelenkerscheinungen die häufigsten Symptome (75%); sie können als Arthralgien oder als Schwellung der Gelenke mit Rötung und Überwärmung auftreten. Die Schmerzen sind häufig schwerer, als der klinische Befund vermuten lässt. Charakteristisch ist das rasche Wechseln der betroffenen großen Gelenke.
Eine Herzbeteiligung (40%) kann sich in Tachykardie, Auftreten von Geräuschen (holosystolisches Geräusch als Zeichen einer Mitralklappenbeteiligung, Diastolikum bei Aorteninsuffizienz), Herzvergrößerung, Perikarditis oder Herzversagen äußern.
Die Chorea minor (10–15%) beginnt als psychische Labilität, Verschlechterung der Schulleistungen und geht über in choreatische Bewegungsstörungen der Extremitäten, des Rumpfs und der Gesichtsmuskulatur.
Rheumaknötchen sind subkutan gelegene erbsgroße, derbe, schmerzlose Knoten, die bevorzugt über Knochenvorsprüngen liegen.
Charakteristisch ist das flüchtige, blassrosa Erythema marginatum (5%).
Es finden sich eine erhöhte Konzentration von C-reaktivem Protein (CRP), eine beschleunigte Blutsenkungsgeschwindigkeit und eine Leukozytose.
Diagnose
Die Diagnose ist wahrscheinlich, wenn die in Tab. 12.6 genannten Kriterien vorhanden sind und der vorangehende Streptokokkeninfekt nachgewiesen wurde.
| Hauptkriterien | Nebenkriterien |
|---|---|
| Karditis | Fieber |
| Polyarthritis | Arthralgie |
| Chorea minor | Vorausgegangenes rheumatisches Fieber |
| Erythema marginatum | CRP oder BSG entzündlich verändert |
| Subkutane Knötchen | PR-Verlängerung im EKG |
Die Diagnose „akutes rheumatisches Fieber“ erfordert das Vorhandensein von 2 Hauptkriterien oder einem Haupt- und 2 Nebenkriterien und den Nachweis der vorangegangenen Streptokokkeninfektion (Antistreptolysintiter erhöht, Rachenabstrich mit A-Streptokokken, oder Scharlach)
Therapie
Man gibt ein nichtsteroidales Antirheumatikum und bei schwerer Herzbeteiligung Kortikosteroide. Stark erregten Kindern mit Chorea minor helfen Diazepam oder Tiaprid.
Auch wenn keine Streptokokken nachweisbar sind, erfolgt eine orale Penicillintherapie (50.000 E/kgKG für 10 Tage). Um Rezidive zu verhindern, ist anschließend eine Antibiotikaprophylaxe mit i.m.-Gabe von 1,2 Mio. Einheiten Benzathinpenicillin alle 3–4 Wochen indiziert. Die Dauer der Prophylaxe ist umstritten.
Prognose
Die Prognose des akuten rheumatischen Fiebers wird durch die Ausprägung der Karditis und ein nachfolgendes Mitral- oder Aortenvitium bestimmt.
Familiäres Mittelmeerfieber
Diese autosomal-rezessiv vererbte Multisystemerkrankung betrifft meist Kinder aus der Türkei, Griechenland oder Israel. Es kommt zu rezidivierenden, wenige Tage anhaltenden Fieberschüben mit starken Bauchschmerzen (sterile Peritonitis), Brustschmerzen (Pleuritis), Arthritiden oder erysipelartigen Hautausschlägen. Während des Fiebers bestehen eine Granulozytose und CRP-Erhöhung. Im Intervall sind die Kinder unauffällig. Häufig wird vor Stellung der richtigen Diagnose eine Appendizitis vermutet, die histologische Untersuchung der Appendix zeigt jedoch bei unauffälliger Mukosa eine Entzündung der Serosa.
Die Diagnose kann durch den Nachweis von homozygoten oder compound heterozygoten Mutationen im MEFV-Gen, das in Granulozyten das Pyrin kodiert, bestätigt werden. Durch die Gabe von Kolchizin können die schmerzhaften Fieberschübe unterdrückt und die Entwicklung einer Nierenamyloidose mit nachfolgendem nephrotischem Syndrom verhindert werden.
Bei rezidivierenden Fieberschüben ohne Erregernachweis sind neben dem familiären Mittelmeerfieber weitere periodische Fiebererkrankungen in Erwägung zu ziehen (Tab. 12.7).
| Syndrom | Besonderheiten |
|---|---|
| Familiäres Mittelmeerfieber (FMF) | Text; Fieberdauer bis 2–3 Tage |
| Hyper-IgD-Syndrom (HID) | IgD im Serum während Fieber und im Intervall nicht immer erhöht; Fieberdauer 4–6 Tage; autosomal-rezessiv vererbter partieller Defekt der Mevalonatkinase |
| Familiäre Kälteurtikaria (FCAS)a | Beginn im 1. Lebensjahr, kälteinduziert, nicht juckendes Exanthem, Schüttelfrost, Fieber, Arthralgien, Myalgien, Kopfschmerzen |
| Muckle-Wells-Syndrom (MWS)a | Kälte triggert Symptome nicht, sensoneurale Schwerhörigkeit, sonst wie FCAS |
| CINCA- oder NOMID-Syndrom (chronisches infantiles neurologisch-kutan-artikuläres Syndrom)a | Beginn bereits in den ersten Lebensmonaten; aseptische Meningitis, Papillenödem, Retardierung, Uveitis, destruktive epiphysäre Veränderungen, sonst wie MWS |
| Tumornekrosefaktorrezeptor assoziiertes periodisches Fiebersyndrom (TRAPS) | Fieberdauer länger 1 Woche; wandernde Myalgien, Erytheme und periorbitales Ödem; autosomal dominant vererbt |
| Periodisches Fieber – aphthöse Stomatitis-Pharyngitis – Lymphadenitis-colli-Syndrom (PFAPA) | Symptome verschwinden nach einigen Jahren; Abortierung der Attacken mit Steroiden möglich; Behandlungsversuch mit Cimetidin oder Tonsillektomie |
| Zyklische Neutropenie | Alle 3–4 Wochen Fieber mit oralen Ulzera; mit G-CSF behandelbar |
a FCAS, MWS und CINCA stellen ein Spektrum zunehmender Krankheitsschwere dar auf der Basis von Gain-of-function-Mutationen des NLRP3-Gens für Cryopyrin, einem Rezeptor, der das angeborene Immunsystem und Entzündungsmediatoren stimuliert. IL-1-Blocker wie Canakinumab sind wirksam, ebenso wie bei HID, TRAPS und refraktärem FMF
Erythema nodosum
Das Erythema nodosum ist eine Pannikulitis und zeigt sich als überwärmte, rote, schmerzhafte Hautinduration, 3–5 cm im Durchmesser, häufig symmetrisch angeordnet über den Extensorenseiten der Unterschenkel. Fieber und Arthralgien können die Hauterscheinungen begleiten. Im Verlauf der Erkrankung verändern sie ihre Farbe zu lila bis blau. Sie verschwinden gewöhnlich nach 3 Wochen, können aber wieder aufflackern.
Auslösende Faktoren sind Infektionen mit Streptokokken, Mycobacterium tuberculosis, Yersinien, Pilzen und Medikamente sowie orale Kontrazeptiva oder eine chronisch-entzündliche Darmerkrankung. Häufig findet sich keine Ursache. Eine Therapie ist manchmal nicht notwendig, Glukokortikoide sind wirksam.
Juveniles Sjögren-Syndrom
Das Sjögren-Syndrom (SjS) ist eine Autoimmunerkrankung, die u. a. durch Autoantikörper gegen die Ro- (SS-A) und La- (SS-B) Antigene vermittelt wird und vornehmlich in exokrinen Drüsen und drüsenähnlichen Geweben zu einer chronischen, vorwiegend mononukleären Entzündung führt. Die Autoimmunität kann sich jedoch auch gegen andere Organe (Pankreatitis, Nephritis, Pneumonitis, Thyreoiditis, Vaskulitis, Neuritis N. optici, Myelitis transversa) richten. Vom primären SjS als autoimmune Grunderkrankung wird das sekundäre SjS im Rahmen einer anderen Autoimmunerkrankung, wie z. B. SLE oder JIA unterschieden. Das primäre juvenile SjS betrifft Mädchen 7- bis 10-fach häufiger als Jungen.
Klinik
Das häufigste Leitsymptom ist eine rezidivierende beidseitige Parotitis, oft mit regionaler Lymphknotenschwellung. Das echte Sicca-Syndrom ist beim juvenilen SjS jedoch seltener als beim adulten SjS. Starke Karies kann sich bei juvenilem SjS auch ohne schwere Sicca-Symptomatik bilden. Renale tubuläre Azidose kann akut durch Hypokaliämie zu Schwäche und Lähmung führen und chronisch zu Minderwuchs und Skelettveränderungen.
Diagnose
Die diagnostischen SS-A/SS-B-Autoantikörper liegen bei ca. 70% der pädiatrischen Patienten vor. Häufig, aber weniger spezifisch, sind auch hochtitrige antinukleare Antikörper, positiver Rheumafaktor, und erhöhte Serumspiegel von Immunglobulin G und Amylase. Am objektivsten ist der Nachweis von chronischen mononuklearen Entzündungsinfiltraten in einer Biopsie der kleinen Speicheldrüsen, die an der Innenseite der Unterlippe leicht durchgeführt werden kann.
Therapie
Künstliche Tränen und Mundbefeuchtung sind hilfreich. Auf gute Zahnpflege ist zu achten. Systemische Symptome werden primär mit Kortikosteroiden und Hydroxychloroquin behandelt. Bei schwerem Organbefall erfolgt die Behandlung wie beim SLE.
Sarkoidose
Die Sarkoidose ist eine chronische granulomatöse Erkrankung unbekannter Genese. Die frühkindliche Form („Vorschulsarkoidose“) besteht oft aus der Trias Exanthem, Uveitis und Arthritis. Im späteren Kindes- und Jugendalter ähnelt die Erkrankung mehr der adulten Verlaufsform, bei welcher hiliäre Lymphadenopathie und eine restriktive Lungenerkrankung im Vordergrund stehen. Als Löfgren-Syndrom bezeichnet man das Zusammentreffen von hilärer Lymphadenopathie mit Erythema nodosum und Polyarthritis. Neben organspezifischen Komplikationen kann es bei Sarkoidose auch zu Hyperkalziämie kommen. Im Serum sind ACE und/oder Lysozym bei 60–75% der Patienten erhöht. Die Behandlung erfolgt primär mit Kortikosteroiden und Methotrexat. Die Prognose ist meist gut, bis zu 80% der pädiatrischen Patienten gehen in permanente Remission.
Nichtbakterielle Osteomyelitis
Definition, Pathophysiologie
Die nichtbakterielle Osteomyelitis (NBO), syn. chronisch rezidivierende multifokale Osteomyelitis (CRMO), ist eine oft multifokal verlaufende Osteomyelitis unbekannter Ätiologie und vermutlich autoinflammatorischer Pathogenese. Am Ort der Entzündung können keinerlei Krankheitserreger nachgewiesen werden. Bei etwa 10% der Patienten mit CRMO finden sich an Hand- und Fußflächen Hauterscheinungen im Sinne einer palmoplantaren Pustulose und bei 3% eine Psoriasis. Außerdem werden in den Familien der Betroffenen gehäuft Autoimmunerkrankungen, wie z. B. M. Crohn, Hashimoto-Thyreoiditis oder Sarkoidose beobachtet.
Klinik
Führendes Symptom sind lokalisierte Schmerzen, auch mit Schwellung und häufig ohne Fieber. An mehreren Stellen des Skelettsystems finden sich entzündliche Herde, überwiegend in den Metaphysen der großen Röhrenknochen und Wirbelkörpern. Prinzipiell kann aber jeder Knochen betroffen sein, so z. B. auch typischerweise die Klavikula (Abb. 12.11). Auch ein unilokulärer Befall ist möglich.


Diagnose
Die Diagnose wird aufgrund der Klinik, Bildgebung (MRT) und oft Biopsie mit lymphozytärer Infiltration gestellt. Schwierig ist die Diagnosestellung bei unilokulären Befall. Hier kann die Abgrenzung zu einer chronisch bakteriellen Osteomyelitis schwierig sein kann. Dies ist auch die häufigste Differenzialdiagnose, zu der auch der Ausschluss maligner Knochenprozesse gehört.
Therapie
Mittel der Wahl sind nichtsteroidale Antirheumatikaund Kortikoide. Refraktäre Fälle bessern sich unter TNF-α-Antagonisten. Bei Wirbelsinterungsfrakturen werden Bisphosphonate versucht.
Prognose
Die Prognose der NBO ist im Allgemeinen gut. Der Verlauf kann jedoch sehr unterschiedlich sein und reicht von Spontanremission zu immer wieder rezidivierenden Verläufen. Das Knochenwachstum ist nicht beeinträchtigt, Wirbelkörperfrakturen sind möglich.