

Abstract
Rezidivierende harmlose Infektionen, insbesondere der Atemwege sind im Kindesalter häufig. Gesunde Kleinkinder erkranken bei Betreuung in Kinderkrippe oder Kindergarten im Durchschnitt 8-mal pro Jahr an fieberhaften, viralen Infektionen, die nur wenige Tage andauern und die Kinder nicht schwer beeinträchtigen. Von dieser physiologischen Infektanfälligkeit unterscheidet sich eine pathologische Anfälligkeit bei angeborenen Immundefekten: Schwere Infektionen treten durch Erreger auf, die vom Immungesunden kontrolliert werden (Pneumonie durch Pneumocystis jirovecii oder Aspergillus spp, Organabszesse durch S. aureus, disseminierte Lymphadenitis durch atypische Mykobakterien, Candidiasis über den Windelbereich und das erste Lebensjahr hinaus und ähnliche Erkrankungen). Auch autoinflammatorische/autoimmunologische Erkrankungen mit Granulombildung, rezidivierendem Fieber, ausgedehnten Ekzemen, Lymphoproliferation oder chronischer Darmentzündung müssen Anlass zur Suche nach angeborenen Ursachen für die immunologische Fehlregulation sein. Bei über 300 molekular definierten Defekten sind die im Folgenden beschriebenen Krankheitsbilder klassische Beispiele für angeborene Immundefekte aus den verschiedenen Bereichen unseres Immunsystems.
Phagozytendefekte
Septische Granulomatose
Definition
Diese Erkrankung („chronic granulomatous disease“, CGD) wird durch den Defekt eines in der Phagozytenmembran befindlichen Enzymsystems (NADPH-Oxidase, enthält u. a. gp91-phox) verursacht. Sie tritt mit einer Häufigkeit von 1:100.000 bis 1:300.000 auf.
Pathogenese
Betroffenen Zellen (besonders Granulozyten und Monozyten, aber auch T-, B- und weiteren Zellen) fehlt die Fähigkeit, reaktive Sauerstoffmetabolite mit Hilfe der NADPH-Oxidase zu bilden. Ihre Produktion ist für die Abwehr bestimmter Bakterien (z. B. S. aureus) und Pilze (Aspergillen) und für normale Entzündungsregulationen wichtig.
Aus dem Defekt resultieren schwere, oft abszedierende Infektionen und überschießende, oft granulomatöse Entzündungsreaktionen, häufig auch ohne Infektion. In etwa 2/3 der Fälle ist das auf dem X-Chromosom kodierte gp91-phox betroffen. Bei autosomalen Formen sind p22-phox, p40-phox, p47-phox, oder p67-phox betroffen.
Klinik
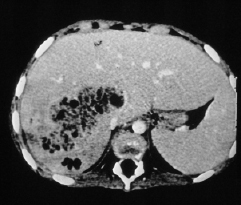
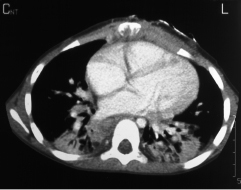
Die Klinik ist sehr komplex und initiale Fehldiagnosen sind häufig. Einerseits treten eitrige, zum Teil abszedierende Infektionen von Lymphknoten, Lunge, Leber (Abb. 10.1) (und weiteren inneren Organen), Weichteilen, Haut und Knochen (Abb. 10.2) auf. Andererseits kommen klinische Bilder vor, die mit Mykobakteriosen, Sarkoidose, exogen allergischer Alveolitis, idiopathischer Lungenfibrose, Morbus Crohn, „Tumoren“ (in Wirklichkeit Granulome) z. B. der ableitenden Harnwege, rheumatischen Erkrankungen (Fieberschübe) usw. verwechselt werden.


Cave
Besonders gefährliche Keime: Aspergillus nidulans, Burkholderia cepacia
Die häufigste Erreger ist S. aureus. Burkholderia cepacia kann eine gramnegative, rasch tödliche Sepsis verursachen. Aspergillusinfektionen der Lunge können bei Inhalation sporenhaltiger Luft sowohl hochakut als auch bei allmählicher Gewebsinfiltration schleichend verlaufen und ebenfalls tödlich verlaufen. Weitere opportunistische Erreger (z. B. S. marcescens, Nocardien) kommen vor, es besteht jedoch keine allgemeine Abwehrschwäche gegenüber Bakterien oder Viren. Autoinflammatorische Entzündungen können zu Organversagen (Lungenfibrose) und schwerer Beeinträchtigung (Kolitis) führen. Fisteln (z. B. perianal) und gestörte Wundheilung sind häufig.
Cave
Auch dysregulierte Entzündungen können zu Organversagen führen, z. B. Lungenfibrose.
Erste Symptome können sehr früh im Säuglingsalter („Late-onset-Sepsis“) auftreten, aber – besonders bei Restaktivität der NADPH-Oxidase – auch erst später im Jugendlichen- oder Erwachsenenalter.
Histologisch findet man in betroffenen Organen Granulome mit Epitheloidzellen und Langerhans-Riesenzellen, was häufig zur Fehldiagnose „Tuberkulose“ führt. Andererseits können Patienten mit CGD auch an Tuberkulose erkranken; besonders typisch ist das Vorkommen einer BCG-itis, einer Infektion nach Impfung gegen Tuberkulose mit BCG-Bakterien. Durch Granulome in der Wand von Hohlorganen wie Ureter, Ösophagus, Magen u. a. können Stenosierungen auftreten. In Familien mit X-chromosomalem rezessivem Erbgang können Heterozygote bei ungünstiger X-Inaktivierung (Lyonisierung) klinische Auffälligkeiten, wie rezidivierende aphthöse Stomatitiden, einen diskoiden Lupus erythematodes oder schwere CGD-typische Infektionen zeigen.
Diagnose
Die Diagnose ist biochemisch durch die In-vitro-Messung reaktiver Sauerstoffmetabolite zu stellen. Dazu stehen mehrere Testverfahren zur Verfügung, wobei der durchflusszytometrische Dihydrorhodamin-123 (DHR)-Test die meisten Vorteile auf sich vereinigt. Die Diagnose muss durch zwei unabhängige Verfahren aus zwei Blutproben bestätigt werden (z. B. DHR-Test zu Messung von H2O2 und Chemolumineszenz oder Nitroblautetrazoliumreduktion zur Messung von O2-).
Eine pränatale Diagnostik ist aus Chorionzottenbiopsat mit molekulargenetischen Techniken möglich. Diese Techniken sind auch für die genetische Beratung und in Zukunft eventuell vor einer Gentherapie wichtig. Genetische oder serologische Analysen erfassen auch, ob durch eine große Deletion eine zusätzliche Anomalie der Erythrozytenmembran (Akanthozytose bei McLeod-Phänotyp) vorliegt.
Cave
Kell-positives Blut darf den Patienten mit McLeod-Phänotyp wegen der Gefahr der Antikörperbildung nicht gegeben werden (vor geplantem Eingriff Einfrieren der eigenen Erythrozyten – Eigenblutspende!).
Therapie
So komplex die klinische Präsentation ist, so komplex gestaltet sich die Therapie. Grundsätzlich müssen Patienten mit CGD in einem spezialisierten Zentrum bzw. in Zusammenarbeit mit einem solchen behandelt werden. Patienten benötigen eine prophylaktische Behandlung (meist Cotrimoxazol und Posaconazol, dazu bei zusätzlicher Notwendigkeit IFN-γ s.c.). Alle 3 Monate sollte eine Kontrolluntersuchung stattfinden, die einen Ultraschall des Abdomen und bei entsprechendem Alter eine Lungenfunktionsprüfung mit einschließt. Entzündliche Infiltrate sollten, wenn immer vertretbar bioptisch und mikrobiologisch evaluiert werden (z. B. durch CT-gesteuerte Nadelbiopsie bei Infiltraten der Lunge). Infektionen werden lange und aggressiv mit Antibiotikakombinationen behandelt, wobei mindestens eins besonders gut zell- und gewebegängig sein sollte. Bei unbekanntem Erreger ist an Burkholderia cepacia zu denken, der auf viele häufig verwendete Antibiotika nicht anspricht, aber in wenigen Tagen zum Tode führen kann. Bei Aspergillusinfektionen ist Voriconazol wirksamer und nebenwirkungsärmer als Amphotericin B. Eine solche Therapie ist lange, z. B. über 6 Monate, durchzuführen. In schwerwiegenden Fällen kann die Therapie durch Granulozytentransfusionen ergänzt werden. Bei Autoinflammation mit und ohne Granulombildung ist unter gewissen Vorsichtsmaßnahmen die Therapie mit Kortikosteroiden (in einigen Fällen sogar längerfristig und hochdosiert (2 mg/kgKG Prednisolon)) und anderen Immunsuppressiva notwendig. Ferner können chirurgische Interventionen notwendig sein.
Eine kurative Therapieoption ist die Stammzelltransplantation, die von einem wenigstens 9/10-HLA-identischen Spender erfolgen sollte. Diese Option sollte grundsätzlich mit Nachdruck bereits bei Diagnosestellung angeboten werden. Die feinere Auflösung der HLA-Merkmale und die individualisiert dosierte, reduzierte (nicht-myeloablative) Konditionierung haben die Komplikationen einer Stammzelltransplantation deutlich vermindert. Ebenso ist die Stammzelltransplantation vor Eintreten schwerer infektiöser (Aspergillose) und autoinflammatorischer (Colitis) Komplikationen risikoärmer.
Prognose
Bei der klassischen Form der CGD war die Prognose früher sehr schlecht. Sie hat sich aber nach Einführung der Prophylaxe (s. oben) und der intensiven Überwachung deutlich gebessert. 40- bis 50-jährige Patienten sind zwar keine extreme Ausnahme mehr, aber die sich mit fortschreitendem Lebensalter häufenden und dann oft doch noch lebensbegrenzenden Komplikationen sprechen für die frühe Stammzelltherapie.
Defekte in der Interferon-γ-Interleukin- 12-Achse und verwandte Störungen
Definition
Bei Defekten in der Interferon-γ-Interleukin-12-Achse handelt es sich um erbliche Störungen der Abwehr von Mykobakterien und weiteren intrazellulären Keimen. Überwiegend liegen dieser Krankheitsgruppe molekular nachweisbare Defekte des γ-Interferon/ IL-12-Signalwegs in Monozyten/Makrophagen zugrunde, die entweder zu einer verminderten Bildung oder zu einer verminderten Wirkung von IFN-γ führen.
Pathophysiologie
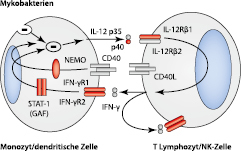
Normalerweise stimulieren Makrophagen spezifische T-Zellen parallel zur Antigenpräsentation mit Interleukin-12/23 (IL-12/23). Die T-Zellen antworten u. a. durch Proliferation und Produktion von Interferon-γ (IFN-γ), das wiederum Makrophagen über den Jak/Stat-Signalweg aktiviert und dadurch die Abtötung von Mykobakterien ermöglicht (Abb. 10.3). Krankheitsauslösende genetische Veränderungen wurden u. a. in den Genen für die Rezeptoren von IFN-γ und IL-12, für STAT1 und IL-12p40 gefunden. Am häufigsten ist der Interleukin-12-Rezeptor-β1-Defekt, gefolgt vom partiellen Interferon-γ-Rezeptor-1-Defekt. Außerhalb des skizzierten Systems können auch Defekte in NEMO, CYBB, IRF8, GATA2 und ISG15 und weiteren noch unbekannten Genen zu einer erhöhten Anfälligkeit für disseminierte Mykobakteriosen führen. Störungen werden autosomal-dominant oder X-chromosomal-rezessiv autosomal-rezessiv vererbt und können partiell oder vollständig sein. Bei partiellen Formen tritt überschießende Granulombildung auf, während bei vollständigen Abwehrschwächen die Granulombildung gestört ist.

Klinik
Wegen der variablen Pathophysiologie sind die klinischen Erscheinungsbilder trotz des relativ einheitlichen und begrenzten Erregerspektrums vielgestaltig, sodass die Erregerisolation wegweisend ist. Im Vordergrund stehen lokale und systemische Infektionen mit nichttuberkulösen Mykobakterien (NTM), und eine BCG-itis nach Impfung. Auch bei disseminiert verlaufender Tuberkulose, invasiven Infektionen durch S. enteritidis und Candidainfektionen muss an diese Defekte gedacht werden. Listerien und Virusinfektionen (VZV, CMV) wurden nur in Einzelfällen beschrieben.
Lokale Infektionen mit vorhandener Granulombildung deuten auf partielle Defekte oder Defekte des IL-12-Rezeptors, ausgedehnte, auch systemische Infektionen mit Fieberschüben, Hepatosplenomegalie, Pneumonie, Knochenläsionen (Abb. 10.4) und Darmbefall deuten auf vollständige Defekte des IFN-γ-Rezeptors hin. Lymphknotenbefall ist häufig, wobei aber der isolierte, oft anzutreffende einseitige Befall von Halslymphknoten durch NTM bei ansonsten gesundem Kleinkind selten auf eine erbliche Störung zurückzuführen ist. Partielle Defekte mit erhaltener Granulombildung ähneln anderen Erkrankungen mit Granulombildung wie eosinophiles Granulom, chronisch rekurrierende multifokale Osteomyelitis und Morbus Crohn.


Diagnose
Ein funktionelles Screening auf zelluläre Reaktionen nach Rezeptorstimulationen im Vollblutansatz gefolgt von genetischen Analysen ist zu empfehlen.
Therapie
Die antimikrobielle Therapie muss danach ausgerichtet werden, ob es sich um atypische Mykobakterien oder echte Tuberkulose bzw. andere Erreger handelt. Zusätzliche pharmakologische Dosen von IFN-γ sind außer beim kompletten IFN-γ-Rezeptordefekt wirksam; bei kompletten IFN-γ Rezeptordefekten ist dagegen höchstens IFN-α mäßig effektiv (wegen sich überschneidender Signalwege von IFN-α und -γ; Einzelfallbeobachtungen).
Bei kompletten Defekten der IFN-γ-Achse ist eine Knochenmarktransplantation mit myeloablativer Konditionierung ohne T-Zelldepletion von einem HLA-identischen Spender (möglichst Geschwister) die einzige therapeutische Option, wobei besondere Komplikationsmöglichkeiten zu beachten sind: gehemmtes Engraftment der Spenderzellen durch hohe IFN-γ-Spiegel im Empfänger, Exazerbation persistierender Mykobakterieninfektionen, überschießende Granulombildung z. B. im ZNS unter Rekonstitution mit Spenderzellen.
Prognose
Bei Defekten, die von einer IFN-γ-Gabe profitieren, ist die Prognose, in Abhängigkeit von der Schwere der Ersterkrankung, meist gut, bei vollständigen IFN-γ Rezeptordefekten nur bei gelungener Knochenmarktransplantation günstig.
Seltene Störungen der Phagozyten
Seltene Störungen der Phagozyten sind in Tab. 10.1 aufgeführt.
| Krankheit | Mutierte Gene | Vererbung | Klinisches Bild |
|---|---|---|---|
| Kostmann-Syndrom und andere angeborene schwere Neutropenien |
ELANE, GFl1 CSF3R, WAS HAX1, SLC37A4, G6PC3, MAPBPIP |
AD (XL) AR |
Schwere Neutropenie <500/µl Peridontitis/Gingivitis Erhöhtes Risiko für AML/MDS |
| Zyklische Neutropenie | ELANE | AD |
Zyklische Schwankungen neutrophiler Granulozyten (Retikulozyten, Thrombozyten und Leukozyten) Erhöhtes Risiko für AML/MDS |
| Retikuläre Dysgenesie | AK2 | AR |
Sehr schwere Neutropenie <200/µl Lymphopenie Innenohrschwerhörigkeit |
| Leukozyten-Adhäsions-Defekte LAD 1, LAD 2, LAD 3 | INTGB2, SLC35C1, FERMT3 | AR |
Leukozytose, chronische Hautulzera, verzögerte Wundheilung, Peridontitis (LAD 1–3) Verspäteter Abfall der Nabelschnur (bei LAD 1 und LAD 3) Bombay-Blutgruppe (LAD 2) |
AD autosomal-dominant; AR autosomal-rezessiv; XL X-linked-rezessiv
Störungen der humoralen Immunität
X-chromosomal vererbte Agammaglobulinämie
Definition, Pathophysiologie
Die häufigste angeborene Form eines vollständigen Antikörpermangelsyndroms wird X-chromosomal rezessiv vererbt (XLA, M. Bruton). Sie wird mit einer Häufigkeit von 1:200.000 beobachtet. Der Defekt besteht in einer Störung der B-Lymphozytendifferenzierung, sodass fast keine reifen, sondern nur Prä-B-Lymphozyten gefunden werden. Der Mangel an reifen B-Lymphozyten wird durch Mutation einer für diese Zellen spezifischen Tyrosinkinase (Btk) verursacht. In vivo und in vitro lässt sich ein Defekt der Antikörperbildung aller Immunglobulinklassen nachweisen. Schwachformen mit Spätmanifestation kommen bei Restaktivität der Btk vor. Auch autosomal-rezessiv vererbte Formen einer Agammaglobulinämie kommen vor.
Klinik
Die Patienten fallen nach einem infektionsfreien Intervall von einigen Monaten bis zu über einem Jahr (nach Verschwinden der diaplazentar übergetretenen mütterlichen IgG-Antikörper; Abb. 10.5) mit bakteriell bedingten Pneumonien, chronischen Bronchitiden, Sinusitiden, Otitis media und später durch Bronchiektasen (Abb. 10.6) und gastrointestinalen Erkrankungen, u. a. ausgelöst durch Lamblien, auf.




Auch bestimmte Virusinfektionen (Enteroviren) verlaufen schwer. Nach einigen, heute nicht mehr verwendeten Impfungen mit Lebenderregern wurden schwere Komplikationen (Impfpoliomyelitis) beobachtet. Bei älteren Patienten wird eine Echovirusinfektion gesehen, die sich als dermatomyositisähnliches Bild äußert, aber auch als persistierende Meningoenzephalitis auftritt. Gelenkentzündungen, die wie eine leichte rheumatoide Arthritis wirken, und Malignome werden (trotz optimaler Therapie) im Verlauf der Erkrankung beobachtet.
Bei jedem Symptom einer Gelenkentzündung (Schwellung, Rötung, Schmerz, Überwärmung) ist eine bakterielle Ursache umgehend auszuschließen!
Diagnose
Zur Diagnose trägt die Messung der Immunglobuline bei, deren Konzentration in allen Klassen erniedrigt ist. Durchflusszytometrisch sind im peripheren Blut keine oder nur wenige reife B-Lymphozyten nachweisbar. Im Knochenmark sind Plasmazellen stark vermindert. Spezifische Antikörper, wie Isoagglutinine, Candida- und Impfantikörper fehlen. Impfungen mit totem Impfstoff sind möglich.
Für Lebendimpfstoffe (z. B. Masern, Mumps, Röteln, Varizella Zoster) muss ausgeschlossen werden, dass dem Immunglobulinmangel ein T-Zelldefekt zugrunde liegt.
Eine Antikörperantwort auf die Impfungen bleibt aus, während eine T-Zell-Antwort möglich ist, sofern die Impfstoffe nicht vollständig durch substituierte Immunglobuline neutralisiert werden. Die Wertigkeit der Impfungen bei Immunglobulinmangel wird kontrovers diskutiert.
Differenzialdiagnostisch muss bei Fehlen von IgG, IgA und IgM v. a. ein kombinierter T-Zelldefekt ausgeschlossen und bei erniedrigtem IgG, IgA und IgM bei Säuglingen und Kleinkindern an einen transitorischen Antikörpermangel (Abschn. 10.2.5) gedacht werden.
Therapie
Die Therapie der Wahl ist die Immunglobulinsubstitution entweder mit intravenös zu applizierenden Präparaten (mindestens 400 mg/kgKG alle 3 Wochen) oder als subkutane Gabe (100 mg/kgKG einmal wöchentlich bzw. mit Hyaluronidase alle 3–4 Wochen). Der Talspiegel im Serum sollte über 7 g/l liegen, da hochdosierte Substitution die Entwicklung von Bronchiektasen hinauszuzögern oder evtl. ganz zu verhindern scheint. Ein höherer Serumtalspiegel kann bei Vorliegen von Bronchiektasen sinnvoll sein. Jedoch können auch unter optimaler Substitution Infektionen im Bereich der Schleimhäute (Sinusitis) oft nicht vermieden werden, da das hier besonders wichtige Immunglobulin A (IgA) nicht ersetzt werden kann. Infektionen müssen intensiv antibiotisch behandelt werden.
Prognose
Die Prognose wurde früher durch das Auftreten nicht mehr beherrschbarer Infektionen und sekundärer pulmonaler Veränderungen durch die rezidivierenden Entzündungen bestimmt. Mit der heute durchführbaren Therapie stehen erhöhte Malignomneigung und chronische Infektionen mit Enteroviren im Vordergrund. Bronchiektasen, die selbst unter regelrechter Immunglobulinsubstitution auftreten können, stellen eine für die Prognose entscheidende Herausforderung dar.
Hyper-IgM-Syndrome
Definition, Pathogenese
Die X-chromosomal-rezessiv vererbte Form dieser Erkrankung umfasst etwa 70% der Hyper-IgM-Syndrome und ist durch rezidivierende Infektionen ähnlich denen bei X-chromosomal vererbter Agammaglobulinämie mit einer zusätzlichen Anfälligkeit für Infektionen durch Cryptosporidium spp. gekennzeichnet. Mit der Ausnahme von IgM (normal bis erhöht) finden sich ebenfalls erniedrigte Immunglobulinspiegel und erniedrigte Impfantikörpertiter.
Ursache sind Mutationen des Gens für den CD-40-Liganden (CD40L), ein Membranprotein, das auf aktivierten T-Lymphozyten exprimiert wird und mit CD40 auf antigenpräsentierenden Zellen interagiert. Diese Interaktion ist für die Differenzierung von B-Lymphozyten und den Isotypen-Klassenwechsel von IgM zu IgG von Bedeutung.
Klinik
Lymphknoten und Tonsillen können spärlich entwickelt, manchmal aber auch hyperplastisch sein. Die Patienten leiden an erhöhter Anfälligkeit für respiratorische Infekte bakterieller Genese (Otitis, Pneumonie), es kommen aber auch Pneumocystis-jiroveci-Pneumonien, Durchfälle (Kryptosporidien?) und aufsteigende Infektionen der Gallenblase vor. Wahrscheinlich durch verstärkte Autoantikörperbildung bedingt, beobachtet man Neutrozytopenien (in 50% der Fälle), Thrombozytopenien und Anämien.
Diagnose
Die Diagnose wird durch den Nachweis von Mutationen des Gens für CD40 L oder durch fehlende Expression dieses Proteins auf aktivierten T-Lymphozyten gestellt.
Therapie, Prognose
Die Therapie ist zunächst symptomatisch und besteht aus Immunglobulinsubstitution und der Gabe von Antibiotika. In den ersten Lebensjahren ist eine Prophylaxe gegen Pneumocystis-jiroveci-Infektionen mit Trimethoprim/Sulfamethoxazol indiziert.
Da die Langzeitprognose bei CD40L-Mangel durch gehäufte Gallengangs- und andere Malignome schlechter ist als bei der X-chromosomal vererbten Agammaglobulinämie, wird eine frühzeitige Stammzelltransplantation empfohlen.
Es gibt eine Reihe weiterer genetischer Defekte, die zu einem Hyper-IgM-Syndrom führen, die sich aber in Klinik und Prognose vom CD40L-Mangel zum Teil unterscheiden können. Dazu gehören Mutationen in den Genen für NEMO („NFk-B essential modulator“), CD40, AID („activation-induced cytidine deaminase“), AID C terminal, UNG („uracile DNA glycosylase“), PMS2( „postmeiotic segregation increased 2“) und in weiteren Genen. Auch Erkrankungen mit eingeschränkter Reparaturkapazität von DNA-Doppelsträngen wie Ataxia telangiectatica und Nijmegen-Breakage-Syndrom sowie das Aktivierte-PI3K-Delta-Syndrom können mit einer Hyper-IgM-Konstellation einhergehen. Allen gemeinsam ist ein defekter Klassenwechsel von IgM zu anderen Immunglobulinen.
Variables Immundefektsyndrom (CVID)
Definition
Diese Form eines Immundefektes (Synonym: „common variable immunodeficiency“, CVID) ist charakterisiert durch das meist späte Auftreten der Symptome (typisch im jungen Erwachsenenalter, gelegentlich bei Schulkindern), die neben den üblichen Zeichen der Agammaglobulinämie beobachteten gastrointestinalen Symptome (Diarrhö, Malabsorption), die hohe Rate von Autoimmunerkrankungen.
Im Gegensatz zur X-chromosomal vererbten Agammaglobulinämie sind phänotypisch reife B-Lymphozyten vorhanden. Das variable Immundefektsyndrom gehört mit einer Inzidenz von 1:10.000–1:50.000 zu den häufigsten Immundefekten.
Ätiopathogenese
Ätiologie und Pathogenese sind nach wie vor meist unbekannt. Befunde bei verschiedenen Patienten zeigen ein heterogenes Bild. So werden sowohl Störungen der B-Lymphozytendifferenzierung als auch Defekte von T-Lymphozytenfunktionen beobachtet. Eine Einteilung des CVID nach Vorhandensein oder Fehlen von CD27+IgM-IgG-B-Gedächtniszellen und anderer B-Zellpopulationen im peripheren Blut, nach In-vitro-Immunglobulinproduktion durch B-Zellen und/oder nach T-Lymphozyten-Zahlen, ist möglich. In einer Minderzahl von CVID-Patienten wurden inzwischen Mutationen gefunden, u. a. in den folgenden Genen: PIK3CD, PIK3R1, LRBA, CTLA4, NFKB1, NFKB2, ICOS, CD19, CD20, CD21, CD27 und CD81. Die pathophysiologische Bedeutung heterozygoter Mutationen im Gen für TACI für die Ausbildung eines CVID ist umstritten.
Klinik
Neben Bronchitiden, Sinusitiden, Otitiden, Pneumonien und Lamblieninfektionen kommen vor: rheumatoide Arthritis, autoimmunhämolytische Anämie, Neutropenie und Thrombozytopenie, Hypothyreose (durch Autoantikörper bedingte Thyreoiditis), Vitiligo (Autoantikörper gegen Melanozyten) und perniziöse Anämie. Granulome und lymphoide Hyperplasie in Lunge, Milz, Leber und Haut ohne fassbare infektiöse Ursache und eine noduläre lymphoide Hyperplasie des Darms sind mögliche zusätzliche Symptome.
Diagnose
Die Diagnose CVID bleibt, wenn sie nicht molekulargenetisch durch einen Defekt in den oben genannten Genen abgesichert werden kann, eine Ausschlussdiagnose. Ein CVID ist wahrscheinlich wenn
der IgG-Spiegel 2 Standardabweichungen unter der Altersnorm liegt und IgM oder IgA deutlich vermindert sind,
die Erkrankung jenseits des 2. Lebensjahres beginnt,
schwache oder abwesende Impfantikörper/Isohämagglutinine vorliegen und
sekundäre Gründe für einen Antikörpermangel (insbesondere renaler oder enteraler Verlust) ausgeschlossen sind.
Differenzialdiagnose
Differenzialdiagnostisch ist ein transitorisches Antikörpermangelsyndrom des Kleinkindalters definitionsgemäß erst jenseits des 5. Lebensjahres vom CVID abgrenzbar. Ebenso muss ein kombinierter T-B-Zelldefekt ausgeschlossen werden.
Therapie, Prognose
Die Dauersubstitution mit i.v. oder s.c. zu applizierenden Immunglobulinpräparaten senkt nicht nur die Infektionsrate, sondern scheint auch die autoantikörperbedingten Symptome zu bessern. Die Prognose wird durch chronische Lungenveränderungen und durch später auftretende Lymphome bestimmt.
Selektiver Mangel von Immunglobulinen
IgA-Mangel
Definition
Der selektive IgA-Mangel ist mit 1:400 bis zu 1:1300 häufig. Ihm kommt nur selten Krankheitswert zu. In fast allen Fällen ist auch das sekretorische IgA vermindert. Andere Störungen des Immunsystems finden sich in der Regel nicht (Kombinationen von IgA-Mangel mit IgG-Subklassendefekten.
Diagnose
Wegen der physiologisch niedrigen Immunglobulinwerte in den ersten Lebensjahren kann eine sichere Diagnose erst später gestellt werden. Der IgA-Mangel kann in einen CVID übergehen und kommt bei der familiären Form des CVID bei einigen Familienmitgliedern als Minimalvariante dieser Erkrankung vor.
Klinik
Personen mit einem selektiven IgA-Mangel sind meist gesund. Man findet in dieser Gruppe aber statistisch gehäuft Patienten mit rezidivierenden respiratorischen Infektionen einschließlich Bronchiektasen, Zeichen von Atopie (allergische Rhinitis, Asthma, Urtikaria und Ekzem), gastrointestinalen Symptomen (Malabsorption, Zöliakie, Morbus Crohn, Colitis ulcerosa) und Autoimmunerkrankungen (juvenile rheumatoide Arthritis, systemischer Lupus erythematodes, Thyreoiditis).
Therapie
Patienten mit dem viel selteneren vollständigen IgA-Mangel können gegen zugeführtes IgA, das in Spuren auch in IgG-Präparaten enthalten ist, Antikörper bilden, die zu schweren Unverträglichkeitsreaktionen führen. Dieser Mechanismus erklärt allerdings nur einen Teil der seltenen IgG-Unverträglichkeiten, die fast nur bei i.v.- aber kaum bei s.c.-Gabe auftreten. Liegt ein zusätzlicher IgG-Subklassendefekt vor und besteht eine klinische Indikation, können IgG-Präparate unter entsprechenden Vorsichtsmaßnahmen gegeben werden. Der IgA-Mangel allein rechtfertigt keine IgG-Substitution.
Cave
Da auch durch eine Bluttransfusion und andere Blutprodukte IgA übertragen wird, ist diese Maßnahme bei Patienten, die auf IgA reagieren, nur bei strenger Indikation und unter größtmöglicher Vorsicht statthaft.
Hyperimmunseren oder Antikörperpräparate (z. B. zur Postexpositionsprophylaxe von Varizellen oder Hepatitis A) dürfen nur unter stationärer Beobachtung verabreicht werden.
IgG-Subklassendefekte
Definition
Immunglobulin G besteht aus 4 Subklassen, die sich in der Zusammensetzung der schweren Ketten unterscheiden. Die Verteilung der IgG-Subklassen bei gesunden Erwachsenen ist:
IgG1 61%,
IgG2 30%,
IgG3 5%,
IgG4 4% des Gesamt-IgG.
Der postnatale Anstieg in der Synthese der einzelnen Immunglobulin-G-Klassen verläuft mit Ausnahme von IgG4 parallel zum Gesamt-IgG.
Ist der Serumspiegel für eine oder mehrere IgG-Subklassen erniedrigt, liegt ein Subklassenmangel vor. Die erniedrigten IgG-Subklassenkonzentrationen scheinen nicht als solche zur Infektanfälligkeit zu führen, sondern sind eher Zeichen einer gestörten Immunregulation.
Klinik
Mangel an verschiedenen Subklassen des Immunglobulins G kann mit rezidivierenden Infektionen verbunden sein. Der häufige IgG4-Mangel ist in den ersten Lebensjahren als isolierter Befund klinisch irrelevant. Bei einzelnen Patienten kann ein IgG2-Mangel (evtl. kombiniert mit IgG4-Mangel und ggf. mit einem IgA-Mangel) für rezidivierende respiratorische Infektionen mit bekapselten Erregern verantwortlich gemacht werden. Auch ein isolierter IgG3-Mangel kann mit pulmonalen Infektionen verbunden sein.
Diagnose
Vor dem 2. Lebensjahr können isolierte Subklassendefekte nicht diagnostiziert werden. Wichtig ist, dass jenseits des 2. Lebensjahres trotz normaler Gesamt-IgG-Spiegel Subklassendefekte vorliegen können. Zu bedenken ist auch, dass sich bei Kleinkindern diagnostizierte Subklassendefekte z. T. im Schulalter normalisieren können. Eine diagnostische Impfung mit nichtkonjugiertem Pneumokokkenimpfstoff kann die Fähigkeit, Antikörper gegen Kapselpolysacharide zu bilden, überprüfen. Bei der Bewertung der Relevanz eines im Labor gefundenen Subklassendefekts spielt die Klinik, in erster Linie Infektionen der oberen und unteren Atemwege, eine entscheidende Rolle.
Therapie
Wurde ein IgG-Subklassendefekt nachgewiesen, so ist bei entsprechendem klinischem Bild eine Substitution mit einem IgG-Präparat indiziert. Bei begleitendem vollständigem IgA-Mangel ist, wie bereits betont, bei den ersten Gaben eine besondere Überwachung notwendig.
Transitorischer Antikörpermangel
Definition
Es handelt sich um einen vorübergehenden Mangel an Immunglobulinen.
Klinik
Das transitorische Antikörpermangelsyndrom dauert gewöhnlich bis in das 2. oder 3. (selten bis zum Ende des 5.) Lebensjahr, ist selten mit vermehrter Infektionsanfälligkeit verbunden und scheint auf einer Verzögerung der eigenen Immunglobulinproduktion zu beruhen. B-Lymphozyten sind nachweisbar. Schon vor Normalisierung der Immunglobulinspiegel sind die Kinder in der Lage Isohämagglutinine und Antikörper gegen Impfstoffe zu bilden. Wahrscheinlich handelt es sich insgesamt um eine Extremvariante der physiologischen Hypogammaglobulinämie des Säuglings. Zu beachten ist, dass trotz vorhandener Impfantikörper die erniedrigten Immunglobulinspiegel sehr selten auf einen T-Zelldefekt oder eine frühe CVID-Variante hindeuten können. Bei sehr niedrigen Immunglobulinspiegeln ist daher initial der Ausschluss eines T-Zelldefekts und die Kontrolle der Immunglobulinspiegel im Verlauf nötig.
Während bei reifgeborenen Kindern das Krankheitsbild selten ist, besteht ein transitorischer Antikörpermangel bei fast allen sehr kleinen Frühgeborenen.
Prognose
Die Prognose des transitorischen Antikörpermangelsyndroms ist ausgezeichnet. Die Notwendigkeit einer Immunglobulinsubstitution ist eine Rarität.
T-Lymphozytendefekte
Schwere kombinierte Immundefekte
Definition, Epidemiologie
Unter dem Begriff versteht man kombinierte Störungen, die sowohl B- wie T-Zellfunktionen betreffen und sich im ersten Lebensjahr manifestieren („severe combined immunodeficiency“, SCID). Das Spektrum der möglichen Infektionen ist sehr breit. Ohne adäquate Stammzelltransplantation führen solche Infektionen meist schon innerhalb des ersten Lebensjahres zum Tode. Die Häufigkeit des SCID wird auf 1:50.000 geschätzt, variiert aber geographisch und in Abhängigkeit von Konsanguinität.
Pathogenese
Allen SCID gemeinsam ist der schwere T-Zell-Defekt, der abhängig vom molekularen Defekt, in unterschiedlichem Maß T-Zellen, B-Zellen und NK-Zellen betrifft. Orientierend können so vier immunologische Phänotypen unterschieden werden: T-B-NK-SCID, T-B+NK-SCID, T-B-NK+SCID und T-B+NK+SCID. Am häufigsten sind Mutationen, die keine normale Expression der γ-Kette der Rezeptoren für IL-2, -4, -7, -9, -15 und -21 erlauben („common γ-chain deficiency“, X-chromosomal). Dies verhindert die Ausreifung von T- und NK aber nicht die von B-Zellen (T-B+NK-SCID). Autosomale Vererbung findet man bei Fehlen der Adenosindeaminaseaktivität (ca. 20% aller SCID-Fälle), bei Jak3-Mangel, Il-7-Rα-Mangel, RAG1- und -2-Mangel, Artemis-Defekt, Cernunos-Defekt, Ligase-IV-Defekt, CD3-Ketten-Defekten, ORAI-Defekt, STIM1-Defekt, CD45-Mangel, Coronin-1a- und ZAP70-Defekt.
Klinik
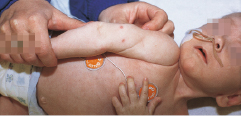
Häufig ist die Familienanamnese positiv für unklare frühe Todesfälle und/oder Konsanguinität. Beim Vollbild des SCID sind die Kinder postnatal unauffällig und entwickeln dann innerhalb von Wochen eine charakteristische Trias aus
hartnäckigen durchfälligen Stühlen mit zunehmender Gedeihstörung (Abb. 10.7),
schwerer Pneumonie (oft durch Pneumocystis jirovecii),
sich ausbreitendem mukokutanem Candida-Befall bzw. ausgeprägter Bronchitis.


Durch Übertritt mütterlicher T-Zellen kommt es nicht selten zu einer Graft-versus-Host-Reaktion der Haut, die sich durch ein morbiliformes Exanthem oder eine Erythrodemie vor dem 2. Lebensmonat unter Beteiligung der Fußsohlen und Handinnenflächen zeigt. Vielfältige polytope und persistierende oder rezidivierende Infektionen sind möglich. Sepsis, Meningitis, Mastoiditis, eitrige Konjunktivitis und Hautabszesse kommen ebenso vor, wie Infektionen mit Viren, insbesondere der Herpesgruppe (CMV, EBV, VSV; Abb. 10.8). Die Pneumocystis-jiroveci (früher: P. carinii)-Pneumonie ist akut lebensbedrohlich.

Ommen-Syndrom
Zu den Symptomen, die auch sonst beim SCID auftreten, kommen ein generalisiertes schuppiges Ekzem mit Eosinophilie und hohem IgE hinzu. T-Zellen sind vorhanden, aber funktionell inadäquat. Häufig findet man Defekte im RAG1- oder RAG2-Gen (Rekombinase-aktivierendes Gen 1 oder 2).
Weitere Varianten
Mutationen, die eine gewisse Restaktivität der betroffenen Struktur erlauben, können zu seltenen Varianten führen, die diagnostisch schwierig zu erfassen sind. So können SCID-typische Symptome später auftreten (u. a. auch beim Adenosindesaminasemangel) oder das klinische Bild kann sich wandeln: Die Infektanfälligkeit kann zumindest initial in den Hintergrund treten und das Krankheitsbild kann durch diverse Autoimmun- oder autoinflamatorische Phänomene, wie z. B. Granulombildung, gekennzeichnet sein. Sogar Impfantikörper können vorhanden sein und die Erkrankung kann sich erst im Kleinkindsalter oder noch später manifestieren. Hinweise geben Veränderungen der Zahl oder Funktion von T-Zellen.
Diagnose
Das lymphatische Gewebe kann vermindert sein. Röntgenologisch und sonographisch erscheint der Thymus stark verkleinert (und ist histopathologisch atrophisch). Parameter wie Temperatur, Akutphaseproteine und Blutbildveränderung der Granulozyten verhalten sich wie bei Gesunden. Eine schwere Lymphopenie ist typisch, fehlt aber bei etwa 1/3 der Fälle. Die Phänotypisierung der Lymphozyten mit Hilfe der Durchflusszytometrie ist fast immer stark auffällig. Wenn Lymphozyten vorhanden sind, muss geprüft werden, ob sie funktionsfähig sind und ob es sich um mütterliche Zellen handelt.
Ungeachtet der verschiedenen Formen des SCID findet man eine verminderte Funktion der T-Lymphozyten, d. h. eine in vitro verminderte Proliferation auf Mitogene (Phythämagglutinin, Concanavalin A und Pokeweed-Mitogen) und Antigen (Candida) sowie das Fehlen von Antikörpern (Isoagglutinine, Candidaantikörper). Nach Impfung mit z. B. Diphtherie- oder Tetanusantigen kommt es meist nicht zur Antikörperbildung (nur Totimpfstoffe sind erlaubt). Die Impfung mit attenuierten lebenden Rotaviren kann zu persistierenden, tödlichen Durchfällen führen.
Differenzialdiagnose
Differenzialdiagnostisch sind pränatale Infektionen und Erkrankungen durch HIV-1 in Erwägung zu ziehen.
Therapie
Die einzige kurative Therapie ist die Stammzelltransplantation.
Eine überbrückende intensive antibakterielle, antimykotische und antivirale Therapie ist unerlässlich. Gegen Pneumozystis jirovecii und Candida sollte eine Prophylaxe mit Cotrimoxazol und Fluconazol schon bei Verdacht auf einen schweren kombinierten Immundefekt begonnen werden. Weitere prophylaktische Maßnahmen sind: keimarme Umgebung, Immunglobulingabe und Aciclovir. Die schwere Gedeihstörung ist mit parenteraler Ernährung zu behandeln.
Die Transplantation von Stammzellen ist die Therapie der Wahl. Sie darf nicht verzögert werden und sollte bei Fehlen eines HLA-identen Spenders entweder haploident von einem Elternteil oder im Rahmen kontrollierter Studien mittels Gentherapie durchgeführt werden. Trotz Prophylaxe ist jederzeit eine Infektion möglich, die die Prognose dramatisch verschlechtert. Daher wird empfohlen, umgehend Kontakt zu einem transplantierenden Zentrum aufzunehmen. Es ist noch nicht beurteilbar, ob die Gentherapie der haploidenten Stammzelltransplantation bezüglich immunologischer Rekonstitution und Gesamtprognose überlegen ist. Die Leukämiegefahr der Gentherapie war bei den viralen Vektoren der ersten Studien erheblich und scheint nur bei ADA-Defizienz mit diesen Vektoren nicht vorhanden. Bei allen anderen SCID-Entitäten werden gentherapeutisch inzwischen modifizierte Vektoren im Rahmen klinischer Studien eingesetzt. Beim ADA-Mangel ist auch die Substitutionsbehandlung mit Adenosindesaminase eine Option.
Wegen der Gefahr einer Graft-versus-Host-Reaktion müssen Blutpräparate vor Transfusion mit 30 Gy bestrahlt werden. Impfungen mit lebenden Erregern (Rotaviren, VZV) sind kontraindiziert.
T-Lymphozytendefekte mit gestörter Proliferation
Autoimmun-lymphoproliferatives Syndrom (ALPS)
Pathophysiologie
Dem Krankheitsbild (im eigentlichen Sinne keine Immundefizienz) liegen verschieden verursachte Störungen des programmierten Zelltodes (Apoptosedefekte) zu Grunde. Am häufigsten sind Mutationen im Gen für Fas/Apo1/CD95 (TNFRSF6). Ist die sog. intrazelluläre „death domain“ betroffen, ergibt sich ein dominant negativer Effekt. Dagegen haben Defekte der extrazellulären und Transmembranregion eine geringere Penetranz und führen bei somatischer Zweitmutation oder aufgrund einer Haploinsuffizienz zur Erkrankung. Die Zellen reagieren vermindert oder gar nicht auf bestimmte Apoptosesignale, sodass unbrauchbare und autoreaktive Lymphozyten überleben.
Klinik
Durch benigne Lymphoproliferation kommt es zu Splenomegalie und zu langsam an- (und ab-) schwellenden, unterschiedlich stark vergrößerten Lymphknoten (Abb. 10.9). In diesen Lymphknoten und im peripheren Blut finden sich häufig vermehrt sog. doppelt negative T-Zellen (TCRαβ+, CD3+, CD4-, CD8-, die neben erhöhten Serumbiomarkern (sFASL und Vitamin B12) einen diagnostischen Stellenwert haben. Von Autoimmunphänomenen ist am häufigsten das hämatopoetische System mit Thrombo- und Neutropenien sowie hämolytischen Anämien betroffen. Auch Glomerulonephritis und andere Organmanifestationen treten auf. Das Risiko für ein malignes Lymphom ist deutlich erhöht.


Therapie
Sirolimus und Mykophenolat-Mofetil sind die effektivsten, steroidsparenden Therapeutika, für die die meiste Erfahrung vorliegen und sollten initial eingesetzt werden. Wegen der Gefahr eines nachhaltigen Immunglobulinmangels sollte Rituximab gemieden werden. Die Indikation für eine Splenektomie ist sehr zurückhaltend zu stellen und eine Prophylaxe gegen bekapselte Erreger gründlich durchzuführen, da beim ALPS die Splenektomie besonders häufig zu plötzlichen überwältigenden systemischen Infektionen (selbst unter Prophylaxe) geführt hat.
Lymphoproliferatives (Purtilo-)Syndrom (XLP1)
Pathophysiologie
Dieses X-chromosomal-rezessiv vererbte Syndrom ist gekennzeichnet durch einen selektiven Immundefekt gegen Epstein-Barr-Virusinfektionen. Das für die Erkrankung verantwortliche Gen kodiert ein Protein (SAP), das die Signaltransduktion in T-Lymphozyten und in NK-Zellen reguliert. Auf EBV reagieren die Patienten mit einer unkontrollierten Antwort der zytotoxischen (CD8+) T-Lymphozyten, die zu einer schweren Destruktion des lymphatischen Gewebes, der Leber und des Knochenmarkes führt.
Klinik
Die Patienten versterben an der ersten EBV-Infektion oft durch Lebernekrose. Daneben sind aplastische Anämien, Hämophagozytose, Hypogammaglobulinämien und B-Zelllymphome beschrieben worden, die in der Regel tödlich verlaufen. Wegen der gestörten Antikörperproduktion ist der Nachweis der EBV-Infektion durch serologische Methoden teilweise schwierig aber die PCR wichtig. Anfänglich besteht eine Dysgammaglobulinämie, im weiteren Verlauf kommt es zum Absinken der Immunglobulinkonzentrationen im Serum. Fast immer besteht eine ausgeprägte Lymphozytose mit atypischen Lymphozyten.
Therapie
Als einzige kurative Möglichkeit waren Knochenmarktransplantationen bei einigen Patienten erfolgreich.
Differenzialdiagnostisch müssen lymphohistiozytäre Erkrankungen (Kap. 23), z. B. die familiäre Lymphohistiozytose, erwogen werden.
T-Lymphozytendefekte kombiniert mit anderen Störungen
Wiskott-Aldrich-Syndrom
Das X-chromosomal vererbte Wiskott-Aldrich-Syndrom ist charakterisiert durch die Trias Thrombozytopenie, Ekzeme und Infektanfälligkeit aufgrund gestörter B- und T-Lymphozytenfunktionen.
Pathophysiologie
Das bei diesen Patienten defekte Gen kodiert ein Protein (WASP), das bei der Aktinpolymerisierung und bei der Bildung von Mikrovesikeln in Lymphozyten und Megakaryozyten eine regulatorische Rolle spielt. Die meist abnorm kleinen Thrombozyten haben eine verkürzte Überlebenszeit. Die Serumspiegel von IgA und IgE sind deutlich erhöht, wogegen die IgM-Werte erniedrigt sind. Typisch ist ein polysacharidspezifischer Immundefekt. Im Verlauf der Erkrankung entwickeln sich eine Lymphopenie und eine Funktionseinschränkung der T-Lymphozyten.
Klinik
Bei einigen Patienten kommt es durch die Thrombozytopenie zu schweren Blutungen (Abb. 10.10a). Trotzdem ist die Indikation für eine Splenektomie wie beim ALPS zurückhaltend zu stellen. Infektionen werden u. a. durch bekapselte Erreger, Pneumocystis jirovecii und später durch Viren der Herpesgruppe verursacht. Das Ekzem wird leicht mit Neurodermitis verwechselt (Abb. 10.10b). Autoimmunphänomene, wie Arthritiden, Vaskulitiden und Kolitis können sehr hartnäckig sein. Die Malignomgefahr wächst mit zunehmendem Alter.


Therapie
Immunglobulingaben und prophylaktische antibiotische und antivirale Therapie können die Rate der Infektionen deutlich senken. Thrombozyten sind vor Transfusion mit 30 Gy zu bestrahlen. Mit einer HLA-identischen Stammzelltransplantation kann ein großer Teil der Patienten geheilt werden. Die haploidentische Transplantation war bisher wenig erfolgreich, wird aber neu evaluiert. Die Leukämiegefahr der Gentherapie war bei den erstmalig eingesetzten viralen Vektoren erheblich, eine Gentherapie mit modifizierten Vektoren ist bei fehlendem Stammzellspender in kontrollierten Studien möglich.
Prognose
Die Prognose ohne Transplantation oder Gentherapie wird im Wesentlichen durch das erhöhte Risiko (100-fach höher) an lymphoretikulären Tumoren zu erkranken und durch Hirnblutungen bestimmt.
Ataxia telangiectatica (Louis-Bar-Syndrom)
Charakteristika, Pathophysiologie
Dieses autosomal-rezessiv vererbte Syndrom ist durch Ataxie ab etwa dem 2. Lebensjahr, durch meist später hinzutretende Teleangiektasien der Haut und der Konjunktiven (Abb. 10.11) sowie durch eine zunehmende neurologische Symptomatik gekennzeichnet. Ein B- und T-Zellen betreffender Immundefekt führt nur bei einem Teil der Patienten schon früh zu sinopulmonalen Infektionen. Mutationen im ATM-Gen verursachen die Erkrankung. Die funktionell intakte Version dieses Gens verlangsamt den Zellzyklus bis alle eventuell aufgetretenen DNA-Schäden repariert sind, während die mutierte Version die Weitergabe von veränderter DNA auf Tochterzellen zulässt.


Klinik, Diagnose
Neben den aufgeführten Symptomen kann es zu schweren bronchopulmonalen Infektionen, Bronchiektasen, endokrinen Störungen und Störungen der Leberfunktion kommen. Mit zunehmendem Alter vermindern sich die intellektuellen Fähigkeiten und es wächst das Malignomrisiko. Auf Grund der erhöhten Strahlenempfindlichkeit sind Röntgenstrahlenexpositionen zu vermeiden. IgG-Mangel ist häufig; Hyper-IgM-Konstellationen und IgA/IgG-Subklassenmangel kommen vor. Ein erhöhtes α-Fetoprotein trägt neben der charakteristischen Klinik zur raschen Diagnose bei.
Therapie, Prognose
Die Therapie ist symptomatisch (Immunglobulingaben, Antibiotika). Blutprodukte sind zu bestrahlen. In einer sehr hohen Rate treten lymphoretikuläre Malignome auf. Mit starken immunologischen Veränderungen korreliert eine besonders schlechte Prognose. Auch Heterozygote (u. a. Eltern) sind gefährdet (z. B. durch Malignome, kardiovaskuläre Erkrankungen), sodass eine Beratung erfolgen muss. Eine Stammzelltransplantation ist nicht indiziert.
Bei der Ataxia teleangiectatica ist Folgendes zu beachten:
Graft-versus-Host-Reaktion durch Transfusion unbestrahlter Blutprodukte sind möglich,
Lebendimpfungen nur in Abhängigkeit vom Ausprägungsgrad des Immundefekts durchführen,
Reaktionen auf i.v.-Immunglobulinsubstitution bei Subklassendefekt mit IgA-Mangel sind möglich,
Röntgen nur in absolut zwingenden Fällen durchführen, MRT bevorzugen,
gesamte Familie beraten.
Komplementdefekte
Primäre Komplementdefekte
Pathophysiologie
Das Komplementsystem besteht aus einer Anzahl von Proteinen, die sich kaskadenartig, ähnlich der Blutgerinnung, aktivieren. Viele Komplementfaktoren sind Proteasen, die von der jeweils nachfolgenden Komponente ein Protein abspalten, das zusätzliche biologische Aktivität entfaltet. Das Komplementsystem kann auf 3 Wegen aktiviert werden, über den klassischen, den alternativen und den MBL-Weg (MBL, Mannose-bindendes Lektin).
Biologische Funktionen des Komplementsystems sind u. a. die Veränderungen der Gefäßwandpermeabilität, die Aktivierung von Entzündungszellen (u. a. im Sinne einer gesteigerten Chemotaxis), die Fähigkeit zur Opsonierung (d. h. der Verbesserung der Phagozytose von zu eliminierenden Partikeln) und die Zelllyse durch Membranporenbildung. Ebenfalls wichtig sind Funktionen bei der Eliminierung von Immunkomplexen, von apoptotischen Zellen und bei der Regulierung von B-Zellen.
Kongenitale Defekte fast aller Komponenten des Komplementsystems sind beschrieben worden. Der Tendenz nach zeigen Patienten mit funktionellem Verlust der in der Nummerierung niedrigen Komplementkomponenten eher autoimmunologische Symptome bis hin zu einem SLE-ähnlichen Krankheitsbild und seltener schwere bakterielle Infektionen, während es sich bei Verlust der höheren Komponenten umgekehrt verhält; (rezidivierende) Meningokokkeninfektionen sind hier am Häufigsten. Andere bekapselte Erreger (z. B. Pneumokokken) kommen ebenfalls vor. Patienten mit Fehlen von bestimmten Komplementfaktoren können durchaus gesund sein (z. B. beim C2- oder MBL-Mangel).
Diagnose
Mit dem Test für die totale hämolytische Komplementaktivität CH50 besteht die Möglichkeit, alle 9 Komponenten des klassischen Aktivierungswegs funktionell zu messen. Defekte im alternativen Aktivierungsweg können durch eine der CH50-analoge Untersuchungsmöglichkeit der AP50 entdeckt werden. Ein Defekt des Mannose-bindenden Lektins ist nur in Kombination mit anderen Störungen klinisch relevant. Bei Verdacht auf hereditäres Angioödem kann die Diagnose nur durch Messung der Konzentration und Funktion des C1-Esteraseinhibitors sicher gestellt werden auch wenn eine C4-Verminderung deutlich hinweisend ist.
Therapie
Die therapeutischen Möglichkeiten sind mit Ausnahme des hereditären Angioödems beschränkt, da die Faktoren kurze Halbwertszeiten haben und bei Serumgabe die Gefahr der Sensibilisierung gegen allogene Komplementkomponenten besteht. Dennoch sollten Komplementdefekte möglichst frühzeitig diagnostiziert werden. Als Konsequenz sollte dann bei akuten, hochfieberhaften Erkrankungen sofort Meningokokken- bzw. Pneumokokken-wirksam behandelt werden.
Hereditäres Angioödem (C1-Esteraseinhibitormangel)
Abschn. 30.6.
Sekundäre Veränderungen des Komplementsystems
Manche Komplementkomponenten (z. B. C3) reagieren wie (träge) Akute-Phase-Proteine, was bei systemischen Entzündungen zu ihrer Erhöhung führen kann, aber nicht primär pathologisch ist. Eindeutige Erniedrigungen haben jedoch immer eine gewisse pathologische Bedeutung und deuten meist auf einen Komplementverbrauch hin. Beispiele für Erkrankungen mit einem solchen Verbrauch sind: Lupus erythematodes, bestimmte Formen der Glomerulonephritis, Verbrennungen und Sepsis.
Weitere angeborene Immundefekte
Weitere Immundefekte, die kombiniert mit anderen Störungen auftreten, sind in Tab. 10.2 beispielhaft aufgeführt (Abb. 10.12 und Abb. 10.13).
| Syndrom | Mutierte Gene | Vererbung | Klinisches Bild | Therapie |
|---|---|---|---|---|
| Chronisch mukokutane Candidiasis |
CARD9 STAT1 IL17RA IL17F |
AR AD AR AD |
Candidainfektionen der Haut, Schleimhäute, Nägel | Symptomatisch Azole (Prophylaxe nur in schweren Fällen) |
| DiGeorge-Sequenz |
Thymushypoplasie Nur in <5% mit relevantem Immundefekt vergesellschaftet, dann eingeschränkte zelluläre und humorale Immunität Mit und ohne Hypoparathyreoidismus, Vitium cordis, Gesichtsdysmorphie (Abb. 10.12) |
Transplantation von fetalem Thymus | ||
| Hyper-IgE-Syndrome |
STAT3 TYK2 DOCK8 |
AD AR AR |
Dermatitis, Staphylokokkeninfektionen, Pneumatozelen (Abb. 10.13), IgE meist erhöht | Antibiotische Prophylaxe gegen S. aureus |
AD autosomal-dominant; AR autosomal-rezessiv




Sekundäre Immundefekte
Haben Kinder ein primär intaktes Immunsystem, das durch ein Ereignis beeinträchtigt wird, spricht man von einem sekundären Immundefekt. Sekundäre Immundefekte sind sehr unterschiedlich ausgeprägt, polyätiologisch und daher schlechter klassifizierbar als die primären. Beispiele sind (chronische Unterernährung, z. B. Kwashiorkor; Kap. 2), Verbrennungen und Zustand nach Splenektomie (Kap. 22).
Eine Reihe von Viren induzieren eine verminderte Immunabwehr, wie z. B. konnatale Röteln, Zytomegalie, Masern und natürlich HIV (Kap. 14). Erhebliche Eiweißverluste z. B. bei exsudativer Enteropathie können zu einem klinisch relevanten Immunglobulinmangel führen. Lymphangiektasien und Verletzungen des Ductus thoracicus mit ausgeprägtem Verlust an Lymphflüssigkeit können über den Immunglobulinmangel hinaus einen Abfall der Lymphozytenzahl zur Folge haben und so eine erhebliche Abwehrschwäche verursachen. Sekundäre Schädigungen des Knochenmarks und des lymphatischen Systems (z. B. Strahlung, Chemikalien) sowie Autoimmun- und maligne Erkrankungen induzieren ebenfalls sekundäre Immundefekte. Schließlich seien als weitere Beispiele Chemotherapie und antientzündliche Therapien einschließlich Steroidtherapie als iatrogen verursachte Immunsuppression erwähnt. Manche Autoimmunstörungen verursachen eine „Phänokopie“ eines angeborenen Immundefektes, z. B. Infektionen mit Mykobakterien bei Vorliegen von anti-IFNgamma-Autoantikörpern.
Praktische Hinweise bei Immundefekten
Abklärung rezidivierender Infektionen zum Ausschluss eines Immundefektes
Rezidivierende Infektionen bei Kindern sind für den Pädiater ein alltägliches Problem. In die Bewertung der Infektionen gehen Schwere, die Lokalisation und die Häufigkeit ein. Vielen Eltern ist nicht bekannt, dass ein Kleinkind physiologischerweise, besonders bei erstmaliger massiver Exposition (z. B. Besuch des Kindergartens) durchschnittlich 8 fieberhafte Infektionen pro Jahr haben kann.
Wird auf Grund der Anamnese ein Immundefekt vermutet, so können Art der Infektion und Erreger Hinweise auf die zugrunde liegende Störung geben (Tab. 10.3) In einer weiteren Tabelle sind einfache, in der pädiatrischen Praxis mögliche und weiterführende Untersuchungsmethoden für die einzelnen Komponenten des Immunsystems zusammengestellt (Tab. 10.4).
| System | Ort der Infektion | Erreger |
|---|---|---|
| Phagozyten | Oberflächen (Haut, Schleimhaut), Monozyten-/Makrophagen-System (Leber, Lymphozyten) | Staphylococcus aureus, Escherichia coli, Aspergillus, Burkholderia cepacia, Serratia marcescens |
| Humoral | ||
| - Antikörper | Schleimhaut (Sinusitis), Pneumonie, Gastrointestinaltrakt | Bakterien, Viren |
| - Komplement | Meningitis, Sepsis, Nephritis | Neisserien (Meningokokken), Streptokokken (Pneumokokken) |
| T-Lymphozyten | Gedeihstörung (schwere Dystrophie), Lungen- und Hautinfektionen | Bakterien, Pilze, Viren, Pneumocystis jirovecii |
| System | Einfach | Erweitert |
|---|---|---|
| Phagozyten | Zahl (absolut) der Granulozyten, Morphologie im Ausstrich, reaktive Sauerstoffmetabolite (Dihydrorhodamintest) | Chemotaxis, Bakterizidie, Adhäsionsproteine, Rezeptoranalyse (z. B. Interferon-γ-Rezeptor) |
| Humoral | ||
| - Antikörper | Immunglobuline, Zahl der B-Lymphozyten, spezifische Impfantikörper | IgG-Subklassen, sekretorisches IgA, Isohämaglutinintiter, Antikörper gegen Kapselpolysacharide |
| - Komplement | CH50, C1-Inhibitor | AP/CH50, Einzelkomponenten, (MBL) |
| T-Lymphozyten | Zahl (absolut) der Lymphozyten, Hauttests | Subpopulationen, HLA-Antigene, Lymphozytenproliferation, Zytokinproduktion, Enzyme (z. B. Adenosindesaminase) |
Alarmzeichen, die möglicherweise auf einen primären Immundefekt hindeuten, sind:
positive Familienanamnese für angeborene Immundefekte (unklare, insbesondere frühkindliche, Todesfälle und Konsanguinität),
zwei oder mehr schwere Sinusitiden pro Jahr,
zwei oder mehr Pneumonien innerhalb eines Jahres,
antibiotische Therapie über zwei oder mehr Monate ohne Effekt,
Impfkomplikationen bei Lebendimpfungen (insbesondere BCG und frühere Polioimpfungen nach Sabin),
Gedeihstörung im Säuglingsalter mit und ohne chronische Durchfälle,
rezidivierende Lymphknoten- oder Organabszesse,
zwei oder mehr viszerale Infektionen (Meningitis, Osteomyelitis, septische Arthritis, Empyem, Sepsis),
über das normale Maß hinausgehende Candidainfektionen an Haut oder Schleimhaut,
chronische Graft-versus-Host-Reaktion (z. B. unklare Erytheme bei kleinen Säuglingen),
rezidivierende systemische Infektionen mit bestimmten einzelnen Erregern (z. B. mit atypischen Mykobakterien).
Das Akronym ELVIS für Erreger, Lokalisation, Verlauf, Intensität und Summe stellt eine gute Zusammenfassung dar.
Solche klassischen Hinweise auf einen Immundefekt gelten durchaus weiterhin für den größeren Teil der Patienten. Daneben werden jedoch zunehmend Immundefekte beschrieben, die sich völlig anders präsentieren, wie z. B.:
Erstmanifestation von schweren Infektionen erst im Schulkind- oder Erwachsenenalter,
Erstmanifestation mit Autoimmun- oder autoinflammatorischen Symptomen oder mit einer Organfibrose,
Auftreten nur einer schweren Infektion (z. B. Herpesenzephalitis) bei unauffälliger Vorgeschichte,
Anfälligkeit gegenüber nur einem Erregertyp (z. B. Mykobakterien),
Mimikry der Symptomatik anderer Erkrankungen, z. B. Sarkoidose usw.
Hier ist das Akronym GARFIELD für Granulome, Autoimmunität, rezidivierendes Fieber, ungewöhnliche Ekzeme, Lymphoproliferation, chronische Darmentzündung hilfreich.
Pauschaltests, die eine immunologische Störung ausschließen, gibt es nicht. In den letzten Jahren ist durch Nutzung des sog. „next generation sequencing“, NGS (Kap. 1), die Zahl der molekular definierten Immundefekte bzw. von Störungen, die auch einen Immundefekt umfassen, erheblich angestiegen. Darüber hinaus verursachen einige Gendefekte ein breites Spektrum an klinisch-phänotypischen Manifestationen, während ähnliche Phänotypen z. T. durch verschiedene Gendefekte verursacht werden. Auch die therapeutischen Optionen werden subtiler. Die Konsequenz aus der zunehmenden Komplexität kann nur sein, die Zusammenarbeit mit pädiatrisch immunologischen Spezialisten zu suchen. Der diagnostische Stellenwert des NGS wird sicher weiter wachsen und durch automatische Quervernetzung zu bekannten Gendefekten weiter verbessert werden.
Impfungen bei Immundefekten
Primäre Immundefekte
Früher wurden Lebendimpfstoffe wegen teilweise tödlich verlaufender Impfinfektionen nach BCG-, Masern-, Pocken- und oraler Poliomyelitisimpfung als absolut kontraindiziert angesehen. Inzwischen wurden Richtlinien erarbeitet, in welchen Fällen Lebendimpfungen doch möglich oder sinnvoll sind. So sollten z. B. Patienten mit septischer Granulomatose normal geimpft werden, nur die BCG-Impfung ist kontraindiziert. Bei T-Zelldefekten ist die Gefährdung durch Lebendimpfstoffe vom Ausmaß abhängig. Die frühere Poliomyelitis-Lebendimpfung ist bei ausgeprägten B-Zelldefekten kontraindiziert. Mit Totimpfstoffen ist eine Impfung immer ohne Gefährdung möglich, aber je nach Art des Immundefekts ist der Impferfolg zweifelhaft. Erfolgreich sind Impfungen mit Totimpfstoffen bei selektiven Immundefekten (z. B. IgG-Subklassendefekt, IgA-Mangel und polysaccharidspezifischer Immundefekt). Mit Totimpfstoffen kann die Kapazität und Funktionalität des Immunsystems überprüft werden, z. B. bei inkompletten Immundefekten. Bei guter Immunantwort ist dann fast immer auch die Impfung mit Lebendimpfstoffen möglich.
Sekundäre Immundefekte
Für sekundäre Immundefekte, insbesondere bei HIV, nach Chemotherapie und Knochenmarktransplantationen, sowie bei anhaltender iatrogener Immunsuppression gibt es Richtlinien für Lebend- und Totimpfstoffe, die beachtet werden müssen.
Contributor Information
H. von Bernuth, Email: Horst.Von-Bernuth@charite.de
J. Roesler, Email: Joachim.Roesler@uniklinikum-dresden.de