

Abstract
Herzfehler gehören zu den häufigsten angeborenen Fehlbildungen. Heute erleben fast 90% dieser Kinder das Erwachsenenalter. Dennoch sind angeborene Herzfehler weiterhin mit einer hohen Morbidität und Mortalität vergesellschaftet, insbesondere wenn ein kritischer Herzfehler vorliegt. Dem Pädiater obliegt nicht nur die rechtzeitige Erkennung eines Herzfehlers, sondern auch im zunehmenden Maße die Mitbetreuung dieser Patienten im prä-und postoperativen Stadium. Das Verständnis für die zugrundeliegende Hämodynamik der jeweiligen Herzfehler sowie die physiologischen Veränderungen der Kreislaufverhältnisse im Laufe des Lebens sind wichtig für die Diagnosestellung und die klinische Verlaufsbeurteilung. Daher wird im folgenden Kapitel besonderen Wert auf die hämodynamischen Zusammenhänge am Beispiel ausgewählter Herzfehler gelegt. Neben primär azyanotischen und zyanotischen Herzfehlern gehören entzündliche Herzerkrankungen sowie Herzrhythmusstörungen zum Spektrum der Kinderkardiologie. Diese Felder werden in Grundzügen beschrieben und sollen dem Kliniker eine Hilfestellung für das diagnostische und therapeutische Vorgehen geben.
Angeborene Herzfehler
Epidemiologie und Ätiologie
Epidemiologie
Die Prävalenz angeborener Herzfehlern beträgt etwa 0,8% aller Lebendgeborenen und gehört damit zu den häufigsten angeborenen Fehlbildungen (Tab. 20.1). Die Prognose der betroffenen Kinder hat sich in den letzten 50 Jahren kontinuierlich verbessert. So erleben heute fast 90% der Kinder das Erwachsenenalter. Viele Herzfehler können heute pränatal diagnostiziert werden. Dies ermöglicht neben einer optimalen medizinischen Versorgung des Neugeborenen auch den betroffenen Eltern sich auf die Situation einzustellen.
| Ventrikelseptumdefekt (VSD) | 30% |
| Atriumseptumdefekt (ASD) | 7% |
| Persistierender Ductus Arteriosus (PDA) | 7% |
| Pulmonalklappenstenose (PST) | 7% |
| Aortenisthmusstenose (COA) | 5–8% |
| Aortenklappenstenose (AST) | 3–6% |
| Fallot-Tetralogie (TOF) | 5–6% |
| Atrioventriukärer Septumdefekt (AVSD) | 4–5% |
| Transposition der großen Gefäße (TGA) | 4% |
| Hypoplastisches Linksherzsyndrom (HLHS) | 1–2% |
| Pulmonalatresie mit VSD | 2–3% |
| Pulmonalatresie ohne VSD | 2% |
| Trikuspidalatresie | 1–2% |
| Truncus arteriosus communis | <1% |
Nichtsdestotrotz gehören angeborene Herzfehler weiterhin zu den Fehlbildungen, die mit einer hohen Morbidität und Mortalität vergesellschaftet sind. In Deutschland ist die pränatale Detektionsrate niedrig und sehr heterogen verteilt, da sie im hohen Maße von der Erfahrung des Untersuchers abhängig ist. Umso wichtiger ist die sorgfältige Durchführung der Vorsorgeuntersuchungen durch den Pädiater, um mögliche angeborene Herzfehler frühzeitig zu detektieren.
Ätiologie
Die Ätiologie angeborener Herzfehler ist komplex und noch nicht vollständig aufgeklärt. Der größte Anteil tritt als isolierte Fehlbildung auf. In den meisten Fällen geht man von einem multifaktoriellen Geschehen aus. Insbesondere die Untersuchung von Familienmitgliedern betroffener Patienten lässt den Schluss auf eine polygenetische oder multifaktorielle Genese zu. Das Wiederholungsrisiko für ein erneutes Auftreten angeborener Herzfehler beträgt für Verwandte ersten Grads 2–5%. Dies korrespondiert mit theoretischen mathematischen Wahrscheinlichkeitsmodellen für multifaktorielle Mechanismen. Multifaktoriell bedeutet hier, dass kardiale Fehlbildungen durch die Wechselwirkung einer oder mehrerer Gene mit Umweltfaktoren verursacht werden.
Ungefähr 25–30% der angeborenen Herzfehler sind mit extrakardialen Anomalien wie Chromosomenanomalien, genetischen Punktmutationen oder komplexen Fehlbildungssyndromen assoziiert (Tab. 20.2).
| Syndrom | Relative Häufigkeit von Vitien | Typische Vitien |
|---|---|---|
| Trisomie 21 (Down-Syndrom) | 40–50% | AVSD,VSD |
| Trisomie 18 (Edwards-Syndrom) | >90% | Septumdefekte, Klappendefekte, PDA, PS |
| Trisomie 13 (Pätau-Syndrom) | >80% | Septumdefekte, Klappendefekte, PDA, Dextrokardie |
| Monosomie X0 (Ullrich-Turner-Syndrom) | 30–40% | COA, im Verlauf Aortendissektion |
| Mikrodeletion 22q11 (DiGeorge-Syndrom) | >75% | TOF, Truncus arteriosus communis, unterbrochener Aortenbogen, VSD |
| Williams-Beuren-Syndrom | Häufig | Supravalvuläre AST, periphere PST |
| Deletion-5p-Syndrom (Cri-du-chat-Syndrom) | 25% | Variabel, VSD, ASD |
| Alagille-Syndrom | >80% | Periphere PST mit und ohne komplexen Vitium |
| Noonan-Syndrom | Häufig | PST, LV-Hypertrophie |
| Cornelia-de-Lange-Syndrom | Ca. 30% | VSD |
| VACTERL-Assoziation | >50% | VSD, variabel |
| Goldenhar-Syndrom | >30% | TOF, VSD |
| CHARGE-Assoziation | >60% | TOF, Truncus arterius communis, Aortenbogenfehlbildungen |
TOF Fallot-Tetralogie, AST Aortenstenose, PST Pulmonalstenenose, COA Aortenisthmusstenose, VSD Ventrikelseptumdefekt, ASD Vorhofseptumdefekt
Zu den Chromosomenanomalien, die durch eine Karyotypisierung detektiert werden, gehören z. B. Trisomie 21 (Down-Syndrom), Trisomie 18 (Edwards-Syndrom) und Monosomie X0 (Ullrich-Turner-Syndrom). Jede dieser Chromosomenanomalien ist mit einer Häufung eines spezifischen Herzfehlers assoziiert, so z. B. Trisomie 21 mit AVSD oder Ullrich-Turner-Syndrom mit der Aortenisthmusstenose. Die häufigeren chromosomalen Syndrome sind auf Mikrodeletionen zurückzuführen und können mittels In-situ-Hybridisierung (FISH) oder arraybasierter „Comparative Genom-Hybridisierung“ (CGH-Array) dargestellt werden. Diese sind v. a. die Mikrodeletion 22q11.2 (Di George-Syndrome, syn. Shprintzen-Syndrom, velokardiofaziales Syndrom) mit conotrunkalen Herzfehlbildungen (Fallot-Tetralogie, „double outlet right ventricle“, Truncus arteriosus, Aortenbogenanomalien) und das Williams-Beuren-Syndrom (v. a. supravalvuläre Aortenstenosen und periphere Pulmonalstenosen).
Monogene Syndrome, die mit Herzfehlern assoziiert sind, sind u. a. RASopathien (z. B. Noonan-Syndrom,) und JAG-Mutationen (z. B. Alagille-Syndrom). Zu den komplexen Fehlbildungssyndromen gehören die VACTERL-Assoziation, CHARGE-Assoziation und das Goldenhar-Syndrom (Tab. 20.2).
Ein kleiner Teil der angeborenen Herzfehler kann auf teratogene Noxen wie Infektionen (z. B. Röteln), Stoffwechselerkrankungen (z. B. Diabetes mellitus Typ I), Systemerkrankungen (z. B. Kollagenosen, Lupus erythematodes) Medikamenteneinnahme (z. B. Antikonvulsiva) oder Drogenabusus (z. B. Alkohol) während der Schwangerschaft zurückgeführt werden.
Klinische Evaluation
Symptome angeborener Herzfehlern können unterschiedlich ausgeprägt sein. Leitsymptome sind neben Herzgeräuschen, die typischen Merkmale der Herzinsuffizienz und/oder Zyanose. Die Erstuntersuchung eines Kindes mit Verdacht auf das Vorliegen eines angeborenen Herzfehlers hat zum Ziel diesen Verdacht auszuschließen oder eine möglichst genaue Verdachtsdiagnose zu stellen. Wie bei jeder klinischen Evaluation sollte man auch hier so systematisch wie möglich vorgehen (Tab. 20.3).
| Primär azynotische Herzfehler | Primär zyanotische Herzfehler | ||
|---|---|---|---|
| Vermehrte LPerf | Normale LPerf | Vermehrte LPerf | Verringerte LPerf |
| VSD | AST | TGA | TOF |
| ASD | PST | HLHS | PAT |
| AVSD | COA | TruncArt | TAT mit PST |
| PDA | hKMP | TAT ohne PST/PAT | TAT mit PAT |
| PAPVR | TAPVR | ||
LPerf Lungenperfusion, VSD Ventrikelseptumdefekt, ASD Aortenstenose, TGA Transposition der großen Gefäße, TOF Fallot-Tetralogie, ASD Vorhofseptumdefekt, PST Pulmonalstenose, HLHS Hypoplastisches Linksherzsyndrom, PAT Pulmonalatresie, AVSD Atrioventrikulärer Septumdefekt, COA Aortenisthmusstenose, TruncArt Truncus arteriosus communis, TAT Trikupidalatresie, PDA Persistierender Ductus arteriosus, hKMP hypertrophe Kardiomyopathie, PAPVR Partielle Lungenvenenfehlmündung, TAPVR Totale Lungenvenenfehlmündung
Herzfehler können in primär azyanotische und zyanotische Herzfehler eingeteilt werden. Eine Zyanose kann bei der körperlichen Untersuchung festgestellt werden und sollte durch die Durchführung einer Pulsoxymetrie bestätigt werden, insbesondere da eine gering ausgeprägte Zyanose bei einer Inspektion nicht erkennbar sein muss. Sowohl azyanotische als auch zyanotische Vitien können weiter unterteilt werden in Vitien mit verminderter bzw. normaler oder vermehrter Lungenperfusion. Eine Röntgenaufnahme des Thorax zeigt dabei entsprechend entweder eine verminderte, normale oder vermehrte Lungengefäßzeichnung (Tab. 20.3). Die Auskultation mit der Charakterisierung der Herztöne bzw. Herzgeräusche sowie deren Lokalisation grenzt die Differenzialdiagnosen weiter ein. Ein EKG kann eine rechts-, links- oder biventrikuläre Hypertrophie aufzeigen oder durch einen pathologischen Lagetyp Hinweise auf spezifische Herzfehler geben. Schließlich wird die definitive Diagnose durch eine Bildgebung, meistens reicht eine Echokardiographie aus, gestellt.
Azyanotische Herzfehler
Azyanotische Herzfehler können vereinfacht nach Volumen- und Druckbelastung des Herzens eingeteilt werden.
Am häufigsten findet sich eine Volumenbelastung des Herzens. Diese entsteht in der Regel durch Defekte mit einem Links-rechts-Shunt. Volumenbelastungen durch Klappeninsuffizienzen oder durch eine eingeschränkte kardiale Pumpleistung sind deutlich seltener.
Die häufigsten Vitien, die zu einer Volumenbelastung des Herzens führen sind der Ventrikelseptumdefekt (VSD), Vorhofseptumdefekt (ASD), persistierender Ductus arteriosus (PDA) und der atrioventrikuläre Septumdefekt (AVSD). Bei all diesen Herzfehlern besteht ein Links-rechts-Shunt, d. h. oxygeniertes Blut wird durch den Defekt zurück in den Lungenkreislauf gepumpt. Es kommt zu einer Rezirkulation. Dieser Shunt kann quantifiziert werden, indem man das Verhältnis von pulmonalen Fluss (Qp) zu systemischen Fluss (Qs) berechnet. Ein Qp-Qs-Verhältnis von 2:1 bedeutet, dass doppelt so viel Blut durch die Lunge fließt als durch den Körper. Das Shuntvolumen ist abhängig von der jeweiligen Defektgröße, der Compliance der Herzkammern sowie dem pulmonalvaskulären Widerstand. All diese Stellgrößen sind variabel, eine Shuntverbindung kann sich verkleinern, die Compliance der Kammern und die Gefäßwiderstände verändern sich. So fällt z. B. der pulmonalvaskuläre Widerstand in den ersten Lebenswochen stark ab.
Das Shuntvolumen ist im Lauf der Zeit dynamischen Veränderungen unterworfen und somit variieren auch die damit verbundenen Symptome.
Daher muss der jeweilige Befund immer auch im Kontext der körperlichen Entwicklung beurteilt werden. So kann z. B. bei einem Neugeborenen trotz großen VSD das Shuntvolumen durch den noch erhöhten pulmonalvaskulären Widerstand begrenzt sein und das Neugeborene symptomfrei sein. Mit Abfall des pulmonalvaskulären Widerstands am Ende der Neugeborenenperiode kommt es zu einer deutlichen Zunahme des Shuntvolumens mit der Manifestation einer Herzinsuffizienz (Abschn. 20.1.3). Entsprechend engmaschig muss das Neugeborene nach der Erstdiagnose im Verlauf untersucht werden und die Eltern über mögliche Herzinsuffizienzzeichen aufgeklärt werden.
Liegt eine eingeschränkte Herzfunktion vor, z. B. bei einer Kardiomyopathie, oder liegt eine ausgeprägte AV-Klappeninsuffizienz vor, besteht ebenfalls eine Volumenbelastung des Herzens. Die resultierende Herzinsuffizienz ist aber durch ein Vorwärtsversagen und nicht wie bei den Shuntläsionen durch eine Rezirkulation bedingt. Die klinischen Zeichen unterscheiden sich wenig; auch hier fallen die Kinder durch eine Herzinsuffizienz auf.
Azyanotische Herzfehler mit Druckbelastung des Herzens sind am häufigsten durch eine Aortenklappenstenose, Pulmonalklappenstenose oder Aortenisthmusstenose bedingt. Seltener sind Mitral-, Trikuspidalklappenstenose oder eine hypertrophe obstruktive Kardiomyopathie. Solange es sich nicht um kritische Stenosen handelt sind die Patienten relativ symptomarm. Bei kritischen Stenosen ist die Obstruktion so stark ausgeprägt, dass eine Ductusabhängigkeit besteht. Die kritischen Stenosen manifestieren sich somit bereits im Neugeborenalter und werden im Abschn. 20.1.5 weiter beschrieben.
Ein Herzgeräusch kann das einzige Symptom sein. Das Herz reagiert auf eine Stenose mit einer konzentrischen Hypertrophie und später mit einer Dilatation, um die vermehrte Druckbelastung zu bewältigen und das Herz-Zeit-Volumen (HZV) aufrecht zu erhalten. Das HZV kann dann jedoch nur noch begrenzt gesteigert werden. Besteht eine hochgradige Obstruktion kann das erste Symptom eine Synkope oder plötzlicher Herztod während körperlicher Belastung sein.
Jede Synkope unter körperlicher Belastung muss schnellstmöglich auf ihre Ursache hin abgeklärt werden.
Zyanotische Herzfehler
Zyanotische Herzfehler können mit verminderter Lungenperfusion oder vermehrter Lungenperfusion einhergehen. Dies ist insofern von Bedeutung, da eine Zyanose mit vermehrter Lungenperfusion weniger ausgeprägt ist und daher klinisch übersehen werden kann.
Bei zyanotischen Herzfehlern mit verminderter Lungenperfusion liegt eine Obstruktion des pulmonalen Blutflusses auf pulmonaler, ventrikulärer oder atrialer Ebene vor. Zusätzlich besteht eine Shuntverbindung (Foramen ovale/ASD, VSD, PDA), die O2-armes Blut an der Lunge vorbei in den Systemkreislauf leitet. Typische zyanotische Herzfehler sind die Fallot-Tetralogie, Trikuspidalatresie, Vitien mit singulärem Ventrikel und die Pulmonalstenose. Säuglinge mit diesen Vitien haben keine Herzinsuffizienzzeichen und daher in der Regel keine Trinkschwierigkeiten oder Gedeihstörung. Der Grad der Zyanose ist dabei abhängig von der Ausprägung der Obstruktion.
Ist die Obstruktion so ausgeprägt, dass die pulmonale Perfusion nur durch einen offenen Ductus arteriosus gewährleistet wird, handelt es sich um einen kritischen Herzfehler (Abschn. 20.1.5).
Bei zyanotischen Herzfehler mit vermehrter Lungenperfusion liegt keine Obstruktion des pulmonalen Blutflusses vor. Die Zyanose entsteht durch eine vollständige Durchmischung von venösen und arteriellen Blut oder Fehlstellungen der großen Gefäße (Aorta, Pulmonalarterie). Am häufigsten ist dies durch eine Transposition der großen Gefäße bedingt. Eine vollständige Mischung von arteriellen und venösen Blut findet sich ebenfalls bei komplexen Herzfehlern mit univentrikulären Herzen, bei der totalen Lungenvenenfehlmündung oder dem Truncus arteriosus. Liegt keine pulmonale Obstruktion vor, zeichnen sich diese Herzfehler durch ein klinisches Mischbild aus Zyanose und Herzinsuffizienz aus. Aufgrund der vermehrten Lungenperfusion kann die Zyanose gering ausgeprägt sein und klinisch wenig imponieren.
Herzinsuffizienz
Herzinsuffizienz ist ein klinisches Symptom und kein eigenständiges Krankheitsbild. Eine Herzinsuffizienz liegt vor, wenn das Herz nicht in der Lage ist ein ausreichendes Herz-Zeit-Volumen (HZV) zu erbringen, um den metabolischen Bedürfnissen des Körpers gerecht zu werden.
Das HZV ist das Produkt aus Schlagvolumen und Herzfrequenz (HZV = SV×HF). Diese Parameter sind wiederum abhängig von:
der Vorlast,
der myokardialen Kontraktilität,
der Nachlast,
einem synchronen Rhythmus,
der interventrikulären Interaktion,
der atrioventrikulären Kopplung.
Eine Herzinsuffizienz kann Folge einer angeborenen oder erworbenen Herzerkrankung sein, die zu einer Volumen- oder Druckbelastung des Herzens führt.
High-output-Failure
Im Kindesalter beruht die Herzinsuffizienz meistens auf einer erheblichen Volumenbelastung. Primär sind dies Vitien mit bedeutsamen Links-Rechts Shunt (VSD, AVSD, PDA). Der Begriff „Herzinsuffizienz“ ist in diesem Fall missverständlich, da die Auswurfleistung des linken Ventrikels deutlich erhöht ist, auch wenn ein Großteil dieses ausgeworfenen Volumens ineffektiv ist, da es direkt zur Lunge zurückgeführt wird ohne den Körperkreislauf zu passieren. Man spricht dann von einem sog. High-output-Failure im Gegensatz zum Low-output-Failure. Der erhöhte Blutfluss in der Lunge führt zu einer Widerstandserhöhung und erhöht die Atemarbeit. Um die erhöhte Auswurfleistung aufrecht zu erhalten, wird durch eine Erhöhung des Sympathiktotonus sowohl das Schlagvolumen als auch die Herzfrequenz gesteigert. Die vermehrte Ausschüttung von Katecholaminen sowie die gesteigerte Atemarbeit führen zu einer Erhöhung des O2-Verbrauchs.
Dies führt schließlich zu den klinischen Symptomen einer Herzinsuffizienz. Bei Kindern entspricht dies meist einer globalen Herzinsuffizienz. Eine Unterscheidung in Rechts- und Linksherzinsuffizienz, wie bei Erwachsenen, ist meist nicht möglich.
Säuglinge und Kleinkinder fallen durch Tachypnoe, Tachykardie, vermehrtem Schwitzen und Trinkschwäche auf. Daraus resultiert im weiteren Verlauf eine Gedeihstörung.
Unter Belastung (insbesondere bei der Nahrungsaufnahme bei Säuglingen) kommt zu einer Zunahme dieser Herzinsuffizienzzeichen. Weitere Symptome sind eine Belastungsintoleranz, die sich bei größeren Kindern anamnestisch oder mittels Belastungsuntersuchungen erschließen lässt, evtl. blasse und kühle Extremitäten, verlängerte Rekapillarisierungszeit sowie eine Hepatomegalie. Halsvenenstauung und Ödeme sind seltener als bei Erwachsenen.
Low-output-Failure
Bei einem Low-output-Failure z. B. bei einer Kardiomyopathie liegt ein Vorwärtsversagen vor und nicht wie bei den Shuntläsionen eine durch eine Rezirkulation bedingte Herzinsuffizienz.
Die klinischen Zeichen unterscheiden sich wenig; auch hier besteht eine Tachykardie, Tachypnoe, evtl. thorakale Einziehungen und Husten v. a. wenn ein Lungenödem vorliegt, verlängerte Rekapillarisierungszeit, Blässe, Hepatomegalie und in der Thoraxröntgenaufnahme eine Kardiomegalie. Aufgrund des Lungenödems kann es auch zu einer pulmonal bedingten Zyanose kommen.
Eine Druckbelastung führt erst zu einer Herzinsuffizienz, wenn eine erhebliche Obstruktion vorliegt. Diese demaskiert sich häufig erst unter Belastung, wenn es sich nicht um eine extrem hochgradige (kritische) Obstruktion handelt. Weiterhin kann eine Herzinsuffizienz natürlich auch bei einer Schädigung des Herzmuskels, z. B. nach einer Myokarditis oder nach herzchirurgischen Eingriffen entstehen.
Diagnose
Die Diagnose der Herzinsuffizienz wird in erster Linie anhand der bestehenden Symptome klinisch gestellt. Eine klinische Schweregradeinteilung kann nach dem modifizierten Ross-Score durchgeführt werden. Dabei werden vermehrtes Schwitzen, Tachypnoe, Atemfrequenz, Herzfrequenz sowie die Lebergröße berücksichtigt.
Echokardiographisch kann im Falle eines Low-output-Failure die eingeschränkte ventrikuläre Funktion dargestellt werden bzw. das ursächliche Vitium diagnostiziert werden. Im Thoraxröntgenbild kann eine Kardiomegalie, eine vermehrte Lungengefäßzeichnung, eine Stauung oder ein Lungenödem auffallen. Laborchemisch ist das natriuretische Peptid (BNP) ein guter kardialer Marker zur Verlaufsbeurteilung der Herzinsuffizienz.
Adaptionsmechanismen
Wie schon oben beschrieben reagiert der Körper sowohl bei verminderter Pumpleistung des Herzens als auch bei zwar erhöhter – aber ineffektiver – Auswurfleistung mit einer Erhöhung des Sympathikotonus und einer vermehrten Ausschüttung von Katecholaminen, die zu einer Erhöhung der Herzfrequenz und Kontraktilität führt. Es kommt im Verlauf zu einer Steigerung des Systemgefäßwiderstands und damit der Nachlast. Zusätzlich wird das Renin-Angiotensin-Aldosteronsystem (RAAS) weiter aktiviert, und führt über eine gesteigerte Vasokonstriktion zur Nachlaststeigerung, über Natrium- und Wasserretention kommt es zur Erhöhung der Vorlast. Die wichtigsten Adaptionsmechanismen einer Herzinsuffizienz sind:
eine neurohumorale Aktivierung,
der Frank-Starling-Mechanismus,
eine Herzfrequenzerhöhung,
eine myokardiale Hypertrophie und
ein Anstieg natriuretischer Peptide.
Diese Mechanismen sind geeignet um eine akute Herzinsuffizienz zunächst zu kompensieren, im chronischen Verlauf aber kommt es zu einem Circulus vitiosus mit Zunahme einer myokardialen Hypertrophie bzw. Dilatation und Funktionsstörung.
Therapie
Bei der Therapie der Herzinsuffizienz muss daher zwischen einer akuten und chronischen Herzinsuffizienz unterschieden werden. Wenn möglich muss die zugrunde liegende Ursache beseitigt werden. Ist dies nicht unmittelbar möglich ist eine medikamentöse Therapie zur Überbrückung bis zu einer endgültigen Behandlung bzw. zur Verbesserung der Symptome angezeigt.
Bei der medikamentösen Behandlung der akuten Herzinsuffizienz stehen die Steigerung der myokardialen Kontraktilität, vorsichtiger Nachlastsenkung zur Steigerung des Auswurfs und Aufrechterhaltung eines ausreichenden Blutdrucks im Vordergrund, während die medikamentöse Behandlung der chronischen Herzinsuffizienz auf die Antagonisierung der Kompensationsmechanismen abzielt.
Bei Säuglingen, die Trinkschwierigkeiten und Gedeihstörungen aufweisen, sollte auf eine ausreichende Kalorienzufuhr geachtet werden. Da diese Patienten pro Mahlzeit weniger Volumen zu sich nehmen können, sollte die Nahrung angereichert werden bzw. hochkalorische Nahrung verabreicht werden. Sind die Kinder nicht in der Lage genug Kalorien zu sich zu nehmen, kann die Nahrung per nasogastraler Sonde verabreicht werden.
ACE-Hemmer stellen die Grundlage der medikamentösen Therapie der chronischen Herzinsuffizienz dar. Sie inhibieren die Aktivierung des RAAS und verbessern die Hämodynamik und Ventrikelfunktion durch eine Nachlastsenkung.
β-Blocker schützen das Herz vor einer chronischen adrenergen Stimulation, senken die Herzfrequenz und verbessern das Verhältnis von O2-Verbrauch und -angebot.
Aldosteronantagonisten haben in mehreren Studien (bei Erwachsenen) prognostisch günstige Effekte gezeigt, die über die diuretische Wirkung hinausgehen. Zusätzlich besteht hier ein Remodeling-Effekt.
Diuretika sollten vorsichtig angewendet werden und sind bei einer Flüssigkeitsretention indiziert. Der längere Einsatz von Diuretika sollte vermieden werden, da sie zu einer ungünstigen Stimulation des RAAS führen.
Herzglykoside werden heutzutage seltener eingesetzt. Im Vergleich zu einem adulten Herzen können Herzglykoside bei Kindern die Inotropie deutlich stärker steigern. Die häufigste Form der Herzinsuffizienz im Kindesalter ist durch eine massive Rezirkulation bei Shuntvitien, also eine High-output-Failure bedingt. Es liegt keine eingeschränkte Ventrikelfunktion per se vor und Herzglykoside sind in diesen Fällen nicht indiziert. Indiziert ist der Einsatz von Herzglykosiden zur Behandlung von Herzrhythmusstörungen bzw. zur Frequenzoptimierung.
Eine weitere Therapieoption bei eingeschränkter Ventrikelfunktion ist eine kardiale Resynchronisationstherapie (CRT). Bei der CRT werden im Gegensatz zu einem herkömmlichen Schrittmacher beide Herzkammern durch entsprechend implantierte Sonden stimuliert. Besteht eine dyssynchrone Kardiomyopathie (z. B. bei Schrittmacher induzierter Kardiomyopathie oder bei symptomatischen Patienten mit eingeschränkter ventrikulärer Funktion (EF <35% und Vorliegen eines ausgeprägten Links- oder Rechtsschenkelblocks) kann mittels CRT eine Resynchronisierung angestrebt werden.
Bei therapierefraktärer Herzinsuffizienz sollte der Patient stationär aufgenommen werden. Dann ist ggf. eine Intensivierung der Therapie durch intravenöse Medikamente wie z. B. Phosphodiesterasehemmer (Milrinon) oder Kalziumsensitizer (Levosimendan) notwendig.
Fetaler und neonataler Kreislauf
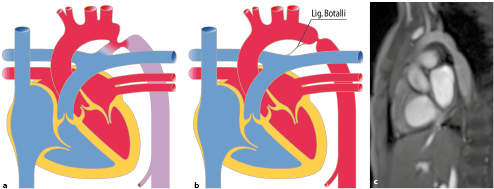
Die Kenntnis des fetalen Kreislaufs (Abb. 20.1) und ihrer postnatalen Anpassung ist eine wichtige Voraussetzung, die Pathophysiologie und die klinischen Erscheinungsbilder angeborener Herzfehler zu verstehen. Die fetale Zirkulation unterscheidet sich von der postnatalen Zirkulation in mehrfacher Hinsicht. Diese Unterschiede sind v. a. auf den unterschiedlichen Gasaustausch zurückzuführen, der beim postnatalen Kreislauf in der Lunge, beim Fetus durch die Plazenta stattfindet.

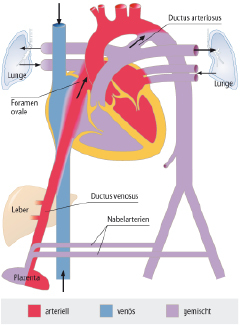
Im fetalen Kreislauf gibt es vier Kurzschlussverbindungen (Abb. 20.1):
Die Plazenta,
den Ductus venosus arantii,
das Foramen ovale und
den Ductus arteriosus botalli.
Da das Blut in der Plazenta oxygeniert wird, findet sich in der Nabelvene der höchste O2-Partialdruck. Somit findet sich in der unteren Hohlvene eine deutlich höhere Sättigung (ca. 70%) als in der oberen Hohlvene (ca. 40%). Ca. 1/3 dieses höher oxygenierten Bluts aus der unteren Hohlvene wird über das Foramen ovale direkt in den linken Vorhof umgeleitet, während der Rest in den rechten Ventrikel und die Pulmonalarterie geleitet werden. Das führt dazu, dass insbesondere das Gehirn und die Koronarien Blut mit einer höheren O2-Sättigung erhalten als die untere Körperhälfte. Weniger oxygeniertes Blut in der Pulmonalarterie fließt durch einen weit offenen Ductus arteriosus in die deszendierende Aorta, und dann zur Plazenta zur Reoxygenierung. Da die Lunge nur 15% des gesamten Herz-Zeit-Volumens (HZV) erhält, sind die Pulmonalarterienäste klein. Der rechte Ventrikel ist präpartal größer und dominanter als der linke Ventrikel, da der rechte Ventrikel mehr als die Hälfte des gesamten HZV stemmen muss. Der Druck in der rechten Herzkammer entspricht dem der linken Herzkammer (im Gegensatz zu einem adulten Herzen). Dies ist u. a. auch am EKG eines Neugeborenen erkennbar, das ein „Rechtsherzüberwiegen“ zeigt.
Während ein adultes Herz sein Schlagvolumen bei abfallender Herzfrequenz steigert, ist das fetale Herz nicht in der Lage das Schlagvolumen zu steigern. Das fetale Herzzeitvolumen ist somit abhängig von der Herzfrequenz. Wenn die Herzfrequenz abfällt, z. B. im Rahmen von fetalen Stresssituationen, führt dies zu einem bedeutsamen Abfall des HZV.
Nach der Geburt übernimmt die Lunge die Funktion des Gasaustauschs. Mit Durchtrennung der Nabelschnur wird der Blutfluss in der Umbilikalvene beendet und führt zu einem Verschluss des Ducuts venosus arantii.
Die ersten Atemzüge führen zu einer Lungenexpansion mit einer deutlichen Reduktion des pulmonalarteriellen Widerstands und einer Erhöhung des Lungenblutflusses. Aufgrund des vermehrten Lungenblutflusses kommt es entsprechend zu einem vermehrten Rückfluss über die Lungenvenen und damit zu einem erhöhten Druck im linken Vorhof, der letztendlich den Druck im rechten Vorhof überschreitet. Das Foramen ovale stellt vereinfacht ausgedrückt einen Schlitz oder Tunnel zwischen zwei sich überlappenden Wänden dar, dem Septum primum und dem Septum secundum. Durch den Druckanstieg im linken Vorhof wird nun das Septum primum an das Septum secundum gepresst und die dazwischenliegende Lücke verschlossen. Dies führt zu einem funktionellen Verschluss des Foramen ovale. Bei ca. 25% aller Menschen bleibt das Foramen ovale offen und ist als isolierter Befund nicht als pathologisch zu werten.
Mit der Beendigung der plazentalen Zirkulation kommt es zu einer Erhöhung des systemischen Widerstands. Da sich gleichzeitig der pulmonalvaskuläre Widerstand durch die Lungenexpansion verringert, übersteigt der systemische Druck den pulmonalarteriellen, d. h. höher oxygeniertes Blut aus der Aorta fließt über den Ductus arteriosus botalli in die Pulmonalarterie, es besteht ein Links-rechts-Shunt. Die höhere O2-Konzentration im Blut sowie der postpartal abfallende endogene Prostaglandinspiegel führen zum Verschluss des Ductus arteriosus botalli. Dieser verschließt sich zunächst durch Kontraktion, im weiteren Verlauf durch Obliteration.
Intrauterin wird der Ductus arteriosus botalli durch die endogene Prostaglandin-E2-Produktion offengehalten. Prostaglandinsynthesehemmer wie ASS oder Ibuprofen während der Schwangerschaft können zu einem vorzeitigen intrauterinen Verschluss des Ductus arteriosus mit der Folge einer schweren Rechtsherzbelastung führen.
Prostaglandin E1 dient zum Offenhalten oder Wiedereröffnen des Ductus arteriosus beim Neugeborenen mit ductusabhängigen Vitium (Abschn. 20.1.5). Es muss aufgrund seiner kurzen Halbwertszeit kontinuierlich gegeben werden. Unerwünschte Nebenwirkungen sind insbesondere Apnoen, Hypotonie und Ödeme.
Der funktionelle Verschluss ist im Normalfall nach 10–15 Stunden, nicht selten erst nach einigen Tagen vollzogen. Der anatomische Verschluss, d. h. die Obliteration und Umwandlung des Ductus in das Ligamentum botalli benötigt mehrere Wochen. Bei einigen Herzfehlern ist ein Überleben nach der Geburt nur durch einen offenen Ductus arteriosus möglich, es handelt sich in diesen Fällen um ein sog. ductusabhängiges Vitium. Der klinische Zustand bei diesen Neugeborenen verschlechtert sich parallel zum zunehmenden Verschluss. Bei dem geringsten Verdacht auf einen solchen Herzfehler ist der großzügige Beginn einer Prostaglandin-E1-Therapie zum Offenhalten bzw. Wiedereröffnen des Ductus arteriosus indiziert (Abschn. 20.1.5).
Sauerstoff kann zum Absenken des Lungenwiderstands sowie zum Verschluss des Ductus arteriosus führen. Bei V. a. ein ductusabhängiges Vitium sollte die O2-Applikation vorsichtig erfolgen. Unkritische O2-Gaben können zum Ductusverschluss führen und insbesondere bei einer ductusabhängigen Systemperfusion zu einer Dekompensation führen.
Der pulmonalvaskuläre Widerstand ist kurz nach der Geburt fast so hoch wie der systemische Widerstand. Mit der Lungenexpansion und dem daraus resultierenden Anstieg der O2-Sättigung in den Alveolen kommt es initial zu einem deutlichen Abfall des pulmonalvaskulären Widerstands, der trotzdem kurz nach der Geburt immer noch deutlich höher ist als am Ende der Neugeborenperiode.
Eine inadäquate peripartale oder postpartale Oxygenierung (z. B. durch Mekoniumaspiaration, Pneumonie, Lungenhypoplasie, Zwerchfellhernie) oder peri-/postpartale Stresssituationen (z. B. Sepsis, Hypoglykämie) können zu einer persistierenden pulmonalen Hypertension (PPHN; syn. persistierende fetale Circulation, PFC-Syndrom) führen. Es kommt zu einem Rechts-links-Shunt über das Foramen ovale und über den weiterhin offenen Ductus arteriosus bei eingeschränkter Lungendurchblutung und resultiert in einem schweren Krankheitsbild mit respiratorischer und metabolischer Azidose sowie einer zentralen Zyanose.
Die persistierende pulmonale Hypertension ist eine Ausschlussdiagnose. Ein Vitium als Ursache der Zyanose muss ausgeschlossen werden.
In den ersten 2–8 Wochen postpartal kommt es zu einem weiteren langsamen aber stetigen Abfall des pulmonalvaskulären Widerstands.
Somit besteht in der Neugeborenenperiode noch ein erhöhter pulmonalarterieller Druck. Dies ist insbesondere im Hinblick auf Shuntvitien wichtig. So können Vitien, die normalerweise zu einem Links-rechts-Shunt führen, wie ein Ventrikelseptumdefekt, initial übersehen oder unterschätzt werden. Der erhöhte Lungenwiderstand führt dazu, dass der Druckunterschied zwischen den Kammern kleiner ist. Herzgeräusche werden durch turbulente Blutströmungen hervorgerufen, die in diesem Fall durch das Druckgefälle bedingt sind. Durch den geringen Druckunterschied v. a. kurz nach der Geburt wird der Shunt begrenzt, es kommt u. U. nicht zu einem Herzgeräusch und auch echokardiographisch kann ein Shunt ggf. noch nicht erkennbar sein.
Mit dem fortschreitenden Abfall des pulmonalvaskulären Widerstands, vergrößert sich auch der Druckunterschied zwischen den Herzkammern. Das Shuntvolumen kann zunehmen und damit auch der pulmonalarterielle Blutfluss. Entsprechend kann ein initial asymptomatisches Shuntvitium wie z. B. ein Ventrikelseptumdefekt im Verlauf der Neugeborenperiode zu einer Herzinsuffizienz führen.
Cave
Der pulmonalvaskuläre Widerstand fällt in den ersten 2–8 Wochen des Lebens ab. Herzfehler mit einem großen Links-rechts-Shunt werden häufig erst symptomatisch, wenn der Lungenwiderstand abgefallen ist. Einige Shuntvitien führen daher kurz nach der Geburt noch zu keinem Herzgeräusch und können so bei den Vorsorgeuntersuchungen U1 und U2 (v. a. wenn sie frühzeitig durchgeführt werden) undetektiert bleiben.
Kritische Herzfehler
Ein kritischer Herzfehler ist postnatal der häufigste Grund für eine akute kardiale Dekompensation. Etwa 15% aller Herzfehler sind kritische Herzfehler, die bereits für das Neugeborene zu einem lebensbedrohlichen Zustand führen können. Um zu überleben benötigen Kinder mit diesen Herzfehlern einen herzchirurgischen oder katheterinterventionellen Eingriff. Die Sterblichkeitsrate liegt auch heute noch bei bis zu 25%.
Kritische Herzfehler werden trotz verbesserter Pränataldiagnostik und postnataler klinischer Untersuchungen immer noch unzureichend detektiert. Insbesondere die Sensitivität der klinischen Untersuchung in den ersten Lebenstagen beträgt aufgrund eines möglichen symptomfreien Intervalls („diagnostische Lücke“) durch die verzögerte Umstellung der fetalen auf die neonatale Kreislaufphysiologie nur ca. 50%. Die meisten Neugeborenen zeigen eine unterschiedlich ausgeprägte Hypoxämie, die jedoch zu diesem Zeitpunkt häufig nicht mit einer klinisch erkennbaren Zyanose einhergeht. Mittels Pulsoxymetriescreening kann die postnatale „diagnostische Lücke“ verkleinert werden, insbesondere wenn ein Rechts-links-Shunt auf Ductus-arteriosus-Ebene besteht, der zu einer differenziellen O2-Sättigung führt mit niedrigeren Sättigungen am Bein im Vergleich zum rechten Arm.
Cave
Eine akute Verschlechterung des Neugeborenen mit kritischem Herzfehler ist am häufigsten mit einem raschen Verschluss des Ductus arteriosus assoziiert, seltener mit einer zunehmenden Restriktion des Shunts auf Vorhofebene.
Die Lokalisation und die Ausprägung der kardialen Fehlbildungen bestimmen das klinische Bild in der postnatalen Phase. Die kritischen angeborenen Herzfehler lassen sich in 4 Gruppen unterteilen:
- Herzfehler mit ductusabhängiger Systemperfusion (Linksherzobstruktion):
- kritische Aortenstenose,
- kritische Aortenisthmusstenose,
- unterbrochener Aortenbogen,
- hypoplastisches Linksherzsyndrom (HLHS).
- Herzfehler mit ductusabhängiger Pulmonalperfusion (Rechtsherzobstruktion):
- kritische Pulmonalstenose,
- Pulmonalatresie mit intaktem Septum oder mit VSD,
- ausgeprägte Form der Fallot-Tetralogie,
- Trikuspidalatresie mit Pulmonalstenose/-atresie,
- Ebstein-Anomalie bzw. Trikuspidalklappendysplasie mit Pseudoatresie der Pulmonalklappe und ductusabhängigen Pulmonalfluss.
- Komplette Transposition der großen Gefäße (d-TGA):
- falls keine kompetente Mischung auf Vorhof-, Ventrikel- oder Ductusebene besteht.
- Sonstige:
- totale Lungenvenenfehlmündung mit Lungenvenenobstruktion,
- Trucus arteriosus communis bei Truncusklappeninsuffizienz,
- univentrikuläres Herz mit Imbalance der Kreislaufzirkulation.
Herzfehler mit ductusabhängiger Systemperfusion (Linksherzobstruktion)
Bei Herzfehlern mit ductusabhängiger Systemperfusion (Linksherzobstruktion) liegt eine hochgradige Obstruktion des linken Herzens oder der Aorta vor. Eine ausreichende Systemperfusion kann nur über einen offenen Ductus arteriosus gewährleistet werden. Die Obstruktion kann auf unterschiedlichen Ebenen bestehen; auf Ventrikelebene: kritische Aortenstenose, hypoplastisches Linksherzsyndrom, auf Aortenebene: kritische Aortenisthmusstenose, unterbrochener Aortenbogen. Je nachdem, wo die Obstruktion liegt und wie ausgeprägt sie ist, versorgt der rechte Ventrikel über einen offen Ductus arteriosus den pulmonalen und systemischen Kreislauf vollständig oder partiell.
Da durch die Obstruktion meist, zumindest zu Beginn ein Vorwärtsversagen des linken Ventrikels besteht, ist der Druck im linken Vorhof erhöht und entsprechend liegt über eine Vorhofkommunikation ein Links-rechts-Shunt vor. Falls ein signifikanter atrialer Links-rechts-Shunt vorliegt ist die O2-Sättigung in der Pulmonalarterie und konsekutiv in der Aorta descendens relativ hoch. Eine Zyanose ist klinisch nicht unbedingt erkennbar. Fehlt ein Shunt oder liegt eine restriktive Vorhofkommunikation vor, staut sich Blut zurück in die Lunge und kann so zu einem passiven Lungenödem führen. Eine Restriktion auf Vorhofebene kann unmittelbar nach der Geburt zu einer Hypoxämie mit Schocksymptomatik führen.
Die ductusabhängige Systemperfusion zeigt bei Verschluss des Ductus arteriosus das klinische Bild eines kardiogenen Schock, das sich nur wenig von dem Bild einer foudroyanten Sepsis unterscheidet.
Leitsymptome
Leitsymptome der ductusabhängigen Systemperfusion sind Herzinsuffizienzzeichen:
Tachypnoe, Dyspnoe, Hüsteln,
Tachykardie, vermehrte präkordiale (rechtsventrikuläre) Palpitation,
Hepatomegalie,
Trinkschwäche,
grau-blasses Hautkolorit, verlängerte Rekapillarisierungszeit,
schwache oder fehlende Pulse an den unteren Extremitäten,
O2-Sättigungsdifferenz zwischen oberer und unter Extremität,
Oligurie,
ein Herzgeräusch liegt häufig nicht vor.
Bei einem Neugeborenen, dessen Zustand sich in den ersten Lebenstagen klinisch verschlechtert, muss daher neben der Verdachtsdiagnose Sepsis immer an einen kritischen Herzfehler gedacht werden. Im Zweifel kann immer eine Therapie mit Prostaglandin E1 in niedriger Dosis begonnen werden!
Eine Echokardiographie ist so schnell wie möglich zur Diagnosestellung als auch zur begleitenden Schocktherapie durchzuführen.
Therapie
Grundsätzliche Therapieziele sind die Stabilisierung des Patienten mit weitgehender Wiederherstellung der fetalen, parallel geschalteten Kreislaufphysiologie sowie der Optimierung der pulmonalarteriellen und systemischen Widerstände.
Der Ductus arteriosus wird mit Prostaglandin-E1-Infusion offengehalten bzw. wiedereröffnet. Die Dosierung hängt vom klinischen Zustand und dem Öffnungszustand des Ductus arteriosus ab. Idealerweise sollte die Indikation und Steuerung der Prostaglandintherapie mittels Echokardiographie erfolgen.
Prostaglandin führt u. a. zu einer Absenkung des Lungenwiderstands. Daher kann eine zu hohe Dosierung nicht nur den Ductus weit öffnen, sondern auch den pulmonal-vaskulären Widerstand so weit absenken, dass der pulmonale Blutfluss auf Kosten der systemischen Perfusion erhöht wird.
Daher sollte bei offenen Ductus arteriosus eine niedrige Prostaglandindosis z. B. von 5–10 ng/kgKG/min angestrebt werden.
Ein Absenken des pulmonalen Widerstands sollte wie schon weiter oben beschrieben unbedingt vermieden werden, d. h. keine inadäquate O2-Therapie. Wenn Sauerstoff appliziert werden muss, nur bis zu einer Zielsättigung zwischen 75–85%, dann kann von einer balancierten Situation mit einem Qp/Qs ≈1 ausgegangen werden. Volumengaben sollten vorsichtig erfolgen.
Vitien mit ductusabhängiger Pulmonalperfusion (Rechtsherzobstruktion)
Bei Vitien mit ductusabhängiger Pulmonalperfusion (Rechtsherzobstruktion) kann die Obstruktion auf mehreren Ebenen existieren. Eine ausreichende Lungenperfusion kann immer nur durch einen Ductus arteriosus oder größere aortopulmonale Kollateralen erreicht werden.
Bei der einfachen Transposition der großen Gefäße (TGA) liegen zwei parallel geschaltete, aber getrennte Kreisläufe vor. Ein Überleben ohne Shuntverbindungen ist nicht möglich. Über einen offenen Ductus wird der pulmonale Blutfluss erhöht, der Rückfluss über die Pulmonalvenen zum linken Vorhof steigt und damit der Druck im linken Vorhof. Dies begünstigt eine bessere Mischung von O2-reichen und -armen Blut über das Foramen ovale. Nur über einen Links-recht-Shunt auf Vorhofebene kann das Systemblut mit Sauerstoff angereichert werden. Entsprechend führt ein restriktives Foramen ovale neben einer ausgeprägten Zyanose zu Tachy- und Dyspnoe bis hin zum Lungenödem.
Leitsymptom der ductusabhängigen Pulmonalperfusion und der TGA ist die O2-resistente Zyanose.
Typischerweise findet sich trotz generalisierter Zyanose ein vitales Neugeborenes. Mit Fortschreiten der Zyanose oder wenn ein restriktives Foramen ovale vorliegt, kommt es zu einer zunehmenden Tachypnoe und Tachykardie.
Ein zyanotisches Neugeborenes ohne pulmonale Ursache (und in relativ guten, vitalen Allgemeinzustand) sollte in erster Linie an eine TGA oder an Vitien mit ductusabhängiger Pulmonalperfusion denken lassen.
Der Ductus arteriosus wird mit Prostaglandin-E1-Infusion offengehalten bzw. wiedereröffnet. Die Dosierung hängt vom klinischen Zustand und dem Öffnungszustand des Ductus arteriosus ab, kann aber bei ductusabhängiger Pulmonalperfusion großzügiger als bei ductusabhängiger Systemperfusion eingesetzt werden.
Nach einer Wiedereröffnung des Ductus arteriosus sollte auch hier eine balancierte Situation zwischen pulmonalen und systemischen Kreislauf angestrebt werden.
Der pulmonale Widerstand kann gesenkt, der systemische Widerstand bei schlechter Lungenperfusion erhöht werden. Eine Zielsättigung um 80% sowie eine milde Alkalose sollten angestrebt werden. Eine intensive Volumentherapie ist meistens notwendig, sofern keine Restriktion auf Vorhofebene besteht.
Bei V. a. das Vorliegen eines kritischen Herzfehlers sollte die Verlegung in ein kinderkardiologisches Zentrum so schnell wie möglich erfolgen.
Bei Vorliegen eines restriktiven Foramen ovale kann eine Ballonatrioseptostomie nach Rashkind als Notfallprozedur durchgeführt werden.
Bei Vorliegen einer kritischen Aortenklappenstenose besteht zudem die Möglichkeit einer Ballonvalvuloplastie.
Liegt eine prostaglandinrefraktäre Ductusstenose vor, kann ein Stent in den Ductus arteriosus implantiert werden.
Neben den Vitien mit ductusabhängiger Systemperfusion und Pulmonalperfusion oder der TGA gibt es noch eine Reihe komplexer Herzfehler, die ebenfalls in der postnatalen Phase zu einer akuten Dekompensation führen können. Bei diesen komplexen Herzfehlern liegt im Prinzip meistens eine Konstellation wie bei der ductusabhängigen System- oder Pulmonalperfusion vor. Anhand der Leitsymptome kann man sich hinsichtlich der ersten Therapiemaßnahmen orientieren, bis die Diagnose echokardiographisch gestellt wird und eine spezifische Therapie (operativ oder katherinterventionell) möglich ist.
Primär azyanotische Herzfehler
Herzfehler mit Links-rechts-Shunt
Persistierender Ductus arteriosus (PDA)
Der Ductus arteriosus botalli ist eine Gefäßverbindung zwischen Aorta und Pulmonalarterie. Der Ductus verschließt sich normalerweise innerhalb von 48 Stunden nach der Geburt. Bleibt der Verschluss des Ductus arteriosus aus, spricht man von einem persistierenden Ductus arteriosus (PDA). PDA haben eine Anteil von ca. 7% aller angeborener Herzfehler (Tab. 20.1).
Abhängig vom Gestationsalter bleibt bei Frühgeborenen aufgrund der Unreife des Gewebes der Ductus arteriosus häufiger und länger offen als bei Reifgeborenen.
Morphologie/Hämodynamik
Die Morphologie des PDA kann in Bezug auf Länge, Durchmesser und Form erheblich variieren.
Kurz nach der Geburt besteht bei offenem Ductus arteriosus botalli noch ein Gleichgewicht zwischen aortalem und pulmonalarteriellem Druck. Mit zunehmendem Abfall des Lungengefäßwiderstands kommt es zu einem zunehmenden Links-rechts-Shunt. Ähnlich wie beim VSD ist die Hämodynamik abhängig vom Shuntvolumen.
Bei einem kleinen Shuntvolumen ist der PDA hämodynamisch unbedeutsam. Liegt ein größeres Shuntvolumen vor, resultiert eine Volumenbelastung des linken Vorhofs und des linken Ventrikels mit konsekutiver Vergrößerung dieser Strukturen. Durch den Abstrom des Bluts, in der Systole als auch in der Diastole, entsteht eine große Blutdruckamplitude mit hohem systolischen und niedrigem diastolischen Druck. Dies kann zu einer Minderdurchblutung der peripheren Kreislaufregionen führen, insbesondere bei unreifen Frühgeborenen kann dies das Risiko für die Entwicklung einer Niereninsuffizienz oder nekrotisierenden Enterokolitis deutlich erhöhen (Kap. 4).
Bei einem großen PDA mit nicht restriktivem Shunt kann sich eine pulmonale Hypertonie entwickeln, die ab einem bestimmten Punkt nicht mehr reversibel ist und dann zu einer Eisenmenger-Reaktion (Abschn. 20.2.1.2) führt.
Klinik
Bei einem kleinen Shunt bleiben die betroffenen Kinder meist asymptomatisch.
Bei großem PDA kann sich eine Herzinsuffizienz in unterschiedlicher Ausprägung präsentieren.
Diagnostik
Auskultatorisch findet sich ein typisches Maschinengeräusch (systolisch/diastolisches Herzgeräusch) mit punktum maximum (p.m.) über dem 2./3. ICR links. Echokardiographisch lässt sich sowohl doppler- als auch farbdopplersonographisch ein systolisch-diastolischer Shunt in die Pulmonalarterie darstellen. Bei Früh- und Neugeborenen findet sich aufgrund des noch erhöhten Lungengefäßwiderstandes nur ein reines Systolikum.
Ist der PDA sehr klein ist kein Herzgeräusch auskultierbar, hierbei handelt es sich dann um einen sog. silenten PDA.
Die Diagnose wird echokardiographisch gestellt. Neben der Größe des PDA wird auch der Druckgradient gemessen, um eine ggf. vorliegende pulmonale Hypertension zu erfassen. Zusätzlich wird die Volumenbelastung des linken Vorhofs und Ventrikels beurteilt. Häufig wird hier das Verhältnis von linkem Vorhof zur Aorta (LA/AO) kalkuliert. Ein Quotient >1,5 deutet auf eine hämodynamische Bedeutsamkeit hin. Weiterhin kann insbesondere bei Neu- und Frühgeborenen durch die Doppleruntersuchung peripherer Arterien (Truncus coeliacus, A. cerebri media) die hämodynamische Bedeutsamkeit beurteilt werden. Je bedeutsamer der PDA, umso niedriger ist der diastolische Fluss bis hin zum Nullfluss oder diastolischer Flussnegativierung.
Therapie
Kleine PDA werden heutzutage nicht mehr verschlossen. Als klein werden sie dann bezeichnet, wenn keine Volumenbelastung der linken Herzstrukturen darstellbar ist und wenn es sich um einen silenten PDA handelt. PDA können mit gutem Ergebnis interventionell verschlossen werden, z. B. mit ablösbaren Spiralen (Cook-Coils) oder entsprechenden Devices. Ein pharmakologischer Verschluss eines PDA ist bei reifgeborenen Kindern in der Regel nicht erfolgreich (bei Frühgeborenen Kap. 4).
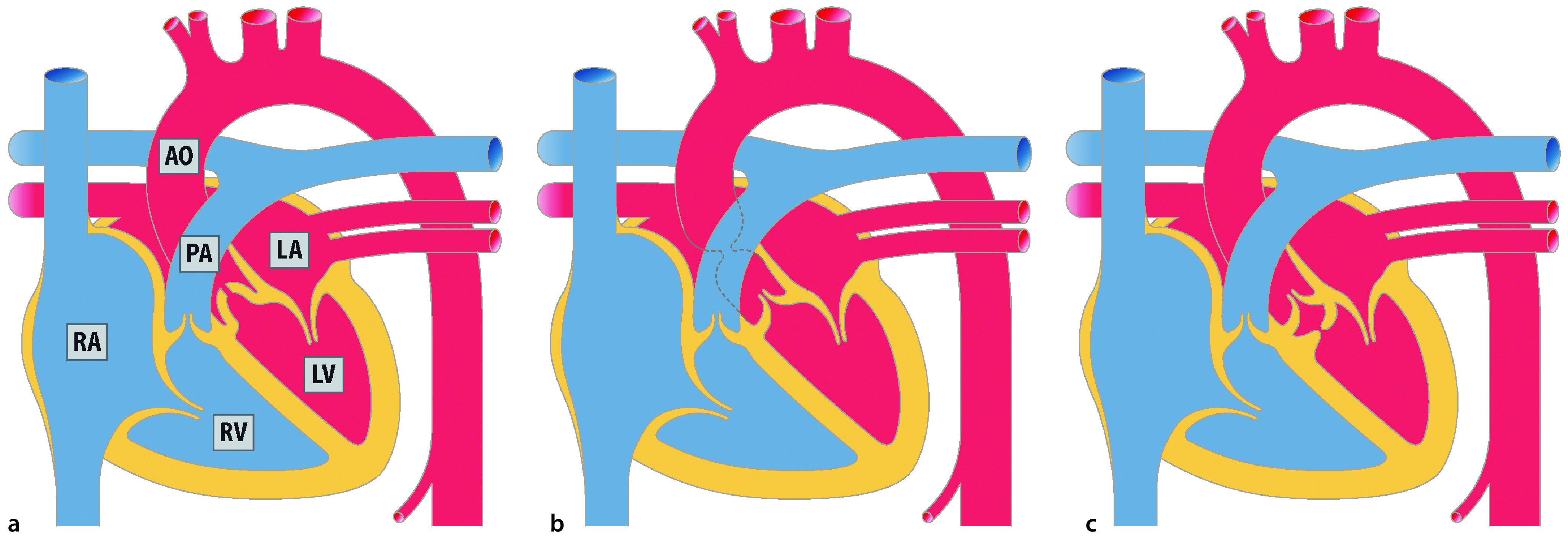
Ventrikelseptumdefekt
Der Ventrikelseptumdefekt (VSD; Abb. 20.2) ist als isolierte Fehlbildung der häufigste angeborene Herzfehler mit einem Anteil von 15–20% aller angeborenen Herzfehler. Als Begleitfehlbildung bei komplexen Herzfehlern macht der VSD einen Anteil bis zu 40% aller Herzfehler aus.

Morphologie und Hämodynamik
Der Ventrikelseptumdefekt wird nach Lokalisation und Größe beurteilt. Je nach Lage im Septum unterscheidet man folgende Typen:
Perimembranöse Defekte, im membranösen Septum unterhalb der Aortenklappe liegend, sind am häufigsten (ca. 70%).
Muskuläre Defekte können im apikalen oder mittleren Septumbereich liegen. Muskuläre Defekte treten häufig auch als multiple kleine Defekte auf, und werden dann als „Swiss-Chesse“-Typ bezeichnet.
Infundibuläre Ventrikelseptumdefekte liegen im Auslassseptum unterhalb der Aorten- und Pulmonalklappe.
Inlet-VSD liegen im Einlassseptum und werden am häufigsten bei einem AVSD vorgefunden.
Beim Malalignment-VSD besteht eine Verlagerung des Ausflusstraktseptums, die dazu führt, dass eine Taschenklappe, z. B. die Aortenklappe über dem VSD „reitet“, wie z. B. bei der Fallot-Tetralogie. Malalignment-VSDs kommen niemals isoliert vor. Sie sind immer mit anderen Herzfehlern assoziiert.
Sowohl infundibuläre als auch Inlet-VSD kommen als isolierte VSD im Vergleich zu perimembranösen und muskulären Defekten relativ selten vor, mit einem Anteil von 5–8% aller VSD.
Bei einem isolierten VSD liegt ein Links-rechts-Shunt vor. Die Größe des Shunts ist abhängig von der Größe des Defekts und dem pulmonalvaskulären Widerstand. Die Lokalisation des Defekts hat keinen Einfluss auf das Shuntvolumen. Allerdings kann die Lage des VSD das Therapiekonzept beeinflussen.
Im Neugeborenenalter ist der pulmonalvaskuläre Widerstand noch erhöht, sodass zwischen den Ventrikel noch ein annähernd gleicher Druck besteht. Dementsprechend ist das Shuntvolumen deutlich reduziert. Dies ist auch der Grund, warum man VSD kurz nach der Geburt sowohl auskultatorisch als auch echokardiographisch unterschätzen oder sogar übersehen kann.
Häufig werden Ventrikelseptumdefekte in kleine, mittelgroße und große Defekte eingeteilt: Bei einem kleinen Defekt besteht eine Restriktion des Shuntvolumens. Es besteht eine Drucktrennung zwischen den Ventrikel. Diese Ventrikelseptumdefekte sind hämodynamisch unbedeutsam und haben eine Tendenz sich spontan zu verschließen, v. a. wenn sie im muskulären oder perimembranösen Septum liegen.
Bei mittelgroßen Defekten überwiegt die Volumenbelastung des linken Ventrikels! Es resultiert eine linksventrikuläre Vergrößerung. Der rechte Ventrikel wird von der Volumenbelastung nicht betroffen. Bei einem VSD liegt ein systolischer Shunt vor. Der rechte Ventrikel kontrahiert sich gleichzeitig mit dem linken Ventrikel. Das Shuntvolumen wird somit fast direkt in die Pulmonalarterie weitergeleitet anstatt im rechten Ventrikel zu verbleiben und dort zu einer Volumenbelastung zu führen. Der rechte Ventrikel ist daher bei einem mittelgroßen VSD nicht volumenbelastet, während sowohl der linke Vorhof als auch der linke Ventrikel durch die Volumenbelastung vergrößert sind.
Liegt ein großer Defekt vor, kommt es zum Druckangleich zwischen beiden Herzkammern. Der rechte Ventrikel ist in diesem Fall auch betroffen und entsprechend vergrößert und hypertrophiert. Das Shuntvolumen wird nur noch vom pulmonalvaskulären Widerstand reguliert, der durch die Druck- und Volumenbelastung im Verlauf zunimmt. Ist diese Erhöhung des pulmonalvaskulären Widerstands an einem Punkt angelegt, an dem sie nicht mehr reversibel ist und den Druck in der rechten Herzkammer übersteigt, kommt es zu einer sog. Shuntumkehr (Eisenmenger-Reaktion). Resultat ist ein Rechts-links-Shunt mit Zyanose. Ein Verschluss des VSD ist nicht mehr möglich. Es würde zu einem Rechtsherzversagen kommen, da der rechte Ventrikel alleine nicht in der Lage ist, den erhöhten pulmonalvaskulären Widerstand zu überwinden.
Cave
Ein irreversibler Anstieg des pulmonalarteriellen Drucks kann bereits nach dem 6. Lebensmonat beginnen.
Liegt ein VSD in der Nähe der Aortenklappe, kann es durch das Prolabieren eines Segels in den Defekt zu einer Schädigung der Aortenklappe mit einer konsekutiven Insuffizienz kommen. Auch wenn der VSD an sich klein und hämodynamisch wenig bedeutsam ist, stellt dies eine Indikation zum operativen Verschluss dar.
Klinik
Entsprechend der beschriebenen Hämodynamik ist die Symptomatik abhängig von der Defektgröße und dem pulmonalen Widerstand. Kinder mit kleinen, hämodynamisch unbedeutenden VSD sind asymptomatisch. Kleine restriktive Defekte zeichnen sich durch ein lautes Systolikum aus („viel Lärm um nichts“).
Sind die Defekte etwas größer, kann sich echokardiographisch bereits eine Vergrößerung der linken Herzhöhlen zeigen, ohne dass diese Kinder symptomatisch werden. Das bedeutet, dass ein nicht unerhebliches Shuntvolumen besteht. In diesem Fall ist eine Verschlussindikation gegeben. Bei größeren Defekten stehen die Zeichen einer Herzinsuffizienz im Vordergrund. Da der pulmonale Widerstand nach der Geburt erst langsam abfällt, nimmt der Links-rechts-Shunt in den ersten Lebenswochen zu, sodass Symptome oft erst nach 2–8 Wochen auftreten oder deutlich zunehmen können. Bei Neugeborenen müssen die Eltern über das Auftreten von Herzinsuffizienzzeichen aufgeklärt werden, damit ggf. eine vorzeitige Wiedervorstellung mit Einleitung einer medikamentösen Therapie erfolgen kann. Liegt ein großer VSD vor, ohne dass Zeichen einer Herzinsuffizienz vorhanden sind, sollte dies ein Warnsignal sein. In diesem Fall liegt bereits eine deutliche pulmonale Widerstandserhöhung vor, die den Patienten zwar vor einer Lungenüberflutung und damit auch einer Herzinsuffizienz schützt, aber auch insofern gefährdet, dass diese Widerstandserhöhung bald nicht mehr reversibel ist. Eine baldige Korrektur des Defekts ist in diesem Fall indiziert.
Diagnostik
Leitsymptom ist bei kleineren und auch mittelgroßen Defekten ein typisches Systolikum. Bei kleinen restriktiven VSD imponiert ein lautes holosystolisches Geräusch mit p.m. über dem 3./4. ICR links.
Aufgrund des noch erhöhten Lungenwiderstands kurz nach der Geburt, kann ein Herzgeräusch bei den Vorsorgeuntersuchungen U1 und U2 oft noch fehlen.
Im Echokardiogramm kann der Defekt sowohl im B-Bild, als auch farbdopplersonographisch dargestellt und anatomisch zugeordnet werden. Der Druckgradient über dem VSD wird mittels Doppler bestimmt. Damit kann beurteilt werden, ob ein drucktrennender restriktiver Defekt besteht oder es bereits zu angehobenen Druckverhältnissen im rechten Ventrikel und im kleinen Kreislauf gekommen ist, also eine pulmonale Hypertension besteht. Weiterhin wird die Größe des linken Vorhofs und des Ventrikels bestimmt als Hinweis auf eine linksventrikuläre Volumenbelastung.
Therapie
Bei symptomatischen Defekten steht die medikamentöse Therapie der Herzinsuffizienz im Vordergrund. Kommt es im Verlauf zu einer Verkleinerung des VSD bzw. zu Abnahme der hämodynamischen Bedeutsamkeit kann die Medikation evtl. abgesetzt werden.
Eine Verschlussindikation besteht bei großen VSD mit klinischer Symptomatik, die medikamentös nicht beherrschbar sind. Hier wird der Verschluss bereits im Säuglingsalter durchgeführt, ebenso bei großen Ventrikelseptumdefekten bei denen eine pulmonale Hypertension droht. Hier sollte die Korrektur-OP ebenfalls im ersten Lebenshalbjahr durchgeführt werden, um eine irreversible pulmonale Hypertension zu vermeiden. Weiterhin besteht eine Operationsindikation bei VSD, die nah an der Aortenklappe liegen und durch ihre Lage eine Aorteninsuffizienz verursachen, da sonst im weiteren Verlauf ein Klappenersatz droht. Bei etwas älteren, asymptomatischen Kindern besteht eine Indikation wenn ein erhebliches Shuntvolumen besteht, erkennbar an einer deutlichen Vergrößerung des linken Vorhofs und des linken Ventrikels. In der Regel werden die meisten Defekte operativ verschlossen. In einigen Fällen ist ein katheterinterventioneller Verschluss möglich.
Prognose und Verlauf
Die Spontanverschlussrate ist bei kleinen muskulären und perimembranösen Defekten hoch. Kleine muskuläre Defekte verschließen sich häufig in den ersten 2 Lebensjahren. Perimembranöse Defekte verschließen sich zu 50%. Wichtig ist zu wissen, dass VSD im Verlauf nicht größer werden. Es besteht also eine relative Verkleinerungstendenz.
Infundibuläre Inlet- und Malalignment-VSD verschließen sich nicht spontan.
Bei nicht korrigierten, hämodynamisch relevanten isolierten VSD entwickelt sich eine fixierte pulmonale Hypertonie und somit eine Eisenmenger-Reaktion. Um festzustellen, ob diese pulmonale Hypertension irreversibel ist, kann die pulmonale Widerstandserhöhung im Herzkatheter mittels Sauerstoff-NO-Beatmung und Prostaglandininfusion untersucht werden. Besteht hier eine Reversibilität kann der VSD auch in höherem Alter noch verschlossen werden, ansonsten ist eine Korrektur nicht mehr möglich.
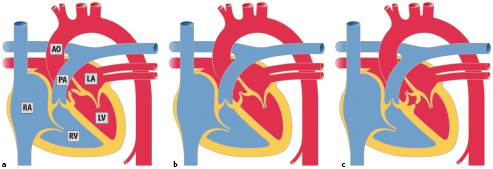
Vorhofseptumdefekt (ASD)
Der Vorhofseptumdefekt (ASD) ist einer der häufigsten angeborenen Herzfehler und hat als isolierter Defekt einen Anteil von ca. 7–10% aller angeborenen Herzfehler. Mädchen sind häufiger betroffen als Jungen.
Morphologie und Hämodynamik
Abhängig von ihrer Lage im Vorhofseptum werden verschiedene Vorhofseptumdefekte unterschieden (Abb. 20.3):

Vorhofseptumdefekt vom Sekundumtyp (Abb. 20.3) ist der häufigste ASD. Es findet sich hier ein Defekt im Bereich der Fossa ovalis, relativ zentral gelegen.
Vorhofseptumdefekt vom Primumtyp (ASD I) liegt im unteren, klappennahen (AV-Klappen) Anteil des Vorhofseptums. Es handelt sich in um eines Defekt des atrioventrikulären Kanals (Abschn. 20.2.1.5).
Sinus-venosus-Defekte liegen an der Einmündung der Hohlvenen in den rechten Vorhof. Entsprechend unterscheidet man einen oberen Sinus-venosus-Defekt (an der Einmündung der oberen Hohlvene) und einen unteren Sinus-venosus-Defekt (selten). Ein Sinus-venosus-Defekt ist den meisten Fällen mit einer partiellen Lungenvenenfehlmündung assoziiert.
Sinus-coronarius-Defekt ist ein seltener ASD. Normalerweise hat der Sinus coronarius seine Öffnung im rechten Vorhof. Bei dem vorliegenden Defekt liegt zusätzlich eine Öffnung im Bereich des linken Vorhofs vor, sodass Blut vom linken Vorhof zum rechten Vorhof gelangen kann.
Persistierendes Foramen ovale (PFO): findet sich in bis zu 25% der Normalbevölkerung. Es liegt kein echter Defekt vor. Die Verschmelzung des Septum primum mit dem Septum secundum ist unvollständig. Unter bestimmten Umständen (z. B. Valsalva-Manöver, pulmonale Hypertension) kann sich das PFO ventilartig öffnen und so einen Rechts-links Shunt und damit Embolien vom venösen ins arterielle System (sog. paradoxe Embolien) ermöglichen.
ASD führen zu einem Links-rechts-Shunt auf Vorhofebene. Durch den Shunt kommt es zu einer Volumenbelastung des rechten Ventrikels und letztendlich auch des Lungenkreislaufs. Das Ausmaß des Links-rechts-Shunts ist abhängig von der Dehnbarkeit (Compliance) der Ventrikel. Der rechte Ventrikel weist im Normalfall eine bessere diastolische Compliance als der muskelstarke linke Ventrikel auf. Allerdings unterscheiden sich im ersten Lebensjahr die Compliance der Ventrikel nur wenig, entsprechend kann das Shuntvolumen auch bei großen Defekten in diesem Alter noch unbedeutend sein.
Klinik
ASD führen zunächst zu keiner ausgeprägten Symptomatik. Die meisten Säuglinge und Kleinkinder mit einem ASD sind asymptomatisch und der Defekt wird als Zufallsbefund entdeckt. Defekte mit großem Shunt können häufig als einziges Symptom im Kindesalter durch eine erhöhte pulmonale Infektanfälligkeit auffallen.
Sehr selten kann es bei sehr großem Shunt schon im Säuglingsalter zu Herzinsuffizienzzeichen kommen. Eine verminderte Belastbarkeit oder Herzrhythmusstörungen aufgrund einer zunehmenden rechtsventrikulären Funktionsstörung treten typischerweise erst im Jugend- bis Erwachsenalter auf. Mit jedem weiteren Lebensjahrzehnt steigt bei großen Defekten die Wahrscheinlichkeit für die Entwicklung einer pulmonalen Hypertension.
Diagnostik
Der ASD selbst führt zu keinem Herzgeräusch, aber aufgrund der Volumenbelastung des rechten Ventrikels kommt es zu einer relativen Pulmonalstenose und somit zu einem Systolikum über dem 2. ICR links und typischerweise zu einem weit fixierten gespaltenen 2. Herzton.
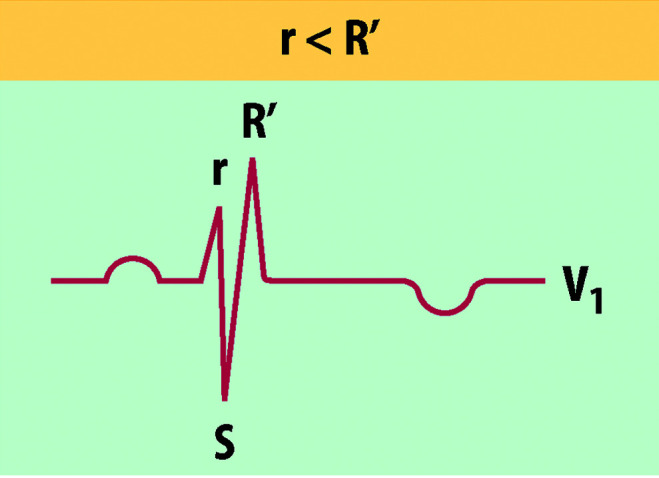
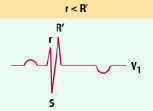
Im EKG findet sich bei größeren Defekten als Ausdruck der rechtsventrikulären Volumenbelastung mit Erregungsausbreitungsstörung eine typische rsRʹ-Konfiguration in der Ableitung V1 (sog. inkompletter Rechtsschenkelblock; Abb. 20.4).
Mittels Echokardiographie können Vorhofseptumdefekte, v. a. bei Kleinkindern, morphologisch sehr gut dargestellt werden. Dabei werden Defekttyp und Defektgröße bestimmt. Mittels Farbdoppler kann ein Shunt nachgewiesen werden. Zusätzlich kann eine Vergrößerung des rechten Vorhofs, des rechten Ventrikels und der Pulmonalarterie als Ausdruck der Volumenbelastung dargestellt werden. Assoziierte Herzfehler wie eine partielle Lungenvenenfehlmündung sollte ausgeschlossen werden. Am besten wird das Vorhofseptum von subcostal visualisiert. Bei Jugendlichen und Erwachsenen ist das Vorhofseptum häufig nur noch mittels transösophagealer Echokardiographie (TEE) gut darstellbar.
Therapie
Kleine, hämodynamisch unbedeutsame Defekte erfordern keine Therapie und sollten nur beobachtet werden. Insbesondere Defekte mit einem Durchmesser <6 mm haben die Tendenz sich spontan zu verschließen. Da mit zunehmendem Lebensalter die Compliance des rechten Ventrikels zunimmt, kann auch das Shuntvolumen im Verlauf zunehmen und so an hämodynamischer Bedeutsamkeit gewinnen. Daher sollten auch hämodynamisch unbedeutsame Defekte regelmäßig kontrolliert werden.
Eine Verschlussindikation besteht wenn sich Zeichen einer rechtsventrikulären Volumenbelastung zeigen, klinische Symptome bestehen oder nach einer paradoxen Embolie.
Cave
ASD I, Sinus-venosus-Defekte und Sinus-coronarius-Defekte verschließen sich nie spontan und können nur operativ verschlossen werden.
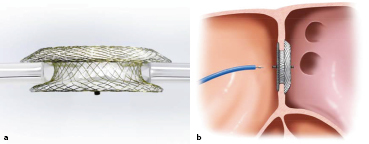
Für die meisten Sekundum-ASD ist ein katheterinterventioneller Verschluss die Therapie der Wahl. Hierbei wird transvenös ein Doppelschirmsystem implantiert, das den Defekt verschließt. Voraussetzung hierfür sind ausreichende Defektränder, damit der Schirm stabil verankert werden kann (Abb. 20.5).

Ist ein katheterinterventioneller Verschluss nicht möglich, kann der ASD mittels Direktnaht oder durch einen Patch operativ verschlossen werden.
Liegen keine anderen zwingenden Gründe vor, werden ASD in der Regel im Vorschulalter verschlossen.
Partielle Lungenvenenfehlmündung
Partielle Lungenvenenfehlmündungen treten selten isoliert auf, sondern häufig in Kombination mit anderen kardialen Anomalien.
Bei der partiellen Lungenvenenfehlmündung drainiert eine oder ein Teil der Lungenvenen (aber nicht alle) anstatt in den linken Vorhof in den rechten Vorhof. Die Hämodynamik der partiellen Lungenvenenfehlmündung entspricht der eines Vorhofseptumdefekts (Abschn. 20.2.1.3). O2-reiches Blut gelangt in den rechten Vorhof. Es ergibt sich eine Volumenbelastung und Dilatation der rechten Herzhöhlen. Sinus-venosus-Defekte gehen anatomisch fast immer mit einer partiellen Lungenvenenfehlmündung einher.
Atrioventrikulärer Septumdefekt (AVSD, syn. AV-Kanal)
Der AVSD hat einen Anteil von ca. 4–5% an allen angeborenen Herzfehlern, und kommt gehäuft bei Patienten mit Trisomie 21 vor (40–50%).
Morphologie und Hämodynamik
Es besteht eine embryonale Entwicklungsstörung des Endokardkissens mit Fehlentwicklung der AV-klappennahen Anteile des Vorhof- und Ventrikelseptums sowie der AV-Klappen. Ein Vorhofseptumdefekt mit fehlendem Vorhofgewebe oberhalb der AV-Klappen ist ein Vorhofseptumdefekt vom Ostium-Primum-Typ (ASD I). Statt Mitral- und Trikuspidalklappe liegt eine gemeinsame angelegte AV-Klappe mit einer variablen Anzahl von Segeln und Öffnungen vor.
Ein partieller AVSD liegt vor, wenn ein ASD vom Ostium-Primum-Typ und eine gemeinsame AV-Klappe mit 2 Öffnungen (je eine zum rechten und linken Ventrikel) bestehen. Beim kompletten AVSD liegt zusätzlich ein Defekt des Ventrikelseptums im Inlet-Septumbereich vor und eine AV-Klappe mit einer Öffnung (Abb. 20.6).

Partieller AVSD: ASD I + gemeinsame AV-Klappe mit 2 Öffnungen.
Kompletter AVSD: ASD I + Inlet-VSD + gemeinsame AV-Klappe mit 1 Öffnung.
Bei einem partiellen AVSD liegt die gleiche Hämodynamik wie bei einem ASD vom Sekundum-Typ vor. Es besteht ein Links-rechts-Shunt auf Vorhofebene mit einer entsprechenden Volumenbelastung des rechten Ventrikels. Eine zusätzliche AV-Klappeninsuffizienz kann die Volumenbelastung noch verstärken.
Bei einem kompletten AVSD liegt ein Links-rechts-Shunt sowohl auf atrialer als auch auf ventrikulärer Ebene vor. Zusätzlich liegt häufig noch eine Insuffizienz der gemeinsamen AV-Klappe vor, welche die Volumenbelastung einer oder beider Ventrikel noch erhöhen kann. Aufgrund des ausgeprägten Shunts besteht eine erheblich vermehrte Lungendurchblutung, die schon im frühen Säuglingsalter zu einer Herzinsuffizienz führen kann. Eine pulmonale Hypertonie entwickelt sich bei einem kompletten AVSD relativ früh. Insbesondere wenn zusätzlich eine Trisomie 21 vorliegt besteht die Gefahr, dass es zu einer frühen irreversiblen pulmonalen Hypertension kommt, die im weiteren Verlauf zu einer Eisenmenger-Reaktion führt. Daher sollte ein kompletter AVSD vor dem 6. Lebensmonat korrigiert werden, insbesondere bei Vorliegen einer Trisomie 21. Eine Zyanose besteht bei erhöhtem pulmonalarteriellen Widerstand oder einer Obstruktion des rechtsventrikulären Ausflusstrakts.
Klinik
Kinder mit partiellem AVSD und nur geringer AV-Klappeninsuffizienz sind im Säuglings- und Kleinkindalter wenig symptomatisch. Die Klinik ähnelt der eines großen Shunts bei ASD II (Abschn. 20.2.1.3).
Beim kompletten AVSD kommt es mit Abfall des Lungengefäßwiderstands rasch zu den typischen Zeichen einer Herzinsuffizienz und evtl. zu rezidivierenden pulmonalen Infekten.
Fehlen bei einem Säugling mit einem kompletten AVSD Herzinsuffizienzzeichen, erfordert dies besondere Aufmerksamkeit. Hier liegt meist bereits eine pulmonale Hypertonie vor, die zwar eine Lungenüberflutung verhindert, aber schon zu einer irreversiblen Schädigung der Lungenstrombahn führen kann.
Diagnostik
Beim partiellen AVSD entspricht der Auskultationsbefund dem eines ASD vom Sekundum-Typ. Es liegt, wenn nicht zusätzlich eine AV-Klappeninsuffizienz vorliegt, eine relative Pulmonalstenose vor mit einem Systolikum mit p.m. über dem 2. ICR links.
Beim kompletten AVSD hört man häufig ein lautes, holosystolisches Geräusch mit Absenken des Lungengefäßwiderstands als Folge eines Links-rechts-Shunts über dem Ventrikelseptumdefekt. Es besteht häufig eine ausgeprägte AV-Klappeninsuffizienz mit hörbarem Systolikum.
Im EKG zeigt sich sowohl beim partiellen als auch kompletten AVSD ein überdrehter Linkstyp, der pathognomonisch für den Defekt ist.
Die Diagnose wird letztendlich echokardiographisch gestellt. Hier kann der Defekt, seine Ausdehnung, die Klappenmorphologie, der Shunt und das Ausmaß der AV-Klappeninsuffizienz dargestellt werden.
Therapie
Beim kompletten AVSD besteht die initiale Therapie in einer medikamentösen Behandlung der Herzinsuffizienz.
Die operative Korrektur sollte im ersten Lebenshalbjahr erfolgen, insbesondere wenn eine Trisomie 21, vorliegt, da hier frühzeitig die Gefahr einer irreversiblen pulmonalen Hypertonie besteht.
Kann das Kind aus verschiedenen Gründen (z. B. niedriges Gewicht, syndromale Grunderkrankung mit weiteren Fehlbildungen) in diesem Zeitraum noch nicht operiert werden oder die Herzinsuffizienz medikamentös nicht kompensiert werden, besteht die Möglichkeit einen operativen Zwischenschritt durchzuführen, indem man ein zentrales pulmonales Banding durchführt. Hierbei wird ein Bändchen um die Pulmonalarterie gelegt und das Lumen eingeengt. Durch diese Maßnahme wird eine artifzielle Pulmonalstenose verursacht und damit eine Lungenüberflutung und Herzinsuffizienz vermieden. Eine Korrektur-OP kann dann auch später als im ersten Lebenshalbjahr erfolgen, ohne dass ein erhöhtes Risiko für eine pulmonale Hypertension besteht.
Die eigentliche Korrekturoperation besteht aus einem Patchverschluss des Vorhof- und Ventrikelseptums und einer Rekonstruktion der AV-Klappen.
Beim partiellen AVSD besteht prinzipiell eine Operationsindikation. Bestehen keine zusätzlichen Herzfehler kann die Korrektur-OP erst im Vorschulalter erfolgen.
Azyanotische Herzfehler ohne Shunt
Aortenisthmusstenose
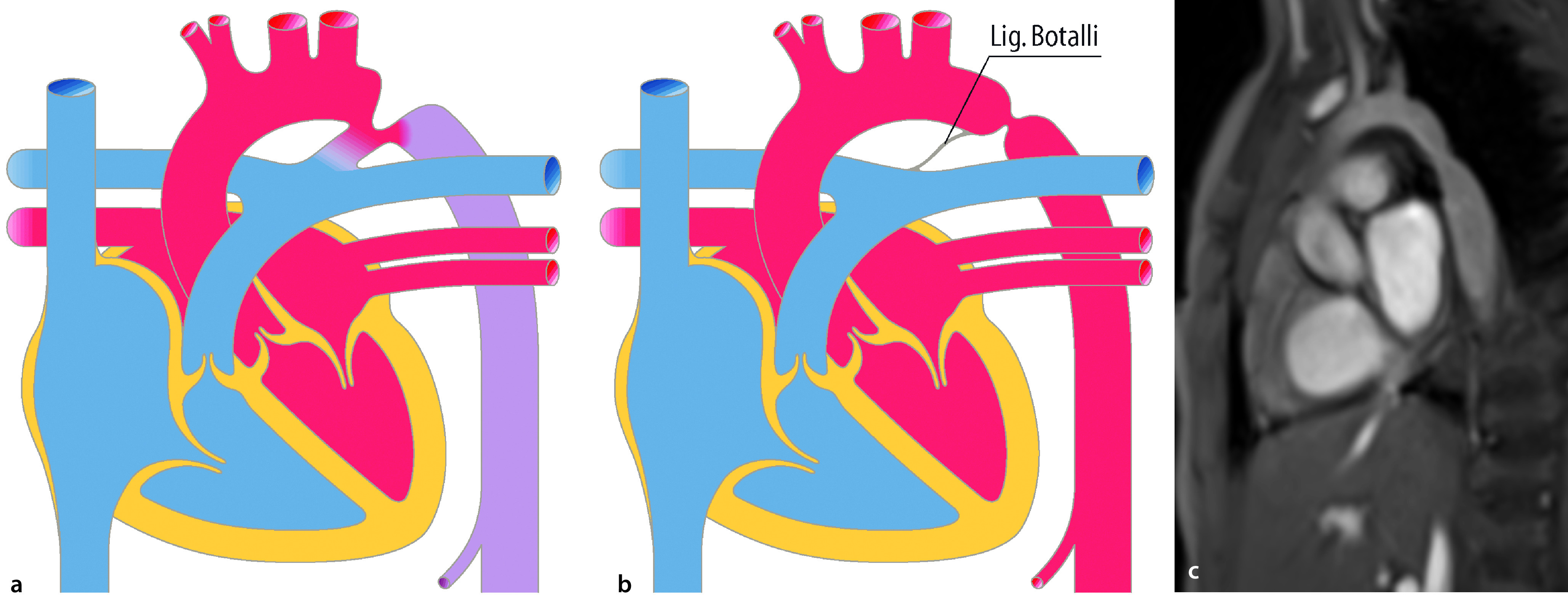
Bei der Aortenisthmusstenose handelt es sich um die Einengung der Aorta im Bereich des Isthmus, also dem Übergang des Aortenbogens zur Aorta descendens (Abb. 20.7). Die relative Häufigkeit der isolierten Aortenisthmusstenose beträgt ca. 5–8% aller angeborenen Herzfehler. Jungen sind etwa doppelt so häufig betroffen wie Mädchen. Das Ullrich-Turner-Syndrom ist in ca. 20–30% mit einer Aortenisthmusstenose assoziiert.
Morphologie und Hämodynamik
Die frühere Einteilung der Aortenisthmusstenosen von prä- und postduktal wurde weitgehend verlassen. Eigentlich liegt immer eine juxtaduktale Aortenisthmusstenose vor (Abb. 20.7), d. h. die Einengung liegt unmittelbar auf Höhe des Ductus arteriosus. Morphologisch gibt es umschriebene Aortenisthmusstenosen und tubuläre Aortenisthmusstenosen. Bei letzterer liegt eine längerstreckige tubuläre Hypoplasie des Bogens vor, evtl. mit zusätzlicher Hypoplasie des Aortenbogens.
Es müssen die kritische Aortenisthmusstenose des Neugeborenen und die weniger ausgeprägte Aortenisthmusstenose, die erst zu einem späteren Zeitpunkt zu Symptomen führt und einer Therapie bedarf, unterschieden werden.
Bei der kritischen Aortenisthmusstenose des Neugeborenen ist die ausreichende Durchblutung der unteren Körperhälfte abhängig vom Offenbleiben des Ductus arteriosus botalli. Über diesen versorgt der rechte Ventrikel über die Pulmonalarterie die untere Körperhälfte mit Blut. Entsprechend ist die O2-Sättigung an den Beinen (postduktale Sättigung), im Vergleich zur Sättigung am rechten Arm (präduktale Sättigung) erniedrigt. Mit Verschluss des Ductus wird die untere Körperhälfte nur noch unzureichend perfundiert. Es kommt zu einer akuten Nachlaststeigerung mit akuter Dekompensation und entsprechenden klinischen Zeichen einer Herzinsuffizienz bis hin zum kardiogenen Schock.
Bei einer weniger ausgeprägten Aortenisthmusstenose besteht eine chronische Nachlaststeigerung mit konsekutiver Hypertrophie des linken Ventrikels. Zwischen der oberen und unteren Körperhälfte besteht ein Blutdruckgradient. Der Blutdruck der oberen Körperhälfte ist hyperton, der Blutdruck in der unteren Körperhälfte entsprechend hypoton. Mit der Zeit entstehen Kollateralkreisläufe.
Klinik
Die klinischen Symptome einer Aortenisthmusstenose variieren je nach Ausprägungsgrad der Stenose. Sie können von der kritischen Aortenisthmusstenose des Neugeborenen bis zur asymptomatischen Aortenisthmusstenose reichen, die erst im Erwachsenenalter diagnostiziert wird.
Entsprechend der Hämodynamik steht bei einer kritischen Aortenisthmusstenose eine rasch progrediente Herzinsuffizienz im Vordergrund (Abschn. 20.1.5). Aufgrund der verminderten Durchblutung der unteren Körperhälfte drohen Niereninsuffizienz, nekrotisierenden Enterokolitis, Azidose und schließlich der kardiogene Schock.
Bei der nicht kritischen Aortenisthmusstenose sind die Leitsymptome eine Hypertonie der oberen Körperhälfte mit evtl. häufigen Kopfschmerzen und Nasenbluten, abgeschwächte Leistenpulse, Wadenschmerzen und eine Claudicatio intermittens.
Die Aortenisthmusstenose kann auch lange asymptomatisch verlaufen und als Zufallsbefund bei der Abklärung eines arteriellen Hypertonus oder nach stattgehabtem Schlaganfall in Folge eines lange bestehenden Hypertonus diagnostiziert werden.
Die Aortenisthmusstenose ist die häufigste, nicht renale Ursache der sekundären Hypertonie.
Diagnose
Führendes Symptom ist die Differenz von Puls und Blutdruck zwischen oberer und unterer Extremität. Trügerischerweise kann ein Blutdruckgradient zwischen oberer und unterer Körperhälfte bei Neugeborenen mit noch offenem Ductus arteriosus fehlen. Eine niedrigere O2-Sättigung der unteren Extremitäten ist zu diesem Zeitpunkt ggf. das einzige klinische Symptom. Das Pulsoxymetriescreening im Rahmen der Vorsorgeuntersuchung U2 ist daher – neben einer sorgfältigen Inspektion – sehr hilfreich, um diese dezenten klinischen Indizien wahrzunehmen.
Auch bei einem älteren Kind oder Erwachsenen muss die Höhe der Blutdruckdifferenz nicht gleichbedeutend mit dem Schweregrad der Stenose sein, da ein gut ausgebildeter Umgehungskreislauf eine relativ gute Durchblutung der unteren Körperhälfte zur Folge haben kann.
Auskultatorisch findet sich evtl. ein Systolikum mit p.m. zwischen den Schulterblättern.
Die Diagnose wird in der Regel echokardiographisch gestellt. Hierbei wird der Aortenisthmus von suprasternal dargestellt. Es findet sich eine Flussbeschleunigung im stenotischen Bereich mit einer typischen diastolischen Ausziehung. Kann der Aortenisthmus nicht gut dargestellt werden, ist auch die Dopplermessung im Truncus coeliacus hilfreich. Eine verminderte systolisch/diastolische Amplitude kann ein Hinweis für eine Aortenisthmusstenose sein.
Mittels MRT ist eine gute Darstellung des Aortenisthmus und der gesamten Aorta möglich, insbesondere bei älteren Kindern oder jungen Erwachsenen ist das MRT hervorragend zur Darstellung der Aortenisthmusregion sowie des gesamten Aortenbogens geeignet.
Therapie
Bei Vorliegen einer kritischen Aortenisthmusstenose besteht eine systemabhängige Ductusperfusion mit Rechts-links-Shunt in der Echokardiographie (Abschn. 20.1.5). Entsprechend muss eine Therapie mit Prostaglandin E1 zum Offenhalten des Ductus arteriosus botalli begonnen werden. Ist der Ductus arteriosus botalli bereits vorschlossen, lohnt es sich trotzdem eine Prostaglandintherapie zu beginnen, da evtl. versprengtes Ductusgewebe im Isthmusbereich noch auf das Protaglandin reagiert und eine Stenose abgemildert werden kann.
Die Therapie besteht im Neugeborenenalter in der operativen Behandlung. Dabei wird die Aortenisthmusstenose reseziert und eine End-zu-End-Anastomose durchgeführt. Bei längerstreckigen Stenosen wird die deszendierende Aorta aufgeschnitten, und mittels Patch erweitert. Eine katheterinterventionelle Therapie im Neugeborenenalter ist nur in Ausnahmefällen indiziert, wenn eine kritische Aortenisthmusstenose vorliegt. Dann wird mittels einer Ballondilatation eine Angioplastie durchgeführt. Da bei Neugeborenen die Rate an Restenosierungen sehr hoch ist, ist die Ballonangioplastie als Überbrückungsmaßnahme bis zur Korrekturoperation oder als Notfallindikation zur Stabilisierung eines kritisch kranken Neugeborenen zu werten.
Jugendliche und Erwachsene werden in der Regel im Herzkatheter mit einer Stentimplantation im stenotischen Bereich behandelt.
Assoziiert mit einer Aortenistmusstenose ist eine arterielle Hypertonie. Je später die Aortenisthmusstenose behandelt wird, desto höher ist das Risiko, dass eine arterielle Hypertonie persistiert. Da es unabhängig vom Eingriff zu Restenosen kommen kann, sollten regelmäßige kardiologische Untersuchungen erfolgen. Obligatorisch ist eine Blutdruckmessung an Arm und Bein. Eine arterielle Hypertonie muss medikamentös frühzeitig angegangen werden. Weiterhin sollte bei Patienten nach operativer Korrektur oder Intervention in regelmäßigen Abständen eine Aneurysmabildung ausgeschlossen werden.
Aortenstenose
Die relative Häufigkeit einer Aortenstenose (AST) liegt bei ca. 4–6% aller angeborenen Herzfehler. Das männliche Geschlecht überwiegt dabei deutlich.
Morphologie und Hämodynamik
Die Einteilung der Aortenstenose orientiert sich an ihrer Lokalisation (Abb. 20.8). Neben der valvulären Aortenstenose, die am häufigsten ist, gibt es die supravalvuläre und subvalvuläre Aortenstenose. Subvalvuläre Aortenstenosen entwickeln sich häufig erst im Verlauf des Lebens oder nach einer Herzoperation. Supravalvuläre Aortenstenosen sind sehr häufig mit einem angeborenen Syndrom assoziiert (z. B. Williams-Beuren-Syndrom).
Eine stenosierte Aortenklappe kann trikuspide, bikuspide oder selten auch monokuspide sein. Gelegentlich findet sich eine dysplastische Klappe mit verdickten und myxomatös verquollenen Segeln. Die rein bikuspide Aortenklappe ohne Stenose ist relativ häufig und findet sich in ca. 0,5–2% der Gesamtbevölkerung.
Der Schweregrad einer Aortenstenose wird anhand des Druckgradienten festgelegt:
milde Stenose: max. systolischer Druckgradient in der Echokardiographie <40 mmHg,
mittelschwere Stenose: Druckgradient 40–60 mmHg,
schwere Stenose: Druckgradient ab >60 mmHg.
Abhängig vom Druckgradienten über der Klappe entsteht eine konzentrische Myokardhypertrophie des linken Ventrikels. Insbesondere bei einer hochgradigen Aortenklappenstenose im Neugeborenenalter kann der linke Ventrikel dilatieren und in seiner systolischen Funktion eingeschränkt sein.
Klinik
Es wird zwischen der kritischen oder hochgradigen Aortenklappenstenose unterscheiden, die bereits im Neugeborenen- und Säuglingsalter zu Symptomen führt, sowie der weniger ausgeprägten Aortenklappenstenose, die bei Kindern in den ersten Lebensjahren meist asymptomatisch verläuft.
Bei der kritischen Aortenklappenstenose ist das Neugeborene kurz nach der Geburt symptomatisch und schwer krank. Das Krankheitsbild ähnelt einem septischen Krankheitsbild mit Herzinsuffizienzsymptomen bis hin zur Schocksymptomatik (Abschn. 20.1.5).
Bei milder oder mäßiger Aortenklappenstenose sind die Kinder meist asymptomatisch. Bei Vorliegen eines höheren Gradienten können unter körperlicher Belastung synkopale Zustände mit dem Risiko eines plötzlichen Herztods auftreten.
Eine Aortenklappenstenose ist immer eine progrediente Erkrankung, d. h. mit zunehmendem Alter kommt es zu einer Verschlechterung der Klappeneigenschaften. Besteht ein Defekt an der Aortenklappe, so bedeutet dies, dass diese Klappe mit hoher Wahrscheinlichkeit im Laufe des Lebens ersetzt werden muss.
Diagnostik
Auskultatorisch ist ein typisches Systolikum mit p.m. über dem 2. ICR rechts mit Fortleitung in die Karotiden zu hören. Das EKG ist bei der Aortenklappenstenose nicht wegweisend, kann jedoch auf eine linksventrikuläre Hypertrophie verweisen. Die Diagnose wird echokardiographisch gestellt. Hier kann der Schweregrad der Stenose als auch die Morphologie der Klappe dargestellt werden, begleitende Probleme wie eine Aortenklappeninsuffizienz, eine Hypertrophie des linken Ventrikels und eine mögliche Funktionseinschränkung können ebenso beurteilt werden.
Bei der kritischen Aortenklappenstenose besteht eine hochgradig eingeschränkte systolische Funktion. Der Ventrikel kann kein ausreichendes HZV mehr aufbauen, der Gradient über die Klappe ist aufgrund der schlechten Funktion falsch niedrig und somit nicht aussagekräftig.
Therapie
Besteht eine kritische Aortenklappenstenose sollte versucht werden, den Ductus arteriosus mittels Prostaglandin-E1-Infusion offen zu halten bzw. wieder zu eröffnen.
Durch die Wiederherstellung der fetalen Kreislaufphysiologie kann der rechte Ventrikel die Systemperfusion mittels Rechts-links-Shunt über den offenen Ductus arteriosus aufrechterhalten Eine frühe interventionelle Behandlung mittels Ballondilatation der Aortenklappe ist notwendig und stellt bei der kritischen Aortenklappenstenose eine Notfallindikation dar.
Weitere Indikationen zur katheterinterventionellen Therapie sind symptomatische Patienten, Patienten mit einer schweren Aortenklappenstenose (systolischer Gradient >60 mmHg) sowie Patienten mit eingeschränkter Ventrikelfunktion.
Die Ballonvalvuloplastie ist die Therapie der Wahl bei valvulären Aortenklappenstenosen ohne begleitende Klappeninsuffizienz.
Die Ergebnisse der Ballonvalvuloplastie sind vergleichbar mit denen einer chirurgischen Rekonstruktion (Kommisurotomie). Ein operativer Ansatz wird bei Vorliegen einer Aortenklappeninsuffizienz bevorzugt. Lässt sich die Aortenklappe chirurgisch nicht mehr rekonstruieren, kommt die Implantation einer Kunstklappe, eines Homografts oder bevorzugt eine Ross-Operation in Frage. Bei der Ross-Operation wird die körpereigene Pulmonalklappe in Aortenposition implantiert, und die Pulmonalklappe durch eine biologische Klappe (Homograft oder Xenograft) ersetzt. Vorteil der Ross-Operation ist, dass die neue Aortenklappe, die ein Autograft ist, deutlich länger haltbar ist als ein Xeno- oder Homograft in Aortenposition und evtl. mitwächst. Das Homo-/Xenograft in Pulmonalisposition muss bei Bedarf durch eine erneute Operation oder eine Herzkatheterintervention ersetzt werden.
Pulmonalstenose
Die Pulmonalklappenstenose (PST) ist die häufigste angeborene Klappenstenose und macht ca. 7% aller angeborenen Herzfehler aus.
Morphologie und Hämodynamik
Die Einteilung der Pulmonalstenose orientiert sich an ihrer Lokalisation (Abb. 20.9). Neben der valvulären Pulmonalstenose, die am häufigsten ist, gibt es die supravalvuläre und die subvalvuläre Pulmonalstenose. Subvalvuläre Pulmonalstenosen kommen als isolierte Fehlbildung selten vor, häufig sind sie mit einem Malalignment-VSD (z. B. wie bei der Fallot-Tetralogie) assoziiert.

Supravalvuläre Pulmonalstenosen können als periphere Pulmonalstenosen, als isolierte Stenosen der zentralen Pulmonalarterie oder als multiple periphere Stenosen vorkommen. Sie sind sehr häufig mit einem angeborenen Syndrom assoziiert (Alagille-Syndrom, Williams-Beuren-Syndrom, Noonan-Syndrom).
Im Falle einer Pulmonalklappenstenose sind die Kommissuren entweder einfach verschmolzen, oder nicht angelegt. Dysplastische Klappen bestehen aus verdicktem, ungleichmäßigem Gewebe, häufig von einer Dysplasie des Klappenanulus begleitet.
Der Schweregrad einer Pulmonalstenose wird anhand des Druckgradienten festgelegt:
milde Stenose: max. systolischer Druckgradient in der Echokardiographie <40 mmHg,
mittelschwere Stenose: Druckgradient 40–60 mmHg,
schwere Stenose: Druckgradient ab >60 mmHg.
Durch die Pulmonalstenose kommt es zu einer rechtsventrikulären Druckbelastung mit Ausbildung einer rechtsventrikulären Hypertrophie. Da der rechte Ventrikel im Rahmen der fetalen Kreislaufverhältnisse an höhere Drücke adaptiert und somit „trainiert“ ist, führt eine Pulmonalstenose postpartal selten zu Symptomen. Nimmt eine Stenose erst im Verlauf des Lebens zu, ist der rechte Ventrikel weniger daran adaptiert. In diesen Fällen besteht häufig eine Belastungsintoleranz, da der rechte Ventrikel aufgrund der Obstruktion durch die Pulmonalklappe das HZV nur begrenzt steigern kann.
Ein Sonderfall ist die kritische Pulmonalstenose des Neugeborenen: Hier liegt eine hochgradige Pulmonalstenose vor, die zu einer lebensbedrohlichen Situation führt. Auf Grund eines Rechts-links-Shunts auf Vorhofebene kommt es zu einer Zyanose. Der rechtsventrikuläre Druck ist mindestens systemisch. Die Lungendurchblutung ist in diesem Fall ductusabhängig, entsprechend benötigen diese Patienten eine Prostaglandin-E1-Infusion, um eine ausreichende Lungendurchblutung durch einen Links-rechts-Shunt zu gewährleisten.
Klinik
Aufgrund der oben beschriebenen Symptomatik sind Kinder mit einer isolierten Pulmonalklappenstenose in der Regel asymptomatisch.
Bei einer höhergradigen Pulmonalstenose kann eine eingeschränkte körperliche Belastbarkeit vorliegen.
Diagnostik
Der Defekt wird aufgrund des Auskultationsbefunds meistens kurz nach der Geburt gestellt. Es findet sich ein typisches systolisches spindelförmiges Austreibungsgeräusch mit p.m. im 2. ICR links mit einem typischen Ejektionsgeräusch. Das EKG kann bei der Diagnose hilfreich sein. Bei ausgeprägter rechtsventrikulärer Hypertrophie findet sich typischerweise ein positiv konkordantes T in V1. Physiologischerweise finden sich bei Säuglingen bis zum Schulalter negative T-Wellen in den rechtspräkordialen Ableitungen. Liegt ein positiver Kammerkomplex mit einer positiven T-Welle vor, spricht man von einer positiven Konkordanz. Dies ist in diesem Alter typisch für eine Rechtsherzhypertrophie.
Die Diagnose wird echokardiographisch gestellt. Dabei wird die Stenoselokalisation dargestellt. Aufgrund der dopplersonographisch abgeschätzten systolischen Gradienten, lässt sich die Pulmonalstenose in verschiedene Schweregrade einteilen und die Klappenmorphologie kann beurteilt werden.
Therapie
Subvalvuläre Stenosen werden in der Regel chirurgisch therapiert. Supravalvuläre Stenosen werden häufig im Herzkatheter mit Ballondilatation und Stentimplantation behandelt.
Handelt es sich um eine isolierte Pulmonalklappenstenose, besteht die Therapie typischerweise in einer Herzkatheteruntersuchung mit einer Ballonvalvuloplastie. Die Indikation zur Katheteruntersuchung wird ab einem Gradienten von >60 mmHg gestellt. Bei dysplastischen Pulmonalklappen ist die Erfolgsrate gering. Liegen nur verschmolzene Kommissuren vor, kann mit einem sehr guten Ergebnis gerechnet werden. Postinterventionell kommt es idealerweise zu einer geringen Insuffizienz mit einer deutlichen Reduktion des Stenosegradienten. Bei dysplastischer Pulmonalklappe ist die chirurgische Kommissurotomie indiziert.
Zyanotische Herzfehler
Zyanotische Herzfehler mit verminderter Lungenperfusion
Fallot-Tetralogie
Die Fallot-Tetralogie (TOF) hat einen relativen Anteil von 5–6% aller angeborenen Herzfehler und stellt den häufigsten zyanotischen Herzfehler dar.
Morphologie und Hämodynamik
Entsprechend der Erstbeschreibung durch Fallot ist die TOF durch das Zusammenkommen folgender Merkmale definiert (Abb. 20.10):
Ein großer Ventrikelseptumdefekt (Malalignment-VSD),
eine den VSD überreitende Aorta,
eine valvuläre und/oder infundibuläre Pulmonalstenose mit unterschiedlich ausgeprägter Hypoplasie der zentralen Pulmonalarterien sowie
eine Hypertrophie des rechten Ventrikels.

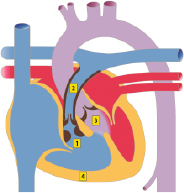
Häufig liegt auch noch ein ASD vor. Dann spricht man von einer Fallot-Pentalogie. In ca. 30% findet sich ein rechtsseitiger Aortenbogen.
Eine Assoziation mit einer Mikrodeletion 22q11 ist häufig, insbesondere bei rechtsseitigem Aortenbogen.
Die Hämodynamik bei einer TOF kann sehr unterschiedlich ausfallen und ist im Wesentlichen durch den Grad der Obstruktion des rechtsventrikulären Ausflusstrakts und der Hypoplasie der Pulmonalarteriengefäße bedingt. Im Prinzip liegt eine Fehlbildung des infundibulären Septums vor. Das Septum deviiert nach rechts, und führt dadurch zu einer Obstruktion des rechtsventrikulären Ausflusstrakts mit konsekutiver Hypoplasie der Pulmonalklappe und einer Verlagerung der Aortenwurzel. Die Aorta „reitet“ nun über dem Defekt. Zwischen beiden Ventrikel besteht über den großen Malalignement-VSD ein Druckausgleich. Dies führt zur rechtsventrikulären Hypertrophie. Bei ausgeprägter Obstruktion des rechtsventrikulären Ausflusstrakts kommt es zu einem überwiegenden Rechts-links-Shunt mit deutlicher Zyanose. Hierbei fließt das Blut – dem geringsten Widerstand folgend – vom rechten Ventrikel unter Umgehung der Pulmonalarterie über den VSD direkt in die Aorta. Das Ausmaß der rechtsventrikulären Ausflusstraktobstruktion bestimmt den Schweregrad der Zyanose. Liegt nur eine geringgradige Pulmonalstenose vor, ist klinisch keine Zyanose erkennbar. In diesem Fall spricht man von einem „Pink Fallot“.
Klinik
Entsprechend der Hämodynamik sind die klinischen Symptome abhängig vom Obstruktionsgrad des rechtsventrikulären Ausflusstrakts. Die Zyanose ist im Neugeborenenalter häufig noch nicht sehr ausgeprägt, nimmt aber in den ersten Lebenswochen zu. Bei Vorliegen einer hochgradigen Pulmonalstenose kommt es mit dem Verschluss des Ductus arteriosus botalli in den ersten Lebenstagen zu schweren (auch lebensbedrohlichen) Zyanosen. In diesem Fall muss die Lungenperfusion durch Wiedereröffnung des Ductus arteriosus mit Hilfe von Prostaglandin E1 gewährleistet werden. Es handelt sich dann um einen kritischen Herzfehler mit ductusabhängiger Pulmonalperfusion.
Bei ausgeprägter infundibulärer Stenose kann es zu sog. hypoxämischen Anfällen kommen. Es handelt sich dabei um einen anfallsartigen Spasmus der hypertrophierten Infundibulummuskulatur, der v. a. durch sympathikotone Reaktionen ausgelöst wird, z. B. beim Schreien und bei Unruhezuständen. Mit der plötzlichen Zunahme der Obstruktion im rechtsventrikulären Ausflusstrakt kommt es zu einer starken Zunahme der Zyanose, die auch zu einem Bewusstseinsverlust führen kann. Früher waren hypoxämische Anfälle eine häufige Todesursache bei TOF-Patienten.
Liegt eine balancierte Situation vor, also eine moderate Obstruktion des rechtsventrikulären Ausflusstrakts, sind die Kinder bis auf eine mäßige Zyanose relativ symptomfrei. Sie haben keine Herzinsuffizienz und gedeihen in der Regel normal.
In Industrieländern wird die TOF typischerweise im Säuglingsalter korrigiert. In Entwicklungsländern ist eine Korrektur häufig erst spät oder überhaupt nicht möglich. Entsprechend findet man bei diesen älteren Kindern, die operativ nicht korrigiert worden sind, die typischen Zeichen einer chronischen Zyanose mit Trommelschlegelfingern und -zehen sowie Uhrglasnägel. Diese Kinder nehmen insbesondere nach körperlicher Belastung eine typische „Hockstellung“ ein. Dies war früher auch in unseren Breiten pathognomonisch für die TOF. Durch die Hockstellung wird der systemische Gefäßwiderstand erhöht und dadurch der Rechts-links-Shunt vermindert.
Diagnostik
Es besteht ein lautes Herzgeräusch bedingt durch die Pulmonalstenose mit einem rauhen systolischen Herzgeräusch mit p.m. über dem 2./3. ICR links. Leitsymptom ist, soweit kein „Pink Fallot“ vorliegt, die Zyanose.
Echokardiographisch zeigen sich die klassischen Befunde mit dem großen subaortal gelegenen Malalignment-VSD, der überreitenden Aorta, die rechtsventrikuläre Ausflusstraktobstruktion mit (sub)valvulärer Pulmonalstenose sowie die rechtsventrikuläre Hypertrophie. In der Echokardiographie sollten die Größe der Pulmonalklappe und die mögliche Hypoplasie der Pulmonalarterien eingehend dargestellt werden. Der Aortenbogenverlauf (links- oder rechtsdeszendierend) sowie weitere mögliche Defekte werden ebenfalls erfasst. Dopplersonographisch kann der Druckgradient im rechtsventrikulären Ausflusstrakt erfasst werden.
Therapie
Liegt eine balancierte Situation vor, also eine moderate Obstruktion des rechtsventrikulären Ausflusstraktes mit einer mäßig ausgeprägten Zyanose (transkutane Sättigung >85%) besteht zunächst kein Handlungsbedarf.
Liegt bereits eine ausgeprägte Zyanose beim Neugeborenen vor, muss von einer ductusabhängigen Pulmonalperfusion ausgegangen werden. Entsprechend besteht die initiale Therapie in der Infusion von Prostaglandin E1 zum Aufhalten oder Wiedereröffnen des Ductus arteriosus botalli.
Bei Kindern mit hochgradiger Infundibulumstenose und zunehmender Zyanose kann eine β-Blockertherapie, z. B. mit Propranolol zur Prophylaxe des Infundibulumspasmus vorgenommen werden, um einen hypoxämischen Anfall zu verhindern.
Im Falle eines hypoxämischen Anfalls kann als erste Maßnahme ein sog. „Taschenmessergriff“ durchgeführt werden, d. h. das Knie des Kindes gegen seine Brust gepresst werden, um den Systemwiderstand zu erhöhen um damit den Rechts-links-Shunt zu vermindern.
Akuttherapie des hypoxämischen Anfalls
Sofortige Sedierung, z. B. Morphin (0,1 mg/kgKG i.v.), Steigerung des Widerstands im Körperkreislauf (Pressen des gebeugten Knies des Kindes gegen die Brust), evtl. Infusion von Vasokonstriktoren (z. B. Noradrenalin). Volumenbolus 20–40 ml/kgKG, β-Blocker. Zur Rezidivprophylaxe ist eine Therapie mit einem β-Blocker p.o. erforderlich (z. B. Propranolol 2–3 mg/kgKG/Tag in drei Einzeldosen).
Das Therapieziel bei der TOF ist die operative Korrektur, die heute in der Regel zwischen dem 3. und 6. Lebensmonat durchgeführt wird. Besteht eine hochgradige Pulmonalstenose mit hypoplastischem Pulmonalgefäßsystem, die mit einer hochgradigen Zyanose einhergeht, kann bei Neugeborenen oder jungen Säuglingen (<3Monate) ein aortopulmonaler Shunt (ein sog. modifizierter Blalock-Taussig-Shunt) in Form eines Gore-Tex-Tubes zwischen rechter A. subclavia und rechter Pulmonalarterie geschaffen werden. Die Korrektur-OP besteht im Verschluss des Ventrikelseptumdefekts mittels Patch, der Muskelresektion der infundibulären Pulmonalstenose sowie der operativen Korrektur der Pulmonalklappe in Form einer Kommissiurotomie oder transanulären Patcherweiterung.
Cave
Positiv inotrope Medikamente sind bei der TOF kontraindiziert, da sie eine Infundibulumstenose verstärken können. Ebenso sollten Diuretika nicht eingesetzt werden, da ein Volumenmangel ebenfalls einen hypoxämischen Anfall induzieren kann.
Prognose
Die Prognose ist abhängig vom Schweregrad der TOF und insbesondere vom Ausprägungsgrad der rechtsventrikulären Ausflusstraktobstruktion. Ist das Pulmonalgefäßsystem gut entwickelt und besteht postoperativ nur eine geringe Pulmonalklappeninsuffizienz sowie keine höhergradige Stenose, besteht eine gute Prognose mit einer geringen Wahrscheinlichkeit für die Notwendigkeit weiterer Operationen.
Bei Vorliegen eines hypoplastischen Lungengefäßsystems mit transanulärer Patcherweiterung ist es wahrscheinlich, dass im weiteren Verlauf mehrere Operationen und Katheterinterventionen notwendig werden, um evtl. Stenosen zu behandeln oder die Pulmonalklappe mittels Homo-/Xenograft zu ersetzen.
Heutzutage bietet sich als Alternative zum operativen Klappenersatz der perkutane Klappenersatz mittels Herzkatheter an. Bei geeigneter Anatomie kann der perkutane Klappenersatz in der Regel ab einem Gewicht von 25 kg durchgeführt werden (Abb. 20.11, Abb. 20.12).

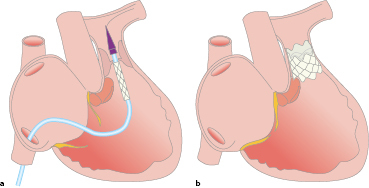
Wichtige Spätkomplikationen bei der TOF sind v. a. Herzrhythmusstörungen. Risikofaktoren für Rhythmusstörungen sind eine schlechte rechtsventrikuläre Funktion und ein verbreiterter Kammerkomplex im EKG (>180 ms). Ventrikuläre Arrhythmien gehen mit einem erhöhten Risiko für einen plötzlichen Herztod einher. Eine lebenslange kardiologische Nachsorge ist auch Jahre nach der Korrektur-OP erforderlich.
Pulmonalatresie mit VSD
Die Pulmonalatresie mit Ventrikelseptumdefekt kann praktisch als Extremform der TOF bezeichnet werden. Durch die komplette Unterbrechung des Blutflusses vom rechten Ventrikel in die Pulmonalarterie besteht eine ductusabhängige Lungendurchblutung. Entsprechend der Morphologie besteht schon beim Neugeborenen eine Zyanose, die mit zunehmender Ductusenge aggraviert. Die Therapie besteht bei Neugeborenen in der Prostaglandin-E1-Infusion zur Offenhaltung des Ductus. Je nach Zentrum wird dann, als zunächst palliativer Schritt, ein aortopulmonaler Shunt entweder operativ oder interventionell mittels Stentimplantation in den Ductus arteriosus geschaffen. Eine operative Korrektur mit Verschluss des Ventrikelseptumdefekts und Einsatz eines klappentragenden Conduits, also einer klappentragenden Kunststoffrohrs, zwischen rechtem Ventrikel und Pulmonalarterie, wird bei entsprechend ausreichender Größe der Pulmonalgefäße im Laufe des ersten Lebenshalbjahres durchgeführt.
Zyanotische Herzfehler mit vermehrter Lungendurchblutung
Komplette Transposition der großen Arterien (d-TGA)
Die Häufigkeit der kompletten Transposition der großen Arterien (d-TGA) liegt bei ca. 3–4%. Es handelt sich um den zweithäufigsten zyanotischen Herzfehler.
Morphologie und Hämodynamik
Bei der TGA sind die großen Arterien vertauscht. Die Aorta entspringt aus dem rechten Ventrikel, der Pulmonalarterie aus dem linken Ventrikel. Es liegt also eine ventrikuloarterielle Diskordanz und eine atrioventrikuläre Konkordanz vor (Abb. 20.13). Die Aorta liegt dabei anterior und rechts der Pulmonalarterie. Es liegt also eine Dextroposition der Aorta vor, daher d(extro)-TGA. Die Hämodynamik der d-TGA charakterisiert sich durch zwei parallel – und nicht hintereinander – geschaltete Kreisläufe. Ein Überleben ist nur möglich, wenn ein ausreichender Austausch zwischen den beiden Kreisläufen erfolgen kann. Dies sind bei der einfachen d-TGA (sog. simple TGA = Transposition der großen Arterien ohne weitere komplexe Fehlbildungen) eine intraatriale Kommunikation wie PFO oder ASD und der Ductus arteriosus botalli.

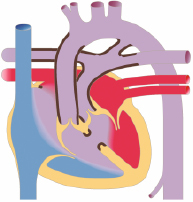
Die Aorta entspringt aus dem rechten Ventrikel. Die Pulmonalarterie entspringt aus dem linken Ventrikel. Der rechte Ventrikel versorgt also den Systemkreislauf, während der linke Ventrikel den Lungenkreislauf versorgt. Ist der ASD, bzw. das offene Foramen ovale zu klein, kann nicht ausreichend O2-reiches Blut vom linken zum rechten Vorhof und somit in den Körperkreislauf gelangen. Daher kommt es bei restriktiver interatrialer Kommunikation zu einer ausgeprägten Zyanose.
Klinik
Leitsymptom ist eine Zyanose, die bereits wenige Stunden nach der Geburt auffällt. Besteht ein großer Vorhofseptumdefekt und ist der Ductus arteriosus botalli noch offen, ist die Zyanose geringer ausgeprägt. Bei einer kleinen Vorhofseptumlücke und zunehmendem Verschluss des Ductus arteriosus kommt es zu einer schweren Zyanose. Diese Zyanose lässt sich durch O2-Gabe oder Intubation mit Beatmung nicht verbessern.
Bei einem zyanotischen Neugeborenem ohne pulmonale Ursache und in relativ guten, vitalen Allgemeinzustand sollte in erster Linie an eine TGA gedacht werden.
Diagnostik
Bei der TGA ohne zusätzliche kardiale Fehlbildungen ist normalerweise kein Herzgeräusch auskultierbar. Die Verdachtsdiagnose wird aufgrund der Klinik gestellt und echokardiographisch bestätigt. In der Echokardiographie kann die TGA, eine evtl. Restriktion der interatrialen Kommunikation und der Ductus arteriosus botalli dargestellt werden. Im Thoraxröntgenbild zeigt sich eine charakteristische Konfiguration, die eiförmig mit schmalem Gefäßband imponiert.
Therapie
Sobald der Verdacht auf eine Transposition der großen Arterien besteht, sollte eine Infusion mit Prostaglandin E1 begonnen werden. Besteht eine restriktive interatriale Kommunikation muss zeitnah eine Ballonatrioseptostomie (Rashkind-Manöver) durchgeführt werden. Dabei wird über die Nabelvene oder die V. femoralis, ein Ballonkatheter eingeführt. Dieser wird über das Foramen ovale in den linken Vorhof vorgeschoben, dann mit Flüssigkeit gefüllt und kräftig zurückgezogen. Dadurch wird das Vorhofseptum eingerissen, und die Vorhoflücke deutlich vergrößert. Dieser Eingriff ist in der Regel auf der Intensivstation unter echokardiographischer Kontrolle möglich und muss nicht im Herzkatheterlabor durchgeführt werden.
Bei einfacher Transposition der großen Arterien erfolgt die Korrektur-OP in Form der arteriellen Switch-OP. Dabei werden Aorta und Pulmonalarterie abgesetzt und umgesetzt. Zusätzlich müssen die Koronararterien mit einer Gefäßmanschette ausgeschnitten und in die Neoaorta eingesetzt werden. Die arterielle Switch-Operation sollte in den ersten Lebenstagen, bzw. in den ersten zwei Lebenswochen, durchgeführt werden, da ansonsten der linke Ventrikel nach Abfall des Lungenwiderstands nicht mehr ausreichend für eine Funktion als Systemventrikel trainiert ist.
Bei komplexen Formen der TGA werden u. a. folgende Operationen durchgeführt: Rastelli-Operation, Translokationsoperation, Damus-Kaye-Stansel-Operation und bis in die Mitte der 1980er Jahre die Vorhofumkehroperation nach Mustard oder Senning. (Ausführliche Beschreibungen siehe Lehrbücher der Kinderkardiologie).
Prognose und Verlauf
Unbehandelt versterben 90% dieser Kinder im ersten Lebensjahr. Bei einer simplen TGA nach arterieller Switch-OP ist die Prognose sehr gut, man geht von einer normalen Lebenserwartung aus. Die Komplikationsrate im weiteren Verlauf liegt bei 5–10%. Hierbei sind v. a. Koronararterienstenosen, Stenosen der Pulmonalarterien, die durch das Umsetzen bei der Operation entstehen, zu nennen. Zusätzlich sieht man häufig eine Dilatation der Aortenwurzel mit Entwicklung einer Aortenklappeninsuffizienz.
Totale Lungenvenenfehlmündung
Bei der totalen Lungenvenenfehlmündung (TAPVR) liegt eine fehlerhafte Einmündung aller Lungenvenen vor. Die Lungenvenen drainieren in den rechten statt in den linken Vorhof oder in ein venöses Gefäß, das mit dem rechten Vorhof in Verbindung steht. Der Anteil an angeborenen Herzfehlern beträgt ca. 1%.
Morphologie und Hämodynamik
Die TAPVR wird im Wesentlichen in folgende Formen eingeteilt:
TAPVR vom suprakardialen Typ (55%; Abb. 20.14a): Die Lungenvenen drainieren hier in einen gemeinsamen Konfluenz, der hinter dem linken Vorhof liegt und über eine nach kranial verlaufende Vene (V. verticalis) in die V. anonyma mündet. Über diese strömt das Blut dann über die obere Hohlvene in den rechten Vorhof.
TAPVR vom kardialen Typ (30%; Abb. 20.14b): Die Lungenvenen münden, entweder mit einem kurzem gemeinsamen Stamm oder separat, in den rechten Vorhof oder in den Sinus coronarius.
TAPVR vom infrakardialer Typ (ca. 13%; Abb. 20.14c): Die Lungenvenen münden in einen Konfluenz, der über eine Vene, die hinter dem Herzen nach kaudal verläuft, durch das Zwerchfell tritt und in die Vv. portae oder den Ductus venosus eintritt und von dort in die untere Hohlvene.
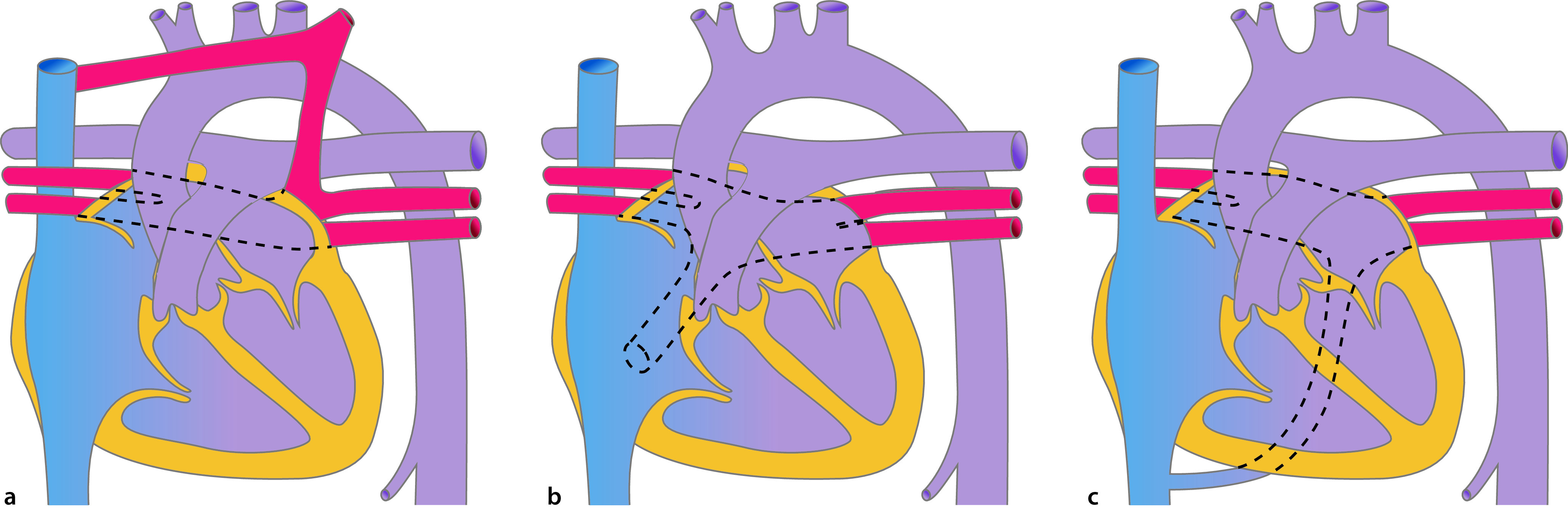
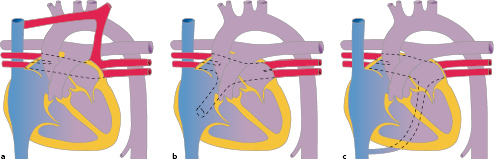
Gemischte Formen sind selten und haben häufig eine schlechtere Prognose. Zusätzlich kann noch eine Lungenvenenobstruktion vorliegen.
Das gesamte Blut des Lungenvenensystems gelangt über unterschiedliche Gefäßverbindungen in den rechten Vorhof. Es liegt also ein Links-rechts-Shunt mit Volumenbelastung der rechtsseitigen Herzhöhlen sowie des Lungenkreislaufs vor. Das Lungenvenenblut kann nur durch einen ASD auf die linke Seite gelangen, d. h. hier gelangt sowohl systemvenöses als auch pulmonalvenöses Blut über den ASD auf die linke Seite und somit in den Körperkreislauf. Es liegt also Mischblut vor.
Klinik
Symptome einer TAPVR sind abhängig davon, ob eine pulmonalvenöse Obstruktion vorliegt oder nicht.
Bei einer TAPVR ohne Lungenvenenobstruktion sind die Patienten oft relativ asymptomatisch. Aufgrund der pulmonalen Rezirkulation ist die Zyanose meistens nur moderat mit transkutanen Sättigungen um 90%. Im Verlauf stehen Herzinsuffizienzzeichen im Vordergrund.
Patienten mit einer TAPVR mit Lungenvenenobstruktion sind schwer erkrankt mit ausgeprägter Zyanose und Dyspnoe, die sich bereits innerhalb der ersten Lebensstunden entwickeln kann. Zusätzlich entwickelt sich bei ausgeprägter Lungenvenenobstruktion schnell ein Lungenödem. Klinisch kann das Krankheitsbild einem ausgeprägten Atemnotsyndrom ähneln und damit die Diagnosestellung verzögern.
Diagnose
Der Auskultationsbefund ist unspezifisch, und nicht richtungsweisend. Im Thoraxröntgenbild zeigt sich bei ausgeprägter Lungenvenenobstruktion eine deutlich vermehrte retikuläre Lungenzeichnung, evtl. mit Zeichen der Lungenvenenstauung und eines Lungenödems. Beim suprakardialen Typ der TAPVR findet sich eine sog. Schneemannform des Herzens, die pathognomonisch für diesen Herzfehler ist. Die Diagnosestellung erfolgt ansonsten in den meisten Fällen echokardiographisch. Auffällig sind ein kleiner linker Vorhof sowie ein deutlich vergrößerter rechter Vorhof und Ventrikel und ein Rechts-links-Shunt auf Vorhofebene. Teils lässt sich hinter dem linken Vorhof ein Sammelgefäß darstellen. Bei der TAPVR vom suprakardialen Typ findet sich hier ein deutlicher Fluss in eine dilatierte obere Hohlvene. Bei einer TAPVR in den Koronarsinus findet sich dieser dilatiert. Bei einer TAPVR vom infrakardialen Typ können sich dilatierte venöse Gefäße in der Leber finden.
Der Nachweis von Lungenvenen kann schwierig sein. Insbesondere bei kleinem linken Vorhof und vergleichsweise großem rechten Vorhof und rechtem Ventrikel sollten die Lungenvenen dargestellt werden. Ist dies echokardiographisch nicht ausreichend möglich, kann eine Herzkatheteruntersuchung oder eine MRT-Untersuchung des Herzens erwogen werden.
Therapie
Besteht keine Lungenvenenobstruktion, steht die konservative Therapie der möglicherweise auftretenden Herzinsuffizienz bis zur operativen Korrektur zunächst im Vordergrund. Die Korrekturoperation sollte innerhalb der ersten drei Lebensmonate durchgeführt werden. Besteht eine restriktive Vorhofkommunikation, kann mittels eines Herzkatheters ein Rashkind-Manöver durchgeführt werden, um einen ausreichenden Shunt auf Vorhofebene zu ermöglichen. Eine TAPVR mit Lungenvenenobstruktion stellt eine kritische Situation dar. Eine umgehende Operation ist indiziert.
Bei der Korrekturoperation ist das Ziel den Lungenvenenkonfluenz breitbasig mit dem linken Vorhof über eine weite Anastomose zu verbinden.
Prognose
Bestehen keine assoziierten Fehlbildungen und besteht keine Lungenvenenobstruktion, ist die körperliche Belastbarkeit und die Prognose sehr gut. Der postoperative Verlauf kann durch neu oder wieder auftretende pulmonalvenöse Obstruktionen kompliziert werden, und damit auf lange Sicht die Prognose verschlechtern.
Truncus arteriosus communis
Der Truncus arteriosus communis (TAC) hat nur einen Anteil von etwa 1% aller angeborenen Herzfehler und ist häufig (70%) mit einer Mikrodeletion 22q11 assoziiert.
Morphologie und Hämodynamik
Beim TAC ist die Trennung zwischen Aorta und Pulmonalarterie unvollständig geblieben, d. h. es entspringt nur ein großes arterielles Gefäß mit einer Taschenklappe (Truncusklappe) aus beiden Ventrikeln. Zusätzlich liegt praktisch immer ein großer Malalignment-VSD vor, der Truncus überreitet sozusagen den Ventrikelseptumdefekt, und versorgt sowohl den systemischen als auch den pulmonalen Kreislauf (Abb. 20.15). Die Truncusklappe ist häufig dysmorph verdickt, weist eine variable Anzahl von Taschen auf und ist häufig insuffizient. Ein rechter Aortenbogen ist häufig.

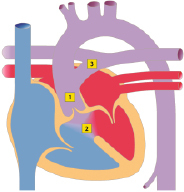
Es gibt unterschiedliche anatomische Variationen des TAC. Am häufigsten entspringen Aorta und Pulmonalarterie aus einem gemeinsamen Gefäß oder Stamm. Die Pulmonalarterie zweigt sich dann kurz nach ihrem Ursprung in ihre Äste auf.
Aufgrund des großen Ventrikelseptumdefekts besteht ein Druckangleich zwischen beiden Ventrikeln. Es kommt zu einer Mischung von system- und pulmonalvenösem Blut. Dieses Mischblut fließt dann sowohl in den System- als auch in den Pulmonalkreislauf. Nach Abfall des Lungenwiderstands in der Neugeborenenzeit kann es durch den geringeren Widerstand im Lungenkreislauf zu einem Überwiegen der Lungendurchblutung kommen. Klinisch imponiert in diesen Fällen die Herzinsuffizienz. Die arterielle O2-Sättigung hängt vom Grad der Lungendurchblutung ab.
Klinik
Die Klinik ist, entsprechend der Hämodynamik, abhängig von der Lungendurchblutung. Bestehen gut ausgebildete Pulmonalarterien, sind die Leitsymptome eine zunehmende Herzinsuffizienz als Folge der Lungenüberflutung. Ist die Lungendurchblutung vermindert, durch z. B. vorliegende Stenosen oder hypoplastische Pulmonalarterien, steht die Zyanose im Vordergrund.
Diagnostik
Der zweite Herzton ist singulär. Das EKG ist meist unspezifisch. Es zeigt sich eine biventrikuläre Hypertrophie. Im Thoraxröntgenbild zeigt sich schon früh eine Kardiomegalie, evtl. mit vermehrter Pulmonalgefäßzeichnung. Die Diagnose wird meist echokardiographisch gestellt. Dabei zeigt sich ein einzelnes, großes Gefäß, das einen großen Ventrikelseptumdefekt überreitet. Der Ursprung der Pulmonalarterien aus diesem Truncus ist, je nach Typ, nicht immer einfach zu visualisieren. Die Truncusklappe kann mit Anzahl der Taschen, Stenose und Insuffizienzkomponente dargestellt werden. Zusätzlich wird der Aortenbogen dargestellt mit evtl. begleitenden Anomalien wie einem rechtsdeszendierenden Aortenbogen, Aortenisthmusstenose oder einem unterbrochenen Aortenbogen. Sind die Pulmonalarterien nicht adäquat darstellbar, sollte eine erweiterte Diagnostik in Form einer MRT-Untersuchung oder eine Herzkatheteruntersuchung erwogen werden.
Therapie
Die Therapie besteht initial in der Behandlung der Herzinsuffizienz. Liegt ein unterbrochener Aortenbogen vor, muss zum Offenhalten des Ductus arteriosus mit Prostaglandin E1 begonnen werden. Da in den meisten Fällen eine übermäßige Lungendurchblutung überwiegt, sollte trotz vorliegender Zyanose kein Sauerstoff appliziert werden, da dadurch der Lungenwiderstand weiter gesenkt und die Herzinsuffizienz verstärkt wird. In der Regel wird eine Korrekturoperation in den ersten Lebensmonaten angestrebt, da es schon früh zu einer fixierten pulmonalen Hypertension kommen kann. Dabei wird der Ventrikelseptumdefekt so verschlossen, dass der Truncus arteriosus zur Aorta wird, also ausschliesslich aus dem linken Ventrikel entspringt. Die Pulmonalarterien werden vom Truncus abgesetzt und mit einem klappentragenden Conduit mit dem rechten Ventrikel verbunden. Liegt zusätzlich eine Aortenbogenanomalie oder ein unterbrochener Aortenbogen vor, wird dieser meist in der gleichen Operation mit korrigiert.
Trikuspidalatresie
Die Trikuspidalatresie (TAT) zählt zu den seltenen, komplexen Vitien und hat einen Anteil von ca. 1–2% aller angeborenen Herzfehler. Jungen sind etwas häufiger betroffen.
Morphologie und Hämodynamik
Es besteht eine Agenesie bzw. Atresie der Trikuspidalklappe. Es gibt somit keine direkte Verbindung zwischen dem rechten Vorhof und dem rechten Ventrikel. Die Einteilung der Trikuspidalatresie erfolgt nach Stellung der großen Gefäße (Normal- oder Transpositionsstellung), dem Vorhandensein eines Ventrikelseptumdefekts und dem Grad der Pulmonalarteriendurchblutung (ohne Pulmonalstenose, Pulmonalstenose, Pulmonalatresie; Abb. 20.16).

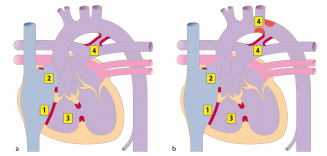
Entsprechend der Morphologie ist die Hämodynamik definiert. Das gesamte Herz-Zeit-Volumen muss vom rechten Vorhof über eine Vorhoflücke in den linken Vorhof und den linken Ventrikel gelangen. Hier liegt nun das gesamte system- und pulmonalvenöse Blut als Mischblut vor und kann nur über einen VSD in den rechten Ventrikel gelangen. Die Lungen- und Systemperfusion ist abhängig von der Stellung der großen Arterien sowie vom Grad der Pulmonalstenose. Liegt eine Normalstellung der großen Arterien vor, wird die Lunge über einen Ventrikelseptumdefekt durchblutet, das Mischblut gelangt über den rechten Ventrikel und die Pulmonalarterien in den Lungenkreislauf. Beim Vorliegen einer Pulmonalatresie mit intaktem Ventrikelseptum ist die Lungendurchblutung ductusabhängig.
Klinik
Entsprechend der Hämodynamik ist auch die Klinik abhängig von der Morphologie:
Es besteht durch den Rechts-links-Shunt auf Vorhofebene bereits im Neugeborenenalter eine Zyanose. Diese ist v. a. bei Vorliegen einer Pulmonalatresie/-stenose ausgeprägt.
Besteht keine Pulmonalstenose stehen durch die Lungenüberflutung Herzinsuffizienzzeichen im Vordergrund.
Bei restriktivem ASD können venöse Stauungszeichen mit Hepatosplenomegalie und Ödemen hinzukommen.
Diagnostik
Je nach Vorhandensein einer Pulmonalstenose kann entsprechend ein Systolikum mit p.m. über dem 2./3. ICR vorliegen. Im EKG zeigt sich häufig ein Linkstyp bis überdrehter Linkstyp.
Ein Neugeborenen-EKG mit Linkstyp sollte immer den Verdacht auf eine Trikuspidalklappenatresie lenken.
Zusätzlich kann auch ein P-dextrokardiale bestehen (hohes, spitzes P).
Im Thoraxröntgenbild zeigt sich je nach Grad der Pulmonalstenose eine vermehrte oder verminderte Lungendurchblutung.
Die Diagnose der Trikuspidalklappenatresie wird echokardiographisch gestellt. Dabei imponieren offensichtlich die Trikuspidalklappenatresie sowie ein auffällig kleiner rechter Ventrikel und ein obligater Rechts-links-Shunt auf Vorhofebene. Echokardiographisch muss geklärt werden, ob der ASD ausreichend groß ist. Weiterhin wird die Stellung der großen Gefäße, Vorhandensein und Größe eines Ventrikelseptumdefekts und v. a. die Beurteilung der Pulmonalklappe (liegt eine Atresie, Stenose oder keine Stenose vor) beurteilt. Eine Herzkatheteruntersuchung ist zur Diagnostik nicht routinemäßig notwendig. Bei restriktivem ASD wird eine Ballonatrioseptostomie durchgeführt.
Therapie
Besteht eine ductusabhängige Lungenperfusion, wird eine Infusion mit Prostaglandin E1 durchgeführt. Besteht eine Herzinsuffizienz, wird eine antikongestive Therapie begonnen. Ähnlich wie bei der Fallot-Tetralogie kann es auch bei einer Trikuspidalatresie zu hypoxämischen Anfällen kommen. Dies kann insbesondere bei einem kleinen bzw. restriktiven VSD und Normalstellung der großen Gefäße vorkommen. Es kommt im Prinzip zu einer dynamischen Zunahme der Pulmonalstenose bzw. Abnahme des pulmonalarteriellen Blutflusses. Das Vorgehen hier entspricht dem Vorgehen bei einen hypoxämischen Anfalls bei einer Fallot-Tetralogie (Abschn. 20.3.1.1). Das längerfristige Therapieziel ist die univentrikuläre Kreislauftrennung nach Fontan.
Besteht hingegen ein vermehrter Lungendurchfluss, kann zunächst durch ein sog. pulmonalarterielles Banding, der Fluss gedrosselt werden und so das Lungengefäßbett vor Überflutung und letztendlich auch pulmonaler Hypertension geschützt werden, um dann die Kreislauftrennung nach Fontan vorzunehmen.
Fontan-Zirkulation
Die Fontan-OP wird immer dann angestrebt, wenn eine Korrektur im eigentlichen Sinne nicht möglich ist. Neben der Trikuspidalatresie gibt es eine Gruppe heterogener, komplexer Herzfehler, die einem funktionell univentrikulären Herzfehler entsprechen. Diese Herzfehler können sich morphologisch stark voneinander unterscheiden. Bei der sog. Fontanisierung wird eine Trennung von Lungen- und Systemkreislauf erreicht. Die Lungen werden danach passiv ohne Unterstützung eines Ventrikels durchblutet. Die Kreislauftrennung wird in der Regel mindestens zweizeitig vorgenommen. Besteht eine ductusabhängige Pulmonalperfusion oder eine hochgradig verminderte Lungenperfusion ist eine zusätzliche Operation notwendig. Dann wird noch im Neugeborenalter ein aortopulmonaler Shunt angelegt.
Im Alter von 4–6 Monaten wird zunächst eine Glenn-Anastomose (V. cava superior anastomosiert an die Pulmonalarterie,) durchgeführt (Abb. 20.17a).

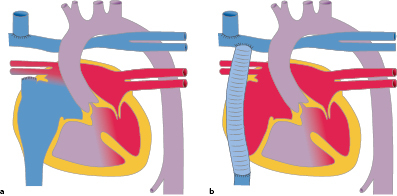
Schließlich wird im Alter von 2½ und 3 Jahren die Komplettierung der Fontan-Zirkulation durchgeführt. Hierbei wird das Blut der unteren Hohlvene über einen lateralen oder extrakardialen Tunnel direkt zum Pulmonalarteriensystem geleitet (Abb. 20.17b). Nach diesem Schritt fließt das gesamte venöse Blut passiv in die Lungenarterien, die Fontan-Zirkulation ist komplett. Häufig wird noch eine kleine Fenestrierung als Überlaufventil vom Fontan-Tunnel zum Vorhof geschaffen. Ohne Fenestrierung besteht nach Fontan-Komplettierung in der Regel keine Zyanose mehr.
Die Fontan-Operation und ihre Modifikation werden nun seit fast 50 Jahren durchgeführt. Nach einer Fontan-Operation besteht zunächst eine gute körperliche Belastbarkeit, allerdings erreichen diese Patienten im Vergleich zu einem gesunden Kollektiv nur ca. 60–70% der körperlichen Leistungsfähigkeit. Mit der Zeit kommt es zu einer abnehmenden Leistungsfähigkeit, evtl. mit erneut auftretender Zyanose, Ödemen sowie Pleura- oder Perikardergüsse. Die Prognose ist im Wesentlichen auch davon abhängig, ob der Systemventrikel ein rechter oder linker Ventrikel ist. Der linke Systemventrikel kann die hämodynamischen Anforderungen besser erfüllen und ist somit mit einem besseren Outcome assoziiert. Langzeitkomplikationen nach der Komplettierung der Fontan-Zirkulation sind ein erhöhtes Thromboserisiko, Herzrhythmusstörungen und eine zunehmende Funktionseinschränkung des Systemventrikels. Eine schwere, jedoch seltene Komplikation ist das sog. Eiweißverlustsyndrom. Hierbei kommt es zu Eiweißverlust über die Darmwand. Der Mechanismus hierfür ist immer noch nicht vollständig verstanden. Durch den Verlust von Eiweiß kommt es zur Ausbildung von Ergüssen (Pleura- und Perikardergüsse) und Aszites. Durch den Verlust von Immunglobulinen haben diese Patienten ein erhöhtes Risiko für gehäufte Infekte bis hin zu letal verlaufenden Septitiden. Die Fontan-Zirkulation stellt somit keine Korrektur, sondern nur eine Palliation dar. Mit nachlassender systemventrikulärer Funktion, zunehmender Verschlechterung der Fontan-Zirkulation sowie wiederholt auftretenden Komplikationen bleibt nur noch die Herztransplantation als ultima ratio.
Hypoplastisches Linksherzsyndrom
Das hypoplastische Linksherzsyndrom (HLHS) ist selten mit ca. 1–2% aller angeborenen Herzfehler. Es ist unbehandelt die häufigste Todesursache in der ersten Lebenswoche. Jungen sind deutlich häufiger betroffen.
Morphologie und Hämodynamik
Das HLHS beschreibt eine Gruppe von Fehlbildungen bei denen die Aorten- und Mitralklappe entweder atretisch oder stenotisch sind, und die mit einer Hypoplasie des linken Ventrikels einhergehen (Abb. 20.18). Teils werden auch andere Herzfehler, bei denen der linke Ventrikel hypoplastisch ist, zu dieser Gruppe gezählt, z. B. der imbalancierte AV-Kanal. Das Ausmaß der Hypoplasie der linksseitigen Strukturen kann stark variieren.

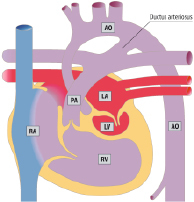
Wichtig ist, dass bereits intrauterin eine ausreichend große Vorhofkommunikation besteht, damit es nicht zu einer pulmonalvenösen Stauung kommt. Beim hypoplastischen Linksherzsyndrom liegt eine systemabhängige Ductusperfusion vor.
Klinik
Die Diagnose wird heutzutage in den meisten Fällen pränatal gestellt, sodass bereits kurz nach der Geburt mit einer Prostaglandin-E1-Applikation begonnen wird. Ansonsten kommt es mit Verschluss des Ductus arteriosus botalli zu einer akuten Verschlechterung innerhalb der ersten Lebenstage (Tachykardie, Tachypnoe, blass-graues Hautkolorit). Es imponieren neben einer zunehmenden Zyanose Zeichen einer globalen Herzinsuffizienz bis hin zum kardiogenen Schock.
Cave
Die O2-Gabe führt zur pulmonalen Vasodilatation. Unkritische O2-Gaben führen zu einem ausgeprägten Links-rechts-Shunt mit Verschlechterung der ductusabhängigen Systemperfusion und triggern den Ductusverschluss. Dies kann beim HLHS letale Folgen haben und sollte daher strikt vermieden werden!
Diagnostik
Die Diagnose wird echokardiographisch gestellt. Dabei wird der kleine hypoplastische linke Ventrikel dargestellt. Die Aorten- und Mitralklappe sind hypoplastisch oder atretisch. Es besteht ein Links-rechts-Shunt auf Vorhofebene. Dabei muss festgestellt werden, ob eine Restriktion auf Vorhofebene besteht. Wichtig ist es, den Ductus arteriosus botalli darzustellen, um zu sehen, ob hier eine Stenose besteht. Abhängig von der Morphologie des Ductus muss die Prostaglandin-E1-Dosierung gesteuert werden.
Zusätzliche Fehlbildungen wie ein Ventrikelseptumdefekt oder eine Aortenisthmusstenose sollten ebenso erkannt und erfasst werden.
Therapie
Das Therapieziel beim HLHS ist, wie bei anderen funktionell univentrikulären Herzen, die Fontan-Zirkulation. Allerdings ist das hypoplastische Linksherzsyndrom der Herzfehler mit der ungünstigsten Prognose und höchsten Letalität bis zum Erreichen einer Kreislauftrennung.
Die klassische Vorgehensweise besteht darin das Kind zunächst mit Prostaglandin E1 zu stabilisieren und eine evtl. restriktive Vorhoflücke mittels Ballonatrioseptostomie zu behandeln, um dann in der ersten oder zweiten Lebenswoche die sog. Norwood-Operation I durchzuführen. Bei der Norwood-I-Operation wird der Stamm der Pulmonalarterie mit der hypoplastischen Aorta ascendens anastomosiert und so eine Neoaorta geschaffen. Der Aortenbogen wird mittels Patch erweitert und die Lungenperfusion wird entweder über einen aortopulmonalen Shunt oder über einen sog. Sano-Shunt (Implantation eines Conduits zwischen rechtem Ventrikel und Pulmonalarterie) sichergestellt. Als zweiter Schritt erfolgt mit 4–6 Monaten die sog. Norwood-II-Operation, die einer Glenn-Operation entspricht (Abschn. 20.3.2.4) durchgeführt, um dann im Alter von 2½–3 Jahren die Kreislauftrennung (Norwood-III-Operation) zu komplettieren.
Alternativ kann das sog. Hybridverfahren (Katheterintervention plus Operation) durchgeführt werden, sodass keine Herzoperation mit Herz-Lungen-Maschine im Neugeborenenalter notwendig ist (Abb. 20.19). Erhoffte Vorteile dieses Verfahrens sind neben der damit geringeren perioperativen Letalität, die beim klassischen Vorgehen bis zur Kreislauftrennung um die 30–50% beträgt, Vermeidung von Hirnschäden und Verbesserung der neurologischen Entwicklung, wenn eine Operation mit Herz-Lungen-Maschine in der vulnerablen Neugeborenenzeit vermieden werden kann. Das Outcome der unterschiedlichen Operationsarten ist aktuell noch Gegenstand mehrerer Studien. (Weitere Informationen Lehrbücher Kinderkardiologie).

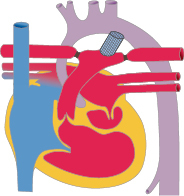
Prognose und Verlauf
Beim HLHS handelt es sich um den Herzfehler mit der schlechtesten Prognose und der höchsten postoperativen Letalität. Nach einer Fontan-Operation muss davon ausgegangen werden, dass der rechte Ventrikel als Systemventrikel früher zu einer Dysfunktion führen wird – mit den entsprechenden begleitenden Komplikationen.
Entzündliche Herzerkrankungen
Myokarditis
Eine Myokarditis ist eine Entzündung des Herzmuskels, die im Kindesalter am häufigsten durch eine Virusinfektion ausgelöst wird. Bei einer Myokarditis kommt es durch die Entzündung zu einer Schädigung der Kardiomyozyten und damit verbunden zum Untergang von Herzmuskelgewebe. Da die Myokarditis asymptomatisch verlaufen kann, wird sie wahrscheinlich häufig nicht erkannt. Daher ist die Inzidenz im Kindesalter unklar. Bei autoptischen Untersuchungen bei Kindern und Jugendlichen die am plötzlichen Herztod verstarben fanden sich bei bis zu 21% entzündliche Veränderungen im Myokard im Sinne einer Myokarditis.
Pathogenese
Als Folge einer Infektion kommt es zu einer Entzündung mit Infiltration von Entzündungszellen. Die akute Entzündungsreaktion zerstört myokardiale Zellen. Die meisten Myokarditen heilen folgenlos ab. Resultat einer narbigen Defektheilung kann eine funktionelle Schädigung des Herzmuskels sein, z. B. eine chronisch dilatative Kardiomyopathie (DCM) mit entsprechend schwerem Krankheitsverlauf.
Die Myokarditis zeichnet also durch 3 Stadien aus:
Phase: Akutphase → Entzündung mit Nekrose und Apoptose (mononukleäre Infitlration),
Phase: Subakutphase → Aktivierung des Immunsystems,
Phase: Ausheilungsphase mit Fibrosierung, bei Persistenz → DCM.
Klinik
Das klinische Bild einer akuten Myokarditis kann von asymptomatischen Fällen bis hin zum fulminanten Verlauf mit kardiogenem Schock und plötzlichem Herztod reichen. Neugeborene und junge Säuglinge zeigen häufig das Bild einer Herzinsuffizienz mit unspezifischen Symptomen einer Infektion, Trinkschwäche, Husten als Hinweis auf ein Lungenödem und deutlich vermehrtes Schwitzen. Bei älteren Kindern und Jugendlichen findet sich anamnestisch häufig eine Virusinfektion, die dem Krankheitsbeginn 10–14 Tage vorausging. Die Patienten klagen über einen Leistungsknick, evtl. über thorakale Schmerzen mit präkordialem Stechen, z. T. zeigen sich neu aufgetretene Herzrhythmusstörungen wie supraventrikuläre oder ventrikuläre Extrasystolen, Arrhythmien und AV-Blockierungen.
Diagnostik
Die Diagnose wird aufgrund der laborchemischen Befunde, des EKG, der Echokardiographie und evtl. weiterer bildgebender Untersuchungen sowie letztendlich durch histologische und immunhistologische Befunde gestellt. In der Labordiagnostik findet sich als Biomarker der myokardialen Schädigung evtl. noch eine Erhöhung des Troponins oder der Kreatinkinase (CK). Normwertige myokardiale Enzyme schließen eine Myokarditis allerdings nicht aus.
EKG
Am häufigsten zeigen sich neben einer Sinustachykardie Repolarisationsstörungen. In schweren Fällen können auch Infarktzeichen vorhanden sein.
Bei neu aufgetretenen Herzrhythmusstörungen, z. B. ventrikuläre Extrasystolen sollte eine Myokarditis in die differenzialdiagnostischen Überlegungen eingeschlossen werden.
Echokardiographie
Häufig findet sich nur eine geringfügig verschlechterte ventrikuläre Funktion, die noch nicht zu einer Dilatation des Ventrikels geführt hat. Die Unterscheidung zwischen einer dilatativen Kardiomyopathie und einer akuten Myokarditis kann nicht immer sicher gestellt werden.
MRT
Mittels MRT mit Kontrastmittel, der sog. Late-enhancement-Technik, kann neben der Funktionseinschränkung evtl. das Ausmaß der Entzündung im Herzmuskel dargestellt werden.
Herzkatheteruntersuchung
Die Hauptindikation für eine Herzkatheteruntersuchung ist die Durchführung einer Endomyokardbiopsie. Mit dieser ist die Diagnose einer Myokarditis möglich. Ergänzend sollte zusätzlich noch eine Koronaranomalie als Ursache einer eingeschränkten Funktion ausgeschlossen werden.
Therapie
Bisher liegen keine klinischen Studien zur spezifischen Therapie der Herzinsuffizienz vor. Am häufigsten wird eine initiale Bettruhe und anschließend die körperliche Schonung für 3–6 Monate empfohlen. Weiterhin sollte eine evtl. bestehende Herzinsuffizienz medikamentös behandelt werden. In der Akutphase wird häufig eine Therapie mit Immunglobulinen (1–2 g/kgKG über 48 h) empfohlen, da es Hinweise für eine Verbesserung der linksventrikulären Funktion bei akuter Myokarditis gibt. Der Einsatz von Immunsuppressiva hat sich bisher nicht durchgesetzt, da bisher kein Vorteil gegenüber einer rein symptomatischen Therapie nachgewiesen werden konnte.
Prognose
Die Mehrzahl der Myokarditen heilt folgenlos aus. Bei Neugeborenen scheint die Mortalität am höchsten zu sein. Ein Teil der Myokarditen kann in eine dilatative Kardiomyopathie übergehen, die mit einem schweren Krankheitsverlauf und hoher Mortalität assoziiert ist.
Infektiöse Endokarditis
Die infektiöse Endokarditis ist eine überwiegend bakteriell bedingte Infektion der endokardialen Strukturen des Herzens. Sie kann in akuter oder subakuter Form vorliegen. Eine Endokarditis kann auch Fremdmaterial wie chirurgisches Patchmaterial, künstliche Gefäßprothesen und künstliche Herzklappen betreffen. Die Endokarditis kommt fast ausschließlich bei Patienten mit angeborenem Herzfehler oder erworbener struktureller Läsion (wie Patienten nach chirurgischen Eingriffen) vor. Endokarditiden betreffen zum überwiegenden Teil die Klappen des Systemkreislaufs. Pulmonal- und Trikuspidalklappe können bei i.v.-Drogenabhängigen befallen sein.
Pathogenese
Bei vielen angeborenen oder erworbenen Herzfehlern bestehen durch Stenosen oder Kurzschlussverbindungen turbulente Blutströmungen, die eine Endokardläsion verursachen können. Sekundär kommt es zur Apposition von Thrombozyten und Fibrin, auf die sich im Laufe einer Bakteriämie Erreger ansiedeln können. Diese sog. endokarditischen Vegetationen können dann durch eine fortschreitende Entzündungsreaktion zu ulzerativen Gewebeeinschmelzungen führen. Ferner kann es zur Embolisation dieser Vegetation mit nachfolgenden Gewebeinfarkten und septischen Absiedlungen im ganzen Körper kommen. Besondern gefürchtet sind Hirnembolien mit sekundärer Hämorrhagie. Klinisch kann die infektiöse Endokarditis akut oder subakut verlaufen. Die häufigsten Erreger der akut verlaufenden Form sind Staphylococcus aureus, Enterobakterien, Pneumokokken und gramnegative Erreger. Die subakut verlaufende Form wird häufig durch Streptokokken der Viridansgruppe, koagulasenegative Staphylokokken sowie Enterokokken verursacht. Ein Erregernachweis gelingt in bis zu 25% der Fälle nicht.
Klinik
Leitsymptome einer akuten infektiösen Endokarditis sind ein septisches Krankheitsbild mit rascher Entwicklung einer Herzinsuffizienz und eines neuen Herzgeräuschs. Die Symptome einer subakuten infektiösen Endokarditis sind deutlich unspezifischer: Fieber ohne identifizierbaren Fokus, Verschlechterung des Allgemeinzustands mit Abgeschlagenheit, Gewichtsverlust, evtl. klinische Zeichen einer Herzinsuffizienz. Spezifische klinische Symptome sind selten. Diese sind ein neu aufgetretenes Herzgeräusch, Osler-Knötchen (linsengroße, schmerzhafte rötliche Knötchen vorwiegend an Fingern und Zehen) Janeway-Läsionen (hämorrhagische nicht schmerzhafte Hautveränderungen an Händen und Füßen). Weitere klinische Befunde können sein: Splenomegalie, Mikrohämaturie, Anämie und Embolie.
Bei Kindern mit angeborenen Herzfehlern muss bei anhaltendem oder rekurrierendem Fieber das Vorliegen einer infektiösen Endokarditis in Erwägung gezogen werden.
Diagnostik
Ein mikrobiologischer Erregernachweis (einschließlich Resistenzbestimmung) hat neben dem echokardiographischen Nachweis von Vegetationen den größten Stellenwert bei der Diagnosestellung. Vor Beginn einer Antibiotikatherapie sollten mehrere (mindestens drei) aerobe und anaerobe Blutkulturen vor Therapiebeginn, bzw. wenn möglich nach drei Tagen Antibiotikapause, abgenommen werden. Weitere Befunde in der Labordiagnostik, die auf eine infektiöse Endokarditis hindeuten können, sind eine Anämie, eine Leukozytose mit Linksverschiebung sowie eine Thrombozytopenie.
Echokardiographie
Durch die Echokardiographie ist evtl. der Nachweis von endokarditischen Vegetationen, Klappenläsionen, Abszessen sowie neu aufgetretenen Klappendysfunktionen möglich.
Ein negativer Echokardiographiebefund schließt eine infektiöse Endokarditis nicht aus.
Therapie
Zur vollständigen Eradikation des Erregers ist eine mehrwöchige bakterizide antibiotische Behandlung notwendig. Die Therapiedauer beträgt meist 4–6 Wochen. Bei einer Prothesenendokarditis verlängert sich die Gesamttherapiedauer auf mindestens 6 Wochen. Bei persistierenden Infektionszeichen trotz adäquater Therapie muss die Indikation zur chirurgischen Sanierung gestellt werden.
Endokarditisprophylaxe
Patienten, die über ein hohes Risiko für eine bakterielle Endokarditis verfügen, sollten daher in erster Linie eine Prävention der Endokarditis anstreben. Dazu gehören neben einer kausalen Therapie des angeborenen Herzfehlers, die Etablierung eines guten Zahnstatus und einer frühzeitigen Zahnsanierung sowie eine sorgfältige Haut- und Nagelpflege. Zusätzlich wird bei Patienten mit hohem Endokarditisrisiko eine Endokarditisprophylaxe empfohlen. Diese besteht darin, dass bei diesen Patienten bei diagnostischen oder therapeutischen Eingriffen eine antibiotische Prophylaxe durchgeführt werden sollten. Dazu gehören zahnärztliche und HNO-ärztliche Eingriffe.
Kardiomyopathien
Kardiomyopathien sind eine Herzmuskelerkrankung, die nach WHO Klassifikation in dilatative, restriktive und hypertrophe Kardiomyopathien unterteilt werden. Es gibt zahlreiche spezifische Erkrankungen, die mit einer Herzmuskelerkrankung einhergehen können. Diese können klassifiziert werden als infektiologisch, metabolisch, systemisch, toxisch, familiär. Die häufigsten Formen sind idiopathisch.
Dilatative Kardiomyopathie
Die häufigste Kardiomyopathie ist die dilatative Kardiomyopathie (DCM). Diese ist durch eine Dilatation des linken Ventrikels mit einer erheblichen Störung der systolischen und diastolischen Funktion assoziiert (Abb. 20.20). In einigen Fällen kann auch die rechte Herzkammer betroffen sein. Die DCM ist im Kindesalter zwar selten, doch ist sie die häufigste Form der kindlichen Kardiomyopathie. Die Inzidenz wird mit 0,5–2,6/100.000 Kindern angegeben. Am häufigsten manifestiert sich die Krankheit innerhalb der ersten zwei Lebensjahre.
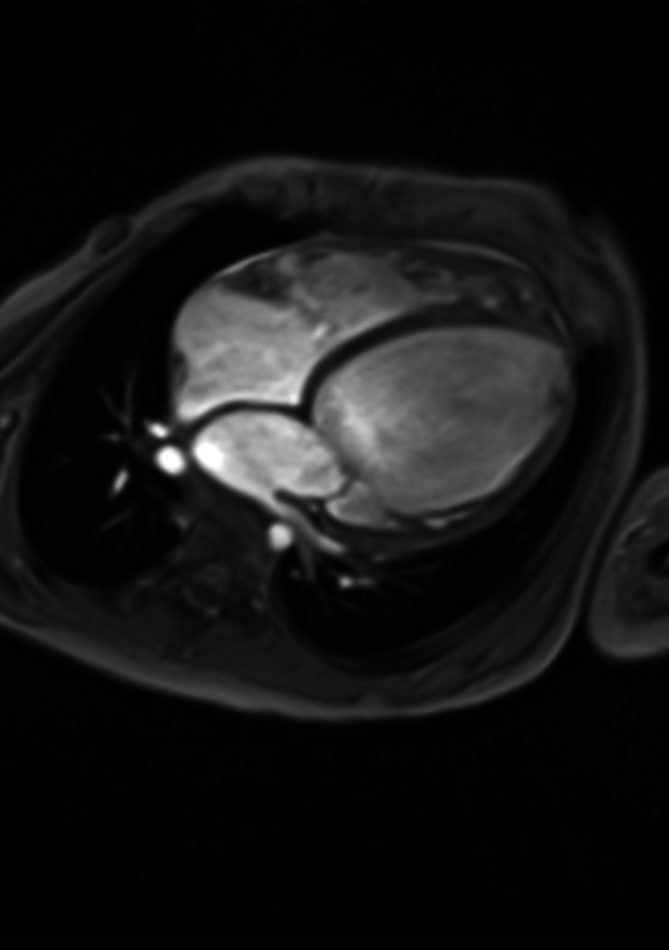
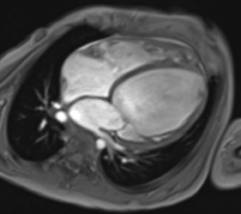
Ätiologie
Die Ätiologie der Erkrankung ist häufig unklar. Die häufigste Ursache ist wahrscheinlich eine Myokarditis. Andere mögliche Ursachen sind v. a. metabolische Störungen sowie Z. n. Chemotherapie mit Anthracycline. Ischämische Myokardschäden, z. B. bedingt durch einen Fehlabgang der linken Koronararterie aus der Pulmonalarterie (Bland-White-Garland-Syndrom) oder andere Erkrankungen des koronararteriellen Systems, sollten ausgeschlossen werden. Weiterhin kann eine familiäre Form der dilatativen Kardiomyopathie vorliegen.
Klinik
Bei der DCM stehen die Zeichen einer ausgeprägten Herzinsuffizienz im Vordergrund, die im Verlauf zunehmen können. Es kann zu akuten Herzversagen im Rahmen von einfachen Infekten kommen.
Diagnostik
Häufig wird ein Thoraxröntgenbild bei zunehmender Dyspnoe durchgeführt. Dabei zeigt sich eine Kardiomegalie mit Stauungszeichen. Im EKG können sich Erregungsrückbildungsstörungen zeigen. Letztendlich wird die Diagnose echokardiographisch gestellt mit Darstellung eines dilatierten, schlecht kontrahierenden linken Ventrikels häufig assoziiert mit einer erheblichen Mitralklappeninsuffizienz. Zur Abklärung einer möglichen Ursache sollte eine Herzkatheteruntersuchung durchgeführt werden mit Darstellung der Koronararterien und Biopsieentnahme.
Therapie
Die Therapie der DCM besteht derzeit hauptsächlich in der Behandlung der Symptome. Die Herzinsuffizienzbehandlung steht im Vordergrund. Herzrhythmusstörungen sollten frühzeitig therapiert werden. Ein biventrikulärer Schrittmacher kann zur Resynchronisation beitragen und dadurch die ventrikuläre Funktion bei einigen Patienten verbessern. Ultima ratio und einzige palliative Therapie ist schließlich die Herztransplantation.
Die DCM ist im Kindesalter der häufigste Grund für eine Herztransplantation.
In einigen Fällen ist zur Überbrückung bis zur Transplantation ein Assist-Device oder Kunstherz notwendig. In Anbetracht der geringen Verfügbarkeit von Spenderorganen und der begrenzten Überlebenszeit des transplantierten Organs (mittlere Überlebenszeit 10–12 Jahre) werden alternative Therapieoptionen intensiv erforscht. Therapieansätze mit Stammzellen, Wachstumshormonen oder die Anlage eines zentralen pulmonalen Bandings sind im klinischen Alltag bislang noch nicht etabliert und z. T. aktuell Gegenstand multinationaler Studien.
Prognose
Die Prognose ist insgesamt schlecht, insbesondere wenn kein kausaler Zusammenhang gefunden wird. In ca. 20–25% kommt es zu einer spontanen Remission.
Hypertrophe bzw. hypertroph obstruktive Kardiomyopathie
Bei diesem Krankheitsbild handelt es sich um eine Hypertrophie, bei der v. a. das interventrikuläre Septum und die linksventrikuläre Hinterwand betroffen sind. Es handelt sich um eine genetisch bedingte Myokardhypertrophie, die nicht durch andere Ursachen wie eine Klappenstenose bedingt ist. Durch die muskuläre Hypertrophie kann es zu einer Obstruktion im Bereich des Ausflusstrakts kommen, die von sehr variabler Ausprägung ist. In diesem Fall spricht man von einer hypertroph obstruktiven Kardiomyopathie (HOCM); ohne Obstruktion liegt entsprechend eine hypertrophe nicht obstruktive Kardiomyopathie (HCM) vor. Die hypertroph obstruktive Kardiomyopathie ist eine der häufigsten Ursachen für den plötzlichen Herztod bei Kindern und Erwachsenen.
Klinik
Die meisten Patienten sind asymptomatisch, daher bleibt die Erkrankung häufig lange unerkannt. Der plötzliche Herztod kann das erste Symptom sein. Durch die Hypertrophie kommt es zu einer zunehmenden Restriktion des Ventrikels mit einer funktionellen Einflussbehinderung in den Ventrikel mit Dyspnoe und Belastungsintoleranz. Synkopen unter Belastung können aufgrund der linksventrikulären Ausflusstraktobstruktion oder auch durch das Auftreten von Herzrhyhmusstörungen unter Belastung verursacht werden. Synkopen sind assoziiert mit einem höheren Risiko für einen plötzlichen Herztod. Angina-pektoris-Beschwerden können in einigen Fällen vorhanden sein.
Synkopen unter Belastung müssen zeitnah abgeklärt werden. Eine hypertrophe Kardiomyopathie muss ausgeschlossen werden.
Diagnostik
Bei einer Obstruktion des linksventrikulären Ausflusstrakts lässt sich ein Systolikum mit p.m. über dem 4. ICR links auskultieren. Im EKG zeigen sich unspezifische Veränderungen mit Zeichen der linksventrikulären Hypertrophie, Erregungsrückbildungsstörungen und evtl. Herzrhythmusstörungen. Die Diagnose wird echokardiographisch gesichert. Dabei zeigt sich die typische Hypertrophie des interventrikulären Septums und/oder der linksventrikulären Hinterwand. Zusätzlich kann eine bestehende Obstruktion des linksventrikulären Ausflusstrakts dargestellt werden. Ein genetischer Nachweis der zugrundeliegenden Mutation gelingt in ca. 30% der Fälle.
Therapie
Therapieziele sind die Hypertrophieentwicklung zu verlangsamen und relevante Rhythmusstörungen zu verhindern. Für Patienten mit hypertropher Kardiomyopathie sind Einschränkungen der körperlichen Belastung empfohlen, für Kinder und Jugendliche bedeutet dies ein Sportverbot. Medikamentös werden die Patienten meistens mit einem β-Blocker behandelt, um das Verhältnis von O2-Verbrauch und O2-Angebot zu ökonomisieren sowie das Auftreten von Herzrhythmusstörungen zu verringern. Häufig wird ein ICD als Primärprophylaxe bei Hochrisikopatienten oder als Sekundärprophylaxe nach stattgefundenem, überlebtem Herzstillstand implantiert.
Positiv inotrope Substanzen sind kontraindiziert, da sie die Obstruktion verstärken. Auch Nachlastsenker sollten vermieden werden, weil sie den Gradienten über dem linksventrikulären Ausflusstrakt verstärken, und die koronare Perfusion verschlechtern können. Therapeutisch ist eine Ablation der Septumhypertrophie durch Alkoholinjektion in einen septalen Koronararterienast möglich. Eine operative Therapie mit Myektomie ist v. a. bei erfolgloser konservativer Therapie symptomatischer Patienten sowie bei einer erheblichen Obstruktion indiziert.
Herzrhythmusstörungen
AV-Block
Beim atrioventrikulären Block (AV-Block) liegt eine Störung der Überleitung der elektrischen Impulse von den Vorhöfen auf die Ventrikel vor.
Der AV-Block wird in drei Grade unterteilt, wobei der AV-Block II° wiederum in einen Typ Mobitz I (syn. Typ Wenckebach) und Typ Mobitz II (Typ Mobitz) unterteilt wird.
Der AV-Block I° ist definiert durch eine isolierte Verlängerung der PQ-Zeit. Der AV-Block II° Typ Mobitz I zeigt eine zunehmende Verlängerung der PQ-Zeit bis der Kammerkomplex ausfällt. Beim AV-Block Typ Mobitz II wird die Vorhoferregung periodisch nicht übergeleitet. Die PQ-Zeit ist bei den übergeleiteten Schlägen gleich lang. Es findet sich meistens eine typische 2:1- oder 3:1-Überleitung. Beim AV-Block III° ist die atrioventrikuläre Leitung komplett blockiert. Vorhöfe und Ventrikel schlagen unabhängig voneinander. Da die Kammern durch das tertiäre Erregungsleitungszentrum erregt werden, besteht ein langsamer Kammerrhythmus. Liegt das Erregungsleitungszentrum weit vom AV-Knoten entfernt, zeigen sich langsame, schenkelblockartige also verbreiterte Kammerkomplexe. Liegt das Erregungszentrum in der Nähe des AV-Knotens am HIS-Bündel, können die Kammerkomplexe schmal sein.
Klinik und Therapie
Der AV-Block I° wie auch der AV-Block II° Typ Mobitz I kann bei Sportlern oder bei erhöhten Vagotonus (z. B. im Schlaf) physiologisch auftreten. Er führt in der Regel zu keinen Symptomen und erfordert keine Therapie.
Beim AV-Block II° Typ Mobitz II liegen meist keine Symptome vor. Ist die Kammerfrequenz jedoch deutlich reduziert, kann es zu Schwindel oder Synkopen kommen. In jedem Fall muss eine weitere Abklärung und Verlaufsuntersuchung erfolgen, da ein Übergang in einen AV-Block III° möglich ist.
Am häufigsten findet sich ein kompletter AV-Block im Kindesalter postoperativ nach Herzoperationen. Selten führen infektiöse (Borreliose, Diptherie, Mykoplasmen oder Chlamydien) oder entzündliche (Myokarditis, Endokarditis) Prozesse zu einem AV-Block.
Ein kongenitaler AV-Block III° kann intrauterin bei einer Autoimmunerkrankung der Mutter (z. B. Lupus erythematodes, Sjörgen-Syndrom) auftreten. Mütterliche Antikörper zerstören dabei den AV-Knoten. Intrauterin kann sich durch die Herzinsuffizienz ein Hydrops fetalis bilden. Postnatal ist die Therapie abhängig vom klinischen Zustand und der Kammerfrequenz des Neugeborenen. Ist die Herzfrequenz ausreichend (60–80 Schläge/Minute, schmale Kammerkomplexe und kein Herzfehler) und liegen keine Herzinsuffizienzzeichen vor, kann zunächst abgewartet werden. Liegt eine symptomatische Bradykardie vor, ist eine Schrittmacherimplantation indiziert. Überbrückend kann versucht werden, die Kammerfrequenz durch Sympathomimetika zu akzelerieren.
Extrasystolen
Man unterscheidet zwischen supraventrikulären und ventrikulären Extrasystolen. Supraventrikuläre Extrasystolen nehmen ihren Ausgang oberhalb des His-Bündels, während ventrikuläre Extrasystolen im oder unterhalb des His-Bündels entspringen.
Supraventrikuläre Extrasystolen sind definiert durch eine vorzeitige P-Welle und einem schmalen Kammerkomplex (vorwiegend) sowie eine postextrasystolische Pause, die nicht vollständig kompensiert ist. Einzelne isoliert auftretende supraventrikuläre Extrasystolen erfordern in der Regel keine Therapie. Sie sind u. a. gehäuft im Neugeborenenalter zu beobachten. Der Befund kann nach 1–2 Wochen mittels EKG kontrolliert werden, evtl. sollten die Elektrolyte bestimmt werden. Finden sich gehäuft auftretende supraventrikuläre Extrasystolen in Form von Couplets und Triplets, sollte eine weitere kinderkardiologische Abklärung erfolgen.
Sog. blockierte supraventrikuläre Extrasystolen treten fast ausschließlich im Neugeborenenalter auf. Die Extrasystole trifft auf eine noch refraktäre Kammer und wird daher nicht übergeleitet. Dies führt zu einer deutlichen Reduzierung der Kammerfrequenz. Eine Therapie ist in der Regel nicht erforderlich, die Extrasystolen verschwinden meistens im Verlauf.
Ventrikuläre Extrasystolen sind definiert durch den vorzeitigen Einfall einer Kammererregung. Die Kammerkomplexe sind meist verbreitert und deformiert. Eine P-Welle ist in der Regel nicht erkennbar. Bei einer erstmalig auftretenden ventrikulären Extrasystolie, auch im Neugeborenenalter, sollten eine strukturelle Herzerkrankung und eine Myokarditis ausgeschlossen werden, Elektrolyte sollten kontrolliert werden und die Einnahme von arrhythmogenen Medikamenten ausgeschlossen werden. Kommt es unter körperlicher Belastung zum Verschwinden der isoliert auftretenden VES, handelt sich meist um eine benigne Form der Extrasystolie, die keiner weiteren Therapie bedarf.
Liegen polymorphe VES, gehäuft auftretende VES in Form von Couplets oder Salven oder kommt zu einem vermehrten Auftreten der VES unter Belastung oder in der Erholungsphase nach Belastung auf, muss eine umgehende weitere Abklärung erfolgen. In diesen Fällen besteht das Risiko, dass die VES eine ventrikuläre Tachykardie oder Kammerflimmern auslösen können. Insbesondere wenn eine strukturelle Herzerkrankung, eine Myokarditis oder Kardiomyopathie vorliegt.
Schmalkomplextachykardie
Supraventrikuläre Tachykardien (SVT)
Die Inzidenz von supraventrikulären Tachykardien reicht von 1:250 bis zu 1:1000. SVT sind die häufigsten tachykarden Rhythmusstörungen im Kindesalter. 30–40% der SVT können in den ersten Lebenswochen schon manifest sein. Tachykardien auf der Basis von akzessorischen Leitungsbahnen sind häufig. Bei 20% dieser Patienten besteht häufig ein angeborener Herzfehler. Supraventrikuläre Tachykardien im Kindesalter zeigen meist einen schmalen Kammerkomplex. Liegt ein Schenkelblock vor oder kommt es während der Tachykardie zu einer antegraden Leitung über eine akzessorische Bahn, liegen breite Kammerkomplexe vor.
Atrioventrikuläre Tachykardie (AVRT)
Atrioventrikuläre Leitungsbahnen sind die häufigste Form von angeborenen, supraventrikulären Tachykardien im Kindesalter. Findet sich eine Deltawelle im Oberflächen-EKG, so liegt eine Präexzitation vor. Eine Präexzitation in Kombination mit einer Tachykardie wird Wolff-Parkinson-White-Syndrom genannt. Es handelt sich bei der AVRT um eine Reentry-Tachykardie, d. h. es liegt eine kreisende Erregung vor. Die AVRT zeichnet sich durch ein paroxysmales Auftreten mit plötzlichem Beginn und Ende aus. Sie ist häufig mit einer Präexzitation assoziiert. Bei genauer Inspektion des EKG während der Tachykardie lässt sich evtl. ein retrogrades P abgrenzen. Am häufigsten findet sich eine orthodrome atrioventrikuläre Reentry-Tachykardie, d. h. die Erregung verläuft während der Tachykardie antegrad über den AV-Knoten und retrograd über die akzessorische Leitungsbahn. Supraventrikuläre Tachykardien, die im Säuglingsalter auftreten, können im ersten Lebensjahr rezidivierend auftreten, und können zunächst medikamentös (z. B. mit einem β-Blocker oder einem Antiarrhythmikum der Klasse 1c) behandelt werden. Die Therapie kann dann bei fehlenden Symptomen im Alter von einem Jahr beendet werden. Ca. 80% der im Säuglingsalter auftretenden Tachykardien zeigen kein Rezidiv.
Wolff-Parkinson-White-Syndrom (WPW)
Bei Vorliegen einer Deltawelle im Oberflächen-EKG spricht man von einer Präexzitation (Abb. 20.21). Kommt es zu einer Tachykardie, spricht man von einem WPW-Syndrom. Das WPW-Syndrom hat eine Prävalenz von ca. 0,2%, und ist mit einem höheren Risiko für einen plötzlichen Herztod assoziiert. Bei Patienten mit WPW kommt es im Vergleich zur Normalbevölkerung häufiger zu Vorhofflimmern. Besteht nun eine schnelle akzessorische Leitungsbahn, kann es bei Vorhofflimmern zu einer schnellen antegraden Überleitung von den Vorhöfen auf die Kammern kommen und so zu einer lebensbedrohlichen ventrikulären Tachykardie führen. Dieses Risiko ist insgesamt niedrig, und liegt je nach Studie bei ca. 0,1–0,15%. Aufgrund des insgesamt erhöhten Risikos besteht die Empfehlung, bei Persistenz der Deltawelle im Rahmen einer Belastungsuntersuchung und einem Alter über 8 Jahre eine elektrophysiologische Untersuchung durchzuführen. Dabei wird die akzessorische Leitungsbahn elektrophysiologisch charakterisiert. Besteht eine potenziell lebensbedrohliche Gefährdung durch die Eigenschaften dieser Bahn, wird dann eine Ablationsbehandlung empfohlen und ggf. durchgeführt.

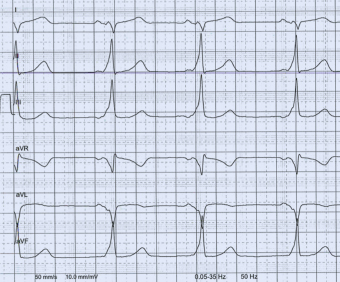
AV-Knoten-Reentry-Tachykardie (AVNRT)
Die AV-Knoten-Reentry-Tachykardie ist die zweithäufigste Tachykardie im Kindesalter. Es liegen zwei separate Leitungsbahnen innerhalb des AV-Knotens vor. Eine von diesen Bahnen hat eine schnelle Überleitungsgeschwindigkeit, während die andere deutlich langsamer leitet. Die AVNRT ist im ersten oder zweiten Lebensjahr selten und tritt ebenso wie die AVRT paroxysmal auf. Im EKG lässt sich während der typischen AVNRT kein retrogrades P abgrenzen. Aufgrund der Leitungseigenschaften besteht kein erhöhtes Risiko für einen plötzlichen Herztod. Eine Indikation zur Behandlung ergibt sich aus der Rezidivhäufigkeit, den Symptomen während der Tachykardie und dem Leidensdruck des Patienten. Dem gegenüber zu stellen ist das Risiko, bei der Ablation der langsam leitenden Bahn einen AV-Block III° zu verursachen. Das Risiko dafür liegt ca. bei 1%. Das Risiko wurde durch das Einsetzen von Kryoenergie minimiert und hat sich auch im Kindesalter als ein sehr sicheres Verfahren etabliert.
Vorhofflattern
Vorhofflattern findet sich zu 80% postoperativ nach kardiochirurgischen Eingriffen. Es kann aber auch selten präpartal und in der Neugeborenenperiode stattfinden. Bei Vorhofflattern liegt ein Reentry-Kreislauf im rechten Vorhof vor, d. h. der AV-Knoten ist nicht Bestandteil des Tachykardiemechanismus. Die Applikation von Adenosin führt zu einem kurzfristigen AV-Block, dadurch wird die Tachykardie zwar nicht terminiert, aber das Vorhofflattern wird demaskiert. Im EKG erkennt man die charakteristische Sägezahnform der Vorhoferregung. Die Vorhoffrequenz kann zwischen 240 und 400/min. liegen, das Überleitungsverhältnis beträgt meistens 2:1 oder 3:1, selten 1:1. Die Vorhoferregung ist regelmäßig. Die Therapie besteht in der Regel in einer elektrischen Kardioversion. Bei isoliertem neonatalen Vorhofflattern ist die Prognose sehr gut und das Risiko eines erneuten Auftretens gering. Eine medikamentöse Prophylaxe ist in der Regel nicht notwendig.
Therapie einer Schmalkomplextachykardie
Liegt eine Tachykardie mit schmalen Kammerkomplexen vor und ist der Patient in einem klinisch stabilen Zustand, sollte zunächst eine Sinustachykardie ausgeschlossen werden. Weiterhin sollten behandelbare Ursachen wie Volumenmangel, Fieber oder Stress ausgeschlossen werden. Zur Terminierung der supraventrikulären Tachykardie können zunächst vagale Manöver versucht werden, d. h. ein Valsalva-Manöver oder bei Säuglingen Eisbeutel auf das Gesicht legen. Führt dies zu keinem Erfolg, kann Adenosin verabreicht werden. Obligat sind dabei eine kontinuierliche Aufzeichnung des EKG sowie eine Defibrillatorbereitschaft. Durch die Adenosingabe kommt es häufig zur Terminierung der Tachykardie. Falls die Tachykardie trotz Adenosingabe persistiert, sollte eine antiarrhythmische Therapie eingeleitet werden. Bei hämodynamisch stabilen Patienten kann mit einem β-Blocker oder Antiarrhythmika der Klasse Ic wie z. B. Propafenon begonnen werden. Bei hämodynamischer Instabilität oder schlechter Ventrikelfunktion ist eine synchronisierte elektrische Kardioversion und/oder antiarrhythmische Medikation mit Amidoaron indiziert.
Ventrikuläre Tachykardie (VT)
Ventrikuläre Tachykardien sind im Kindesalter selten. Eine ventrikuläre Tachykardie zeichnet sich in der Regel durch eine Folge von mindestens 5 verbreiterten Kammerkomplexen aus, die häufig charakterisiert sind durch eine AV-Dissoziation. Es besteht eine komplette Dissoziation von Vorhöfen und Kammer. Die VT wird als nicht anhaltend (non-sustained) bei einer Dauer <30 s, als anhaltend (sustained) bei einer Dauer >30 s bezeichnet. Analog zur Nomenklatur der Extrasystolie kann sie monomorph oder polymorph sein. Polymorphe ventrikuläre Tachykardien neigen im Vergleich zu monomorphen Tachykardien häufig zur Degeneration und Kammerflimmern. Folgende Ursachen können zu einer ventrikulären Tachykardie führen:
Kardiomyopathie,
Myokarditis,
angeborene Herzfehler v. a. nach Herzoperationen,
Ionenkanalerkrankungen,
idiopathische.
Differenzialdiagnostisch kann es sich bei einer Breitkomplextachykardie um eine antidrome AVRT handeln: während der Tachykardie verläuft die Erregung antegrad über die akzessorische Leitungsbahn vom Vorhof auf die Ventrikel und zurück über den AV-Knoten. Alternativ kann ein vorbestehender Schenkelblock bestehen.
Im Kindesalter ist bei einer Breitkomplextachykardie das Vorliegen einer supraventrikulären Tachykardie mit Schenkelblock wahrscheinlicher als eine ventrikuläre Tachykardie.
Im Zweifel muss jedoch immer von einer ventrikulären Tachykardie ausgegangen werden, v. a. bei hämodynamischer Instabilität des Patienten oder wenn angeborene Herzfehler vorliegen.
Therapie
Im Falle einer instabilen Hämodynamik sollte entsprechend eine Kardioversion durchgeführt werden. Ist der Patient in einem klinisch stabilen Zustand, kann korrespondierend zum Vorgehen bei einer Schmalkomplextachykardie zunächst Adenosin gegeben werden. Adenosin dient dabei als Therapeutikum, um die Tachykardie zu terminieren und als diagnostisches Mittel um die Tachykardie zu demaskieren/differenzieren. Besteht die Tachykardie weiter, müssen weitere Antiarrhythmika erwogen werden, z. B. das Klasse-III-Antiarrhythmikum Amiodaron.
Long-QT-Syndrom
Das Long-QT-Syndrom (LQTS) ist eine angeborene Ionenkanalerkrankung, die mit einer Verlängerung der QT-Zeit einhergeht. Die Prävalenz beträgt je nach Studie zwischen 1:2.000 bis 1:5.000. Aufgrund des jeweiligen Ionenkanaldefekts kommt es zu einer myokardialen Repolarisationsstörung, die sich im EKG typischerweise in einer verlängerten QT-Dauer zeigt, und mit einem erhöhten Risiko für potenziell lebensbedrohliche ventrikuläre Arrhythmien einhergeht. Diese treten typischerweise als polymorphe ventrikuläre Tachykardien (sog. Torsade-de-Pointes) auf, und sind pathognomonisch für die Erkrankung. 50% der betroffenen Patienten haben ein klinisches Ereignis vor dem 15. Lebensjahr, 40% davon in Form einer Synkope, 10% in Form eines Reanimationsereignisses. Inzwischen sind zahlreiche Gene und damit auch unterschiedliche Long-QT-Typen identifiziert worden. Das Long-QT-1 ist die häufigste Form des Long-QT-Syndroms. Synkopen treten bei dieser Form v. a. unter körperlicher Belastung auf. Eine besondere Häufung von Synkopen tritt beim Schwimmen auf.
Reanimationsereignisse beim Schwimmen sollten immer zu einer Abklärung eines Long-QT-Syndroms führen!
Patienten mit Long-QT-2 beschreiben Synkopen häufig nach emotionalem Stress. Patienten mit Long-QT-3 erleben ein kardiales Ereignis häufig in Ruhe oder im Schlaf.
Long-QT-Syndrome werden, sofern keine weiteren Fehlbildungen vorliegen, auch als Romano-Ward-Syndrom zusammengefasst, und werden autosomal dominant vererbt. Liegt zusätzlich eine Innenohrschwerhörigkeit vor, liegt ein Jervell-Lange-Nielsen-Syndrom vor. Die Vererbung ist autosomal rezessiv. Die Diagnose eines Long-QT-Syndroms wird aufgrund der Befunde im EKG (Ruhe- sowie Belastungs-EKG), der Eigen- und Familienanamnese gestellt. Im EKG sind dabei folgende Befunde wegweisend:
eine verlängerte QTc-Zeit (Frequenz korrigierte QT-Zeit),
eine auffällige T-Wellen-Morphologie mit gekerbten T-Wellen oder T-Wellen mit wechselnder Morphologie,
eine nicht altersentsprechende Bradykardie und
dokumentierte Torsade-de-Pointes.
Im Rahmen der Eigenanamnese sind v. a. Synkopen abzufragen, die beim Long-QT-Syndrom (bei körperlicher oder emotionaler Belastung) auftreten können.
Zusätzlich zur Eigenanamnese ist die Familienanamnese für die Diagnosestellung wichtig:
Gibt es Familienmitglieder mit LQTS?
Plötzliche Todesfälle bei Familienmitgliedern <50 Jahre?
Die Befunde sind in einem Risikoscore zusammengefasst, der 1993 von Schwartz publiziert wurde. Dieser Risikoscore soll eine Entscheidungshilfe sein, inwiefern das Vorliegen eines Long-QT-Syndroms (un)wahrscheinlich ist.
Die molekulargenetische Analyse führt in ca. 80% der betroffenen Patienten zu einem Ergebnis. 20% der Genloci sind dementsprechend noch unbekannt.
Therapie
Beim Long-QT-Syndrom besteht, abhängig von der erlebten Symptomatik und des molekulargenetischen Befunds (falls dieser vorliegt), folgende Empfehlung:
Generell wird allen Patienten, insbesondere Patienten mit LQTS-1, empfohlen, auf Sport mit Wettbewerbscharakter zu verzichten.
Zusätzlich wird eine Tachykardieprophylaxe in Form von β-Blocker empfohlen, die insbesondere beim LQTS-1 sehr effektiv ist, und das Risiko eines kardialen Ereignisses von 80% auf unter 5% senken kann. Bei den anderen LQTS-Typen ist der β-Blocker als Tachykardieprophylaxe leider weniger wirksam. Kommt es trotz β-Blocker zu einem kardialen Ereignis ist die Implantation eines ICD indiziert.
Synkopen unter körperlicher Belastung oder nach emotionalem Stress sollten immer abgeklärt werden und an eine Ionenkanalerkrankung denken lassen. Das EKG ist dabei immer in Verbindung mit einer detaillierten Eigen- und Familienanamnese zu beurteilen.