Abstract
Funktionsstörungen der verschiedenen Komponenten motorischer Einheiten führen zu neuromuskulären Erkrankungen (NME), bei denen das zentrale, sensible oder auch autonome Nervensystem und andere Organsysteme mitbetroffen sein können. Allen NME gemein ist eine Verminderung der groben Kraft mit meist progredienter degenerativer Muskelatrophie.
Wichtige neuromuskuläre Erkrankungen (NME)
Die wichtigsten NME sind von proximal nach distal solche
der motorischen Vorderhornzelle → spinale Muskelatrophien,
des Axons oder der Schwann-Zelle → Neuropathien,
der motorischen Endplatte → Myasthenien,
der Ionenkanäle der Muskelzelle → Myotonien,
der Muskelzelle → Muskeldystrophien, Myopathien,
der Mitochondrien → Mitochondriopathien.
Weitere Informationen zu den einzelnen Erkrankungen sowie Querverweise auf hier nicht aufgeführte Differenzialdiagnosen können im Internet gefunden werden (www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM und www.treat-nmd.eu). Myopathien können auch im Rahmen von Systemerkrankungen auftreten. Die Dermatomyositis mit entzündlich veränderter Muskelzelle wird im Kapitel der rheumatologischen Erkrankungen (Kap. 12), Mitochondriopathien werden in Abschn. 9.9 abgehandelt.
Cave
Bei Patienten mit einer Muskelerkrankung besteht ein erhöhtes Risiko einer malignen Hyperthermie, das bei jeder Narkose zu beachten ist. Die Patienten sind mit einem Muskelpass zu versorgen.
Spinale Muskelatrophien
Definition
Spinale Muskelatrophien (SMA) sind in erster Linie degenerative Erkrankungen der motorischen Vorderhornzellen im Rückenmark und ihrer peripheren Axone. Häufig ist der N. hypoglossus mitbetroffen mit dem Symptom einer Zungenfaszikulation.
Epidemiologie
Die klassische autosomal-rezessive SMA ist in der mitteleuropäischen Bevölkerung mit einer Inzidenz von 1:10.000 Lebendgeburten und einer Überträger- oder Heterozygotenfrequenz von 1:50 die zweithäufigste autosomal-rezessiv erbliche Erkrankung des Kindesalters nach der zystischen Fibrose.
Pathogenese
Die autosomal-rezessive SMA wird in ca. 96% der Fälle durch homozygote Deletionen/Genkonversionen des telomeren SMN1-Gens (Chromosom 5q13) verursacht, das das Survival-motor-neuron-1-Protein kodiert. Dies führt zu einem Verlust des SMN1-Produkts und zu einer SMA, deren Verlaufsform statistisch durch eine variable Anzahl zentromerer SMN2-Genkopien bestimmt wird. SMN2 unterscheidet sich von SMN1 durch einen stillen Basenaustausch im Exon 7 und nur wenige SMN2-Transkripte entsprechen einem funktionsfähigen SMN1-Protein. In der Regel gilt, je mehr SMN2-Kopien ein Patient besitzt, desto milder ist der Verlauf seiner SMA (Gendosiseffekt).
Spinale Muskelatrophien Typ 1–3 (SMA1–3)
Klinik, Verlauf
Die autosomal-rezessive SMA des Kindes- und Jugendalters wird nach Zeitpunkt der Manifestation, Schweregrad und Prognose in 3 Verlaufsformen (SMA1–3) unterteilt. Im Folgenden wird der natürliche Verlauf der Erkrankung vor Einführung unten genannter medikamentöser Therapie im Jahre 2017 aufgeführt.
SMA1
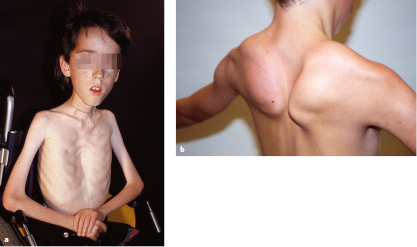
Bei der frühinfantilen schweren Verlaufsform SMA1 (Morbus Werdnig-Hoffmann) berichten die Mütter oft von schwachen Kindsbewegungen während der Schwangerschaft. Die Kinder haben bereits bei Geburt eine generalisierte Muskelschwäche („floppy infant“, Abb. 9.1) mit proximaler Betonung oder zeigen bis zum 6. Lebensmonat erste Symptome. Eine symmetrische, proximal betonte Muskelschwäche, bei der die Beine mehr als die Arme betroffen sind, eine schlechte, meist fehlende Kopfkontrolle, eine wache aufmerksame Mimik, die auf eine relative Aussparung der fazialen Muskulatur weist, eine Atrophie der Interkostalmuskulatur mit Tachypnoe und paradoxer Schaukelatmung, bei der bei Inspiration sich die Rippen senken, Zungenfaszikulation und Polyminimyoklonien oder Tremor der Hände sind typische Symptome. Oft können die Säuglinge nur noch ihre Finger und Zehen bewegen, und das Trinken fällt ihnen schwer.
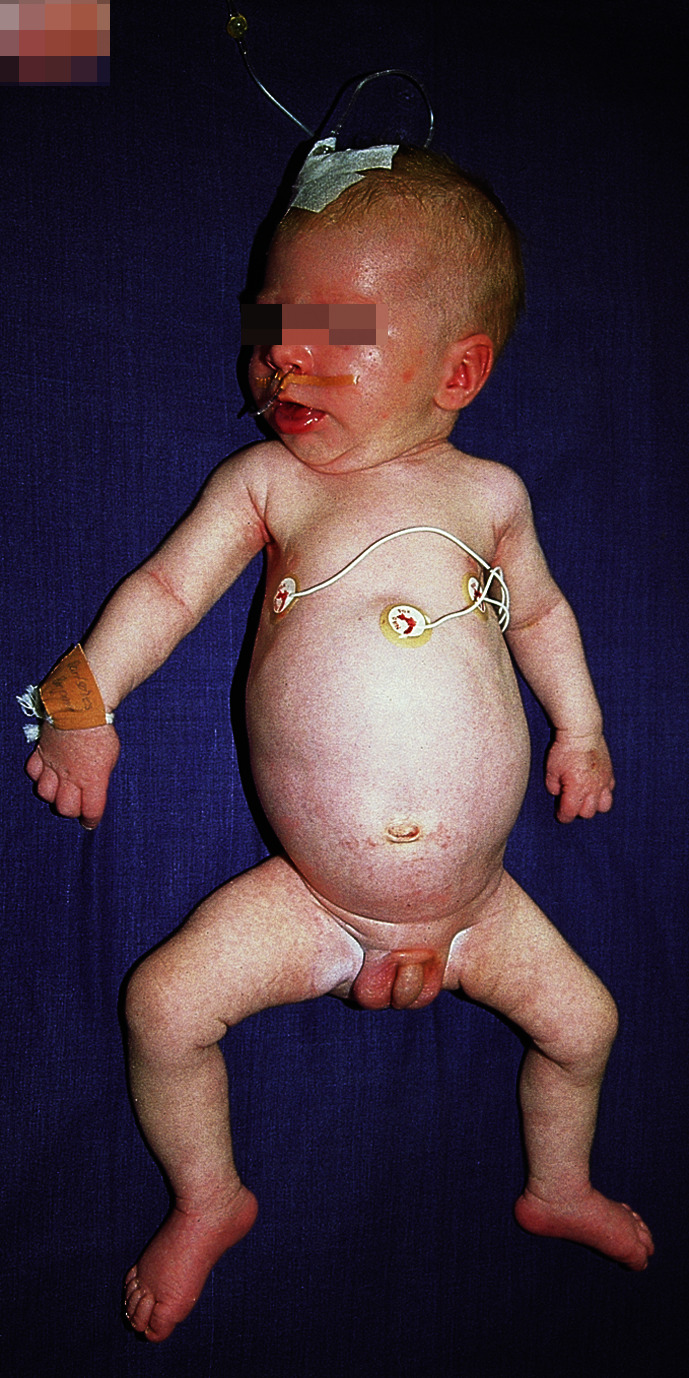
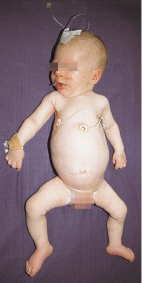
Das charakteristische Bild eines Glockenthoraxes beruht auf einer Atrophie der Interkostal- und Schultergürtelmuskulatur, die typische sog. Froschschenkelhaltung auf einer ausgeprägten Muskelschwäche der Beine (Abb. 9.1). Die Muskeleigenreflexe sind erloschen. Den betroffenen Kindern ist freies Sitzen auch im fortgeschrittenen Alter nicht möglich. Untherapiert überleben aufgrund einer fortschreitenden respiratorischen Insuffizienz und Bulbärparalyse nur 1/3 aller Kinder die ersten 2 Lebensjahre.
Auch eine Arthrogryposis multiplex congenita kann Folge einer pränatal manifesten SMA1 sein.
SMA2
Bei der spätinfantilen intermediären SMA2 treten erste Symptome mit etwa 8, spätestens 18 Lebensmonaten auf. Die Patienten erreichen den motorischen Meilenstein des kurzzeitigen freien Sitzens, können aber nicht frei laufen. Die Muskelschwäche führt oft im Verlauf zu Skoliosen, Trichterbrust, Gelenkkontrakturen und einer eingeschränkten Lungenfunktion. Fast alle Patienten erreichen das 11. und viele (ca. 80%) das 20. Lebensjahr.
SMA3
Die milde Verlaufsform SMA3 (Morbus Kugelberg-Welander) beginnt jenseits des 18. Lebensmonats und vor dem 30. Lebensjahr mit einer großen Variabilität der klinischen Symptome. Alle Patienten lernen, frei zu laufen, manche verlieren ihre Gehfähigkeit; viele führen ein unabhängiges Leben. Insbesondere die proximale Oberschenkelmuskulatur ist atroph.
Diagnose
Entscheidend für die Diagnose einer SMA sind das typische klinische Bild, eine normale oder nur geringfügig erhöhte Kreatinkinaseaktivität (CK) im Serum und der Nachweis einer homozygoten SMN1-Deletion/Genkonversion (Untersuchung einer EDTA-Blutprobe). Ist der Befund negativ, so ist eine SMA zwar noch nicht ausgeschlossen, doch sind Missense-Mutationen selten (ca. 5%). Degenerationen der großen ventromedialen Vorderhornzellen mit Waller-Degeneration der Axone führen zu einem progredienten Ausfall der motorischen Einheiten und einer kompensatorischen kollateralen Aussprossung intakter Motoneurone. Im Elektromyogramm (EMG) entspricht dies einem gelichteten Interferenzmuster (Lattenzaunmuster) und hoch- und breitamplitudigen, oft polyphasischen Aktionspotenzialen. Die histologische Untersuchung einer Muskelbiopsie zum Nachweis einer felderförmigen Muskelfaseratrophie und -hypertrophie und das EMG sind in aller Regel obsolet.
Differenzialdiagnose
Insbesondere kongenitale Muskeldystrophien, die kongenitale myotone Dystrophie, hereditäre kongenitale Neuropathien, das Prader-Willi-Syndrom, Stoffwechselerkrankungen und weitere Formen der SMA kommen in Betracht. Symptome wie Diaphragmaschwäche (Zwerchfellhochstand im Röntgen-Thoraxbild), (olivo)pontozerebelläre Hypoplasie (MRT) und eine deutlich erhöhte Kreatinkinaseaktivität (CK) im Serum (>10-facher Normalwert) schließen die Diagnose einer SMA sicher und eine deutlich erniedrigte Nervenleitgeschwindigkeit (NLG, <70% der Norm) in der Regel aus. Schwere Muskelhypotonie, Trinkschwäche und Zungenfaszikulationen werden auch bei der infantilen Verlaufsform der Glykogenose Typ II, Morbus Pompe, beobachtet.
Therapie
Seit Juli 2017 ist das Medikament Spinraza (Nusinersen) für alle Subformen der SMA als intrathekale Injektion zugelassen. Der Wirkmechanismus beruht auf einer Hochregulation der SMN2-Genkopien was zu einer vermehrten Bildung von SMN-Protein führt. Weitere Medikamente werden derzeit in Studien untersucht. Die Therapie der SMA ist symptomatisch und sollte multidisziplinär angelegt sein. Wichtig sind die physiotherapeutische und orthopädische Therapie (Behandlung von Skoliose und Kontrakturen, Gehhilfe- und Rollstuhlversorgung) sowie eine intermittierende Maskenbeatmung bei den spätinfantilen Verlaufsformen. Die von der STIKO empfohlenen Impfungen sollten gerade wegen der erhöhten Gefährdung durch Atemwegsinfektionen wahrgenommen werden. Den Eltern des Patienten sollte eine genetische Beratung durch einen Humangenetiker angeboten werden.
Distale spinale Muskelatrophie Typ 1 (DSMA1)
Pathogenese
Die autosomal-rezessive DSMA1 (SMA mit „respiratory distress“ Typ 1, SMARD1) beruht auf Mutationen des IGHMBP2-Gens, das das „immunoglobulin μ binding protein 2“ kodiert.
Klinik
Eine oft kongenitale, immer distal betonte Muskelschwäche (Kontrakturen der Füße), inspiratorischer Stridor, leises Schreien, Trinkschwäche und rezidivierende Bronchopneumonien gehen einer in den ersten Lebensmonaten abrupt einsetzenden, rasch progredienten Atemnot voraus, die irreversibel zur Beatmungspflichtigkeit führt. Im Säuglingsalter kann es zu Apnoen mit plötzlichem Kindstod kommen. Während bei der SMA1 eine fortschreitende Atrophie der Interkostalmuskulatur und des proximalen Schultergürtels zu dem Bild eines Glockenthoraxes führt, fehlen diese Symptome bei der DSMA1, da hier das Diaphragma und die distalen Muskeln primär betroffen sind. Typisch ist ein Hochstand des atrophen Zwerchfells im Thorax-Röntgenbild.
Andere spinale Muskelatrophien
Eine fetale Manifestation einer spinalen Muskelatrophie kann, ähnlich wie bei den fetalen Formen einer Myasthenie, zu einer Arthrogryposis multiplex congenita führen mit typischen Begleitsymptomen wie fetaler Hydrops und intrauterine Akinesie, Mikrognathie, tiefsitzende Ohren, multiple Pterygien, perinatale Frakturen, Lungenhypoplasie oder kongenitale Atemnot („lethal congenital contracture syndrome“, LCCS). Andere spinale Muskelatrophien gehen mit einer pontozerebellären Hypoplasie oder einer Myoklonus-Epilepsie einher.
Die X-chromosomal-rezessive SMAX1 oder „spinale und bulbäre Muskelatrophie“ (Kennedy disease) hat ihre Ursache in einer Expansion von CAG-Repeats im Androgenrezeptor-Gen (AR), die zu der Bildung eines neurotoxischen Polyglutaminfragments führen. Neben einer Muskelschwäche der Beine, Muskelkrämpfen und einer Dysarthrie, leiden die männlichen Patienten auch unter endokrinen Störungen wie persistierende Gynäkomastie, Impotenz, Hodenatrophie und Infertilität. Im Unterschied zu anderen spinalen Muskelatrophien ist die CK im Serum oft massiv erhöht. Manifest wird die Erkrankung im jungen Erwachsenen- selten im Jugendalter.
Polyradikuloneuritis Guillain-Barré
Epidemiologie
Die Inzidenz liegt bei etwa 1–2:100.000 pro Jahr.
Pathogenese
Das Guillain-Barré-Syndrom (GBS) ist eine postinfektiöse Autoimmunerkrankung. In 80% der Fälle gehen eine Infektion des Respirations- oder Gastrointestinaltrakts voraus. CMV, EBV, Mykoplasmen, Haemophilus influenzae und insbesondere Campylobacter jejuni sind häufige Erreger. Die Immunantwort richtet sich nicht nur gegen Epitope der Erreger, sondern auch gegen solche des Axons und der Schwann-Zelle (molekulares Mimikry). Bei einem Teil der Patienten werden Antikörper gegen Ganglioside gefunden. Sehr selten kann ein GBS auch nach einer Impfung auftreten (z. B. Influenza, Hepatitis).
Klinik
Neben Rücken- und Beinschmerzen ist das erste Symptom eine aufsteigende, meist symmetrische Muskelschwäche, die oft in den Beinen beginnt. Diese Muskelschwäche kann zu einer vollständigen Lähmung der Extremitäten sowie Atemlähmung führen. Die Muskeleigenreflexe sind schwach oder nicht auslösbar. Störungen der Sensibilität mit Kribbelparästhesien oder Taubheitsgefühl, Herzrhythmusstörungen und erhebliche Schwankungen des Blutdrucks sind Ausdruck einer Mitbeteiligung des autonomen Nervensystems. Ein schwerer Verlauf mit Beatmungspflicht wird bei ca. 15% aller Patienten beobachtet und erfordert eine entsprechende Überwachung der Patienten. Bis zum Erreichen des Krankheitsgipfels der initialen progredienten Phase vergehen durchschnittlich 10 Tage (1–35 Tage), und nach ca. 5–10 Wochen können die Patienten in der Regel wieder frei laufen. Sonderformen des GBS sind das Miller-Fischer-Syndrom mit Ophthalmoplegie, Areflexie und Ataxie sowie die chronisch inflammatorische demyelinisierende Polyneuropathie.
Diagnose
Diagnostische Symptome sind eine rasch progrediente (≤4 Wochen), meist symmetrische Muskelschwäche der Extremitäten sowie eine Abschwächung aller und Verlust wenigstens der distalen Muskeleigenreflexe, oft auch einhergehend mit Dys- oder Parästhesien. Weitere Befunde unterstützen die Diagnose: Fazialisschwäche und bulbäre Symptome, autonome Dysregulation, im Liquor erhöhte Eiweißkonzentration bei normaler Glukosekonzentration und Zellzahl (<10 mononukleäre Zellen/mm3; zytalbuminäre Dissoziation) oder Pleozytose (<50 Zellen/mm3), initial kein Fieber und normale Nervenleitgeschwindigkeit (NLG) sowie im Krankheitsverlauf Zeichen einer proximalen Demyelinisierung mit verlangsamter NLG, multifokalen Leitungsblöcken und Anomalien der F- und H-Antwort.
Differenzialdiagnose
Blasen-/Mastdarmfunktionsstörungen, Restharn in der Blase und/oder fehlender Sphinkterreflex sind Symptome, die auch an einen spinalen raumfordernden oder entzündlichen Prozess (Myelitis transversa) denken lassen. Eine deutliche, anhaltende Asymmetrie der Paresen sind Symptome spinaler Tumoren, einer Syrinx, eines Tethered-cord-Syndroms oder auch einer Poliomyelitis. Eine Ptosis lässt an Botulismus denken. Liquorbefunde mit höherer Zellzahl oder sehr hohen Eiweißwerten weisen auf ZNS-Infektionen hin. Eine protrahierte initiale progrediente Phase (>4 Wochen) lenkt den Verdacht auf eine chronisch-entzündliche demyelinisierende Polyradikuloneuritis. Borreliose, Diphtherie, Mykoplasmeninfektion, spinale Form der multiplen Sklerose, systemischer Lupus erythematodes, Porphyrie und Lymphom (M. Hodgkin) sind weitere Differenzialdiagnosen.
Therapie
Die Therapie der Wahl ist die Gabe von 7S-Immunglobulin G. Sicher profitieren die Patienten von dieser Therapie im Sinne einer rascheren Rekonvaleszenz, die Langzeitprognose ist aber durch die Therapie nicht beeinflussbar. Eine Behandlungsalternative ist besonders bei älteren Kindern die Plasmapherese oder Immunadsorption. Kortikosteroide sind in der Regel ohne Wirkung, ausgenommen in Fällen, in denen der Verdacht auf eine chronisch-entzündliche demyelinisierende Polyradikuloneuritis aufkommt oder der einer Myelitis transversa nicht ausgeschlossen werden kann. Antithrombose- und Physiotherapie sind oft notwendige Supportivmaßnahmen.
Prognose
Die Prognose ist in der Kindheit meist gut. Nach 6 Monaten sind 92% der Patienten weitgehend beschwerdefrei und alle in der Lage, ohne Hilfen zu laufen. Ungünstig für die Langzeitprognose sind ein Manifestationsalter von <9 Jahren und eine rasche Progredienz der maximal erreichten Muskelschwäche. Ca. 20% aller Patienten zeigen nach 5 Jahren leichte neurologische Defizite.
Hereditäre Neuropathien
Definition
Hereditäre motorisch-sensible Neuropathien (HMSN), auch Charcot-Marie-Tooth-(CMT)-Neuropathien genannt, bilden eine heterogene Gruppe von Erkrankungen der von den Schwann-Zellen gebildeten Markscheiden oder des Axons. Nach der historischen Klassifikation unterscheidet man 7 HMSN-Formen nach elektrophysiologischen und histopathologischen Kriterien (HMSN1 = demyelinisierende, HMSN2 = axonale Neuropathie), nach besonderem Krankheitsverlauf (HSMN3 = schwere demyelinisierende Form des frühen Kindesalters) und nach Zusatzsymptomen (HMSN4–7). Der genetischen Klassifikation folgend, werden die CMT-Neuropathien zusätzlich nach dem Erbgang eingeteilt.
Epidemiologie
Einer von 2.500 Menschen leidet unter einer Form der heterogenen HSMN. Die häufigste Form ist die autosomal-dominante demyelinisierende CMT1A (= HMSN1A).
Pathogenese
Verschiedene Mutationen in für Myelinbestandteile kodierenden Genen wie dem Peripheral-myelin-protein-22kD-Gen (PMP22), dem Myelin-protein-zero-Gen (MPZ) und dem Gap-junction-protein-β1-Gen (GJB1) sind als Ursache verschiedener HSMN oder CMT beschrieben worden. Unterschiedliche Mutationen im selben Gen können zu unterschiedlichen Phänotypen führen. So wird das klinische Bild der PMP22-Mutationen von einem Gendosiseffekt bestimmt.
Bei einer Deletion der Region (Gendosis = 50%) leidet der hemizygote Patient unter einer Neuropathie mit Neigung zu Druckläsionen (HNPP).
Eine heterozygote Duplikation (Gendosis = 150%) ist Ursache der autosomal-dominanten CMT1A. Die Duplikation führt zu einer Überexpression des Proteins in den Schwann-Zellen, histologisch zu chronischen De- und Remyelinisierungen mit Zwiebelschalenformationen und verdickten Nervensträngen und elektrophysiologisch zu einer reduzierten NLG.
Eine homozygote Duplikation (Gendosis = 200%) und auch Punktmutationen im PMP22-Gen führen zu einer schwereren Verlaufsform aus der Gruppe der hypertrophen Neuropathien Déjérine-Sottas (DSS, HMSN3).
Charcot-Marie-Tooth-Neuropathie Typ 1 (CMT1)
Die Hauptsymptome der 4 autosomal-dominanten, meist demyelinisierenden CMT1-Formen sind eine langsam progrediente, symmetrische, distal ausgeprägte Muskelschwäche und -atrophie mit dünnen Unterschenkeln (Storchenbeine). Früh betroffen sind die Handmuskeln mit einer Palmaratrophie (verstrichener Daumenballen), die Fußhebermuskeln, wodurch der Patient nicht auf den Hacken stehen oder laufen kann, die peronäale Muskelgruppe mit der Folge eines Steppergangs sowie die Zehenstrecker mit Ausbildung eines Hohlfußes. Erste Symptome treten bei 40–75% aller betroffenen Kinder bis zum 11., selten vor dem 4. Lebensjahr auf. Die Muskeleigenreflexe sind abgeschwächt oder erloschen. Die motorischen NLG aller Nerven ist auch bei noch gut erhaltener Muskeltrophik reduziert (N. medianus ≤38 m/s; normal >52 m/s). Weitere seltenere Symptome sind verdickt tastbare Nervenstränge (N. retroauricularis), leichter Tremor, propriozeptive Ataxie, distal symmetrisch ausgeprägte Sensibilitätsstörungen, vasomotorische Störungen wie Kältegefühl der Beine oder livide Marmorierung der Haut, Dysästhesien und Kyphoskoliose.
Déjérine-Sottas-Syndrom (DSS, HMSN3)
Die diagnostischen Kriterien der demyelinisierenden Neuropathie DSS sind ähnlich denen der CMT1, unterscheiden sich aber von dieser durch eine frühe Manifestation im Säuglings- und Kleinkindesalter (meist im 2. Lebensjahr) und folgende Symptome: Skelettdeformitäten, verzögertes Erreichen der motorischen Meilensteine, häufig verdickte Nervenstränge, stark reduzierte motorische NLG (N. medianus ≤10 m/s), erloschene Muskeleigenreflexe, Ataxie und eine erhöhte Eiweißkonzentration im Liquor. Seltenere Symptome sind Sensibilitätsstörungen, Dysästhesien oder Schmerzen und Pupillenanomalien.
Charcot-Marie-Tooth-Neuropathie Typ 2 (CMT2)
Der Phänotyp der axonalen, meist autosomal-dominant (selten rezessiv) vererbten CMT2-Formen ähnelt klinisch dem der CMT1-Formen. Die CMT2-Formen sind seltener, und die Patienten sind in der Regel älter und zeigen eine normale oder leicht verminderte NLG (N. medianus >35 m/s).
Hereditäre Neuropathie mit Neigung zu Druckläsionen (HNPP)
Bei dieser autosomal-dominanten „hereditary neuropathy with liability to pressure palsies“ (HNPP) kommt es durch geringe stumpfe Traumata, Druck oder Zug zu rezidivierenden, meist komplett reversiblen Lähmungen und Parästhesien (selten Schmerzen) peripherer Nerven (Schulter-, Arm- und Unterschenkelbereich – z. B. nach längerem Sitzen auf einer Bank).
Prognose
Die Prognose der Muskelatrophien vom Typ CMT1 und CMT2 sind bei einem langsam progredienten Verlauf meist gut, nur wenige Patienten sind schwer behindert oder berufsunfähig. Die Prognose der Neuropathie Déjérine-Sottas (DSS, HMSN3) reicht von der einer schweren Behinderung bis zu einer ähnlich langsamen Progredienz wie bei CMT1.
Diagnose
Die Familienanamnese, das Manifestationsalter und der Schweregrad bieten Hinweise auf den Vererbungsmodus und den Subtyp. Elektrophysiologische Untersuchungen (NLG auch der Eltern) erlauben zum einen eine Zuordnung zur Gruppe der demyelinisierenden HMSN1/CMT1- oder axonalen HMSN2/CMT2-Neuropathien und zu dominanten oder rezessiven Vererbungsmodi.
Die im Kindes- und Jugendalter häufigste Neuropathie ist die autosomal-dominante demyelinisierende CMT1A und wird bei entsprechend passenden klinischen, elektrophysiologischen und genetischen Befunden in aller Regel molekulargenetisch als erste abgeklärt. Ist der Befund negativ, so schließen sich weitere molekulargenetische Untersuchungen an. Bei der schweren demyeliniserenden Neuropathie DSS (HMSN3) wird zunächst nach Punktmutationen im PMP22- und MPZ-Gen gesucht, es folgen Untersuchungen des EGR2-Gens und weiterer Kandidatengene. Die HNPP wird durch Punktmutationen und Deletionen des PMP22-Gens verursacht.
Differenzialdiagnose
Neben Polyneuropathien toxischer Genese verursacht eine Reihe von Stoffwechselerkrankungen eine Neuropathie. Hierzu gehört die familiäre isolierte Vitamin-E-Defizienz, die durch eine Bestimmung der α-Tocopherol-Konzentration im Plasma leicht abgeklärt werden kann und behandelbar ist.
Therapie, Beratung
Allein eine molekulargenetisch gesicherte Diagnose erlaubt die Einschätzung der Prognose und ermöglicht damit die Familien- und Berufsberatung. Eine Heilung ist nicht möglich. Physiotherapie zur Kontrakturprophylaxe und orthopädische Maßnahmen zur Korrektur und Vorbeugung von Fehlstellungen plegischer Extremitätenabschnitte und einer Skoliose sind indiziert.
Myasthenien
Ein Aktionspotenzial bzw. Nervenimpuls öffnet an der präsynaptischen Membran spannungsabhängige Kalziumkanäle. Die schlagartig in den Endkolben des Axons eindringenden Kalziumionen führen zu einer Freisetzung des Neurotransmitters Azetylcholin (ACh) aus seinen präsynaptischen Speichervesikeln. ACh bindet an die postsynaptischen nikotinischen ACh-Rezeptoren (AChR), die aus je 5 Untereinheiten (adulter Typ: 2α, β, ε, δ; fetaler Typ: γ statt ε) einen Kationenkanal bilden. Diese Kanäle werden durch ACh geöffnet und bauen Endplattenpotenziale auf, deren Amplitude proportional der Anzahl der mobilisierten ACh-Moleküle und angeregten AChR ist. Ab einer kritischen Amplitudenhöhe führen diese Endplattenpotenziale zur Muskelkontraktion. Eine kontrolliert rasche Beendigung der ACh-Wirkung auf den Rezeptor gewährleistet die ACh-Esterase (AChE), die im synaptischen Spalt ACh spaltet.
Myasthenien beruhen auf prä-, intra- oder postsynaptischen Defekten der neuromuskulären Erregungsübertragung.
Myasthenia gravis
Epidemiologie
Die Prävalenz der Myasthenia gravis liegt bei 3–10:100.000 Einwohnern, und rund 10–15% aller Patienten sind jünger als 16 Jahre. Die bereits im präpubertären Alter bestehende Geschlechtswendigkeit von w:m = 2:1 steigt nach der Pubertät auf w:m = 14:1.
Pathogenese
Bei dieser postsynaptischen Autoimmunerkrankung beeinträchtigen Antikörper (AK) der IgG-Subklasse, die gegen Bestandteile des adulten AChR gerichtet sind, die neuromuskuläre Erregungsübertragung. Es wird ein Zusammenhang zwischen den bei 90% der Patienten vorhandenen Thymusveränderungen und der Entstehung der Myasthenia gravis (MG) diskutiert. Die AChR-AK-Titer korrelieren nicht mit der Schwere, jedoch häufig mit dem Verlauf der Erkrankung und dem Ansprechen auf eine Therapie. 10–20% aller MG-Patienten sind seronegativ für AChR-AK. In der Mehrzahl dieser Patienten (60–70%) lassen sich Antikörper gegen die postsynaptische muskelspezifische Rezeptor-Tyrosinkinase (MUSK) nachweisen, die eine essenzielle Rolle bei der Entwicklung des AChR spielt. Sehr viel seltener liegen Autoantikörper gegen Titin, Low-density-lipoprotein-receptor-related-Protein 4 (LRP4) oder Agrin vor.
Während der Entwicklung werden von der 8. bis zur 33. Schwangerschaftswoche (SSW) fetale AChR (γ-Untereinheit) synthetisiert, die mit der Entwicklung schrittweise durch den adulten Typ des AChR (ε-Untereinheit) ersetzt werden. Durch den diaplazentaren Übertritt mütterlicher IgG-AK gegen den adulten Typ des AChR kann es zu einer transienten neonatalen MG des Kindes kommen. In seltenen Fällen sind mütterliche AK gegen den fetalen Typ des AChR gerichtet, was klinisch eine Arthrogryposis multiplex congenita zur Folge hat.
Klinik, Verlauf
Kardinalsymptom der MG ist eine abnorme Ermüdbarkeit der Muskulatur, die im Tagesverlauf zunimmt und sich nach Ruhepausen bessert. Ptosis und Doppelbilder, die auf einer Schwäche der äußeren Augenmuskeln beruhen, sind typische Frühsymptome, nasale Sprache und Schluckstörungen sind Zeichen einer Schwäche der Kau- und Schlundmuskulatur. Im Verlauf schreitet die Erkrankung über die Rumpfmuskulatur (Atemprobleme) bis zu den Extremitätenmuskeln fort.
Cave
Infektionen, eine abrupte Beendigung einer Immunsuppression, muskelrelaxierende Substanzen und Medikamente wie Antibiotika können zu einer myasthenen Krise mit lebensbedrohlicher Ateminsuffizienz und hoher Mortalität führen.
Die Hauptsymptome der transienten neonatalen Myasthenia gravis sind Saug-/Fütterprobleme, generalisierte Muskelhypotonie, Atembeschwerden, leises Schreien, Gesichtsmuskelschwäche und seltener eine Ptosis. Ähnlich der fetalen Manifestation einer spinalen Muskelatrophie ist die oft letale „fetale Myasthenia gravis“ neben einer Arthrogryposis multiplex congenita charakterisiert durch fetalen Hydrops und intrauterine Akinesie, Mikrognathie, tief sitzende Ohren sowie Lungenhypoplasie und kongenitale Atemnot, nicht aber durch eine Myasthenie, da jenseits der 33. SSW die mütterlichen, gegen den fetalen AChR gerichteten AK ihre Antigene und damit Wirkung verloren haben.
Diagnose
Der klinischen Untersuchung folgen die Elektromyographie (EMG) unter niederfrequenter repetitiver Stimulation eines Nerven mit 3 Hz zum Nachweis eines pathologischen Dekrement des muskulären Summenaktionspotenzials, die Bestimmung der zirkulierenden AChR-AK und bei seronegativem Befund die der MUSK-AK. Ein zellbasierter AChR-Cluster-Test kann trotz negativem AChR-AK-Test positive Ergebnisse liefern. Eine Testung mit dem AChE-Hemmer Edrophoniumchlorid (Camsilon-Test) ist dann sinnvoll, wenn Symptome vorliegen, die sich rasch verbessern können wie eine Ptose. Diese Untersuchung sollte unter Intubationsbereitschaft und nach Legen eines venösen Zugangs und Aufziehen von Atropin zur Antagonisierung von Nebenwirkungen (Brady-/Arrhythmie, Hypotonie) erfolgen (Abb. 9.2).

Differenzialdiagnose
Zu denken ist an Krankheiten wie kongenitale myasthene Syndrome, Schilddrüsenerkrankungen, Kearns-Sayre-Syndrom und die okulopharyngeale Muskeldystrophie. Dem Lambert-Eaton-Myasthenie-Syndrom liegen AK gegen die präsynaptischen Kalziumkanäle zugrunde, die zu einer verminderten ACh-Freisetzung führen. Eine Manifestation im Kindesalter ist selten und kann paraneoplastisch bedingt sein.
Therapie
Die Therapie basiert zum einen auf der Verbesserung der neuromuskulären Übertragung durch Hemmung der AChE an der neuromuskulären Synapse und zum anderen auf der Inhibition der Autoimmunreaktion durch Immunsuppressiva. Der AChE-Hemmer Pyridostigminbromid ist das Medikament der ersten Wahl. In der Regel sollte zeitnah eine kombinierte mit Prednisolon und steroidsparendem Azathioprin angeschlossen werden. Es ist zu bedenken, dass sich die Symptomatik in den ersten 10 Tagen der Steroidtherapie verschlechtern kann. Ciclosporin A oder Cyclophosphamid werden wegen des Nebenwirkungsprofils als Langzeittherapeutika nicht mehr gerne eingesetzt, eine Alternative ist Mycophenolatmofetil, allerdings im Kindes- und Jugendalter als „off label use“. 7S-Immunglobulin-G-Infusionen helfen bei bulbärer Symptomatik, i.v.-Immunglobulingaben und Plasmapherese in Krisen und werden auch präoperativ eingesetzt. Eine Thymektomie ist bei kindlichen und jugendlichen Patienten sinnvoll und zeigt bessere Ergebnisse, wenn sie früh im Verlauf durchgeführt wird. Beschwerdefrei werden 2/3 aller Patienten, eine Besserung innerhalb von 10 Jahren erfährt 1/3.
Kongenitale myasthene Syndrome (CMS)
Pathogenese
Den heterogenen CMS liegen Mutationen synapsenassoziierter Gene zugrunde. Die präsynaptische autosomal-rezessive CMS mit episodischer Apnoe beruht auf Mutationen des CHAT-Gens, dessen Produkt, die Cholin-Acetyl-Transferase, die Biosynthese von ACh katalysiert. Die autosomal-rezessive synaptische CMS mit Endplatten-Acetylcholinesterase-Defizienz ist verursacht durch Mutationen des COLQ-Gens, dessen Produkt die AChE mit der Basalmembran verankert. Postsynaptische CMS und autosomal-dominante Slow-channel-Syndrome haben ihre Ursachen in Mutationen der Gene, die die Untereinheiten des adulten Typs des AChR kodieren (CHRNA1/B1/D/E). Andere postsynaptische CMS beruhen auf Mutationen des MUSK- (Abschn. 9.4.1, Pathogenese) oder Rapsyn-Gens (RAPSN), deren Produkte an der Entwicklung des AChR beteiligt sind. Mutationen des CHRNG-Gens, das die den fetalen AChR determinierende γ-Untereinheit kodiert, führen zu einer Arthrogryposis multiplex congenita mit multiplen Pterygien (Escobar-Syndrom) und den gleichen Symptomen, die auch mütterliche Antikörper gegen den fetalen AChR hervorrufen können (Abschn. 9.4.1).
Klinik, Verlauf
Typisch sind eine Manifestation im Neugeborenen- bis Kleinkindesalter unterschiedlicher Schweregrade, die von einer leichten Muskelschwäche bis zur schweren Verlaufsform eines „floppy infant“ reichen. Die Begleitsymptome sind wechselnd ausgeprägte Ptosis, Facies myopathica, leises Schreien sowie Schluck- und Saugschwächen. Im Säuglingsalter kann es zu Apnoen mit plötzlichem Kindstod oder bei respiratorischen Infekten zu einer lebensbedrohlichen Ateminsuffizienz kommen. Im Jugendalter treten eine abnorme Ermüdbarkeit der Muskulatur bei Belastung mit Ptosis mit oder ohne Augenmuskellähmung und eine meist proximal betonte Muskelschwäche der Extremitäten in den Vordergrund. Bei einigen CMS kann es auch jenseits des Kleinkindesalters zu krisenhaften Verschlechterungen mit lebensbedrohlichen Atemmuskellähmungen kommen.
Diagnose
Neben der typischen klinischen Symptomatik mit Manifestation im Säuglings- oder Kindesalter sind der fehlende Nachweis von AChR- und MUSK-Antikörpern im Serum Voraussetzung für die Diagnose eines CMS. Im EMG zeigt sich unter der repetitiven Stimulation (Abschn. 9.4.1, Diagnose) ein pathologisches Dekrement. Derzeit sind >30 Gene kausal für CMS verantwortlich. RAPSN- und CHRNE-Mutationen sind relativ häufig und zuerst abzuklären.
Therapie, Prognose
In der Regel ist die Prognose gut. In vielen Fällen sprechen die Patienten auf den AChE-Hemmer Pyridostigminbromid an (Abb. 9.2), dies gilt nicht für Patienten mit einer COLQ-Mutation, bei denen Ephedrin oder Salbutamol positive Effekte zeigen und AChE-Hemmer kontraindiziert sind. Eine Behandlungsalternative bietet das 3,4-Diaminopyridin, durch das die ACh-Freisetzung erhöht wird und das auch in Kombination mit Pyridostigminbromid gegeben werden kann. Patienten mit einem Slow-channel-Syndrom profitieren von einer Therapie mit Chinidin. Eine solche Therapie kann bei anderen Formen der CMS zu einer Verschlechterung der Symptomatik führen. So ist die molekulargenetische Abklärung wichtige Voraussetzung für eine langfristig sinnvolle Therapie.
Myotonie, periodische Paralyse und Paramyotonie
Grundlagen
Endplattenpotenziale (Abschn. 9.4, Grundlagen) öffnen Natriumkanäle des Sarkolemm. Deren Aktionspotenziale wiederum öffnen an der Triade des T-tubulären Systems und sarkoplasmatischen Retikulums Kalziumkanäle, Kalzium wird freigesetzt und die Muskelkontraktion findet statt.
Definition
Ionenkanaldefekte der Muskelzelle führen zu einer Myotonie (hyperexzitables Sarkolemm) mit verzögerter Erschlaffung der Muskulatur oder zu einer Paralyse (hypoexzitables Sarkolemm) mit temporär eingeschränkter Muskelfunktion. Durch wiederholte Muskelkontraktionen nimmt die Myotonie bei Chlorid- und weniger ausgeprägt bei Natriumkanaldefekten ab (Warm-up-Phänomen), während sie bei der Paramyotonie „paradox“ zunimmt. Die periodischen Paralysen zeichnen sich durch spontan auftretende Episoden schlaffer Muskelschwäche aus.
Pathogenese
Die Myotonia congenita vom autosomal-rezessiven Typ Becker und vom dominanten Typ Thomsen sind verursacht durch Mutationen eines Chloridkanals (CLCN1-Gen), seltener auch des Natriumkanals. Nach willkürlicher Kontraktion kommt es bei diesen Patienten zu weiteren Muskelaktionspotenzialen, weil die vorwiegend stabilisierende Funktion des Chloridkanals eingeschränkt ist. Diese Dauererregung imponiert klinisch als Muskelstarre. Die hyperkaliämische periodische Paralyse und die Paramyotonia congenita sind verursacht durch Mutationen der α-Untereinheit eines Natriumkanals (SCN4A-Gen), die primäre hypokaliämische periodische Paralyse durch Mutationen der α1-Untereinheit eines Kalziumkanals (CACNA1S-Gen), eines spannungsabhängigen Kaliumkanals (KCNE3-Gen) oder des SCN4A-Gens.
Eine Ausnahme bildet die seltene chondrodystrophische Myotonie (Schwartz-Jampel-Syndrom), die mit Kleinwuchs und Skelettfehlbildungen einhergeht und bei der die Myotonie nicht primär auf einem Ionenkanaldefekt beruht, sondern auf Mutationen des Perlecan-Gens.
Cave
Ionenkanaldefekte der Muskelzelle gehen mit einem erhöhten Narkoserisiko einher.
Myotonia congenita Becker und Thomsen
Die generalisierte, autosomal-rezessive Myotonia congenita vom Typ Becker wird zwischen dem 3. und 30. Lebensjahr klinisch manifest. Die ersten Beschwerden beginnen in den Beinen und breiten sich in den folgenden Jahren auf die Arm-, Gesichts- und Nackenmuskulatur aus. Bei der Mehrzahl der Patienten folgt nach einer transienten Muskelschwäche der Arme oder Hände über mehrere Sekunden die myotone Muskelversteifung. Letztere hindert die Betroffenen an der Ausführung einer Bewegung und ist von einer anhaltenden Inaktivität gefolgt (Unfähigkeit, Hände nach repetitivem Faustschluss rasch zu öffnen). Viele Patienten entwickeln eine deutliche Hypertrophie ihrer Muskulatur. Typisch sind das Warm-up-Phänomen und sog. „myotonic runs“ im Elektromyogramm (EMG). Die Symptome verstärken sich bei Kälte, in Stresssituationen und nach Ruhe.
Der autosomal-dominante Typ Thomsen stellt eine mildere Verlaufsform dar. Das betroffene Neugeborene kann nach dem Schreien oder nach dem Waschen mit kaltem Wasser nicht die Augen öffnen. Im späteren Lebensalter bleibt bei Blickwendung nach unten das Augenweiß sichtbar, da das Oberlid verzögert mitgeht („lid lag“, Graefe-Zeichen).
Hypokaliämische periodische Paralyse
Die hypokaliämische periodische Paralyse manifestiert sich meist vor dem 16. Lebensjahr. Während es bei der milden Form nur vereinzelt zu Episoden mit generalisierten Muskelschwächeanfällen kommt, sind bei der schweren Form Anfälle so häufig, dass zwischen den Episoden keine normale Muskelkraft erlangt wird. Die mit einer Hypokaliämie verbundenen Anfälle treten typischerweise in der zweiten Nachthälfte und beim Aufstehen auf. Auslösende Faktoren sind hohe körperliche Belastung oder kohlenhydrat- bzw. natriumreiche Nahrung am Vortag. Unabhängig von der Häufigkeit der Anfälle kann sich eine chronisch progrediente Myopathie entwickeln. Myotone EMG-Veränderungen findet man in der Regel nicht. Ist der Kaliumspiegel im Serum auch zwischen den Anfällen erniedrigt, so kann es sich um eine sekundäre hypokaliämische periodische Paralyse mit renalem oder gastrointestinalem Kaliumverlust oder auch um eine Thyreotoxikose handeln.
Hyperkaliämische periodische Paralyse
Das klinische Bild der hyperkaliämischen periodischen Paralyse ist variabler als das der hypokaliämischen Form. Patienten können von einer Myotonie mit entsprechenden Veränderungen im EMG oder einer Paramyotonie betroffen sein. Erste Schwächeanfälle treten innerhalb der ersten 10 Lebensjahre auf und nehmen typischerweise im Alter wieder ab. Die morgens auftretenden Schwächeanfälle sind häufiger und milder als bei der hypokaliämischen Form. Anfälle können durch kaliumhaltige Nahrung, Stress und Glukokortikoide provoziert werden. Im Anfall kann der Kaliumspiegel bis auf ca. 6 mmol/l ansteigen oder auch normal bleiben.
Paramyotonia congenita
Hauptsymptom der Paramyotonia congenita sind das Auftreten bei Kälteexposition und die „paradoxe“ Zunahme der Myotonie während einer muskulären Belastung. Prädilektionsorte sind Gesicht (Amimie bei Kälte) und distale obere Extremitäten. Die paramyotonen Symptome sind kongenital (Nichtöffnen der Augen des Neugeborenen nach dem Schreien oder Waschen mit kaltem Wasser) oder spätestens bis zum 10. Lebensjahr sichtbar, in der Regel nicht progredient und persistieren lebenslang. Muskelschwächeanfälle manifestieren sich während der Adoleszenz.
Diagnose
Die Reihenfolge der zu untersuchenden Gene richtet sich nach der Klinik, die allerdings nur erste Anhaltspunkte bieten kann, da die Symptome sich überlappen. Die häufigste Verlaufsform im Kindesalter ist die Myotonia congenita Becker, die durch Sequenzierung des CLCN1-Gens abgeklärt wird. Wenn dies negativ bleibt, empfiehlt sich eine Untersuchung des SCNA4-Gens.
Therapie
Die molekulargenetische Abklärung ist eine Voraussetzung einer jeden Therapie, da in Abhängigkeit vom betroffenen Ionenkanal die verschiedenen Medikamente die Myotonie verschlimmern oder verbessern können. Eine Therapie der Myotonia congenita ist unter Berücksichtigung der potenziell erheblichen Nebenwirkungen vorhandener Substanzen nur bei schweren Verläufen indiziert. Das Medikament der ersten Wahl ist der Natriumkanalblocker Mexiletin. Medikamente der zweiten und dritten Wahl sind Carbamazepin und Phenytoin. Bei der hypokaliämischen periodischen Paralyse können Schwächeanfälle durch die perorale Einnahme ungesüßten Kaliumchlorids abgemildert werden. Kohlenhydratreiche Mahlzeiten sind ebenso zu vermeiden wie körperliche Belastung. Natrium, Antiphlogistika oder Lokalanästhetika können schwere Anfälle auslösen. Die Therapie mit Acetazolamid und anderen Medikamenten ist Aufgabe des Spezialisten.
Bei der hyperkaliämischen periodischen Paralyse wirken zahlreiche kohlenhydratreiche, kaliumarme Mahlzeiten präventiv. Bariumhaltige Kontrastmittel sind kontraindiziert. Schwächeanfälle können durch die perorale rasche Einnahme von Traubenzucker abgefangen werden. Präventive Maßnahmen zur Stabilisierung der Körpertemperatur und des Kaliumserumspiegels sind bei jedem Eingriff mit Narkose indiziert. Bei der Paramyotonia congenita sollten depolarisierende Muskelrelaxanzien gemieden und eine Auskühlung des Körpers vermieden werden; eine Therapie ist meist überflüssig.
Myotone Dystrophien
Definition
Myotone Dystrophien (DM) sind Multisystemerkrankungen, bei denen neben dem Muskel (Myotonie und Muskeldystrophie) weitere Organsysteme beteiligt sind. Man unterscheidet zwei Formen, die myotone Dystrophie Typ 1 (DM1; Curschmann-Steinert-Krankheit) und die DM Typ2 (DM2; proximale myotone Myopathie, PROMM).
Epidemiologie
Die Inzidenz liegt bei 1:8.000 (DM1) und 1:20.000 (DM2).
Pathogenese
Die beiden Formen der DM sind autosomal-dominant und beruhen auf Repeat-Expansionen. Genetische Ursache der DM1 ist die Verlängerung eines CTG-Trinukleotid-Repeats auf >50 Repeats in der 3’-nicht-translatierten Region des Myotonin-Protein-Kinase-Gens (DMPK) und bei der DM2 die eines CCTG-Repeats (ca. 5.000 Repeats, normal: 10–30 Repeats) im 1. Intron des Zinc-finger-protein-9-Gens (ZNF9). Die expandierten CUG- und CCUG-(RNA-)Repeats nehmen Einfluss auf die RNA-Prozessierung und Expression verschiedener Gene. So ist bei der DM1 die Myotonie durch eine defekte RNA-Prozessierung des intakten Chloridkanal-Gens CLCN1 verursacht (Abschn. 9.5, Pathogenese). Expandierte CTG-Repeats sind instabil und können von Generation zu Generation weiter zunehmen. Entsprechend können bei der DM1 das Manifestationsalter ab- und die Schwere der Erkrankung zunehmen (Antizipation). Eine solche Genotyp-Phänotyp-Korrelation ist bei der DM2 deutlich weniger ausgeprägt.
Myotone Dystrophie Typ 1 (DM1)
Die schwerste und kongenitale Form der DM1 mit >1.000 CTG-Repeats ist immer vererbt und dies überwiegend von der Mutter, möglicherweise gehen Spermatozyten mit sehr hoher Repeatzahl zugrunde. Die Neugeborenen zeigen eine Facies myopathica, meist mit hochgezogener Oberlippe, und eine generalisierte Hypotonie („floppy infant“) mit oft lebensbedrohlicher Ateminsuffizienz in den ersten Lebenswochen. Später halten diese Kinder den Mund geöffnet, was den Eindruck der vorhandenen mentalen Retardierung noch verstärkt (Abb. 9.3). Die Myotonie tritt meist erst nach dem 10. Lebensjahr auf.

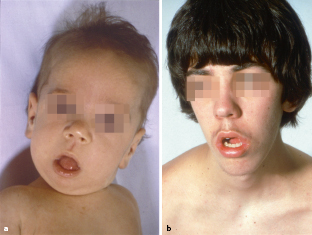
Hauptsymptome der juvenilen Form sind Antriebsarmut, distal betonte Muskelschwäche, Myotonie, ausgeprägte Facies myopathica, Ptosis, Atrophie der Gesichtsmuskulatur, durch die der Patient einen abgezehrten, hageren Eindruck vermittelt. Die Myotonie kann durch eine Perkussion der Zunge und durch ein Elektromyogramm (EMG) nachgewiesen werden. Weitere Symptome der kongenitalen wie der juvenilen Formen sind Katarakt und Innenohrschwerhörigkeit, Schluckstörung und Dysarthrie, Symptome einer Schwäche der glatten Muskulatur mit Cholelithiasis, Obstipation oder Megakolon, eine Skoliose, periphere Neuropathien, Hypersomnie und Intelligenzminderung sowie eine Stirnglatze bei Männern. Kardiologisch sind Kardiomyopathie, Mitralklappenprolaps und Herzreizleitungsstörungen, die zum plötzlichen Herztod führen können, möglich und sorgfältig abzuklären. Häufige endokrine Störungen sind Hodenatrophie, Amenorrhö und erhöhte FSH- und Insulinspiegel im Serum, selten ein Diabetes mellitus.
Bei der milden Form, die sich meist im mittleren Alter manifestiert, stehen nicht die muskulären Symptome, sondern eine Dysarthrie und eine Katarakt im Vordergrund.
Myotone Dystrophie Typ 2 (DM2)
DM2-Patienten leiden wie DM1-Patienten unter einer progredienten Muskelschwäche, Myotonie, kardialen Arrhythmie, Katarakt, Hodenatrophie und Insulinresistenz. Im Unterschied zur DM1 ist bei der DM2 bisher nur ein Fall mit kongenitaler Manifestation und verzögerter mentaler Entwicklung bekannt. Antriebsschwäche, Hypersomnie und distal betonte und faziale Muskelschwäche führen den DM2-Patienten selten zum Arzt führen. Vielmehr sind Muskelschmerzen oder -versteifung oder proximal betonte Muskelschwäche der unteren Extremitäten isolierte erste Symptome. Die DM2 wird selten vor dem zweiten Lebensjahrzehnt manifest.
Diagnose
Bei der kongenitalen und infantilen DM steht neben der des Kindes die Abklärung der nahezu immer betroffenen Mutter im Vordergrund (Facies myopathica, Dysarthrie, Katarakt). Eine molekulargenetische Untersuchung der CTG-Repeats im DMPK-Gen (DM1) und danach der CCTG-Repeats im ZNF9-Gen (DM2) schließen sich an.
Differenzialdiagnose
Die Hypersomnie der DM1 wird leicht verwechselt mit einer Narkolepsie. Die Zeichen einer Myotonie im Säuglingsalter weisen auf eine Paramyotonia congenita oder eine Myotonia congenita Typ Thomsen, nicht auf eine kongenitale myotone Dystrophie, bei der die Myotonie erst später manifest wird. Eine Ptosis im Säuglingsalter findet man häufiger beim Möbius-Syndrom, bei einer Myasthenie oder auch einigen kongenitalen Myopathien.
Therapie
Eine kausale Therapie ist nicht möglich. Bei Herzreizleitungsstörungen ist eine Indikation einer Versorgung mit einem Herzschrittmacher zu klären. Vor jeder Narkose muss der Anästhesist über die Diagnose und den pulmonalen und kardialen Befund informiert werden.
Cave
Depolarisierende Relaxanzien und Neostigmin sind zu meiden. Barbiturate, Opiate und Benzodiazepine können zu einer schweren Apnoe führen.
Muskeldystrophien
Definition
Muskeldystrophien sind eine heterogene Gruppe hereditärer Erkrankungen, die durch progrediente Muskelschwäche gekennzeichnet sind und deren primärer Defekt in der Muskelzelle zu finden ist. Es sind mehr als 30 verschiedene Genorte für Muskeldystrophien bekannt. Allen Muskeldystrophien gemein sind eine Muskelfaserdegeneration und -regeneration und bereits im frühen Stadium eine endo- und perimysiale Fibrose.
Diagnose
Die Diagnostik umfasst eine (Familien)anamnese (inkl. Stammbaum), eine kardiologische und neurologische Untersuchung und die Bestimmung der Kreatinkinaseaktivität (CK) im Serum und in manchen Fällen und insbesondere bei kongenitalen Muskeldystrophien eine Untersuchung des ZNS (MRT) inkl. der Augen. In der Regel schließt sich eine Muskelbiopsie zur weiterführenden Diagnostik an. Die Analysen der Histopathologie inkl. Immunhistochemie und der Proteine (Western-Blot) und ggf. eine molekulargenetische Analyse aus EDTA. Eine Ausnahme bildet die Abklärung der Muskeldystrophie Duchenne, bei der meist auf eine Muskelbiopsie verzichtet werden kann.
Therapie
Eine kausale Therapie besteht nicht. Experimentelle Ansätze zur Gen- und Stammzelltherapie sind klinisch nicht etabliert. Im Vordergrund stehen eine symptomatische Therapie und die Behandlung von Komplikationen. Mit dem Ziel, Therapiemaßnahmen rechtzeitig einzuleiten, sollten regelmäßig neurologische, kardiologische (EKG, Echo), pulmonologische (Lungenfunktion, nächtliche Pulsoxymetrie/Polysomnographie) und orthopädische Untersuchungen erfolgen. Eine Physiotherapie dient der Vorbeugung von Gelenkkontrakturen und Verbesserung der respiratorischen Situation. Eine fortschreitende Kardiomyopathie oder Herzrhythmusstörungen sind frühzeitig zu behandeln. Die Indikation einer Heimbeatmung ist abzuklären bei anamnestischen Hinweisen auf eine nächtliche Hypoventilation wie Schnarchen, Mundtrockenheit beim Aufstehen, Kopfschmerzen und Tagesmüdigkeit. Eine psychologische Betreuung des Kindes und/oder der Familie sowie eine Beratung der Eltern durch einen Humangenetiker sind oft sinnvoll.
Dystrophinopathien
Epidemiologie
Die beiden Verlaufsformen der Dystrophinopathie sind die Duchenne- (DMD) und die Becker-Muskeldystrophie (BMD), sehr milde Formen, die nur mit einer erhöhten CK im Serum oder Myalgien einhergehen, sind selten. Die DMD ist die häufigste Muskeldystrophie und hat eine Inzidenz von ca. 1:3.500 männlichen Lebendgeburten und eine Prävalenz von 32:1 Mio. Kinder. Die BMD ist mit einer Inzidenz von ca. 1:5.000 Neugeborenen seltener.
Pathogenese
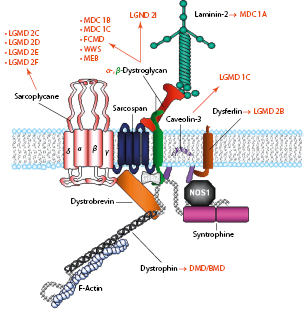
Ursache der X-chromosomal-rezessiven und damit in erster Linie das männliche Geschlecht betreffenden DMD und BMD sind Mutationen im Dystrophin-Gen (DMD; Xp21). Am häufigsten liegen Deletionen (60–65%), seltener Duplikationen (5–10%) oder Punktmutationen vor. In der Regel führen DMD-Mutationen, die den Leserahmen des Gens verschieben (Nonsense- oder Out-of-frame-Mutationen), zur schweren Verlaufsform der DMD und DMD-Mutationen ohne Auswirkungen auf den Leserahmen (Missense- oder In-frame-Mutationen) zur milderen BMD. In der Skelettmuskelzelle verbindet der Dystrophin-Glykoprotein-Komplex (DGC; Abb. 9.4) die extrazelluläre Matrix mit dem Zytoskelett. Dabei fungiert Dystrophin als Kettenglied zwischen F-Actin und Dystroglycan. Fehlt Dystrophin, so kommt es wahrscheinlich während der Kontraktions- und Relaxationsphasen zu Schäden der Muskelfasern.

Klinik, Verlauf
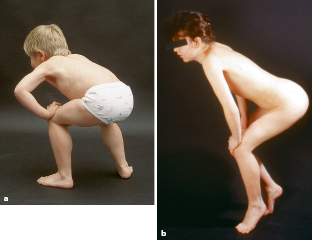
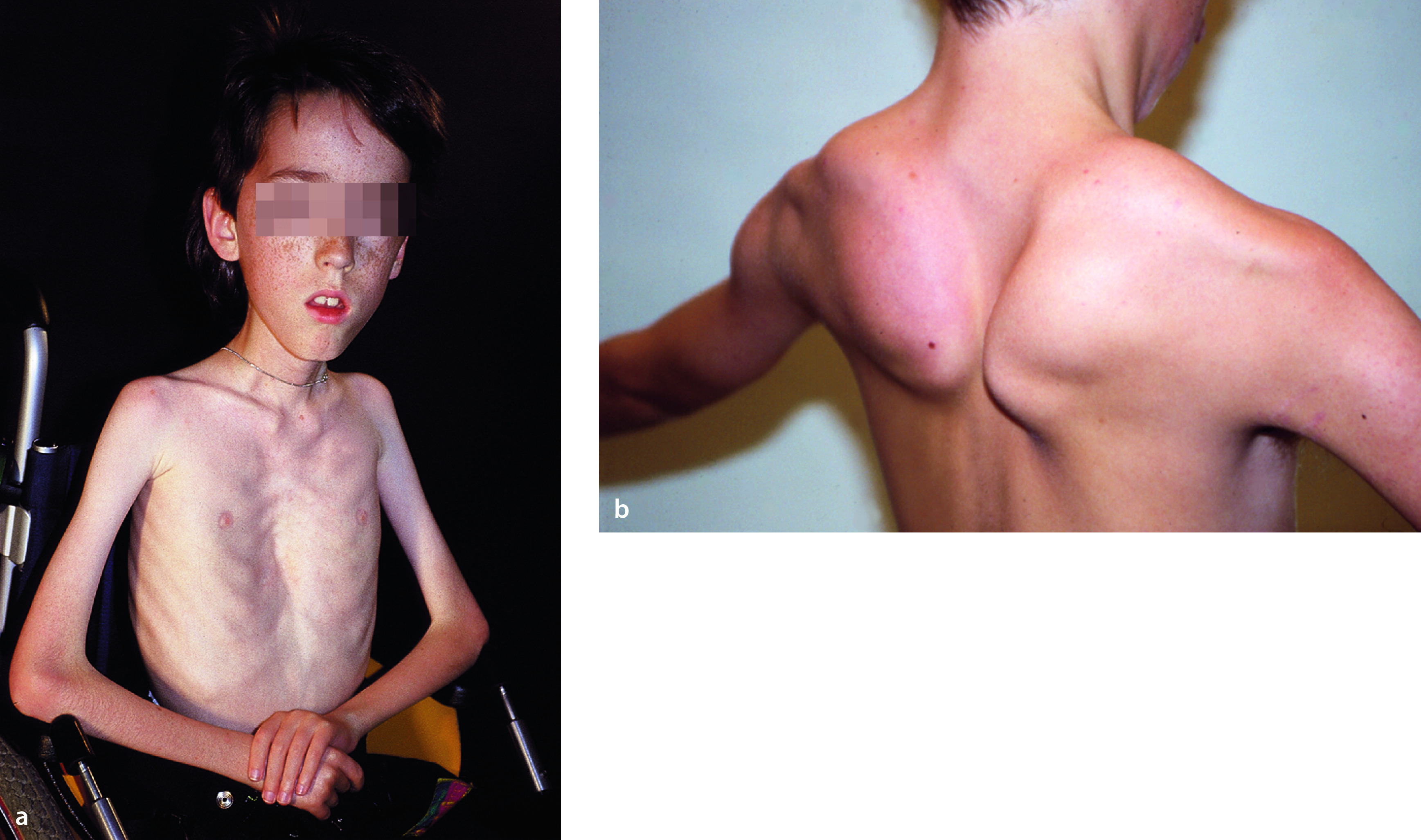
Erste Symptome werden bei der DMD meist im Kleinkindesalter beobachtet (bei >50% verzögerte statomotorische Entwicklung). Eindeutig manifest wird die zunächst symmetrisch proximal betonte Muskelschwäche meist erst ab einem Alter von 3–5 Jahren. Die Jungen fallen durch eine Fallneigung und Schwierigkeiten beim Rennen und Treppensteigen auf. Diagnostische Zeichen der Beckengürtelschwäche ist das Gowers-Phänomen, das „Hochklettern“ an den eigenen Beinen beim Aufrichten aus dem Sitzen (Abb. 9.5a). Durch die fortschreitende Verfettung und Fibrosierung der Muskulatur kommt es zu einer Induration und Pseudohypertrophie der Muskeln, die sich an den Waden leicht tasten lässt und später zu einer Spitzfußneigung und Kontraktur der Sprunggelenke führt. Mit fortschreitenden Lebensjahren entwickeln die Kinder einen Watschelgang, eine deutliche Schultergürtelschwäche mit abstehenden Schulterblättern und eine Hyperlordose. Jenseits des 7. Lebensjahres nehmen die Fähigkeiten wie Treppensteigen und Laufen rasch ab, und die Kinder werden zwischen dem 10. und 13. Lebensjahr rollstuhlpflichtig. Komplikationen entwickeln sich durch Beugekontrakturen, eine Skoliose, eine zunehmende respiratorische Insuffizienz und eine Mitbeteiligung des Herzmuskels (Kardiomyopathie) und Reizleitungsstörungen. In 1/3 der Fälle ist eine nicht progrediente kognitive Teilleistungsschwäche festzustellen. Das gelegentliche Auftreten von Blasenentleerungsstörungen, paralytischem Ileus und Magendilatation spricht für eine Beteiligung der glatten Muskulatur. Das Versagen der Atmung oder des Herzmuskels führen bei unbehandelter DMD meist vor dem 20. Lebensjahr zum Tod.

BMD-Patienten können je nach Mutation auch eine normale Lebenserwartung haben; sie bedürfen einer lebenslänglich regelmäßigen kardiologischen Untersuchung aufgrund der potenziellen Entwicklung einer Kardiomyopathie. Die Mütter der Patienten sind als Überträgerinnen überwiegend klinisch unauffällig, zeigen aber in 70% eine Erhöhung der CK im Serum und können eine Kardiomyopathie entwickeln.
Diagnose
Muskelschwäche und eine deutlich erhöhte CK im Serum lenken bei einem männlichen Kleinkind schnell den Verdacht auf das Vorliegen einer DMD. Bei dieser Konstellation ist der erste diagnostische Schritt die genetische Analyse (MLPA-Methode), womit 80% der Mutationen im DMD-Gen entdeckt werden können. Es folgt bei fehlendem Mutationsnachweis die Gesamtsequenzierung des Dystrophin-Gens. Zur differenzialdiagnostischen Abklärung kann die Durchführung einer Muskelbiopsie sinnvoll sein. Bleibt die Diagnose nach der immunhistochemischen Analyse von Dystrophin auf Gefrierschnitten unklar, so schließt sich eine Western-Blot-Untersuchung an. Es werden Muskelproteine der Größe nach aufgetrennt und mit spezifischen Antikörpern detektiert, wie z. B. auf Nonsense-Mutationen beruhende verkürzte Dystrophine.
Differenzialdiagnose
Eine Schwäche der Schulter- und Beckengürtelmuskulatur inkl. Gowers-Phänomen zeigen auch Patienten mit einer Gliedergürteldystrophie (LGMD) und Patienten mit einer SMA3, letztere geht aber mit einer nicht oder nur gering erhöhten CK im Serum einher. Weitere Differenzialdiagnosen sind die Emery-Dreifuss- und die fazioskapulohumerale Muskeldystrophie und einige metabolische Myopathien, insbesondere die Glykogenosen Typ II und V (Kap. 3.8).
Therapie
Durch eine Behandlung mit Prednisolon ab dem 5. Lebensjahr kann die Progredienz der Muskelschwäche temporär verzögert werden. Es müssen Nebenwirkungen berücksichtigt werden, besonders Gewichtszunahme, vermindertes Längenwachstum und Kataraktentwicklung. Für Patienten mit DMD und zu einem Stopp-Kodon führenden Punktmutationen steht zusätzlich Translarna (Ataluren) in einigen Ländern zur Verfügung, welches dazu führt, dass das vorzeitige Stopp-Kodon überlesen wird.
Es gibt bis heute keine Therapieform, durch die das Fortschreiten der Krankheit wesentlich beeinflusst wird. Durch eine multiprofessionelle Betreuung und Therapie können Lebenserwartung und -qualität verbessert werden.
Gliedergürtelmuskeldystrophien
Epidemiologie
Die Prävalenz der „limb-girdle muscular dystrophies“ (LGMD) liegt bei etwa 1:100.000.
Pathogenese
Die LGMD folgen einem autosomal-dominanten (LGMD1) oder rezessiven (LGMD2) Erbgang und sind verursacht durch Mutationen von Genen, die zum großen Teil Proteine der extrazellulären Matrix, des Sarkolemms und des subsarkolemmalen Zytoskeletts kodieren (Abb. 9.4) oder an der posttranslationalen Modifikation und Prozessierung von Proteinen beteiligt sind.
Klinik, Verlauf
Leitsymptom ist ähnlich wie bei der DMD eine progrediente Muskelschwäche insbesondere im Bereich der Schulter- und Beckengürtelmuskulatur („limb-girdle muscular dystrophy“, LGMD; Abb. 9.5b). Die Muskelschwäche kann sich im Verlauf auf die distalen Abschnitte der Extremitäten ausdehnen. Es besteht eine große inter- und intrafamiliäre Variabilität des Phänotyps. Die LGMD nehmen meist einen milderen Verlauf als die DMD.
Diagnose
Eine Manifestation im Kleinkindesalter und eine dilatative Kardiomyopathie kennzeichnen die Gruppe der autosomal-rezessiven Sarkoglykanopathien (LGMD2C-F). Eine Manifestation im 2. Lebensjahrzehnt und eine Pseudohypertrophie der Waden sind typisch für die autosomal-rezessive LGMD2A mit Mutation des CAPN3-Gens, das die muskelspezifische Protease Calpain 3 kodiert (Abb. 9.5b). Ein der DMD ähnliches klinisches Bild in Verbindung mit einer Makroglossie, die man bei DMD-Patienten nur selten sieht, sind Leitsymptome einer autosomal-rezessiven LGMD2I, die auf Mutationen des FKRP-Gens beruht. Kardiomyopathien mit atrioventrikulärer Reizleitungsstörung lenken den Verdacht auf eine autosomal-dominante LGMD1B. Schmerzhafter Zehenspitzengang oder Muskelkrämpfe und (durch den Reflexhammer leicht auslösbare) rasche, wellenartig sich ausbreitende Muskelkontraktionen („rippling“) am M. quadriceps femoris, die von myotonen Reaktionen abzugrenzen sind, sind Leitsymptome einer autosomal-dominanten LGMD1C. In der Regel führt nicht allein der Phänotyp zum zugrundeliegenden Genotyp und damit zur Diagnose, sondern Analysen einer Muskelbiopsie und ggf. eine Kopplungsanalyse, denen sich die Sequenzierung der in Frage kommenden Kandidatengene anschließt.
Differenzialdiagnose
Neben den unter der DMD aufgeführten sind weitere Differenzialdiagnosen die sicher auszuschließenden Dystrophinopathien und kongenitale Muskeldystrophien.
Kongenitale Muskeldystrophien (MDC)
Epidemiologie
Die geschätzte Prävalenz der MDC in der kaukasischen Bevölkerung unter 16 Jahren beträgt 2–3:100.000.
Pathogenese
Die kongenitalen Muskeldystrophien („muscular dystrophy congenital“, MDC) sind eine heterogene Gruppe autosomal-rezessiv vererbter Erkrankungen. α- und β-Dystroglycan sind Schlüsselelemente im Dystrophin-Glykoprotein-Komplex (Abb. 9.4). Beide Proteine werden von einem Gen transkribiert, posttranslational modifiziert und ubiquitär exprimiert. Das stark glykosylierte α-Dystroglycan dient als Rezeptor für Basalmembranproteine wie Laminine. Mutationen des Laminin-α2-Gens führen zu der häufigsten MDC in der kaukasischen Bevölkerung (MDC1A; Abb. 9.6a). Mutationen in Glykosyltransferasen verursachen die „congential disorders of O-glycosylation“ (CDG-Syndrome) oder α-Dystroglykanopathien (MDC1C, Walker-Warburg-Syndrom, muscle-eye-brain disease, Fukuyama MDC). Weitere MDC wie z. B. solche mit proximal betonten Kontrakturen beruhen auf Defekten extrazellulärer Proteine.
Klinik
MDC zeichnen sich durch eine in der Regel frühe Manifestation (muskuläre Hypotonie, respiratorische Insuffizienz, Trinkschwierigkeiten, Kardiomyopathie, „floppy infant“) bei Geburt oder in den ersten Lebensmonaten aus. Sowohl eine Arthrogryposis multiplex congenita als auch eine abnorme Überstreckbarkeit der Gelenke werden beobachtet. Nicht selten sind das periphere oder zentrale Nervensystem (demyelinisierende Neuropathien, mentale Retardierung, Epilepsie, Migrationsstörungen wie Lissenzephalie, Pachy- oder Polymikrogyrie, Hydrozephalus, Leukodystrophie, Hirnstammhypotrophie, zerebelläre Zysten) und die vorderen und hinteren Augenabschnitte mitbeteiligt (Kararakt, Glaukom, retinale Dysplasien, Myopie, Optikusatrophie, Megakornea, Nystagmus, Ophthalmoplegie). Es besteht eine ausgeprägte Variabilität des Phänotyps, soweit bekannt, zeigen aber alle MDC-Formen einen progredienten Verlauf.
Diagnose
Neben der neurologischen stehen augenärztliche und kardiologische Untersuchungen im Vordergrund. Es folgen eine Bestimmung der CK im Serum, elektrophysiologische Untersuchungen, eine Schädel-MRT, die immunhistochemische Analyse einer Muskelbiopsie und eine Sequenzierung der in Frage kommenden Kandidatengene. Die häufige MDC1A (Abb. 9.6a) ist meist mit einer Leukenzephalopathie vergesellschaftet. Eine ausgeprägte Makroglossie ist ein Leitsymptom der MDC1C, Fehlbildungen des Hirns oder der Augen lassen an eine MDC1C, „muscle eye brain disease“, kongenitale Muskeldystrophie Fukuyama oder das Walker-Warburg-Syndrom denken, eine starre Wirbelsäule an eine „Rigid-spine“-Muskeldystrophie, und Kontrakturen der proximalen und Überstreckbarkeit der distalen Gelenke an eine MDC vom Typ Ullrich.
Fazioskapulohumerale Muskeldystrophie (FSHD)
Epidemiologie
Die Prävalenz der FSHD beträgt ca. 1:20.000.
Pathogenese
Die FSHD wird autosomal-dominant vererbt. Die deutlich höhere Penetranz bei Männern kann innerhalb einer Familie einen X-chromosomalen Erbgang vortäuschen. Ca. 10% der Patienten haben De-novo-Mutationen. Zur Erkrankung trägt eine Verkürzung eines extragen gelegenen Tandem-Repeats auf Chromosom 4q35 (D4Z4-Repeats) bei, die aber nicht allein für die Pathogenese der FSHD verantwortlich ist.
Klinik und Verlauf
Die FSHD betrifft vorwiegend Jugendliche und Erwachsene. Die klinische Abgrenzung zu anderen Muskeldystrophien gelingt über die typische Verteilung der (oft initial asymmetrischen) Muskelschwäche und -atrophie im Bereich des Gesichts (Facies myopathica), des Schultergürtels (Scapula alata) und der Oberarme (Abb. 9.6b). Wegen der Schwäche der Gesichtsmuskulatur können die Patienten oft nicht pfeifen und haben Schwierigkeiten beim Augenschluss. Im weiteren Verlauf kommt es auch zur Schwäche der Unterarm-, Unterschenkel- und Beckengürtelmuskulatur. Eine Hochtonschwerhörigkeit (bei 2/3 der Patienten) und retinale Teleangiektasien können assoziiert sein, sind aber selten symptomatisch. Die Patienten gehen oft zum Arzt auf Grund ihrer Muskelschmerzen, erheblichen Probleme, die Arme über den Kopf zu heben (Abb. 9.6b), und einer Fußheberschwäche. Der Verlauf ist mit Ausnahme seltener schwer verlaufender infantiler Formen langsam progredient. Ca. 20% aller Patienten werden im fortgeschrittenen Alter rollstuhlabhängig, die Lebenserwartung ist fast normal.
Diagnose
Die klinische Verdachtsdiagnose wird durch den molekulargenetischen Nachweis von Deletionen auf Chromosom 4q35 gesichert.
Emery-Dreifuss-Muskeldystrophien (EDMD)
Grundlagen
Die Gruppe der heterogenen EDMD werden X-chromosomal, autosomal-dominant oder selten auch autosomal-rezessiv vererbt. Ursache aller EDMD sind Mutationen von Genen (X-chromosomales EMD, autosomale LMNA, SYNE-1/2), deren Proteine alle miteinander interagieren und bei der Verknüpfung des Kern- mit dem Zytoskelett eine Rolle spielen (Emerin, Lamin-A/C, Nesprin-1 und -2).
Klinik
Die heterogenen EDMD sind durch eine langsam progrediente Muskelschwäche, die häufig im Kindesalter beginnt, und durch früh auftretende Kontrakturen und eine Kardiomyopathie gekennzeichnet. Die Muskelschwäche zeigt ein humeroperonäales Verteilungsmuster, d. h. eine proximale Beteiligung im Bereich der oberen und eine distale im Bereich der unteren Extremitäten. Die Gehfähigkeit bleibt meist lange erhalten. Kontrakturen betreffen in erster Linie die Ellenbogen, die oberen Sprunggelenke und die Wirbelsäule („rigid spine“). Eine dilatative Kardiomyopathie mit Reizleitungsstörungen und später lebensbedrohlichen ventrikulären Tachyarrhythmien (plötzlicher Herztod) wird meist im frühen Erwachsenenalter, seltener im Kindesalter beobachtet.
Diagnose
Treten zu einem X-chromosomalen oder autosomal-dominanten Erbgang die Symptome humeroperonäale Muskelschwäche und kardiale Reizleitungsstörungen, ist die Diagnose suggestiv. Die CK ist oft leicht erhöht. Immunhistochemisch lassen sich das Fehlen einer Emerin-Synthese, nicht aber einer Lamin-A/C-Expression (da dominant vererbt und somit immer auch eine normale Kopie des Gens vorhanden) im Muskelgewebe nachweisen. Die Emerin- Synthese kann auch in Zellen der Mundschleimhaut oder an Hautzellen durch Western-Blot-Untersuchungen überprüft werden. Bei der X-chromosomalen Form können auch die weiblichen, meist klinisch unauffälligen Überträgerinnen an Herzrhythmusstörungen erkranken.
Therapie
Es gibt keine kausale Therapie. Wichtig ist eine stringente Diagnosestellung zur rechtzeitigen Versorgung mit Herzschrittmacher, bei autosomal-dominanten LMNA-Mutationen inkl. implantierbarem Kardioverter-Defibrillator. Schwere dilatative Kardiomyopathien, die insbesondere bei LMNA-Mutationen auftreten, können eine Herztransplantation erfordern.
Kongenitale Strukturmyopathien
Grundlagen
Kongenitale Strukturmyopathien sind heterogene Erkrankungen der Muskulatur, die oft kongenital mit autosomal-rezessivem oder dominantem Erbgang auftreten und in der Regel nicht oder nur gering progredient sind. Die X-chromosomale rasch progrediente myotubuläre Myopathie bildet hier eine Ausnahme.
Klinik
Das klinische Spektrum dieser Erkrankungen ist geprägt von dem Bild eines „floppy infant“ mit Atmungs- und Fütterungsproblemen. Manifestationen im späteren Alter sind möglich. Zu einer oft generalisierten Muskelschwäche und -hypotrophie können Symptome wie Kardiomyopathie, respiratorische Insuffizienz, Muskelschmerzen (nach Belastung), Kontrakturen, Skoliose, Skelettdeformitäten, kongenitale Hüftluxation und Ophthalmoplegie hinzutreten.
Diagnose
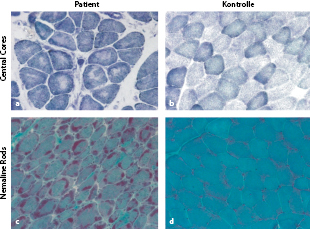
Die Kreatinkinaseaktivität (CK) im Serum ist nicht oder nur leicht erhöht. Der muskelhistologische Befund ermöglicht eine erste Zuordnung (Abb. 9.7), der sich Elektronenmikroskopie und genetische Diagnostik anschließen. Eine primär molekulargenetische Gen-Panel-Diagnostik kann gerechtfertigt sein.

Contributor Information
A. M. Kaindl, Email: angela.kaindl@charite.de
U. Schara, Email: ulrike.schara@uk-essen.de
M. Schülke-Gerstenfeld, Email: markus.schuelke@charite.de