

Abstract
In der allgemeinen Sprechstunde wird der Kinderarzt häufig mit Hauterkrankungen konfrontiert. Dabei stehen in bestimmten Altersstufen verschiedene Dermatosen im Vordergrund. Neugeborene und Säuglinge werden wegen Genodermatosen, atopischem oder seborrhoischem Ekzem, Pigmentmalen und Hämangiomen vorgestellt. Kleinkinder leiden oft an Hautinfektionen durch Bakterien (Pyodermien), Viren (Masern, Röteln, Warzen), Parasiten (Skabies, Pedikulose) und an atopischem Ekzem. In der Adoleszenz tritt bei fast allen Jugendlichen eine Akne auf, kosmetisch störende Hautveränderungen gelangen zunehmend ins Blickfeld der Betroffenen. Das klinische Erscheinungsbild, die Diagnostik und Therapie häufiger Hautkrankheiten weisen im Kindesalter oft Besonderheiten auf, die in diesem Kapitel besprochen werden.
Effloreszenzenlehre und Untersuchung der Haut
Die Haut ist das größte und am leichtesten zugängliche Körperorgan des Menschen. Kindliche Haut zeigt eine geringere Keratinisierung und daher eine höhere Rate an entzündlichen Affektionen. Auch Blasenbildung oder Erytheme zeigen sich aufgrund der eher lockeren Hautstruktur häufiger.
Die Untersuchung des Kindes mit einer Hauterkrankung erfordert nach Erhebung der Anamnese die gründliche Inspektion der Haut, Schleimhaut und der Hautanhangsgebilde (Haare und Nägel).
Die Inspektion, das Sehen, das Erkennen und Tasten der Einzeleffloreszenzen und die Bestimmung des Verteilungsmusters kann häufig schon zur Diagnose führen.
Die häufigsten Effloreszenzen sind in Tab. 30.1 dargestellt. Primäre Effloreszenzen sind die direkt als Folge der Erkrankung aufgetretenen Hauterscheinungen, während sich Sekundäreffloreszenzen im Anschluss an eine primäre Effloreszenz durch Fortschreiten der krankhaften Veränderungen oder durch äußere Schädigung der Haut entwickeln.
| Primäreffloreszenzen | |
| Makula (Fleck) | Vorübergehende oder bleibende Farbveränderung der Haut ohne Konsistenz- oder Niveauänderung |
| Papula (Papel) | Umschriebene Verdickung oder Auftreibung der Haut bis zu einer Größe von 5 mm |
| Nodulus (Knötchen) | Umschriebene, solide, gut von der Umgebung abgesetzte Substanzvermehrung, meist kutan bis subkutan gelegen von mehr als 5 mm Größe |
| Vesicula (Bläschen) | Mit Flüssigkeit gefüllter bis erbsgroßer, über das Hautniveau erhabener, in der Regel mehrkammriger Hohlraum |
| Bulla (Blase) | Mit Flüssigkeit gefüllter, in der Regel einkammeriger Hohlraum über Erbsgröße |
| Pustula (Eiterbläschen) | Mit Eiter gefüllter Hohlraum |
| Urtica (Quaddel) | Flüchtige, scharf begrenzte Erhabenheit, bedingt durch ein Ödem in der oberen Dermis |
| Sekundäreffloreszenzen | |
| Squama (Schuppe) | Lose oder festaufsitzende, scheibenförmige Hornauflagerung |
| Crusta (Kruste) | Auf Erosionen oder Ulzera eingetrocknetes Sekret |
| Erosion | Substanzdefekt des Epithels bis zur Basalmembran. Abheilung ohne Narbe |
| Exkoriation | Gewebedefekt, der bis ins Stratum papillare reicht. Dadurch entstehen punktförmige Blutaustritte |
| Rhagade | Spaltförmiger tiefer Einriss der Epidermis und der Dermis |
| Ulkus | Tiefgreifender Substanzdefekt, der stets narbig abheilt |
| Atrophie | Schwund von Epidermis und Anhangsorganen, von Dermis oder der Subkutis isoliert oder gemeinsam |
| Cicatrix (Narbe) | Bindegewebiger Ersatz von Substanzverlusten in der Dermis |
Hautveränderungen bei Neugeborenen
Harlekin-Farbphänomen
Besonders bei Frühgeborenen entsteht eine harmlose, lageabhängige, halbseitige, blasse bis tiefrote Verfärbung am Kopf und am ganzen Körper. Ursache ist wahrscheinlich eine gestörte Regulation der Gefäßwandinnervation. Dieses Phänomen verschwindet mit zunehmendem Alter.
Erythema neonatorum toxicum
Die Pathogenese des Erythema neonatorum toxicum ist unbekannt. Es tritt v. a. beim 2. Kind auf. In den ersten Lebenstagen entstehen bei bis zu 50% der Neugeborenen am Stamm und an den Extremitäten kleine rote Maculae, Papeln, Vesikeln und Pusteln. Im Pustelausstrich finden sich zahlreiche Eosinophile. Differenzialdiagnostisch in Frage kommende staphylogene Infektionen zeigen im Blasengrundausstrich neutrophile Leukozyten. Als Therapie empfiehlt sich Lotio zinci. Es folgt in der Regel eine spontane Abheilung.
Transiente neonatale pustulöse Melanose
Bei wenigen Neugeborenen finden sich bereits bei der Geburt v. a. an Gesäß, Rücken und Kopfbereich kleine Vesikel oder Pusteln auf pigmentierten Maculae. Nach Aufplatzen bilden sich braune Krusten. Es erfolgt eine spontane Abheilung innerhalb von 3 Monaten.
Neugeborenensklerem und Fettgewebsnekrose
Beiden Erkrankungen ist eine Verhärtung des Unterhautfettgewebes gemeinsam. Das Neugeborenensklerem tritt bei kranken Früh- und Neugeborenen auf. Die Haut über den beinahe steinharten Veränderungen ist nicht betroffen. Mit Besserung des Allgemeinzustands verschwinden die Erscheinungen.
Die über der Fettgewebsnekrose des Neugeborenen liegende Haut zeigt braune bis violette Veränderungen. Die knotigen Verhärtungen bilden sich im Laufe von Wochen bis Monaten zurück. Sie kommen bei gesunden Neugeborenen vor, gelegentlich findet sich eine belastende Geburtsanamnese.
Aplasia cutis congenita
Sehr selten befinden sich bei Neugeborenen v. a. am Kopf, aber auch am Stamm 1,5–2 cm große, scharf abgegrenzte Ulzerationen unterschiedlicher Tiefe, die selten sogar den Knochen erreichen. Diese Defekte können familiär gehäuft vorkommen; außerdem sind sie ein häufiges Symptom bei der Trisomie 13. Gelegentlich können auch andere Fehlbildungen assoziiert sein. Nur bei größeren oder sehr tiefen Defekten ist eine chirurgische Therapie erforderlich, sonst epithelisiert sich der Defekt allmählich.
Sonstige Hauterkrankungen des Neugeborenen
Weitere Hauterkrankungen im Neugeborenenalter werden in den folgenden Kapiteln abgehandelt: Gefäßnävi und Hämangiome (Abschn. 30.12.4), Mongolenfleck (Abschn. 30.12.1), Acne neonatorum (Abschn. 30.11).
Genodermatosen
Keratosen
Grundlagen
Ein gestörtes Verhältnis von Neubildung und Abschilferung der Hornschicht führt zu vermehrter Horn- und Schuppenauflagerung. Ist die Epidermopoese verstärkt, werden zu viele Hornzellen gebildet. Man spricht hier von einer Proliferationshyperkeratose. Werden zu wenige Hornzellen an der Hautoberfläche abgeschilfert, so spricht man von einer Retentionshyperkeratose.
Nach der klinischen Lokalisation werden folgende Keratosen unterschieden:
Ichthyosen (diffuse Keratosen),
palmoplantare Keratosen,
Erythrokeratodermien,
follikuläre Keratosen,
umschriebene Keratosen.
Häufig liegt einer gestörten Hornbildung eine genetische Störung zugrunde. Auf die im Kindesalter wichtigsten Genodermatosen gehen wir in diesem Kapitel genauer ein.
Ichthyosis vulgaris
Genetik
Bei der Ichthyosis vulgaris liegt eine autosomal-dominante Vererbung mit ausgeprägter intrafamiliärer Variabilität der klinischen Ausprägung vor. Mutationen im Gen von Filaggrin führen bei Ichthyosis vulgaris zu Störungen der Keratinisierung im unterschiedlichen Ausmaß und zur Assoziation mit atopischer Diathese. Mit einer Morbiditätsrate von 1:1.000 ist sie die häufigste Genodermatose.
Pathogenese, Klinik
Aufgrund Verminderung oder Fehlens von Filaggrin entsteht eine Retentionshyperkeratose.
Bereits im 1.–2. Lebensjahr fallen helle, mittel- bis feinlamellöse Schuppen an den Streckseiten der Extremitäten und des Rumpfes auf (Abb. 30.1). Hals und seitliches Gesicht sind von der Schuppung weniger betroffen. Gelenkbeugen und Schleimhäute bleiben immer ausgespart. Follikuläre Keratosen an den proximalen Extremitäten führen zu einer „Reibeisenhaut“. Als Ichthyosishand wird ein vergröbertes Handfurchenrelief mit vermehrter Linienzeichnung bezeichnet. Da die Haut der Patienten insgesamt sehr trocken ist, beeinflussen eine feuchte und warme Umgebung die Krankheit günstig.

Diagnose, Therapie, Prognose
Die Hautbiopsie zeigt ein Fehlen der Granulazellschicht in der Epidermis. Wichtig ist eine symptomatische Therapie der Haut mit rückfettenden Bädern und fettenden kochsalz- oder harnstoffhaltigen (5–10%) Emulsionen bzw. Salben. Eine Besserung kann mit interner Gabe von Acitretin (Neotigason) erreicht werden, hilft aber leider nur für die Dauer der Therapie. Häufig liegt gleichzeitig ein atopisches Ekzem vor. Prognostisch verschlechtert sich der Zustand bis zur Pubertät und bessert sich im Sommer und im Alter.
X-chromosomal-rezessiv erbliche Ichthyosis vulgaris
Genetik, Pathogenese
Dieser X-chromosomal-rezessiv vererbten Form der Ichthyose liegt ein Mangel des Enzyms Steroidsulfatase zugrunde. Genort ist Xp 22.32. Mit ca. 1:4.000 Neugeborenen ist sie die zweithäufigste Ichthyose. Nur beim männlichen Geschlecht findet sich die volle Ausprägung.
Klinik
Bereits in den ersten Lebensmonaten entwickelt sich eine polygonale, gelbbraune Keratose der Streckseiten der Extremitäten und des Rumpfs, wobei die großen Beugen im Gegensatz zur Ichthyosis vulgaris mitbefallen sind. Die Ichthyosishand und die follikulären Keratosen fehlen. Stark betroffen sind hingegen Kopfhaut, Ohren und Hals.
Diagnose
Anamnese, klinisches Bild und direkte Messung der Enzymaktivität der Steroidsulfatase oder indirekte durch Lipidelektrophorese (beschleunigte Wanderungsgeschwindigkeit der β-Lipoproteine) ermöglichen die Diagnose. Eine pränatale Diagnostik (Steroidsulfataseassay, DNA-Analyse) ist möglich.
Therapie
Eine symptomatische örtliche Therapie und Hautpflege wie bei der Ichthyosis vulgaris sind ausreichend.
Komplikationen, Prognose
Bis zu 50% der Kinder entwickeln Hornhauttrübungen, die in der Regel symptomlos bleiben, und bei ca. 20% findet sich ein Kryptorchismus mit Hypogonadismus. Nach progredientem Verlauf bis zur Pubertät bleibt der Schweregrad der Erkrankung weitgehend stationär mit deutlicher Besserung im Sommer.
Hereditäre Epidermolysen
Grundlagen
Die Haut von Kindern mit hereditären Epidermolysen neigt zur Blasenbildung, ausgelöst durch mechanische und thermische Reize. Es können verschiedene Formen mit Unterschieden in der klinischen Ausprägung (von Spontanheilung über Defektheilung mit Gelenkkontrakturen, Nagelverlust und Verstümmelung bis zu letalem Ausgang) und im Erbgang (autosomal-dominant und -rezessiv) voneinander abgegrenzt werden.
Außer bei den schweren Formen lernen die Patienten bzw. deren Eltern die auslösenden Mechanismen zu meiden, sodass sie ein relativ normales Leben führen können.
Nach der Lokalisation der Spaltbildung lassen sich die Epidermolysen einteilen in:
Epidermolysis-bullosa-simplex-Formen: Blasenbildung in der Epidermis,
junktionale Epidermolysen: Blasenbildung in der Basalmembranzone,
dystrophische Epidermolysen: Blasenbildung in der Dermis.
Epidermolysis bullosa simplex (EBS)
Genetik, Epidemiologie
Die Häufigkeit dieser autosomal-dominanten Genodermatose ist etwa 1:50.000 Lebendgeburten, wobei eine Präferenz für das männliche Geschlecht beobachtet wird. Die lokalisierte Form der EBS (Weber-Cockayne) tritt im 1.–3. Lebensjahrzehnt, die generalisierte Form (Köbner) bereits bei Geburt oder in den ersten Lebensjahren auf. In vielen Fällen erfolgt der Nachweis von Mutationen der Gene von Keratin 5 (12q) oder 14 (17q), eine pränatale Diagnostik ist möglich.
Klinik
An mechanisch belasteten Stellen treten bei der Geburt, oder wenn das Kind sich zu bewegen beginnt, kleine einkammerige Blasen mit serösem Inhalt auf. Die Blasen liegen innerhalb der Epidermis und heilen narbenfrei ab (Abb. 30.2). Gewöhnlich sind Haare, Nägel und Zähne nicht von einer Entwicklungsstörung betroffen, häufig sind zusätzlich plantare Keratosen und Hyperhidrose zu beobachten.
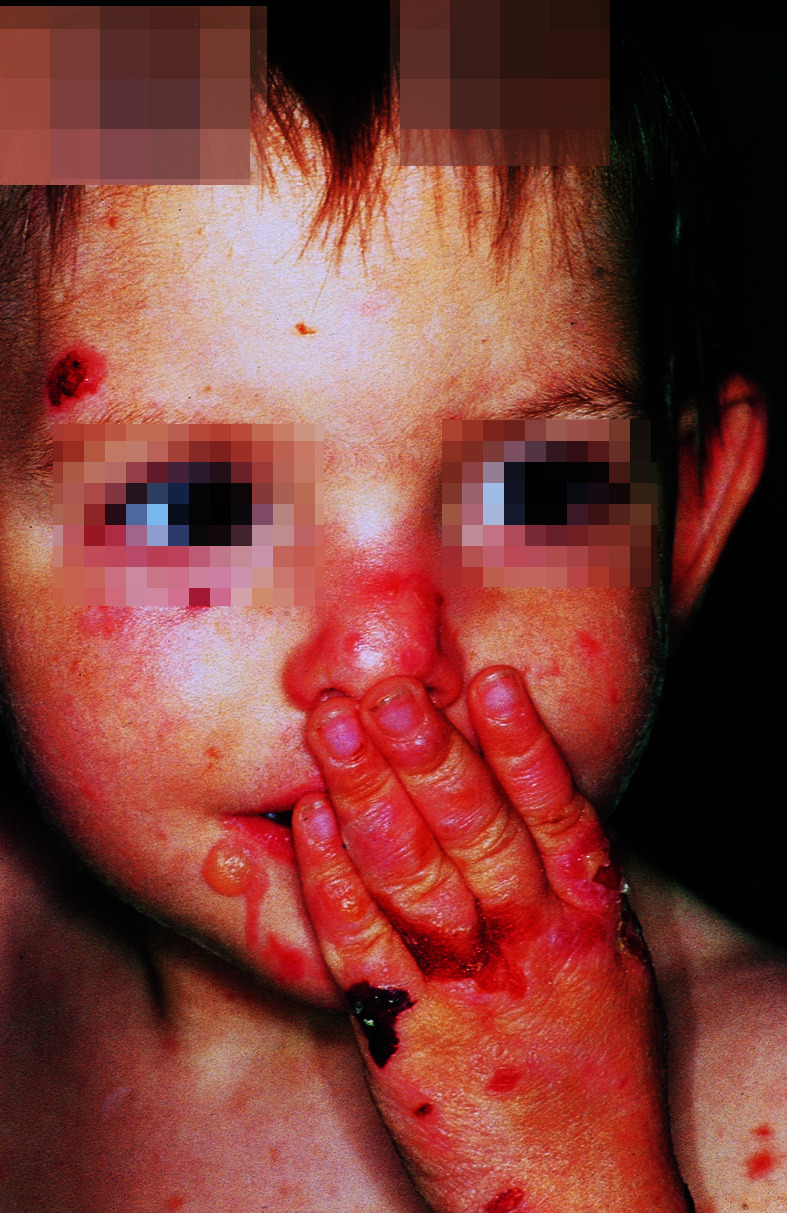
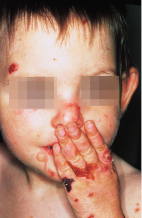
Therapie
Eine kausale Therapie steht nicht zur Verfügung. Eventuell helfen Antiperspiranzien und konsequenter Hautschutz mit rückfettenden Salben. Blasen sollten eröffnet oder abpunktiert werden, wobei das Blasendach erhalten bleibt.
Prognose
Eine Besserung der Symptomatik tritt meist in der Pubertät ein. Die Neigung zur Blasenbildung kann aber auch lebenslang anhalten und belastet die Patienten stark.
Junktionale Epidermolysis bullosa
Genetik, Pathogenese
Dieser autosomal-rezessiven Genodermatose liegt eine Spaltbildung innerhalb der Lamina lucida der Basalmembranzone zugrunde, ausgelöst durch verschiedene Mutationen im Genort der 3 Lamininsubklassen.
Klinik
Bereits bei Geburt oder kurz danach finden sich an allen mechanisch belasteten Stellen große, teilweise hämorrhagische Blasen, die zerplatzen und erodieren. Auch Mundschleimhaut, Trachea und Bronchien können mitbetroffen sein. Paronychialer Befall führt zu Nageldystrophien oder Nagelverlust. Bakterielle Sekundärinfektionen sind die Haupttodesursache häufig schon in früher Kindheit.
Therapie
Wichtig ist eine lokale Pflege zur Vermeidung von bakteriellen und mykotischen Sekundärinfektionen. Bei stärkerer Ausprägung kommen Glukokortikoide (initial 0,5–1,0 mg/kgKG Prednisolon, später minimale Erhaltungsdosis) zur Anwendung.
Prognose
Die Blasen heilen in der Regel narbenfrei ab. Bei geringer Ausprägung bleibt lediglich die Neigung zur Blasenbildung erhalten. Bei ausgeprägtem Befall sterben die Neugeborenen bereits in den ersten Lebensmonaten.
Bei allen Epidermolysen ist eine konsequente externe Therapie wichtig. Mechanische Druckstellen sollten geschützt und Sekundärinfektionen vermieden werden.
Dystrophe Epidermolysis bullosa
Genetik, Pathogenese
Man unterscheidet eine autosomal-dominante und eine autosomal-rezessive Form. Subepidermale Blasenbildung führt zu Narben und Deformationen. Genort ist das Kollagen-Typ-VII-Gen (3p21), die pränatale Diagnostik erfolgt durch DNA-Analyse.
Klinik
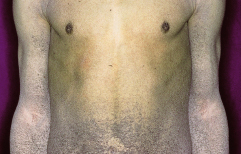
Nicht nur an mechanisch belasteten Stellen entstehen Blasen, teilweise auch spontan. Die Abheilung erfolgt mit Hautatrophie, Hyper- und Depigmentierungen. Typischerweise finden sich im Bereich der abgeheilten Blasen zahlreiche postbullöse Milien. Finger- und Zehennägel fallen aus, die Haare sind dünn. Zähne sind häufig mitbetroffen. Bei ca. 20% der Kinder sind auch die Schleimhäute befallen. Folge sind u. a. Heiserkeit, Schluckbeschwerden, Ösophagusstrikturen und konjunktivale Synechien. Synechienbildung der übrigen Haut führt zu dermatogenen Kontrakturen und Mutilationen (Abb. 30.3).

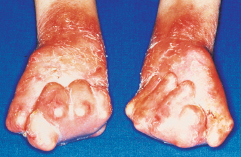
Therapie
Wichtig wie bei allen blasenbildenden Erkrankungen ist die Vermeidung von Sekundärinfektionen. Synechienbildung kann evtl. ein chirurgisches Vorgehen erfordern.
Prognose
Durch narbige Synechienbildung v. a. an Fingern, Zehen und im Ösophagusbereich können die Kinder außerordentlich beeinträchtigt sein. Im Verlauf ist die Inzidenz von spinozellulären Karzinomen erhöht und ein regelmäßiges Hautkrebsscreening notwendig.
Neurofibromatosis generalisata (von Recklinghausen)
Grundlagen
Bei den Neurofibromatosen handelt es sich um eine neuroektodermale Systemerkrankung. Es lassen sich mehrere Typen klinisch und genetisch unterscheiden. Die wichtigsten sind die Neurofibromatose vom peripheren Typ (NF-1) (viel häufiger) und die vom zentralen Typ (NF-2) mit Akustikusneurinomen.
Genetik
Beide Neurofibromatosen werden autosomal-dominant vererbt, wobei der Gendefekt bei der NF-1 auf Chromosom 20 im Gen von Neurofibromin, dessen wichtigste Funktion die Inaktivierung des ras-Onkogens ist, bei der NF-2 im Schwannomin-Gen auf Chromosom 22 liegt. Insbesondere bei der NF-1 finden sich bei 50% Spontanmutationen und die klinische Expressivität variiert stark. Eine pränatale Diagnostik von NF-1 und NF-2 ist möglich.
Klinik
NF-1
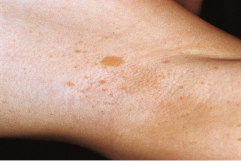
Wegweisend zur Diagnose sind multiple (mehr als 5) Café-au-lait-Flecken ab dem ersten Lebensjahr, im Laufe des Lebens auftretende zahlreiche Neurofibrome (Abb. 30.4; hautfarbene, weiche, breit oder gestielt aufsitzende Knoten) sowie sommersprossenartige Pigmentierungen (kleinfleckige Hyperpigmentierungen) axillär (Abb. 30.5) oder inguinal ab dem 3. Lebensjahr. Nur bei der NF-1 treten in der Pubertät sog. Lisch-Knötchen (Irishamartome) auf, die mit der Spaltlampe sichtbar sind. Neurofibrome am zentralen Nervensystem können zu Krampfanfällen führen. Bekannt sind auch maligne Tumoren des lymphatischen oder hämatopoetischen Systems. Neben gelegentlich auftretenden Lernbehinderungen finden sich ZNS-Störungen, Malignome, Skoliose, gastrointestinale Neurofibrome, Nierenarterienstenosen u. a.


NF-2
Es treten nur bei 42% der Patienten Café-au-lait-Flecke und bei 19% Neurofibrome auf. Hingegen manifestiert sich immer im Laufe des Lebens ein uni- oder bilaterales Akustikusneurinom, welches durch Druck auf den 8. Hirnnerv zu Hörverlust führt.
Cave
Wegen des Auftretens von Akustikusneurinomen sind regelmäßige audiologische und CT-Untersuchungen notwendig, die eine frühe chirurgische Intervention ermöglichen.
Therapie
Die chirurgische Exzision oder eine laserchirurgische Abtragung störender Neurofibrome ist v. a. bei Druck auf Nerven oder bei schnell wachsenden Tumoren indiziert. Akustikusneurinome sollten frühzeitig entfernt werden. Nicht zu vergessen ist eine genetische Beratung der Patienten.
Prognose
Im Einzelfall ist die Prognose nicht vorhersehbar. Zahl und Größe der Neurofibrome nehmen im Laufe des Lebens zu und können zu einer psychischen Belastung des Patienten führen. Selten gibt es maligne Entartungen.
Tuberöse Hirnsklerose (Morbus Bourneville-Pringle)
Genetik
Die Vererbung erfolgt autosomal-dominant mit intrafamiliärer Expressivitätsschwankung; bei mehr als 2/3 der Fälle tritt die Erkrankung sporadisch durch Mutationen der Gene für Tuberin (auf Chromosom 9) und Hamartin (auf Chromosom 16) auf.
Klinik
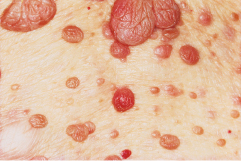
Charakteristische Veränderungen finden sich bei allen Patienten zunächst an der Haut. Zu sehen sind bereits in den ersten Lebensjahren eschenlaubartige oder konfettiartige Hypopigmentierungen. Später treten im Gesicht kleine knotige, schmutzig-braune bis rote Fibroangiome in symmetrischer, schmetterlingsförmiger Aussaat an Nase und Wangen auf, die als Adenoma sebaceum bezeichnet werden (Abb. 30.6).
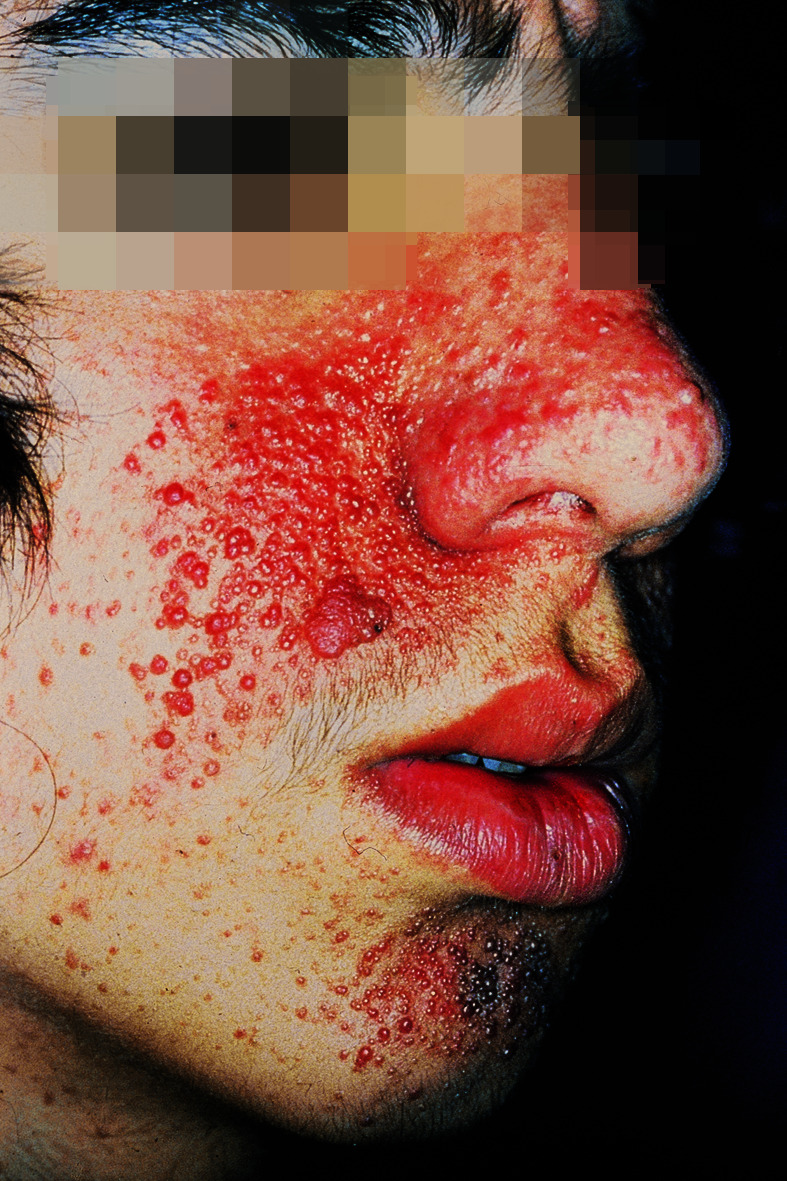
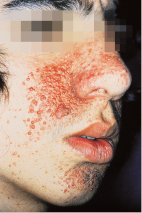
Weiterhin finden sich Fibrome an Nagelfalz und Zahnfleisch (Koenen-Tumoren), die pathognomonisch für diese Erkrankung sind. Sakral zeigen sich häufig typische flächenhafte Bindegewebsnävi (Pflastersteinnävi). Gliawucherungen im zentralen Nervensystem führen zu epileptiformen Anfällen, geistiger Retardierung und spastischen Lähmungen.
Therapie
Im Vordergrund steht die antiepileptische Behandlung. Das kosmetisch störende Adenoma sebaceum ist einer laserchirurgischen Therapie, einer Dermabrasio und seit kurzem auch durch eine topische Behandlung mit Sirolimus zugänglich, Koenen-Tumoren können exzidiert werden.
Komplikationen, Prognose
Intrakranielle Verkalkungen, Sehnervenatrophie, Stauungspapille, Netzhauttumoren, Zystennieren, Fibrosarkome am Herz und Spongiosaresorption v. a. in den kleinen Röhrenknochen von Händen und Füßen sind als Komplikationen zu beobachten. Gesamt gesehen ist die Prognose wegen der zentralen Symptome schlecht. In 5–10% der Fälle treten Fibrosarkome an Herz oder Niere auf.
Dermatomykosen
Grundlagen
Hautmykosen werden durch Fadenpilze (Dermatophyten) oder durch Sprosspilze (Blastomyzeten, Hefen) verursacht. Dermatophyten bilden aus den Sporen ein echtes Hyphenmyzel, während sich die Sprosspilze durch Aneinanderlagerung von Sporen in Form von Pseudomyzelen ausbreiten.
Typische Hautzeichen, die auf eine Mykose hinweisen, sind:
runde bis ovale Läsionen mit randständiger Schuppung,
weiße, abwischbare Beläge auf Schleimhäuten,
erosiv-nässende Erytheme mit Rhagaden in intertriginösen Bereichen (z. B. Zehenzwischenräume),
schuppende, nässende Bereiche auf der Kopfhaut.
Fast immer jucken die durch Pilzinfektionen ausgelösten Läsionen.
Diagnostische Maßnahmen sind neben dem klinischen Aspekt die Inspektion befallener Haut mit UVA-Licht (Wood-Lampe), der mikroskopische Nachweis von Hyphen und Sporen im Nativpräparat und die Pilzkultur nach Entnahme von Hautschuppen.
Hautmykosen durch Dermatophyten
Grundlagen
Aus klinischer Sicht werden sie in Epidermomykosen, Trichomykosen und Onychomykosen (bei Kindern sehr selten) eingeteilt. Der häufigste Vertreter ist Trichophyton rubrum, gefolgt von Trichophyton mentagrophytes und Epidermophyton. Infektionen werden mit dem Wort „Tinea“, unabhängig von der genauen Art des Erregers, bezeichnet (Abb. 30.7). Hinter das Wort „Tinea“ wird ein lokalisatorischer Begriff gesetzt: Tinea capitis, Tinea faciei, Tinea manum, Tinea pedis, Tinea corporis, Tinea unguium.
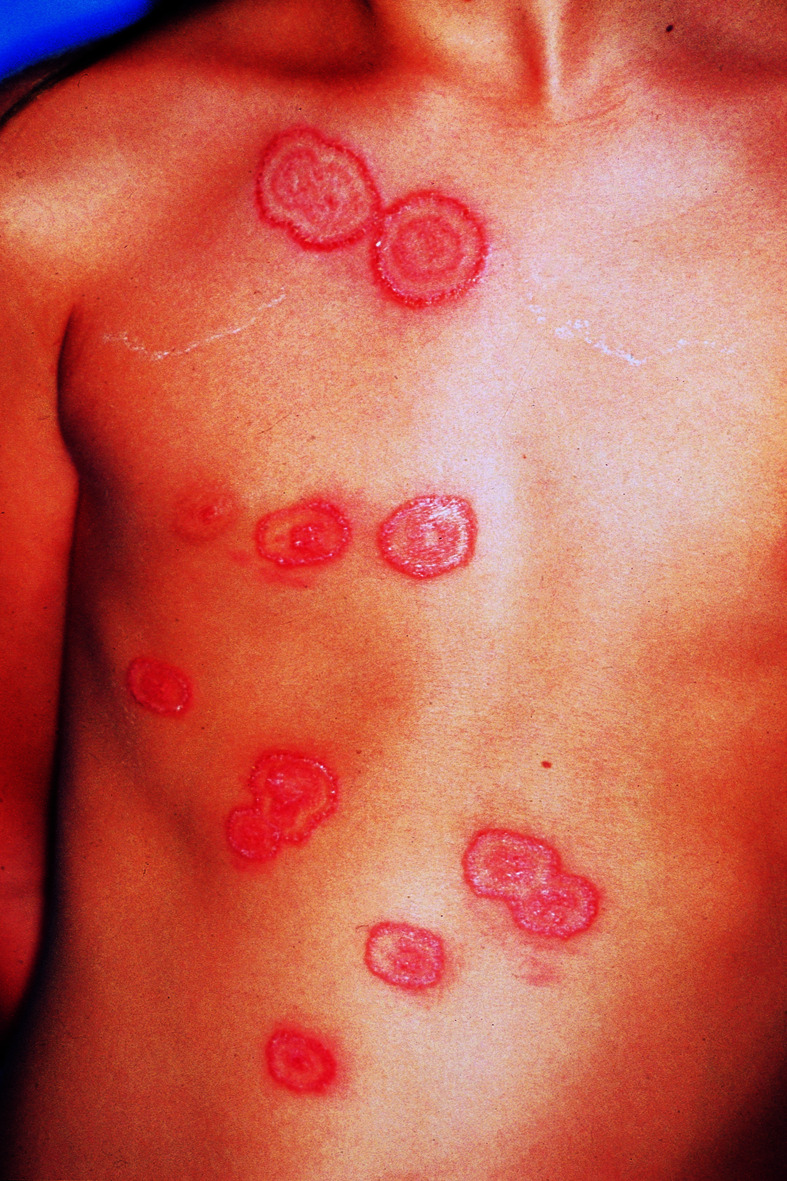
Die häufigsten Formen von Tinea, ihre Klinik und deren Therapie sind in Tab. 30.2 zusammengestellt.
| Erreger | Klinik | Therapie | |
|---|---|---|---|
| Tinea capitis | Microsporum audouinii, Microsporum canis | Zirkuläre, schuppende Läsionen mit Abbrechen der Haare, Juckreiz, Alopezie | Griseofulvin oral (10 mg/kgKG/Tag) für 6–8 Wochen |
| Trichophyton tonsurans | Multiple, kleine Bezirke, Alopezie, Entstehung von weichen, knotigen Granulomen (Kerion celsi) | Äußerlich: 10%ige Schwefelzinkpaste, Chinosolumschläge | |
| Trichophyton schoenleinii (Favus) | Graugelbe 0,5–1 cm große, schüsselförmige (eingedellte) Läsionen (Mäuseuringeruch), Alopezie | Salicylsäurehaltige Cremes | |
| Tinea corporis | Trichophyton rubrum, Trichophyton mentagrophytes, Microsporum canis | Blassrote, sich ausbreitende, schuppende, flache Herde mit Betonung des Rands oder in Gruppen angeordnete Pusteln; Tinea profunda (granulomatöse Entzündung) | Lokale Antimykotika (Clotrimazol, Ciclopiroxolamin), bei tiefem oder ausgedehntem Befall Griseofulvin oral (10 mg/kgKG/Tag) für 6–8 Wochen |
| Tinea pedis | Trichophyton rubrum, Trichophyton mentagrophytes, Epidermophyton floccosum | Rötung, Fissuren, Rhagaden, Schuppung, Bläschen (T. mentagrophytes), sich abschälende Haut; besonders 3. und 4. Interdigitalraum, häufig gleichzeitig Infektion mit Candida albicans | Bei mildem Befall: sorgfältige Trocknung der Interdigitalräume. Bei stärkerem Befall: lokale Antimykotika (Ciclopiroxolamin, Clotrimazol) |
Hautmykosen durch Sprosspilze
Candidainfektionen
Klinik
Candida albicans ist der häufigste Erreger (Soormykose), der auf gesunder Haut nicht anzutreffen ist, sich aber bei warmer Feuchtigkeit oder Veränderung der physiologischen, bakteriellen Besiedlung auf Haut und Schleimhäuten ausbreitet. Auf Letzteren (Mundhöhle, Vaginalbereich) ist eine Candidainfektion durch weiße, im Gegensatz zu z. B. Milchresten nur schwer entfernbare Beläge mit Rötung der Umgebung gekennzeichnet. Erythematöse, nässende Partien mit Bläschen, die sich teilweise mit bogenförmigem Rand abgelöst haben, sind die Manifestation einer Candidamykose auf der Haut.
Ein häufiges Problem ist die mit Candida superinfizierte Windeldermatitis. Die Erreger stammen in der Regel aus dem Darm; eine Soorinfektion der Mundhöhle kann gleichzeitig bestehen. Daher ist eine nur lokale Therapie der Windeldermatitis häufig nicht erfolgreich.
Therapie
Die betroffenen Hautbereiche sollten durch Pasta zinci im Wechsel mit antimykotischen Pasten abgedeckt werden. Häufiges Wechseln der Windeln ist ebenfalls wichtig. Bei einigen Immundefekten (chronisch mukokutane Candidiasis und weitere) kann eine interne Therapie mit Fluconazol notwendig sein. Im Falle von Mund- und Vaginalschleimhautbefall besteht die Behandlung in lokaler Applikation von Nystatin oder anderen antimykotischen Substanzen (Ketoconazol, Terbinafin) in Form von Lösungen, Tabletten und Suppositorien.
Hauterkrankungen durch Parasiten
Skabies
Pathogenese
Skabies wird durch die Krätzmilbe Acarus siro va. hominis hervorgerufen. Die weibliche Milbe ist etwa 0,4 mm lang. Sie bohrt sich in die Hornschicht der Epidermis und legt dort ihre Eier ab. So entstehen die blind endenden Gänge. Aus den Eiern entwickeln sich Larven, Nymphen und schließlich geschlechtsreife Milben, die an der Hautoberfläche von männlichen Milben befruchtet werden können. Die Übertragung erfolgt durch engen körperlichen Kontakt.
Klinik
Nach Sensibilisierung gegen die Milbenantigene beginnt der äußerst quälende Juckreiz, besonders in der Bettwärme. Bevorzugte Hautregionen sind Interdigitalräume (auch Füße), Handgelenke, Gelenkbeugen, Achselfalten, Schulter- und Nabelregion. Bei Säuglingen sind auch Handflächen, Fußsohlen (Abb. 30.8), Gesicht und Kopf befallen. Hier bestehen 1–2 cm lange, leicht aufgeworfene Gänge und Papeln unterschiedlicher Größe. Durch Exkoriationen und Sekundärinfektionen entsteht ein buntes Bild mit Ekzem- und Pustelbildung.
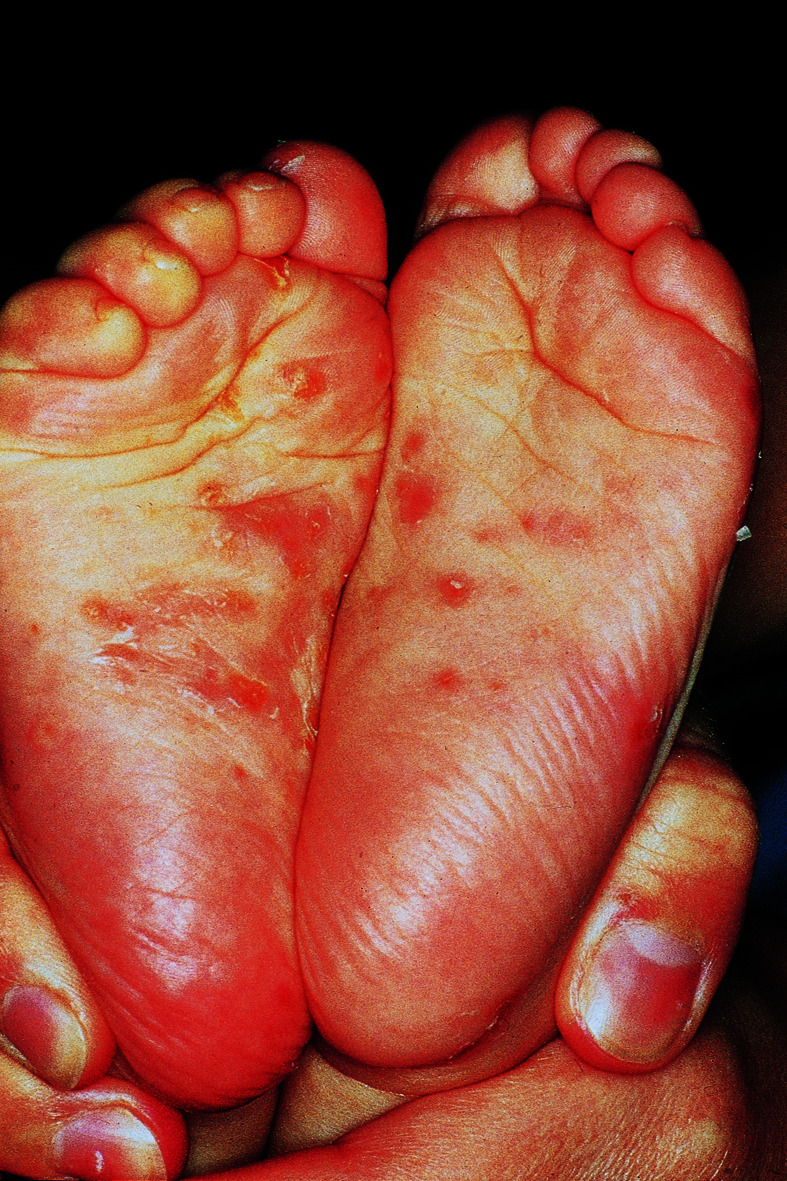
Diagnose
Die Diagnose wird durch den Nachweis der Milben gestellt. Nach Herausheben mit einer Nadel aus ihrem Gang oder durch Auftropfen von Öl und Abkratzen der Epidermis können die Milben unter dem Mikroskop gesehen werden.
Therapie
Die Therapie der Wahl ist die äußerliche Anwendung von Permethrin 5% in Cremegrundlage (z. B. Infectoscab-5%-Creme). Die Haut des Patienten wird einmalig vom Hals abwärts mit der Creme eingerieben. Nach 8–12 h erfolgt ein Reinigungsbad oder eine Dusche. Sind die Handinnenflächen oder Fußsohlen befallen, sollte aufgrund der Dicke der Hornschicht nach einer Woche erneut behandelt werden. Bei Kindern unter 3 Jahren sollte zusätzlich der Kopf unter Aussparung des Mund- und Augenbereichs eingecremt werden.
Indikationen für eine orale Therapie mit Ivermectin, welches erst vor kurzem in Deutschland zugelassen wurde, sind ein ausgeprägter Befall, Immunsuppression oder geringe Adhärenz.
Eine gleichzeitige Untersuchung und Behandlung von Kontaktpersonen ist wegen der häufigen Rezidive erforderlich.
Pedikulose
Grundlagen
Die Kopflaus (Pediculus humanis capitis), die Kleiderlaus (Pediculus humanis corporis) und die Filzlaus (Phthirus pubis) sind Parasiten des Menschen. Bei Kindern sind Infektionen durch Kopfläuse am häufigsten. Nur die Kleiderlaus kann andere Erkrankungen wie Rickettsiosen, Fleckfieber, Wolhynisches Fieber und Rückfallfieber übertragen. Die Kopf- und Kleiderlaus ist 3 bzw. 4 mm, die Filzlaus nur 2 mm lang. Weibliche Läuse legen pro Tag mehrere Eier (Nissen), die nach 2 Wochen ausgewachsen sind. Jede Laus saugt etwa 10-mal pro Tag Blut um zu überleben, dabei wird Speichel übertragen. Dies und die Ablage von Fäzes auf der Haut rufen den Juckreiz hervor.
Klinik, Therapie
Alle Läuse werden durch direkten Kontakt oder über Kleidung übertragen. Während die Prädilektionsstellen der Kopflaus das Capillitium und insbesondere retroauriculär ist, findet sich die Kleiderlaus unter der Kleidung und die Filzlaus in Pubeshaaren, Wimpern und Barthaaren. Häufig finden sich Exkoriationen. Die Behandlung erfolgt mit Dimeticon oder Permethrinlösung und Auskämmen der Haare bzw. Auskochen der Wäsche (Tab. 30.3).
| Kopflaus | Kleiderlaus | Filzlaus | |
|---|---|---|---|
| Größe (mm) | 2–3,5 | 3–4,5 | 1,5–2 |
| Übertragung | Direkter Kontakt, schlechte Hygiene | Direkter Kontakt, Kleidung | Direkter Kontakt, Kleidung, Handtücher |
| Prädilektion | Kapillitium, hinter den Ohren | Unter Kleidung | Pubes, Wimpern, Bart |
| Eiablage | Am Haarschaft | Kleidung | Pubes, Wimpern |
| Juckreiz | Ausgeprägt | Mäßig | Mäßig |
| Klinik | Urtikarielle Papeln, Exkoriationen | Rötung, Urticae, Papeln, Exkoriationen | Maculae coeruleae, keine Exkoriationen |
| Therapie | Dimeticon und Auskämmen der Haare. Alternativ: Permethrinlösung, Spülung mit verdünntem Essigwasser. | Auskochen der Wäsche, Hygiene | Wie bei Kopfläusen |
Allergische Krankheiten und Ekzemerkrankungen
Grundlagen
Allergisch bedingte Hauterkrankungen sind ausschließlich Folgen einer überschießenden bzw. fehlgeleiteten Immunreaktion, meistens ausgelöst durch als Antigen (Allergen) wirkende Substanzen. Sofortreaktionen (überwiegend Typ-I-Reaktionen, IgE-vermittelt) und solche vom Spättyp (Typ-IV-Reaktionen, T-Lymphozyten-abhängig) können unterschieden werden. Beispiele einer Typ-I-Reaktion sind Urtikaria, Angioödem, allergische Rhinitis und Insektengiftallergien. Das klinische Bild einer Spättypallergie entspricht dem eines Ekzems.
Hautreaktionen vom Soforttyp
Urtikaria
Die Urtikaria gehört zu den häufigsten Hauterkrankungen, die über 30% der Menschen einmal im Leben erleidet. Man unterscheidet eine akute Form (bis maximal 6 Wochen anhaltend) von einer selten bei Kindern auftretenden chronischen und rezidivierenden Form (über 6 Wochen Dauer). Die Quaddel ist vorwiegend Folge einer Histaminfreisetzung aus den Mastzellen und Bildung eines umschriebenen Ödems in der oberen Dermis.
Auch bei Kindern gibt es zahlreiche allergische und nichtallergische Auslösefaktoren, wie Nahrungsmittel, Arzneimittel, Parasiten, bakterielle und virale Infekte und physikalische Auslöser wie z. B. Kälte. Häufig lässt sich allerdings kein Auslöser ermitteln.
Klinik
Die Urtikaria ist durch flüchtige, quaddelförmige, juckende, erythematöse, gut abgegrenzte Hautveränderungen gekennzeichnet (Abb. 30.9). Quaddeln entwickeln sich rasch innerhalb von wenigen Minuten und sind nach Größe und Form verschieden. In der Regel handelt es sich um eine flüchtige, nur wenige Stunden anhaltende Erscheinung. Entsteht die Quaddel nicht in der oberen Dermis, sondern in der Tiefe, so entsteht eine hautfarbene Schwellung, wie sie für das Quincke-Ödem und das hereditäre Angioödem charakteristisch ist. Auch Schleimhäute (Glottisödem oder Larynxödem) und innere Organe (Asthmaanfälle, Abdominalschmerzen, Durchfälle und Schocksymptome bis hin zum allergischen Herz-Kreislauf-Versagen) können beteiligt sein. Auch physikalische Einwirkungen (Wärme, Kälte, Druck) können eine Urtikaria verursachen.
Diagnose
Bei Auftreten einer akuten Urtikaria ist besonders an Infekte, Arzneimittel, Nahrungs- oder Genussmittel, Inhalationsantigene, Insektenstiche oder Insektenbisse zu denken. Dauern Urtikariaschübe länger als 6 Wochen, sollte eine ausführliche Diagnostik erfolgen. Diese umfasst neben einer sorgfältigen Erhebung der Anamnese und allgemeinen klinischen Untersuchung Fokussuche, Hauttestungen (Reibe-, Prick- und Intrakutantest), Radioimmun-, Enzymimmuntests und Provokationstests (z. B. mit Nahrungsmitteln, Nahrungsmittelzusatzstoffen, Acetylsalicylsäure).
Therapie
Bei einer akut auftretenden Urtikaria stehen oral oder bei Bedarf i.v.-verabreichte Antihistaminika an erster Stelle. Sie unterdrücken sehr erfolgreich die Quaddeln. Lokal können kortikosteroidhaltige Externa in Form von kühlender Hautmilch eingesetzt werden.
Cave
Bestehen anaphylaktische Reaktionen mit Atemnot, Schluckbeschwerden, Blutdruckabfall, Pulsanstieg und drohender Bewusstlosigkeit, müssen Kortikosteroide i.v., evtl. auch Adrenalin gegeben und eine intensivmedizinische Betreuung angeschlossen werden.
Bekannte Noxen sind zu vermeiden. Bei chronischer Urtikaria sollte eine umfangreiche Diagnostik erfolgen.
Allergische Krankheiten und Ekzeme
Grundlagen
Ekzemerkrankungen sind sehr häufig. Sie sind nichtinfektiös und nichtkontagiös. Die pathologischen Veränderungen in der Epidermis und im oberen Korium prägen das klinische Bild. Bei akutem Verlauf stehen exsudativ-entzündliche Hautveränderungen mit Rötung, Schwellung, Bläschen, Nässen und Krusten im Vordergrund, bei chronischem Verlauf Rötung, Epidermisverdickung, Schuppung, Lichenifikation und Rhagadenbildung. Bei akutem Auftreten spricht man eher von Dermatitis, bei chronischen Veränderungen eher von Ekzemen.
Im Folgenden soll auf die im Kindesalter wichtigen Ekzemformen näher eingegangen werden.
Allergisches Kontaktekzem
Pathogenese
Dem allergischen Kontaktekzem liegt eine T-zellvermittelte Immunreaktion vom Typ IV zugrunde. Die Sensibilisierung wird durch antigenpräsentierende Langerhans-Zellen in der Epidermis eingeleitet, an deren Oberfläche das Antigen den T-Lymphozyten präsentiert wird. Allergenspezifische T-Zellen gelangen nach Proliferation in den Lymphknoten, schließlich über den Blutkreislauf in die Haut zurück und rufen nach erneutem Allergenkontakt eine Ekzemreaktion hervor. Begünstigend für eine Allergenisierung ist eine gestörte Hautbarriere (z. B. durch häufiges Waschen mit Seifen, vorbestehende Ekzeme) und eine hohe Potenz des Allergens (z. B. Nickelionen).
Klinik
Je nach Sensibilisierungsgrad, Kontakthäufigkeit und Lokalisation kann sich ein sehr variables klinisches Bild zeigen. Akute Hautveränderungen treten bei hoher Sensibilisierung auf (Rötung, Nässen, Papulovesikel). Häufiger Kontakt mit Stoffen bei niedriger Sensibilisierung führt eher zu chronischen Hautveränderungen mit Schuppenbildung, Lichenifikation und Rhagadenbildung.
Diagnose
Nach sorgfältiger Anamnese kann durch einen Epikutantest der Auslöser der allergischen Reaktion aufgedeckt werden. Dabei werden Teststreifen, die verschiedene Allergene enthalten, auf normale, nicht entzündliche Haut aufgeklebt und für 24 h belassen. Nach Entfernung der Pflaster kann in den folgenden 48 h die Reaktion abgelesen werden. Schwierig ist die Abgrenzung irritativ-toxischer Reaktionen.
Therapie
Oberstes Gebot ist die Vermeidung der auslösenden Substanz. Bei akuten Hautveränderungen kommen externe Glukokortikosteroide in einer wässrigen Grundlage in Betracht.
Cave
Bei großflächiger äußerlicher Behandlung, insbesondere unter Okklusion, ist daran zu denken, dass größere Glukokortikosteroidmengen resorbiert werden können.
Bei zusätzlicher bakterieller oder mykotischer Besiedlung kommen antibakterielle und antimykotische Externa zum Einsatz. Geeignet sind z. B. feuchte Umschläge mit Chinosol.
Bei chronischen Ekzemen sind wegen der trockenen Hautveränderungen fettende Salben angezeigt. Harnstoff besitzt keratolytische und wasserbindende Eigenschaften, kann aber die Haut von Kleinkindern irritieren. Besser geeignet ist hier eine milchsäurehaltige Salbe. Günstige Einflüsse bei chronischen Ekzemen mit Lichenifizierung der Haut können auch mit farblosen Schieferölpräparaten wie Ichthyol erzielt werden. Steinkohlenteer wird bei Kindern in der Regel nicht eingesetzt.
Akute toxische Kontaktdermatitis und chronisch-kumulativ-toxisches Kontaktekzem
Pathogenese
Hierbei handelt es sich um eine direkte, nichtallergische Schädigung der Haut durch externe Faktoren. Bei Kindern spielen Speichel, Urin, Nahrungssäfte, Detergenzien, Seifen, Badezusätze und Spülmittel die Hauptrolle.
Klinik
Speichel verursacht eine Kontaktdermatitis im Gesicht und im Nacken (bei Säuglingen und retardierten Kindern). Die sog. Windeldermatitis (Ammoniakdermatitis) ist die häufigste Kontaktdermatitis im Säuglingsalter. Als Reaktion auf den chronischen Kontakt mit Urin und Stuhl, verstärkt durch Wärme und Feuchtigkeit, entwickeln sich nässende Erytheme mit Bläschen, Pusteln und Mazerationen der Haut. Sekundärinfektionen (bakteriell und mit Candida albicans) sind fast immer nachzuweisen.
Therapie
Häufiger Wechsel der Windeln, gründliche Reinigung der Haut mit warmem Wasser und anschließender Anwendung von Zinköl oder Pasta zinci mollis führen meistens zur raschen Abheilung der Windeldermatitis. Bei therapieresistenten Verläufen sollte die mögliche bakterielle Infektion durch spezifische, lokal wirkende Salben bekämpft werden.
Bei den anderen Formen der toxischen Kontaktdermatitis kann meistens das toxische Agens eliminiert oder die betroffene Hautpartie durch entsprechende Salben geschützt werden.
Seborrhoisches Ekzem des Säuglings
Pathogenese
Vermutlich durch eine erhöhte Talgdrüsenaktivität (Seborrhö) und eine Besiedlung mit Malassezia-Hefen und deren Stoffwechselprodukten kommt es bei Säuglingen bereits in den ersten drei Lebensmonaten zu typischen Hautveränderungen.
Klinik
An den Prädilektionsstellen am Kopf im Scheitelbereich, aber auch am Stamm oder diffus über das ganze Integument verteilt, treten gelbe, fettglänzende Schuppen ohne entzündliche Rötung (Gneis) auf. In intertriginösen Bereichen befinden sich nässende oder trockene Erytheme, die ebenfalls mit gelb-fettigen Schuppen bedeckt sein können.
Therapie
Stärkere Schuppen können mit Olivenöl allein oder mit Zusatz von 2%iger Salicylsäure abgelöst werden. Danach können kurzfristig kortikosteroidhaltige Cremes oder Lotiones der Stärkeklasse 1 aufgetragen werden. Zur Behandlung intertriginöser Bereiche hat sich Vioform-Zinköl (0,5%) bewährt. Gebadet werden sollte in handwarmen Bädern mit entzündungshemmenden Zusätzen (z. B. Weizenkleieextrakt) und Zusätzen von Öl.
Atopisches Ekzem
Definition
Ein atopisches Ekzem ist eine chronisch entzündliche Hauterkrankung bei Kindern mit einer genetisch bedingten Disposition zu allergischen Reaktionen wie Rhinitis allergica und Asthma bronchiale. Die Ätiologie ist ungeklärt. Bei der Mehrzahl der Patienten ist die IgE-Konzentration im Serum erhöht. Allerdings gibt es auch Patienten, die trotz atopischer Dermatitis normale IgE-Serumspiegel zeigen. Die Funktion von T-Lymphozyten kann vermindert sein. Außer diesen, nicht konstant nachweisbaren immunologischen Auffälligkeiten zeigt der atopische Patient eine gesteigerte Empfindlichkeit der Haut, z. B. eine erniedrigte Juckreizschwelle, paradoxe Schweißsekretion und abnorme Gefäßreaktion (weißer Dermographismus).
Pathogenese
Die Pathogenese des atopischen Ekzems ist multifaktoriell; bei 1/3 der betroffenen Kinder lassen sich Mutationen im Gen von Filaggrin, einem Hauptbestandteil der Hornschicht, nachweisen. Die Genmutation führt zu einem Verlust der wichtigen Funktionen von Filaggrin für die Aufrechterhaltung der mechanischen Hautbarriere. Kinder mit Filaggringenmutation haben zudem ein höheres Risiko, ein allergisches Asthma und Nahrungsmittelallergien zu entwickeln. Neben der endogenen genetischen Veranlagung spielen auch zahlreiche exogene Faktoren bei der Manifestation eine Rolle. Dazu gehören eine gestörte humorale und zelluläre Immunabwehr, Hautfunktionsstörungen, erhöhte Hautirritabilität, Nahrungsmittelunverträglichkeiten, Allergien, Stress und trockenes und kaltes Klima.
Klinik
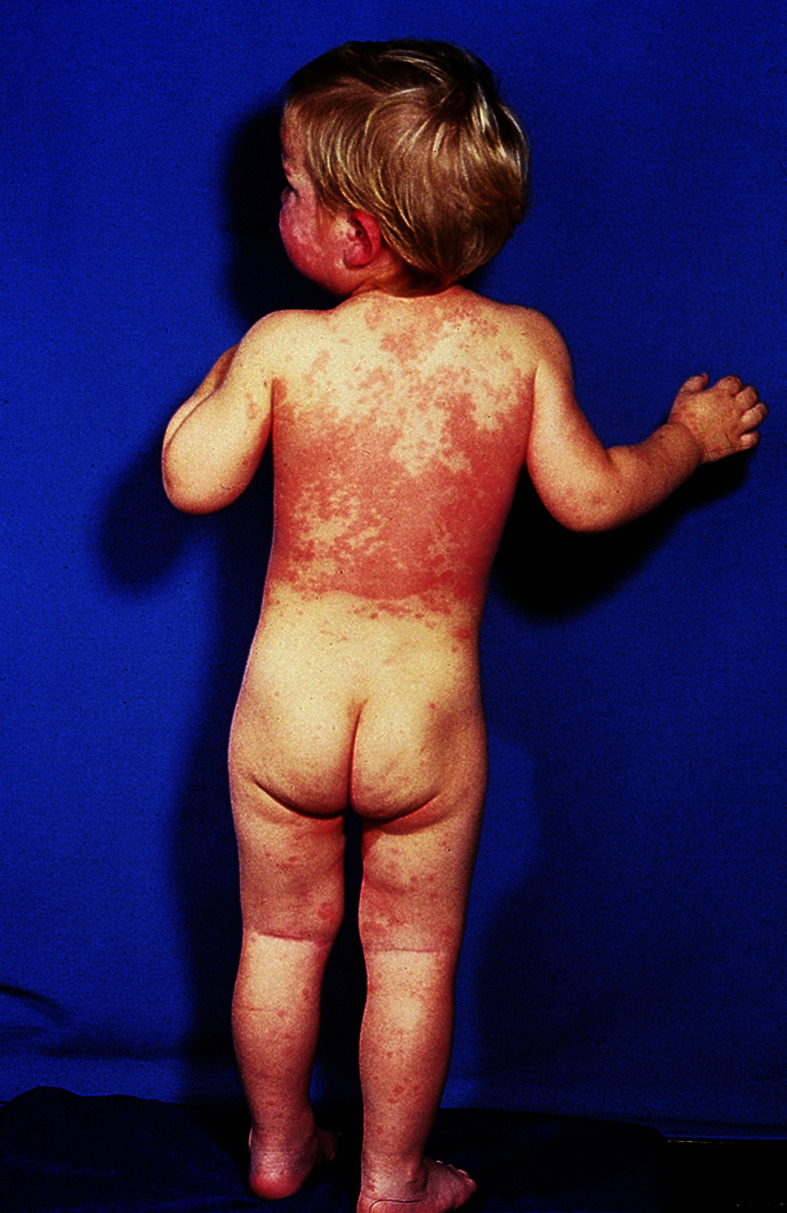
Die klinischen Erscheinungen reichen von nässenden, krustösen, stark juckenden, geröteten Effloreszenzen bis zu trockenen, schuppenden oder lichenifizierten Ekzemherden. Bei Säuglingen und Kleinkindern stehen die erstgenannten Veränderungen im Vordergrund. Die Erkrankung manifestiert sich ab dem 2. Lebensmonat, häufig lokalisiert an Wangen und Gesicht (Abb. 30.10), Nacken und Streckseiten der Extremitäten.
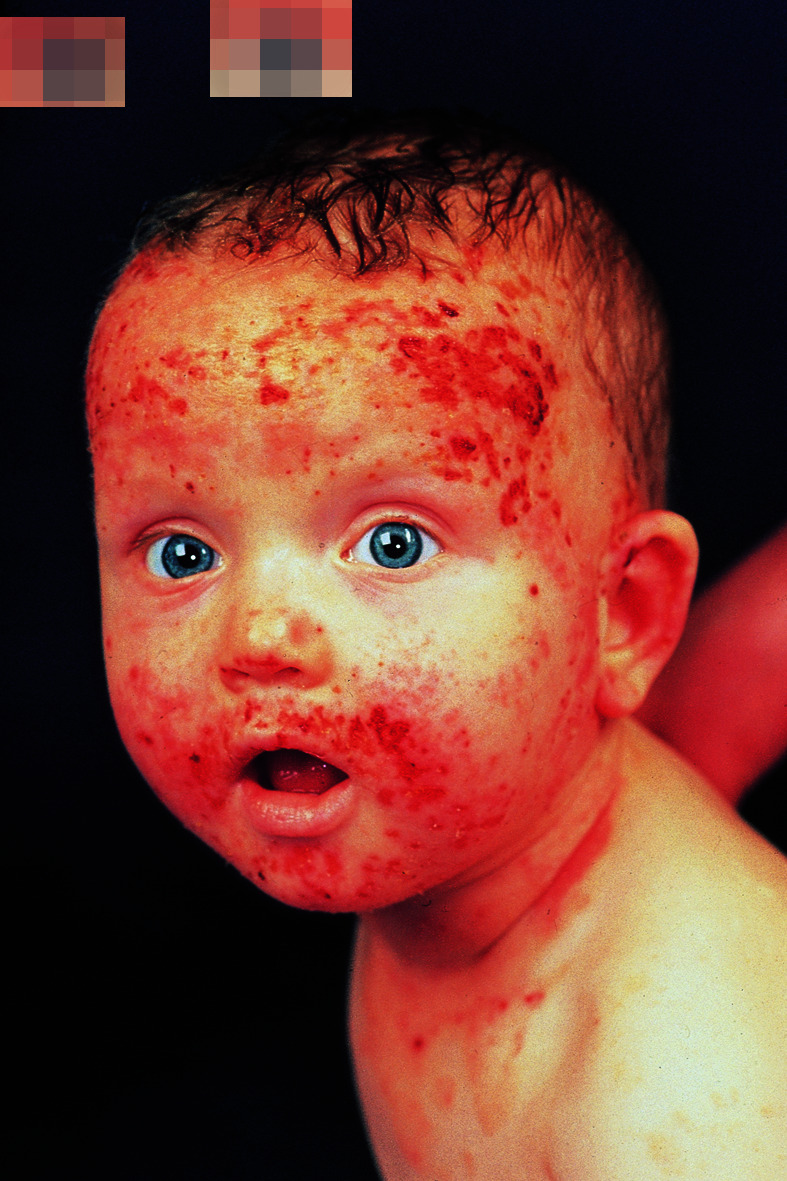
Später, bei Jugendlichen, entwickelt sich dann das typische Erwachsenenmuster des atopischen Ekzems, nämlich Befall der Gelenkbeugen mit erythematös-squamösen Veränderungen oder flächigen, zentral grobstrukturierten Lichenifizierungen (Abb. 30.11). Im Bereich der Finger oder Zehen befindliche chronisch schuppende Hautveränderungen bei Jugendlichen können Symptom eines atopischen Ekzems sein. In dieser Altersstufe nimmt die Haut, besonders im Gesicht, eine weiße Färbung an, die die Patienten vorgealtert aussehen lässt.
Häufig finden sich zusätzlich zahlreiche atopische Stigmata wie: Xerodermie, Dennie-Morgan-Infraorbitalfalte (Atopiefalte im Bereich der Unterlider), tiefer Haaransatz, Hertoghe-Zeichen (Ausfall der lateralen Augenbrauen), trockene Lippen, Perlèche, Mamillenekzem, Pulpitis sicca, Keratokonus, Neigung zu Hautinfektionen, Pityriasis alba, Ichthyosis vulgaris, Wollempfindlichkeit, Nahrungsmittelunverträglichkeit (v. a. gegen Kuhmilch, Eiweiß, Nüsse, Fisch, Soja).
Immer wieder betroffene Hautpartien können Hyperpigmentationen und vermehrte Rauigkeit wie bei Ichthyose aufweisen („dirty neck“). In den meisten Fällen verschwinden die Symptome vor dem Schulalter oder nehmen doch erheblich an Intensität ab.
Diagnose
Pruritus, typische Lokalisation der Ekzeme, atopische Eigen- oder Familienanamnese und der chronische Verlauf der Erkrankung sind für die Diagnose wegweisend. Atopische Stigmata können bei geringer Ausprägung der Ekzeme die Diagnose stützen. Weiterhin führen ein weißer Dermographismus und ein erhöhtes Gesamt-IgE zur richtigen Diagnose.
Therapie
Nässende Ekzemflächen sollten mit feuchten Kompressen behandelt werden. Für trockene ekzematöse Hautpartien eignen sich kortikoidhaltige Salben und besonders die Durchführung eines sog. „Fettbettes“. Dabei werden die Kinder mit fetthaltigen Salben eingerieben und für 2 h in feuchte Handtücher gewickelt. Zwischen den akuten Phasen der Erkrankung sollte die Haut mit wasserhaltigen Cremes und Kühlsalben gefettet und vor Austrocknung geschützt werden. Bei der an sich schon trockenen Haut des Ekzematikers sind austrocknende Seifen und häufiges Baden nicht angebracht. Sinnvoll ist längerer Kontakt mit Wasser nur dann, wenn Feuchtigkeitsverlust durch Zugaben von rückfettenden Badeölen vermieden und die noch feuchte Haut nach dem Bad sofort gesalbt wird. Normale Seifen werden im Allgemeinen schlecht vertragen, besser geeignet sind Syndets.
In der Akuttherapie des atopischen Ekzems sind kortikosteroidhaltige Cremes oder Salben der Stärkeklasse 1 oder 2 häufig nicht zu vermeiden, sollten aber nicht länger als 2–3 Wochen kontinuierlich angewendet werden. Die seit mehreren Jahren verfügbaren topischen Calcineurininhibitoren Tacrolimus und Pimecrolimus bringen einen großen Fortschritt gerade in der Langzeittherapie des atopischen Ekzems, da unerwünschte Wirkungen wie Hautatrophie und Bildung von Teleangiektasien nicht beobachtet werden. Bei längerfristiger Anwendung kann die Zahl neuer Ekzemschübe verringert und dadurch der Verbrauch von kortikoidhaltigen Externa reduziert oder vermieden werden.
Eine Infektion mit Herpes simplex bei Kindern mit manifestem atopischem Ekzem kann zu einer Generalisierung von Herpesbläschen und zu hohem Fieber führen (Ekzema herpeticatum).
Prophylaxe
Hitzestau und Situationen mit starkem Schwitzen sollten vermieden werden. Ideal ist ein warmes Klima mit mäßiger Luftfeuchtigkeit und hoher Luftkonvektion. Mäßige Sonnenbestrahlung und salzhaltiges Wasser führen in den meisten Fällen zur Besserung der Hautsymptome (Ostsee, Mittelmeer). Am besten wird baumwollhaltige Kleidung vertragen. Zu vermeiden sind synthetische Fasern und Wolle.
Nur wenn ein eindeutiger Zusammenhang zwischen der Einnahme von Nahrungsbestandteilen und der Exazerbation der atopischen Dermatitis besteht, sind diese zu meiden. In der Regel ist ein solcher Zusammenhang aber nicht zu eruieren.
Arzneimittelexantheme
Grundlagen
Die häufigsten Nebenwirkungen nach Medikamenteneinnahme sind bei Kindern Hautreaktionen. Inwieweit es sich um allergische, pseudoallergische oder toxische Reaktionen handelt, ist meistens nicht zu klären.
Klinik
Das klinische Bild des Arzneimittelexanthems ist nicht einheitlich. Es können urtikarielle, papulöse, bullöse oder am häufigsten morbiliforme und scarlatiniforme Exantheme auftreten. Sie sind vorwiegend symmetrisch lokalisiert und häufig stammbetont. Arzneimittel und andere Substanzen können auch zum Bild eines Erythema exsudativum multiforme führen, dessen schwere Verlaufsform (Beteiligung von Konjunktiven und Schleimhäuten) Stevens-Johnson-Syndrom genannt wird.
Beim Lyell-Syndrom (toxische epidermale Nekrolyse) handelt es sich um eine bei Kindern sehr seltene Hypersensitivitätsreaktion auf unterschiedliche Faktoren, die unter dem Bild einer Verbrennung (Erythem, Blasenbildung und Hautnekrose) verläuft. Auslösende Faktoren sind v. a. Medikamente (Pyrazolone, Barbiturate, Antibiotika, Sulfonamide) oft in Zusammenhang mit Infektionen oder Impfungen. Nach Prodromalerscheinungen (Fieber, reduzierter Allgemeinzustand, Hauterythem) entwickeln sich auf gesundem Integument Blasen, die sich rasch vergrößern und großflächig ablösen.
Therapie
Die Therapie des Lyell-Syndroms entspricht der von Verbrennungen. Man muss mit einer hohen Letalität rechnen (altersabhängig bis 40%).
Differenzialdiagnose
Abgegrenzt werden muss bei Kleinkindern das staphylogene Lyell-Syndrom („staphylococcal scalded skin syndrome“), eine durch Staphylokokkenexotoxin ausgelöste lebensbedrohliche Erkrankung mit blasiger Ablösung der Haut. Schleimhäute werden nicht befallen. Histologisch ist hier eine akantholytische Blasenbildung subkorneal zu finden. Eine frühzeitige Therapie mit penicillinasefesten Penicillinen ist einzuleiten.
Autoimmunkrankheiten
Chronisch diskoider Lupus erythematodes
Pathogenese, Epidemiologie
Der Pathomechanismus der Hautveränderungen beim chronisch diskoiden Lupus erythematodes (CDLE) ist wenig erforscht. Für eine lokale Immunkomplexvaskulitis wie bei systemischem Lupus erythematodes oder für eine Ro(SSA)-antikörperabhängige zytotoxische Reaktion wie bei dem subakuten Lupus erythematodes finden sich wenig Anhaltspunkte. Weniger als 2% der Patienten entwickeln einen CDLE vor dem 10. Lebensjahr. Im Gegensatz zu Erwachsenen sind Jungen genauso häufig wie Mädchen betroffen; Angaben über vermehrte Lichtempfindlichkeit fehlen häufig.
Klinik
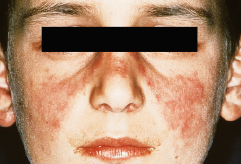
Die lokalisierte Form des CDLE zeigt sich häufig in Schmetterlingsform an Nase und Wangen (Abb. 30.12), an den Ohren und am Kapillitium. Seltener sind Augenlider, Lippenregion oder Schleimhäute befallen. Bei der disseminierten Form zeigen sich die scharf begrenzten, scheibenförmigen Erytheme mit festhaftender, weißlicher Schuppung auch im Schulterbereich, an den Streckseiten der Arme und Handrücken. Bei Kindern können auch frostbeulenartige, knotige Veränderungen an Finger- und Zehenrücken auftreten (sog. „Chilblain“-Lupus). Bei der seltenen Form des familiären Chilblain-Lupus führen Mutationen des Gens für die Exonuklease TRX1 zu einer Abbauhemmung intrazellulärer Nukleinsäuren mit nachfolgender Interferon-α-vermittelter Entzündung.

Diagnose
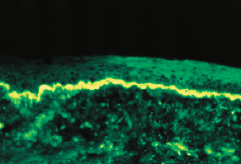
Mittels Immunfluoreszenzmikroskopie erfolgt der Nachweis von bandförmigen Niederschlägen von Immunglobulinen IgG oder IgM häufig zusammen mit dem Komplementspaltprodukt C3 entlang der Basalmembranzone (sog. Lupusband, Abb. 30.13).

Therapie, Prognose
Im Vordergrund steht eine äußerliche Therapie mit glukokortikosteroidhaltigen Cremes oder Salben bei gleichzeitigem Lichtschutz; bei Nichtansprechen kann eine innerliche Gabe von Glukokortikosteroiden, Antimalariamitteln oder Dapson erwogen werden. Der Übergang in einen systemischen Lupus erythematodes ist bei Kindern häufiger als bei Erwachsenen.
Zirkumskripte Sklerodermie (Morphea)
Die umschriebene Sklerose der Haut und des Unterhautgewebes manifestiert sich häufig bei Kindern und Jugendlichen; in diesen Lebensaltern wird fast nie ein Übergang in eine systemische Sklerodermie mit ausgeprägter Organbeteiligung beobachtet.
Pathogenese
Ursache ist unbekannt; Verletzungen, Impfungen, Infektionen, Bestrahlungen werden als Provokationsfaktoren diskutiert. Es besteht auffällige Ähnlichkeit mit der chronischen Graft-versus-Host-Krankheit nach allogener Knochenmark- oder Stammzelltherapie. Nicht selten besteht gleichzeitig v. a. im Genital- oder Analschleimhautbereich ein Lichen sclerosus et atrophicus (LSA), der, meist bei Mädchen, auch isoliert vorkommt. Hier zeigen sich stark juckende weißlich-atrophische Herde an der Vulva oder perianal, teilweise mit Hämorrhagien oder Blasen die zu Schrumpfung und Atrophie der Schleimhäute führen können.
Klinik
In Abhängigkeit von Größe, Ausprägung und Lokalisation werden 5 klinische Formen der zirkumskripten Sklerodermie unterschieden:
herdförmige zirkumskripte Sklerodermie (Morphea),
subkutane zirkumskripte Sklerodermie,
kleinfleckige Morphea,
generalisierte zirkumskripte Sklerodermie,
lineare zirkumskripte Sklerodermie.
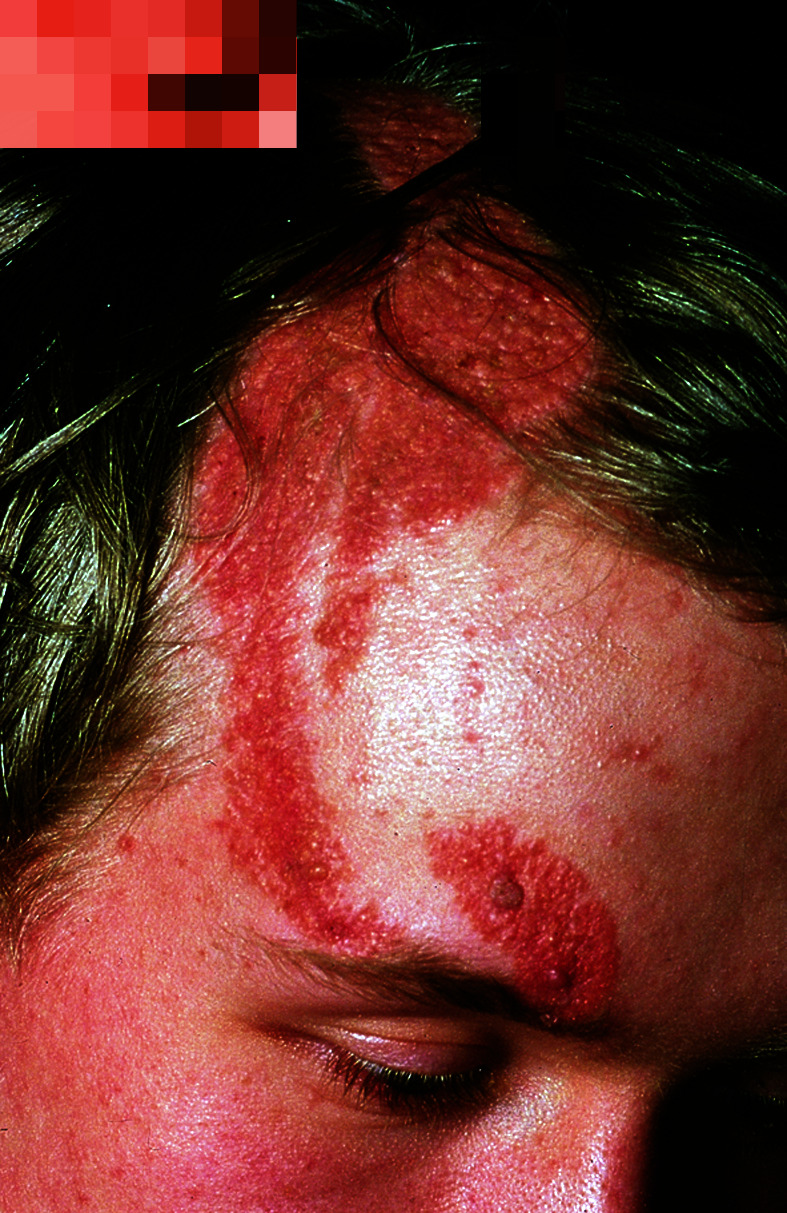
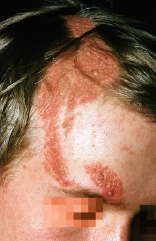
Bei Kindern ist die lineare zirkumskripte Sklerodermie am häufigsten: unilateral an einer Extremität oder seltener am Rumpf besteht eine strangförmige Hautsklerose mit Beteiligung des subkutanen Gewebes, die zu Muskelatrophie, ossären Wachstumsstörungen und Gelenkkontrakturen führen kann. Charakteristisch ist die lineare zirkumskripte Sklerodermie im Stirnbereich (Abb. 30.14) als „Coup des sabre“, aus der sich eine lebenslang bestehende Hemiatrophia faciei entwickeln kann.
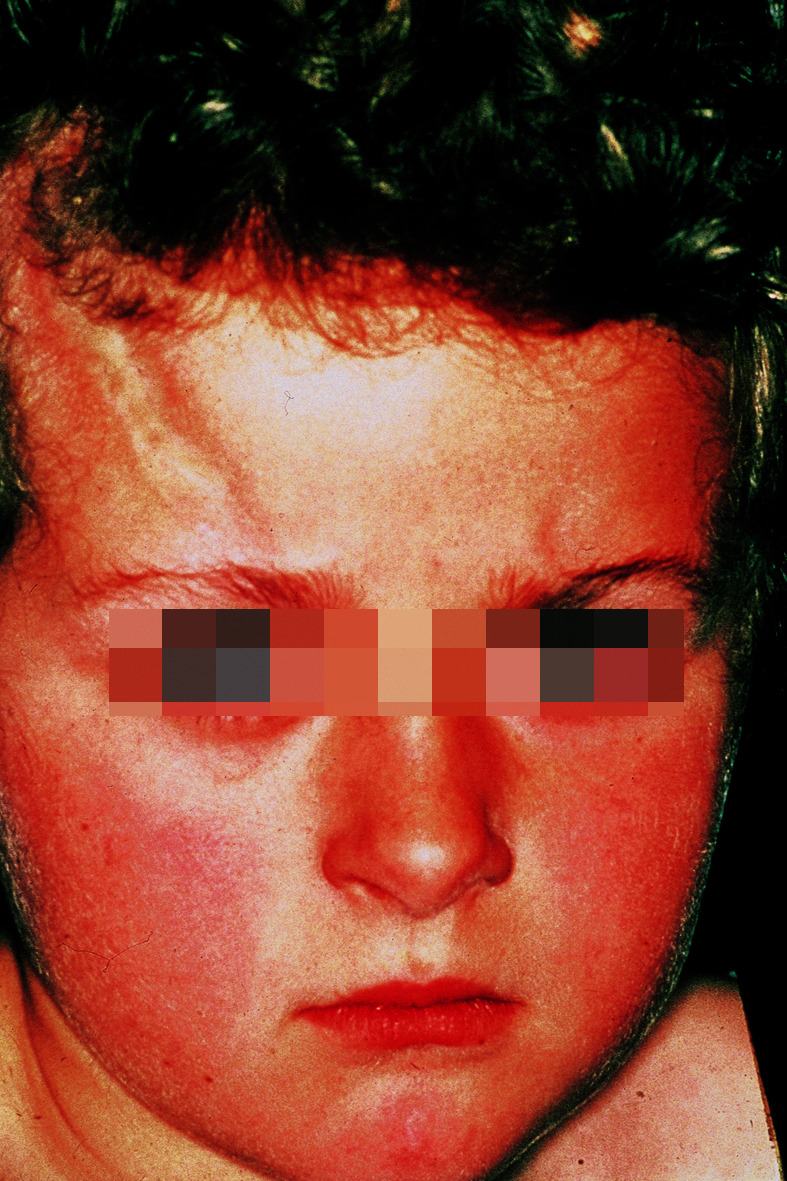
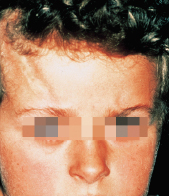
Diagnose
Meist durch das typische klinische Bild möglich, Bestätigung durch histologische Untersuchung; immunserologisch bei etwa 40% der Kinder Nachweis von antinukleären Antikörpern im Serum; für systemische Sklerodermie spezifische Antikörper (z. B. gegen Scl-70 oder Zentromerproteine) kommen bei zirkumskripter Sklerodermie nicht vor.
Therapie
Konsequente äußere Behandlung mit fettenden Salben, evtl. mit Glukokortikosteroid- und Heparinoidzusatz. Bei chronischen Verläufen auch Photochemotherapie mit UVA1 oder UVA nach vorherigem Bad in 8-MOP-haltigem Wasser oder nach Auftragen einer 8-MOP-haltigen Creme (Bade- bzw. Creme-PUVA). Im entzündlichen Frühstadium kann eine hochdosierte i.v.-Therapie mit Penicillin versucht werden Bei Lichen sclerosus et atrophicus werden Salben oder Cremes mit hochpotenten Glukokortikoiden oder Calcineurininhibitoren empfohlen. Bei ausgeprägtem Juckreiz sind intraläsionale Injektionen einer Glukokortikoidkristallsuspension (Triamcinolonacetonid 1:3–1:5 mit Lokalanästhetikum verdünnt) gut wirksam.
Bei linearer zirkumskripter Sklerodermie besteht die Gefahr bleibender Atrophien im Muskel- und Knochenbereich sowie von Gelenkkontrakturen.
Juvenile Dermatomyositis
Kap. 12.5.
Blasenbildende Autoimmunerkrankungen
Pathogenese
Die blasenbildenden Autoimmunkrankheiten führen zu einer chronischen oder schubweise verlaufenden Entzündung von Haut- und Schleimhäuten mit Blasenbildung und sekundär mit Erosionen und Krustenbildung. Ausgelöst wird die Entzündung durch spezifische Autoantikörper der Immunglobulinklasse IgG oder IgA, die mit definierten Strukturproteinen im Bereich der Epidermis, der Basalmembranzone oder der Dermis reagieren; je nach Lokalisation der spezifischen Antigen-Antikörper-Reaktionen entsteht eine:
intraepidermale Spaltbildung (z. B. beim Pemphigus),
subepidermale Spaltbildung (z. B. bei der linearen IgA-Dermatose) oder
dermale Spaltbildung (z. B. bei der Dermatitis herpetiformis).
Epidemiologie
Diese Erkrankungen können in allen Lebensaltern auftreten; bei Kindern und Jugendlichen ist die lineare IgA-Dermatose und die Dermatitis herpetiformis am häufigsten. Durch diaplazentare Übertragung spezifischer Antikörper von einer erkrankten Schwangeren auf das sich entwickelnde Kind können einige dieser Krankheiten auch neonatal auftreten; dazu gehören Pemphigus vulgaris und das Schwangerschaftspemphigoid (Herpes gestationis). Die Geschlechtsverteilung ist annähernd ausgewogen.
Lineare IgA-Dermatose (LAD)
Definition
Durch Autoantikörper der IgA-Klasse gegen Adhäsionsproteine im Bereich der Basalmembranzone ausgelöste blasenbildende Dermatose, die überwiegend bei Kindern und Jugendlichen beginnt.
Pathogenese
Die Autoantikörper bei LAD sind v. a. gegen BPAG-2 (Kollagentyp XVII) gerichtet, ein transmembranöses Adhäsionsmolekül, das die basalen Keratinozyten der Epidermis mit der Basalmembranzone verbindet. Bei LAD stellt vorwiegend der extrazelluläre Anteil von Kollagentyp XVII mit einem Molekulargewicht von 120 kD (LAD1) das Autoantigen dar.
Klinik
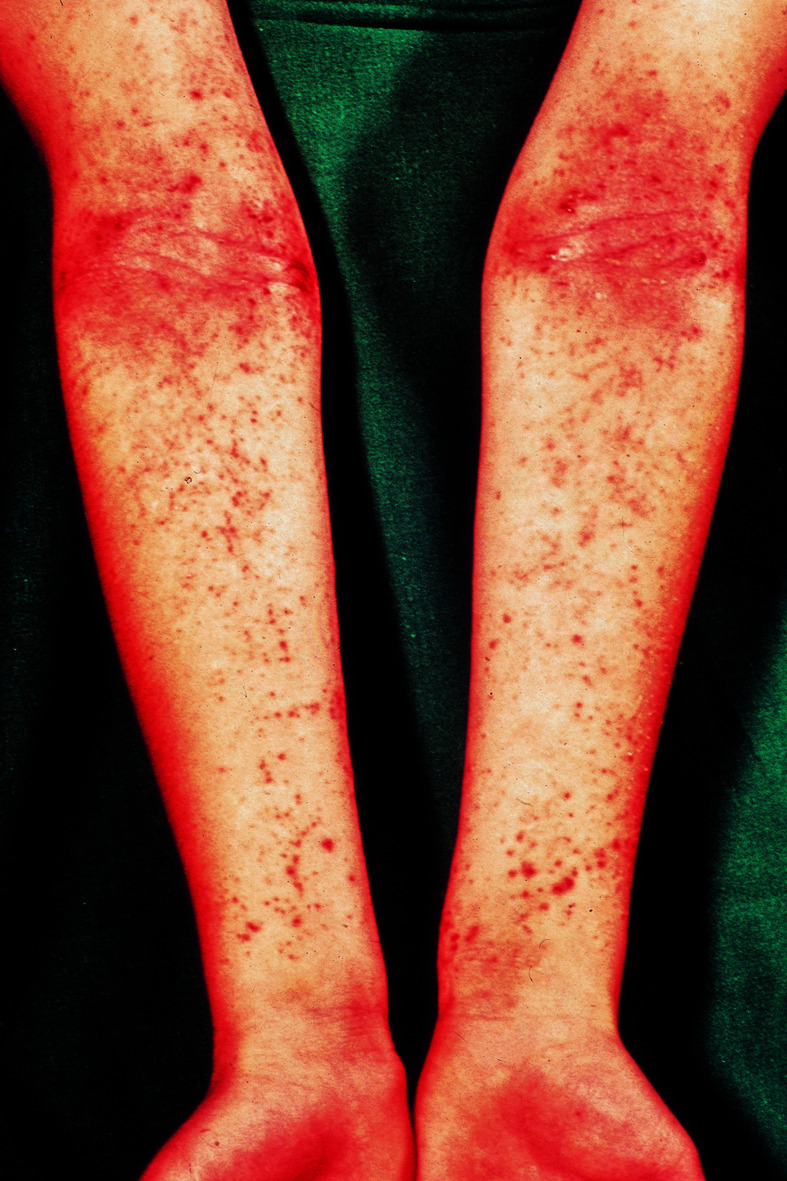
Charakteristisch sind ring- oder kranzförmig angeordnete Blasen auf Erythem, die häufig perioral, perigenital, perianal, an Händen und Füßen sowie im Gesicht lokalisiert sind (Abb. 30.15). In 40–60% Befall der Schleimhaut, meist in Form von Erosionen. Eine glutensensitive Enteropathie wie bei Dermatitis herpetiformis kommt bei LAD nicht vor.
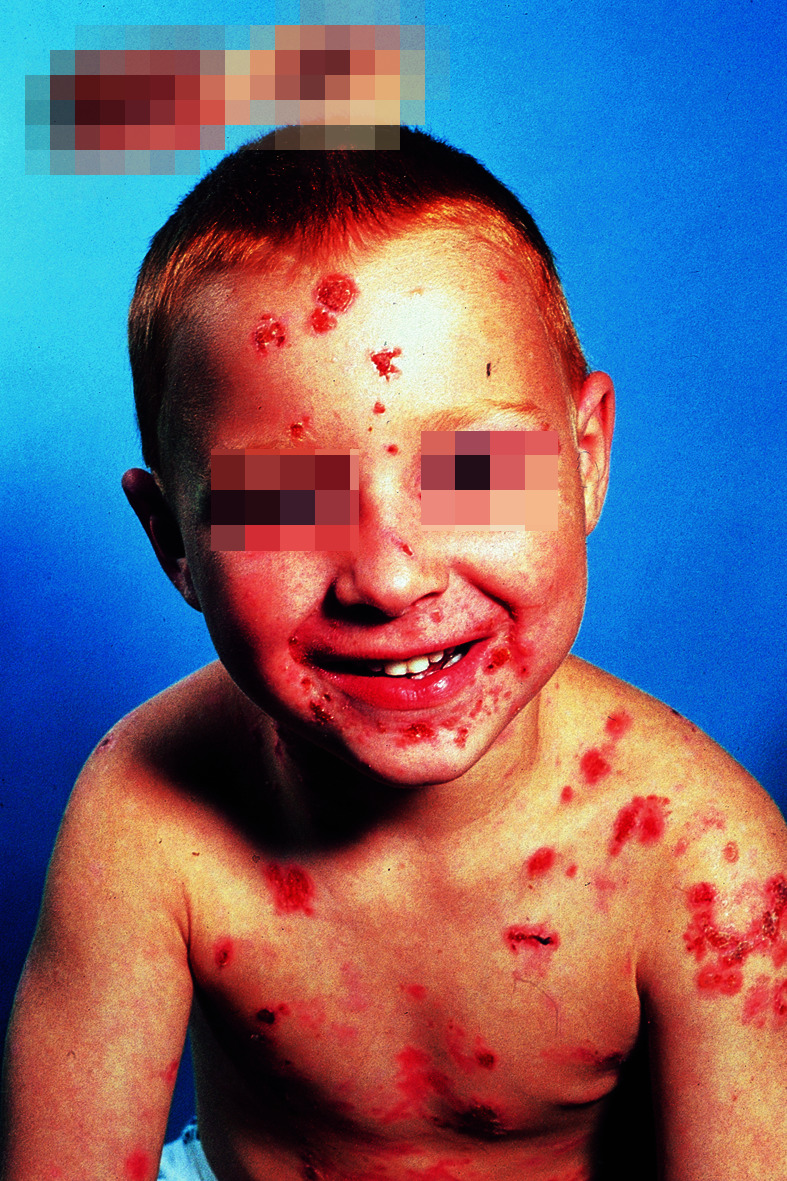
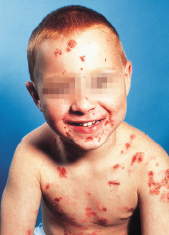
Diagnose
Wegweisend ist die direkte Immunfluoreszenzuntersuchung mit Nachweis von linearen Niederschlägen von IgA-Antikörpern entlang der Basalmembranzone (Abb. 30.16); im Serum ist der Nachweis von Antikörpern gegen Kollagentyp XVII und/oder das 120-kD-Fragment (LAD1) durch indirekte Immunfluoreszenzuntersuchung bzw. Immunoblot möglich. Histologisch Nachweis einer subepidermalen Blasenbildung.
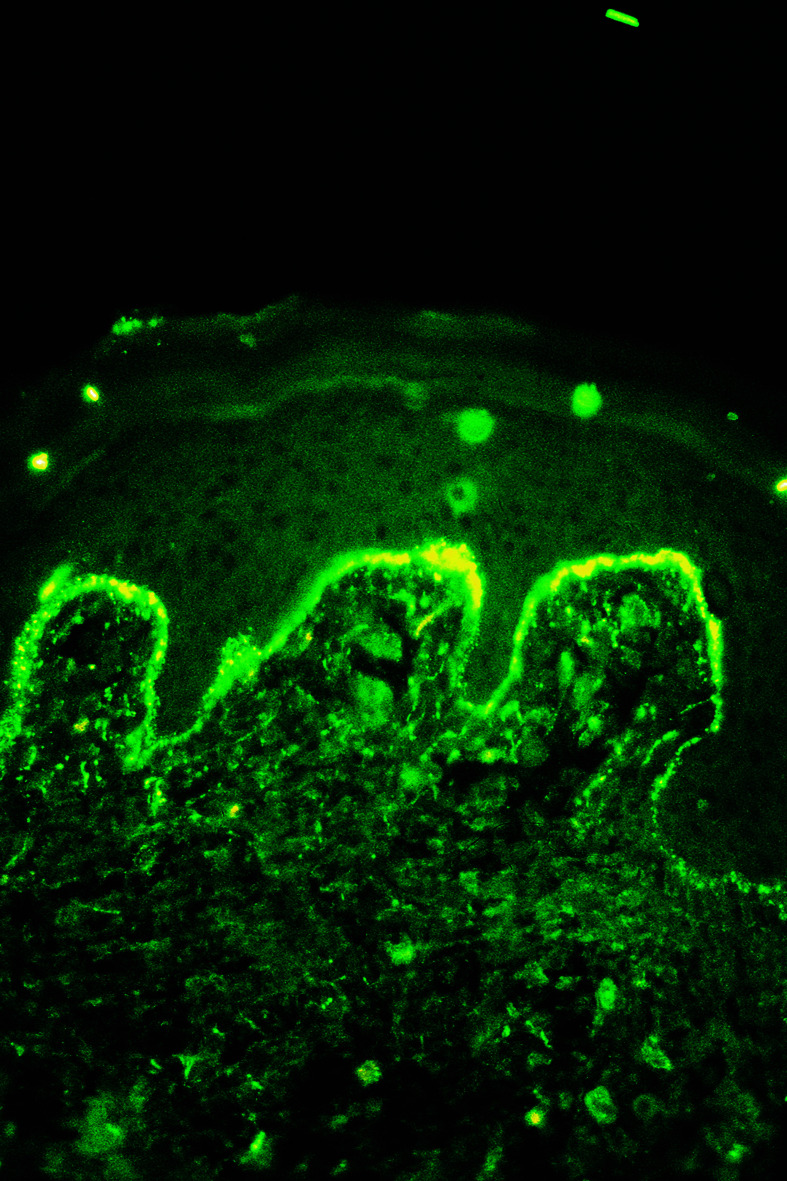
Therapie, Prognose
Bewährt hat sich eine systemische Kombinationstherapie aus Glukokortikosteroiden (z. B. 0,5 mg/kgKG Prednisonäquivalent) und Dapson (z. B. 1–1,5 mg/kgKG täglich), äußere Behandlung mit glukokortikosteroidhaltigen Cremes, Hautpflege mit soja- oder mandelölhaltigen Bädern. Auf Besiedelung der Haut mit Bakterien oder Candida albicans ist zu achten. Der Verlauf ist schubweise und hochchronisch; nicht selten Abheilung in der Pubertät.
Dermatitis herpetiformis
Definition
Chronisch rezidivierende blasenbildende Dermatose, die meist im Kindes- oder frühen Erwachsenenalter auftritt und immer mit einer Zöliakie assoziiert ist; nur in etwa 20–40% zeigen sich klinisch manifeste Zeichen der Enteropathie (Kap. 22).
Pathogenese
Wie bei Zöliakie besteht eine enge genetische Koppelung an die HLA-Antigene DR3 und DQ2. Auffällig ist die Überempfindlichkeit gegen Halogenide, insbesondere gegen jodidhaltige Medikamente und Nahrungsmittel (z. B. Seefisch), welche die Hauterscheinungen auslösen können.
Klinik
Charakteristisch sind herpetiform gruppierte Bläschen mit starkem, z. T. brennendem Juckreiz. Die Bläschen können an beliebiger Stelle lokalisiert sein, bevorzugt an den Ellenbogen, gluteal, am behaarten Kopf und den Streckseiten der Extremitäten. Bei Kindern ist auch eine exanthematische Aussaat von papulovesikulösen und sekundär verkrusteten Effloreszenzen möglich, die an Ekzeme erinnern können.
Diagnose
Histologischer Nachweis einer Spaltbildung unterhalb der Lamina densa der Basalmembran (dermolytische Blase) mit abszessartiger Ansammlung von neutrophilen Granulozyten in den dermalen Papillen. Charakteristisch ist der Nachweis von IgA-Niederschlägen in den Papillenspitzen mithilfe der direkten Immunfluoreszenzmikroskopie.
Im Serum Nachweis von Antikörpern gegen Gliadin der IgG- oder IgA-Klasse möglich; charakteristisch ist der Nachweis von Antikörpern der IgA-Klasse gegen Gewebstransglutaminase mittels indirekter Immunfluoreszenz oder ELISA; diese Anti-tTGase-Antikörper stellen serologische Marker für die glutensensitive Enteropathie dar.
Therapie
Pathognomonisch ist das schlagartige Ansprechen auf Dapson (bis 2 mg/kgKG/Tag).
Cave
Dapson ist bei Vorliegen einer Glukose-6-Phosphat-Dehydrogenase-Defizienz wegen der Gefahr einer hämolytischen Anämie kontraindiziert.
Wichtig ist eine gleichzeitige und konsequent durchgeführte glutenfreie Diät; darunter kann es – auch nach Absetzen von Dapson – innerhalb von 6–12 Monaten zur vollständigen Rückbildung der blasigen Hautveränderungen kommen.
Granulomatöse Erkrankungen
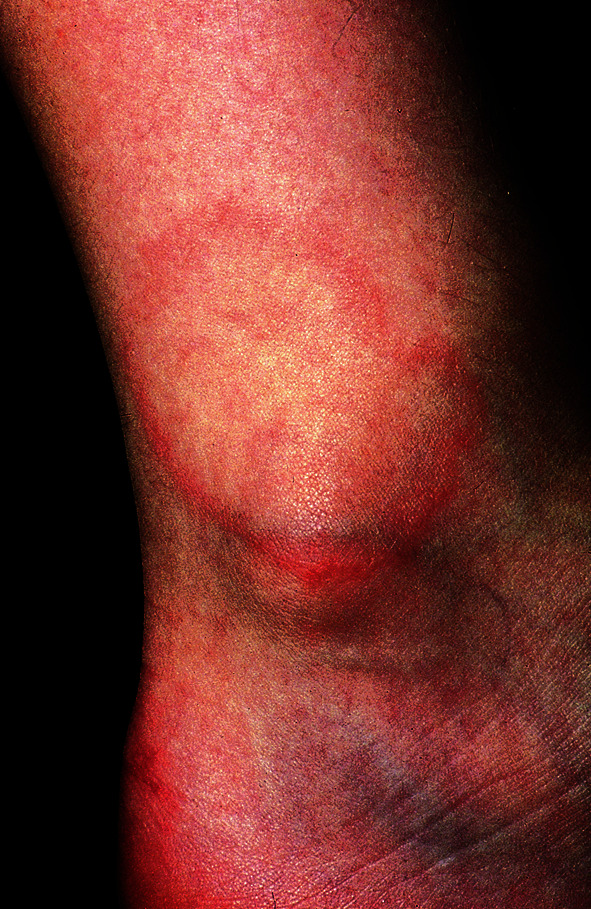
Granuloma anulare
Pathogenese
Bislang ist die Ursache des Granuloma anulare unbekannt. Anscheinend spielt eine T-zellmediierte Immunreaktion bei der Entstehung eine Rolle. Hinweise auf eine infektiöse oder toxische Genese liegen vor, das Antigen ist aber noch nicht identifiziert. Meist erkranken Kinder und Jugendliche.
Klinik
An den Prädilektionsstellen (Handrücken, Fußrücken und Finger, v. a. in der Nähe der Knöchel sowie an Glutäen und im Gesicht) finden sich zunächst kleine, scharf begrenzte und wenig gerötete Papeln, die nicht jucken. Die Herde breiten sich nach peripher aus und heilen zentral ohne Narbenbildung ab. So entstehen polyzyklisch begrenzte, randbetonte Herde von bis zu Handtellergröße (Abb. 30.17). Selten kann es auch einmal zu einer disseminierten Aussaat von aggregierenden Papeln und Knötchen am gesamten Integument kommen (Granuloma anulare disseminatum).
Therapie, Prognose
Häufig wird eine Spontanheilung ohne Therapie beobachtet. Möglich ist eine externe Therapie mit Glukokortikoiden unter einem Okklusivverband. Auch eine Kryotherapie (flüssiger Stickstoff) und intraläsionale Injektion von Triamcinolonacetonid-Kristallsuspension kommen zur Anwendung. Bei Therapieresistenz oder disseminierter Ausbreitung kann auch eine PUVA-Therapie oder Balneophotochemotherapie (PUVA-Bad) sowie die interne Gabe von Glukokortikoiden, Dapson oder Hydroxychloroquin versucht werden. Insgesamt ist die Prognose günstig.
Erythematosquamöse und papulöse Hauterkrankungen
Psoriasis vulgaris
Epidemiologie
Mit einer Morbidität von 1–2% der Bevölkerung ist die Psoriasis eine der häufigsten Hauterkrankungen. Sie tritt familiär gehäuft auf, was eine genetische Veranlagung wahrscheinlich macht. Man geht von einer multifaktoriellen bzw. polygenen Vererbung aus, bei der auch Umweltfaktoren für die Manifestation der Erkrankung eine Rolle spielen. Es findet sich eine Assoziation mit den HLA-Antigenen A2, B13, B27, Bw57, Cw2, Cw6 und DR7. Kinder von Eltern mit Psoriasis erkranken zu 10–20%, sind beide Elternteile betroffen, so erhöht sich die Wahrscheinlichkeit auf bis zu 50%. Eineiige Zwillinge zeigen eine Konkordanz von 90% für Psoriasis. Die Erkrankung kann in jedem Lebensalter beginnen. Eine Erstmanifestation in früher Kindheit und bei alten Menschen ist aber eher selten.
Man unterscheidet 2 Typen der Psoriasis. Die Typ-I-Psoriasis (Assoziation mit HLA-Cw6) beginnt bereits früh (<40. Lebensjahr) und zeigt eine familiäre Häufung. Der Krankheitsverlauf ist schwerer als bei der Typ-II-Psoriasis, die sich meist erst nach dem 40. Lebensjahr manifestiert.
Klinik
Die Primäreffloreszenz ist ein erythematöser, umschriebener, scharf begrenzter, mit silberweißer Schuppung bedeckter Herd (Abb. 30.18).
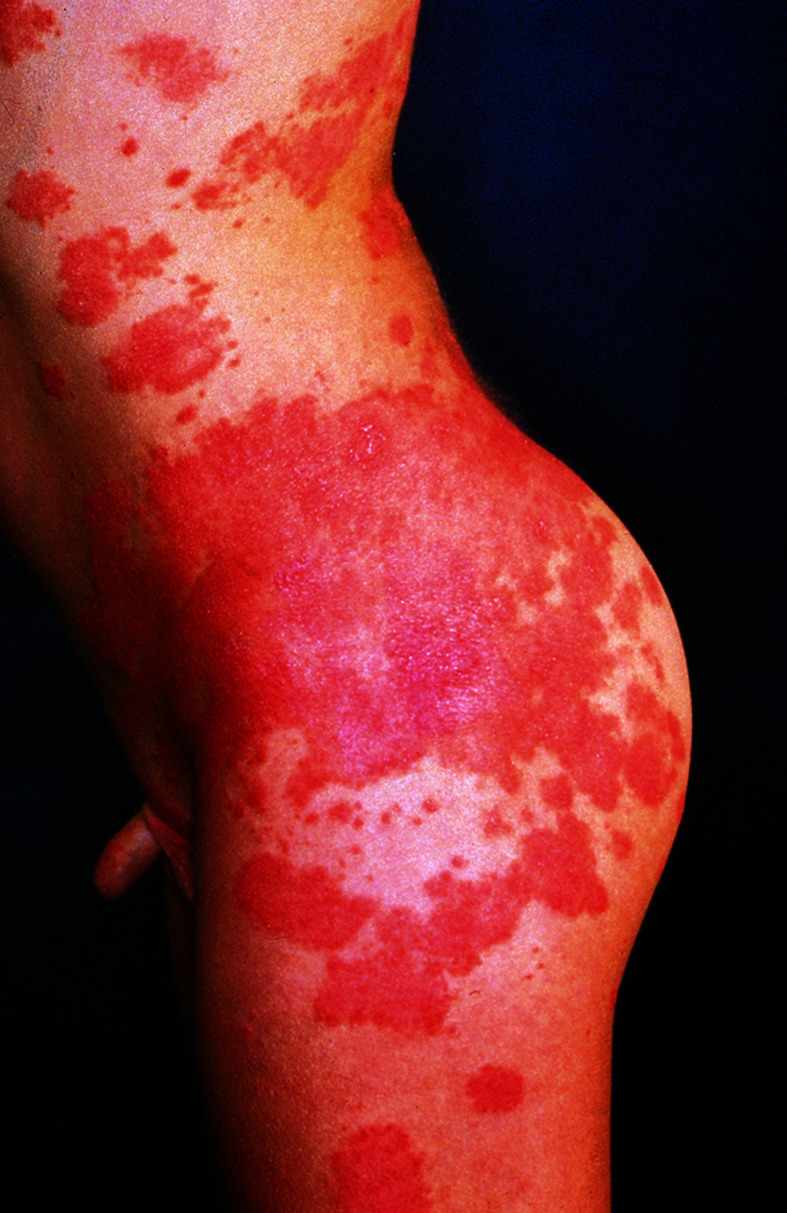
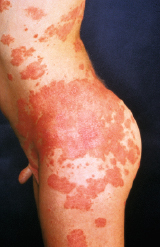
Prädilektionsstellen sind die Streckseiten der Extremitäten, v. a. im Bereich der Ellbogen und der Knie, die Lendengegend und der behaarte Kopf. Bei Neugeborenen sind psoriatische Hauterscheinungen ausgesprochen selten. Bei Auftreten im Kindesalter dominiert die Psoriasis guttata, eine Psoriasisform mit ovalären, kleineren Herden lokalisiert am Stamm und im Gesicht. Häufig manifestiert sich die Psoriasis bei Kindern im Anschluss an eine Streptokokkeninfektion. Wachsen die Herde weiter, so können größere, zusammenliegende Herde entstehen: Psoriasis nummularis und Psoriasis geographica.
Nagelveränderungen
Eine Nagelpsoriasis kommt bei etwa 50% der Patienten vor. Am häufigsten sind Tüpfelnägel zu beobachten. Dabei handelt es sich um stecknadelkopfgroße, grübchenförmige Einsenkungen in der Nagelplatte in Folge punktförmiger Psoriasisherde in der Nagelmatrix. Erkrankt das Nagelbett, so schimmern die Psoriasisherde gelblich durch den Nagel hindurch wie ein Ölfleck (psoriatischer Ölfleck). Durch die Schuppen unter dem Nagel kann die Nagelplatte abgehoben werden, es kommt zur distalen Onycholyse (Onycholysis psoriatica).
Sonderformen
Als Psoriasis inversa wird ein Vorkommen von Psoriasisherden in intertriginösen Räumen bezeichnet. Sind Handflächen und Fußsohlen von der Psoriasis betroffen, spricht man von einer Psoriasis palmaris et plantaris. Durch Zunahme von exsudativen Veränderungen können sich histologisch sichtbare Mikroabszesse in der Hornschicht bilden (Munro-Mikroabszesse). Bei stärkerer Ausprägung sind auch klinisch Pusteln sichtbar: Psoriasis pustulosa.
Therapie
Die festhaftenden Schuppen können durch lokale Anwendung von Salicylsäure (3–5%) gelöst werden. Auch Harnstoff und kochsalzhaltige Bäder haben sich dabei bewährt.
Die externe Therapie der Psoriasis erfolgt mit verschiedenen Externa: Dithranol (Anthralin, Cignolin), Calcipotriol und UV-B wirken der epidermalen Hyperproliferation entgegen und fördern die Differenzierung der Keratinozyten, während Glukokortikosteroide und UV-A-Licht primär antientzündlich wirken. Glukokortikosteroide können aber wegen der Gefahr der Entstehung einer Epidermisatrophie nur zeitlich begrenzt angewendet werden. Wegen der möglichen Einwirkung von Calcipotriol auf den Kalziumstoffwechsel ist die Anwendung bei Kindern nicht zugelassen. Für mittelschwere bis schwere Verläufe sind die TNF-α-Antagonisten Etanercept und Adalimumab sowie der Interleukin-12/23-Antikörper Ustekinumab zugelassen.
Cave
Bei einer großflächigen Anwendung von salicylsäurehaltigen Salben bei Kindern ist die resorptiv-toxische Wirkung zu beachten.
Lichen ruber planus
Epidemiologie
Diese Erkrankung, deren Ursache unbekannt ist, tritt meist zwischen dem 3.–6. Lebensjahrzehnt auf, aber auch Kinder können bereits erkranken.
Klinik
Klinisch imponiert eine rötlich-braune, polygonale, stecknadelkopf- bis reiskorngroße, derbe Papel, die im Gegenlicht weißlich schimmert. Auf der planen Oberfläche ist eine netzförmige weiße Zeichnung zu erkennen (Wickham-Phänomen), hervorgerufen durch eine Verdickung des Stratum granulosum.
Prädilektionsstellen der Erkrankung sind Beugeflächen der Handgelenke und Unterarme, seitliche Halsregion, Glutealgegend, Knöchelgegend der Fußgelenke und Penis. Die Mundschleimhaut ist bei etwa 50% der Patienten mit befallen (Lichen ruber mucosae). Da sich hier keine Papeln ausbilden können, ist das Wickham-Phänomen sehr deutlich mit netzartig verzweigten, weißlichen, nicht abstreifbaren Zeichnungen an den Wangenschleimhäuten zu erkennen.
Therapie
Im Vordergrund steht eine symptomatische, entzündungs- und juckreizhemmende Lokaltherapie. Nur in schweren Fällen kommen orale Steroide oder Retinoide zur Anwendung. Günstig hat sich auch eine Balneophotochemotherapie (Bade-PUVA) bewährt.
Infantile papulöse Akrodermatitis
Pathogenese
Diese relativ seltene, auch als Gianotti-Crosti-Syndrom bekannte Erkrankung des Kleinkindesalters gilt als kutane Manifestation verschiedener viraler Infekte, v. a. durch Hepatitis-Viren, Coxsackie-Viren und Epstein-Barr-Viren.
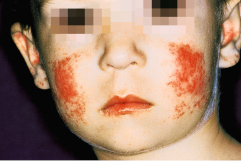
Klinik
An den Extremitäten, Wangen (Abb. 30.19), Handflächen, Fußsohlen und am Rumpf treten in symmetrischer Verteilung akut papulöse, teilweise papulovesikulöse Effloreszenzen mit hämorrhagischer Note auf. Die Schleimhäute bleiben frei, Allgemeinerscheinungen fehlen.

Therapie, Prognose
Die Behandlung erfolgt symptomatisch, die Abheilung nach bis zu 2 Monaten.
Akne
Epidemiologie
Die Akne beginnt bereits vor der Pubertät, erreicht ihr Maximum in der Jugend und bildet sich im frühen Erwachsenenalter langsam zurück.
Die Inzidenz der Akne beträgt nahezu 100%. Bei fast jedem Jugendlichen entstehen in der Adoleszenz Komedonen, Papeln und Pusteln, nur der Schweregrad ist unterschiedlich.
Pathogenese
Bei der Akne handelt es sich um eine entzündliche Talgdrüsenerkrankung auf seborrhoischer Grundlage. Die Ursache ist unbekannt. Neben genetischen Faktoren sind weitere Veränderungen für die Entstehung der Akne entscheidend. Durch vermehrte Bildung von Androgenen in den Gonaden und Nebennieren vergrößern sich die Talgdrüsen und bilden mehr Talg, welches für die Ausbildung der Symptome erforderlich ist. Im Talg, innerhalb der Follikelkanals, vermehren sich Propionibakterien (Propionibacterium acnes), deren Stoffwechselprodukte die Bildung von Komedonen und die entzündliche Umwandlung fördern.
Klinik
Prädilektionsstellen der Akne sind v. a. Gesicht, Brust und Rücken. Charakteristisch sind Komedonen, Papeln, Pusteln und knotige Läsionen. Durch eine Retentionshyperkeratose im Akroinfundibulum des Follikels bildet sich ein Hornpfropf, bestehend aus Hornzellen und Talg. Klinisch imponiert der so entstandene Komedo als kleines, weißliches, festes Knötchen mit kaum sichtbarem Ausführungsgang (geschlossener Komedo). Durch weitere Talg- und Hornproduktion vergrößert sich der Komedo, die Follikelöffnung wird ausgeweitet. Es entsteht ein dunkelpigmentierter, 2–3 mm großer Hornpfropf (offener Komedo). Eine nur aus Komedonen bestehende Akne wird als Acne comedonica bezeichnet.
Durch partiellen Untergang der Komedonen, Infektion mit Propionibakterien und Freisetzung entzündungsfördernd wirkender Lipide entstehen Papeln und Pusteln (Acne papulopustulosa). Die schwere Form der Akne (Acne conglobata) ist durch konfluierende, tief liegende Abszesse gekennzeichnet (Abb. 30.20).
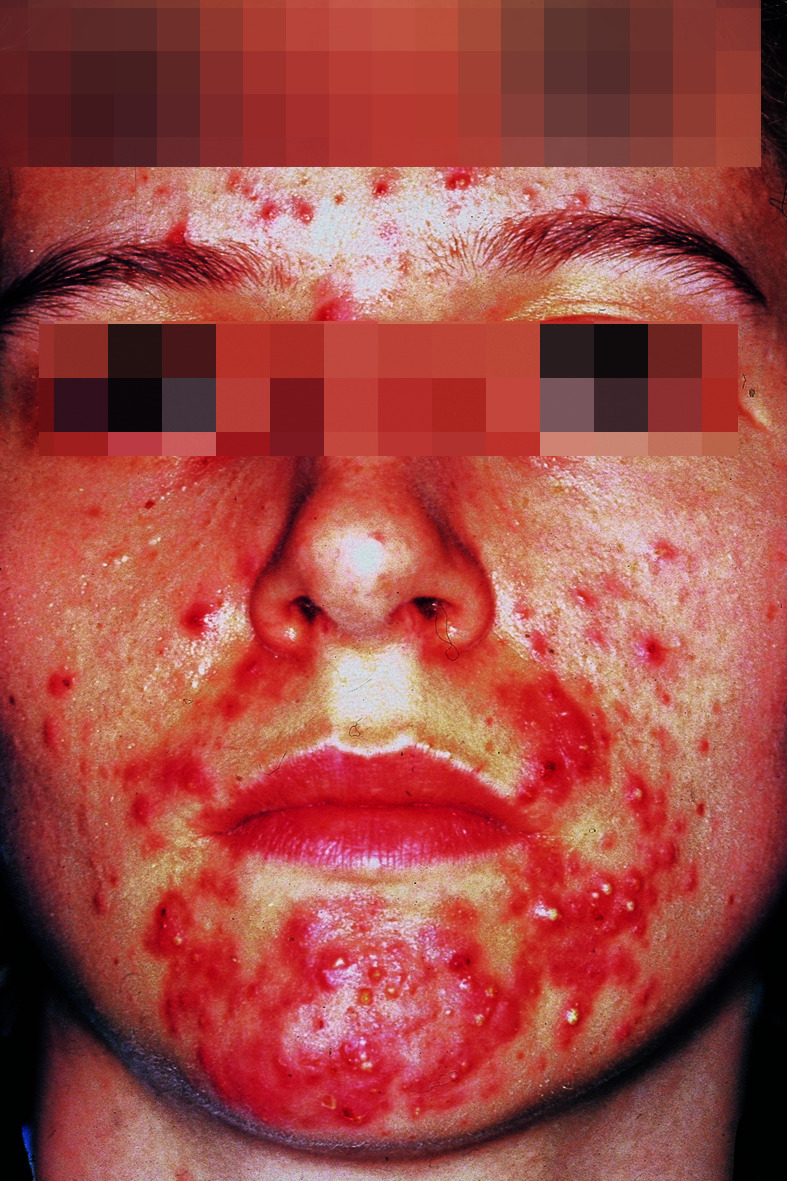
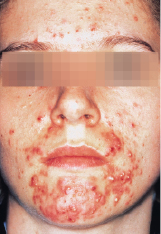
Bei manchen Kindern treten bereits nach der Geburt oder in den ersten Lebenswochen vereinzelte Akneeffloreszenzen auf (Acne neonatorum), die sich innerhalb weniger Monate spontan zurückbilden. Ursache scheinen erhöhte Androgenbildung der Nebennieren und erhöhte Androgenempfindlichkeit der Talgdrüsen zu sein.
Therapie
Bei einer leichten Form der Akne werden lokale Vitamin-A-Säure-Derivate eingesetzt. Bei stärkerer Ausprägung werden diese bevorzugt mit Benzoylperoxid oder auch mit Clindamycin in Fixkombinationen angewendet. Ebenfalls bewährt hat sich die Kombination aus Benzoylperoxid und Clindamycin. Monotherapien mit topischen Antibiotika sollten vermieden werden.
Systemisch haben sich Doxycyclin 100 mg/Tag oder Minocyclin 50 mg/Tag bewährt.
In schweren Formen, v. a. bei der Acne conglobata, kann Isotretinoin die Talgproduktion bis zu 90% senken und zu einer anhaltenden Verkleinerung der Talgdrüsen führen.
Cave
Die Teratogenität von Isotretinoin ist dabei zu beachten und erfordert eine sichere Antikonzeption bei jungen Frauen.
Benigne Tumoren und Nävi
Nävi sind umschriebene Fehlbildungen oder Hamartome der Haut, die durch besonderes Aussehen auffallen. Sie stammen von unterschiedlichen Hautbestandteilen (Epithel, Talg- und Schweißdrüsen, Haare und Gefäße) ab, die sich lokal atypisch entwickelt haben, können bereits bei Geburt vorhanden sein oder entwickeln sich im Laufe des Lebens. Man unterscheidet Pigmentzellnävi, bestehend aus Melanozyten und organoide Nävi.
Pigmentzellnävi
Pigmentzellnävi bestehen aus epidermalen Melanozyten, die Melanin bilden und an die Keratinozyten abgeben. Sie entstammen der Neuralleiste und wandern während der Embryonalzeit in die Epidermis ein, können aber auch unterhalb liegen bleiben.
Café-au-lait-Fleck
Es finden sich auf der Haut kleine bis handtellergroße, milchkaffeefarbene, runde, meist scharf begrenzte Makulae. Sie sind harmlos.
Bei Vorliegen von mehr als 5 größeren Café-au-lait-Flecken ist an eine Neurofibromatose zu denken.
Melanosis naeviformis
Synomym: Becker-Nävus, Naevus pigmentosus et pilosus). Es handelt sich um einen gleichmäßig hell- bis dunkelbraunen Herd von bizarrer Form, in dem gröbere und kosmetisch störende Haare wachsen.
Naevus spilus
Meist seit Geburt findet sich eine hellbraune Makula, in die in späteren Jahren Pigmentflecke einstreuen.
Mongolenfleck
Bei nahezu allen Mongolen sind über dem Kreuzbein graublaue Verfärbungen der Haut zu sehen, die sich bis zur Pubertät wieder zurückbilden. Auch bei weißrassigen Menschen kommt diese Hautveränderung vor.
Naevus coeruleus
Aufgrund in der Dermis liegen gebliebener Melanozyten erscheint dieser kleine Knoten blauschwarz. Die Konsistenz ist meist derb.
Nävuszellnävi
Nävuszellen stammen ebenfalls aus der Neuralleiste und entsprechen den Melanozyten. Sie können ebenfalls Melanin produzieren und liegen meist in Nestern unterhalb der Epidermis.
Die klinische Erscheinungsform ist sehr variabel. Sie können punktförmig, aber auch flächenhaft sein, makulös bis papulös und nehmen alle Schattierungen von braun bis schwarz an. Je nach Lage unterscheidet man Junktionsnävi, epidermodermale und dermale Nävi.
Wegen der Möglichkeit der malignen Entartung sollten Nävi regelmäßig kontrolliert werden.
Die in Tab. 30.4 aufgeführte ABCDE-Regel kann bei der Beurteilung der Dignität helfen. Treffen mehrere Merkmale zu, so sollte eine prophylaktische Exzision erfolgen.
| A | Asymmetrie (normal sind Nävi symmetrisch) |
| B | Begrenzung (normal ist eine scharfe Begrenzung) |
| C | Color (sollte einheitlich sein) |
| D | Durchmesser (<0,5 cm ist normal) |
| E | Erhabenheit (bei Änderung dieser ist Vorsicht geboten) |
Organoide Nävi
Organoide Nävi sind seit Geburt vorhanden und bestehen aus Zellen normaler Hautstruktur.
Epidermale Nävi
Hierbei handelt es sich um meist streifig angeordnete, hautfarbene bis graue, weiche, scharf begrenzte, papillomatöse Hautveränderungen durch eine Akanthose der Epidermis, begleitet gelegentlich von Hyperkeratose und entzündlicher Reaktion.
Naevus sebaceus
Ausgehend von Talgdrüsen finden sich meist am Kapillitium flache, scharf begrenzte, rötlich-gelbliche, papillomatöse oder beetartige Gebilde (Abb. 30.21).
Wegen der Möglichkeit der Entstehung von Tumoren aus Talgdrüsennävi, am häufigsten Trichoblastome und Basaliome, sollte die Exzision eines Naevus sebaceus angestrebt werden.
Gefäßnävi und Hämangiome
Naevus flammeus
Klinik
Man unterscheidet den medianen Naevus flammeus (Storchenbiss) vom selteneren lateralen Naevus flammeus. Histologisch liegt eine Erweiterung und Vermehrung der Kapillaren vor. Mediane Naevi flammei sind blassrosa, im Niveau der Haut liegende Makulae verschiedener Größe, die bei ca. 50% aller Neugeborenen gesehen werden. Sie sind am häufigsten im Bereich der Glabella, der oberen Augenlider und im Nacken (Storchenbiss) lokalisiert. Im Gesichtsbereich verschwinden sie spontan in den ersten Monaten, während die nuchalen persistieren können. Laterale Naevi flammei sind scharf begrenzt, rosa bis dunkelrot, von unterschiedlicher Größe (bloß eine Körperhälfte befallend) und asymmetrisch angeordnet. Es gibt keine Rückbildungstendenz.
Gelegentlich ist ein Naevus flammeus nur ein Symptom einer Systemerkrankung. Beim Sturge-Weber-Syndrom finden sich neben einem Naevus flammeus im Trigeminusbereich Gefäßfehlbildungen im Gehirn mit Glaukom und zentralnervösen Störungen. Das Klippel-Trénaunay-Weber-Syndrom zeichnet sich durch einen Naevus flammeus der gesamten Extremität mit varikösen Venektasien und partiellem Riesenwuchs aus. Beim Kasabach-Merritt-Syndrom kann es durch Sequestration und vermehrten Verbrauch von Thrombozyten im Hämangiom, aggraviert durch eine Verbrauchskoagulopathie, zu einer Thrombozytopenie kommen.
Therapie
Flache Naevi flammei lassen sich mit einem Farbstofflaser in mehreren Sitzungen aufhellen. Möglich ist auch das Abdecken mit Spezialkosmetika.
Infantile Hämangiome
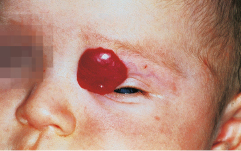
Diese Läsionen treten in den ersten Lebenswochen an jeder Stelle der Haut oder Schleimhaut auf. Sie sind hell- bis kräftig rot, können im Hautnieveau liegen, erhaben sein oder auch subkutan liegen (Abb. 30.22). Meist treten sie einzeln und lokalisiert auf, können aber auch multipel auftreten. Nach einer Phase der weiteren Vergrößerung im ersten Lebensjahr bilden sie sich während der nächsten 2–6 Jahre spontan zurück. Der Beginn der Rückbildung wird durch Auftreten von zentralen weißen Bezirken angezeigt. Neben den lokalisierten Hämangiomen gibt es auch segmental auftretende Hämangiome, bei denen an assoziierte Fehlbildungen zu denken ist und in seltenen Fällen auch multiple als Hämangiomatose mit möglichem extrakutanem Organbefall.

Therapie
Bei unkomplizierte Verläufen und Hämangiomen am Stamm oder Extremitäten ist eine Therapie nicht zwingend erforderlich, kann aber frühzeitig angeboten werden, da die Entwicklung dieser in den ersten Lebensmonaten nicht abgeschätzt werden kann und die Therapie von den Säuglingen gut vertragen wird. Für flache Hämangiome eignet sich eine Farbstofflasertherapie oder eine Kryokontakttherapie. Auch mittels Propranolol in einer Gelgrundlage unter Okklusion lassen sich flache Hämangiome am Körperstamm gut behandeln. Selten sind wegen ungewöhnlicher Lokalisation und Wachstumstendenz mit Beeinträchtigung lebenswichtiger Organe aktivere Maßnahmen notwendig. Hier hat sich eine systemische Therapie mit Propranolol in niedriger Dosierung bewährt.
Störungen der Melaninpigmentierung
Zwischen den Basalzellen liegen die in der Neuralleiste gebildeten Melanozyten, die mit Hilfe des Enzyms Tyrosinase Melanin bilden und es an die Keratinozyten abgeben.
Es gibt keine geschlechts- oder rassespezifischen Unterschiede in der Melanozytenzahl. Lediglich die genetisch determinierte, individuelle Aktivität der Melanozyten bestimmt die Bräunung der Haut.
Störungen der Melaninpigmentierung können durch Veränderung der Melanozytenzahl, Funktionsstörung der Melaninsynthese und Störung des Melanosomentransfers bedingt sein.
Epheliden
Pathogenese
Sommersprossen werden autosomal-dominant vererbt und kommen v. a. bei Menschen mit rötlich-blondem Haar vor. Ursache der stärkeren Pigmentierung im Sommer ist die Fähigkeit einiger Melanozyten, rascher und mehr Melanin zu bilden als die Melanozyten in der Umgebung.
Klinik, Therapie
Symmetrisch im Bereich der Wangen und der Nase finden sich hellbräunliche Pigmentflecke. Auch an Ober- und Unterarmen kommen sie vor. Eine Therapie dieser Pigmentveränderung ist nicht möglich. Weder Lichtschutzpräparate noch verschiedene Bleichcremes zeigen Erfolge.
Incontinentia pigmenti (Bloch-Sulzberger-Syndrom)
Pathogenese
Diese Erkrankung ist x-chromosomal-dominant vererbt und kommt fast nur bei Mädchen vor. Bei männlichen Merkmalsträgern verläuft sie meist letal.
Klinik
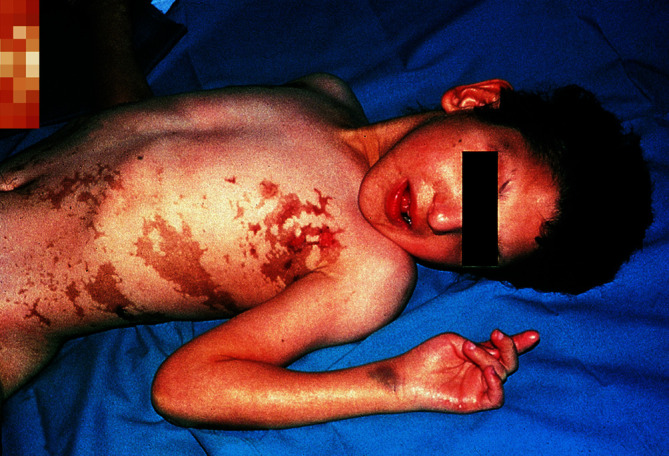
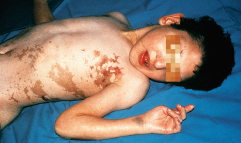
Die Erkrankung geht mit Hautsymptomen, Fehlbildungen an Augen, am Zentralnerven- und Skelettsystem einher. Bereits beim Neugeborenen finden sich entlang der Blaschko-Linien entzündliche Erytheme mit Bläschen und Blasen. Nach kurzer Zeit entstehen lichenoide Papeln sowie keratotische Hautveränderungen. Nach Abheilung treten bizarr geformte oder lineäre gräuliche Pigmentierungen entlang der Blaschko-Linien auf (Abb. 30.23). Sind diese bereits beim Neugeborenen zu sehen, so ist das ein Zeichen dafür, dass die Erkrankung bereits in utero begonnen hat.
Komplikationen
Zahnentwicklungsstörungen, Augenanomalien mit Optikusatrophie, Strabismus, Katarakt, ZNS-Entwicklungsstörungen, Skelettmissbildungen und Herzfehler treten bei einem Großteil der Betroffenen auf.
Therapie
Lediglich eine externe Therapie im Entzündungsstadium zur Vermeidung von Sekundärinfektionen, ggf. innerliche Anwendung von Glukokortikoiden, ist möglich. Eine genetische Beratung sollte angestrebt werden.
Vitiligo
Pathogenese, Klinik
Diese häufige Erkrankung die bereits im Kindesalter vorkommen kann, führt als Folge des Untergangs von Melanozyten zur Depigmentierung der Haut. Meist tritt die scharf begrenzte Depigmentierung zwischen dem 10. und 30. Lebensjahr auf, häufig symmetrisch um Körperöffnungen und an Körperstellen stärkerer Pigmentierung. Im Laufe des Lebens nehmen die Herde an Größe und Zahl zu.
Komplikationen
An assoziierten Erkrankungen finden sich gleichzeitig Autoimmunthyreoditiden, perniziöse Anämie, Diabetes mellitus, Alopecia areata und eine atopische Diathese mit erhöhten Serumkonzentrationen von IgE.
Therapie
Eine Heilung der Vitiligo ist nicht möglich. Verschiedene Ansätze (topische Glukokortikoide und Calcineurininhibitoren, Bestrahlung mit UV-B im Schmalspektrumsbreich von 311 nm bei generalisierter Vitiligo ab dem 16. Lebensjahr; bei segmentaler umschriebener Vitiligo auch mittels einer monochromatischen UVB-Lampe mit 308-nm-Wellenlänge) können zu einer teilweisen Repigmentierung der Hautareale führen.
Wichtig ist ein konsequenter Lichtschutz der der Sonne ungeschützt ausgesetzten Areale.
Sonstige Hauterkrankungen
Pityriasis rosea
Klinik
Die Pityriasis rosea ist eine häufige, mild verlaufende Hauterkrankung, die bei Schulkindern und Adoleszenten vorkommt. Sie beginnt mit einer 2–6 cm großen, ovalen Einzelläsion, die zentral abgeblasst ist und am leicht erhabenen Rand eine „Schuppenkrause“ aufweist. Nach 5–10 Tagen entwickelt sich ein generalisiertes, symmetrisches Exanthem mit in Richtung der Hautspaltlinien angeordneten, runden bis ovalen, weniger als 1 cm großen, ganz leicht erhabenen, blassrosa bis bräunlichen Effloreszenzen, die mit einer feinen, kleieförmigen Schuppung bedeckt sind. Selten besteht leichter Juckreiz.
Therapie
Nach 2–4 Wochen kommt es zur Spontanheilung, sodass sich eine Therapie häufig erübrigt. Eventuell äußere Therapie mit Cremes oder Salben, die Glukokortikosteroide niedriger Wirkstärke (z. B. Hydrocortison) enthalten.
Alopecia areata
Diese Krankheit tritt v. a. bei Kindern und Jugendlichen, 20% familiär und häufig gleichzeitig mit einer atopischen Diathese auf.
Pathogenese
Die Ursache der entzündlichen, herdförmigen, reversiblen Alopezie ist unklar. Eine Autoimmunreaktion vom Spättyp wird diskutiert, wobei exogene und endogene Faktoren wie psychischer Stress, Trisomie 21, Schilddrüsenerkrankungen und Atopie eine Rolle spielen. Haarfollikelkeratinozyten bilden Zytokine, die T-Lymphozyten und Makrophagen anlocken, die ihrerseits wieder Zytokine freisetzen und zu einem entzündlichen Infiltrat im Bereich des Haarbulbus und dermaler Haarpapille führen. Dies endet in Haarausfall, Matrixdystrophie und -degeneration.
Klinik
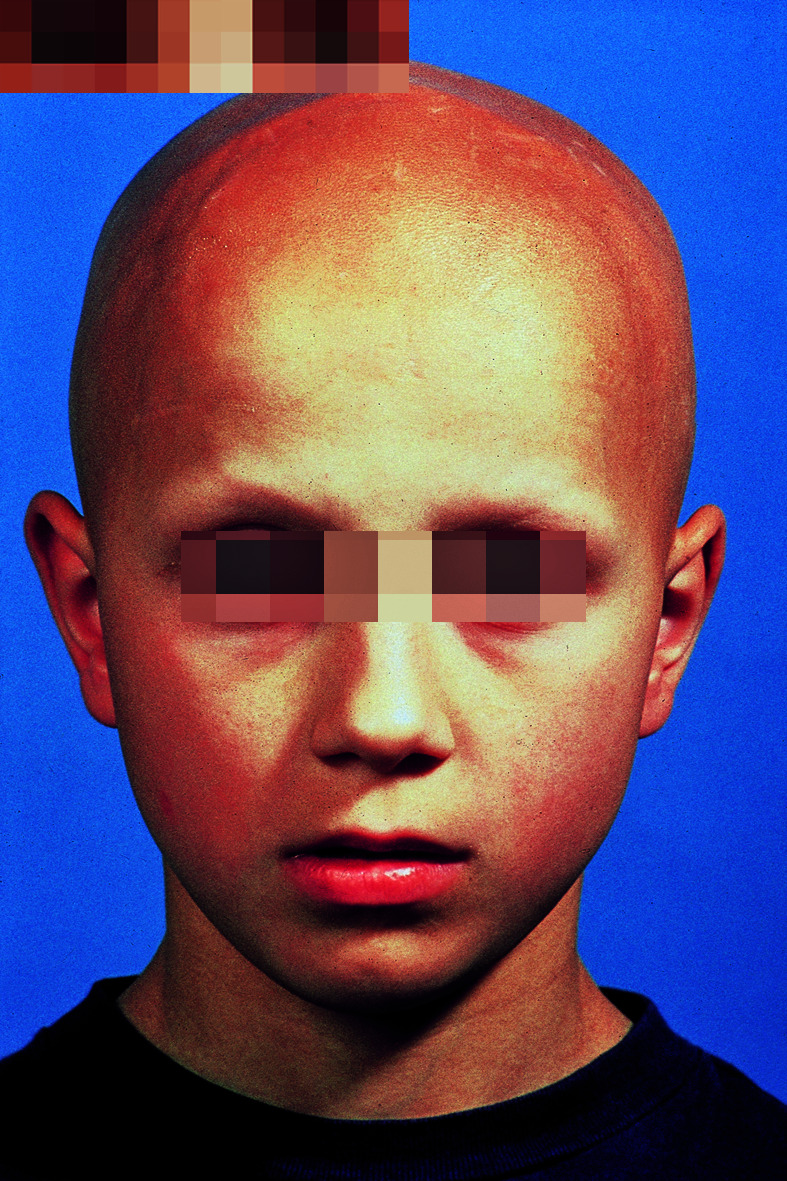
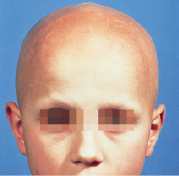
Meist im Temporalbereich oder okzipital innerhalb kurzer Zeit ein oder mehrere scharf begrenzte, runde oder ovale haarlose Stellen ohne Atrophie der Haut. Follikelmündungen immer vorhanden. Epilierte Haare im Randbereich sind zugespitzt und dystroph. Hier auch 0,2–0,7 cm lange, wenig pigmentierte Haare (Peladehaare, Kommahaare) und schwarze komedoartige Follikelverschlüsse (Kadaverhaare, Ausrufungszeichenhaare). Auch Augenbrauen, Wimpern und sogar die übrigen Körperhaare können vom Ausfall betroffen sein. Nagelveränderungen bei 20%.
Sonderformen sind:
Ophiasis (Randgebiet des Kapillitiums betroffen),
Alopecia areata totalis (gesamte Kopfhaare) und
Alopecia areata universalis (sämtliche Körperhaare; Abb. 30.24).
Diagnose, Therapie
Die Diagnose erfolgt über Anamnese und Klinik. Für die Behandlung sind Mittel der Wahl Glukokortikosteroide lokal, okklusiv und evtl. intrakutan bei einzelnen Lokalisationen. Bei Nichtansprechen, v. a. bei Alopecia areata totalis, ist eine topische Immuntherapie mit DCP (Diphencyprone) möglich. Interne Glukokortikosteroide kommen selten zum Einsatz.
Prognose
Gut. Abheilung bei 50% innerhalb eines Jahres, allerdings häufig Rezidive; 1/3 rezidivfreie Abheilung. Ungünstig bei Ophiasis, Alopecia areata totalis und universalis.
Mastozytosen
Pathogenese
Gemeinsames Merkmal der Mastozytosen ist eine Vermehrung von Mastzellen in der Haut oder in anderen Geweben ohne andere zugrunde liegende Erkrankung oder Entzündung. Die Erkrankung kann bei 15% der Betroffenen bereits bei Geburt vorhanden sein. Bei weiteren 30% manifestiert sie sich innerhalb der ersten 6 Lebensmonate und bis zum 2. bzw. 15. Lebensjahr bei jeweils weiteren 10%. Die bei Erwachsenen mit Mastozytose häufig gefundenen c-kit-Mutationen kommen im Kindesalter nur bei schwersten Verlaufsformen vor. Die Klinik der Mastozytosen ist sehr variabel, wobei bei Kindern 2 Hauptformen unterschieden werden können: die noduläre oder Plaqueform, die als Mastozytom häufig isoliert auftritt, und die diffuse makulo-papulöse Form, die auch als Urticaria pigmentosa bezeichnet wird, obwohl sie nicht zum Formenkreis der Urtikaria zählt.
Durch verschiedene Reize kommt es zur Freisetzung präformierter Mediatoren, hauptsächlich Histamin:
physikalische Faktoren: Hitze, Kälte, Druck, Wasser u. a.,
Arzneimittel: Azetylsalizylsäure, nichtsteroidale Antiphlogistika, Kodein, Morphin, Muskelrelaxanzien, Röntgenkontrastmittel u. a.,
biologische Auslöser: Bienen- und Wespengift, Bakterientoxine, Alkohol.
Klinik
Mastozytose
Die meisten Patienten sind jünger als 2 Jahre. Typischerweise finden sich ein oder mehrere rötliche bis rotbraune Plaques oder Knoten von 1–3 cm Durchmesser, die auf mechanischen Reiz (z. B. Reiben mit Holzspatel) urtikariell anschwellen und jucken (Darier-Zeichen). Histologisch zeigt sich ein umschriebenes dichtes Infiltrat von Mastzellen in der Dermis.
Urticaria pigmentosa
An den Prädilektionsstellen (Stamm, Oberarme und Oberschenkel) finden sich zahlreiche bis Hunderte hellbraun hyperpigmentierte, etwa stecknadelkopfgroße Maculae, teilweise auch Papeln. Nur bei Kindern bis zum 2. Lebensjahr können Blasen vorhanden sein (bullöse Urticaria pigmentosa). Bei tief dermal liegenden Läsionen wirken die klinischen Erscheinungen gelblich (peau d‹orange) und erinnen an Xanthelasmen. Durch mechanische Reize wie Reiben und Kratzen werden bei nahezu allen Patienten Urticae provoziert (Darier-Zeichen). Juckreiz ist bei 50% aller Erwachsenen vorhanden, aber nur bei 10% der Kinder.
Neben der Haut können auch innere Organe durch Mastzellen infiltriert sein. Eine Beteiligung des Gastrointestinaltrakts führt zu Diarrhö und abdominellen Schmerzen. Ebenso können Leber, Milz, Lymphknoten, Skelettsystem und Knochenmark Proliferationen von Mastzellen enthalten.
Therapie
Als symptomatische Therapie kommen H1-Rezeptor-Antagonisten systemisch zum Einsatz. Die zusätzliche Gabe von H2-Rezeptor-Antagonisten kann in einigen Fällen den Effekt verstärken. Bei nicht ausreichender Wirkung kann zusätzlich Ketotifen gegeben werden. Ein Notfallausweis sollte mitgeführt werden.
Cave
Wegen der Gefahr der generalisierten Mastzelldegeneration mit Anaphylaxie sollten thermische Reize, wie z. B. ein Sprung ins kalte Wasser oder heißes Duschen oder Baden gemieden werden.
Prognose
Im Gegensatz zur Manifestation im Erwachsenenalter ist die Prognose im Kindesalter gut. Die Erkrankung nimmt mit den Jahren an Intensität ab und heilt meist während der Pubertät ab. Eine invasive Diagnostik ist in der Regel nicht notwendig.
Borreliose
Epidemiologie
Diese durch Zecken (Ixodes ricinus) beim Saugakt übertragene Infektion durch gramnegative Spirochäten kommt weltweit, v. a. in gemäßigten Klimazonen vor. Erreger ist Borrelia burgdorferi. 4–60% der Zecken sind infiziert.
Klinik
Die Erkrankung wird in 3 Stadien eingeteilt:
In der Frühphase (StadiumI) ruft sie nach einer Inkubationszeit von ungefähr 10 Tagen häufig ein Erythema migrans hervor, das bei Kindern häufig im Kopf- und Nackenbereich auftritt und leicht übersehen werden kann. An der Bissstelle der Zecke bildet sich ein sich peripher ausbreitendes, randbetontes Erythem. Zentrale Abblassung, an Bissstelle kleine Papel. Bei 2/3 grippeähnliche Symptome, flüchtige Gelenkschmerzen und Myalgien.
Im StadiumII disseminierte Aussaat der Erreger mit multipler Organbeteiligung: u. a. Polyarthritiden, Nervenbeteiligung (bei Kindern relativ häufig mit Fazialisparese), AV-Block, Myokarditis und Augenbeteiligung. In allen Stadien, v. a. im Stadium II bei Kindern, Jugendlichen und Frauen lymphoproliferative knotige Reaktion überwiegend an Ohren, Brustwarzen, Achselhöhlen und Skrotum möglich (Lymphadenosis cutis benigna). Klinisch weicher, dunkelroter bis livider Knoten, bei Diaskopie gelblichbraunes Infiltrat.
Das chronische InfektionsstadiumIII spielt bei Kindern keine Rolle.
Diagnose
Die Serologie (ELISA, Westernblot) führt zur richtigen Diagnose. Zu beachten ist, dass beim Erythema migrans nur bei ca. 50% der Erkrankten Antikörper nachweisbar sind.
Therapie
Im Stadium I Amoxicillin (20–40 mg/kgKG/Tag), Doxycyclin 200 mg/Tag (nicht bei Kindern <8 Jahre!) oder alternativ Erythromycin 30 mg/kgKG/Tag über 14 Tage. In späteren Stadien Therapie parenteral über 21 Tage wegen der langen Generationszeit der Erreger (Kap. 15).
Contributor Information
M. Meurer, Email: michael.meurer@uniklinikum-dresden.de
R. Aschoff, Email: roland.aschoff@uniklinikum-dresden.de
M. Gahr, Email: manfred.gahr@uniklinikum-dresden.de