

Abstract
Die Weltgesundheitsorganisation und europäische Union haben die Prävention und Behandlung genetischer und seltener Krankheiten („orphan diseases“) als zentrale Herausforderung für die Gesundheitsversorgung des 21. Jahrhunderts in den Mittelpunkt ihrer Aufmerksamkeit gestellt. Innerhalb dieser Gruppe sind die mehr als 700 angeborenen Stoffwechselerkrankungen von besonderer Relevanz. Sie sind relativ häufig (ca. 1% der Bevölkerung), viele ursächlich behandelbar, oder es sind in absehbarer Zukunft wesentliche therapeutische Fortschritte realistisch. Dabei handelt es sich v. a. um neue medikamentöse Ansätze, Enzymersatztherapien und Organtransplantationen. Am Anfang stehen z. T. mutationsspezifische molekulare Therapien. Das Neugeborenenscreening und die damit mögliche Frühbehandlung gut behandelbarer Erkrankungen sind entscheidende Maßnahmen der Sekundärprävention.
Neugeborenenscreening
Bei vielen genetisch bedingten Stoffwechselkrankheiten oder hormonellen Erkrankungen bestehen bei möglichst kurz nach der Geburt eingeleiteter Therapie sehr gute Aussichten, schwere Krankheitsmanifestationen zu verhindern. In den 1960er Jahren wurde durch die Initiative des Pädiaters Horst Bickel in der Bundesrepublik Deutschland und des klinischen Genetikers Alwin Knapp in der ehemaligen DDR das Neugeborenenscreening auf angeborene Stoffwechselstörungen und Endokrinopathien für alle Neugeborenen eingeführt. Es ist inzwischen die erfolgreichste Sekundärprävention gesundheitlicher Beeinträchtigungen überhaupt.
Ziel des Neugeborenenscreenings ist die frühzeitige und vollständige Diagnosestellung wichtiger behandelbarer Erkrankungen bei präsymptomatischen Neugeborenen.
Inzwischen wurden in Deutschland ca. 38 Mio. Neugeborene untersucht und mehr als 15.000 Patienten rechtzeitig diagnostiziert und erfolgreich behandelt.
Am Beginn eines derartigen Programms muss die Frage stehen, nach welchen Erkrankungen bei allen Neugeborenen gefahndet werden soll. Für diese Festlegung hat die WHO 1968 Kriterien definiert:
ausreichende Schwere und Häufigkeit der Erkrankung in der untersuchten Population,
symptomfreies Intervall nach der Geburt, in dem die Diagnose anhand klinischer Symptome nicht möglich ist,
nachgewiesener Nutzen einer präsymptomatisch eingeleiteten Therapie für das betroffene Kind,
einfache, an großen Probenzahlen (möglichst Trockenblutproben) mit geringen Kosten durchführbare Nachweismethode mit hoher Sensitivität und Spezifität.
Der Umfang sowie die Durchführung des Neugeborenenscreenings wurden 2005 durch den „Gemeinsamen Bundesausschuss der Ärzte und Krankenkassen“ in den Kinderrichtlinien festgelegt und im März 2011 aktualisiert. Für alle Neugeborenen wurde die Untersuchung auf 12 Stoffwechselerkrankungen sowie 2 Endokrinopathien empfohlen (Tab. 3.1).
| Zielkrankheit | Prävalenz |
|---|---|
|
Hyperphenylalaninämien - Klassische Phenylketonurie - Hyperphenylalaninämie |
1: 5300 1:10.300 1:11.600 |
| Mittelkettiger-Acyl-CoA-Dehydrogenase (MCAD)-Mangel | 1:10.200 |
| Weitere Tandem-MS-Erkrankungen (Ahornsiruperkrankung, Glutarazidurie Typ I, Isovalerianazidurie und sonstige Fettsäurenoxidationsdefekte) | 1:28.800 |
| Klassische Galaktosämie | 1:69.500 |
| Biotinidasemangel | 1:22.900 |
| Kongenitale Hypothyreose | 1:3500 |
| Adrenogenitales Syndrom, 21-Hydroxylasemangel | 1:13.700 |
| Kumulativ | Ca. 1:1340 |
Prävalenzen nach der Statistik der Deutschen Gesellschaft für Neugeborenenscreening 2004–2012 (n=6.112.987 Neugeborene, DGNS http://www.screening-dgns.de/screeningregister-2i.htm)
Eine wesentliche Erweiterung erfuhr das Neugeborenenscreening durch Einführung der ESI-Tandemmassenspektrometrie (kurz Tandem-MS). Erkannt werden damit Störungen
des Aminosäurenstoffwechsels (Aminoazidopathien),
des Abbaus organischer Säuren (Organoazidopathien),
der Fettsäurenoxidation (Fettsäurenoxidations- und Carnitinstoffwechseldefekte).
Im Jahr 2016 wurde die Mukoviszidose (zystische Fibrose, CF) als 15. Zielkrankheit eingeführt. Mit dem Screening und Diagnose von CF-Patienten kurz nach der Geburt können effektive Therapiemaßnahmen chronische Veränderungen hinauszögern oder ganz verhindern.
Praxis des Neugeborenenscreenings
Von jedem Neugeborenen soll im Alter von 36 bis 72 Lebensstunden aus der Ferse Blut auf eine Spezialfilterpapier (Guthrie-Karte) aufgetropft werden (Abb. 3.1). Diese Blutprobe wird getrocknet und täglich zur Untersuchung mit der Post an das Screeninglabor versandt.


Die Screeninglaboratorien sind verantwortlich für die sofortige Bearbeitung der Proben und die zeitgerechte Weitergabe des Befunds an die Einsender. Der Einsender ist verantwortlich für die sachgerechte Durchführung der Probenentnahme, den Probenversand, die vollständige Dokumentation einschließlich des Befundrücklaufs und die Einleitung der erforderlichen Maßnahmen bei pathologischem Screeningergebnis (Information der Eltern, Organisation von Wiederholungsuntersuchungen und/oder Veranlassung einer Behandlung).
Spezialdiagnostik angeborener Stoffwechselerkrankungen
Entscheidend für eine erfolgreiche Diagnostik von Stoffwechselerkrankungen sind die vom behandelnden Arzt zu treffende Auswahl der Patienten und eine gute Kommunikation mit dem Stoffwechselspeziallaboratorium, um die richtige Kombination biochemischer und genetischer Untersuchungen zu veranlassen.
Indikation zur weiterführenden Diagnostik
In Zeiten begrenzter finanzieller Ressourcen und Kapazitäten sowie ständig steigender Vielfalt und Komplexität diagnostischer Verfahren sollten mehr denn je nur Patienten untersucht werden, bei denen die Synopsis (familien)anamnestischer und klinischer Befunde, ergänzt um eine laborchemische und radiologische Basisdiagnostik eine angeborene Stoffwechselerkrankung möglich erscheinen lassen. So sollte bei isoliert vorhandener unspezifischer Symptomatik, wie Entwicklungsverzögerung, Epilepsie oder gehäuften Infekten, auf komplexe und teure Spezialuntersuchungen verzichtet werden.
Ungewöhnlich schwere Krankheitsverläufe, z. B. bei interkurrente Infekten, v. a. zusätzliche neurologische Symptome wie Wesensveränderung, Ataxie, extrapyramidale Bewegungsstörungen, Krampfanfälle oder Koma, müssen Anlass für eine Basisdiagnostik auf angeborene Stoffwechselstörungen sein (Ammoniak, Blutgasanalyse, Blutzucker, Laktat und Ketonkörper im Urin). Einen besonderen Hinweis verdient die einfache Stixbestimmung der Ketone im Urin. Insbesondere bei Neugeborenen ist eine Ketonurie ein entscheidender Hinweis auf eine Stoffwechselerkrankung. Es kann nicht genug betont werden, dass die Bestimmung aller Laborparameter bei der Erstversorgung erfolgen soll. Für Spezialuntersuchungen sollten gleichzeitig weitere Urin- und Serumproben asserviert werden.
Hyperammonämie und Harnstoffzyklusdefekte
Definition und Einteilung
Harnstoffzyklusdefekte sind genetisch bedingte Stoffwechselstörungen der Stickstoffentgiftung, die mit dem Leitsymptom Hyperammonämie einhergehen. Insgesamt sind 6 Enzymdefekte des Harnstoffzyklus bekannt (Abb. 3.2):
N-Azetylglutamatsynthetase (NAGS)-Mangel,
Carbamylphosphatsynthetase (CPS)-Mangel,
Ornithintranskarbamylase (OTC)-Mangel,
Argininosuccinatsynthetasemangel (Zitrullinämie),
Argininosuccinatlyasemangel (Argininbernsteinsäurekrankheit),
Arginasemangel (Hyperargininämie).


Der Harnstoffzyklus wird ferner durch genetische Defekte des Membrantransports von Aminosäuren gestört oder der mitochondrialen Carboanhydrase VA verursacht. Es resultieren wiederum Hyperammonämien. Folgende autosomal-rezessive vererbte Membrandefekte sind bekannt:
Hyperammonämie-Hyperornithinämie-Homozitrullinurie (HHH)-Syndrom,
lysinurische Proteinintoleranz (LPI),
Citrinmangel.
Epidemiologie
Harnstoffzyklusdefekte sind seltene Erkrankungen mit einer kumulativen Häufigkeit von ca. 1:40.000. Der OTC-Mangel ist mit ca. 1:56.000 Kindern der häufigste Defekt.
Klinik
Die initialen Symptome Nahrungsverweigerung, Lethargie, Apathie, Atmungsstörungen und zerebrale Krampfanfälle werden oft schon im Neugeborenenalter manifest und lassen im Einzelfall zunächst an häufigere Erkrankungsursachen, wie Infektionen, Herzfehler oder intrakranielle Blutungen infolge von Geburtsverletzungen oder Vitamin K-Mangel denken. Es entwickelt sich das klinische Bild einer systemischen Intoxikation mit schweren Enzephalopathie bis zum Koma oder Multiorganversagen. Gerinnungsstörungen können zu zerebralen Blutungen erheblichen Ausmaßes führen.
Ältere Kinder und Erwachsene zeigen eine fluktuierende und häufig progrediente neurologische oder psychiatrische Symptomatik (Zephalgien, Epilepsie, Ataxie, Verwirrtheitszustände, mentaler Abbau).
Cave
Aufgrund der hohen Neurotoxizität des Ammoniaks ziehen schon 2(–3) Tage eines hyperammonämischen Komas schwere irreversible Gehirnschäden nach sich!
Erstmanifestationen bis zum tödlichen Koma infolge von Harnstoffzyklusdefekten können bis ins Erwachsenenalter auftreten. Die häufigste Störung des Harnstoffzyklus, der OTC-Mangel, wird nicht autosomal-rezessiv, sondern X-chromosomal vererbt. Entsprechend sind Jungen zumeist schwerer betroffen. Hemizygote Mädchen und Frauen zeigen in Abhängigkeit von der genetischen Heterogenität passagere Hyperammonämien und nur gelegentlich schwere Stoffwechselentgleisungen. Im Vordergrund stehen bei ihnen progrediente neurologische und psychiatrische Symptome.
Bei der Abklärung jeder akuten unklaren neurologischen Symptomatik muss auch eine Ammoniakbestimmung durchgeführt werden. Nur dann haben Patienten mit Harnstoffzyklusdefekten eine Chance auf einen günstigen Krankheitsverlauf.
Hyperammonämie
Die Hyperammonämie ist der einzig wegweisende Parameter für Harnstoffzyklusdefekte. Eine eindeutig abklärungsbedürftige Hyperammonämie liegt beim Neugeborenen ab Ammoniakkonzentrationen von 150 µmol/l (260 µg/dl), jenseits des Neugeborenenalters ab 100 µmol/l (175 µg/dl) vor.
Erhöhten Ammoniakwerten muss sofort nachgegangen werden, da die Erkrankungen einen dramatischen Verlauf nehmen können, und die Zeitspanne vom Erkrankungsbeginn bis zu irreversiblen Schäden oder Hirntod kurz ist.
Differenzialdiagnose
In die Differenzialdiagnose der Hyperammonämie müssen zahlreiche, insbesondere andere metabolische Erkrankungen einbezogen werden. Entscheidend ist die rasche quantitative Bestimmung der Aminosäuren im Plasma und Urin, der Acylcarnitine sowie der organischen Säuren und der Orotsäure im Urin.
Differenzialdiagnose der Hyperammonämie
Harnstoffzyklusdefekte
Störungen des Transports von Harnstoffzyklusmetaboliten (HHH: Hyperammonämie-Hyperornithinämie-Homozitrullinurie-Syndrom), lysinurische Proteinintoleranz (LPI), Citrinmangel
Defekt der mitochondrialen Carboanhydrase VA
Hyperinsulinismus-Hyperammonämie-Syndrom, Hypoprolinämie, Glutaminsynthasemangel
Organoazidopathien (z. B. Propionazidämie, Methylmalonazidurie)
Genetische Lebererkrankungen (z. B. konnatale Hepatiditiden, Tyrosinämie Typ I, Atmungskettendefekte, Gallensäurensynthesedefekte, klassische Galaktosämie, α1-Antitrypsinmangel)
Passagere Hyperammonämie des Frühgeborenen (persistierender Ductus venosus Arantii; NH3 meist <180 µmol/l)
Andere sekundäre Ursachen (z. B. Gefäßmissbildungen, Shunt, Valproat)
Molekulargenetische Diagnostik
Die Diagnose eines Harnstoffzyklusdefekts wird durch Mutationsnachweis gesichert. Für die molekulare Diagnostik sollten 10 ml EDTA-Vollblut asserviert werden.
Mutationsanalysen mit eingehender Familienuntersuchung erlauben auch eine sichere Pränataldiagnostik.
Cave
Eiweißbelastungstests sollten wegen der Gefahr einer Hyperammonämie mit Stoffwechselentgleisung nicht durchgeführt werden.
Therapie
Generell müssen bei allen Zuständen mit Hyperammonämie schon vor Abschluss der speziellen Untersuchungen und einer endgültigen Diagnose alle zur Verfügung stehenden Möglichkeiten zur Senkung des Ammoniakspiegels genutzt werden. Zur Notfalltherapie müssen die Kinder so rasch wie möglich in ein Stoffwechselzentrum mit Erfahrung in extrakorporalen Entgiftungsverfahren verlegt werden.
Notfalltherapie
Die initiale Therapie zielt darauf ab, den Ammoniakspiegel rasch zu senken, da die Dauer des hyperammonämischen Komas eng mit dem Ausmaß einer bleibenden Hirnschädigung korreliert. Bei Ammoniakwerten zwischen 200 µmol/l (350 µg/dl) und 400 µmol/l (700 µg/dl) wird die Notfallbehandlung entsprechend den folgenden Prinzipien durchgeführt:
Stopp der Eiweißzufuhr,
hochkalorische Ernährung (Kohlenhydrate, Insulin, Fett),
ausreichende Flüssigkeitszufuhr,
forcierte Diurese,
L-Argininmalat/-hydrochlorid i.v.,
medikamentöse Entgiftung des Ammoniaks mit Na-Benzoat und/oder Na-Phenylbutyrat,
antiemetische Behandlung (z. B. Zofran 0,15 mg/kg KG i.v.),
L-Carnitin (100 mg/kg KG und Tag i.v.),
vor Kenntnis der spezifischen Diagnose ggf. Therapieversuch mit Vitaminen (Biotin, Riboflavin, Thiamin, Vitamin B12).
Extrakorporale Entgiftung
Bei Ammoniakwerten über 400 µmol/l (700 µg/dl) müssen Hämofiltration, Hämodialyse oder Hämodiafiltration eingesetzt werden.
Spätestens nach 48 h muss wegen der Gefahr eines Katabolismus infolge eines Mangels an essenziellen Aminosäuren wieder Eiweiß zugeführt werden.
Langzeitbehandlung
Die Ernährung der Patienten mit Harnstoffzyklusdefekten erfolgt entsprechend den Vorgaben der Deutschen Gesellschaft für Ernährung. Zusätzlich werden folgende Medikamente eingesetzt, welche Ammoniak alternativ zum Harnstoffzyklus entgiften (Natriumbenzoat und Natriumphenylbutyrat) bzw. eine Teilfunktion des Harnstoffzyklus in Gang halten (Arginin und Zitrullin).
Notfallregime bei interkurrenten Infekten
Bei interkurrenten Infekten muss ein spezielles Notfallregime befolgt werden. Eckpfeiler des Notfallregimes ist wie in der Akuttherapie (s. oben) die Vermeidung einer katabolen Stoffwechselsituation bzw. die Aufrechterhaltung einer Anabolie durch eine ausreichende Glukose- und Flüssigkeitszufuhr.
Wichtig ist für jeden Patienten mit einem Harnstoffzyklusdefekt ein Notfallausweis bzw. ein Notfallmedaillon mit den wichtigsten Telefonnummern sowie Angaben über die ersten unverzüglich durchzuführenden Maßnahmen!
Verlauf, Prognose
Patienten mit Harnstoffzyklusdefekten und neonataler Symptomatik haben eine ernste Prognose. In der ersten Krise versterben 30–50%. Auch nach erfolgreicher Therapie der initialen Stoffwechselkrise entstehen über die Jahre fast regelhaft schwere Entwicklungsstörungen infolge rezidivierender Hyperammonämien, in denen weitere 10% bis zum 6. Lebensjahr versterben. Eine realistische therapeutische Alternative mit guter Langzeitprognose ist die Lebertransplantation.
Ein in der Neonatalperiode durchgemachtes hyperammonämisches Koma führt fast immer zu schweren neurologischen Folgeschäden. Ebenfalls beeinträchtigen milde chronische Hyperammonämien die psychomotorische Entwicklung. Der Schweregrad des neurologischen Krankheitsbilds von Patienten mit intermittierenden oder chronischen Krankheitsverläufen, insbesondere hemizygoter Mädchen und Frauen mit OTC-Mangel, hängt von der klinischen Symptomatik zum Zeitpunkt der Diagnosestellung und Beginn und Konsequenz der spezifischen Therapie ab. Verlaufsuntersuchungen konnten belegen, dass neurologische Folgeschäden und Retardierung unter konsequenter Therapie nicht fortschreiten.
Laktatazidose und Mitochondriopathien
Grundlagen
Erhöhte Laktatkonzentrationen, bzw. Laktatazidosen, sind ein entscheidender Hinweis auf unterschiedliche angeborene Stoffwechselerkrankungen und müssen differenzialdiagnostisch verfolgt werden. Laktaterhöhungen sind das primäre Leitsymptom erblicher Störungen der oxidativen Phosphorylierung, die einen Mangel an energiereichen Phosphaten in Form von ATP bedingen. Die Häufigkeit von Mitochondriopathien wird auf etwa 1:3000 bis zu 1:10.000 geschätzt, wobei man davon ausgeht, dass 50% der betroffenen Kinder in den ersten 5 Lebensjahren symptomatisch werden.
Synonyme
Folgende Begriffe werden synomym verwendet:
Atmungskettendefekte,
erbliche Laktatazidosen,
Mitochondriopathien,
mitochondriale Erkrankungen,
mitochondriale Zytopathien,
mitochondriale (Enzephalo)myopathien,
OXPHOS-Erkrankungen.
Definition
Mitochondriopathien verursachen vielgestaltige Multisystemerkrankungen. Sie sind durch große klinische Variabilität ihrer Symptome wie auch biochemische Heterogenität gekennzeichnet. Ein wichtiger Hinweis auf Mitochondriopathien sind Kombinationen (progredienter) Symptome unterschiedlicher Organe (Tab. 3.2). Oft steht eine neuromuskuläre Symptomatik, bei Kindern insbesondere Anfälle, bei Erwachsenen Muskelschwäche, im Vordergrund. Krankheitsspektren und -symptome haben sich seit 2010 noch wesentlich erweitert, da jetzt zusätzlich zu den früher schon diagnostizierenden Defekten der mitochondrialen DNA mehrere 100 nukleäre kodierte Defekte als mitochondriale Krankheiten aufgedeckt werden konnten.
| Symptome, Primärdiagnosen | Wegweisende Zusatzsymptome (zusätzlich zu Laktaterhöhungen) | Diagnose |
|---|---|---|
| Autismus | Anfälle | Deletionen und Duplikationen der mtDNA |
| Diabetes mellitus Typ 2 | Dystrophie; Taubheit | MELAS |
| Epilepsie | Abrupter infektassoziierter Beginn; nächtliche Anfälle; generalisierte EEG-Veränderungen | Deletionen der mtDNA |
| Erblindung | Optikusatrophie; Dystonie | Lebersche Optikusatrophie |
| Ertaubung | Jugendliche und junge Erwachsene | MELAS |
| Herzinsuffizienz | Hypertrophe Kardiomyopathie bei Jugendlichen und jungen Erwachsenen | Deletionen der mtDNA |
| Leberversagen | Fehlender Virusnachweis, zerebrale Anfälle | Deletionen der mtDNA, mitochondriale DNA-Depletionssyndrome |
| Leukämie | Maternal vererbte Thrombozytopenie | Deletionen der mtDNA |
| Leukodystrophie | Muskelhypotonie | Deletionen der mtDNA |
| Migräne | Diabetes; Schlaganfälle; Taubheit | MELAS |
| Multiple Sklerose | Anfälle | Mutationen der mtDNA |
| (Chronische) Pankreatitis | Schlaganfälle | MELAS |
| Reflux im Säuglingsalter | Karnitinmangel; Dystrophie | GA II, LCHAD, MELAS |
| Renale tubuläre Azidose | Muskelhypotonie | Defekte der Komplexe I und IV, Depletion der mtDNA |
| Schizophrenie | Anfälle | MELAS |
| Sprachentwicklungsverzögerung | Muskelhypotonie | MELAS |
| Zerebralparese | Verschlechterung bei interkurrenten Infekten | MELAS |
mtDNA mitochondriale DNA; MELAS mitochondriale Enzephalopathie, Laktatazidose, Schlaganfälle; GA II Glutarazidurie Typ II; LCHAD 3-Hydroxyazyl-CoA-Dehydrogenasemangel
Pathogenese, Pathologie
Entscheidend für die Pathogenese der Mitochondriopathien ist der intrazelluläre Energiemangel. Am stärksten betroffen sind besonders energieabhängige Gewebe: Gehirn, Retina, Muskel, Herz, Endokrinium, Leber, Niere.
Infolge des Energiemangels akkumulieren im ZNS freie Radikale, welche die oxidative Belastung sowie andere, aus Energiemangel entstehende zelluläre Fehlfunktionen, wie Störungen des Membranpotenzials, vermehren. Es resultieren spongiforme Degenerationen, Verluste an Neuronen, Gliosen und Demyelinisierung. Sowohl generalisierte als auch umschriebene Lokalisationen werden beobachtet. Besonders häufig sind Basalganglien, Hirnstamm und Kleinhirn betroffen.
In der Muskulatur finden sich neben den pathognomonischen, aber nicht obligaten „ragged red fibers“, Faseratrophie und -dysproportion sowie feintropfige Fettspeicherung.
Genetik
Mitochondriopathien liegen alle vorstellbaren Vererbungsmodi zu Grunde: Spontanmutationen, autosomal-rezessive, dominante, X-gebundene oder maternale Vererbungen. Die meisten Erkrankungen werden durch nukleär kodierte Defekte verursacht und folgen den klassischen Mendel-Regeln.
Einigen Mitochondriopathien liegen primäre Defekte der mitochondrialen DNA zugrunde, mit folgenden Charakteristika:
Mitochondriale DNA wird ausschließlich maternal, unabhängig vom Geschlecht auf alle Kinder vererbt.
Mitochondriale DNA hat eine sehr hohe Mutationsrate, sowohl für Punktmutationen als auch für Deletionen/Insertionen.
Während Zellteilungen werden defekte Mitochondrien in Abhängigkeit von vorliegenden Mutationen über bislang noch unbekannte Mechanismen entweder zufällig auf die Tochterzellen verteilt, oder positiv oder auch negativ selektioniert. Der Anteil defekter Mitochondrien innerhalb und zwischen einzelnen Geweben kann sowohl zu- als auch abnehmen und wechselnde Symptomkonstellationen und Krankheitsverläufe verursachen.
In der Oogenese werden nur wenige bis einzelne Kopien mitochondrialer DNA an die Folgegeneration weitergegeben. Sowohl Genotyp als auch Phänotyp können daher innerhalb einer Familie sehr unterschiedlich sein (Bottleneck-Theorie).
In der genetischen Beratung von Familien mit maternaler Vererbung muss besprochen werden, dass in der Nachkommenschaft bis zu 100% der Kinder betroffen sein können.
Klinisch wichtig ist, dass die Mütter häufig keine oder nur eine minimale Symptomatik wie Schwerhörigkeit, Kleinwuchs oder Hormonstörungen aufweisen können. In vielen Fällen sind maternal vererbte Defekte erst in den Keimbahnen oder in den frühen Entwicklungsstadien entstanden, mit erheblich geringeren Wiederholungsrisiken.
Klinik
Neonatalzeit
Während der Neonatalzeit dominieren folgende Symptomkombinationen:
Hypotrophie, ketoazidotisches Koma, Apnoen, zerebrale Anfälle, Muskelhypotonie, Lebervergrößerung, Tubulopathie,
rasch progrediente, konzentrische hypertrophe Kardiomyopathie und muskuläre Hypotonie,
konzentrische hypertrophe Kardiomyopathie und Neutropenie (Barth-Syndrom) mit X-chromosomaler Vererbung (Xq28),
schwere Leberfunktionsstörung sowie renale Tubulopathie.
Säuglinge und Kleinkinder
Unter den v. a. in dieser Altersgruppe sehr vielfältigen Organmanifestationen können die im Folgenden dargestellten charakteristischen Symptomkombinationen abgegrenzt werden. Im Einzelfall muss bei den Patienten gezielt nach einer Beteiligung weiterer Organsysteme gesucht werden. Mischformen sind häufig.
Rasch progrediente Enzephalomyopathie mit schwerer Hypotonie, zerebellärer Ataxie, Pyramidenbahnzeichen, häufig assoziert mit einer hypertrophen Kardiomyopathie und Tubulopathie.
Subakut nekrotisierende Enzephalomyopathie (Morbus Leigh): klinisch progredienter Verlauf in mehreren Schüben mit psychomotorischer Retardierung und Hirnstammdysfunktion; neuroradiologisch und -pathologisch finden sich symmetrische Nekrosen in Stammhirn, Thalamus, Nucleus subthalamicus, Basalganglien, Hintersträngen und Zerebellum, Kortex und Hippocampus bleiben ausgespart, unterschiedliche Vererbungen.
Rezidivierende Rhabdomyolysen mit Myoglobinurie und muskulärer Hypertonie.
Sideroblastische Anämie mit Neutropenie, Thrombopenie und exokriner Pankreasinsuffizienz (Pearson-Syndrom), mt-DNA Erkrankung.
Gedeihstörung infolge einer Darmzottenatrophie.
Proximale Tubulopathie (De-Toni-Debré-Fanconi-Syndrom) oder interstitielle Nephritis mit Niereninsuffizienz und rezidivierenden Durchfällen, fleckige Pigmentierungen nach Lichtexposition sowie Enzephalomyopathie.
Zwergwuchs mit hypertropher Kardiomyopathie, Innenohrschwerhörigkeit und Retinitis pigmentosa.
Diabetes mellitus und Diabetes insipidus mit Optikusatrophie und Taubheit (Wolfram-Syndrom), autosomal-rezessiv.
Progressive sklerosierende Poliodystrophie (Anfälle und Degeneration der grauen Substanz) und Leberversagen, cave Valproinsäure (MorbusAlpers).
Kinder und Erwachsene
Im Vordergrund stehen meist neuromuskuläre Symptome. Sie können bei bis dato völlig unauffälligen Personen in jedem Lebensalter oligosymptomatisch beginnen (Tab. 3.2):
(progrediente) Muskelschwäche, evtl. mit externer Ophthalmoplegie,
progrediente externe Ophthalmoplegie bis zum Kearns-Sayre-Syndrom (Trias: progrediente externe Ophthalmoplegie, Retinadegeneration plus mindestens eines der folgenden Symptome: kompletter AV-Block, Eiweißerhöhung im Liquor oder zerebelläre Ataxie), mt-DNA Erkrankung,
- progrediente Enzephalomyopathien mit jeweils charakteristischen Zusatzsymptomen:
- MERRF: Myoklonus, Epilepsie, „ragged red fibers“, Muskelschwäche, Ataxie, Hörverlust, mt-DNA Erkrankung,
- MELAS: mitochondriale Enzephalopathie, Laktatazidose, Schlaganfälle (auch hier finden sich „ragged red fibers“), mt-DNA Erkrankung,
- NARP: neurogene Myopathie, Ataxie, Retinitis pigmentosa und fakultativ sensible Polyneuropathie, Anfälle, Demenz, mt-DNA Erkrankung,
- MNGIE: myoneurogastrointestinale Enzephalopathie (intermittierende Diarrhö und Pseudoobstruktionen, Enzephalopathie, Myopathie und periphere Neuropathie), autosomal rezessiv,
Leber-kongenitale Optikusatrophie: Beginn 12–30 Jahre; schnelle Erblindung; evtl. Herzrhythmusstörungen, mt-DNA Erkrankung.
Diagnose
Leitsymptom der Mitochondriopathien ist die Laktaterhöhung. Sie ist je nach Organbefall in unterschiedlichen Kompartimenten nachzuweisen. Bei generalisierter Symptomatik bzw. Leberbeteiligung im Blut, bei Nierenbeteiligung im Urin und bei ausschließlich neurologischer Symptomatik evtl. nur im Liquor und ZNS. Die meisten im klinischen Alltag gefundenen Laktaterhöhungen sind allerdings Folge falscher Abnahmetechnik (u. a. Stauen) oder durch Herz-Kreislauf-Insuffizienz (z. B. bei Sepsis) bedingt.
Cave
Durch normale Laktatkonzentrationen im Blut werden Mitochondriopathien nicht ausgeschlossen.
Grenzwertige Laktaterhöhungen, bzw. der klinische Verdacht auf einen Defekt der oxidativen Phosphorylierung erfordern ggf. wiederholte prä- und postprandiale Bestimmungen von Laktat, Pyruvat, 3-Hydroxybutyrat, Azetoacetat und Alanin, eine Glukosebelastung mit 1–2 g/kgKG. Nach Glukosebelastung steigt Laktat beim Stoffwechselgesunden um höchstens 20%. Besonders aussagekräftig ist ein „paradoxer“ postprandialer Ketonkörperanstieg (normal: Abfall). Bei pathologischen Basiswerten erübrigen sich Belastungsteste. Hilfreich sind ferner wiederholte Bestimmungen des Laktat/Kreatinin-Quotienten im Urin und insbesondere bei neurologischer Symptomatik Bestimmungen von Laktat, Pyruvat und Alanin im Liquor und spektroskopisch von Laktat im Gehirn.
Vor allem bei älteren Kindern, Jugendlichen und Erwachsenen kann der Nachweis klassischer Punktmutationen bzw. Deletionen der mitochondrialen DNA (häufiger nukleäre Defekte) gelingen. Entscheidende diagnostische Bausteine liefern Muskelhistologie, Histochemie und ggf. Elektronenmikroskopie mit dem Nachweis von „ragged red fibers“ oder abnorm strukturierter Mitochondrien.
Die biochemische Aufarbeitung einer frischen Muskelbiopsie stellt den Goldstandard zum Nachweis einer Mitochondriopathie dar, ist aber nur in einigen Speziallaboratorien möglich. Neue genetische Verfahren zur parallelen Untersuchung hunderter Gene bzw. des Exoms sollten gleichzeitig initiiert werden.
Therapie
Erfolgreiche rationale Therapieansätze sind nur für die Q10-Supplementierung bei spezifischem Q10-Mangel, Thiamin-Supplementierung und eine ketogene Diät bei einigen Patienten mit Defekten im PDH-Komplex sowie für die Argininsupplementierung beim MELAS-Syndrom nachgewiesen. Für alle anderen Mitochondriopathien sind kausale Therapien nicht gesichert. Die symptomatische Behandlung beinhaltet neben unterstützenden Maßnahmen die Vermeidung von Medikamenten, welche die Atmungskette hemmen (u. a. Valproinsäure, Tetrazykline, Aminoglykoside, Chloramphenicol, Metformin, Propofol).
Überwachung
Regelmäßig müssen diejenigen Organsysteme kontrolliert werden, welche im Verlauf von Mitochondriopathien häufig betroffen werden: Skelettmuskel, Herz (Reizleitung!), ZNS, Auge, Gehör, Niere und Leber.
Vermeidung kataboler Stoffwechselsituationen
Entscheidend ist eine ausreichende Zufuhr von Energie (Glukose, Fett), Flüssigkeit und Elektrolyten. Oft ist eine Sonden-PEG-Ernährung sehr hilfreich. Je nach Lokalisation des Defekts müssen die zuführenden Energieträger sorgfältig abgewogen werden. Bei Defekten der Pyruvatdehydrogenase kann die Stoffwechselsituation durch die Zufuhr größerer Mengen an Glukose krisenhaft verschlechtert werden.
Vitamine und Kofaktoren
Bei einigen Mitochondriopathien wurden durch die Gabe von speziellen Vitaminen und Kofaktoren individuelle Verbesserungen berichtet. Bei Erstmanifestation einer schweren Laktatazidose können folgende Substanzen versuchsweise kombiniert eingesetzt werden:
L-Carnitin (50 mg/kgKG/Tag),
Biotin (2-mal 10 mg/Tag),
Riboflavin (3–20 mg/kgKG/Tag, bis zu 400 mg/Tag),
Thiamin (25–100 mg/kgKG/Tag, bis zu 300 mg/Tag),
Koenzym Q10 (5–15 mg/kgKG/Tag).
Therapie der akuten ketoazidotischen Krise
Akute ketoazidotische Krisen erfordern den Einsatz z. T. großer Mengen an Natriumbikarbonat.
Diäten und weitere Maßnahmen
Bei vielen Patienten mit Mitochondriopathien werden Therapieversuche v. a. mit unterschiedlichen Vitaminen begonnen. Nur bei Defekten der Pyruvatdehydrogenase und des Komplex I der Atmungskette ist eine ketogene Diät evtl. plus Succinat sinnvoll, da diese Substrate via Komplex II verstoffwechselt werden. Bei Patienten mit Komplex-III-Defekten kann die Einnahme von Vitamin K3 plus Vitamin C hilfreich sein. Die Therapieeffekte sind im Einzelfall nicht vorhersehbar, sodass ein Versuch in Abwägung der Schwere der Krankheitsbilder und bei meist dringend vorgetragenem Wunsch der Familie oft gerechtfertigt ist, aber zeitlich limitiert bzw. kritisch evaluiert werden muss.
Verlauf, Prognose
Erschwert werden eine zusammenfassende Beurteilung von Krankheitsverläufen sowie von Therapiestudien bei Patienten mit Mitochondriopathien durch die relative Seltenheit der individuellen Krankheitsbilder und die große Variabilität und Fluktuation einzelner Krankheitsverläufe. Speziell im Kindesalter sind individuelle Vorhersagen nicht möglich. Die Symptomatik ist häufig rasch progredient. Jederzeit kann es aber zu Phasen der Stabilität und auch zu einer Verbesserung der klinischen und biochemischen Befunde kommen. Diese Erfahrungen müssen in die Beratung einzelner Patienten bzw. ihrer Familien, in die Therapieplanung und auch in die Beurteilung ihrer Erfolge bzw. Misserfolge immer wieder eingebracht werden.
Hypoglykämie
Grundlagen
Hypoglykämien sind im Kindesalter häufig. Sie resultieren aus einem Missverhältnis zwischen Angebot und Verbrauch an Glukose.
Pathogenese
Ursächlich sind Enzymdefekte und Regulationsstörungen der Glykogenolyse (Glykogenosen), der endogenen Glukoseproduktion (Glukoneogenesedefekte) und der Ketonkörperbildung (Fettsäurenoxidationsdefekte und Ketonkörperbildung) sowie endokrinologische Störungen. Hypoglykämien gefährden v. a. das Gehirn, welches seine Energie unter physiologischen Bedingungen ausschließlich aus Glukose bezieht. In Mangel- und Fastenperioden kann das Gehirn teilweise auf alternative Substrate zurückgreifen, insbesondere Ketonkörper.
Cave
Das kindliche Gehirn ist durch Hypoglykämien besonders gefährdet. Rezidivierende Hypoglykämien führen zu irreversiblen Schädigungen!
Klinik
Blutglukosekonzentration und klinische Symptomatik korrelieren nicht zuverlässig. Die Schwere der Hypoglykämiesymptome hängt u. a. von der Geschwindigkeit des Blutzuckerabfalls und der Möglichkeit ab, alternative Substrate (z. B. Ketonkörper, Laktat) energetisch zu nutzen. Auch ohne das Vorliegen einer angeborenen Stoffwechselstörung sind Frühgeborene, hypotrophe Neugeborene infolge pränataler Mangelernährung, Neugeborene von Müttern mit Diabetes mellitus und Neugeborene mit erhöhtem Glukoseverbrauch infolge von Hypoxie oder Infektionen besonders gefährdet. Bei Neugeborenen und jungen Säuglingen überwiegen in der Regel uncharakteristische vegetative Symptome, während bei älteren Kindern neurologische Symptome im Vordergrund stehen (Tab. 3.3).
| Neugeborene, junge Säuglinge | Ältere Säuglinge, Kinder |
|---|---|
|
- Trinkschwäche - Zittrigkeit - Blässe - Tachypnoe - Hypotonie - Hyperexzitabilität - Apnoeanfälle - Zyanose, Hypothermie - Krampfanfall - Koma |
- Blässe - Schwitzen - Apathie - Übelkeit, Erbrechen - Hunger, Bauchschmerzen - Kopfschmerzen - Ungewöhnliches Verhalten - Bewusstseins-, Sehstörung - Krampfanfall - Koma |
Diagnose
Altersabhängige Abstufungen der Hypoglykämie wurden inzwischen verlassen. In jedem Alter liegt ab einer Plasmaglukosekonzentration von <2,5 mmol/l (45 mg/dl) eine Hypoglykämie vor. Die Differenzialdiagnose der Hypoglykämie ist aufgrund der zahlreichen metabolischen und endokrinologischen Ursachen vielfältig. Entscheidend für die Diagnosestellung ist eine systematische Untersuchung der Hormone sowie der Metabolitenkonstellation in der Hypoglykämie.
Labor
Folgende Laborparameter sollten in einer unerklärten Hypoglykämie bestimmt werden: Blutglukose, Blutgasanalyse, Insulin, Laktat, Ketonkörper, freie Fettsäuren und Azylkarnitine (aus einer Trockenblutprobe).
Der erste Urin nach einer Hypoglykämie muss asserviert und qualitativ auf Ketonkörper und quantitativ auf organische Säuren untersucht werden.
Darüber hinaus ist es sinnvoll, weitere Laborparameter wie Elektrolyte, Leberwerte, Ammoniak, CK, Harnsäure, Aminosäuren und Karnitin zu bestimmen sowie Serum, Plasma und Urin für Folgeuntersuchungen zu asservieren (z. B. Wachstumshormon, Kortisol etc.). Zusammen mit der Anamnese und dem klinischen Befund ermöglicht die Metabolitenkonstellation der wichtigsten Laborparameter zumeist bereits eine Verdachtsdiagnose (Abb. 3.3).


Therapie
Die Akuttherapie bei Hypoglykämie besteht bei leichteren Formen in der Gabe von schnell verfügbaren Kohlenhydraten (z. B. Dextroenergen/Dextropur in Tee gelöst, Banane, Apfel). Bei schweren Formen muss sofort 20%ige Glukose (ca. 1 ml/kgKG entsprechend 200 mg/kgKG) i.v. verabreicht werden.
Kongenitaler Hyperinsulinismus
Pathogenese
Der kongenitale Hyperinsulinismus ist die häufigste Ursache für rezidivierende Hypoglykämien im frühen Kindesalter (Abschn. 4.12). Ursächlich ist eine übermäßige Insulinausschüttung aufgrund von Regulationsstörungen der Insulinsekretion. Molekularbiologische Studien deckten bei einem Teil der Patienten krankheitsverursachende Mutationen in einem ATP-sensitiven Kaliumkanalprotein auf, welcher die Insulinsekretion reguliert (sog. Sulfonylharnstoffrezeptor (diffus) bzw. der Untereinheit Kir6.2, lokal). Diese sind in der Regel mit einem schweren Krankheitsverlauf assoziiert.
Eine Sonderform stellt der fokale Hyperinsulinismus dar, der sporadisch auftritt. Ursache hierfür ist ein Verlust der Heterozygotie für den kurzen Arm des maternalen Chromosoms 11. Zusätzlich liegt eine begleitende Mutation auf dem paternalen Gen für den Sulfonylharnstoffrezeptor vor.
Wichtig sind ferner aktivierende Mutationen der Glukokinase oder der Glutamatdehydrogenase (autosomal-dominant), die gut auf Diazoxid ansprechen, und das Hyperinsulinismus-Hyperammonämie-Syndrom (autosomal-dominante GLDH-aktivierende Mutationen), welches Leucin-sensitiv und mit zunehmendem Alter rückläufig sind.
Klinik
Kinder mit kongenitalem Hyperinsulinismus werden meist in der Neugeborenenzeit symptomatisch, leichtere Formen können jedoch auch erst später manifest werden.
Neugeborene sind häufig makrosom. Vegetative Symptome stehen im Vordergrund. Bei älteren Kindern finden sich Blässe, Schwitzen, Schwindel, Bewusstseinsstörungen oder Krämpfe (Tab. 3.3).
Diagnose
Entscheidend ist der Nachweis einer gesteigerten Insulinausschüttung durch gleichzeitige Bestimmung von Blutzucker und Insulin während hypoglykämischer Episoden (Abb. 3.3). Ein Hyperinsulinismus führt zu einer hypoketotischen Hypoglykämie mit niedriger Konzentration freier Fettsäuren und Ketonkörper. Der Glukosebedarf zum Erhalten einer Normoglykämie ist größer als die endogene Glukoseproduktionsrate (>10 mg/kgKG/min). Bei Patienten mit Mutationen im Glutamatdehydrogenasegen liegt häufig eine moderate Hyperammonämie (100–200 µmol/l) vor. In der Differenzialdiagnose müssen der transiente Formen des Hyperinsulinismus bei Neugeborenen (diabetische Fetopathie, Asphyxie, Sepsis, Rhesusinkompatibilität, SGA usw.), die Hypophysen- oder Nebenniereninsuffizienz und das Beckwith-Wiedemann-Syndrom abgegrenzt werden.
Vor einer operativen Therapie sollte das Vorliegen einer fokalen Form z. B. durch PET-Untersuchungen oder eine selektive Pankreasvenenkatheterisierung mit gleichzeitigen Insulin- und Glukosebestimmungen ausgeschlossen werden.
Therapie
Eine effektive und frühzeitige Therapie ist gerade bei den neonatalen Formen entscheidend, um bleibende zerebrale Schäden zu vermeiden.
Akuttherapie
Zunächst muss der Blutzucker durch i.v.-Zufuhr von Glukose (bis zu 25 mg/kgKG/min) über einen zentralen Zugang stabilisiert werden. Initial kann eine Erhöhung des Blutzuckers durch Glukagongaben (1 mg/kgKG/Tag kontinuierlich i.v.) oder alternativ durch Somatostatin (1–5 µg/kgKG/h i.v.) erreicht werden.
Kohlenhydratreiche Mahlzeiten
Grundlage einer konservativen Therapie sind häufige kohlenhydratreiche Mahlzeiten mit ungekochter Stärke (Maltodextrin) zur Nacht. In Einzelfällen ist eine kontinuierlich nächtliche Sondenernährung indiziert.
Medikamentöse Therapie
Als medikamentöse Therapie wird Diazoxid (5–15 mg/kgKG/Tag in 3 Einzeldosen, ansprechen in der Regel spätestens nach 5 Tagen) ggf. in Kombination mit Hydrochlorothiazid (1–2 mg/kgKG/Tag in 2 Dosen), eingesetzt. Zur längerfristigen Behandlung ist auch Octreotid, ein Somatostatinanalogon (5–20 mg/kgKG/Tag in 3–4 Dosen s.c.) geeignet. Zusätzlich liegen in Einzelfällen Berichte über erfolgreiche Therapien mit dem Kalziumkanalblocker Nifedipin (0,5–2 mg/kgKG/Tag) vor.
Operative Therapie
Bei Versagen der konservativen Therapieversuche ist eine 90–95%ige Pankreasresektion zu erwägen. Durch die Operation lassen sich Hypoglykämien häufig, jedoch nicht immer beseitigen. Fokale Areale können selektiv reseziert werden. Nur wenige Patienten entwickeln direkt postoperativ einen insulinpflichtigen Diabetes mellitus, später steigt die Zahl der Diabetesmanifestationen bei den operierten Patienten kontinuierlich an. Die iatrogene exogene Pankreasinsuffizienz ist selten symptomatisch und lässt sich gut behandeln.
Die Gesamtprognose hängt v. a. von Häufigkeit und Schweregrad der Hypoglykämien in den ersten Lebensjahren ab.
Defekt des Glukosetransports
Die Passage von Glukose durch die Blut-Hirn-Schranke und in Astrozyten, Neurone und Gliazellen erfolgt mittels erleichterter Diffusion und wird durch ein Glukosetransportprotein (GLUT 1) vermittelt. Ein Defekt dieses Transportes wird als Glukosetransporter(GLUT1)-Defekt oder nach dem erst Erstbeschreiber De-Vivo-Disease bezeichnet.
Die klinischen Merkmale des GLUT1-Defekts sind zerebrale Anfälle, Entwicklungsretardierung, komplexe Bewegungsstörungen (Hypotonie, Ataxie, Dystonie). Hinweisend ist ein ungeklärt niedriger Liquorzucker bei Nüchternlumbalpunktion (<2,7 mmol/l oder 50 mg/dl). Das GLUT1-Gen befindet sich auf dem kurzen Arm von Chromosom 1. Patienten mit GLUT1-Defekt weisen individuelle heterozygote Mutationen auf.
Der Verlauf der Erkrankung kann durch eine ketogene Diät, die dem ZNS eine alternative Energiequelle in Form von Ketonkörpern anbietet, günstig beeinflusst werden.
Störungen des Galaktosestoffwechsels (Galaktosämien)
Die 3 bekannten hereditären Defekte im Galaktosestoffwechsel werden durch autosomal-rezessiv vererbte Enzymdefekte verursacht (Abb. 3.4):
Galaktokinase,
Galaktose-1-Phosphat-Uridyltransferase („klassische Galaktosämie“),
UDP-Galaktose-4-Epimerase.


Während Defekte der Galaktokinase oder Galaktose-1-Phosphat-Uridyltransferase zu Erhöhungen der freien Galaktose im Blut nach Zufuhr von Milch oder Milchprodukten führen, beruht eine Erhöhung von Galaktose-1-Phosphat auf einem Galaktose-1-Phosphat-Uridyltransferase- oder UDP-Galaktose-4-Epimerasemangel.
Galaktokinasemangel
Pathogenese
Beim Galaktokinasemangel, der regional unterschiedlich mit einer Häufigkeit von < 1:100.000 vorkommt, wird aus der sich anstauenden Galaktose mittels des Enzyms Aldosereduktase dessen Zuckeralkohol Galaktitol gebildet, welcher zur Kataraktbildung führt.
Klinik
Führendes Symptom ist die Entwicklung von Katarakten, die bereits um die 3.–5. Lebenswoche manifest werden. Deren Früherkennung ist wichtig, da sie bei sofortiger milchfreier Ernährung reversibel sein können. Leberfunktionsstörungen oder mentale Retardierung entstehen nicht.
Diagnose
Die Galaktosespiegel im Blut sind erhöht (am ausgeprägtesten 30–90 min nach Milchmahlzeiten). Dadurch ist eine frühzeitige Erfassung des Defekts im Neugeborenenscreening möglich. Im Urin ist eine vermehrte Ausscheidung von Galaktose und Galaktitol nachweisbar. Die Diagnose wird durch Nachweis des Enzymdefekts in Erythrozyten gesichert.
Therapie
Die exogene Galaktoseaufnahme muss bei Patienten mit Galaktokinasemangel möglichst gering gehalten werden, d. h. sie müssen lebenslang laktosefrei und galaktosearm ernährt werden. Die Prognose ist bei frühzeitiger Diagnose und Behandlung gut.
Galaktose-1-Phosphat-Uridyltransferasemangel („klassische Galaktosämie“)
Pathogenese
Der ausgeprägte Mangel des Enzyms Galaktose-1-Phosphat-Uridyltransferase in roten und weißen Blutzellen, Hautfibroblasten, Dünndarm und Leber der betroffenen Patienten führt dazu, dass Galaktose-1-Phosphat nicht in Glukose-1-Phosphat umgewandelt und somit nicht in die Glykogensynthese/Glykolyse eingeschleust werden kann (Abb. 3.4). Die hohen Konzentrationen des Phosphatzuckers Galaktose-1-Phosphat führen zur Schädigung von Leber, Niere, Gehirn und Augenlinse.
Epidemiologie
Die Inzidenz der sog. „klassischen Galaktosämie“ mit vollständigem Defekt der Galaktose-1-Phosphat-Uridyltransferase beträgt ca. 1:70.000 (Tab. 3.1).
Klinik
Die Erkrankung manifestiert sich nach Zufuhr laktosehaltiger Milch in den ersten Lebenstagen mit zunehmender Trinkunlust, Erbrechen, Hypoglykämien mit Krampfanfällen, Lethargie und Koma, Nierenschäden mit Ausbildung eines Fanconi-Syndroms sowie v. a. mit einer akuten schweren Leberfunktionsstörung. Aufgrund des akuten Leberversagens mit Gerinnungsstörung, Ikterus gravis, Hautblutungen und Hepatosplenomegalie ist die Letalität hoch. Im späteren Säuglingsalter stehen Gedeihstörungen, Erbrechen, Hepatomegalie und Katarakte im Vordergrund. Ältere Kindern entwickeln eine mentale Retardierung und Leberzirrhose.
Diagnose
Im Neugeborenenscreening können im Trockenblut bei einem Substratscreening die Galaktosekonzentration der Probe als freie Galaktose oder als Gesamtgalaktose (freie Galaktose plus Galaktose nach Abspaltung aus der Phosphatbildung) sowie die Aktivität der Galaktose-1-Phosphat-Uridyltransferase bestimmt werden. Letztere ist unabhängig von der Milchfütterung diagnostisch. Voraussetzung für diagnostische erhöhte Metabolite ist eine begonnene Milchfütterung. In den Erythrozyten ist der Galaktose-1-Phosphatspiegel erhöht. Die vermehrte Galaktose kann auch im Urin als reduzierende Substanz nachgewiesen werden. Die Diagnosesicherung erfolgt durch die Enzymaktivitätsbestimmung der Galaktose-1-Phosphat-Uridyltransferase in Erythrozyten. Zuverlässige molekulargenetische Untersuchungen sind möglich und auch für eine pränatale Diagnostik einsetzbar.
Therapie
Wegen des oftmals foudroyanten Verlaufs der Krankheit müssen Neugeborene bei dem geringsten Verdacht auf eine „klassische Galaktosämie“ sofort laktosefrei (milchzuckerfrei) ernährt werden, d. h. das Stillen muss bis zum Vorliegen der Laborbefunde unterbrochen werden.
Laktosefreie Säuglingsnahrungen sind auf Sojabasis aufgebaut. Bei Bestätigung der Diagnose muss die exogene Galaktoseaufnahme lebenslang möglichst gering gehalten werden, d. h. diese Patienten müssen laktosefrei und galaktosearm ernährt werden. Eine vollständig galaktosefreie Ernährung ist durch den Gehalt an freier und gebundener Galaktose in fast allen Lebensmitteln in der Praxis nicht durchführbar und aufgrund einer hohen endogenen Galaktoseproduktion nicht angemessen. Die endogene Galaktoseproduktion im intermediären Stoffwechsel bewegt sich beim gesunden Erwachsenen wie bei Patienten mit Galaktose-1-Phosphat-Uridyltransferasemangel im Bereich von 1–2 g/Tag. Sie kann derzeit therapeutisch nicht beeinflusst werden. Durch den Verzicht auf Milch und Milchprodukte ist der Kalziumgehalt der Nahrung gering. Weibliche Patienten leiden oft an einer ovariellen Insuffizienz und müssen mit galaktosefreien hormonellen Sequenzpräparaten substituiert werden, um die Ausbildung der sekundären Geschlechtsmerkmale und das Auftreten von Abbruchblutungen zu erzielen. Die Infertilität wird hierdurch nicht korrigiert.
Verlauf, Prognose
Eine frühzeitig begonnene galaktosearme Ernährung führt zu einer Reversibilität bereits aufgetretener Katarakte. Trotz adäquater Behandlung werden bei vielen Patienten mit Galaktose-1-Phosphat-Uridyltransferasemangel ab dem Schulalter Sprachentwicklungsstörungen, eine milde mentale Retardierung, weitere neurologische Spätkomplikationen (Intentionstremor, Ataxie) sowie bei ca. 80% der weiblichen Patienten ein hypergonadotroper Hypogonadismus mit überwiegend fibrotischen Ovarien, stark verzögerter Pubertätsentwicklung und deutlicher Einschränkung der Fertilität offensichtlich.
Störungen des Fruktosestoffwechsels
Fruktose ist Bestandteil des Kochzuckers (Saccharose, Fruktose-Glukose-Disaccharid) und ist in großen Mengen in Obst und diversen Gemüsesorten enthalten. Daneben werden Fruktose, Saccharose oder Sorbit (das v. a. über Fruktose abgebaut wird) häufig Lebensmitteln zugesetzt. Im Fruktosestoffwechsel sind 3 autosomal-rezessiv vererbte Defekte bekannt, und zwar Aktivitätsverluste folgender Enzyme (Abb. 3.5):
Fruktokinase,
Fruktose-1-Phosphat-Aldolase („hereditäre Fruktoseintoleranz“),
Fruktose-1,6-Diphosphatase.


Benigne Fruktosurie
Die benigne Fruktosurie infolge eines Mangels der Fruktokinase ist meist eine Zufallsdiagnose. Ein Teil der zugeführten Fruktose wird im Urin ausgeschieden und kann über eine Reduktionsprobe (Clinitest) nachgewiesen werden. Die Häufigkeit beträgt ca. 1:50.000. Es handelt sich um eine harmlose, nicht behandlungsbedürftige Störung.
Hereditäre Fruktoseintoleranz
Grundlagen
Bei der hereditären Fruktoseintoleranz besteht ein Aktivitätsverlust der Fruktose-1-Phosphat-Aldolase B. Die Inzidenz wird auf 1:20.000 geschätzt. Ein Metabolitenscreeningtest in der Neugeborenenperiode ist nicht möglich.
Klinik
Betroffene Kinder entwickeln Symptome erst mit Aufnahme fruktosehaltiger (saccharosehaltiger) Nahrungsmittel. Dieses geschieht in der Regel im Säuglingsalter bei Übergang auf Beikost (Säfte, Früchte, Gemüse) oder bei Zufütterung einer saccharosehaltigen Folgenahrung. Je jünger ein Kind und je größer die aufgenommene Fruktosemenge ist, desto schwerer sind die klinischen Symptome.
Akute Symptome nach Fruktosezufuhr können gastrointestinale Beschwerden und Hypoglykämien mit Übelkeit, Erbrechen, Blässe, Schwitzen, Zittern, Lethargie und Krampfanfällen sein (Tab. 3.3). Bei fortgesetzter Fruktosezufuhr kommt es zur Gedeihstörung, progredienter Leberfunktionsstörung (Hepatosplenomegalie, Ikterus, schweren Gerinnungsstörungen, Ödemen und Aszites) und immer auch zum renal-tubulären Schaden (De-Toni-Debré-Fanconi-Syndrom). Nach Elimination der Fruktose aus der Nahrung tritt in der Regel eine schnelle Erholung ein. Verantwortlich für Hypoglykämien, Leber- und tubulären Nierenschäden ist die Akkumulation von Fruktose-1-Phosphat.
Einige Säuglinge entwickeln wegen fruktoseinduzierter gastrointestinaler Beschwerden schon sehr früh eine Aversion gegen fruktosehaltige Speisen, was einen Selbstschutz darstellt, aber die frühzeitige Erkennung verhindern kann.
Diagnose
Bei Verdacht auf hereditäre Fruktoseintoleranz muss sofort vollständig fruktosefrei ernährt werden. Ein rascher Rückgang der Symptome innerhalb von Tagen ist dabei eine erste Bestätigung der Verdachtsdiagnose. Zunächst sollte der Mutationsnachweis im Aldolase-B-Gen zur Diagnosesicherung angestrebt werden. In diagnostisch unklaren Fällen kann nach Normalisierung der Leberfunktion die Diagnose durch Aktivitätsmessung der Fruktose-1,6-Diphosphataldolase in einer funktionell normalen Leber oder in einem Dünndarmbiopsat gesichert werden. Eine intravenöse Fruktosebelastung ist obsolet. Eine orale Fruktosebelastung ist nicht evaluiert, unangenehm für den Patienten und kann erhebliche Nebenwirkungen haben.
Therapie
Die Behandlung besteht in der Elimination sämtlicher Fruktose aus der Nahrung. Dieses betrifft alle Nahrungsmittel, welche natürlicherweise Fruktose, Saccharose oder Sorbit enthalten oder denen diese Substanzen zugesetzt wurden.
Cave
Bei Patienten mit hereditärer Fruktoseintoleranz muss bei der Verabreichung von Medikamenten, insbesondere bei Säften, genau auf die Inhaltsstoffe geachtet werden!
Verlauf, Prognose
Bei einigen Kindern bleibt trotz fruktose- und sorbitfreier Ernährung über Monate und Jahre eine Hepatomegalie bestehen. Mit zunehmendem Alter erhöht sich die Toleranz gegenüber Fruktose leicht, sodass Kinder täglich bis zu 0,5–1 g und Erwachsene bis zu 2,5 g Fruktose tolerieren. Die Prognose ist unter kontrollierter Diät gut.
Glykogenosen
Definition
Glykogen wird v. a. in Leber und Muskulatur aus Glukose-6-Phosphat synthetisiert und als wichtigster initialer Energieträger gespeichert. Bei den Glykogenosen handelt es sich um Speicherkrankheiten charakterisiert durch einen abnormen Gehalt an normal oder pathologisch strukturiertem Glykogen mit Funktionsstörungen in Leber und/oder Muskeln sowie eine gestörte Glukosehomöostase. Die Einteilung der Glykogenosen erfolgt in chronologischer Reihenfolge ihrer Erstbeschreibung mit römischen Ziffern und Kleinbuchstaben (Tab. 3.4).
| Typ | Enzymdefekt | Speicherorgan | Hypoglykämie | Symptome |
|---|---|---|---|---|
| Ia | Glukose-6-Phosphatase | Leber, Niere | +++ | Hepatomegalie, Hyperlaktatämie, Hypertriglyzeridämie, Hyperurikämie, Nephromegalie, Nephropathie, Kleinwuchs, Blutungsneigung |
| I b | Glukose-6- Phosphattranslokase | Leber | +++ | Wie Ia, zusätzlich Neutropenie, Infektneigung, Morbus-Crohn-ähnliche Darmerkrankung |
| II | Lysosomale alpha- Glukosidase | Generalisiert | Nein |
Infantile Form: Kardiomegalie, Hepatomegalie, progressive Muskelhypotonie Juvenile/adulte Form: Myopathie |
| III | Amylo-1,6-Glukosidase | Leber, Muskel, Erythrozyten | + | Hepatomegalie, Myopathie |
| IV | Brancher-Enzym | Leber | Nein | Leberzirrhose, Hepatosplenomegalie, Myopathie |
| V | Phosphorylase | Muskel | Nein | Muskelkrämpfe nach Belastung, rasche Ermüdbarkeit, Myoglobinurie |
| VI | Phosphorylase | Leber | + | Hepatomegalie |
| VII | Phosphofruktokinase | Muskel, Erythrozyten | Nein | Wie V, hämolytische Anämie |
| IX | Phosphorylasekinase | Leber, Muskel, Erythrozyten | + | Hepatomegalie, Myopathie, Kleinwuchs |
| 0a | Glykogensynthetase | Leber, keine Speicherung | +++ | Gedeihstörung |
| 0b | Glykogensynthetase | Muskel | Nein | (Kardio)myopathie |
Epidemiologie
Die Inzidenz aller Glykogenosen wird in Europa auf ca. 1:25.000 geschätzt. Sie schwankt im Einzelfall je nach Typ und ethnischer Zusammensetzung der Bevölkerung beträchtlich. Die Glykogenosen Typ Ia II, III und IX kommen am häufigsten vor.
Genetik
Mit Ausnahme der häufigsten Unterform der Glykogenose Typ IX (Phosphorylase-b-Kinasemangel), die X-chromosomal vererbt wird, liegt allen Glykogenosen ein autosomal-rezessiver Erbgang zugrunde. Mittlerweile konnten bei allen beteiligten Enzymsysteme bzw. Transportproteine die entsprechenden Gene lokalisiert und zugrunde liegende Mutationen identifiziert werden.
Klinik
Klinisch lassen sich 3 Manifestationsformen unterscheiden:
überwiegend hepatische Beteiligung (Typ I, IV, VI, IX, 0),
vorwiegend muskuläre Beteiligung (Typ II, V, VII)
gemischte Symptomatik (Typ III).
Diagnose
Infolge der fortschreitenden Entwicklung der diagnostischen Methoden (Enzymbestimmungen in Erythrozyten, Leukozyten, Fibroblasten und v. a. Molekulargenetik) ist in der Regel die quantitative Bestimmung und die histologische Beurteilung des Glykogens in Leber und/oder Muskel nicht mehr erforderlich. Belastungstests mit Glukose, Galaktose oder Glukagon spielen ebenfalls nur noch eine untergeordnete Rolle. Die pränatale Diagnostik unter Zuhilfenahme von Chorionvilli- oder Amnionzellen ist für alle Glykogenosen möglich.
Therapie
Diätetische Maßnahmen sind die Eckpfeiler der Therapie und haben wesentlich zur Verbesserung der Stoffwechselkontrolle geführt, insbesondere bei Patienten mit Glykogenose Typ I. Lebertransplantationen wurden bei einer Reihe von Patienten mit Glykogenosen I und IV aufgrund von Leberadenomen und -karzinomen bzw. Leberzirrhose durchgeführt. Ansätze einer Enzymersatztherapie sind bei Glykogenose II erfolgversprechend.
Glykogenosen Typ I (Morbus v. Gierke)
Pathogenese
Ursache der Glykogenosen Typ I sind Defekte der Glukose-6-Phosphatase. Dieses membrangebundene Enzymsystem, welches ausschließlich in der Leber vorkommt, besteht aus 6 Untereinheiten. Es katalysiert die Umwandlung von Glukose-6-Phosphat in Glukose, die gemeinsame Endreaktion von Glykogenolyse und Glukoneogenese. Bei der Glykogenose Typ I ist daher jegliche Freisetzung von Glukose aus der Leber blockiert und auch keine Glukosebereitstellung aus Fruktose, Galaktose oder Glyzerin möglich. Patienten sind zur Aufrechterhaltung einer Normoglykämie vollständig von der oralen Aufnahme von Glukose oder Glukosepolymeren (Maltodextrin, Stärke) abhängig. Durch die gestörte hepatische Glukoseproduktion kommt es zur Anhäufung von Glukose-6-Phosphat und anderen Glykolyseintermediaten, die vermehrt in Laktat (Laktatazidose) umgewandelt werden und die Glykogen- und Triglyzeridsynthese (Hypertriglyzeridämie) sowie den oxidativen Pentoseweg (Hyperurikämie) stimulieren.
Klinik
Auffällig werden betroffene Kinder zumeist im Alter von 3–6 Monaten mit hypoglykämischen Krampfanfällen, einer ausgeprägten Hepatomegalie (ohne Splenomegalie), weitausladendem Abdomen, Wachstumsrückstand und einem puppenähnlichen Aussehen (Abb. 3.6). Häufig besteht eine ausgeprägte Blutungsneigung mit Nasenbluten und multiplen Hämatomen. Bei der Ultraschalluntersuchung fällt zusätzlich eine beidseitige Nephromegalie auf. Kinder mit den selteneren Glykogenosen Typ Ib leiden zusätzlich an rezidivierenden schweren akuten oder chronischen Infektionen und Entzündungen infolge einer Neutropenie (<1000/µl) und einer gestörten Leukozytenfunktion (Granulozyten und Monozyten).


Diagnose
Laborchemisch bestehen Hypoglykämien bzw. niedrige Blutglukosekonzentrationen im Tagesprofil, eine ausgeprägte Laktatazidose, Hypertriglyzeridämie und evtl. Hyperurikämie sowie meist leichte Transaminasenerhöhungen. Im Rahmen eines oralen Glukosetoleranztests kommt es zu einem Abfall der Laktatwerte. Durch Glukagon (50 µg/kgKG) wird der postresorptive Blutglukoseabfall nicht beeinflusst, nicht die Glukose-, sondern die Blutlaktatwerte steigen an.
Für die Glykogenosen Typ Ia und Typ I non-a sind zahlreiche Mutationen beschrieben worden. Gelingt der Nachweis pathogener Mutationen aus Leukozyten, so kann auf die Leberbiopsie verzichtet werden.
Therapie
Ziel der Behandlung der Glykogenosen Typ I ist die Vermeidung von Hypoglykämien sowie die weitestgehende Verhinderung sekundärer Stoffwechselveränderungen. Dies wird durch häufige oder/und kontinuierliche exogene Zufuhr von Glukose und weitgehende Elimination von Laktose, Saccharose und Fruktose aus der Nahrung erreicht.
Zur Deckung des nächtlichen Glukosebedarfs stehen 2 alternative Strategien zur Verfügung:
die kontinuierliche nächtliche Dauersondierung von Dextrinlösungen im Säuglings- und Kindesalter und
die orale Verabreichung von ungekochter Maisstärke (Mondamin), aus der Glukose verzögert freigesetzt werden kann.
Die Umstellung von der Sondenernährung auf die nächtliche Behandlung mit ungekochter Maisstärke erfolgt im Schulkindesalter.
Bei Hyperurikämie wird Allopurinol eingesetzt. Zur Behandlung der Glykogenose Typ Ib wird zusätzlich rekombinanter humaner Granulozyten-colony-stimulating-Faktor (G-CSF) eingesetzt, wodurch Frequenz und Schweregrad der Infektionen günstig beeinflusst werden.
Verlauf, Prognose
Bei frühzeitiger und konsequenter Therapie ist die geistige Entwicklung normal. Trotz adäquater Stoffwechselkontrolle kann es bei einem Teil der Betroffenen mittelfristig zu Komplikationen kommen, meistens in der 2. oder 3. Lebensdekade in Form von Leberadenomen, Osteoporose und Beeinträchtigung der Nierenfunktion bis zur Niereninsuffizienz. Patienten mit Glykogenose Typ Ib entwickeln häufig eine dem Morbus Crohn ähnliche Darmerkrankung. Berichte über eine maligne Entartung der Leberadenome unterstreichen die Notwendigkeit regelmäßiger Kontrollen.
Glykogenose Typ II (Morbus Pompe)
Im Gegensatz zu den übrigen Glykogenosen kommt es bei der Glykogenose Typ II durch den Mangel an α-1,4-Glukosidase (saure Maltase) zu einer intralysosomalen Speicherung von Glykogen.
Klinik
Klinisch sind 3 Verlaufsformen bekannt, die Übergänge sind fließend. Der klassische Morbus Pompe (infantile Form) zeichnet sich durch Trinkschwäche, muskuläre Hypotonie („floppy infant“), Wachstumsverzögerung und rasch progrediente hypertrophe Kardiomyopathie aus. Die juvenile Form und die adulte Form der Glykogenose Typ II sind in der Regel auf die Skelettmuskulatur begrenzt und unterscheiden sich durch den Schweregrad der Beeinträchtigung und den zeitlichen Verlauf.
Diagnose
Zum Screening eignet sich der Nachweis von vakuolisierten Lymphozyten in einem Blutausstrich. Die Bestimmung der Enzymaktivität kann sowohl in Fibroblasten als auch im Muskelgewebe durchgeführt werden. Molekulargenetische Untersuchungen sind möglich. Zur pränatalen Diagnostik sind Amnion- und Chorionzellen geeignet. Eine Korrelation zwischen Phänotyp und Genotyp bzw. Restaktivität des Enzyms ist nicht immer gegeben. In denselben Familien wurden sowohl infantile als auch juvenile bzw. adulte Verlaufsformen beschrieben.
Therapie
Eine Therapie war lange Zeit nur ansatzweise durch proteinreiche Ernährung bzw. hochdosierte Substitutionen mit Alanin möglich. Inzwischen wurde jedoch eine Enzymersatztherapie etabliert.
Prognose
Die Prognose der infantilen Form ist ohne Enzymersatztherapie infaust. Die Kinder sterben zumeist im 1. Lebensjahr an Herzversagen. Juvenile und adulte Formen haben eine bessere Prognose. Morbidität und Mortalität wird bei ihnen v. a. durch die Beteiligung der Atemmuskulatur bestimmt.
Glykogenosen Typ III (Morbus Cori)
Die Glykogenosen Typ III werden durch einen Mangel von Amylo-1,6-Glukosidase verursacht. Das Debrancherenzym besitzt 2 katalytische Zentren, die der Translokation von 1,6–1,4-glukosidisch verbundenen Glukosemolekülen und der anschließenden Hydrolyse der α1,6-glukosidischen Verbindungen (Verzweigpunkte) dienen. Klinisch und biochemisch unterscheiden sich ein häufigerer Typ IIIa mit Leber-und Muskelbeteiligung und ein seltener Typ IIIb mit ausschließlicher Leberbeteiligung.
Klinik
Leitsymptome sind Hepatomegalie, ketotische Hypoglykämie, Hyperlipidämie und Transaminasenerhöhung (Abb. 3.7). Die laborchemischen Veränderungen und die Neigung zu Hypoglykämien sind weniger ausgeprägt als bei der Glykogenose Typ I. Beim Typ IIIa können zusätzlich eine Kardiomyopathie, Muskelschwäche und Muskelschwund auftreten, häufig längere Zeit ohne klinische Symptome. Einziger Hinweis ist eine Erhöhung der CK im Plasma. Die muskulären Symptome werden in der Regel nach der Pubertät krankheitsbestimmend. Die Enzymaktivität lässt sich in unterschiedlichen Geweben (Leber, Muskel, Fibroblasten) bestimmen, am einfachsten in Erythrozyten. Molekulargenetische Mutationsnachweise sind möglich.

Therapie
Therapeutisch kommen wie bei Glykogenose Typ I nächtliche Glukosepolymerinfusionen und ungekochte Maisstärke zum Einsatz. Zusätzlich wird bei muskulärer Beteiligung eine proteinreiche Diät empfohlen. Die Behandlung muss weniger strikt als bei der Glykogenose Typ I durchgeführt werden. Die Prognose ist individuell verschieden von sehr leichten bis zu schweren myopathischen Verlaufsformen.
Glykogenose Typ V (Morbus McArdle)
Ursache der Glykogenose Typ V ist ein Defekt der Muskelphosphorylase.
Klinik
Zu den typischen Symptomen zählen mangelnde Ausdauer, Muskelschwäche und -krämpfe, die bei kurz anhaltender intensiver oder in der Intensität steigender körperlicher Belastung auftreten. Ein sog. Second-wind-Phänomen mit Verbesserung der Symptomatik unmittelbar nach kurzer intensiver Anstrengung ist typisch. Bei betroffenen Erwachsenen kann eine Myoglobinurie auftreten. Gefürchtete Komplikation ist eine Rhabdomyolyse.
Diagnose
Zur Diagnose kann ein ischämischer Belastungstest durchgeführt werden, mit physiologischem Anstieg des Blutammoniaks aber fehlendem Anstieg des Blutlaktats. CK und LDH sind im Serum erhöht. Die Bestimmung der Enzymaktivität erfolgt im Muskelgewebe. Mutationsanalysen sind möglich.
Der besondere Fall
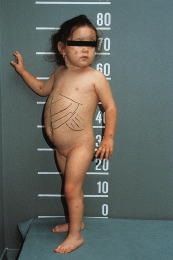
Anamnese. Ein bisher gesunder 6 Monate alter Säugling wird mit einem generalisierten afebrilen zerebralen Krampfanfall in die Klinik eingeliefert. Aus den vergangenen 6 Wochen werden 2 ähnliche Episoden berichtet, die jeweils 1–2 min dauerten.
Befund. Bei der körperlichen Untersuchung fällt ein ausladendes Abdomen mit deutlicher Hepatomegalie (8 cm unter dem Rippenbogen) ohne Splenomegalie auf (Abb. 3.6).
Bei den Laboruntersuchungen zeigt sich eine deutlich erniedrigte Blutglukose (0,6 mmol/l). Abnorm hohe Werte werden für folgende Laborparameter erhoben: Laktat 4,7 mmol/l, Triglyzeride 10,5 mmol/l, Cholesterol 6,1 mmol/l bei leicht erhöhten Transaminasen. Wiederholt durchgeführte Differenzialblutbilder sind unauffällig. Im Ultraschall des Abdomens fällt eine beidseitige Nephromegalie auf.
Verlauf. Diesmal sistierte der Krampfanfall erst nach intravenöser Glukosegabe. Im Verlauf zeigte sich eine stark erniedrigte Fastentoleranz von nur 3 h. Im Rahmen einer Glukosebelastung kam es zu einem Abfall der Blutlaktatwerte. Der Befund eines stark erhöhten Gehalts an Glykogen und einer deutlich erniedrigten Enzymaktivität der Glukose-6-Phosphatase nach durchgeführter perkutaner Leberbiopsie bestätigte den Verdacht auf das Vorliegen einer Glykogenose Typ Ia. Diese Diagnose wurde im weiteren Verlauf auch molekulargenetisch bestätigt.
Therapie. Zur Vermeidung von Hypoglykämien werden häufige Mahlzeiten mit langsam resorbierbaren Kohlenhydraten gegeben. Nachts erfolgt die Glukosegabe kontinuierlich über eine Nahrungspumpe. In der Diät soll die Gabe von Galaktose und Fruktose möglichst vermieden werden. Die Entlassung erfolgte nach knapp 2 Wochen mit Ausstellung eines Notfallausweises und regelmäßiger Anbindung an ein Stoffwechselzentrum.
Beurteilung. Typische klinische und biochemische Manifestation einer Glykogenose Typ Ia. Wichtig ist die Differenzierung zwischen Typ Ia (normale Neutrophilenzahl) und Typ I non-a (Neutropenie).
Glykogenosen Typ IX
Ätiologie, Genetik
Ursache der relativ häufigen Glykogenosen Typ IX sind unterschiedliche Defekte des Phosphorylase/-kinase-Komplexes, der aus 4 unterschiedlichen Proteinen besteht. Der Defekt der α-Untereinheit in der Leber wird X-chromosomal vererbt, 2 weitere Untereinheiten autosomal-rezessiv.
Klinik
Die klinische Symptomatik ist in aller Regel mild. Leitsymptom ist eine Hepatomegalie und ein Kleinwuchs. Eine Muskelbeteiligung und Hypoglykämien können auftreten, sind aber selten und relativ mild. Mit zunehmendem Alter bilden sich die Hepatomegalie und die Hypoglykämieneigung zurück.
Diagnose
Die Diagnose wird durch Nachweis des Enzymmangels im betroffenen Gewebe gestellt (Erythrozyten oder Leukozyten), zumeist wird primär eine Mutationsanalytik durchgeführt.
Therapie
Die Therapie ist bei guter Prognose in der Regel symptomatisch; die meisten Patienten benötigen keine Behandlung.
Aminoazidopathien
Enzymdefekte in der Umwandlung und im Abbau von Aminosäuren (Bausteine der Proteine) verursachen einen Anstau von Stoffwechselzwischenprodukten, die meist neurotoxisch oder auch hepatotoxisch wirken.
Klinik
Die klinische Symptomatik wird bestimmt durch Ausmaß und Dauer der Proteinzufuhr bzw. des endogenen Proteinabbaus (im Rahmen eines Gewebskatabolismus, z. B. bei Operationen, interkurrenten Infekten, Nahrungsverweigerung, Erbrechen oder auch Eiweißexzessen), durch den Schweregrad des Enzymdefekts und die spezifische Toxizität der Metabolite.
Therapie
Eckpfeiler der Behandlung von Störungen des Aminosäurenstoffwechsels sind spezielle Diäten.
Die Zufuhr von nicht oder nicht ausreichend abbaubaren und daher toxischen Aminosäuren wird auf ein Minimum reduziert, ohne dass eine katabole Stoffwechsellage oder eine Mangelernährung auftreten.
Prinzipien der Diätbehandlung
Die Ernährungsbehandlung von Aminoazidopathien basiert auf folgenden Prinzipien:
Verzicht auf eiweißreiche Nahrungsmittel und begrenzte Aufnahme eiweißarmer Nahrungsmittel,
Zufuhr einer mit Vitaminen, Mineralien und Spurenelementen angereicherten semisynthetischen Mischung inkl. der nicht im Abbau gestörten Aminosäuren,
ausreichende Deckung des Energiebedarfs durch eiweißarme Spezialnahrungsmittel sowie reine Fette und Kohlenhydrate,
häufige Kontrollen der Spiegel der betroffenen Aminosäure(n).
Die Kunst der Diätbehandlung besteht darin, unter Berücksichtigung der 3 erstgenannten Komponenten eine abwechslungsreiche und schmackhafte Kost zusammenzustellen, die den Nahrungsbedarf deckt. Eine entscheidende Rolle spielt die richtige Auswahl der Nahrungsmittel. Fleisch, Geflügel, Fisch, Wurst, Milch, Milchprodukte, Getreide und Getreideprodukte, Hülsenfrüchte, Nüsse und Kakao enthalten viel Eiweiß und sind deshalb ungeeignet. Obst und viele Gemüsearten enthalten relativ wenig Eiweiß und sind die wesentliche Quelle natürlichen Proteins. Zu den aus dem normalen Warenangebot entnehmbaren Nahrungsmitteln kommen spezielle eiweißarme Produkte, z. B. Spezialmehl, Brot, Gebäck und Teigwaren, die aus Stärke hergestellt werden (Abb. 3.8).


Überwachung unter Diätbehandlung
Unter der Dauerbehandlung mit der Spezialdiät sind neben regelmäßiger Gedeihkontrolle (Gewicht, Größe, Kopfumfang, Entwicklung) u. a. folgende Laborkontrollen erforderlich: quantitative Bestimmung aller Aminosäuren im Plasma (speziell der eingeschränkten sowie der essenziellen Aminosäuren), Blutbild, Kalzium, Phosphat, Magnesium, Eisen, Transaminasen, alkalische Phosphatase, Gesamteiweiß, Albumin und Präalbumin.
Notfallmaßnahmen
Eiweißexzesse oder interkurrente Erkrankungen (Infekte, Impfungen, Unfälle, Operationen, etc.) mit verminderter Nahrungszufuhr und Abbau des körpereigenen Eiweißes für die Glukoneogenese führen zu einem raschen Anstieg toxischer Metabolite. Bei Erkrankungen mit akuter Toxizität können Patienten innerhalb kürzester Zeit schwerste zerebrale Schädigungen erleiden oder versterben, z. B. bei der Ahornsiruperkrankung.
Entscheidend sind konsequent und zuverlässig durchgeführte Notfallmaßnahmen schon im Frühstadium von Entgleisungen.
Eckpfeiler der Notfallbehandlung sind
die Vermeidung bzw. rasche Umkehrung einer katabolen Stoffwechsellage durch ausreichende Zufuhr von Flüssigkeit, Elektrolyten und Energie (Glukose, Fette) und
die konsequente Fortführung der spezifischen oralen Medikation (z. B. Vitamine, Kofaktoren).
Da Entgleisungen meist zu Hause beginnen, müssen betroffene Familien ausführlich geschult werden, um adäquat reagieren zu können. Die Patienten sollten einen Notfallausweis bzw. -medaillon mit den wichtigsten Erstinformationen und Telefonnummern sowie Angaben über die ersten unverzüglich durchzuführenden Maßnahmen bei sich tragen. Bei Operationen müssen besondere Vorsichtsmaßnahmen getroffen werden. Die spezifische Notfalltherapie muss lebenslang beachtet werden.
Für Erkrankungen, bei denen die Schädigungen auf einer kumulativen chronischen Toxizität beruhen, wie z. B. bei der Phenylketonurie, gelten die gleichen Prinzipien zur Pathophysiologie von Metabolitenerhöhungen; die Notfallmaßnahmen beschränken sich jedoch auf ambulante orale Anpassungen der Therapie.
Phenylketonurie und Hyperphenylalaninämien
Definition
Störung des Aminosäurenstoffwechsels, verursacht durch einen Mangel des hepatischen Enzyms Phenylalaninhydroxylase.
Epidemiologie
Die Phenylketonurie (PKU) ist die häufigste Störung des Aminosäurenstoffwechsels in Mitteleuropa (Inzidenz in Deutschland ca. 1:10.000).
Pathogenese
Infolge des erhöhten Phenylalanins werden alternative Stoffwechselwege aktiviert (Abb. 3.9). Es entstehen eine Vielzahl bei Stoffwechselgesunden nicht vorkommender phenolischer Säuren, u. a. die Phenylessigsäure, die einen „mäuseartigen“ Körpergeruch verursacht. Da diese Stoffwechselwege weniger effektiv arbeiten als die Phenylalaninhydroxylase, bleiben die Spiegel von Phenylalanin hoch (Normbereich für Säuglinge im Plasma <100 µmol/l ≈ 1,7 mg/dl).


Klinik
Bei unbehandelten Patienten mit PKU führen die erhöhten Phenylalaninkonzentrationen zu einer irreversiblen Schädigung des sich entwickelnden Gehirns, insbesondere zu einer Störung der geistigen Entwicklung. Im Säuglingsalter manifestiert sich bei ca. 1/3 der betroffenen Kinder eine schwere Entwicklungsstörung, eine epileptische Enzephalopathie (generalisierte und/oder BNS-Anfälle), die in eine Grand-mal-Epilepsie übergeht. Im Weiteren entwickeln sich Mikrozephalie, extrapyramidale Symptome, psychotische Störungen, häufig mit Episoden von Erregung und Depression, Hyperaktivität, Destruktivität und Autoaggressionen bis zu Selbstverstümmelungen, ekzematoide, stark juckende Dermatitiden und Pigmentarmut der Haut und Haare. Einige Patienten erleiden im Erwachsenenalter zusätzliche neurologische Schäden mit Lähmungen und Pyramidenbahnläsionen. Die Lebenserwartung ist nicht wesentlich eingeschränkt.
Schwere geistige Behinderung, Epilepsie und psychotische Störungen durch PKU sind bei Kindern und jüngeren Erwachsenen durch Neugeborenenscreening und frühzeitige Diättherapie fast unbekannt, können aber bei Patienten vorliegen, die nicht erfasst wurden (z. B. Flüchtlingskindern, Kindern aus der Türkei, arabischen Ländern, deutschen Kindern, die im Ausland geboren wurden oder bei fehlerhaftem Neugeborenenscreening).
Diagnose
Die PKU wird im Neugeborenenscreening erfasst, die Diagnose wird durch Bestimmung der Aminosäuren im Plasma (erhöhtes Phenylalanin und erniedrigtes Tyrosin) bestätigt.
Verschiedene Schweregrade sind durch unterschiedlich schwere Mutationen (variable Restaktivitäten des Enzyms) erklärlich. Unterhalb von 600 µmol/l (10 mg/dl) besteht eine persistierende Hyperphenylalaninämie (milde Hyperphenylalaninämie), die nicht diätpflichtig ist. Eine Mutationsanalyse kann diagnostisch hilfreich sein und Hinweise auf den zu erwartenden Schweregrad der PKU bzw. Hyperphenylalaninämie geben.
Differenzialdiagnose
Von genetischen Defekten der Phenylalaninhydroxylase müssen vor Beginn einer diätetischen Therapie sekundäre, teilweise vorübergehende Erhöhungen des Phenylalaninspiegels und v. a. genetische Defekte in der Synthese oder der Regenerierung von Tetrahydrobiopterin, dem Kofaktor der Phenylalaninhydroxylase, abgegrenzt werden. Dazu müssen bei Neugeborenen mit erhöhten Phenylalaninwerten im Neugeborenenscreening die Pterine sowie die Aktivität der Dihydropteridinreduktase im Guthrie-Kärtchen bestimmt werden.
Differenzialdiagnose der Hyperphenylalaninämien
- Genetische Defekte der Umwandlung von Phenylalanin zu Tyrosin
- Genetische Defekte der Phenylalaninhydroxylase:
- Klassische PKU (Plasmaphenylalanin >1200 µmol/l)
- Milde PKU (Plasmaphenylalanina 600 µmol/l–1200 µmol/l)
- Milde Hyperphenylalaninämie (Plasmaphenylalanina 180 µmol/l–600 µmol/l)
- Genetische Defekte der Tetrahydrobiopterinbildung (BH4-Kofaktor)
- Sekundäre Phenylalaninerhöhungen
- Tyrosinämien
- Frühgeburtlichkeit
- Leber- oder Nierenversagen
- Einnahme von Trimethoprim
- Zytostatikatherapie
a unter altersentsprechender Ernährung
Therapie
Die Phenylalaninspiegel sollten bei Patienten mit behandlungsbedürftigen Hyperphenylalaninämien in den ersten 10 Lebensjahren zwischen 0,7 und 4 mg/dl liegen, zwischen dem 10. und 16. Lebensjahr zwischen 0,7 und 15 mg/dl, danach zwischen 0,7 und 20 mg/dl. In Einzelfällen können bei älteren Patienten psychopathologische (selten neurologische) Alterationen eine strikte diätetische Behandlung erforderlich machen. Neurologisch vorgeschädigte Patienten (etwa nach verspäteter Diagnosestellung) profitieren im späteren Lebensalter oft noch deutlich von einer Ernährungsbehandlung.
Verlauf, Prognose
Eine konsequente Ernährungsbehandlung mit Phenylalaninspiegeln im Zielbereich ermöglicht eine weitgehend normale psychomotorische und intellektuelle Entwicklung.
Maternale Phenylketonurie
In den letzten Jahren wird bei Kindern frühbehandelter und normal entwickelter Frauen mit PKU, die inzwischen das gebärfähige Alter erreicht haben, vermehrt ein neues Krankheitsbild beobachtet: die maternale PKU.
Klinik
Die erhöhten Phenylalaninspiegel einer Mutter mit PKU wirken sowohl embryo- als auch fetotoxisch (Tab. 3.5). Das Krankheitsbild ähnelt der Alkoholembryopathie. Schwangerschaftskomplikationen wie intrauterine Dystrophie, erhöhte Abortraten und Totgeburten treten gehäuft auf. Betroffene Kinder entwickeln eine geistige Behinderung, eine Mikrozephalie, einen Minderwuchs und innere und äußere Fehlbildungen, insbesondere Herzfehler. Weitere, im Zusammenhang mit maternaler PKU beobachtete Fehlbildungen sind Katarakte, Meningomyelozelen, Gaumenspalten (Pierre-Robin-Sequenz), Ösophagusatresien, intestinale Malrotationen, Hiatushernien, Syndaktylien und Hämangiome. Das Ausmaß der geistigen Behinderung reicht von schweren Intelligenzdefekten bis zu einer erhöhten Inzidenz eines hyperkinetischen Syndroms und korreliert mit der kumulativen Erhöhung des mütterlichen Phenylalaninspiegels in der Schwangerschaft.
| Mütterlicher Phenylalaninspiegel >1200 μmol | Normalbevölkerung | |
|---|---|---|
| Geistige Behinderung in % | 92 | 5,0 |
| Mikrozephalie in % | 73 | 4,8 |
| Intrauterine Dystrophie in % (Geburtsgewicht <2500 g) | 40 | 9,6 |
| Herzfehler in % | 12 | 0,8 |
Die kindlichen Schäden bei mütterlicher Phenylketonurie können nur durch das Einhalten einer sehr strengen Diät schon vor der Empfängnis und über die gesamte Schwangerschaft hindurch vermieden werden, d. h. es müssen geplante Schwangerschaften angestrebt werden.
Defekte der Tetrahydrobiopterinbildung
Tetrahydrobiopterin wird nicht nur für die Funktion der Phenylalaninhydroxylase sondern auch für die zweier weiterer Enzymsysteme in der Biosynthese der Neurotransmitter Dopamin und Serotonin benötigt (Tyrosin- bzw. Tryptophanhydroxylase). Die schwere klinische Symptomatik (infantiles Parkinson-Syndrom, Dystonien, Dyskinesien, Myoklonien, therapieresistente Epilepsie ab dem frühen Säuglingsalter) wird weniger durch die oft nur mäßig stark erhöhten Phenylalaninspiegel, sondern durch den Neurotransmittermangel im ZNS hervorgerufen.
Defekte der Tetrahydrobiopterinbildung liegen in Deutschland ca. 2%, in einzelnen Regionen der Türkei und Arabien in bis zu 30% den genetischen bedingten Hyperphenylalaninämien zugrunde.
Therapie
Die Therapie von Defekten der Tetrahydrobiopterinbildung erfordert neben einer Supplementation mit synthetischem Tetrahydrobiopterin oder einer phenylalaninarmen Diät die Gabe von L-Dopa zusammen mit einem Dekarboxylasehemmstoff (Carbidopa) in Kombination mit 5-Hydroxytryptophan zur Supplementierung der Dopamin- und Serotoninsynthese. Die Therapie muss lebenslang eingehalten und über Bestimmungen von Phenylalanin sowie der Neurotransmittermetabolite Homovanillinsäure und 5-Hydroxyindolessigsäure im Liquor gesteuert werden. Bei frühzeitigem Therapiebeginn ist eine gute Entwicklung zu erreichen.
Tyrosinämie Typ I
Grundlagen
Die Tyrosinämie Typ I wird durch einen autosomal-rezessiv vererbten Defekt der Fumarylazetoazetase verursacht, welche am Ende des Abbauwegs von Phenylalanin und Tyrosin die Spaltung von Fumarylazetoacetat in Fumarat und Azetoazetat katalysiert (Abb. 3.9). Es entstehen die hochreaktiven und toxischen Metabolite Fumarylacetoacetat, Maleylazetoazetat, Sukzinylacetoazetat und Sukzinylazeton, welche intrazellulär mit Makromolekülen und Glutathion reagieren sowie die Porphobilinogensynthese hemmen. Die Prävalenz der Tyrosinämie Typ I liegt bei etwa 1:120.000.
Klinik
Die toxischen Metabolite führen zu einem Leberversagen in der Säuglingszeit oder zu einer protrahierteren Hepatopathie mit zirrhotischem Umbau und zu Hepatomen. Häufig entstehen schon im Kindesalter hepatozelluläre Karzinome. Nierenfunktionsstörungen manifestieren sich in einer hypophosphatämischen Rachitis und können bis zum Nierenversagen fortschreiten. Eine erhebliche Morbidität und Mortalität resultiert aus einer peripheren Neuropathie sowie neurologischen Krisen, entsprechend einer akuten Porphyrie infolge der Hemmung der Porphobilinogensynthese.
Diagnose
Der Nachweis von Sukzinylazeton in der Analytik der organischen Säuren beweist das Vorliegen einer Tyrosinämie Typ I. Spezifisch sind ferner Erhöhungen von 5-Aminolävulinsäure im Urin infolge der gehemmten Porphobilinogensynthese. Erhöht finden sich auch die Aminosäuren Tyrosin, Methionin, in geringerem Ausmaße Phenylalanin sowie zahlreiche, über alternative Stoffwechselwege entstandene Metabolite. α-Fetoprotein ist zum Teil exorbitant erhöht. Die letztgenannten Veränderungen können auch bei anderen infektiösen oder genetischen Lebererkrankungen vorkommen. Die Mutationen sollten diagnostisch nachgewiesen oder der Enzymdefekt in Lymphozyten oder Fibroblasten bestätigt werden.
Therapie
Während früher die Leber- bzw. kombinierte Leber-Nieren-Transplantation die einzige Erfolg versprechende Therapieoption war, wurde Anfang der 1990er-Jahre mit 2-(2-Nitro-4-Trifluoromethylbenzoyl)-1,3-Zyklohexadion (NTBC) ein potenter Hemmstoff der 4-Hydroxyphenylpyruvatdioxygenase oberhalb der bei Tyrosinämie Typ I defekten Fumarylazetoazetase als neues Therapieprinzip entwickelt (Abb. 3.9). Unter der Behandlung mit NTBC steigen bei Patienten mit Tyrosinämie Typ I die Tyrosinspiegel noch weiter an, die Bildung der hochreaktiven und toxischen Metabolite Fumarylazetoazetat, Maleylazetoazetat, Sukzinylazetoazetat und Sukzinylazeton wird aber blockiert. Leber- und Nierenfunktion normalisieren sich langsam, ebenso die Porphobilinogensynthese. Erforderlich bleibt eine phenylalanin- und tyrosinarme Diät. Die Prognose hat sich unter dieser Behandlung entscheidend gebessert.
Ahornsirupkrankheit
Grundlagen
Bei der Ahornsirupkrankheit ist der mitochondriale Abbau von Leuzin, Isoleuzin und Valin auf der Stufe der gemeinsamen Oxidierung der durch reversible Transaminierung entstandenen α-Ketosäuren gestört. Die Prävalenz liegt bei etwa 1:150.000.
Klinik
Die Erkrankung verursacht eine progrediente Enzephalopathie, zunächst eine Ataxie, dann Anfälle, Somnolenz, Hirnödem und Koma. Erste Symptome, Lethargie und Trinkschwäche, treten zwischen dem 3. und 5. Lebenstag auf. Die Kinder verschlechtern sich sehr rasch. Oft fällt der typische Geruch von Ahornsirup bzw. Maggi auf. Kinder mit milderen Verlaufsformen infolge einer Restaktivität des Enzyms fallen später durch Entwicklungsverzögerung, neurologische Störungen und rezidivierende ketoazidotische Entgleisungen (Differenzialdiagnose: ketonämisches Erbrechen) auf.
Diagnose
Die Erkrankung ist seit 2005 als Zielkrankheit in das erweiterte Neugeborenenscreening integriert. Die Analyse der Aminosäuren im Plasma zeigt eine massive Erhöhung von Valin, Isoleuzin und Leuzin (deren Metabolit α-Ketoisokapronsäure ist besonders toxisch) sowie von L-Alloisoleuzin. Bei der Analyse der organischen Säuren im Urin finden sich neben den α-Ketosäuren auch die α-Hydroxysäuren erhöht.
Therapie
Akute schwere Entgleisungen sind nach Beginn des Neugeborenenscreenings auf diese Erkraknung selten geworden und erfordern zumeist eine intensivmedizinische Notfalltherapie mit Infusion von Glukose und Insulin, ggf. Dialyse. Die Langzeitbehandlung besteht aus einer eiweißarmen Ernährung mit Supplementierung eines Aminosäurengemisches ohne Leuzin, Isoleuzin und Valin. Die Therapiekontrolle erfolgt über die Leuzinkonzentration im Plasma. Wenige Patienten sprechen auf eine hochdosierte Gabe des Kofaktors Thiamin (5 mg/kgKG/Tag) an.
Die Prognose der Ahornsirupkrankheit ist nur bei rascher Diagnose (vor dem 5. Lebenstag) und konsequenter Therapie befriedigend.
Homozystinurie und Hyperhomozysteinämien
Der Homozysteinstoffwechsel umfasst 3 vitaminabhängige metabolische Sequenzen (Abb. 3.10):
Remethylierung zu Methionin,
Transmethylierung von Methionin zu Homozystein,
Transsulfurierung zu Zystein.


Da Homozystein sehr toxisch ist, wird es im Stoffwechsel rasch in Methionin zurückgeführt oder zu Zystein abgebaut. Alternativ kann Methionin mittels der hepatischen Betain-Homozystein-Methyltransferase (als CH3-Donor fungiert Betain) erneuert werden.
Genetische Defekte aller im Homozysteinstoffwechsel beteiligten Enzymsysteme können, ebenso wie nutritive Vitaminmangelzustände oder genetische Defekte in Aufnahme, Transport oder intrazellulärer Umsetzung der Kofaktoren den Stoffwechsel stören und zu einer Anhäufung von Homozystein führen. Daneben resultiert ein erhöhter Homozysteinspiegel aus einer gesteigerten Aktivität von Makrophagen, z. B. bei Infektionen.
Pathogenese
Erhöhte Homozysteinspiegel beschleunigen die Progredienz der Arteriosklerose und erhöhen das Risiko thromboembolischer Komplikationen. Das Risiko steigt mit dem Plasmaspiegel von Homozystein, ohne dass ein Schwellenwert nachweisbar wäre. Homozystein wurde ferner als Risikofaktor für die Entstehung von Neuralrohrdefekten identifiziert.
Homozystinurie
Die klassische Homozystinurie wird durch einen Defekt der Zystathionin-β-Synthetase verursacht. Dieser führt zu einer schweren Homozysteinämie, einer Homozystinurie sowie einer vermehrten Remethylierung von Homozystein zu Methionin (Abb. 3.10).
In einigen Ländern ist eine Methioninbestimmung zur Identifizierung von betroffenen Neugeborenen ins Neugeborenenscreening integriert (Großbritannien, Irland, Japan, Qatar, USA). Sie leidet allerdings unter einem hohen Anteil falsch-negativer Befunde, sodass die Inzidenz nicht bekannt ist und mindestens 1:100.000 beträgt.
Klinik
Exzessive Homozysteinerhöhungen induzieren zusätzlich zu schweren vorzeitigen Gefäßerkrankungen (Infarkte, Thrombosen, Embolien im Kindes- und Jugendlichenalter) auch Konformationsänderungen am Kollagen und anderen Strukturproteinen. Unbehandelt entwickelt sich bei den bei Geburt unauffälligen Patienten eine charakteristische Multisystemerkrankung:
ZNS: Psychomotorische Retardierung (ca. 80%), Anfälle, psychiatrische Symptome,
Augen: Ectopia lentis (≥90% zwischen dem 3. und 10. Lebensjahr) mit dem Frühsymptom einer rasch progredienten Myopie, Katarakte, Glaukom, Retinadegeneration,
Skelett: marfanoider Habitus (Hochwuchs, Kyphoskoliose, Beinfehlstellungen, Pes cavus), bikonkave Wirbelkörper, Osteoporose (gelegentlich mit pathologischen Frakturen),
arterielle Thromboembolien: Hirninfarkte, Herzinfarkte, periphere arterielle Embolien (häufigste Todesursache),
Beinvenenthrombosen.
Diagnose
Diagnostisch sind Plasmahomozysteinwerte zwischen 100 und 400 µmol/l (normal <12 µmol/l) bei erhöhten Methioninkonzentrationen. Die Enzymaktivität kann in Fibroblasten bzw. pathogene Mutationen aus Blutzellen nachgewiesen werden.
Therapie
Bei etwa 50% der Patienten führen pharmakologische Dosen von Pyridoxin (Vitamin B6) zu einer guten Reduktion des Homozysteinspiegels. Sinkt er nicht unter 50 µmol/l, muss eine strikte methioninarme Diät eingeführt werden. Eine zusätzliche Normalisierung lässt sich durch die alternative Remethylierung von Homozystein mittels Betain (bis zu 3-mal 3 g/Tag) erreichen (Abb. 3.10). Günstig ist eine zusätzlich Supplementierung mit Folsäure (5 mg/Tag) und ggf. von Vitamin B12. Eine lebenslange konsequente Therapie ist notwendig.
Prognose
Die Prognose hängt vom Zeitpunkt des Therapiebeginns sowie dem Ausmaß der Pyridoxinabhängigkeit ab. Sie ist bei einem Behandlungsbeginn in früher Kindheit befriedigend.
Milde Hyperhomozysteinämien
Eine milde Hyperhomozysteinämie ist ein erheblicher Risikofaktor für vorzeitige Gefäßerkrankungen (Infarkte, Thrombosen, Embolien) im 3. und 4. Lebensjahrzehnt (nicht im Kindesalter relevant) sowie für das Auftreten von Spina bifida etc. Sie wird durch verschiedene endogene und exogene Störungen des Folat- und Vitamin-B12-Stoffwechsels verursacht, z. B. nutritive Mangelzustände, oder auch eine Heterozygotie für den Zystathionin-β-Synthasemangel.
Im Gen der 5´10´-Methylentetrahydrofolatreduktase konnte ein häufiger Polymorphismus identifiziert werden (C677T), der als thermolabile Variante die Enzymaktivität beeinträchtigt. Im homozygoten Zustand (ca. 5% der Bevölkerung) führt dieser Polymorphismus zu hochnormalen bis grenzwertig erhöhten Spiegeln von Homozystein im Plasma, deutlicheren Erhöhungen von Homozystein nach Methioninbelastung sowie niedrigen Folsäurespiegeln.
Therapie
Die Behandlung milder Hyperhomozysteinämien ist wegen ihrer Häufigkeit von gesundheitspolitischer Bedeutung. Folsäure in einer Dosierung von 5 mg/Tag halbiert die Homozysteinspiegel. Über 90% werden durch eine kombinierte Therapie mit 5 mg Folsäure plus 100 mg Vitamin B6 pro Tag normalisiert.
Fest etabliert ist inzwischen die perikonzeptionelle Folsäuresupplementation bei allen Frauen mit 0,4 mg/Tag zur Prävention von Neuralrohrdefekten bzw. 4 mg/Tag als Wiederholungsprophylaxe.
Aminosäurentransportstörungen
Bei den Störungen des Aminosäurentransports werden partielle Defekte mit vermehrter renaler Ausscheidung einzelner oder mehrerer Aminosäuren (z. B. Zystinurie) von generalisierten Transportstörungen unterschieden.
Generalisierte Hyperaminoazidurien sind meist sekundäre Folge multisystemischer Stoffwechselerkrankungen, die zu tubulären Funktionsstörungen im Sinne eines De-Toni-Debré-Fanconi-Syndroms führen. Ursächlich sind dabei ein gestörter Energiestoffwechsel der Tubuluszellen oder tubuläre Ablagerung toxischer Substanzen. Neben einer generalisierten Hyperaminoazidurie ist das De-Toni-Debré-Fanconi-Syndrom gekennzeichnet durch Glukosurie, renalen Bikarbonatverlust und Hyperphosphaturie. Es entwickeln sich Polydipsie, Polyurie, Dehydratation, Hypokaliämie, metabolische Azidose und Vitamin-D-resistente Rachitis. Die Therapie besteht in der Entfernung pathologischer Metabolite (z. B. Galaktosämie) bzw. in einer Substitutionstherapie (z. B. Zystinose).
Stoffwechselerkrankungen als Ursache eines De-Toni- Debré-Fanconi-Syndroms
Zystinose
Hereditäre Fruktoseintoleranz
Tyrosinämie Typ I
Glykogenose Typ I
Fanconi-Bickel-Syndrom
Lowe-Syndrom
Mitochondriopathien
Morbus Wilson
Zystinurie
Die Zystinurie beruht auf einer autosomal-rezessiv vererbten Transportstörung von Zystin und den dibasischen Aminosäuren Ornithin, Lysin und Arginin an den Nierentubuli und am Dünndarmepithel. Die Aminosäurekonzentrationen im Blut sind unverändert.
Diagnose
Die Diagnose wird durch das charakteristische Aminosäurenmuster im Urin gestellt. Krankheitswert besitzt die Störung, wenn das schlecht lösliche Zystin auskristallisiert und in den Harnwegen Steine bildet (Urolithiasis, Nephrolithiasis).
Therapie
Die Therapie besteht bei großen Steinen in einer operativen Entfernung oder Lithotripsie. Bei kleineren Steinen kann eine Auflösung mit D-Penicillamin und Alkalisierung des Urins unter verstärkter Diurese versucht werden. Zur Steinprophylaxe empfiehlt sich eine konstant hohe Flüssigkeitszufuhr (auch nachts!), eine konsequente Alkalizufuhr (bei alkalischem pH-Wert ist die Löslichkeit von Zystin erhöht) sowie sulfhydrylgruppenhaltige Medikamente wie D-Penicillamin oder Mercaptopropionylglyzin, die mit Zystin ein besser lösliches Disulfid bilden.
Organoazidopathien
Grundlagen
Organoazidopathien sind genetisch bedingte, autosomal-rezessiv vererbte Stoffwechselstörungen, die sich weder hinsichtlich ihrer Ätiologie noch ihrer Pathogenese grundsätzlich von den Aminoazidopathien unterscheiden. Nachdem Aminosäuren in den späten 1940er-Jahren durch die Ninhydrinreaktion gut zu detektieren waren, ermöglichten in den 1970er und 1980er-Jahren gaschromatographisch-massenspektrometrische Methoden die Entdeckung einer Vielzahl weiterer Defekte, die entsprechend den nachzuweisenden Analyten, den organischen Säuren, als Organoazidopathien bezeichnet werden.
Epidemiologie
Die Häufigkeit der Organoazidopathien liegt in ihrer Gesamtheit bei ca. 1:6.000 Kindern.
Pathogenese
Durch verschiedene Enzym- und Koenzymdefekte ist zumeist der Abbau von Aminosäuren oder Fettsäuren gestört. Vor dem Block liegende und für jede Krankheit charakteristische Metabolite und/oder deren Folgeprodukte stauen sich an und stören empfindlich die Körperhomöostase. Häufig akkumulieren CoA-Derivate und hemmen mehrere zentrale mitochondriale Stoffwechselfunktionen:
Hemmung des Harnstoffzyklus → Hyperammonämie,
Hemmung der Glukoneogenese → Hypoglykämie und Laktatazidose,
Hemmung der Atmungskette → Laktatazidose, Ketose und gelegentlich Hyperurikämie,
Hemmung der Funktion des Nierentubulus → sekundärer Karnitinmangel.
Klinik
Bei vielen Organoazidopathien erfolgen die Schädigungen mit vielen Parallelen zu Mitochondriopathien aber auch zu exogenen Intoxikationen v. a. in energieabhängigen Geweben: Gehirn, Retina, Muskel, Herz, Leber, Niere. Viele Organoazidopathien entwicklen zusätzlich zu langsam progredienten Hirnfunktions- und Entwicklungsstörungen akute Stoffwechselentgleisungen. Letztere können durch Infektionen, Fieber, Impfungen, Verletzungen, Narkosen, Operationen und auch durch längere Nahrungskarenz oder Eiweißexzesse ausgelöst werden. Die Patienten können in kurzer Zeit schwerste zerebrale Schädigungen erleiden oder versterben. Konzeptionell können 3 Manifestationsformen unterschieden werden.
Akute neonatale Stoffwechselkrise
Das primär gesund erscheinende Neugeborene entwickelt wenige Tage nach Nahrungsaufnahme eine ausgeprägte Symptomatik mit Trinkschwäche, rezidivierendem Erbrechen, muskulärer Hypotonie, Myoklonien, Somnolenz und Koma infolge einer schweren metabolischen Enzephalopathie. Klinisch-chemisch findet sich oft eine metabolische Azidose, Laktatazidose, Hypoglykämie, Hyperammonämie und/oder Ketonurie.
Spätere oder intermittierende Manifestationsform
Die Patienten werden zumeist ab dem Kleinkindesalter auffällig durch häufiges Erbrechen, Entwicklungs- und Gedeihstörung, aber auch durch rezidivierende, schwere ketoazidotische Krisen bis hin zum Koma. Fast regelhaft resultiert eine progrediente psychomotorische Retardierung, häufig eine symptomatische Epilepsie.
Neurodegenerativer Krankheitsverlauf
Diese besondere Gruppe von Organoazidurien manifestiert sich ausschließlich mit charakteristischen und progredient verlaufenden neurologischen Symptomen wie Ataxie, Myoklonus, extrapyramidalen Störungen, Epilepsie, rezidivierenden „metabolischen“ Hirninsulten oder Makrozephalie. Akute Stoffwechselkrisen sind selten. Zumeist fehlen richtungweisende laborchemische Befunde wie Hypoglykämie, Laktatazidose oder metabolische Azidose.
Diagnose
Da organische Säuren renal effizient ausgeschieden werden, steht deren Analytik im Urin mittels Gaschromatographie-Massenspektrometrie an erster Stelle der Diagnostik. Das Metabolitenmuster erlaubt in den meisten Fällen eine sichere Diagnosestellung, die durch enzymatische und molekularbiologische Untersuchungen präzisiert werden sollte. Im erweiterten Neugeborenenscreening (Abschn. 3.1) können 80% der Patienten früh erfasst werden, sodass jetzt zunehmend Kinder betreut werden, die nicht von vornherein eine schwere Stoffwechselentgleisung mit potenziellen Folgeschäden durchleben mussten.
Pränataldiagnostik
Für fast alle Organoazidopathien sind zuverlässige Methoden der Pränataldiagnostik etabliert. Nach eingehender Familienuntersuchung können molekulargenetische Methoden eingesetzt werden. Die gezielte quantitative Bestimmung pathognomonisch erhöhter Metabolite aus der Amnionflüssigkeit mittels stabiler Isotope-Verdünnungstechniken ist ab der 11. Schwangerschaftswoche möglich. Bei vielen Enzymdefekten kann eine fehlende Aktivität in Amnionzellen und/oder Chorionzotten nachgewiesen werden. Voraussetzung ist in jedem Fall die exakte Diagnose des Indexpatienten.
Therapie
Da die meisten Organoazidopathien durch Enzymdefekte im Abbau von Aminosäuren bzw. Fettsäuren verursacht werden, gestaltet sich ihre Behandlung oftmals analog der Therapie der Aminoazidopathien und Harnstoffzyklusdefekte:
Karenz von Substanzen, deren Abbau gestört ist (zumeist spezifische Diätbehandlung),
Vermeidung einer katabolen Stoffwechsellage,
Herstellung und Aufrechterhaltung des Stoffwechselanabolismus,
spezifische Entgiftungsmaßnahmen,
Substitution von Vitaminen oder Kofaktoren, insbesondere L-Karnitin.
Alle empfohlenen Impfungen sollten konsequent durchgeführt werden, zusätzlich sollte auch gegen Pneumokokken, Varizellen und jährlich gegen Influenza geimpft werden.
Notfallbehandlung
Entscheidend für die Prognose sind der Zeitpunkt der Diagnosestellung und kompetent, rasch und zuverlässig durchgeführte Notfallmaßnahmen schon im Frühstadium interkurrenter Erkrankungen (Infekte, Impfungen, Operationen, etc.). Die Familien müssen ausführlich geschult werden, um die Therapie bereits zu Hause anpassen zu können.
Jeder Patient mit einer Organoazidopathie sollte einen Notfallausweis bzw. ein Notfallmedaillon mit den wichtigsten Erstinformationen und Telefonnummern bei sich tragen!
Dauert die interkurrente Erkrankung (insbesondere Erbrechen) an, muss der Patient sofort in der behandelnden Klinik vorgestellt und ggf. mit einer hochdosierten Zufuhr von Energie (Glukose, Fett), Flüssigkeit und Elektrolyten über eine Magensonde oder parenteral versorgt werden. Relevante laborchemische Parameter sind v. a. Blutzucker, Blutgasanalyse, Elektrolyte, Gerinnung, Laktat, Transaminasen und Ammoniak. Die spezifische Behandlung und Notfallmaßnahmen sind nicht auf das Kindesalter beschränkt und müssen lebenslang angewandt werden.
Propionazidurie
Grundlagen
Der Propionazidurie liegt ein Defekt des biotinabhängigen Enzyms Propionyl-CoA-Karboxylase zugrunde. Propionyl-CoA wird beim Abbau von Isoleuzin, Valin, Methionin, Threonin, ungeradzahligen Fettsäuren, der Cholesterolseitenketten und auch von Darmbakterien gebildet. Die Häufigkeit liegt bei ca. 1:200.000.
Klinik
Die Propionazidurie manifestiert sich zumeist nach wenigen Lebenstagen mit zunächst unspezifischen Symptomen wie Appetitlosigkeit, Trinkschwäche, rezidivierendem Erbrechen, Dehydratation, Gewichtsverlust und Muskelhypotonie. Rasch entwickelt sich eine ausgeprägte neurologische Symptomatik mit Dyspnoe, Somnolenz, Apathie, Krampfanfällen und Koma sowie eine schwere metabolische Azidose mit Hyperammonämie. Länger überlebende Patienten entwickeln häufig Dystonien, eine schwere Chorea und Läsionen des pyramidalen Systems.
Diagnose
Die Propionazidurie ist keine Zielkrankheit des erweiterten Neugeborenenscreenings. Die pathognomonischen Metabolite (u. a. Methylzitrat, 3-Hydroxypropionsäure) können im Urin nachgewiesen werden. Mutationen oder die Aktivität der Propionyl-CoA-Karboxylase werden in Leukozyten oder Fibroblasten bestimmt.
Therapie
Entscheidend für die Prognose sind neben dem Zeitpunkt der Diagnosestellung die Dauer und Qualität der Therapie der metabolischen Entgleisung zwischen Erstmanifestation und Diagnosestellung und eine Verhinderung bzw. zuverlässige rasche Behandlung späterer Stoffwechselentgleisungen.
Kontinuierlich erfolgen Karnitinsubstitution und Proteinrestriktion mit spezifischer Reduktion der Vorläuferaminosäuren Isoleuzin, Valin, Methionin und Threonin. Zur Hemmung der enteralen Bildung von Propionsäure kann intermittierend Metronidazol (z. B. Clont) eingesetzt werden.
Methylmalonazidurien
Pathogenese
Ursache dieser Gruppe von Erkrankungen sind Störungen der Methylmalonyl-CoA-Mutase, ein mitochondriales Vitamin B12-abhängiges Enzym. Vitamin B12 ist auch Kofaktor der Remethylierung des Homozysteins zum Methionin (Abb. 3.10). Entsprechend betreffen Defekte entweder die Methylmalonyl-CoA-Mutase direkt (mut0 ohne Restaktivität oder mut- mit Restaktivität) oder die Bereitstellung von Adenosylkobalamin aus Vitamin B12. Bei den Defekten im Kobalaminstoffwechsel kann wiederum ausschließlich der Methylmalonatstoffwechsel oder zusätzlich die Remethylierung des Homozysteins zum Methionin betroffen sein. Im letzteren Fall resultieren sowohl eine Akkumulation von Methylmalonsäure als auch von Homozystein.
Eine vegane Fehlernährung oder atrophische Gastritis einer stillenden Mutter kann im Säuglingsalter einen alimentären Vitamin-B12-Mangel verursachen und zu einer den genetischen Defekten vergleichbaren Gedeihstörung und neurodegenerativer Symptomatik, Methylmalonazidurie, Homozystinurie, Hyperhomozysteinämie mit Hypomethioninämie und megaloblastärer Anämie führen.
Epidemiologie
Die Inzidenz aller Methylmalonazidurien wird auf ca. 1:140.000 geschätzt.
Klinik
Entsprechend den heterogenen biochemischen Ursachen kommen unterschiedliche klinische Verlaufsformen vor. Foudroyante Verläufe ähneln der Propionazidurie (Abschn. 3.11.1) mit schweren ketoazidotischen Entgleisungen, Erbrechen, Dehydratation, muskulärer Hypotonie, Lethargie und Koma. Klinisch-chemisch bestehen eine metabolische Azidose, Hyperammonämie, Hypoglykämie und Leuko- bzw. Thrombozytopenie oder Anämie. Mildere Verläufe sind u. a. durch Entwicklungsretardierung, Gedeihstörung und progrediente neuropsychiatrische Probleme gekennzeichnet.
Diagnose
Die Methylmalonazidurien sind keine Zielkrankheit des erweiterten Neugeborenenscreenings. Im Urin können zumeist massiv erhöhte Ausscheidungen von Methylmalonsäure und Begleitmetaboliten (3-Hydroxypropionsäure, Methylzitrat) nachgewiesen werden. Im Plasma sind die Konzentrationen von Alanin und Glyzin erhöht und von Karnitin erniedrigt. Bei den kombinierten Defekten, die auch die Remethylierung des Homozysteins zum Methionin betreffen, bestehen zusätzlich eine Homozystinurie, Hyperhomozysteinämie mit Hypomethioninämie sowie eine megaloblastäre Anämie. Untersuchungen an kultivierten Fibroblasten (14C-Propionatfixation, Methylmalonyl-CoA-Mutasebestimmung, Komplementierungsanalysen) oder Mutationsanalysen sind für die endgültige Differenzierung erforderlich.
Therapie
Initial muss ein Behandlungsversuch mit Vitamin B12 (1–5 mg Hydroxycobalamininjektionen i.m. über mehrere Tage) durchgeführt werden; ansonsten erfolgen Notfall- und Dauerbehandlung analog zur Propionazidurie (Abschn. 3.11.1).
Verlauf, Prognose
Bei schwerer Stoffwechselstörung (fehlende Restaktivität bzw. fehlende Vitamin-B12-Abhängigkeit) entwickeln sich trotz frühzeitigem Therapiebeginn oftmals Komplikationen mit psychomotorischer Retardierung, extrapyramidale Bewegungsstörungen, Osteoporose und progressive Niereninsuffizienz.
Defekte des Biotinstoffwechsels (Biotinidasemangel und Holokarboxylasesynthetasemangel)
Pathogenese
Die Karboxylierungen von 3-Methylkrotonyl-CoA, Propionyl-CoA, Azetyl-CoA und Pyruvat sind biotinabhängig. Ein multipler Karboxylasemangel kann durch fehlende Aktivierung der Apoenzyme (Holokarboxylasesynthetasemangel), durch mangelnde Bereitstellung von Biotin aus Biozytin und proteingebundenem Biotin (Biotinidasemangel) sowie sehr selten durch erworbenen Biotinmangel (Darmsterilisation, Ernährung mit rohem Eiweiß) verursacht werden (Abb. 3.11).


Epidemiologie
Die Häufigkeit des schweren Biotinidasemangels (Restaktivität <10%) liegt bei etwa 1:30.000.
Klinik
Zeitpunkt und Ausprägung der Symptomatik ist abhängig von der Schwere der Enzymdefekte und äußeren Faktoren (z. B. Ernährung). Oft manifestiert sich der Holokarboxylasesynthetasemangel schon im Neugeborenenalter, ähnlich wie andere akute Organoazidopaxthien, mit Atemproblemen (Tachypnoe, Hyperventilation, Kussmaulsche Atmung), Erbrechen, Muskelhypotonie, Lethargie, Koma und Krämpfen.
Ein Biotinidasemangel wird zumeist erst im Verlaufe von Wochen bis Jahren klinisch relevant, im Mittel mit 3 Monaten. Oft dominieren unspezifische neurologische Symptome wie Muskelhypotonie, Lethargie, (myoklonische) Anfälle, Entwicklungsretardierung und Sprachstörungen. Später können sich Ataxie und ein irreversibler sensorineuraler Hörverlust, Optikusatrophie und Amaurose einstellen. Weniger als die Hälfte der Patienten entwickeln die charakteristischen Hautveränderungen und Alopezie (auch Verlust der Augenbrauen) sowie respiratorische Probleme (Hyperventilation, Stridor, Apnoe).
Diagnose
Laborchemisch bestehen während metabolischer Entgleisungen zusätzlich zur Azidose profunde Hyperammonämien sowie Laktatazidosen. Wechselnde Erhöhungen von Ammoniak und Laktat können auch im Intervall bestehen, insbesondere Laktaterhöhungen im Liquor.
Im akuten Stadium der Erkrankung sind zusätzlich zu den typischen Metaboliten der Propionazidurie (Störung der Propionyl-CoA-Karboxylase), Laktat (Störung der Pyruvatkarboxylase) und 3-Hydroxyisovaleriansäure sowie 3-Methylkrotonylglyzin (Störung der 3-Methylkrotonyl-CoA-Karboxylase) im Urin nachweisbar. Bei chronischem Verlauf ist die 3-Hydroxyisovaleriansäure der empfindlichste Marker.
Die Biotinidaseaktivität kann direkt in Guthrie-Karten bestimmt werden (diese Untersuchung ist im Neugeborenenscreening integriert). Der Holokarboxylasesynthetasemangel wird durch Mutationsnachweis oder Bestimmung der Enzymaktivitäten der einzelnen Karboxylasen in Fibroblasten oder Lymphozyten gesichert. Im Gegensatz zum Biotinidasedefekt ist der Holokarboxylasesynthetasemangel keine Zielkrankheit des erweiterten Neugeborenenscreenings.
Therapie
Tägliche orale Supplementation mit 5–10 mg Biotin. Bei frühzeitiger Diagnose und Therapiebeginn ist die Prognose sehr gut.
Glutarazidurie Typ I
Grundlagen
Die Glutarazidurie Typ I wird durch eine Störung im Abbau der Aminosäuren Lysin, Hydroxylysin und Tryptophan verursacht. Der zugrunde liegende Defekt, der Glutaryl-CoA-Dehydrogenasemangel, führt zum Anstau von Glutaryl-CoA und zu einer vermehrten Ausscheidung von Glutarsäure, Glutarylkarnitin, 3-Hydroxyglutarsäure und Glutakonsäure im Urin. Die Häufigkeit liegt bei ca. 1:130.000.
Klinik
Häufig zeigen die Kinder in den ersten Lebensmonaten eine geringe Muskelhypotonie, Irritabilität und Zittrigkeit (Abb. 3.12a). Bei den meisten besteht bei Geburt ein Makrozephalus, oder es entwickelt sich in den folgenden Monaten ein Kreuzen der Kopfumfangsperzentilen. In der Neuroradiologie zeigen sich frontotemporale Atrophien mit oder ohne subdurale Hygrome oder Hämatome und eine verzögerte Myelinisierung. Um den ersten Geburtstag herum manifestiert sich zumeist plötzlich im Rahmen eines Infekts eine schwere Hirnschädigung. Oft wird relativ isoliert das Striatum zerstört. Es entwickeln sich schwere Dyskinesien und Dystonien (Abb. 3.12b).


Diagnose
Nachweis der in allen Körperflüssigkeiten meist reichlich zu findenden Glutarsäure und 3-Hydroxyglutarsäure. Der Enzymdefekt kann in Fibroblasten oder Leukozyten nachgewiesen werden, Mutationen aus Vollblut. Die Glutarazidurie ist eine Zielkrankheit des erweiterten Neugeborenenscreenings.
Therapie
Rasche, effektive Behandlung von Stoffwechselentgleisungen, Karnitinsubstitution, lysinarme und tryptophanreduzierte Diät. Frühzeitig diagnostizierte und behandelte Kinder entwickeln sich meist normal, während sich das Vollbild der neurologischen Schädigung nach Striatumnekrose auch unter Behandlung nicht wesentlich bessert.
Defekte der Fettsäurenoxidation und Ketogenese
Grundlagen
Die langkettigen Fettsäuren (C16–C20) sind zentraler Bestandteil der Triglyzeride, die der Organismus im Fettgewebe speichert. Während längerer Fastenperioden werden den Organen durch die mitochondriale Fettsäurenoxidation Ketonkörper als Energieträger zur Verfügung gestellt. Relevante Krankheiten können durch Gendefekte 10 verschiedener Enzyme der Fettsäurenoxidation und Ketogenese sowie dreier Transportproteine verursacht werden (Tab. 3.6). Die Enzymdefekte verursachen einen Mangel an freiem CoA im Mitochondrium, und, je nach Lokalisation, eine Akkumulation toxischer Azylkarnitine. Alternativ wird ein Teil der sich anstauenden Fettsäuren zu Dikarbonsäuren oxidiert, die renal eliminert werden. Außer beim Karnitin-Palmitoyl-Transferase-I-Mangel führen alle Defekte zu einem generalisierten Karnitinmangel durch den Verlust pathologischer Azylkarnitine.
| Defekt | Charakteristische Befunde (zusätzlich zu hypoketotischen Hypoglykämien) |
|---|---|
| Karnitintransporter | Reye-Syndrom, Muskelschwäche, Kardiomyopathie, Koma, plötzlicher Tod |
| Karnitin-Palmitoyl-Transferase I | Hepatische Symptomatik (Reye-Syndrom, Hepatomegalie, Leberversagen), renale tubuläre Azidose |
| Karnitin/Azylkarnitin-Translokase | Anfälle, Koma, Leberversagen, schwere Kardiomyopathie, Arrhythmien |
| Karnitin-Palmitoyl-Transferase II |
Infantile Form: hepatische Symptomatik (Reye-Syndrom, Hepatomegalie, Leberversagen), Kardiomyopathie, Koma, schlechte Prognose Adulte Form: muskuläre Symptomatik (Belastungsintoleranz, Myalgien, Rhabdomyolysen) |
| Azyl-CoA-Dehydrogenase überlangkettiger Fettsäuren (VLCAD) |
Infantile Form: hepatische Symptomatik (Reye-Syndrom, Hepatomegalie, Leberversagen), Kardiomyopathie, Koma, schlechte Prognose Adulte Form: muskuläre Symptomatik (Belastungsintoleranz, Myalgien, Rhabdomyolysen) |
| Trifunktionelles Protein | Muskelschwäche, Kardiomyopathie, Koma, früher Tod |
| 3-Hydroxyazyl-CoA-Dehydrogenase langkettiger Fettsäuren (LCHAD) | Muskuläre Symptomatik (Belastungsintoleranz, Myalgien, Rhabdomyolysen), Kardiomyopathie, hepatische Symptomatik, Retinitis pigmentosa, Neuropathie, Koma, plötzlicher Tod |
| Azyl-CoA-Dehydrogenase mittelkettiger Fettsäuren (MCAD) | Reye-Syndrom, Muskelschwäche, Anfälle, Lethargie, Koma, plötzlicher Tod |
| Azyl-CoA-Dehydrogenase kurzkettiger Fettsäuren (SCAD) | Muskelschwäche, Lethargie, Dystrophie, psychomotorische Retardierung |
| Kurzkettige-Hydroxyazyl-CoA-Dehydrogenase (SCHAD) | Hyperinsulinämische Hypoglykämien |
| Multiple Azyl-CoA-Dehydrogenase (Glutarazidurie Typ II) |
Infantile Form: Reye-Syndrom, Erbrechen, Anfälle, Dysmorphien, renale Zysten, früher Tod Adulte Form (häufig riboflavinabhängig): Hepatopathie, Muskelschwäche, Lethargie, Dystrophie, psychomotorische Retardierung |
| 3-Hydroxy-3-methylglutaryl-(HMG-)CoA-Synthase | |
| 3-Hydroxy-3-methylglutaryl-(HMG-)CoA-Lyase | Hepatopathie, oft letal im Rahmen Reye-artiger Krise |
Klinik
Pathophysiologisch, klinisch und biochemisch überlappen sich die Krankheitsbilder (Tab. 3.6). Leitsymptom sind sich rasch entwickelnde hypoketotische Hypoglykämien (Abb. 3.3) mit akuter neurologischer Symptomatik bis zum Koma (Tab. 3.3). Ursache sind eine vermehrte Zufuhr exogener oder Mobilisation endogener Fettsäuren, z. B. bei verlängerter Nüchternphase, Unfällen, Operationen oder Infekten. Zusätzlich finden sich Zeichen von Leberfunktionsstörungen mit erhöhten Transaminasen und Hyperammonämie bis zum Leberausfall. Bei einigen Störungen (v. a. bei Defekten im Bereich der Oxidation langkettiger Fettsäuren) können sich Neuropathien und Retinopathien entwickeln. Muskuläre Symptome sind häufig, von einer relativ unspezifischen Muskelschwäche bis zur Rhabdomyolyse sowie akuten oder chronischen Kardiomyopathien und Arrhythmien. Histologische Untersuchungen zeigen eine Leberverfettung oder eine Lipidmyopathie.
Wegen der Häufigkeit in Mitteleuropa (1:10.200 im erweiterten Neugeborenenscreening) besonders hervorzuheben ist der mittelkettige Azyl-CoA-Dehydrogenase(MCAD)-Defekt. Er manifestiert sich klinisch meist zwischen dem 4. Lebensmonat und dem 3. Lebensjahr mit rezidivierenden hypoketotisch-hypoglykämischen Attacken, die einem Reye-Syndrom ähnlich sein können. Bis zu 25% der Kinder versterben während solcher foudroyant verlaufender metabolischer Krisen unerwartet, wobei in Einzelfällen die Diagnose nicht erkannt und z. B. als plötzlicher Kindstod fehlgedeutet werden kann. Andererseits kennt man Betroffene, die lebenslang asymptomatisch bleiben. In Deutschland findet sich bei über 85% der Patienten mit MCAD-Mangel die Mutation A985G, sodass bei entsprechendem klinischem und/oder biochemischem Verdacht auch eine primäre molekulare Diagnostik möglich ist.
Diagnose
Die wesentlichen Festtsäurenoxidationsdefekte sind Zielkrankheiten des erweiterten Neugeborenenscreenings. Am aussagekräftigsten ist die Labordiagnostik aus der akuten Entgleisung (Serum, Vollblut und Urin). Im Vordergrund steht die Hypoglykämie mit begleitender (Laktat)azidose. Auffällig ist die stark verminderte bis fehlende Ketonkörperbildung bei hohen Werten der freien Fettsäuren im Serum (Abb. 3.3). Hinweisend können ferner Erhöhungen von Transaminasen, Kreatinkinase und Ammoniak sowie eine Myoglobinurie sein.
In der spezialisierten Stoffwechselanalytik zeigen Defekte der Fettsäurenoxidation
eine teilweise exzessive Urinausscheidung pathologischer Dikarbonsäuren,
starke Erniedrigung des Gesamtkarnitins, insbesondere des freien Karnitins (Ausnahme Karnitin-Palmitoyl-Transferase-I-Mangel),
relative Erhöhungen der Azylkarnitine,
Nachweis spezifischer pathologischer Azylkarnitine.
Die Gesamtheit dieser Befunde kann die exakte Lokalisation eines zu vermutenden Fettsäurenoxidationsdefektes gut eingrenzen. Eine molekulare oder enzymologische Bestätigung ist allerdings unabdingbar.
Therapie
Alle Formen der Fettsäurenoxidationsdefekte müssen in der akuten Krise durch umgehende hochdosierte Glukosezufuhr behandelt werden. Vorbeugend sollten häufigere, kohlenhydratreiche und fettarme Mahlzeiten gegeben werden. Die Fastentoleranz muss individuell ermittelt werden. Karnitin wird regelhaft nur beim Karnitintransporterdefekt substituiert. Bei Defekten im Abbau langkettiger Fettsäuren werden mittelkettige Triglyzeride in der Ernährungstherapie eingesetzt, da sie unterhalb vom Enzymdefekt in die Fettsäurenoxidation eingeschleust werden. Defekte der kurzkettigen Azyl-CoA-Dehydrogenase und des Elektronenflusses in Richtung Atmungskette durch das Elektronentransferflavoprotein (ETF) sowie die ETF-Dehydrogenase (Synonym: multipler Azyl-CoA-Dehydrogenasemangel oder Glutarazidurie Typ II) können positiv auf pharmakologische Dosen von Riboflavin reagieren.
Der besondere Fall
Anamnese. Ein bisher gesunder 11 Monate alter Säugling erkrankt an einer Gastroenteritis mit heftigem Durchfall und Erbrechen. Er verweigert jegliche Flüssigkeitszufuhr. Innerhalb von wenigen Stunden wird er zunehmend lethargisch.
Befund. Beim Eintreffen in der Klinik präsentiert sich der Säugling in schwer reduziertem Allgemeinzustand mit blass-grauem Hautkolorit, einer deutlichen Tachydyspnoe (Atemfrequenz 100/min) und einer Hepatomegalie. Kurz darauf tritt ein zerebraler Krampfanfall auf, der nach intravenöser Glukosegabe sistiert. Bei den initialen Laborwerten fanden sich eine metabolische Azidose, erniedrigte Glukosewerte und eine erhöhte Harnsäure. Die Quantifizierung der organischen Säuren im Urin zeigte fehlende Ketonkörper bei einer massiv erhöhten Ausscheidung von Dikarbonsäuren.
Verlauf und Therapie. Unter intensivmedizinischen Maßnahmen einschließlich parenteraler Glukose- und Elektrolytzufuhr und mehrfachem Azidoseausgleich mit Natriumbikarbonat stabilisiert sich der klinische Zustand innerhalb von 2 Tagen. Durch den Urinbefund der erhöhten Ausscheidung von Dikarbonsäuren wurde der dringende Verdacht auf das Vorliegen eines MCAD-Mangels gestellt. Die molekulargenetische Bestätigung der Diagnose erfolgte durch homozygoten Nachweis der Punktmutation A985G.
Für den Säugling wurde ein Notfallausweis mit Verhaltensregeln für Infekte mit Erbrechen oder Nahrungsverweigerung ausgestellt. Er wurde nach einer Woche mit der Maßgabe entlassen, auf regelmäßige, häufige und kohlenhydratreiche sowie fettreduzierte Mahlzeiten ohne längere Pausen zu achten. Im Alter von 3 Jahren war noch einmal eine stationäre Behandlung mit intravenöser Glukosezufuhr bei Noro-Viren-Gastroenteritis erforderlich.
Beurteilung. Typische Präsentation eines MCAD-Mangels, der häufigsten Fettsäurenoxidationsstörung. Der foudroyante Verlauf wird durch Fasten oder eine interkurrente Erkrankung ausgelöst. Charakteristisch sind die hypoketotische Hypoglykämie und die Dikarbonazidurie. Da die erste Krise nicht letal und ohne Residualschäden verlief, ist die Prognose im geschilderten Fall bei Vermeiden von rezidivierenden Entgleisungen als sehr gut einzuschätzen.
Peroxisomale Erkrankungen
Peroxisomen sind kleine, von einer Membran umgebene Organellen. Sie spielen eine wichtige Rolle in der β-Oxidation von gesättigten unverzweigten überlangkettigen Fettsäuren und verwandten Substraten (u. a. zur Bereitstellung von Azetyl-CoA für Biosynthesen) sowie in der Synthese von Ätherlipiden (z. B. Plasmalogene) und Gallensäuren. Viele O2-abhängige Oxidationen laufen (zum Schutz der Zelle gegen Sauerstoffradikale) in den Peroxisomen ab, das entstehende H2O2 wird mittels einer Katalase abgebaut.
Störungen der Peroxisomenbildung und peroxisomalen β-Oxidation
Ein kombinierter Mangel peroxisomaler Enzyme wird durch Störungen der Peroxisomenbildung verursacht. Diese Krankheitsbilder werden autosomal-rezessiv vererbt und als generalisierte peroxisomale Erkrankungen bezeichnet. Alle peroxisomalen Enzymsysteme sind in unterschiedlichem Ausmaß gestört.
Klinik
Leitsymptome generalisierter peroxisomaler Erkrankungen sind beim Neugeborenen Dysmorphien (sehr große Fontanellen, niedrige Orbitalränder, hohe Stirn, Epikanthus, Vierfingerfurche, antevertierte Nares, dysplastische Ohren und Knorpelverkalkungen), schwere neurologische Störungen (Hypotonie, Epilepsie), und hepatointestinale Funktionsstörungen (Hepatomegalie, konjugierte Hyperbilirubinämie, Fütterungsschwierigkeiten). Im Kleinkindesalter kommen Gedeihstörungen, Osteoporose, Spastizität, sensoneuraler Hörverlust (Frühsymptom: pathologisches Elektroretinogramm) und Erblindung (Frühsymptom: Retinitis pigmentosa) hinzu.
Das Zusammenkommen von psychomotorischer Retardierung und Hypotonie mit nur einem weiteren Symptom, wie Epilepsie, kraniofaziale Dysmorphien, Seh- oder Hörstörung, sollte eine biochemische Untersuchung auf peroxisomale Erkrankungen veranlassen.
3 Krankheitsbilder
Das Vollbild der geschilderten Multisystemerkrankung wird als Zellweger-Syndrom bezeichnet. Die betroffenen Kinder versterben zumeist im 1. Lebensjahr. Der infantile M. Refsum verläuft protrahierter mit variabler Überlebenszeit. Patienten können sogar, wenn auch verspätet, das Gehen mit ataktisch breitbasigem Gang erlernen. Der Verlauf der neonatalen Adrenoleukodystrophie ist häufig rasch progredient mit fortschreitender Leukodystrophie und demyelinisierender peripherer Neuropathie. Zwischen den 3 Krankheitsbildern bestehen Überlappungen und Mischbilder.
Klinisch ähnlich verlaufen Einzelenzymdefekte der peroxisomalen β-Oxidation. Sie werden als Pseudo-Zellweger-Syndrom bezeichnet.
Diagnose
Hinweisend auf peroxisomale Erkrankungen sind ein Ikterus prolongatus, mäßige Hypoglykämien und Hypocholesterolämien sowie variable Erhöhungen von Transaminasen, Serumeisen und Transferrin. Entscheidend und spezifisch ist der Nachweis erhöhter überlangkettiger Fettsäuren (VLCFA: „very long chain fatty acids“). Diagnostisch bedeutsam ist ferner der Nachweis verminderter Plasmalogene im Plasma und Geweben (defekte Biosynthese) sowie einer erhöhten Phytansäure (defekter Abbau der aus der Nahrung stammenden Säure). Bei generalisierten peroxisomalen Erkrankungen kann ein Fehlen oder die starke Verminderung der Peroxisomen elektronenmikroskopisch in einer Leberbiopsie oder in kultivierten Fibroblasten nachgewiesen werden. Die Diagnose muss durch den Nachweis der spezifischen Enzymdefekte in Fibroblasten oder pathologischer Mutationen bestätigt werden.
Generalisierte peroxisomale Erkrankungen werden durch mehr als 12 unterschiedliche Einzelgendefekte verursacht. Bei 2/3 der Familien sind Mutationen im PEX1-Gen die Krankheitsursache, in einigen weiteren Familien Mutationen im PEX5-Gen. In diesen Familien ist nach Mutationsanalyse eine pränatale Diagnostik wesentlich erleichtert.
Therapie
Spezifische therapeutische Ansätze sind nicht vorhanden.
Peroxisomale Einzelenzymdefekte
Während einzelne isolierte Enzymdefekte der peroxisomalen β-Oxidation Phänokopien generalisierter peroxisomaler Erkrankungen verursachen (Abschn. 3.12.1), führen Defekte anderer peroxisomaler Enzyme zu spezifischen Krankheitsbildern, deren Symptome sich auf den Funktionsverlust des betroffenen Enzyms zurückführen lassen. Klinische Überschneidungen mit den generalisierten peroxisomalen Erkrankungen zeigen die rhizomele Chondrodysplasia punctata und die X-chromosomal vererbte Adrenoleukodystrophie.
Rhizomele Chondrodysplasia punctata
Klinik
Genetische Defekte der Rezeptoren des peroxisomalen Zielsignals 2 oder der spezifischen Enzyme Dihydroxyazetonphosphat-Azyltransferase und Alkyl-Dihydroxyazetonphosphat-Synthase verursachen das klinische Bild einer rhizomelen Chondrodysplasia punctata. Oberschenkel und Oberarme sind stark verkürzt (Abb. 3.13). Im Gegensatz zum Zellweger-Syndrom findet sich die Chondrodysplasia punctata (epiphysäre punktförmige Verkalkungen) nicht nur lokalisiert sondern generalisiert. Die gestörte enchondrale Knochenbildung führt zu koronaren Spalten an den Wirbelkörpern. Viele Patienten zeigen neben fazialen Dysmorphien eine Ichthiose und Katarakte. Das Ausmaß der geistigen Retardierung ist zumeist schwer. Es entwickeln sich Spastik und Kontrakturen.


Diagnose
Pathophysiologisch entscheidend ist eine gestörte Biosynthese der Plasmalogene, die diagnostisch am einfachsten in Erythrozytenmembranen nachzuweisen ist. Bei Rezeptordefekten des peroxisomalen Zielsignals 2 (Mutationen im PEX7-Gen), wodurch mehrere unterschiedliche Enzyme nicht ins Peroxisom integriert werden können, ist zusätzlich zur Biosynthese der Plasmalogene die Oxidation der Phytansäure defekt, sodass die Diagnosestellung auch über den Nachweis erhöhter Phytansäurespiegel im Serum erfolgen kann.
X-chromosomale Adrenoleukodystrophie
Die X-chromosomale Adrenoleukodystrophie beruht auf einem Defekt des peroxisomalen ATP-bindenden Membranproteins (ABCD1-Gen) und folgt als einzige peroxisomale Erkrankung einem X-chromosomalen Erbgang.
Klinik
Klinisch kann sich die Krankheit im Schulkindesalter (ca. 40%), oder in unterschiedlichen Varianten später in der Adoleszenz oder bei Erwachsenen manifestieren.
Klassischerweise zeigen betroffene Jungen im Schulalter zunächst Verhaltensstörungen, eine motorische Ungeschicklichkeit und kortikale Empfindungsstörungen. Eine gestörte Körperorientierung, Sehstörungen und epileptische Anfälle können ebenfalls Frühsymptome sein. Die Erkrankung verläuft dann rasch progredient. Die Kinder sind nach wenigen Jahren blind, taub und schließlich dezerebriert.
Es ist unklar, warum bei etwa 20% der Patienten die neurologische Symptomatik sehr viel später beginnt und milder verläuft (Adrenomyeloneuropathie). Im Erwachsenenalter fallen zuerst Steifheit und Ungeschicklichkeit beim Gehen auf, gefolgt von einer generalisierten Muskelschwäche, Gewichtsverlust, Schwindelanfällen und vermehrter Hautpigmentation. Letztere ist Folge erhöhter ACTH-Konzentrationen bei einer Nebenniereninsuffizienz (Addison-Erkrankung), einem weiteren Charakteristikum der Erkrankung. Während im Erwachsenenalter eine Nebenniereninsuffizienzneurologischen Symptomen viele Jahre vorausgehen kann und letztere völlig ausbleiben können, ist im Kindesalter ein isolierter Morbus Addison selten (7% aller Patienten). Dennoch sollte bei Jungen wie Männern mit Morbus Addison immer eine Bestimmung der überlangkettigen Fettsäuren im Serum erfolgen.
Diagnose
Diagnostischer Marker ist ein erhöhter Gehalt an überlangkettigen Fettsäuren in Geweben und Körperflüssigkeiten.
Therapie
Der diätetische Behandlungsansatz (1 Teil Glyzerylerucasäure plus 4 Teile Glyzeryltrioleat) ist als Lorenzos-Öl durch den gleichnamigen Film bekannt geworden. Umfangreiche Nachuntersuchungen haben leider keine Wirksamkeit nachweisen können, wohingegen eine frühe Knochenmarktransplantation Krankheitsverlauf und -schwere positiv beeinflussen kann.
Primäre Hyperoxalurie Typ I
Die primäre Hyperoxalurie Typ I wird durch einen Mangel der Alanin-Glyoxalat-Aminotransferase verursacht und führt über die stark vermehrte Urinausscheidung von Oxalsäure zu Nierensteinen, einer Nephrokalzinose und schließlich zu einem chronischen Nierenversagen.
Cholesterolbiosynthesedefekte
Defekte der Cholesterolbiosynthese verursachen monogene vererbte embryofetale Fehlbildungssyndrome.
Am Anfang des Stoffwechselweges verursacht der Mevalonatkinasemangel die Mevalonazidurie sowie das Hyper-IgD-Syndrom, am Ende u. a. Defekte der 3β-Hydroxysteroid-∆8,∆7-Isomerase das Conradi-Hünermann-Syndrom (X-gebundene Chondrodysplasia punctata), der Sterol-4-Demethylase das Child-Syndrom und der 3β-Hydroxysterol-∆7-Reduktase das Smith-Lemli-Opitz-Syndrom.
Klinisch manifestieren sich die Erkrankungen durch unterschiedliche kraniofaziale Dysmorphien, Organ- und Skelettfehlbildungen sowie schwere körperliche und pychomotorische Entwicklungsverzögerungen.
Smith-Lemli-Opitz-Syndrom
Am besten bekannt ist das Smith-Lemli-Opitz-Syndrom, 1964 von David W. Smith, Luc Lemli und John Marius Opitz erstbeschrieben und mit einer Inzidenz von 1:60.000.
Klinik
Klinisch fallen die Patienten mit Gedeihstörungen, psychomotorischer Retardierung, muskulärer Hypotonie und typischen fazialen Dysmorphien wie Mikrozephalie, Mikrognathie, Ptosis, Katarakte, antevertierte Nares, tiefsitzende, posterior rotierte Ohren sowie multiplen Fehlbildungen der Extremitäten und der inneren Organe auf (Abb. 3.14). Der Schweregrad der Organfehlbildungen und damit die Prognose der Patienten zeigt eine negative Korrelation zur Konzentration des Cholesterols im Serum. Das Spektrum reicht von milden Formen mit leichter Retardierung bis zu letalen Verläufen in den ersten Lebenstagen oder Totgeburten.

Das Hyper-IgD-Syndrom ist durch rezidivierende Fieberschübe charakterisiert.
Diagnose
Die Diagnostik der Erkrankungen erfordert bei der Mevalonazidurie die spezialisierte Analytik der Mevalonsäure im Urin, bei den anderen Erkrankungen die differenzierte quantitative Bestimmung der Sterole im Serum mittels Gaschromatographie-Massenspektrometrie oder molekulare Untersuchungen.
Cave
Erniedrigte Cholesterolkonzentrationen im Serum sind kein verlässlicher diagnostischer Parameter.
Bei der Mevalonazidurie ist eine pränatale Diagnostik über den Nachweis erhöhter Mevalonsäure in der Amnionflüssigkeit ab der 11. Schwangerschaftswoche, beim Smith-Lemli-Opitz-Syndrom über den Nachweis erhöhten 7-Dehydrocholesterols in Chorionzottenbiopsien ab der 10. Schwangerschaftswoche möglich. In informativen Familien können molekulargenetische Untersuchungen durchgeführt werden.
Therapie
Erfolgversprechende Therapieansätze beim Smith-Lemli-Opitz-Syndrom zielen auf die Erhöhung der abnorm niedrigen Cholesterolkonzentrationen durch Zufuhr von exogenem Cholesterol. Gleichzeitig sollen die erhöhten 7-Dehydrocholesterolspiegel durch Hemmung der endogenen Biosynthese durch Inhibitoren der HMG-CoA-Reduktase (z. B. Simvastatin) gesenkt werden. Pränatale Schädigungen zeigen allerdings naturgemäß keine Besserungstendenz.
CDG-Syndrome („congenital disorders of glycosylation“)
Grundlagen
CDG-Syndrome resultieren aus einer gestörten Glykosylierung, die fehlerhafte Strukturen von Kohlenhydratseitenketten unterschiedlicher Glykoproteine (Membranproteine, Transportproteine, Hormone, Gerinnungs- und Komplementfaktoren, Enzyme oder endogene Enzymhemmstoffe, wie z. B. α1-Antitrypsin) verursacht. Inzwischen sind über 50 unterschiedliche Gendefekte bekannt, die sich sowohl im Muster des als diagnostischen Marker verwandten Serumproteins Transferrin als auch in der klinischen Symptomatik unterscheiden.
PMM2-CDG (früher CDG-Syndrom Typ Ia) wurde bisher am häufigsten gefunden (>1000 Patienten). Die meisten Patienten sind bisher in Skandinavien mit einer Inzidenz von ca. 1:50.000 diagnostiziert worden. Der Erbgang ist autosomal-rezessiv. Die Krankheitszeichen sind geistige und körperliche Behinderung, Muskelschwäche, Ataxie, Schielen, Störung der Leberfunktion, Gedeihstörung, eingezogene Brustwarzen und eine auffällige Fettverteilung. Bei Routineblutuntersuchungen findet sich meist nur eine leichte Erhöhung der Leberwerte (Transaminasen). Bei spezielleren Untersuchungen findet sich darüber hinaus neben einem auffälligen Transferrinmuster (IEF) häufig ein Mangel an verschiedenen Glykoproteinen, wie Gerinnungsfaktoren (z. B. Faktor XI und AT III).
Diagnose
Die Diagnose wird mittels isoelektrischer Fokussierung von Transferrin als Markerprotein gestellt. Komplementär können auch andere Glykoproteine für die Diagnosestellung verwendet werden (z. B. α1-Antitrypsin, AT III, α1-saures Glykoprotein). Infolge der Hypoglykolisierung zeigen sich je nach CDG-Syndrom in der Elektrophorese unterschiedliche Muster der Proteinbanden. Eine Sicherung der Diagnose kann bei den bekannten Defekten in kultivierten Hautfibroblasten, z. T. in Leukozyten bzw. molekulargenetisch erfolgen.
Therapie
Bislang stehen effektive Therapien nur für das MPI-CDG-Syndrom (früher Typ Ib) durch eine regelmäßige orale Zufuhr von Mannose sowie für das SLC35C1-CDG durch orale Zufuhr von Fukose zur Verfügung. Für die anderen CDG-Syndrome, die v. a. mit schweren neurologischen Störungen einhergehen, existiert keine effektive rationale Therapie.
Lysosomale Speicherkrankheiten
Pathogenese
Lysosomen dienen dem Abbau von kleinen bis sehr großen Molekülen und Molekülverbänden. Dazu enthalten sie in einem sauren Milieu (pH 5) eine Vielzahl verschiedener Hydrolasen. Defekte lysosomaler Hydrolasen führen zum Anstau des unvollständig verdauten Substrats. Die Speicherung führt zur Vergrößerung der Lysosomen, die mikroskopisch als Vakuolen sichtbar werden (Abb. 3.15), zu einem Anschwellen der Zellen, Zellvermehrung und letztlich zu Funktionsstörungen. Betroffen sind v. a. mesenchymatöse Organe (Haut, Knorpel, Knochen), parenchymatöse Organe (Leber, Milz) und das Nervensystem.


Auch im Normalfall wird ein Teil der lysosomalen Enzyme als sekretorische Proteine aus den Zellen ausgeschleust, und können relativ einfach im Blut oder Urin nachgewiesen und quantifiziert werden.
Genetik
Die Mukopolysaccharidose Typ II (M. Hunter) und die Sphingolipidose M. Fabry werden X-chromosomal, alle anderen autosomal-rezessiv vererbt.
Klinik
Es sind etwa 50 lysosomale Speicherkrankheiten bekannt, mit einer Gesamtprävalenz von ca. 1:8.000. Sie zählen zu den häufigsten neurometabolischen Erkrankungen und verursachen langsam fortschreitende Organfehlfunktionen ohne akute metabolische Entgleisungen.
Krankheitsbilder und -verläufe zeigen starke Überschneidungen bei unterschiedlichsten Manifestationsaltern. Zumeist entwickelt sich das Neugeborene initial unauffällig; allerdings sind lysosomale Speicherkrankheiten wichtige Differenzialdiagnosen bei nichtimmunologischem Hydrops fetalis und Kardiomegalie im Neugeborenen- und Säuglingsalter.
Vergröberte Gesichtszüge, Skelett- und Hautveränderungen infolge einer mesenchymalen Speicherung entwickeln sich mit unterschiedlicher Dynamik (Abb. 3.16). Sie führen bei starker Ausprägung, ebenso wie eine Viszeromegalie, rasch zur Verdachtsdiagnose einer Speichererkrankung.


Erscheinungsbild und Prognose der meisten Speicherkrankheiten werden durch progrediente neurologische Symptome bestimmt. Bei Beginn im Säuglings- oder frühen Kindesalter imponieren zunächst eine muskuläre Hypotonie und statomotorische Entwicklungsverzögerung. Bald wird eine progrediente psychomotorische Retardierung mit Verlust bereits erworbener Fähigkeiten offensichtlich. Wird die Erkrankung erst im Schulalter oder noch später manifest, können Verhaltensstörungen, emotionale Labilität, Schulversagen oder psychiatrische Symptome im Vordergrund stehen, oft lange Zeit bevor ein offensichtlich fortschreitender demenzieller Abbau, Gangstörungen, Lähmungen oder Urininkontinenz hinzukommen. Wichtige Zusatzsymptome sind Hör- und Sehstörungen, Myoklonien, Pyramidenbahnzeichen oder eine Ataxie. Während neuropathische Erkrankungen mit Beginn im frühen Kindesalter rasch zur Dezerebration und Tod führen, sind Verlauf und Prognose bei den juvenilen und adulten Formen sehr variabel.
Diagnose
Wesentliche Hilfen sind organbezogene Zusatzuntersuchungen. Spezifisch untersucht werden sollten: Skelett (Becken und Wirbelsäule), parenchymatöse Organe (Sonographie), Herz (EKG, Echokardiographie), Augen (Makula, Katarakte), Gehör, ggf. elektrophysiologische Untersuchungen der peripheren Nervenleitung und Gehirn (kernspintomographische Untersuchung).
Pathognomonisch sind ein kirschroter Makulafleck oder eine Dysostosis multiplex.
Ein entscheidender Hinweis ist der histologische Nachweis von Speicherphänomenen. Vakuolisierte Lymphozyten können bei einigen Erkrankungen in peripheren Blutzellen nachgewiesen werden. Erforderlich sind ein direkter Ausstrich auf den Objektträger, nicht erst im Labor aus einem EDTA-Blutbildröhrchen, und eine erfahrene kritische Beurteilung. Sehr viel deutlicher sind Speicherungen in Knochenmarkzellen nachzuweisen (Abb. 3.15). Bei einigen Erkrankungen sind spezifische Speicherungen nur in Biopsien der betroffenen Organe erkennbar, am aussagekräftigsten in einer kombinierten Haut- und Nervenbiopsie (Zeroidlipofuszinosen).
Eine besonders wichtige, da relativ einfache und im positiven Falle diagnostisch spezifische Untersuchung ist die Bestimmung und Differenzierung der Mukopolysaccharide, der Oligosaccharide und der freien Neuraminsäure im Urin, die relativ breit als Screeninguntersuchung eingesetzt werden können. Sind weder pathologische Urinausscheidungen noch Lymphozytenvakuolen nachweisbar, muss in Abhängigkeit von der spezifischen klinischen Symptomatik eine primäre Enzymaktivitätsbestimmung im Serum oder Fibroblasten oder Mutationsanalysen erfolgen. Bei progredientem demenziellen Abbau mit Pyramidenbahnläsionen und Leukodystrophie sind Bestimmungen der Arylsulfatase A (metachromatische Leukodystrophie) und der β-Galaktozerebrosidase (Globoidzell-Leukodystrophie bzw. M. Krabbe) sinnvoll, bei Nachweis einer peripheren Neuropathie ohne Leukodystrophie (ansonsten wiederum primäre Verdachtsdiagnose M. Krabbe) Bestimmungen der Zeramidase (Lipogranulomatose bzw. M. Farber) oder bei normaler Intelligenz und Angiokeratomen der α-Galaktosidase (M. Fabry). Problematisch in der Diagnostik bleiben dann „nur“ die Mukolipidose IV und Patienten aus der Gruppe der Zeroidlipofuszinosen.
Therapie
Die Betreuung von Patienten mit lysosomalen Speicherkrankheiten erfordert komplexe, individuelle, interdisziplinäre und langfristig ausgerichtete Konzepte. Besonders wichtig ist das frühzeitige Erkennen von Seh- und Hörstörungen, problematisch die Behandlung von Myoklonien. Eine kurative Therapie ist bei den meisten Speicherkrankheiten nicht möglich.
Während Versuche mit Organtransplantationen von Milz, Leber und Nieren erfolglos geblieben sind, lassen sich bei einigen präsymptomatischen Patienten Erfolge durch Knochenmarktransplantationen erzielen, z. B. bei der MPS Typ I, dem spätmanifestierendem M. Krabbe und der metachromatischen Leukodystrophie. Erfolgreich sind Enzymersatztherapien beim M. Gaucher mit primär viszeraler Symptomatik, beim M. Fabry, M. Pompe und den MPS Typ I, II und VI. Dieser Ansatz wird auch für andere Erkrankungen ausgearbeitet und befindet sich für die MPS Typ IV (A) und mit intrathekaler Enzymersatztherapie für die CLN2 und die neuronopathischen Formen von MPS II und IIIA (Enzyme passieren nicht die Blut-Hirn-Schranke) in der klinischen Erprobung. Für die Behandlung mäßig schwerer Formen des M. Gaucher kann auch eine Substratreduktion eingesetzt werden. Dabei wird nicht das defekte Enzym substituiert sondern die Bildung der Speichersubstanz gehemmt.
Mukopolysaccharidosen
Mukopolysaccharidosen sind eine Gruppe von Erkrankungen, die durch Defekte einzelner lysosomaler Enzyme im stufenweisen Abbau von Glykosaminoglykanen verursacht werden. Letztere sind lange, aus sulfatierten und azetylierten Aminozuckern bestehende Polysaccharidketten, die an ein Proteinskelett geheftet als Proteoglykane die Grundsubstanz der extrazellulären Matrix bilden.
Kinder mit Mukopolysaccharidosen sind bei Geburt unauffällig. Je nach Restaktivität der defekten Enzyme kommt es auch bei identischen Enzymdefekten zu unterschiedlichen Phänotypen. Gemeinsam sind progrediente Skelettdeformitäten mit typischer Fazies, Knochendysplasien im Sinne einer Dysostosis multiplex (Abb. 3.16) und Gelenkkontrakturen sowie eine Hepato- und/oder Splenomegalie. Abhängig vom Typ finden sich Hornhauttrübungen und Taubheit. Hernien und rezidivierende Atemwegsinfekte sind häufig. Bei Typ IV und Typ VI bleibt die Intelligenz normal.
Die Diagnose erfolgt über den Nachweis pathologischer Fragmente von Glykosaminoglykanen im Urin und wird über gezielte enzymatische Analysen bestätigt.
Oligosaccharidosen
Die Oligosaccharidosen ähneln klinisch den Mukopolysaccharidosen. Sie beruhen auf einem gestörten Abbau der komplexen Kohlenhydratseitenketten von glykosylierten Proteinen (Glykoproteinen), z. B. Membran- und Strukturproteinen, aber auch metabolisch aktiven Proteinen. Zu dieser Gruppe gehört auch die ursprünglich klinisch als Mukolipidose Typ I klassifizierte Sialidose.
Oligosaccharidosen können schon sehr früh symptomatisch werden (Hydrops fetalis, Kardiomegalie und Herzinsuffizienz beim Neugeborenen) und rasch zum Tode führen. Eine fortschreitende psychomotorische Retardierung ist obligat, Krampfanfälle häufige Begleitsymptome. Die Diagnose erfolgt über den Nachweis pathologischer Oligosaccharide im Urin und wird über gezielte enzymatische Analysen bestätigt.
Mukolipidosen
Mukolipidosen wurden primär klinisch als Gruppe lysosomaler Speicherkrankheiten definiert, die klinische Merkmale von Mukopolysaccharidosen und Sphingolipidosen vereinen und biochemisch Zeichen von Oligosaccharidosen und z. T. Sphingolipidosen aufweisen. Sie werden durch Störungen der Phosphorylierung, des Transportes und der Integration lysosomaler Enzyme in den Lysosomen verursacht, z. B. bei der I-Cell-Krankheit. In den Körperflüssigkeiten der Patienten finden sich erhöhte Konzentrationen mehrerer Hydrolasen.
Lipidspeicherkrankheiten
Eine wichtige, biochemisch heterogene Gruppe von Erkrankungen ist durch eine Speicherung unterschiedlicher Lipide charakterisiert. Die klinische Symptomatik ähnelt den Sphingolipidosen; die Diagnostik muss primär über Enzymaktivitätsbestimmungen oder eine molekulare Diagnostik erfolgen. Man unterscheidet folgende Krankheitsformen:
Morbus Niemann-Pick
Beim Morbus Niemann-Pick Typ II (Synonym: Typ C) führt eine Störung im intrazellulärem Transport und Speicherung von Cholesterol zu einer ubiquitären Speicherung von Sphingomyelin, Cholesterol, Glykospingolipiden und Bis(Monoazylglyzero)phosphat.
Morbus Wolman
Beim Morbus Wolman führt ein Defekt der sauren Lipase zur Speicherung von Cholesterolestern und Triglyzeriden. Bei dieser Erkrankung steht oft eine gastrointestinale Symptomatik mit konsekutiver Gedeihstörung im Vordergrund. Als Besonderheit werden ferner vergrößerte und verkalkte Nebennieren beobachtet.
Neuronale Zeroidlipofuszinosen
Eine besonders wichtige Gruppe neurometabolischer Erkrankungen sind die neuronalen Zeroidlipofuszinosen. Sie werden durch eine Speicherung autofluoreszierender Lipidpigmente oder Zeroid verursacht. Aufgrund unterschiedlicher Krankheitsdynamik und elektronenpathologischer Befunde wurden infantile, spätinfantile, juvenile und adulte Manifestationen abgegrenzt. Kürzlich konnte diese Erkrankungsgruppe molekularbiologisch in bis jetzt 10 unterschiedliche primäre Gendefekte differenziert werden (CNL 1–10), wobei die Funktionen der betroffenen Gene und damit die Pathophysiologie teilweise noch ungeklärt sind. Am häufigsten kommt in Mitteleuropa die spätinfantile Form vor, verursacht durch einen Defekt der pepstatininsensitiven Peptidase (CLN 2, M. Jansky-Bielschowsy) und die juvenile Form infolge eines Defekts des Membranproteins CLN 3 (M. Batten, M. Spielmeyer-Vogt).
Klinik
Frühsymptome sind Verhaltensstörungen, schlechte Koordination, Sprachschwierigkeiten sowie Sehstörungen mit Nystagmus und Pigmentverschiebungen in der Retina bis zum Vollbild einer Retinitis pigmentosa. Zunehmend wird ein Verlust erworbener Fähigkeiten offensichtlich; die Patienten erblinden; Ataxie, extrapyramidale Bewegungsstörungen oder Anfälle dominieren zeitweilig das klinische Bild. Patienten mit der spätinfantilen Form versterben zumeist im Schulkinder- oder Adoleszentenalter, die Überlebenszeit bei Patienten mit juveniler Form in einem vegetativen Zustand ist sehr variabel.
Diagnose
Die Diagnosestellung erfolgt durch den mikroskopischen Nachweis der Zeroideinlagerungen in einer Hautbiopsie bzw. Lymphozyten. Während die Speicherungen lichtmikroskopisch gleich erscheinen, ließen sich elektronenmikroskopisch unterschiedliche, teilweise fingerdruckartige Muster differenzieren. Bei CNL 1, 2 und 10 können Enzymaktivitätsbestimmungen durchgeführt werden, ansonsten muss primär eine molekulare Diagnostik erfolgen.
Lysosomale Transportdefekte
Einzelne Abbauprodukte müssen aktiv aus den Lysosomen ins Zytosol transportiert werden. Defekte des Transportsystems für Zystin führen zur nephropathischen Zystinose, der Freisetzung von Vitamin B12 zur kombinierten Methylmalonazidurie und Homozystinurie (cblF-Defekt, Abschn. 3.11.2) und des Transportsystems für Sialinsäure zur Salla-Krankheit. Nur Letztere zeigt die für lysosomale Erkrankugen charakteristische Speicherung und fortschreitenden neurologischen Abbau.
Klinik
Die Zystinose wird im Kleinkindesalter mit einer tubulären Nephropathie unter dem Vollbild eines De-Toni-Debré-Fanconi-Syndroms symptomatisch. Im Vordergrund stehen Rachitis und Minderwuchs. Ohne spezifische Behandlung entsteht rasch ein Nierenversagen. Zystinspeicherungen können ferner endokrine Störungen, eine Hepatosplenomegalie oder Myopathie verursachen. Diagnostisch wegweisend ist der Nachweis von Zystinkristallen in der Kornea. Sie verursachen eine Photophobie.
Diagnose
Die Diagnose wird durch den Nachweis eines stark erhöhten Gehalts an Zystin in Leukozyten gestellt. Die Konzentration an Zystin in der Analytik der Aminosäuren in Körperflüssigkeiten ist demgegenüber normal.
Therapie
Neben der symptomatischen Therapie der Tubulopathie bzw. der Niereninsuffizienz konnte mit der Verabreichung von Cysteamin, systemisch und zusätzlich lokal als Augentropfen, ein effizienter spezifischer Behandlungsansatz entwickelt werden. Prognostisch entscheidend ist der Zeitpunkt der Diagnose und damit verbunden der Beginn und schließlich die Konsequenz der Cysteaminbehandlung. Die beste Prognose haben diejenigen Patienten, die bei Beginn der Behandlung noch über eine gute bis befriedigende Nierenfunktion verfügen.
Lipidstoffwechselstörungen
Grundlagen
Im Blut werden die wasserunlöslichen Lipide als Lipoproteine, d. h. Verbindungen aus Lipiden und Proteinen (sog. Apolipoproteinen) transportiert, wodurch sie wasserlöslich sind. Entsprechend unterschiedlicher Wanderungsgeschwindigkeiten in der Elektrophorese und ihrer Dichte stellen sich die Lipoproteine als Chylomikronen, „very low density lipoproteins“ (VLDL), „intermediate density lipoproteins“ (IDL), „low density lipoproteins“ (LDL) und „high density lipoproteins“ (HDL) dar. Einen vereinfachten Überblick über die Zusammensetzung und Funktionen zeigt Tab. 3.7. Zu den wichtigsten Apoproteinen zählen ApoB-100, welches für die Proteinbindung von VLDL und LDL an den LDL-Rezeptor erforderlich ist, sowie ApoC-II, der Kofaktor der Lipoproteinlipase.
| Bezeichnung | Elektrophoresebanden | Lipidanteil | Hauptlipide | Hauptapoproteine | Entstehungsort | Funktion |
|---|---|---|---|---|---|---|
| Chylomikronen | Auftragsort | 98% | Triglyzeride | A, B48, C, E | Darm | Transport exogener Triglyzeride |
| VLDL | Prä-b | 90% | Triglyzeride | B100, C, E | Darm, Leber | Transport endogener Triglyzeride von der Leber zu extrahepatischen Geweben |
| IDL | Prä-b | 80% | Triglyzeride | B100, C-II, E | Darm, Leber | Entsteht aus VLDL nach Triglyzeridabbau |
| LDL | B | 75% | Cholesterol | B100 | Abbauprodukt des VLDL | Cholesteroltransport zu extrahepatischen Geweben |
| HDL | A | 50% | Cholesterol, Phospholipide | A, C, D, E | Leber, Darm | Cholesterolrücktransport aus Geweben zur Leber |
Diagnose
Bei der Beurteilung der Lipide im Plasma im Kindes- und Jugendalter sind die Cholesterolwerte zur Abschätzung des Atheroskleroserisikos nicht ausreichend.
Besondere Bedeutung kommt der Beurteilung der Plasmaspiegel von LDL und HDL zu. Erhöhtes LDL gilt als atherogener Risikofaktor, während hohes HDL als protektiv gilt. Da die direkte Bestimmung des LDL aufwendig ist, wird dieses bei nüchternen Patienten häufig nach der Friedewald-Formel berechnet:
LDL-Cholesterol = Gesamtcholesterol – HDL-Cholesterol – (Triglyzeride/5)
Werte in mg/dl ×0,0250= mmol/l
Die Normalwerte für Cholesterol, LDL und HDL sowie Triglyzeride sind alters- und geschlechtsabhängig.
Für ein Screening, welches in einem Alter von etwa 5 Jahren empfohlen werden kann, genügt die Bestimmung des Gesamtcholesterols im nicht nüchternen Zustand. Bei erhöhtem Gesamtcholesterol (≥200 mg/dl) wird eine Nüchternblutentnahme mit Bestimmung von Cholesterol, Triglyzeriden, HDL-Cholesterol und Berechnung des LDL-Cholesterols durchgeführt.
Sekundäre Hyperlipidämien sind bei Kindern und Jugendlichen relativ häufig und zunehmend. Die Ursachen sind vielfältig, z. B. Adipositas, Anorexia nervosa, Hepatopathien, endokrinologische Störungen, Alkohol und verschiedene Medikamente (u. a. Kortison, Östrogene, Thiaziddiuretika, β-Blocker).
Therapie
Weglassen der auslösenden Substanz oder Behandlung der Grundkrankheit bewirkt in der Regel eine Besserung. Genetische Hyperlipidämien dagegen bedürfen meist einer spezifischen Therapie, die vor dem Auftreten irreversibler Schäden begonnen werden sollte. Dazu ist eine frühzeitige Diagnosestellung erforderlich.
Hyperlipoproteinämien
Unter den verschiedenen primären genetischen Hyperlipidämien steht im Kindesalter die heterozygote familiäre Hypercholesterolämie (Abb. 3.17) mit einer Häufigkeit von ca. 1:500 Neugeborenen im Vordergrund. Eine Vielzahl von verschiedenen Genmutationen verursacht dabei eine etwa 50%ige Reduktion von funktionellen LDL-Rezeptoren auf Zelloberflächen, sodass die rezeptorvermittelte Aufnahme von LDL beeinträchtigt ist. Eine Hyperchylomikronämie ist dagegen durch seltenere Defekte der Lipoproteinlipase oder dem phänotypisch identischen Defekt des Apoproteins CII, das als Kofaktor das Enzym aktiviert, verursacht. Ein Überblick über diese beiden wichtigsten Hyperlipoproteinämien ist in Tab. 3.8 dargestellt.


| Familiäre Hypercholesterolämie (Hyperlipoproteinämie Typ II) | Hyperchylomikronämie (Hyperlipoproteinämie Typ I) | |
|---|---|---|
| Häufigkeit |
Homozygote: 1:250.000–1:1 Mio. Heterozygote 1:500 |
1:1 Mio. |
| Ursache | Intrazellulärer Rezeptordefekt cholesteroltransportierender Proteine, LDL wird nicht in Zelle aufgenommen | Lipoproteinlipasemangel bzw. Apo-C-II-Mangel |
| Symptome | Xanthome, Xanthelasmen, Arcus corneae, Angina pectoris, Herzinfarkt, Apoplex. Heterozygote werden erst später symptomatisch | Bauchschmerzen, Hepatosplenomegalie, Xanthome, rezidivierende Pankreatitiden, Lipaemia retinalis |
| Diagnose |
Heterozygote: Cholesterol >230 mg/dl, LDL >170 mg/dl Homozygote: Cholesterol 600–1200 mg/dl Familienuntersuchung |
Chylomikronen und Triglyzeride (>1000 mg/dl) erhöht, Cholesterol mäßig erhöht, VLDL normal, LDL und HDL erniedrigt, Messung der Lipoproteinlipase im Plasma nach Heparingabe, isoelektrische Fokussierung des VLDL-Proteins bei Apo-C-II-Mangel |
| Therapie |
Fett- und cholesterolarme, polyensäurereiche Diät, Colestyramin, Statine Homozygot: LDL-Apherese, ggf. portokavale Anastomose, Lebertransplantation |
Fettarme Diät (15–20 g/Tag), mittelkettige Triglyzeride (MCT-Öl und Margarine), Fibrate |
| Prognose | Frühzeitige Atherosklerose, Homozygote können bereits im ersten Lebensjahr versterben | Kein Atheroskleroserisiko, schwere rezidivierende Pankreatitiden |
Hypolipoproteinämien
Die Abetalipoproteinämie und der familiäre HDL-Mangel gehören zu den wichtigsten genetischen Hypolipoproteinämien. Sie sind im Vergleich zu den Hyperlipoproteinämien sehr selten und durch einen gestörten Plasmalipidstoffwechsel ohne obligate Hyerlipidämie charakterisiert (Tab. 3.9).
| Abetalipoproteinämie (Kornzweig-Bassen-Syndrom) | Familiärer HDL-Mangel (Tangier-Krankheit) | |
|---|---|---|
| Ursache | Apolipoprotein-B-Mangel | Fehlen von Apolipoprotein-A-I, gestörte HDL-Bildung |
| Symptome | Fettmalabsorption, Steatorrhö, Gedeihstörung, muskuläre Hypotonie, zerebelläre Ataxie, Wachstumsretardierung, Retinitis pigmentosa, Blutungsneigung, Akanthozytose | Hepatomegalie, orange-gelblich verfärbt hyperplastische Tonsillen, Lymphadenopathie, diffuse Korneatrübung, periphere Neuropathie |
| Diagnose | Cholesterol und Triglyzeride erniedrigt; Apolipoprotein B, Chylomikronen, LDL und VLDL stark vermindert | Apolipo-HDL und Cholesterol stark vermindert, Triglyzeride normal |
| Therapie | Fettarme Diät mit hochgesättigten Fettsäuren und Linolensäure, hohe Dosen Vitamin E, A und K | Diätetische Fettreduktion |
| Prognose | Progredienter Verlauf mit spinozerebellärer Degeneration ab 2. Lebensdekade | Gehäuft kardiovaskuläre Erkrankungen |