Abstract
Im Folgenden werden die derzeit bekannten Transporterdefekte des Gastrointestinaltrakts beschrieben und in 2 Kategorien eingeteilt (Tab. 6.1): - solche, deren genetische Mutation bekannt ist, - jene, bei denen das verantwortliche Chromosom identifiziert wurde, das betroffene Gen aber noch nicht bekannt ist. Allen Krankheitsbezeichnungen gemeinsam ist die zuständige OMIM-Nummer, mit deren Hilfe die klinische und genetische Entität in ausführlicher Beschreibung in der oben genannten Datenbank im Internet abgerufen werden kann. Diese Art der Darstellung erleichtert es dem Leser, auch sehr seltene Krankheiten erwähnt zu finden und weiterführende Information zu erhalten.
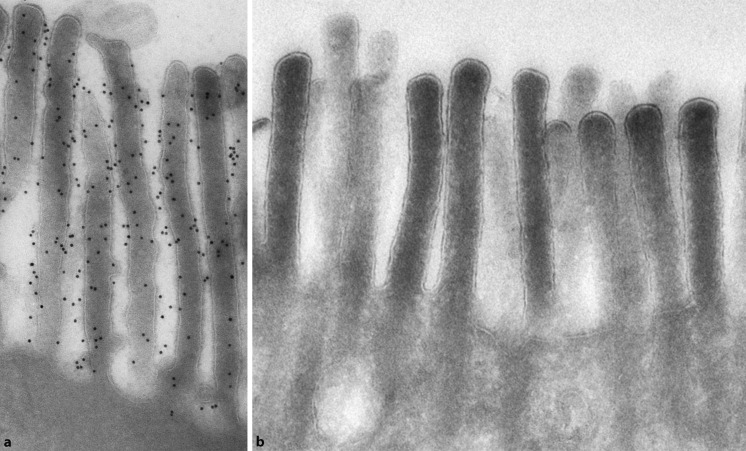
Syndromatische (phänotypische) Diarrhö (tricho-hepato-enterisches Syndrom)
Diese Erkrankung tritt autosomal-rezessiv mit einer geschätzten Inzidenz von 1 : 400.000 bis 1 : 500.000 Lebendgeborenen auf. Bei Patienten, die bei der Geburt hypotroph sind und in den ersten Lebensmonaten Durchfälle entwickeln, können Gesichtsdysmorphien mit prominenter Stirn, breiter Nase und Hypertelorismus ausgebildet sein. Ferner fallen eine Haaranomalie (Trichorrhexis nodosa) und ein Immunmangel in Form einer defekten T-Zell-Funktion und Antikörperreaktion sowie abnormer Befunde antigenspezifischer Hauttests auf.
Molekulargenetisch liegt ein Defekt des Thespin (TTC37), einem Tetratricopeptid-Repeat-Protein, vor, der sich auf verschiedene Bürstensaumtransportproteine auswirkt.
Beim tricho-hepato-enterischen Syndrom steht eine Symptomatik mit therapieresistenter Diarrhö (auch im Sinne einer „Crohn-ähnlichen“ Erkrankung) und Haaranomalien (wollige, wenig pigmentierte Haare, Haarausfall) im Vordergrund. Etwa die Hälfte der Patienten entwickelt eine Hämosiderose mit Leberfibrose /-zirrhose. Ferner wurden bei Patienten mit tricho-hepato-enterischem Syndrom Herzfehler (Fallot-Tetralogie, Atriumseptumdefekt, Ventrikelseptumdefekt), Hypopigmentierungen, Nabel-/ Leistenhernien, eine Sekretionsstörung von α-Granula der Thrombozyten, mentale Retardierung und Pubertas tarda beschrieben. Pränatal fallen neben einer Wachstumsretardierung ein Polyhydramnion und eine Plazentahyperplasie auf. Zudem ist eine Hypermethioninämie nachweisbar.
Es gibt weniger stark betroffene Patienten mit diesem Syndrom, die im Verlauf von der parenteralen Ernährung entwöhnt werden können.
Weitere seltene kongenitale Diarrhöen
Bei der enterischen Anendokrinose (Mutationen des NEUROG3-Gens) tritt eine generalisierte Malabsorption mit Erbrechen, Diarrhö, Gedeihstörung, Dehydrierung und Acidose auf; diagnostisch kann mit Anti-Chromogranin-Antikörpern das Fehlen von neuroendokrinen Zellen in der Darmmukosa nachgewiesen werden.
Bei der kongenitalen Form der intestinalen Lymphangiektasie stehen eine Eiweißverlustenteropathie mit Ödemen und Aszites, eine Malabsorption von Fetten und fettlöslichen Vitaminen mit Steatorrhö und eine Lymphopenie im Vordergrund. Die angeborene Form tritt häufig zusammen mit dem Turner-, Noonan- oder Klippel-Trenaunay-Weber-Syndrom auf. Sekundäre Formen der Lymphangiektasie werden z. B. bei konstriktiver Perikarditis, Herzinsuffizienz, abdominaler Tuberkulose und retroperitonealer Fibrose und Malignombildung beobachtet.
Der Proprotein-Konvertase-1 / 3-Mangel verursacht eine chronische Diarrhö mit postpartalem Beginn. Die betroffenen Kinder besitzen einen Hyperinsulinismus mit Hypoglykämien (erhöhtes Proinsulin bei niedrigem Insulin), einen hypogonadotropen Hypogonadismus und Hypokortisolimus. Die Patienten entwickeln eine Hyperphagie und bereits im Kleinkindesalter eine Adipositas. Proprotein-Konvertase 1 / 3 ist in die Prozessierung von Prohormonen und Neuropeptiden (Proinsulin, Proglukagon, Proopiomelanocortin) involviert.
Das Krankheitsbild der zerebrotendinösen Xanthomatose ist gekennzeichnet durch neonatale Cholestase, juvenile Katarakt, Entwicklungsverzögerung, frühe Atherosklerose und zerebelläre Ataxie. Die chronische Diarrhö beginnt neonatal. Charakteristisch sind Xanthome der Sehnen, insbesondere der Achillessehne und Cholesterinspeicherungen in Sehnen, Gehirn und Lungen. Bedingt durch den Mangel an Sterol-27-Hydroxylase, einem mitochondrialen Zytochrom P450, liegt ein Defekt der Gallensäurensynthese vor. Therapeutisch ist die Gabe von Pravastatin und Chenodesoxycholsäure angezeigt.
Bei einer therapieresistenten Diarrhö sollte differenzialdiagnostisch an eine Mitochondropathie, einen Mangel an Heparansulfatproteoglykanen in Enterozyten und den Phosphomannose-Isomerase-Mangel ( „carbohydrate-deficient glycoprotein syndrome 1b“) gedacht werden; Letzterer ist durch orale Zufuhr von Mannose therapierbar. Ferner wurde eine familiäre Form einer akuten sekretorischen Diarrhö bei IgG2-Subklassen-Mangel beschrieben.
Auch bei angeborenen selektiven Malabsorptionssyndromen, z. B. Hartnup-Krankheit (Malabsorption neutraler Aminosäuren), Smith-Strang-Syndrom (Methionin-Malabsorption), lysinurische Proteinintoleranz (Malabsorption dibasischer Aminosäuren), Tangier-Krankheit (Analphalipoproteinämie) und Folsäuremalabsorption, kann es im Verlauf der Erkrankung zu Manifestation einer Diarrhö kommen (▶ Abschn. 6.1). Chronische Durchfälle bestehen ferner bei der Abetalipoproteinämie (Bassen-Kornzweig-Syndrom), der Chylomikronen-Retentionskrankheit (Anderson-Krankheit) und dem M. Wolman mit einem Mangel an lysosomaler saurer Lipase.
Beim Trypsinogen- und Enteropeptidasenmangel entstehen die chronische Diarrhö, Gedeihstörung und Eiweißmangelödeme auf der Grundlage einer Maldigestion. Beide werden durch Substitution mit Pankreasenzymen, Proteinhydrolysatnahrung und mittelkettigen Triglyceriden (MCT) behandelt. Die Enteropeptidase ist eine Serinprotease des Bürstensaums und für die Aktivierung von Trypsinogen in Trypsin verantwortlich.
Literatur
Literatur zu Abschn. 6.1
- Booth IW, Stange G, Murer H, Fenton TR, Milla PJ. Defective jejunal brush-border Na+//H+ exchange: a cause of congenital secretory diarrhoea. Lancet. 1985;I:1066–1068. doi: 10.1016/S0140-6736(85)92369-4. [DOI] [PubMed] [Google Scholar]
- Hoglund P, Holmberg C, de la Chapelle A, Kere J. Paternal isodisomy for chromosome 7 is compatible with normal growth and development in a patient with congenital chloride diarrhea. Am J Hum Genet. 1994;55:747–752. [PMC free article] [PubMed] [Google Scholar]
- Heinz-Ehrian P, Muller T, Krabichler Mutations in SPINT2 cause a syndromic form of congenital sodium diarrhea. Am J Human Genet. 2009;84:188–196. doi: 10.1016/j.ajhg.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg C, Perheentupa J, Launiala K. Colonic electrolyte transport in health and in congenital chloride diarrhea. J Clin Invest. 1975;56:302–310. doi: 10.1172/JCI108094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg C, Perheentupa J, Launiala K, Hallman N. Congenital chloride diarrhea. Clinical analysis of 21 Finnish patients. Arch Dis Child. 1977;52:255–267. doi: 10.1136/adc.52.4.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kury S, Kharfi M, Kamoun R, et al. Mutation spectrum of human SLC39A4 in a panel of patients with acrodermatitis enteropathica. Hum Mutat. 2003;22:337–338. doi: 10.1002/humu.9178. [DOI] [PubMed] [Google Scholar]
- Moynahan EJ. Acrodermatitis enteropathica: a lethal inherited human zinc deficiency disorder. Lancet. 1974;II:399–400. doi: 10.1016/S0140-6736(74)91772-3. [DOI] [PubMed] [Google Scholar]
- Pahari A, Milla PJ, van't Hoff WG. Neonatal nephrocalcinosis in association with glucose-galactose malabsorption. Pediatr Nephrol. 2003;18:700–702. doi: 10.1007/s00467-003-1184-3. [DOI] [PubMed] [Google Scholar]
- Simon DB, Lu Y, Choate KA, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science. 1999;285:103–106. doi: 10.1126/science.285.5424.103. [DOI] [PubMed] [Google Scholar]
- Tasic V, Slaveska N, Blau N, Santer R. Nephrolithiasis in a child with glucose-galactose malabsorption. Pediatr Nephrol. 2004;19:244–246. doi: 10.1007/s00467-003-1327-6. [DOI] [PubMed] [Google Scholar]
- Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes acopper-transporting ATPase. Nat Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- Wasserman D, Hoekstra JH, Tolia V, et al. Molecular analysis of the fructose transporter gene (GLUT5) in isolated fructose malabsorption. J Clin Invest. 1996;98:2398–2402. doi: 10.1172/JCI119053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MH, Rao PN, Pettenati MJ, Dawson PA. Localization of the ileal sodium-bile acid cotransporter gene (SLC10A2) to human chromosome 13q33. Genomics. 1996;33:538–540. doi: 10.1006/geno.1996.0233. [DOI] [PubMed] [Google Scholar]
- Wright EM, Turk E, Martin MG. Molecular basis for glucose-galactose malabsorption. Cell Biochem Biophys. 2002;36:115–121. doi: 10.1385/CBB:36:2-3:115. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschnitt 6.2.1
- Croft NM, Howatson AG, Ling SC, et al. Microvillous inclusion disease: an evolving condition. J Pediatr Gastroenterol Nutr. 2000;31(2):185–189. doi: 10.1097/00005176-200008000-00019. [DOI] [PubMed] [Google Scholar]
- Goulet O, Phillips AD, et al. Congenital enteropathy involving intestinal mucosa developement. In: Walker WA, Goulet O, Kleinman RE, et al., editors. Pediatric gastrointestinal disease. Hamilton: Decker; 2004. pp. 922–928. [Google Scholar]
- Goulet O, Kedinger M, Brousse N, et al. Intractable diarrhea of infancy with epithelial and basement membrane abnormalities. J Pediatr. 1995;127(2):212–219. doi: 10.1016/S0022-3476(95)70297-0. [DOI] [PubMed] [Google Scholar]
- Müller T, Hess MW, Schiefermeieer N, et al. MYO5B mutatins cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40(10):1163–1165. doi: 10.1038/ng.225. [DOI] [PubMed] [Google Scholar]
- Phillips AD, Schmitz J. Familial microvillous atrophy: a clinicopathological survey of 23 cases. J Pediatr Gastroenterol Nutr. 1992;14(4):380–396. doi: 10.1097/00005176-199205000-00003. [DOI] [PubMed] [Google Scholar]
- Phillips AD, Szafranski M, Man LY, Wall WJ. Periodic acid-Schiff staining abnormality in microvillous atrophy: photometric and ultrastructural studies. J Pediatr Gastroenterol Nutr. 2000;30(1):34–42. doi: 10.1097/00005176-200001000-00015. [DOI] [PubMed] [Google Scholar]
- Reinshagen K, Naim HY, Zimmer KP. Autophagocytosis of the apical membrane in microvillus inclusion disease. Gut. 2002;51(4):514–521. doi: 10.1136/gut.51.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Literatur zu Abschn. 6.2.2
- Cameron DJ, Barnes GL. Successful pregnancy outcome in tufting enteropathy. J Pediatr Gastroenterol Nutr. 2003;36(1):158. doi: 10.1097/00005176-200301000-00033. [DOI] [PubMed] [Google Scholar]
- Goulet O, Kedinger M, Brousse N, et al. Intractable diarrhea of infancy with epithelial and basement membrane abnormalities. J Pediatr. 1995;127(2):212–219. doi: 10.1016/S0022-3476(95)70297-0. [DOI] [PubMed] [Google Scholar]
- Goulet O, Phillips AD, et al. Congenital enteropathy involving intestinal mucosa developement. In: Walker WA, Goulet O, Kleinman RE, et al., editors. Pediatric gastrointestinal disease. Hamilton: Decker; 2004. pp. 922–928. [Google Scholar]
- Sivagnanam M, Müller JL, Lee H, et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology. 2008;135:429–437. doi: 10.1053/j.gastro.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Literatur zu Abschn. 6.3
- Girault D, Goulet O, LeDeist F, et al. Intractable infant diarrhea associated with phenotypic abnormalities and immunodeficiency. J Pediatr. 1994;125(1):36–42. doi: 10.1016/S0022-3476(94)70118-0. [DOI] [PubMed] [Google Scholar]
- Goulet O, Phillips AD, et al. Congenital enteropathy involving intestinal mucosa developement. In: Walker WA, Goulet O, Kleinman RE, et al., editors. Pediatric gastrointestinal disease. Hamilton: Decker; 2004. pp. 922–928. [Google Scholar]
- Hartley JL, Zachos NC, Dawood B, et al. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy) Gastroenterology. 2010;138:2388–2398. doi: 10.1053/j.gastro.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Literatur zu Abschn. 6.4
- Zimmer KP, Branski D. Rare inborn defects causing malabsorption. In: Kliegman RM, Stanton B, St. Geme J, Schor N, Behrman RE, editors. Nelson textbook of pediatrics. 19. Philadelphia: Elsevier; 2011. pp. 1319–1322. [Google Scholar]
Literatur zu Abschn. 6.5
- Bayless TM, Rothfeld B, Massa C, et al. Lactose and milk intolerance: clinical implications. N Engl J Med. 1975;292(22):1156–1159. doi: 10.1056/NEJM197505292922205. [DOI] [PubMed] [Google Scholar]
- Enattah NS, Sahi T, Savilahti E, et al. Identification of a variant associated with adult-type hypolactasia. Nat Genet. 2002;30(2):233–237. doi: 10.1038/ng826. [DOI] [PubMed] [Google Scholar]
- Gugatschka M, Dobnig H, Fahrleitner-Pammer A, et al. Molecularly-defined lactose malabsorption, milk consumption and anthropometric differences in adult males. Qjm. 2005;98(12):857–863. doi: 10.1093/qjmed/hci140. [DOI] [PubMed] [Google Scholar]
- Kuokkanen M, Kokkonen J, Enattah NS, et al. Mutations in the translated region of the lactase gene (LCT) underlie congenital lactase deficiency. Am J Hum Genet. 2006;78(2):339–344. doi: 10.1086/500053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim HY, Zimmer K-P, et al. Congenital disease of dysfunction and absorption. 1. Genetically determined disaccharidase deficiency. In: Kleinmann RE, Goulet O, Mieli-Vergani G, et al., editors. Pediatric gastrointestinal disease. 5. Hamilton: Decker; 2008. pp. 275–289. [Google Scholar]
- Newton T, Murphy MS, Booth IW. Glucose polymer as a cause of protracted diarrhea in infants with unsuspected congenital sucrase-isomaltase deficiency. J Pediatr. 1996;128(6):753–756. doi: 10.1016/S0022-3476(96)70325-6. [DOI] [PubMed] [Google Scholar]
- Sander P, Alfalah M, Keiser M, et al. Novel mutations in the human sucrase-isomaltase gene (SI) that cause congenital carbohydrate malabsorption. Hum Mutat. 2006;27(1):119. doi: 10.1002/humu.9392. [DOI] [PubMed] [Google Scholar]
- Swallow DM. Genetics of lactase persistence and lactose intolerance. Annu Rev Genet. 2003;37:197–219. doi: 10.1146/annurev.genet.37.110801.143820. [DOI] [PubMed] [Google Scholar]
- Zimmer KP. Laktose- und Fruktosemalabsorption. Monatsschr Kinderheilkd. 2007;155:565–576. doi: 10.1007/s00112-007-1540-7. [DOI] [Google Scholar]