

Abstract
Die zystische Fibrose (Synonym: Mukoviszidose) ist auch heute noch eine unheilbare, letal verlaufende Krankheit. Durch die fortlaufende Verbesserung der Therapie ist es in den vergangenen Jahren gelungen, Lebenserwartung und Lebensqualität deutlich zu verbessern. Im Rahmen der Multisystemerkrankung finden sich im Gastrointestinaltrakt wichtige Manifestationen (an Pankreas, Dünndarm, Leber und Gallenwegen), die mit der auch heute noch prognostisch ungünstigen, aber häufig vorkommenden Ernährungsstörung zusammengehen. Frühdiagnose und Frühtherapie können das Auftreten von Komplikationen verhindern bzw. hinauszögern. Neueste Daten zeigen, dass eine mutationsspezifische pharmakologische Therapie auf Ionenkanalebene wirksam ist. Die konventionelle, zentrumorientierte und qualitätsgesicherte Behandlung der zystischen Fibrose erfordert den integrierten Einsatz der pädiatrischen Gastroenterologie.
Die zystische Fibrose (Synonym: Mukoviszidose) ist auch heute noch eine unheilbare, letal verlaufende Krankheit. Durch die fortlaufende Verbesserung der Therapie ist es in den vergangenen Jahren gelungen, Lebenserwartung und Lebensqualität deutlich zu verbessern. Im Rahmen der Multisystemerkrankung finden sich im Gastrointestinaltrakt wichtige Manifestationen (an Pankreas, Dünndarm, Leber und Gallenwegen), die mit der auch heute noch prognostisch ungünstigen, aber häufig vorkommenden Ernährungsstörung zusammengehen. Frühdiagnose und Frühtherapie können das Auftreten von Komplikationen verhindern bzw. hinauszögern. Neueste Daten zeigen, dass eine mutationsspezifische pharmakologische Therapie auf Ionenkanalebene wirksam ist. Die konventionelle, zentrumorientierte und qualitätsgesicherte Behandlung der zystischen Fibrose erfordert den integrierten Einsatz der pädiatrischen Gastroenterologie.
Epidemiologie und Genetik
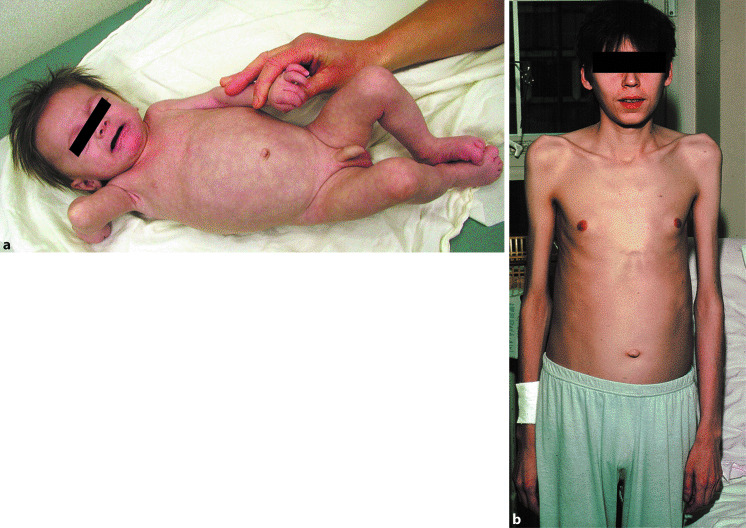
Die zystische Fibrose ist die häufigste letale genetische Multisystemerkrankung in der weißen Weltbevölkerung. Der Erbgang ist autosomal-rezessiv. Die Prävalenz beträgt 1 : 2500 (Kaukasier). Die Genträgerhäufigkeit beläuft sich in Deutschland auf 1 : 20. Die Krankheit ist auch heute noch nicht heilbar; sie ist jedoch multidisziplinär behandelbar. Lebensqualität und Lebenserwartung der Patienten sind gestiegen. Während noch in den 1970er Jahren kaum ein Patient das Erwachsenenalter erreichte, beträgt die mittlere Überlebenswahrscheinlichkeit in Deutschland heute 41,6 Jahre; fast alle Patienten erreichen das junge Erwachsenenalter (◘ Abb. 24.1).

Pathophysiologische Basis für die Erkrankung ist ein defekter Ionentransport an epithelialen Oberflächen. Viele Organe, vor allem der Gastrointestinal- und der Respirationstrakt, sind in individuell unterschiedlicher Ausprägung betroffen. Der Multisystemcharakter der Krankheit wird durch Interaktionen zwischen den Veränderungen auf verschiedenen Ebenen verstärkt.
Ein einziger Genlokus auf dem langen Arm von Chromosom 7 trägt das „Cystic-fibrosis“-(CF-)Gen, welches ein Protein mit einer Länge von 1480 Aminosäuren kodiert. Dieses Protein wird auch CF-Transmembranregulator („cystic fibrosis transmembrane conductance regulator“, CFTR) genannt. Es wird in den Epithelien der Luftwege, des Gastrointestinaltrakts, der Schweißdrüsen und des Urogenitaltrakts exprimiert und hat Ionenkanal- und Regulationsfunktionen. Diese Funktionen sind bei zystischer Fibrose durch verschiedene Mutationen gestört. Hier setzen neue pharmakologische Behandlungswege (Ionenkanalpotenzierung, Korrektur mit Wirkung auf Lungenfunktion, Schweißtest und Gewicht der Patienten) ein. Möglicherweise bedeutet dies eine neue Ära der Behandlung der zystischen Fibrose.
Mehr als 1800 Mutationen des CF-Gens sind bereits bekannt. Die häufigste Mutation (70 %) in Nordeuropa ist Delta F508 . Die Beziehung zwischen Genotyp und Phänotyp ist komplex, und sie wird von zusätzlichen modifizierenden Genen mitbeeinflusst. „Schweren“ Mutationen wie Delta F508, fast immer mit exokriner Pankreasinsuffizienz verbunden, stehen „leichte“ Mutationen gegenüber (z. B. R117H), oft mit klinischer Pankreassuffizienz. Es resultiert ein weites Spektrum klinischer Ausprägungen in den verschiedenen Altersstufen (◘ Tab. 24.1).
| Altersstufe | Symptome |
|---|---|
| Neugeborene und Säuglinge |
- Mekoniumileus - Gedeihstörung - Fettstühle - Hypochlorämische Alkalose - Husten - Lungenentzündung |
| Kinder und Jugendliche |
- Untergewichtigkeit - Chronischer produktiver Husten - Sauerstoffmangel - Uhrglasnägel - Nasenpolypen - Leber- und Milzvergrößerung - Diabetes mellitus |
| Erwachsene |
- Globale Lungeninsuffizienz - Pneumothorax - Bluthusten - Infertilität (männliche Patienten) - Osteoporose - Arthritis |
Pathophysiologie
Die genetisch bedingte CFTR-Dysfunktion führt zu einer Transportstörung von Salz und Wasser durch epitheliale Membranen. In den Atemwegen kommt es zur Produktion eines hochviskösen Sekrets, das die mukoziliäre Clearance behindert. Eine Obstruktion der kleinen Atemwege mit weitreichenden Folgen (chronische Entzündung und Infektion) ist die Folge. In den Pankreas- und Gallengängen sowie im Urogenitaltrakt kommt es in ähnlicher Weise zur Produktion eines hochviskösen, eiweißhaltigen Sekrets und zur Obstruktion. In den Schweißdrüsen beeinträchtigt die gestörte Ionenkanalfunktion die Reabsorption von Chlorid. Dadurch resultiert ein vermehrter Kochsalzgehalt des Schweißes (Nachweis mittels Schweißtest; ▶ Abschn. Diagnostik und Screening).
Während die respiratorischen Manifestationen der zystischen Fibrose mit chronischer Atemwegsinfektion durch Keime wie Haemophilus influenzae, Staphylococcus aureus, Pseudomonas aeruginosa und Burkholderia cepacia geprägt sind, tragen nichtinfektiöse pathologische Mechanismen zur Ausprägung der verschiedenen gastrointestinalen Manifestationen bei: intestinale Obstruktion, Transportstörung, exokrine Pankreasinsuffizienz, Gedeihstörung, Diabetes mellitus, Gallengangobstruktion und Salzverlust (◘ Tab. 24.2).
| Betroffene Organe | Manifestationen |
|---|---|
| Ösophagus und Magen |
- Gastroösophagealer Reflux - Hyperacidität |
| Dünndarm |
- Mekoniumileus - Distale intestinale Obstruktion - Obstipation - Invagination - Bildung von Adenokarzinomen |
| Dickdarm |
- Wandverdickung - Fibrosierende Kolonopathie - Rektumprolaps |
| Pankreas |
- Exokrine Insuffizienz - Diabetes mellitus - Pankreatitis (bei Pankreassuffizienz) |
| Leber |
- Fettleber - Fokale biliäre Zirrhose - Multilobuläre Zirrhose - Pfortaderhochdruck |
| Gallenwege |
- Neonatale Cholestase - Mikrogallenblase - Cholelithiasis - Gallengangstenose - Sklerosierende Cholangitis |
Morphologisch fassbare Veränderungen finden sich am exokrinen Pankreas:
Gangobstruktion durch eosinophiles Sekret,
Verlust von Azini,
Zystenbildung,
Fibrose.
Daraus resultiert der international übliche Krankheitsname „zystische Fibrose“ des Pankreas.
Die Pankreasinseln sind erst ab dem 2. Lebensjahrzehnt betroffen. Bei Mekoniumileus finden sich eine Becherzellhyperplasie und eine mechanische Obstruktion des distalen Ileums. In den Speicheldrüsen zeigt sich eine eosinophile Gangobstruktion, der Ösophagus weist eine Ösophagitis und Varizen auf. Als seltene Komplikation findet sich eine fibrosierende Kolonopathie. Eine Sekretobstruktion zeigt sich auch im biliären System: Cholelithiasis und Mikrogallenblase kommen häufig vor. Fettleber, fokale biliäre Zirrhose und multilobuläre Leberzirrhose sind weitere charakteristische pathologische Befunde.
Klinisches Bild
Pankreas
Exokrines Pankreas
Einjährige Patienten mit zystischer Fibrose zeigen zu 90 % eine exokrine Pankreasinsuffizienz (PI) mit Maldigestion und Malabsorption, welche die wichtigsten Ursachen für die häufig anzutreffende Gedeihstörung darstellen. Es kommt zur massiv verminderten Bereitstellung von Lipase, Kolipase, Trypsin und Chymotrypsin sowie zur verminderten Bicarbonatsekretion. In Einzelfällen kann die Entwicklung der exokrinen Pankreasinsuffizienz allerdings erst Jahre nach der Diagnosestellung einsetzen.
Die wichtigsten Symptome, die sich auf die exokrine Pankreasinsuffizienz zurückführen lassen, sind voluminöse, fettige und übelriechende Stühle sowie eine ab dem Säuglingsalter progrediente Gedeihstörung bis hin zu Eiweißmangel, Ödemen und Anämie. Ein geringer Anteil von Patienten mit zystischer Fibrose weist dauerhaft eine Pankreassuffizienz (PS) auf. Diese besondere Situation ist mit spezifischen selteneren Mutationen verknüpft und hat eine deutlich günstigere Prognose.
Die gestörte Energiebilanz und das Ernährungsdefizit sind im Säuglingsalter zentrale Ansatzpunkte einer erfolgreichen Therapie.
Zum Nachweis der exokrinen Pankreasinsuffizienz dienen die Stuhlmikroskopie (Fetttröpfchen) und die Bestimmung der Elastase-1-Konzentration im Stuhl, außerdem die Stuhlfettbestimmung über 3 Tage, verbunden mit einer Fettbilanz (Ernährungsprotokoll). Der invasive Sekretin-Pankreozymin-Test wird heute kaum noch durchgeführt. Neuere Atemtests auf Basis 13 C-markierter gemischter Triglyceride sind eine wichtige diagnostische Bereicherung, die sich – ebenso wie die Stuhlfettbilanzierung – auch zur Therapiekontrolle eignen. Alle diagnostischen Methoden haben ihre Grenzen. Den einzelnen, idealen Pankreasfunktionstest bei zystischer Fibrose gibt es nicht. Wichtig ist bei initialer Pankreassuffizienz die jährliche Nachkontrolle.
Bei Pankreassuffizienz (PS) kann auch eine Pankreatitis auftreten. Dies ist jedoch überwiegend erst im Erwachsenenalter der Fall. Zudem ist eine monosymptomatische Manifestation der zystischen Fibrose als Pankreatitis möglich.
Endokrines Pankreas: Diabetes mellitus
Ab dem 2. Lebensjahrzehnt kann es zur Entwicklung eines Diabetes mellitus kommen. Die Glukoseintoleranz zeigt sich anfangs meist während pulmonaler Exazerbationen oder Phasen einer Steroidbehandlung. Aufgrund der steigenden Lebenserwartung wird die Diabetesprävalenz bei Patienten mit zystischer Fibrose weiter steigen. Bei 30-jährigen Betroffenen liegt sie derzeit bereits bei 50 %.
Die Anfangssymptome wie Müdigkeit, schlechter Appetit und Gewichtsabnahme sind uncharakteristisch und können übersehen werden. Polyurie und Polydipsie kommen hinzu. Eine Ketoacidose wird nur selten beobachtet.
Ab dem 10. Lebensjahr muss ein Screening auf einen Diabetes mellitus mittels oralem Glukosetoleranztest durchgeführt werden. Die Bestimmung des Anteils an glykiertem Hämoglobin (HbA1c) ist nicht als Screeningtest, sondern nur zur Einstellungskontrolle geeignet. Sowohl im Kontext der Ernährungsprobleme als auch der Begünstigung weiterer pulmonaler Exazerbationen ist der Diabetes mellitus im Rahmen einer zystischen Fibrose prognostisch wichtig, was therapeutisch berücksichtigt werden muss (Fortführung der hyperkalorischen fettreichen Kost, evtl. Gabe oraler Antidiabetika, meist aber Insulintherapie).
Gastrointestinale Manifestationen
CFTR wird in den epithelialen Zellmembranen des Gastrointestinaltrakts exprimiert, vor allem in Dünn- und Dickdarm. Die Entstehung der gastrointestinalen Komplikationen ist darüber hinaus multifaktoriell und weist eine spezifische Altersverteilung auf.
Mekoniumileus
Bei 15–20 % der Neugeborenen mit zystischer Fibrose kommt es innerhalb der ersten 48 Lebensstunden zu einem Mekoniumileus, einer Obstruktion des distalen Dünndarms durch visköses Mekonium. Das Auftreten des Mekoniumileus ist von der exokrinen Pankreasinsuffizienz unabhängig. Symptome sind massive abdominale Vorwölbung, Erbrechen und fehlender Mekoniumtransport. Bereits intrauterin kann es zu Dünndarmobstruktion, Darmperforation, Mekoniumperitonitis und distalem Mikrokolon kommen.
Die Diagnostik umfasst die Röntgenleeraufnahme des Abdomens, evtl. mit anschließendem Kontrastmitteleinlauf (◘ Abb. 24.2).
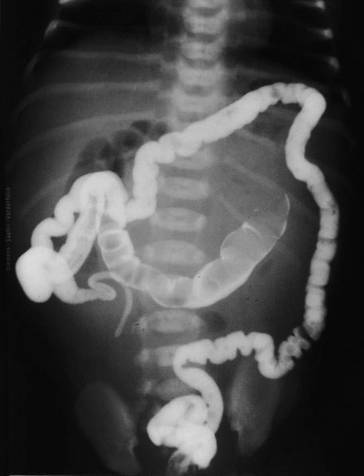

Wichtige Differenzialdiagnosen sind das Mekoniumpfropfsyndrom, intestinale Atresien und M. Hirschsprung.
Distales intestinales Obstruktionssyndrom (DIOS)
Jenseits des Neugeborenenalters bezeichnet man die mechanische Darmobstruktion durch hochviskösen Stuhl als distales intestinales Obstruktionssyndrom (früher: Mekoniumileusäquivalent). Die Häufigkeit beträgt bis zu 30 %; sie nimmt mit steigendem Lebensalter und in besonderen Situationen (Dehydratation, Lungentransplantation) zu. Die Symptomatik ist durch die Trias aus Bauchschmerzen, fehlender Stuhlentleerung und einer tastbaren Masse im rechten Unterbauch geprägt.
Die Ultraschalluntersuchung und die Röntgenleeraufnahme des Abdomens führen zur Diagnose. Die Differenzialdiagnostik ist umfangreich; sie umfasst die häufige Obstipation sowie Invagination, Appendizitis und weitere Erkrankungen.
Differenzialdiagnosen von Bauchschmerzen bei zystischer Fibrose
Distale intestinale Obstruktion
Unzureichende Pankreasenzymsubstitution
Gastritis, gastroösophagealer Reflux
Invagination
Appendizitis, Abszess, Mukozele
Morbus Crohn
Cholelithiasis
Pankreatitis (PS)
Basale Pleuropneumonie
Hypochlorämische Alkalose
Vor allem Säuglinge und Kleinkinder mit zystischer Fibrose können durch Kochsalzverluste bei hohen Außentemperaturen und im Rahmen von Brechdurchfällen so viel Kochsalz verlieren, dass es zu einer schweren hypochlorämischen Alkalose (Pseudo-Bartter-Syndrom) kommt. Die Symptome – Müdigkeit, Fieber, Gewichtsverlust – setzen schleichend ein. Die diagnostische Zuordnung erfolgt mittels Blutgasanalyse und Elektrolytbestimmung (Alkalose, Hypochlorämie, Hypokaliämie).
Gastroösophagealer Reflux
Motilitätsstörungen wie die gastroösophageale Refluxkrankheit und auch eine chronische Obstipation kommen bei Patienten mit zystischer Fibrose häufig vor (25 %). Die gastroösophageale Refluxkrankheit äußert sich vor allem bei jüngeren Kindern als rezidivierendes Schreien und Erbrechen. Später kommen retrosternale Schmerzen und Sodbrennen hinzu. Die Diagnostik umfasst pH-Metrie, Sonographie, Endoskopie sowie die röntgenologische Kontrastmittelpassage von Ösophagus und Magen.
Fibrosierende Kolonopathie
Die fibrosierende Kolonopathie ist eine seltene gastrointestinale Komplikation der zystischen Fibrose mit Obstruktion des Colon ascendens durch Strikturbildung. Sie ist mit einer höchstdosierten Pankreasenzymgabe assoziiert und wird nach der Erstbeschreibung im Jahre 1994 derzeit kaum noch beobachtet. Abzugrenzen sind Darmwandverdickungen, die sich sonographisch häufig nachweisen lassen, ohne dass sie zu klinischen Symptomen oder Problemen führen (◘ Abb. 24.3).


Rektumprolaps
Bei 20 % der noch nicht behandelten Kinder mit zystischer Fibrose unter 5 Jahren kommt es zum akuten Rektumprolaps, der sich fast immer gut reponieren lässt und durch die Pankreasenzymsubstitution wirksam vermieden werden kann.
Weitere gastrointestinale Manifestationen
Weitere gastrointestinale Komplikationen sind die pseudomembranöse Enterokolitis, ausgelöst durch Clostridium-difficile-Toxin, die gehäufte Assoziation mit M. Crohn sowie weitere Enterokolitiden und das Auftreten maligner gastrointestinaler Tumoren jenseits des 20. Lebensjahres (Kolonkarzinom sowie hepatozelluläres Karzinom und Cholangiokarzinom). Die nicht selten beobachtete persistierende Fettmalabsorption unter Pankreasenzymsubstitution weist zusätzlich auf bisher noch unzureichend erforschte gastrointestinale Störungen mit möglicherweise genetischer Basis hin (bakterielle Überbesiedlung des Dünndarms, Entzündung, Störung der Motilität, Störungen von pH-Wert und Gallensalzhaushalt und des Fettsäuremetabolismus).
Leber- und Gallenwegsmanifestationen
Mit steigender Lebenserwartung nimmt der Anteil der Patienten mit chronischer hepatobiliärer Beteiligung zu; im Erwachsenenalter beträgt er 30 %. Zu unterscheiden ist die metabolisch bedingte Fettleber von der biliären Erkrankung, welche durch die CFTR-Expression in den Gallekanalikuli mit nachfolgender Obstruktion geprägt ist. Die schwere Leberbeteiligung bei CF ist mit dem Z-Allel des Serpina-1-Gens verknüpft.
Fettleber
Die Ausprägung einer Fettleber ist in allen Altersgruppen häufig, besonders bei Malnutrition, Mangel an essenziellen Fettsäuren und Diabetes mellitus. Die Leber ist dann vergrößert, die Konsistenz ist jedoch weich. Die Ultraschalldiagnostik ermöglicht die morphologische Abgrenzung gegenüber den biliär bedingten Formen der Leberbeteiligung.
Fokale biliäre Zirrhose
Die fokale biliäre Zirrhose stellt die charakteristische Lebermanifestation der zystischen Fibrose dar. Sie kommt bei 25–50 % der Patienten mit zunehmendem Alter vor.
Meist ist die fokale biliäre Zirrhose asymptomatisch. Spezifische Schmerzen im rechten Oberbauch können jedoch bestehen. Die Diagnostik umfasst die Ultraschalluntersuchung mit Nachweis wechselnder Echogenität sowie die Bestimmung der Enzymaktivitäten von Transaminasen, alkalischer Phosphatase und γ-Glutamyltranspeptidase (γ-GT), welche über einen Zeitraum von mehr als 12 Monaten deutlich erhöhte Werte aufweisen. Leberszintigraphie, Magnetresonanz-Cholangiopankreatikographie (MRCP) und Dopplersonographie des hepatischen Blutflusses sind hilfreich. In besonderen Fällen ist eine Leberbiopsie erforderlich.
Multilobuläre biliäre Zirrhose
Die multilobuläre biliäre Zirrhose ist als schwerste Form der Leberbeteiligung bei zystischer Fibrose mit einer Häufigkeit von 1 % bei Kindern und immerhin 25 % bei Erwachsenen relativ seltener als die fokale biliäre Zirrhose. Es besteht keine Korrelation mit dem Grad der Lungenmanifestation.
Das klinische Bild zeigt eine Hepatomegalie, eine grobhöckrige Leberkante und eine derbe Organkonsistenz. Die Milz ist vergrößert, und es finden sich Zeichen des Hypersplenismus. Laborchemisch fallen erhöhte Transaminasenaktivitäten auf, außerdem Zeichen der Cholestase und der gestörten Lebersyntheseleistung (Gerinnungswerte, Cholinesteraseaktivität, Albumin- und Ammoniakkonzentration).
Eine portale Hypertension tritt bei 2 % der erwachsenen Patienten auf. Klinisch finden sich Ösophagus- und Fundusvarizenblutungen, Splenomegalie, Aszites und portokavale Enzephalopathie. Schwere Spätfolgen der portalen Hypertension sind das hepatopulmonale Syndrom und der Pulmonalarterienhochdruck. Bei Splenomegalie und oberer gastrointestinaler Blutung muss die Diagnostik intensiviert werden; sie umfasst die Ultraschalluntersuchung inklusive Dopplersonographie, die obere gastrointestinale Endoskopie sowie eine MRCP.
Cholelithiasis und Gallenganganomalien
Etwa 10 % der erwachsenen Patienten weisen eine Cholelithiasis auf. Die Steine sind röntgendurchlässig und enthalten Kalziumbilirubinat, Cholesterin und Proteine. Häufig bleibt die Cholelithiasis asymptomatisch. Klassisch ist allerdings die Präsentation mit kolikartigen Schmerzen im rechten Oberbauch mit Ausstrahlung in den Rücken. Zielführend ist die Abdomensonographie. MRCP und gezielt auch die endoskopische retrograde Cholangiopankreatikographie (ERCP) sind wichtige diagnostische Hilfsmethoden.
Zusätzliche Gallengangbesonderheiten sind die benigne Mikrogallenblase (20 %) und die seltene distale Gallengangstenose (1 %). Sekundäre Cholangitiden können den hepatobiliären Verlauf komplizieren.
Ernährungsstörungen und Malnutrition
Eine negative Energiebilanz sowie Ernährungsdefizite im Makro- und Mikronährstoffbereich sind zentrale Probleme in der Behandlung der zystischen Fibrose mit unmittelbarer prognostischer Bedeutung. Die Prävention der Mangelernährung und die frühe Intervention bei Auftreten eines Defizits sind Grundlagen des praktischen Vorgehens. Die rechtzeitige Diagnostik und der frühzeitige Einsatz spezialisierter Ernährungstherapien sind erforderlich. Die Bestimmung des Body-Mass-Indexes und der Perzentilenvergleich dienen der Orientierung. Die Beobachtung des Längenwachstums und der Pubertätsentwicklung können auf zusätzliche Probleme hinweisen. Die Grenzen, ab denen eine therapeutische Intervention erfolgen soll, sind durch Leitlinien definiert (◘ Tab. 24.3).
| Intervention | Altersabhängige Kriterien | ||
|---|---|---|---|
| <2 Jahre | 2–18 Jahre | >18 Jahre | |
|
Normaler Ernährungszustand Präventive Ernährungsberatung |
BMIp ≥50 | BMIp ≥50 | BMI: 18,5–25; kein Gewichtsverlust |
| Spezielle Ernährungsberatung, Supplemente | Gedeihstörung |
BMIp 15–50; Gewichtsverlust 4–6 Monate, Stillstand 6 Monate |
BMI: <18,5; Gewichtsverlust 5 % über 2 Monate |
| Invasiver Ernährungssupport |
Trotz Supplement: Gedeihstörung |
Trotz Supplement: – BMIp <15 – Gewichtsverlust entsprechend ≥2 Perzentilen |
Trotz Supplement: – BMI: <18,5 – Gewichtsverlust ≥5 % über 2 Monate |
BMI Body-Mass-Index; BMIp Body-Mass-Index-Perzentile.
Die negative Energiebilanz resultiert bei zystischer Fibrose aus vielen verschiedenen Faktoren wie Maldigestion, Malabsorption, unzureichender Energiezufuhr, vermehrtem Energieumsatz, hepatobiliärer Beteiligung und Diabetes mellitus. Zusätzlich zum Energiedefizit bestehen Mangelsituationen hinsichtlich fettlöslicher Vitamine, essenzieller Fettsäuren, Antioxidanzien, Mineralien und Spurenelementen. Nicht immer ist allerdings eine biochemisch festgestellte Mangelsituation auch mit klinischen Problemen oder Symptomen verknüpft.
Diagnostik und Screening
Die Diagnose der zystischen Fibrose wird klinisch gestellt und laborchemisch bestätigt. Der Nachweis einer exokrinen Pankreasinsuffizienz, chronischer bronchopulmonaler Infekte, einer doppelseitigen Sinusitis sowie einer Azoospermie müssen zu der Verdachtsdiagnose hinführen. Die Basis der Diagnosestellung ist der Schweißtest ( Pilocarpiniontophorese, Bestimmung der Chloridkonzentration). Chloridwerte ab 60 mmol/l sind beweisend, Werte zwischen 30 und 60 mmol/l liegen im Grenzbereich und erfordern eine zusätzliche Diagnostik. Zur Vermeidung falsch-positiver und falsch-negativer Resultate bleibt die Durchführung des Schweißtests erfahrenen Zentren vorbehalten. Der Nachweis von zwei Zystische-Fibrose-Mutationen oder einer pathologischen nasalen Potenzialdifferenz ist ebenfalls diagnostisch wegweisend. Die In-vivo-Messung der nasalen transepithelialen Potenzialdifferenz bleibt wenigen spezialisierten Zentren überlassen.
Ein Neugeborenenscreening, z. B. mittels immunreaktivem Trypsinogen (IRT) im Fersenblut, gefolgt von der Suche nach den häufigsten Zystische-Fibrose-Mutationen, ist möglich; die Sensitivität beträgt etwa 95 %. Frühe Ernährungsdefizite lassen sich auf diese Weise vermeiden, auch das längerfristige Wachstum kann verbessert werden. Screeningprogramme, verbunden mit Frühtherapie und Qualitätssicherung, werden derzeit in Europa durchgeführt (Frankreich, England, Tschechien, aber noch nicht Deutschland).
Diagnosekriterien der zystischen Fibrose
Typische Symptome oder
Erkrankte Geschwister oder
Positives Screening
Plus
Bei 2-maliger Durchführung des Schweißtests Chloridkonzentration von ≥60 mmol/l oder
Nachweis zweier Zystische-Fibrose-Mutationen oder
Pathologische Nasenpotenzialdifferenz
Therapie
Die Therapie der zystischen Fibrose erfolgt multidisziplinär. Sie muss früh einsetzen, um die Malnutrition mit ihren Folgen und die Entwicklung chronischer Komplikationen zu verhindern oder zumindest wirksam zu verlangsamen. Im Folgenden werden nur die gastroenterologischen Manifestationen der zystischen Fibrose behandelt.
Pankreasmanifestationen
Damit eine hyperkalorische Ernährung möglich wird, muss die exokrine Pankreasinsuffizienz (PI) durch Pankreasenzymsubstitution ausgeglichen werden. Ziel ist die Erreichung eines annähernd normalen Fettresorptionskoeffizienten. Die Pankreasenzymgabe ist auf die Nahrungsfettzufuhr abzustimmen. Unter Kontrolle von Stuhlfettausscheidung und Gewichtsentwicklung ist eine Dosisanpassung bis hin zum Maximum von 10.000 IE Lipase/kg KG/Tag erforderlich. Für die Ermittlung der Nahrungsfettzufuhr sind spezielle Tabellen hilfreich. Wenn mehr als 10.000 IE Lipase/kg KG/Tag benötigt werden und die Steatorrhö noch nicht vollständig kontrolliert ist, muss eine gastroenterologische Zusatzdiagnostik erfolgen. Zöliakie, Laktoseintoleranz, Enteritis, Enterokolitis und chronisch-entzündliche Darmerkrankungen müssen ausgeschlossen werden. Viele Patienten zeigen eine Magensäurehypersekretion und eine verminderte pankreatische Bicarbonatproduktion. Diese Patienten können von einer zusätzlichen medikamentösen Säuresuppression profitieren.
Seltene Nebenwirkungen der Pankreasenzymgabe sind Hyperurikämie und Hyperurikosurie sowie allergische Reaktionen und perianale Irritationen. Bei Überdosierung wurde sehr selten die fibrosierende Kolonopathie als Nebenwirkung beschrieben.
Pankreasenzymsubstitution
Präparate: magensaftresistente Mikropellets oder Mikrotabletten
-
Dosierung:
- Säuglinge: Start mit 2500 IE Lipase/120 ml Säuglingsnahrung
- Kleinkinder unter 4 Jahren: 1000 IE Lipase/kg KG/Hauptmahlzeit
- Kinder ab 4 Jahren: 500 IE Lipase/kg KG/Hauptmahlzeit
- Maximaldosis: 10.000 IE Lipase/kg KG/Tag
- Dosisanpassung: 500–4000 IE Lipase/g Fett
-
Vorgehen bei mangelnder Wirksamkeit:
- Diagnostische Aufarbeitung
- Gabe von H2-Blockern oder Protonenpumpeninhibitoren
Gastrointestinale Manifestationen
Mekoniumileus Ziel der Therapie des Mekoniumileus ist die Beseitigung der intestinalen Obstruktion. Bei unkompliziertem Mekoniumileus ist ein konservativer Therapieversuch mit Gastrografin- und Kochsalzeinlauf angezeigt. In vielen Fällen, vor allem bei Komplikationen wie Volvulus, Atresie, Perforation oder Peritonitis, ist eine operative Therapie indiziert. Die Behandlung der Wahl ist die schonende Enterotomie mit anterograder Spülung. Nach Anwendung dieses gestuften Therapieschemas ist die Prognose des Mekoniumileus deutlich verbessert worden, sie unterscheidet sich nicht mehr von derjenigen der zystischen Fibrose ohne Mekoniumileus. Allerdings sind Komplikationen der chirurgischen Therapie wie Bridenileus und das Syndrom der blinden Schlinge möglich.
Therapie des Mekoniumileus
Konservativ (unkompliziert, Erfolgsrate 60 %): Gastrografin-, NaCl- oder N-Acetylcysteineinlauf
-
Operativ:
- Laparatomie, Enterotomie, Spülung
- Adhäsiolyse, sparsame Darmresektion
- Anastomosierung, Anlage eines Enterostomas
- Cave: Sekundärkomplikationen: Bridenileus, Syndrom der blinden Schlinge
Distales intestinales Obstruktionssyndrom (DIOS)
Die Behandlung des distalen intestinalen Obstruktionssyndroms erfolgt – wenn irgend möglich – konservativ. Zur Prävention ist auf eine ausreichende Pankreasenzymsupplementierung und eine hinreichende Flüssigkeitszufuhr sowie eine ballaststoffreiche Ernährung zu achten. Die Behandlung besteht in der intestinalen Lavage mit Macrogolelektrolytlösung in Kombination mit Einläufen. Zur Langzeitbehandlung ist Macrogol (0,5–1,0 g/kg KG/Tag) geeignet. Bei Versagen der konservativen Therapie (sehr selten) ist eine Laparatomie unumgänglich.
Therapie des distalen intestinalen Obstruktionssyndroms
Ausreichende Pankreasenzymsupplementierung und Flüssigkeitszufuhr
-
Intestinale Lavage mit Macrogolelektrolytlösung (Golytely, KleanPrep, Movicol):
- 20–40 ml/kg KG/h p.o. oder nasogastrisch
- max. 1 l/h über 8 h
-
Einlauf:
- Macrogolelektrolytlösung oder
- 0,9%ige NaCl-Lösung: 3-mal 500–1500 ml/Tag
- Gastrografin
Leber- und Gallenwegsmanifestationen
Eine Fettleber bedarf keiner Behandlung. Hyperkalorische Ernährung, Substitution fettlöslicher Vitamine und Pankreasenzymersatz dienen der Prävention der Malnutrition mit möglicher konsekutiver Leberschädigung. Die Behandlung der fokalen biliären Zirrhose ist immer noch nicht ausreichend evidenzbasiert. Häufig wird Ursodesoxycholsäure in einer Dosierung von 20 mg/kg KG/Tag in 2 Einzeldosen eingesetzt. Ziele sind eine Verbesserung des Galleflusses und eine Normalisierung der Transaminasenwerte; der Langzeiteffekt hinsichtlich der Prävention von multilobulärer Zirrhose und portaler Hypertension ist ungewiss.
Die portale Hypertension mit Ösophagusvarizenblutung, Splenomegalie, Aszites und portokavaler Enzephalopathie wird bei zystischer Fibrose nicht anders behandelt als bei anderen Ursachen. In Einzelfällen kann eine isolierte Lebertransplantation indiziert sein.
Ernährungsstörungen
Zur Prävention der Malnutrition erfolgt die Ernährung anfangs durch Muttermilch mit häufigem Anlegen bzw. durch Kuhmilchformula mit Energiesupplementierung bei Stillhindernis. Nur bei Spezialproblemen sind Proteinhydrolysatformulanahrungen oder mittelkettige Triglyceride einzusetzen. Bei Flüssigkeitsverlusten und hohen Temperaturen ist zusätzlich Kochsalz zu substituieren. Ab dem 4.–6. Lebensmonat wird normale bis fettreiche Beikost, ab dem 2. Lebensjahr normale bis fettreiche Kleinkinderkost verabreicht. Spezifische Supplementprodukte gibt es für Kinder bis 5 Jahren, danach sind energieangereicherte Supplementnahrungen für Erwachsene anwendbar. Die nasogastrische oder durch perkutane endoskopische Gastrostomie (PEG) ermöglichte Gabe hochkalorischer Sondennahrungen ist bei hartnäckiger Malnutrition erfolgreich. Die totale parenterale Ernährung bleibt speziellen Indikationen vorbehalten.
Für junge Erwachsene sind der Einsatz einer hochkalorischen, fettreichen Kost inklusive kaloriendichter oraler Supplemente und die PEG-Sondenernährung sinnvoll. Die Ernährungstherapie muss von einem Muskeltrainingsprogramm und einer psychosozialen Begleitung ergänzt werden. Bronchopulmonale Infekte sind sorgfältig zu behandeln. Vitaminsupplemente sind bei exokriner Pankreasinsuffizienz und ggf. auch bei Pankreassuffizienz für die fettlöslichen Vitamine erforderlich, bei Darmresektion für Vitamin B12; eine generelle Supplementierung mit wasserlöslichen Vitaminen ist nicht erforderlich (◘ Tab. 24.4).
| Vitamin | Monitoring | Substitutionsdosis | |
|---|---|---|---|
| Fettlösliche Vitamine | |||
| A | Serumretinol | 0–12 Monate | 1500 IE/Tag |
| 1–3 Jahre | 5000 IE/Tag | ||
| 4–8 Jahre | 5000–10.000 IE/Tag | ||
| >8 Jahre | 10.000 IE/Tag | ||
| D | Serum-25-(OH-)D | 0–12 Monate | 400 IE/Tag |
| >1 Jahr | 400–1000 IE/Tag | ||
| E | Serum-α-Tocopherol | 0–12 Monate | 40–50 IE/Tag |
| 1–3 Jahre | 80–150 IE/Tag | ||
| 4–8 Jahre | 100–200 IE/Tag | ||
| >8 Jahre | 200–400 IE/Tag | ||
| K | Serumprothrombinzeit | 0–8 Jahre | 0,3–0,5 mg/Tag |
| Erwachsene | 5–10 mg/Woche | ||
| Wasserlösliche Vitamine | |||
| B12 | Cyanocobalamin | 100 μg/Monat i. m. | |
Prognose
Durch die frühzeitige multidisziplinäre Behandlung ist die Prognose der zystischen Fibrose hinsichtlich Lebensqualität und Lebenserwartung deutlich gestiegen. Die mittlere Lebenserwartung beträgt in Deutschland derzeit 41,6 Jahre. Die steigende Lebenserwartung führt zur Verschiebung des Manifestationsspektrums, gerade auch der gastrointestinalen Manifestationen, denen gezielt begegnet werden muss. Das Ernährungsdefizit bleibt auch heute noch prognostisch wichtig. In Deutschland waren im Jahre 2009 noch 26 % der Patienten aller Altersklassen untergewichtig. Das deutsche Projekt „Qualitätssicherung Mukoviszidose“ zeigte im Verlauf der letzten Jahre zwar langsame, aber kontinuierliche Fortschritte. Zukünftige Behandlungsmöglichkeiten umfassen auch für die gastroenterologischen Manifestationen die somatische Gentherapie und die Ionenkanalkorrektur.
Literatur
- Ballmann M, Smaczny C. CF-Manual. 2. Hannover: Solvay Arzneimittel GmbH; 2008. [Google Scholar]
- Bartlett JR, et al. Genetic modifiers of liver disease in cystic fibrosis. JAMA. 2009;302(10):1076–1083. doi: 10.1001/jama.2009.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowitz D, et al. Cystic fibrosis foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr. 2009;155:S73–S93. doi: 10.1016/j.jpeds.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo C. Liver disease in cystic fibrosis. Curr Opin Pulm Med. 2007;13:529–536. doi: 10.1097/MCP.0b013e3282f10a16. [DOI] [PubMed] [Google Scholar]
- Colombo C, et al. Guidelines for the diagnosis and management of distal intestinal obstruction syndrome in cystic fibrosis patients. J Cyst Fibros. 2011;10(2):S24–S28. doi: 10.1016/S1569-1993(11)60005-2. [DOI] [PubMed] [Google Scholar]
- Debray D, et al. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease. J Cyst Fibros. 2011;10(2):29–S36. doi: 10.1016/S1569-1993(11)60006-4. [DOI] [PubMed] [Google Scholar]
- Farrell PM, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics. 2001;107(1):1–13. doi: 10.1542/peds.107.1.1. [DOI] [PubMed] [Google Scholar]
- Gilljam M, et al. GI complications after lung transplantation in patients with cystic fibrosis. Chest. 2003;123(1):37–41. doi: 10.1378/chest.123.1.37. [DOI] [PubMed] [Google Scholar]
- Hodson ME, Geddes DM, editors. Cystic fibrosis. 3. London: Arnold; 2007. [Google Scholar]
- Houwen RH, et al. Defining DIOS and constipation in cystic fibrosis with a multicentre study on the incidence, characteristics, and treatment of DIOS. JPGN. 2009;49:54–58. doi: 10.1097/MPG.0b013e3181a6e01d. [DOI] [PubMed] [Google Scholar]
- Jadin SA, et al. Growth and pulmonary outcomes during the first 2 y of life of breastfed and formula-fed infants diagnosed with cystic fibrosis through the Wisconsin Routine Newborn Screening Program. Am J Clin Nutr. 2011;93:1038–1047. doi: 10.3945/ajcn.110.004119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalnins D, et al. Nutritional management of cystic fibrosis patients. Curr Opin Clin Nutr Metab Care. 2007;10:348–354. doi: 10.1097/MCO.0b013e3280a94f80. [DOI] [PubMed] [Google Scholar]
- Kerem E, et al. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros. 2005;4:7–26. doi: 10.1016/j.jcf.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Maqbool A, Stallings VA. Update on fat-soluble vitamins in cystic fibrosis. Curr Opin Pulm Med. 2008;15(6):574–581. doi: 10.1097/MCP.0b013e3283136787. [DOI] [PubMed] [Google Scholar]
- Moran A, et al. Clinical care guidelines for cystic fibrosis-related diabetes. Diabetes Care. 2010;33(12):2697–2708. doi: 10.2337/dc10-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt D, et al., editors. Cystische Fibrose. Berlin Heidelberg New York: Springer; 2001. [Google Scholar]
- Rowland M, et al. Outcome in cystic fibrosis liver disease. Amer J. Gastroenterology. 2011;106:104–109. doi: 10.1038/ajg.2010.316. [DOI] [PubMed] [Google Scholar]
- Sens B, Stern M, editors. Qualitätssicherung Mukoviszidose – Überblick über den Gesundheitszustand der Patienten in Deutschland 2010. Bad Honnef: Hippocampus; 2011. [Google Scholar]
- Sinaasappel M, et al. Nutrition in patients with cystic fibrosis: a European Consensus. J Cystic Fibros. 2002;1:51–75. doi: 10.1016/S1569-1993(02)00032-2. [DOI] [PubMed] [Google Scholar]
- Sloane PA, Rowe SM. Cystic fibrosis transmembrane conductance regulator protein repair as a therapeutic strategy in cystic fibrosis. Curr Opin Pulm Med. 2010;16:591–597. doi: 10.1097/MCP.0b013e32833f1d00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern M, et al. et al. Mukoviszidose (CF): Ernährung und exokrine Pankreasinsuffizienz. Leitlinien der GPGE, Update. In: Wirth S, et al.et al., editors. Leitlinien Kinder- und Jugendmedizin. München: Elsevier, Urban & Fischer; 2012. p. N176. [Google Scholar]
- Walkowiak J, et al. Indirect pancreatic function tests in children. JPGN. 2005;40:107–114. doi: 10.1097/00005176-200502000-00001. [DOI] [PubMed] [Google Scholar]
- Werlin SL, et al. Evidence of intestinal inflammation in patients with cystic fibrosis. JPGN. 2010;51(3):304–208. doi: 10.1097/MPG.0b013e3181d1b013. [DOI] [PubMed] [Google Scholar]
- Wiedemann B, et al. Evaluation of body mass index percentiles for assessment of malnutrition in children with cystic fibrosis. Europ J Clin Nutr. 2007;61:759–768. doi: 10.1038/sj.ejcn.1602582. [DOI] [PubMed] [Google Scholar]
- Wouthuyzen-Bakker M, et al. Persistent fat malabsorption in cystic fibrosis; lessons from patients and mice. J Cyst Fibros. 2011;10:150–158. doi: 10.1016/j.jcf.2011.03.008. [DOI] [PubMed] [Google Scholar]