

Abstract
Das Pankreas ist sowohl ein exokrines wie auch ein endokrines Organ (Tab. 21.1). Ihm kommt die zentrale Rolle in der Aufschließung der Nahrungsbestandteile sowie in der Regulation des Blutzuckerspiegels zu.
Exokrine und endokrine Funktion
In der Antike wurde die Verdauung als Umwandlung der Nahrung durch Fäulnis (Empedokles von Agrigent) und das Pankreas als Polster mit Schutz- und Stützfunktion anderer Organe (Galenos) angesehen. Erst 1782 wies Lazarro Spallanzini (1729–1799) nach, dass es sich bei der Verdauung nicht um Fäulnis oder Gärung, sondern um einen chemischen Prozess handelt. 1856 beschrieb Claude Bernard (1813–1878) die Bedeutung des Pankreas für die Fettverdauung, dessen Sekret die Fette nicht nur emulgiert, sondern auch zu Fettsäuren und Glycerin spaltet. Er wies erstmals auf die universale Verdauungsfunktion des Pankreas hin, da sein Sekret auf alle drei Nahrungskomponenten (Kohlenhydrate, Eiweiße und Fette) einwirke. Rudolf Peter Heidenhain (1834–1897) postulierte 1875, dass sich im Pankreas kein freies Ferment befände, sondern nur ein Mutterkörper, und schlug für diesen die Bezeichnung „Zymogen“ vor. Im Jahre 1876 führte Wilhelm Friedrich Kühne (1837–1900) den Begriff Enzym als Bezeichnung für chemische Fermente ein; das eiweißspaltende Enzym des Pankreas nannte er Trypsin. Im Jahre 1902 beschrieben William Bayliss (1860–1924) und Ernest Starling (1866–1927) das Hormon Sekretin und die endokrine Regulation des exokrinen Pankreas, und 1910 zeigte Iwan Pawlow (1849–1936) als erster die nervale Stimulierbarkeit der Bauchspeicheldrüse.
Exokrine Funktion
Pankreassekret
Das Pankreas ist sowohl ein exokrines wie auch ein endokrines Organ (◘ Tab. 21.1). Ihm kommt die zentrale Rolle in der Aufschließung der Nahrungsbestandteile sowie in der Regulation des Blutzuckerspiegels zu.
| Zelltyp | Funktion |
|---|---|
| Exokrine Zellen | |
| Azinuszellen | Synthese und Sekretion von Verdauungsenzymen |
| Zentroazinäre Zellen | Sekretion von Elektrolyten, Bicarbonat und Wasser |
| Duktale Zellen | Austausch von Bicarbonat und Chloridionen, Sekretion von Elektrolyten und Wasser |
| Endokrine Zellen | |
| α-Zellen | Synthese von Glukagon |
| β-Zellen | Synthese von Insulin |
| δ-Zellen | Synthese von Somatostatin |
| F-Zellen | Synthese des pankreatischen Polypeptids (PP) |
In Abhängigkeit von der Nahrungszufuhr sezerniert das Pankreas beim Erwachsenen täglich etwa 3 l enzym- und elektrolythaltiges Sekret in das Duodenum. Die Sekretionsrate steigt nach Stimulation von 0,2–0,3 ml/min auf über das 10-Fache der Ruhewerte. Diese Steigerung ist vorwiegend durch eine vermehrte Sekretion der duktalen Epithelzellen bedingt. Auch die Zusammensetzung des Pankreassekrets ändert sich während der Verdauungsphase. Peptidhormone wie Cholezystokinin (CCK) und durch den N. vagus vermittelte Efferenzen erhöhen die Enzymausscheidung aus den Azinuszellen, während das Hormon Sekretin vornehmlich die Bicarbonatsekretion der Gangzellen fördert (◘ Abb. 21.1).


Anorganische Komponenten
Die wesentlichen anorganischen Komponenten des Pankreassaftes sind Wasser sowie Natrium, Kalium, Chlorid und Bicarbonat. Der hohe Bicarbonatgehalt sorgt für einen alkalischen pH-Wert von etwa 8 und bewirkt die Neutralisierung des sauren Mageninhaltes und damit eine optimale Aktivität der Verdauungsenzyme im Darmlumen. Im Gegensatz zu den Konzentrationen der Kationen, die während der Stimulation konstant bleiben, ändern sich die Chlorid- und Bicarbonatkonzentrationen spiegelbildlich zueinander, ohne dass sich aber die Gesamtkonzentration der beiden Anionen ändert. Mit zunehmender Bicarbonatkonzentration steigt auch der pH-Wert des Pankreassekrets.
Pankreasenzyme
Etwa 90 % der Proteine des Pankreassekrets sind Verdauungsenzyme. Quantitativ wie funktionell ist Trypsin das wichtigste Verdauungsenzym. Alle proteolytischen Enzyme werden als inaktive Vorstufen (Zymogene) in den Azinuszellen des Pankreas synthetisiert und ins Duodenum sezerniert. Erst im Darm erfolgt durch das in der Bürstensaummembran lokalisierte Enzym Enteropeptidase (Enterokinase) die Spaltung von Trypsinogen zu aktivem Trypsin. Trypsin vermag sowohl Trypsinogen als auch alle anderen Proenzyme in ihre aktive Form umzuwandeln. Erfolgt eine signifikante Enzymaktivierung schon im Pankreas selbst, kommt es zu einer Selbstandauung des Organs, die sich klinisch als akute Pankreatitis manifestiert.
Zwei Hauptformen des Trypsinogens, anionisches und kationisches Trypsinogen, wurden aus dem Pankreassekret isoliert. Beide besitzen die gleiche enzymatische Aktivität; das kationische Isoenzym zeigt jedoch eine stärkere Neigung zur Selbstaktivierung und eine geringere Inaktivierungstendenz.
Neben Trypsin sind Chymotrypsin und Protease E weitere pankreatische Endopeptidasen, die bestimmte Peptidbindungen innerhalb der Proteine spalten, während die Carboxypeptidase A und B einzelne Aminosäuren vom C-terminalen Ende der Eiweißmoleküle abspalten.
Die α-Amylase spaltet α-1,4-glykosidische Bindungen in Polysacchariden wie Stärke und Glykogen in Oligosaccharide unterschiedlicher Länge. Die Oligosaccharide werden durch verschiedene, in der Bürstensaummembran des Darms lokalisierte, Oligosaccharidasen zu Monosacchariden wie Glukose, Galaktose und Fruktose gespalten.
Die wesentlichen Pankreasenzyme zur Fettverdauung sind die Lipase, die Phospholipase A2 und die Cholesterinesterase. Die Lipase hydrolysiert Triglyceride in freie Fettsäuren, Mono- und Diglyceride. Für ihre Wirkung bedarf sie einer Kolipase, die – im Gegensatz zur Lipase – als inaktives Proenzym (Prokolipase) synthetisiert wird.
Regulation der Pankreassekretion
Die exokrine Sekretion des Pankreas wird nerval und hormonell reguliert. Mehr als ein Dutzend unterschiedlicher gastrointestinaler Peptide wirken auf das exokrine Pankreas (◘ Tab. 21.2). Die durch Nahrungsaufnahme stimulierte Sekretion lässt sich in eine kephale, eine gastrale und eine intestinale Phase unterscheiden.
| Hormon | Syntheseort | Wirkung |
|---|---|---|
| Cholezystokinin (CCK) | Duodenum, Jejunum |
Enzymsekretion ↑ Verstärkt Sekretinwirkung |
| Sekretin | Duodenum, Jejunum |
Bicarbonat- und Wassersekretion ↑ Insulinsekretion ↑ |
| Gastrisches inhibitorisches Polypeptid (GIP) | Duodenum, Jejunum, Ileum (K-Zellen) | Insulinsekretion ↑ |
| Glukagon-like-Peptid 1 (GLP1) | Jejunum, Ileum, Duodenum (L-Zellen) |
Exokrine Sekretion ↓ Insulinsekretion ↑ Glukagonsekretion ↓ |
| Glukagon | Pankreas (α-Zellen) | Exokrine Sekretion ↓ |
| Insulin | Pankreas (β-Zellen) | Exokrine Sekretion ↑ |
| Somatostatin | Pankreas (δ-Zellen), gesamter Intestinaltrakt |
Exokrine Sekretion ↓ Insulin- und Glukagonsekretion ↓ |
| Pankreatisches Polypeptid (PP) | Pankreas (F-Zellen), Magen, Duodenum | Exokrine Sekretion ↓ |
| Gastrin-releasing-Peptid (GRP) | Magen, Duodenum, Jejunum | Steigerung der exokrinen Sekretion ↑ |
| Ghrelin | Magen, Darm, Pankreas |
Enzymsekretion ↓ Insulinsekretion ↓ Glukagonsekretion ↑ |
| Leptin | Fettgewebe, Magen |
Enzymsekretion ↓ Insulinsekretion ↓ |
| Motilin | Duodenum, Jejunum | exokrine Sekretion ↑ |
| Neurotensin | Ileum, Kolon |
Enzymsekretion ↑ Bicarbonat- und Wassersekretion ↑ |
| „Pituitary adenylate cyclase activating polypeptide“ (PACAP) | Gesamter Intestinaltrakt, Pankreas |
Exokrine Sekretion ↑ Insulin- und Glukagonsekretion ↑ |
| Peptid YY | Ileum, Kolon | Exokrine Sekretion ↓ |
| Vasoaktives intestinales Peptid (VIP) | Ösophagus bis Rektum | Exokrine Sekretion ↑ |
↑ Erhöhung; ↓ Verminderung.
Die kephale Phase wird parasympathisch über efferente Fasern des N. vagus vermittelt. Die vagale Freisetzung von Acetylcholin stimuliert über muskarinerge Rezeptoren auf den Azinuszellen die Sekretion von Enzymen ohne jedoch die Bikarbonatsekretion wesentlich zu steigern. Weitere Mediatoren der kephalen Phase sind das vasointestinale Polypeptid (VIP) und das gastrin-releasing Peptid (GRP), die über nervale Rezeptoren die Enzymsekretion der Azinuszellen fördern. An den duktalen Zellen führt VIP zu einer vermehrten Bicarbonat- und Wassersekretion.
Hauptstimulus der gastralen Phase ist die Dehnung des Magens durch Nahrung. Über einen vagovagalen Reflex kommt es zur Ausscheidung eines enzymreichen Sekrets.
Mit dem Eintritt des Nahrungsbreis in das Duodenum beginnt die intestinale Phase der Pankreassekretion, die sowohl humoral als auch nerval vermittelt wird. Die Ansäuerung des proximalen Duodenums durch den Nahrungsbrei setzt Sekretin aus den S-Zellen des Duodenums und Jejunums frei. Sekretin ist der Hauptmediator für die Bicarbonatsekretion der zentroazinären Zellen. Die sekretininduzierte Bicarbonatsekretion wird durch Cholezystokinin und cholinerge Efferenzen verstärkt.
Cholezystokinin (CCK) ist der wichtigste humorale Mediator für die Sekretion von Verdauungsenzymen und wird in der oberen Dünndarmmukosa gebildet. Intraluminale langkettige Fettsäuren und deren Monoglyceride sowie Peptide und Aminosäuren stimulieren die CCK-Freisetzung. Neben der pankreatischen Enzymsekretion bewirkt CCK auch eine Kontraktion der Gallenblase. Humane Azinuszellen besitzen im Gegensatz zu denen anderer Spezies keine CCK-Rezeptoren. Die Wirkung von CCK auf die Pankreassekretion erfolgt bei Menschen somit indirekt über CCK-A-Rezeptoren auf afferenten Neuronen des N. vagus im Sinne einer vagovagalen Schleife, mit Acetylcholin als endgültigem Mediator via muskarinergen Rezeptoren vom Typ M3.
Neben CCK fördern auch Sekretin und VIP die Enzymfreisetzung über Aktivierung des azinären Adenylatzyklasesystems. Beide Hormone verstärken zudem die CCK-Wirkung auf die Azinuszellen.
Endokrine Funktion
Die endokrine Funktion des Pankreas wird von den hormonproduzierenden Zellen des Inselapparates (Langerhans-Inseln) vermittelt. Die Hormone der Inselzellen sind maßgebend beteiligt an der Regulation des Kohlenhydratstoffwechsels. Es werden verschiedene Zelltypen unterschieden, von denen die α-Zellen das Glukagon, die β-Zellen das Insulin, die δ-Zellen das Somatostatin und die F-Zellen das pankreatische Polypeptid bilden (◘ Tab. 21.1). Die Hauptfunktionen der Pankreashormone bestehen in der Speicherung der aufgenommenen Nahrung als Glykogen und Lipide (Insulin), in der Freisetzung dieser Energiereserven während Hungerphasen (Glukagon) sowie in der Regulation des Blutzuckerspiegels und des Wachstums. Die Inselzellhormone wirken auch auf das exokrine Pankreas, indem sie die Sekretion von Bicarbonat und Verdauungsenzymen beeinflussen.
Insulin
Die β-Zellen stellen mit 50–80 % den Hauptzelltyp des Inselapparates. Das in diesen Zellen synthetisierte Insulin ist das bedeutendste blutzuckersenkende Hormon des menschlichen Körpers. Das Peptidhormon Insulin wurde 1923 von Banting und Best aus dem Rinderpankreas isoliert. Insulin wird als inaktives Proenzym gebildet und im Golgi-Apparat und den Granula der β-Zellen durch die insulinkonvertasevermittelte Abspaltung des C-Peptids in die biologisch aktive Form umgewandelt.
Der wichtigste physiologische Stimulus für die Insulinsekretion ist die Erhöhung der extrazellulären Glukosekonzentration. Die Insulinsekretion wird zudem hormonal und nerval reguliert: Enterohormone wie das glukoseabhängige insulinotrope Polypeptid (gastrisches inhibitorisches Polypeptid, GIP) und das glukagonähnliche Peptid 1 (GLP1) stimulieren die Sekretion, wohingegen Somatostatin, Ghrelin, Leptin und Katecholamine die Sekretion hemmen.
Die Hauptwirkung des Insulins ist die gesteigerte Glukoseaufnahme in Muskulatur und Fettgewebe mit konsekutiver Senkung der Glukosekonzentration im Blut. Durch Aktivierung der Glykogensynthase und der Phosphodiesterase bewirkt Insulin eine vermehrte Glykogensynthese und verminderte Glykogenolyse in Leber- und Muskelzellen sowie eine Hemmung der hepatischen Glukoneogenese. Im Weiteren stimuliert Insulin die Proteinbiosynthese durch verstärkte zelluläre Aufnahme von Aminosäuren. Im Fettgewebe hemmt es die Lipolyse und steigert die Triglyceridsynthese. Summa summarum stellt Insulin das wichtigste anabole Hormon des menschlichen Körpers dar.
Glukagon
Das Peptidhormon Glukagon wird in den α-Zellen des Inselapparates aus einem Vorläufermolekül, dem Präproglukagon, gebildet und durch pankreatische Prohormonkonvertasen in das biologisch aktive Glukagon umgewandelt. Präproglukagon enthält neben der Sequenz für Glukagon auch die Aminosäuresequenzen für zwei weitere Peptidhormone, die aufgrund ihrer Strukturähnlichkeit zum Glukagon als „glucagon-like peptide 1“ (GLP1) und GLP2 bezeichnet werden. Neben den α-Zellen bilden auch das Zentralnervensystem und die intestinale Mukosa Präproglukagon (Enteroglukagon). Im Intestinaltrakt sind die wichtigsten Spaltprodukte das GLP1 und GLP2. GLP1 wird nach Nahrungsaufnahme aus der Darmmukosa freigesetzt und stimuliert die Insulinausschüttung der pankreatischen β-Zellen und inhibiert die Glukagonabgabe aus den α-Zellen. Dieser Mechanismus erklärt auch, warum oral verabreichte Glukose zu höheren Insulinspiegeln im Blut führt als parenteral verabreichte.
Wie beim Insulin erfolgt die Glukagonsekretion in Abhängigkeit von der Glukosekonzentration im Blut. Im Gegensatz zum Insulin stimuliert aber ein Abfall der Glukosekonzentration die Glukagonauschüttung, während ein Glukoseanstieg die Glukagonabgabe hemmt. Glukagon wirkt insulinantagonistisch, indem es die Glykogenolyse steigert und die Glukoneogenese hemmt. Glukagon fördert auch die Aufnahme von Aminosäuren in die Leberzelle und wirkt hier gleichsinnig wie Insulin. Neben erhöhten Aminosäurespiegeln regen auch Katecholamine und Cholezystokinin-Pankreozymin die Glukagonausschüttung an.
Somatostatin
Somatostatin wurde erstmalig aus dem Hypothalamus isoliert und hemmt das hypophysäre Wachstumshormon (Somatotropin). Somatostatin findet sich neben dem Zentralnervensystem in vielen weiteren Organen und besitzt vornehmlich inhibitorische Funktionen. Es ist ein Peptidhormon, das in zwei biologisch aktiven Isoformen bestehend aus 14 und 28 Aminosäuren vorkommt, die sich in ihrer biologischen Wirksamkeit unterscheiden: So hemmt das aus 28 Aminosäuren bestehende Hormon die Insulinsekretion wirksamer als das kleinere, aus 14 Aminosäuren bestehende Peptid.
Im Gastrointestinaltrakt wirkt es sowohl auf exokrine als auch endokrine Drüsen. Somatostatin hemmt die Magensäuresekretion wie auch die Ausschüttung von Enzymen und Bicarbonat aus dem Pankreas. Das in den δ-Zellen des Pankreas gebildete Somatostatin wirkt parakrin auf α- und β-Zellen, indem es die Ausschüttung von Glukagon und Insulin drosselt. Dadurch wirkt Somatostatin starken Schwankungen der Sekretion beider Hormone und damit des Blutglukosespiegels entgegen.
Pankreatisches Polypeptid
Pankreatisches Polypeptid besteht aus 36 Aminosäuren und wird überwiegend in den Langerhans-Inseln des Pankreaskopfes von den F-Zellen (auch als PP-Zellen bezeichnet) synthetisiert. Pankreatisches Polypeptid hemmt die Sekretion von Bicarbonat und Enzymen aus dem Pankreas wie auch die Kontraktion der Gallenblase. Zusätzlich ist es über ZNS-Rezeptoren an der Regulation des Appetits beteiligt. Die Freisetzung des pankreatischen Polypeptids unterliegt einer vagalen Kontrolle und kann durch Vagotomie oder Atropin blockiert werden.
Embryonalentwicklung und Pankreasanomalien
Das Pankreas entsteht aus zwei entodermalen Knospen des Vorderdarms, der ventralen und dorsalen Pankreasanlage. Während der Embryonalentwicklung dreht sich die ventrale Anlage nach dorsal (Rotation), um sich anschließend mit der dorsalen Anlage zu vereinigen (Fusion). Die meisten angeborenen Pankreasanomalien lassen sich auf Störungen der drei kritischen Entwicklungsschritte des Pankreas – Gewebsdifferenzierung, Rotation und Fusion – zurückführen. Störungen der Differenzierung und Rotation sind selten, während Fusionsanomalien häufig auftreten, aber in der überwiegenden Mehrzahl der Fälle asymptomatisch bleiben.
Embryonalentwicklung
Das Pankreas entwickelt sich während der 4. Gestationswoche aus zwei Ausstülpungen des primitiven Vorderdarms, der ventralen und dorsalen Pankreasanlage. Die ventrale Anlage ist im ventralen Mesenterium unterhalb der Leberanlage lokalisiert, während die dorsale Anlage leicht oberhalb und gegenüber der Leberanlage entsteht. In den folgenden zwei Gestationswochen wandert die ventrale Anlage zusammen mit der Einmündung des Ductus choledochus um die Rückseite des Vorderdarms, hinter und unter die dorsale Anlage (Rotation). Anschließend verschmelzen sowohl das Parenchym wie auch die Ausführungsgänge beider Anlagen, wobei die dorsale Anlage den Körper und Schwanz und die ventrale den Kopf und Processus uncinatus bilden (Fusion). Der Ductus pancreaticus (Wirsung), der zum Hauptausführungsgang des Pankreas wird, entsteht durch die Fusion des Gangs der ventralen Anlage mit dem distalen Ganganteil der dorsalen Anlage. Der proximale Ganganteil der dorsalen Anlage bildet sich entweder vollständig zurück oder bleibt als akzessorischer Gang (Ductus Santorini) bestehen (◘ Abb. 21.2).
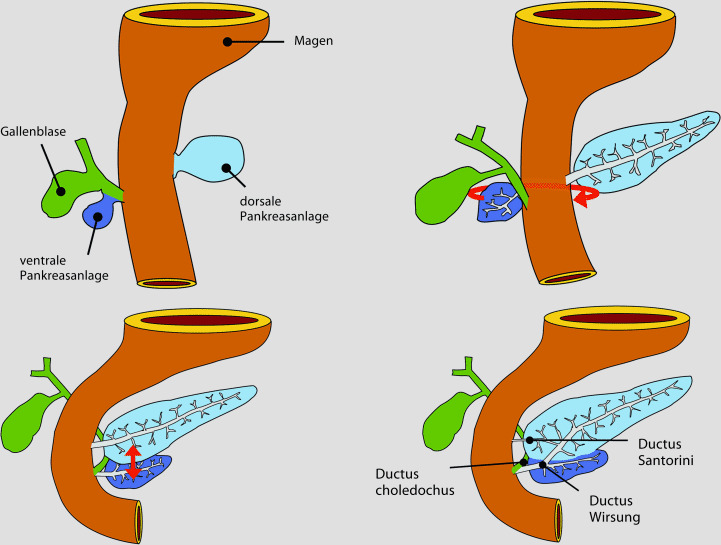

Parallel zur Organentwicklung differenzieren sich die epithelialen Zellen des Pankreas zu azinären, duktalen und endokrinen Zellen. Endokrine Zellen (Inselzellen) sind ab der 12. Gestationswoche und azinäre Strukturen ab der 14.–16. Woche nachweisbar. Die Bildung von Zymogengranula in den Azinuszellen wie auch die Insulinsekretion beginnt etwa ab dem 5. Monat. Die exokrine Pankreasfunktion ist beim reifen Neugeborenen nur unvollständig ausgebildet und unterliegt einem postnatalen Reifungsprozess, der erst im 2. Lebensjahr abgeschlossen ist. Insbesondere beträgt die Sekretion der Amylase und der Lipase bei Geburt weniger als 1 % bzw. 10 % der Sekretion des Erwachsenen.
Differenzierungsanomalien
Das Pankreasgewebe differenziert sich aus dem primitiven Vorderdarm. Eine gestörte Differenzierung kann ein Fehlen (Aplasie) bzw. eine mangelnde Ausbildung (Hypoplasie) des Organs oder die Entstehung ektopen Pankreasgewebes verursachen.
Aplasie und Hypoplasie
Die vollständige wie auch die partielle Nichtanlage des Pankreas sind seltene Ereignisse, die isoliert oder in Kombination mit anderen Defekten wie z. B. einer zerebellären Agenesie auftreten können. Die Hälfte der Patienten weist heterozygote Mutationen im Transkriptionsfaktor GATA6 auf (sog. Haploinsuffizienz). Vereinzelt wurden auch genetische Defekte im Insulinpromotorfaktor 1 (IPF1) oder im pankreatischen Transkriptionsfaktor 1 (PTF1) beschrieben. Der Hypoplasie liegt häufig eine gestörte Anlage einer der beiden Pankreasknospen zugrunde, wobei Störungen der dorsalen Anlage mit daraus bedingter fehlender Ausbildung von Pankreaskörper und -schwanz überwiegen. Klinisch manifestiert sich die Aplasie mit einem neonatalen Diabetes mellitus und schwerer intrauteriner Wachstumsretardierung. Während eine komplette Aplasie üblicherweise mit dem Leben unvereinbar ist, variiert das klinische Bild bei der Hypoplasie von einem asymptomatischen Verlauf bis hin zu einer endokrinen und exokrinen Insuffizienz. Die Diagnose kann mittels Sonographie, abdominaler Computertomographie (CT), Magnetresonanztomographie (MRT) oder endoskopischer retrograder Cholangiopankreatikographhie (ERCP) gestellt werden. Die Therapie besteht je nach klinischer Präsentation in der Substitution von Insulin bzw. Verdauungsenzymen.
Ektopes Pankreas
Ektopes Pankreas , auch als heterotopes, aberrantes oder akzessorisches Pankreas bezeichnet, ist definiert als Pankreasgewebe ohne anatomische Verbindung zur Bauchspeicheldrüse. Pathogenetisch wird eine fehlerhafte Differenzierung pluripotenter entodermaler Stammzellen diskutiert. Die Häufigkeit eines ektopen Pankreas wird in Autopsieserien mit 0,5–15 % angegeben. In der Mehrheit der Fälle ist das ektope Gewebe in der Submukosa des oberen Gastrointestinaltraktes lokalisiert (Magen, Duodenum und Jejunum), wo es als 0,3–3 cm großer Knoten imponiert. Ektopes Pankreas stellt meistens einen Zufallsbefund im Rahmen einer Gastroskopie oder Magen-Darm-Passage dar, kann aber mit Schmerzen, einer gastrointestinalen Blutung oder einer Invagination vergesellschaftet sein. Bei symptomatischem ektopem Pankreas ist die chirurgische Entfernung indiziert.
Rotationsanomalien: Pancreas anulare
Beim Pancreas anulare ist das Duodenum vollständig, seltener teilweise ringförmig von Pankreasgewebe umschlossen. Als ursächlich wird eine Fixierung der ventralen Pankreasanlage vor Einsetzen der Rotation mit daraus bedingter Persistenz des ventralen Pankreas angesehen. Das Pancreas anulare kann in jeder Altersgruppe symptomatisch werden oder asymptomatisch bleiben.
Die Mehrzahl der Fälle manifestiert sich in der ersten Lebenswoche als Duodenalkompression mit galligem Erbrechen und ist häufig mit weiteren Anomalien wie Trisomie 21, intestinaler Malrotation, Duodenal- oder Analatresie oder kardialen Fehlbildungen vergesellschaftet.
Diagnostisch wegweisend ist die Röntgenaufnahme des Abdomens, in der sich zwei Luft-Flüssigkeits-Spiegel im Magen und Duodenum zeigen („double bubble“). Da jede duodenale Obstruktion in dieser Altersgruppe eine chirurgische Intervention erfordert, erfolgt die Bestätigung der Diagnose mittels Laparotomie. Therapie der Wahl ist eine Bypass-Operation mit Duodenoduodeno- oder Duodenojejunostomie. Jenseits der Neonatalzeit ist das Pancreas anulare vorwiegend mit einer Gastritis oder Pankreatitis assoziiert. In diesen Fällen sind Röntgenübersichtsaufnahmen des Abdomens oft diagnostisch unzureichend, so dass Kontrastmitteluntersuchungen oder eine ERCP erforderlich werden. Die Prognose des Pancreas anulare ist abhängig vom Manifestationsalter und weist in der Neugeborenenperiode aufgrund des hohen Prozentsatzes weiterer Organfehlbildungen die höchste Mortalität auf.
Fusionsanomalien
Mit der Fusion der ventralen und dorsalen Pankreasanlage verschmelzen auch die Ausführungsgänge beider Anlagen. Dieser Prozess begünstigt die Entstehung einer Vielzahl anatomischer Varianten des Pankreasgangsystems. In der Mehrzahl der Fälle findet sich ein Hauptausführungsgang (Wirsung), der mit einem akzessorischen Pankreasgang (Santorini) verbunden ist. Der Ductus Wirsung mündet zusammen mit dem Ductus choledochus in die Papilla major (Vateri) ein, über welche die alleinige Drainage des Pankreassekrets erfolgt, während der Ductus Santorini blind endet (eine Papilla minor kann jedoch dennoch angelegt sein; ◘ Abb. 21.3a). In 30 % der Fälle mündet der Ductus Santorini über die Papilla minor ins Duodenum, so dass beide Gänge Pankreassekret ableiten (◘ Abb. 21.3b). Beim Pancreas divisum ist die physiologische Fusion beider Pankreasanlagen ausgeblieben. Es resultiert eine getrennte Einmündung des ventralen Anteils in die Papilla major und des dorsalen Anteils über den Ductus Santorini in die Papilla minor, über die somit 80 % des gebildeten Pankreassekrets drainiert wird (◘ Abb. 21.3c). Gelegentlich stehen beide Gangsysteme über einen kleinen Seitenast miteinander in Verbindung, der in der ERCP nach entsprechender Druckanwendung als zarter kommunizierender Gang dargestellt werden kann (inkomplettes Pancreas divisum) (◘ Abb. 21.3d).
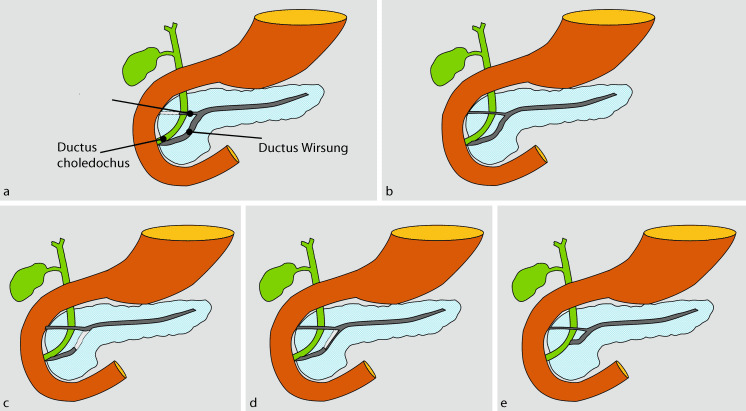

Auch die Verbindung des Pankreashauptausführungsgangs mit dem Gallengang weist eine große Variabilität auf. In der Mehrzahl der Fälle vereinigen sich beide Gänge zu einem kurzen, ca. 5 mm langen, gemeinsamen Gang. Nicht selten bestehen allerdings gesonderte Gangmündungen in der Papilla major oder sogar eine Einmündung in zwei getrennten Papillen. Klinisch bedeutsam sind Anomalien, bei denen Pankreas- und Gallengang außerhalb des Duodenums zusammenlaufen und das gemeinsame Gangsegment länger als 1,5 cm ist (sog. Long common channel) (◘ Abb. 21.3e).
Pancreas divisum
Das Pancreas divisum ist die häufigste Fusionsanomalie des Pankreas. Die Inzidenz wird in Autopsieserien mit 5–10 % und in ERCP-Studien mit 2–5 % angegeben.
Die klinische Bedeutung eines Pancreas divisum wird bis heute kontrovers diskutiert.
Während manche Autoren das Pancreas divisum als unbedeutende Normvariante betrachten, postulieren andere, dass die schmale Öffnung der Papilla minor das Sekret aus dem dorsalen Pankreas nur unzureichend zu drainieren vermag. Folge sei eine relative funktionelle Stenose, die zu einer obstruktiven rezidivierenden Pankreatitis disponiere. Aufgrund der hohen Häufigkeit in der Normalbevölkerung ist davon auszugehen, dass ein Pankreas divisum als alleinige Ursache einer Pankreatitis eher selten in Frage kommt und wahrscheinlich erst in Kombination mit weiteren Umweltfaktoren oder genetischen Risikofaktoren klinische Relevanz erlangt.
Die Diagnose des Pancreas divisum erfolgt mittels ERCP oder Magnetresonanz-Cholangiopankreatikographie (MRCP), wobei die ERCP die sensitivere Untersuchung darstellt und zudem, im Gegensatz zur MRCP, eine gleichzeitige therapeutische Intervention in Form einer Sphinkterotomie der Papilla minor ermöglicht.
Pankreatikobiliäre Mündungsanomalien
Die meisten pankreatikobiliären Anomalien sind Normvarianten. Klinisch bedeutsam sind Anomalien, bei denen Pankreas- und Gallengang ein langes gemeinsames Gangsegment bilden (sog. Long common channel). Derartige Anomalien begünstigen einen Reflux von Pankreassekret in den Ductus choledochus und können zu einer Entzündung des Gallengangs führen. Ein Rückfluss von Gallesekret in das Pankreasgangsystem als Ursache einer Pankreatitis wird in der Literatur kontrovers diskutiert.
Angeborene Choledochuszysten , die häufig mit pankreatikobiliären Maljunktionen assoziiert sind, manifestieren sich häufig mit einer akuten oder rezidivierenden Pankreatitis. Die Diagnose erfolgt mittels MRCP oder ERCP. Die Therapie von Choledochuszysten ist operativ (▶ 10.1007/978-3-642-24710-1_20#Sec4).
Auch die klinische Bedeutung einer Dysfunktion des Sphincter Oddi als kausaler Faktor einer Pankreatitis wird in der Literatur uneinheitlich bewertet.
Angeborene Pankreaszysten
Die meisten Pankreaszysten sind erworbene Pseudozysten entzündlichen Ursprungs und finden sich im Rahmen einer Pankreatitis oder zystischen Fibrose. Angeborene Pankreaszysten sind hingegen sehr selten.
Angeborene Pankreaszysten sind mit Epithel ausgekleidet und können einzeln oder multipel vorkommen. Solitäre Zysten können als asymptomatische abdominale Raumforderung imponieren oder durch Kompression benachbarter intestinaler bzw. biliärer Strukturen epigastrische Schmerzen, Erbrechen und Ikterus hervorrufen. Die Diagnose erfolgt sonographisch. Differenzialdiagnostisch sind Pseudozysten, zystische Pankreastumoren und gastrointestinale Duplikationsanomalien auszuschließen. Symptomatische solitäre Zysten erfordern eine operative Resektion bzw. eine Zystenterostomie. Multiple Zysten sind häufig mit weiteren angeborenen Anomalien vergesellschaftet. So finden sich Pankreaszysten bei etwa 10 % der Patienten mit autosomal-dominanter polyzystischer Nierenerkrankung und bei 40–70 % der Patienten mit einem von-Hippel-Lindau-Syndrom . Bei Letzterem können die Zysten die einzige abdominale Manifestation sein und anderen Manifestationen der Erkrankung um Jahre vorausgehen.
Literatur
Literatur zu Abschn. 21.1
- Chey WY, Chang T. Neural hormonal regulation of exocrine pancreatic secretion. Pancreatology. 2001;1:320–335. doi: 10.1159/000055831. [DOI] [PubMed] [Google Scholar]
- Chey WY, Chang TM. Secretin, 100 years later. J Gastroenterol. 2003;38:1025–1035. doi: 10.1007/s00535-003-1235-3. [DOI] [PubMed] [Google Scholar]
- Konturek SJ, Zabielski R, Konturek JW, Czarnecki J. Neuroendocrinology of the pancreas; role of brain-gut axis in pancreatic secretion. Eur J Pharmacol. 2003;481:1–14. doi: 10.1016/j.ejphar.2003.08.042. [DOI] [PubMed] [Google Scholar]
- Kuhlmann H. Die Zauberstoffe im Wampenbries. Die Geschichte der Erforschung des exokrinen Pankreas und des Wirkstoffes Pankreatin. Hannover: Solvay Arzneimittel GmbH; 1999. [Google Scholar]
- Pandol SJ. Pancreatic physiology and secretory testing. In: Feldman M, Friedman LS, Sleisinger MH, editors. Gastrointestinal and liver disease. Pathophysiology/diagnosis/management. 7. Philadelphia: Saunders; 2002. pp. 871–880. [Google Scholar]
Literatur zu Abschn. 21.2
- Allen HL, Flanagan SE, Shaw-Smith C, et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2011;44:20–22. doi: 10.1038/ng.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambs HJ. Entwicklungsanomalien und kongenitale Erkrankungen des Pankreas. Radiologe. 1996;36:381–388. doi: 10.1007/s001170050086. [DOI] [PubMed] [Google Scholar]
- Cano DA, Hebrok M, Zenker M. Pancreatic development and disease. Gastroenterology. 2007;132:745–762. doi: 10.1053/j.gastro.2006.12.054. [DOI] [PubMed] [Google Scholar]
- Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part I, pathophysiology. Gastrointest Endosc. 2004;60:419–425. doi: 10.1016/S0016-5107(04)01815-2. [DOI] [PubMed] [Google Scholar]
- Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part II, patient selection and treatment. Gastrointest Endosc. 2004;60:585–589. doi: 10.1016/S0016-5107(04)01896-6. [DOI] [PubMed] [Google Scholar]
- Kozu T, Suda K, Toki F. Pancreatic development and anatomical variation. Gastrointest Endosc Clin N Am. 1995;5:1–30. [PubMed] [Google Scholar]
- Newman BM, Lebenthal E, et al. Congenital abnormalities of the exocrine pancreas. In: Go VLW, et al., editors. The exocrine pancreas: biology, pathobiology and diseases. New York: Raven Press; 1986. pp. 773–782. [Google Scholar]