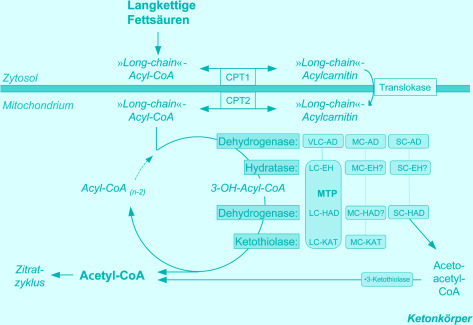
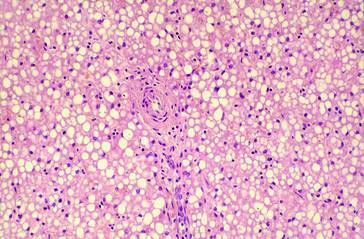
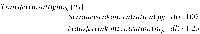Abstract
Entsprechend ihrer Wanderung bei isoelektrischer Fokussierung werden die allelen Varianten des α1-AT als Proteinaseinhibitorphänotypen (Pi) klassifiziert. Die dominierende Isoform ist der normale Phänotyp M, daneben gibt es die Mangelvarianten S und Z sowie eine 0-Variante.
Literatur
Literatur zu Abschn. 17.1
- Bals R. Alpha-1-antitrypsin deficiency. Best Pract Res Clin Gastroenterol. 2010;24:629–623. doi: 10.1016/j.bpg.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Blank CA, Brantly M. Clinical features and molecular characteristics of α1-antitrypsin deficiency. Ann Allergy. 1994;72:105–121. [PubMed] [Google Scholar]
- Chappell S, Hadzic N, Stockley R, et al. A polymorphism of the alpha1-antitrypsin gene represents a risk factor for liver disease. Hepatology. 2008;47:127–132. doi: 10.1002/hep.21979. [DOI] [PubMed] [Google Scholar]
- Doherty DG, Donaldson PI, Whitehouse DB, et al. HLA-phenotypes and gene polymorphisms in juvenile liver disase associated with α1-antitrypsin deficiency. Hepatology. 1990;12:218–223. doi: 10.1002/hep.1840120207. [DOI] [PubMed] [Google Scholar]
- Gadek JE, Hunninghake GW, Zimmerman RL, Crystal RG. Regulation of release of alveolar macrophage-derived neutrophil chemotactic factor. Am Rev Respir Dis. 1980;121:723–733. doi: 10.1164/arrd.1980.121.4.723. [DOI] [PubMed] [Google Scholar]
- Hunninghake GW, Gadek JE, Fales HM, Crystal RG. Human alveolar macrophage-derived chemotactic factor for neutrophils: stimuli and partial characterization. J Clin Invest. 1980;66:473–483. doi: 10.1172/JCI109878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC, Kao FF, Law ML, Woo SLC. Assignment of the α1-antitrypsin gene and sequence-related gene to human chromosome 14 by molecular hybridization. Am J Hum Genet. 1983;35:385–392. [PMC free article] [PubMed] [Google Scholar]
- Mowat AP. Alpha1-antitrypsin deficiency (PiZZ): features of liver involvement in childhood. Acta Paediatr. 1994;393:13–17. doi: 10.1111/j.1651-2227.1994.tb13200.x. [DOI] [PubMed] [Google Scholar]
- Pan S, Huang L, McPherson J, et al. Single nucleotide polymorphism-mediated translational suppression of endoplasmic reticulum mannosidase I modifies the onset of end-stage liver disease in alpha1-antitrypsin deficiency. Hepatology. 2009;50:275–281. doi: 10.1002/hep.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter DH. The cellular basis for liver injury in α1-antitrypsin deficiency. Hepatology. 1991;13:172–185. [PubMed] [Google Scholar]
- Pittschieler K. Heterozygotes and liver involvement. Acta Paediatr. 1994;393:21–23. doi: 10.1111/j.1651-2227.1994.tb13202.x. [DOI] [PubMed] [Google Scholar]
- Rabin M, Watson M, Kidd V, et al. Regional location of α1 chymotrypsin and α1-antitrypsin genes on human chromosome 14. Somat Cell Mol Genet. 1986;12:209–214. doi: 10.1007/BF01560668. [DOI] [PubMed] [Google Scholar]
- Sifers RN, Finegold MJ, Wood SLC. Molecular biology and genetics of α1-antitrypsin deficiency. Semin Liver Dis. 1992;12:301–310. doi: 10.1055/s-2008-1040399. [DOI] [PubMed] [Google Scholar]
- Stockley RA, Parr DG, Piitulainen E, et al. Therapeutic efficacy of α-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respir Res. 2010;11:136. doi: 10.1186/1465-9921-11-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sveger T. Liver disease in α1-antitrypsin deficiency detected by screening of 200.000 infants. N Engl J Med. 1976;294:1316–1321. doi: 10.1056/NEJM197606102942404. [DOI] [PubMed] [Google Scholar]
- Sveger T. Prospective study of children with α1-antitrypsin deficiency. Eight-year-old follow up. J Pediatr. 1984;104:91–94. doi: 10.1016/s0022-3476(84)80599-5. [DOI] [PubMed] [Google Scholar]
- Sveger T. The natural history of liver disease in α1-antitrypsin deficient children. Acta Pediatr Scand. 1988;77:847–851. doi: 10.1111/j.1651-2227.1988.tb10767.x. [DOI] [PubMed] [Google Scholar]
- Sveger T. Screening for alpha1-antitrypsin deficiency. Acta Paediatr. 1994;393:18–20. doi: 10.1111/j.1651-2227.1994.tb13201.x. [DOI] [PubMed] [Google Scholar]
- Sveger T, Eriksson S. The liver in adolescents with α1-antitrypsin deficiency. Hepatology. 1995;22:514–517. doi: 10.1002/hep.1840220221. [DOI] [PubMed] [Google Scholar]
- Sveger T, Thelin T. Four-year-old children with α1-antitrypsin deficiency. Acta Pediatr Scand. 1981;70:171–176. doi: 10.1111/j.1651-2227.1981.tb05537.x. [DOI] [PubMed] [Google Scholar]
- Wu Y, Whitman I, Molmenti E, et al. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc Natl Acad Sci USA. 1994;91:9014–9018. doi: 10.1073/pnas.91.19.9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.2
- Berry GT, Segal S, Gitzelmann R. Disorders of galactose metabolism. In: Fernandes J, Saudubray JM, van den Berghe G, Walter JH, editors. Inborn metabolic diseases: diagnosis and treatment. 5. Berlin Heidelberg New York: Springer; 2011. [Google Scholar]
- Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006;29:516–525. doi: 10.1007/s10545-006-0382-0. [DOI] [PubMed] [Google Scholar]
- Chen YT. Glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 1521–1551. [Google Scholar]
- Chou JY, Raben N (eds) (2002) Glycogen storage diseases. Curr Mol Med 2: 1–227 [DOI] [PubMed]
- Cox TM. The genetic consequences of our sweet tooth. Nat Rev Genet. 2002;3:481–487. doi: 10.1038/nrg815. [DOI] [PubMed] [Google Scholar]
- Franco LM, Krishnamurthy V, Bali D, et al. Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J Inherit Metab Dis. 2005;28:153–162. doi: 10.1007/s10545-005-7500-2. [DOI] [PubMed] [Google Scholar]
- Bundesministerium für Gesundheit Bekanntmachung eines Beschlusses des Gemeinsamen Bundesausschusses über eine Änderung der Kinder-Richtlinien: Anpassung des erweiterten Neugeborenen-Screenings an das Gendiagnostikgesetz (GenDG) Bundesanzeiger. 2011;40:1013. [Google Scholar]
- Jaeken J, Matthijs G, Carchon H, van Schaftingen E. Defects of N-glycan synthesis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2004. pp. 1601–1623. [Google Scholar]
- Kim SY, Jun HS, Mead PA, Mansfield BC, Chou JY. Neutrophil stress and apoptosis underlie myeloid dysfunction in glycogen storage disease type Ib. Blood. 2008;111:5704–5711. doi: 10.1182/blood-2007-12-129114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrune P. Glycogen storage disease type I: indications for liver and/or kidney transplantation. Eur J Pediatr. 2002;161(1):53–55. doi: 10.1007/s00431-002-1004-y. [DOI] [PubMed] [Google Scholar]
- Lee PJ. Glycogen storage disease type I: pathophysiology of liver adenomas. Eur J Pediatr. 2002;161(1):46–49. doi: 10.1007/s00431-002-1002-0. [DOI] [PubMed] [Google Scholar]
- Matern D, Seydewitz HH, Bali D, Lang C, Chen YT. Glycogen storage disease type I: diagnosis and phenotype/genotype correlation. Eur J Pediatr. 2002;161(1):10–99. doi: 10.1007/s00431-002-0998-5. [DOI] [PubMed] [Google Scholar]
- Morava E, Lefeber D. CDG – an update. J Inherit Metab Dis. 2011;34:847–939. doi: 10.1007/s10545-011-9366-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaro F, Andorno E, Basile G, et al. Simultaneous liver-kidney transplantation for glycogen storage disease type IA (von Gierke’s disease) Transplant Proc. 2004;36:1483–1484. doi: 10.1016/j.transproceed.2004.05.070. [DOI] [PubMed] [Google Scholar]
- Rake JP, Visser G, Labrune P, et al. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I) Eur J Pediatr. 2002;161(1):20–34. doi: 10.1007/s00431-002-0999-4. [DOI] [PubMed] [Google Scholar]
- Santer R, Rischewski J, von Weihe M, et al. The spectrum of aldolase B (ALDOB) mutations and the prevalence of hereditary fructose intolerance in Central Europe. Hum Mutat. 2005;25:594. doi: 10.1002/humu.9343. [DOI] [PubMed] [Google Scholar]
- Schweitzer-Krantz S. Early diagnosis of inherited metabolic disorders towards improving outcome: the controversial issue of galactosaemia. Eur J Pediatr. 2003;162(1):50–53. doi: 10.1007/s00431-003-1352-2. [DOI] [PubMed] [Google Scholar]
- Steinmann B, Santer R, van den Berghe G. Disorders of fructose metabolism. In: Fernandes J, Saudubray JM, van den Berghe G, Walter JH, editors. Inborn metabolic diseases – diagnosis and treatment. Berlin Heidelberg New York: Springer; 2011. [Google Scholar]
- Waisbren SE, Potter NL, Gordon CM, et al. The adult galactosemic phenotype. J Inherit Metab Dis. 2011;34:165–171. doi: 10.1007/s10545-011-9372-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.3
- Brewer GJ, Askari FK. Wilson’s disease: clinical management and therapy. J Hepatol. 2005;42(1):13–21. doi: 10.1016/j.jhep.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139–142. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- Lee VD, Northup PG, Berg CL. Resolution of decompensated cirrhosis from Wilson’s disease with zinc monotherapy: a potential therapeutic option? Clin Gastroenterol Hepatol. 2006;4:1069–1071. doi: 10.1016/j.cgh.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Mueller T, Schafer H, Rodeck B, et al. Familial clustering of infantile cirrhosis in Northern Germany: a clue to the etiology of idiopathic copper toxicosis. J Pediatr. 1999;135(2 Pt 1):189–196. doi: 10.1016/s0022-3476(99)70021-1. [DOI] [PubMed] [Google Scholar]
- Nicastro E, Ranucci G, Vajro P, Vegnente A, Iorio R. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology. 2010;52:1948–1956. doi: 10.1002/hep.23910. [DOI] [PubMed] [Google Scholar]
- Sanchez-Albisua I, Garde T, Hierro L, et al. A high index of suspicion: the key to an early diagnosis of Wilson’s disease in childhood. J Pediatr Gastroenterol Nutr. 1999;28:186–190. doi: 10.1097/00005176-199902000-00018. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.4
- Beutler E, Felitti VJ, Koziol JA, Ho NJ, Gelbart T. Penetrance of 845G–A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002;359:211–218. doi: 10.1016/S0140-6736(02)07447-0. [DOI] [PubMed] [Google Scholar]
- Bridle KR, Frazer DM, Wilkins SJ, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361:669–673. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- Fleming RE, Britton RS. Iron imports. VI. HFE and regulation of intestinal iron absorption. Am J Physiol Gastrointest Liver Physiol. 2006;290:G590–G594. doi: 10.1152/ajpgi.00486.2005. [DOI] [PubMed] [Google Scholar]
- Franchini M, Veneri D. Hereditary hemochromatosis. Hematology. 2005;10(2):145–149. doi: 10.1080/10245330500065771. [DOI] [PubMed] [Google Scholar]
- Grabhorn E, Richter A, Burdelski M, Rogiers X, Ganschow R. Neonatal hemochromatosis: long-term experience with favorable outcome. Pediatrics. 2006;118:2060–2065. doi: 10.1542/peds.2006-0908. [DOI] [PubMed] [Google Scholar]
- Olynyk JK, Cullen DJ, Aquilia S, et al. A population-based study of the clinical expression of the hemochromatosis gene. N Engl J Med. 1999;341:718–724. doi: 10.1056/NEJM199909023411002. [DOI] [PubMed] [Google Scholar]
- Pan X, Kelly S, Melin-Aldana H, Malladi P, Whitington PF. Novel mechanism of fetal hepatocyte injury in congenital alloimmune hepatitis involves the terminal complement cascade. Hepatology. 2010;51:2061–2068. doi: 10.1002/hep.23581. [DOI] [PubMed] [Google Scholar]
- Rand EB, Karpen SJ, Kelly S, et al. Treatment of neonatal hemochromatosis with exchange transfusion and intravenous immunoglobulin. J Pediatr. 2009;155:566–567. doi: 10.1016/j.jpeds.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Rodrigues F, Kallas M, Nash R, et al. Neonatal hemochromatosis – medical treatment vs. transplantation: the king‘s experience. Liver Transpl. 2005;11:1417–1424. doi: 10.1002/lt.20497. [DOI] [PubMed] [Google Scholar]
- Wallace DF, Subramaniam VN. Non-HFE haemochromatosis. World J Gastroenterol. 2007;13:4690–4698. doi: 10.3748/wjg.v13.i35.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitington PF, Malladi P. Neonatal hemochromatosis: is it an alloimmune disease? J Pediatr Gastroenterol Nutr. 2005;40:544–549. doi: 10.1097/01.mpg.0000162004.44971.92. [DOI] [PubMed] [Google Scholar]
- Wood MJ, Powell LW, Ramm GA. Environmental and genetic modifiers of the progression to fibrosis and cirrhosis in hemochromatosis. Blood. 2008;111:4456–4462. doi: 10.1182/blood-2007-11-122374. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.5
- Ahmed I. Childhood porphyrias. Mayo Clin Proc. 2002;77:825–836. doi: 10.4065/77.8.825. [DOI] [PubMed] [Google Scholar]
- Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439–450. doi: 10.7326/0003-4819-142-6-200503150-00010. [DOI] [PubMed] [Google Scholar]
- Kauppinen R. Porphyrias. Lancet. 2005;365:241–252. doi: 10.1016/S0140-6736(05)17744-7. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.6
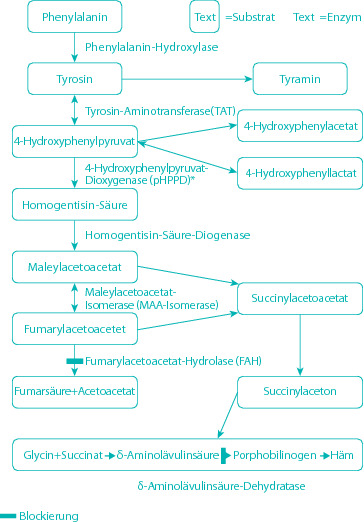
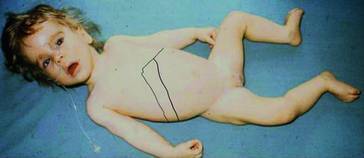
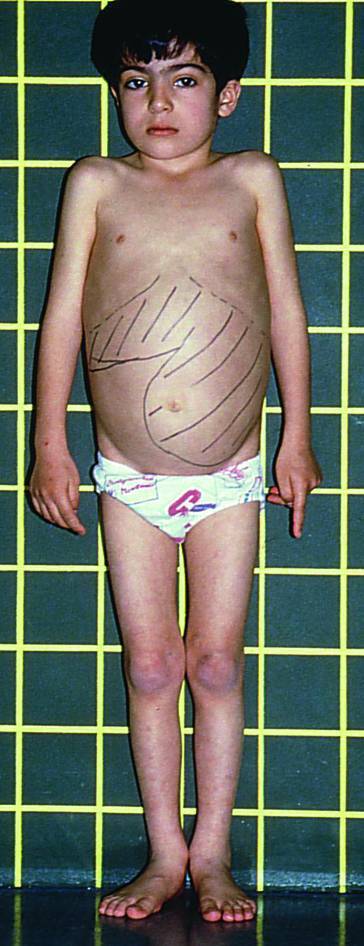
- Halvorsen S, Kvittingen EA, Flatmark A. Outcome of therapy of hereditary tyrosinemia. Acta Paediatr Jpn. 1988;30:425–428. doi: 10.1111/j.1442-200x.1988.tb02532.x. [DOI] [PubMed] [Google Scholar]
- Harms E, Roscher A, Grueters A, et al. Neue Screening-Richtlinien. Monatsschr Kinderheilkd. 2002;150:1424–1440. [Google Scholar]
- Kvittingen EA. Tyrosinaemia type I – an update. J Inherit Metab Dis. 1991;14:554–562. doi: 10.1007/BF01797926. [DOI] [PubMed] [Google Scholar]
- Laberge C. Hereditary tyrosinemia in a French Canadian isolate. Am J Hum Genet. 1969;21:36–45. [PMC free article] [PubMed] [Google Scholar]
- Laberge C, Grenier A, Valet JP, Morissette J. Fumarylacetoacetase measurement as a mass-screening procedure for hereditary tyrosinemia type I. Am J Hum Genet. 1990;47:325–328. [PMC free article] [PubMed] [Google Scholar]
- Lindblad B, Lindstedt S, Steen G. On the enzymic defects in hereditary tyrosinemia. Proc Natl Acad Sci USA. 1977;74:4641–4645. doi: 10.1073/pnas.74.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstedt S, Holme E, Lock EA, Hjalmarson O, Strandvik B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase [see comments] Lancet. 1992;340:813–817. doi: 10.1016/0140-6736(92)92685-9. [DOI] [PubMed] [Google Scholar]
- Mitchell G, Larochelle J, Lambert M, et al. Neurologic crises in hereditary tyrosinemia. N Engl J Med. 1990;322:432–437. doi: 10.1056/NEJM199002153220704. [DOI] [PubMed] [Google Scholar]
- Mitchell GA, Lambert M, Tanguay RM. Hypertyrosinemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 1995. pp. 1077–1106. [Google Scholar]
- Paradis K, Weber A, Seidman EG, et al. Liver transplantation for hereditary tyrosinemia: the Quebec experience. Am J Hum Genet. 1990;47:338–342. [PMC free article] [PubMed] [Google Scholar]
- Poudrier J, Lettre F, Scriver CR, Larochelle J, Tanguay RM. Different clinical forms of hereditary tyrosinemia (type I) in patients with identical genotypes. Mol Genet Metab. 1998;64:119–125. doi: 10.1006/mgme.1998.2695. [DOI] [PubMed] [Google Scholar]
- Sima AA, Kennedy JC, Blakeslee D, Robertson DM. Experimental porphyric neuropathy: a preliminary report. Can J Neurol Sci. 1981;8:105–113. doi: 10.1017/s0317167100042992. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.7
- Beck M. Variable clinical presentation in lysosomal storage disorders. J Inherit Metab Dis. 2001;24(2):45–46. doi: 10.1023/a:1012463605992. [DOI] [PubMed] [Google Scholar]
- Boelens JJ, Prasad VK, Tolar J, Wynn RF, Peters C. Current international perspectives on hematopoietic stem cell transplantation for inherited metabolic disorders. Pediatr Clin North Am. 2010;57:123–145. doi: 10.1016/j.pcl.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Desnick RJ. Enzyme replacement and enhancement therapies for lysosomal diseases. J Inherit Metab Dis. 2004;27:385–410. doi: 10.1023/B:BOLI.0000031101.12838.c6. [DOI] [PubMed] [Google Scholar]
- Ellinwood NM, Vite CH, Haskins ME. Treatment of lysosomal storage disorders: cell therapy and gene therapy. J Inherit Metab Dis. 2004;27:411–415. doi: 10.1023/B:BOLI.0000031170.69676.68. [DOI] [PubMed] [Google Scholar]
- Guo Y, He W, Boer AM, et al. Elevated plasma chitotriosidase activity in various lysosomal storage disorders. J Inherit Metab Dis. 1995;18:717–722. doi: 10.1007/BF02436762. [DOI] [PubMed] [Google Scholar]
- Krivit W. Allogeneic stem cell transplantation for the treatment of lysosomal and peroxisomal metabolic diseases. Springer Semin Immunopathol. 2004;26:119–132. doi: 10.1007/s00281-004-0166-2. [DOI] [PubMed] [Google Scholar]
- Malatack JJ, Consolini DM, Bayever E. The status of hematopoietic stem cell transplantation in lysosomal storage disease. Pediatr Neurol. 2003;29:391–403. doi: 10.1016/j.pediatrneurol.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Marsden D, Levy H. Newborn screening of lysosomal storage disorders. Clin Chem. 2010;56:1071–1079. doi: 10.1373/clinchem.2009.141622. [DOI] [PubMed] [Google Scholar]
- Parkinson-Lawrence EJ, Shandala T, Prodoehl M, et al. Lysosomal storage disease: revealing lysosomal function and physiology. Physiology (Bethesda). 2010;25:102–115. doi: 10.1152/physiol.00041.2009. [DOI] [PubMed] [Google Scholar]
- Staretz-Chacham O, Lang TC, LaMarca ME, Krasnewich D, Sidransky E. Lysosomal storage disorders in the newborn. Pediatrics. 2009;123:1191–1207. doi: 10.1542/peds.2008-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vom Dahl S, Mengel E. Lysosomal storage diseases as differential diagnosis of hepatosplenomegaly. Best Pract Res Clin Gastroenterol. 2010;24:619–628. doi: 10.1016/j.bpg.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Weibel TD, Brady RO. Systematic approach to the diagnosis of lysosomal storage disorders. Ment Retard Dev Disabil Res Rev. 2001;7:190–199. doi: 10.1002/mrdd.1027. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Coppola S, Liu SL. Insights into the diagnosis and treatment of lysosomal storage diseases. Arch Neurol. 2003;60:322–328. doi: 10.1001/archneur.60.3.322. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.8
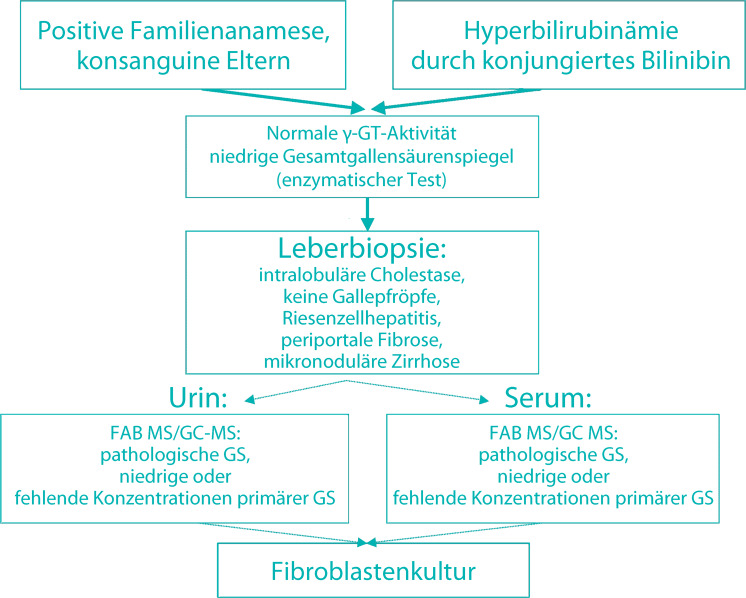
- Balistreri WF. Inborn errors of bile acid biosynthesis and transport. Novel forms of metabolic liver disease. Gastroenterol Clin North Am. 1999;28:145–172. doi: 10.1016/s0889-8553(05)70048-0. [DOI] [PubMed] [Google Scholar]
- Bove KE, Heubi JU, Balistreri WF, Setchel KD. Bile acid synthetic defects and liver disease: a comprehensive review. Pediatr Dev Pathol. 2004;7:315–334. doi: 10.1007/s10024-002-1201-8. [DOI] [PubMed] [Google Scholar]
- Clayton PT. Applications of mass spectrometry in the study of inborn errors of metabolism. J Inh Metab Dis. 2001;24:139–150. doi: 10.1023/a:1010358715835. [DOI] [PubMed] [Google Scholar]
- Setchel KD, Heubi JU, Bove KE, et al. Liver disease caused by failure to racemize trihydroxycholestanoic acid: gene mutation and effect of bile acid therapy. Gastroenterology. 2003;124:217–232. doi: 10.1053/gast.2003.50017. [DOI] [PubMed] [Google Scholar]
- Rand EB, Karpen SJ, Kelly S, et al. Treatment of neonatal hemochromatosis with exchange transfusion and intravenous immunoglobulin. J Pediatr. 2009;155:568–571. doi: 10.1016/j.jpeds.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Gonzales E, Gerhardt MF, Fabre M, et al. Oral cholic acid for hereditary defects of primary bile acid synthesis: a safe and effective long-term therapy. Gastroeneterolgy. 2009;137:1310–1320. doi: 10.1053/j.gastro.2009.07.043. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.9
- Arias IM. Chronic unconjugated hyperbilirubinemia without overt signs of hemolysis in adolescents and adults. J Clin Invest. 1962;41:2233–2245. doi: 10.1172/JCI104682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellodi-Privato M, Aubert D, Pichard V, et al. Successful gene therapy of the Gunn rat by in vivo neonatal hepatic gene transfer using murine oncoretroviral vectors. Hepatology. 2005;42:431–438. doi: 10.1002/hep.20794. [DOI] [PubMed] [Google Scholar]
- Bosma PJ, Chowdhury JR, Bakker C, et al. The genetic basis of the reduced expression of bilirubin UDP- glucuronosyltransferase 1 in Gilbert's syndrome. N Engl J Med. 1995;333:1171–1175. doi: 10.1056/NEJM199511023331802. [DOI] [PubMed] [Google Scholar]
- Clarenburg R, Kao CC. Shared and separate pathways for biliary excretion of bilirubin and BSP in rats. Am J Physiol. 1973;225:192–200. doi: 10.1152/ajplegacy.1973.225.1.192. [DOI] [PubMed] [Google Scholar]
- Crigler JF, Jr, Najjar VA. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics. 1952;10:169–180. [PubMed] [Google Scholar]
- Dennery PA. Metalloporphyrins for the treatment of neonatal jaundice. Curr Opin Pediatr. 2005;17:167–169. doi: 10.1097/01.mop.0000156270.25640.5a. [DOI] [PubMed] [Google Scholar]
- Dimmock D, Brunetti-Pierri N, Palmer DJ, Beaudet AL, Ng P. Correction of hyperbilirubinemia in gunn rats using clinically relevant low doses of helper-dependent adenoviral vectors. Hum Gene Ther. 2011;22:483–488. doi: 10.1089/hum.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox IJ, Chowdhury JR, Kaufman SS, et al. Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation. N Engl J Med. 1998;338:1422–1426. doi: 10.1056/NEJM199805143382004. [DOI] [PubMed] [Google Scholar]
- Galbraith RA, Drummond GS, Kappas A. Suppression of bilirubin production in the Crigler-Najjar type I syndrome: studies with the heme oxygenase inhibitor tin-mesoporphyrin. Pediatrics. 1992;89:175–182. [PubMed] [Google Scholar]
- Gallagher TF, Jr, Mueller MN, Kappas A. Estrogen pharmacology. IV. Studies of the structural basis for estrogen-induced impairment of liver function. Medicine (Baltimore) 1966;45:471–479. [PubMed] [Google Scholar]
- Knoppke B, Vermehren J, Grothues D, et al. Auxiliary liver transplantation in a child with Crigler-Najjar syndrome type I. Transpl Int. 2011;24:40. [Google Scholar]
- Kren BT, Parashar B, Bandyopadhyay P, et al. Correction of the UDP-glucuronosyltransferase gene defect in the gunn rat model of crigler-najjar syndrome type I with a chimeric oligonucleotide. Proc Natl Acad Sci USA. 1999;96:10349–10354. doi: 10.1073/pnas.96.18.10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysy PA, Najimi M, Stephenne X, et al. Liver cell transplantation for Crigler-Najjar syndrome type I: update and perspectives. World J Gastroenterol. 2008;14:3464–3470. doi: 10.3748/wjg.14.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan G, McLellan A, McGeehan A, et al. Gilbert's syndrome is a contributory factor in prolonged unconjugated hyperbilirubinemia of the newborn. J Pediatr. 1999;134:441–446. doi: 10.1016/s0022-3476(99)70201-5. [DOI] [PubMed] [Google Scholar]
- Okolicsanyi L, Ghidini O, Orlando R, et al. An evaluation of bilirubin kinetics with respect to the diagnosis of Gilbert's syndrome. Clin Sci Mol Med. 1978;54:539–547. doi: 10.1042/cs0540539. [DOI] [PubMed] [Google Scholar]
- Olsson R, Bliding A, Jagenburg R, et al. Gilbert's syndrome – does it exist? A study of the prevalence of symptoms in Gilbert's syndrome. Acta Med Scand. 1988;224:485–490. [PubMed] [Google Scholar]
- Regev RH, Stolar O, Raz A, Dolfin T. Treatment of severe cholestasis in neonatal Dubin-Johnson syndrome with ursodeoxycholic acid. J Perinat Med. 2002;30:185–187. doi: 10.1515/JPM.2002.025. [DOI] [PubMed] [Google Scholar]
- Roberts RJ, Plaa GL. Effect of phenobarbital on the excretion of an exogenous bilirubin load. Biochem Pharmacol. 1967;16:827–835. doi: 10.1016/0006-2952(67)90055-x. [DOI] [PubMed] [Google Scholar]
- Shieh CC, Chang MH, Chen CL. Dubin-Johnson syndrome presenting with neonatal cholestasis. Arch Dis Child. 1990;65:898–899. doi: 10.1136/adc.65.8.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinaasappel M, Jansen PLM. The differential diagnosis of Crigler-Najjar disease,types 1 and 2, by bile pigment analysis. Gastroenterol. 1991;100:783–789. doi: 10.1016/0016-5085(91)80026-6. [DOI] [PubMed] [Google Scholar]
- van der Veere CN, Sinaasappel M, McDonagh AF, et al. Current therapy for Crigler-Najjar Syndrome type 1:report of a world registry. Hepatology. 1996;24:311–315. doi: 10.1002/hep.510240205. [DOI] [PubMed] [Google Scholar]
- Wada M, Toh S, Taniguchi K, et al. Mutations in the canilicular multispecific organic anion transporter (cMOAT) gene, a novel ABC transporter, in patients with hyperbilirubinemia II/Dubin-Johnson syndrome. Hum Mol Genet. 1998;7:203–207. doi: 10.1093/hmg/7.2.203. [DOI] [PubMed] [Google Scholar]
- van der Wegen P, Louwen R, Imam AM, et al. Successful treatment of UGT1A1 deficiency in a rat model of Crigler-Najjar disease by intravenous administration of a liver-specific lentiviral vector. Mol Ther. 2006;13:374–381. doi: 10.1016/j.ymthe.2005.09.022. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.10
- DiMauro S. Mitochondrial diseases. Biochim Biophys Acta. 2004;1658:80–88. doi: 10.1016/j.bbabio.2004.03.014. [DOI] [PubMed] [Google Scholar]
- D‘Souza GG, Weissig V. Approaches to mitochondrial gene therapy. Curr Gene Ther. 2004;4:317–328. doi: 10.2174/1566523043346200. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Lamantea E, Donati A, et al. Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-gamma A. Brain. 2005;128:723–731. doi: 10.1093/brain/awh410. [DOI] [PubMed] [Google Scholar]
- Mandel H, Szargel R, Labay V, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- Marriage B, Clandinin MT, Glerum DM. Nutritional cofactor treatment in mitochondrial disorders. J Am Diet Assoc. 2003;103:1029–1038. doi: 10.1016/s0002-8223(03)00476-0. [DOI] [PubMed] [Google Scholar]
- Rogac M, Meznaric M, Zeviani M, Sperl W, Neubauer D. Functional outcome of children with mitochondrial diseases. Pediatr Neurol. 2011;44:340–346. doi: 10.1016/j.pediatrneurol.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Smeitink JA. Mitochondrial disorders: clinical presentation and diagnostic dilemmas. J Inherit Metab Dis. 2003;26:199–207. doi: 10.1023/a:1024489218004. [DOI] [PubMed] [Google Scholar]
- Sperl W, Freisinger P, Mayr H, Burgard P (2010) Diagnostik und Therapieansatze bei Mitochondriopathien im Kindes- und Jugendalter. http://www.awmf.org/uploads/tx_szleitlinien/. Zugegriffen: 19. April 2012
- Spinazzola A, Viscomi C, Fernandez-Vizarra E, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- Zeviani M, Di Donato S. Mitochondrial disorders. Brain. 2004;127:2153–2172. doi: 10.1093/brain/awh259. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.11
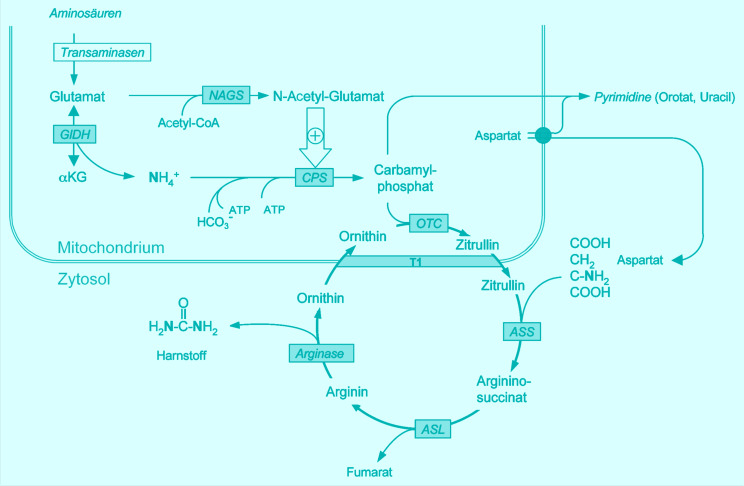
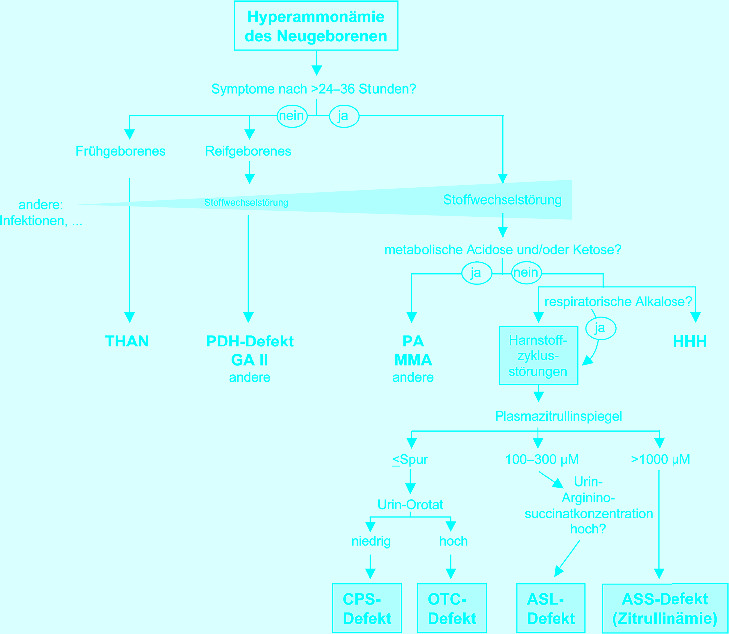
- Bachmann C. Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation. Eur J Pediatr. 2003;162:410–416. doi: 10.1007/s00431-003-1188-9. [DOI] [PubMed] [Google Scholar]
- Bachmann C, Batshaw M, Hammond J, Tuchman M, Wilcken B. New developments in urea cycle disorders. Mol Genet Metab. 2004;81(1):3–91. [Google Scholar]
- Brusilow S, Horwich AL. Urea cycle enzymes. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 1909–1963. [Google Scholar]
- Häberle J, Lindner M, Boddaert N et al. (2012) Guideline for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis (in press) [DOI] [PMC free article] [PubMed]
- Häberle J. Clinical practice: the management of hyperammonemia. Eur J Pediatr. 2011;170:21–34. doi: 10.1007/s00431-010-1369-2. [DOI] [PubMed] [Google Scholar]
- Morioka D, Kasahara M, Takada Y, et al. Current role of liver transplantation for the treatment of urea cycle disorders: a review of the worldwide English literature and 13 cases at Kyoto University. Liver Transpl. 2005;11:1332–1342. doi: 10.1002/lt.20587. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.12
- Belay ED, Bresee JS, Holman RC, et al. Reye’s syndrome in the United States from 1981 through 1997. N Engl J Med. 1997;340:1377–1382. doi: 10.1056/NEJM199905063401801. [DOI] [PubMed] [Google Scholar]
- Bove KE, McAdams AJ, Partin JS, Hug G, Schubert WK. The hepatic lesion in Reye’s syndrome. Gastroenterology. 1975;69:685–697. [PubMed] [Google Scholar]
- Brown JK, Imam H. Interrelationship of liver and brain with special reference to Reye’s syndrome. J Inherit Metab Dis. 1991;14:436–458. doi: 10.1007/BF01797917. [DOI] [PubMed] [Google Scholar]
- Khuroo MoS, Khuroo MeS, Farahat KLC. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: A meta analysis. Liver Transplant. 2004;10:1099–1106. doi: 10.1002/lt.20139. [DOI] [PubMed] [Google Scholar]
- Martin SR, Atkison P, Anand R, Lindblad AS, SPLIT Research Group Studies of pediatric liver transplantation 2002: patient and graft survival and rejection in pediatric recipients of a first liver transplant in the United States and Canada. Pediatr Transplant. 2004;8:273–283. doi: 10.1111/j.1399-3046.2004.00152.x. [DOI] [PubMed] [Google Scholar]
- Pinsky PF, Hurwitz ES, Schonberger LB, Gumn WJ. Reye’s syndrome and aspirin. Evidence for a dose-response effect. J Am Med Assoc. 1988;260:657–661. [PubMed] [Google Scholar]
- Reye RDK, Morgan G, Baral L. Encephalopathy and fatty degeneration of the viscera. A disease entity in childhood. Lancet. 1963;II:749–751. doi: 10.1016/s0140-6736(63)90554-3. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.13
- Chace DH, Kalas TA, Naylor EW. Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin Chem. 2003;49:1797–1817. doi: 10.1373/clinchem.2003.022178. [DOI] [PubMed] [Google Scholar]
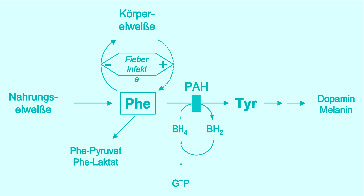
- Ding Z, Harding CO, Thony B. State-of-the-art 2003 on PKU gene therapy. Mol Genet Metab. 2004;81:3–8. doi: 10.1016/j.ymgme.2003.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen N, Bross P, Andresen BS. Genetic defects in fatty acid beta-oxidation and acyl-CoA dehydrogenases. Molecular pathogenesis and genotype-phenotype relationships. Eur J Biochem. 2004;271:470–482. doi: 10.1046/j.1432-1033.2003.03949.x. [DOI] [PubMed] [Google Scholar]
- Grosse SD, Khoury MJ, Greene CL, Crider KS, Pollitt RJ. The epidemiology of medium chain acyl-CoA dehydrogenase deficiency: an update. Genet Med. 2006;8:205–212. doi: 10.1097/01.gim.0000204472.25153.8d. [DOI] [PubMed] [Google Scholar]
- Muntau AC, Roschinger W, Habich M, et al. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med. 2002;347:2122–2132. doi: 10.1056/NEJMoa021654. [DOI] [PubMed] [Google Scholar]
- Roe CR, Ding J. Mitochondrial fatty acid oxidation disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 2297–2326. [Google Scholar]
- Scriver CR, Kaufman S. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 1667–1724. [Google Scholar]
- Spiekerkoetter U, Lindner M, Santer R, et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009;32:488–497. doi: 10.1007/s10545-009-1125-9. [DOI] [PubMed] [Google Scholar]
- van Spronsen FJ, Huijbregts SC, Bosch AM, Leuzzi V. Cognitive, neurophysiological, neurological and psychosocial outcomes in early-treated PKU-patients: a start toward standardized outcome measurement across developmen. Mol Genet Metab. 2011;104:45–51. doi: 10.1016/j.ymgme.2011.09.036. [DOI] [PubMed] [Google Scholar]
- Wilcken B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis. 2010;33:501–506. doi: 10.1007/s10545-009-9001-1. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 17.14
- Harrison SA, Torgerson S, Hayashi P, Ward J, Schenker S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2003;98:2485–2490. doi: 10.1111/j.1572-0241.2003.08699.x. [DOI] [PubMed] [Google Scholar]
- Lavine JE, Schwimmer JB, Van Natta ML, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. 2011;305(16):1659–1668. doi: 10.1001/jama.2011.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EA. Steatohepatitis in children. Best Pract Res Clin Gastroenterol. 2002;16:749–765. doi: 10.1053/bega.2002.0331. [DOI] [PubMed] [Google Scholar]
- Schwimmer JB, Deutsch R, Rauch JB, et al. Obesity, insulin resistance, and other clinicopathological correlates of pediatric nonalcoholic fatty liver disease. J Pediatr. 2003;143:500–505. doi: 10.1067/S0022-3476(03)00325-1. [DOI] [PubMed] [Google Scholar]
- Vairo P, Lenta S, Socha P, et al. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr. 2012;54(4):700–713. doi: 10.1097/MPG.0b013e318252a13f. [DOI] [PubMed] [Google Scholar]